

Summary
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by the polyglutamine (polyQ) expansion in huntingtin (HTT) protein. The challenge of obtaining full-length HTT proteins with high purity limits the understanding of the HTT protein function. Here, we provide a protocol to generate and purify full-length recombinant human HTT proteins with various polyQ lengths, which is key to investigate the biochemical function of HTT proteins and the molecular mechanism underlying HD pathology.
For complete details on the use and execution of this protocol, please refer to Jung et al. (2020).
Subject areas: Protein Biochemistry, Protein expression and purification, Cryo-EM
Graphical abstract
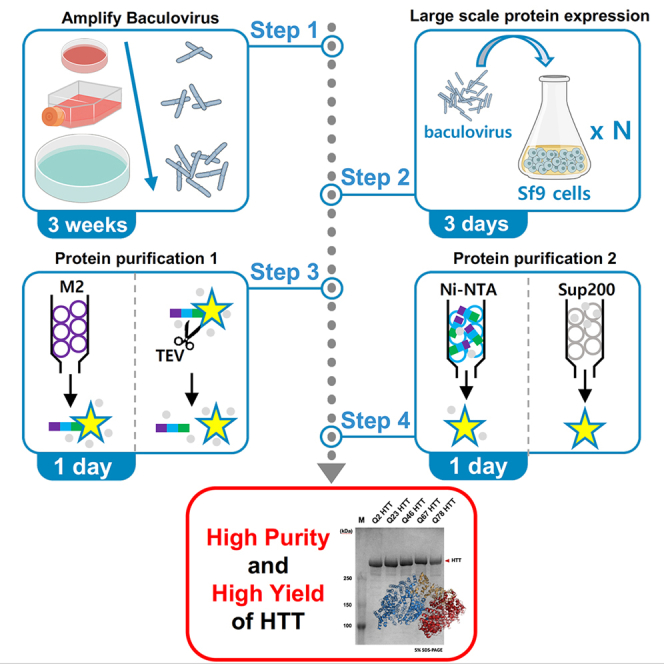
Highlights
-
•
Expression and purification of full-length recombinant human huntingtin (HTT) proteins
-
•
Generation of HTT proteins with allelic series of polyglutamine length
-
•
Purified HTT proteins form complexes with HAP40 or HAP1.
Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by the polyglutamine (polyQ) expansion in huntingtin (HTT) protein. The challenge of obtaining full-length HTT proteins with high purity limits the understanding of the HTT protein function. Here, we provide a protocol to generate and purify full-length recombinant human HTT proteins with various polyQ lengths, which is key to investigate the biochemical function of HTT proteins and the molecular mechanism underlying HD pathology.
Before you begin
Prepare plasmids for generating baculovirus encoding full-length human huntingtin cDNAs
Timing: 3 days
To express HTT proteins in Spodoptera frugiperda (Sf9) insect cells using baculovirus, we modified pFastBac1 vector (Thermo Fisher Scientific) before cloning the Huntingtin (HTT) genes into the vector (Figure 1A) (Seong et al., 2010).
Note: The HTT plasmids with allelic series of CAG (polyQ) lengths are available upon a written request to the corresponding author (J.S.).
-
1.
We inserted a new linker cassette containing a FLAG tag, a 6×His tag, a TEV protease recognition site and new restriction enzyme sites (see the Figure 1A) into the pFastBac1 vector between the BamHI (GGATCC) and KpnI (GGTACC) restriction sites. Before inserting the cassette, the KpnI site in the vector was removed by incubation with a Klenow enzyme and the new linker cassette was ligated with BamHI and the blunt end KpnI site, which eliminates the KpnI site.
-
2.
HTT cDNA fragments encoding amino acids 1–171 (based on polyQ23 human HTT) containing the CAG triplet repeat region were cloned between the NcoI (CCATGG) and XhoI (CTCGAG) restriction sites.
Note: We can generate HTT clones with various CAG repeats with this step.
Optional: We usually add the final 1× Band Doctor (SolGent) to PCR mixtures, which improves PCR when Htt is used as a template.
-
3.
A 9,046 bp fragment HTT cDNA (GenBank accession number L12392) fragments encoding amino acids 172-3,144 was cloned with the newly inserted XhoI (CTCGAG) and SacII (CCGCGG) restriction sites located in the inserted cassette.
Note: The original SacII restriction site in the pFastBac1 vector was deleted. In addition, the KpnI site was removed from the plasmid to utilize the KpnI site (1,761 bp, 587 a.a.) located at the loop region in N-HEAT, which has many phosphorylation sites, for the future usage.
Figure 1.

The cloning strategy of full-length HTT cDNA in a modified pFastBac1 vector
(A) A new linker cassette was inserted in pFastBac1 (Thermo Fisher Scientific, 4775 bp) vector. The linker cassette contains a FLAG tag, a 6×His-tag, a TEV protease recognition site and restriction enzyme sites (NcoI, XhoI, SacII and others, underlined and shown in italic).
(B) Htt cDNA fragment containing huntingtin amino acid 1–171 was inserted into the modified pFastBac1 vector with NcoI and XhoI restriction sites and then the rest of Htt cDNA fragment containing huntingtin amino acid 172-3,144 was cloned into the vector with XhoI and SacII restriction sites to express HTT tagged with FLAG, HIS and TEV sites. Each Htt DNA fragments and vector size are not up to scale.
An overall scheme of the plasmid containing Htt and the cloning steps are described in Figure 1B.
Amplify the HTT plasmids while stably maintaining the number of CAG repeat
When HTT plasmids are amplified, it is difficult to maintain a constant number of CAG repeats in the clones as the CAG repeats are lost during plasmid amplification. In addition, the large size of the HTT plasmid causes a low transformation efficiency and slow growth rate. To resolve these issues, we optimized the plasmid amplification protocol.
-
4.
Remove 50 μL of XL10 Gold ultracompetent cells (Agilent) from the −80°C freezer and place them immediately on ice. Allow the cells to thaw for 5 min.
-
5.
Add 100 ng of the Htt plasmid to be transformed and label the tubes. After incubation on ice for 30 min, incubate the tubes for 1 min at 42°C for heat shock.
-
6.
Incubate for 10 min on ice after the heat shock and then add 900 μL of S.O.C. medium.
-
7.
Incubate the tubes at 37°C for 45 min at a shaking incubator.
-
8.
Centrifuge the cells at 4,000×g for 10 min and decant 700 μL of the supernatant.
-
9.
Resuspend the rest of the supernatant by slowly pipetting.
-
10.
Plate the resuspended cells on an LB plate with 25 μg/mL of ampicillin.
-
11.
Incubate the plate at 30°C for 2 days.
-
12.
After 2 days, pick a single colony and proceed to a liquid culture with 25 μg/mL of ampicillin at 30°C for 2 days for mini-, midi-, or maxiprep (Qiagen).
CRITICAL: To maintain the constant number of CAG triplet repeats, transformation and culture condition should be performed with LB media supplemented with 25 μg/mL of ampicillin and at 30°C for two days. The CAG lengths should be monitored by sequencing with the hHTT R146 reverse primer (see the key resources table) whenever the plasmids are amplified.
The critical points of this protocol
Before starting the experiment, there are several critical points to follow this protocol.
Note: This protocol without any modification can be applied to all allelic series of HTT proteins regardless of the polyQ lengths.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Recombinant DNA | ||
| Htt plasmids with allelic series of CAG (polyQ) lengths (Q2, Q23, Q46, Q67 and Q78) | The plasmids are available upon a written request to the corresponding author (J.S.). | N/A |
| pFastBac1 vector | Invitrogen | V011648 |
| Critical commercial assays | ||
| DNA Polymerase I, Klenow Fragment | NEB | M0210L |
| BamHI restriction enzyme | Enzynomics | CR003M |
| KpnI restriction enzyme | Enzynomics | R014M |
| XhoI restriction enzyme | Enzynomics | R007M |
| SacII restriction enzyme | Enzynomics | R036M |
| NcoI restriction enzyme | Enzynomics | R004M |
| 5×Band Doctor™ | SolGent | SBD41-B10k |
| 2×TOPsimple™ PreMIX-Forte | Enzynomics | P610F |
| 2×TOPsimple™ PreMIX-Tenuto | Enzynomics | P610 |
| Plasmid DNA Purification Kit Mini | LaboPass | CMP0112 |
| HiSPeed Plasmid Midi Kit | QIAGEN | Cat#12643 |
| Bacterial strains | ||
| Escherichia Coli DH5α | Enzynomics | CPO11 |
| DH10Bac | Thermo Fisher Scientific | LS10361012 |
| XL10-Gold Ultracompetent Cells | Agilent | Cat#200314 |
| Experimental models: Cell lines | ||
| Sf9 cells in Sf-900TM II SFM | Thermo Fisher Scientific | Cat#11496015 |
| Oligonucleotides | ||
| M13 forward primer | This paper | GTTTCCCAGTCACGAC |
| hHTT R146 reverse primer | This paper | CCTGACATCTGACTCTGCGTCATCAC |
| Chemicals, peptides,and recombinant proteins | ||
| Tryptone | LPS Solution | CHS-80 |
| Yeast extract | LPS Solution | CHS-02 |
| Sodium chloride (NaCl) | LPS Solution | NACL05 |
| Agar | LPS Solution | AGAR500 |
| Tris | AMRESCO | 0497-5kg |
| Glycerol | LPS Solution | GLYC01 |
| Imidazole | Bio Basic | IB0277 |
| HEPES | Sigma-Aldrich | H3375-550G |
| Hydrochloric acid (HCl) | Samchun | H0255 |
| Ampicillin | Glentham | GA7355-100g |
| Kanamycin sulfate | Bio Basic | KB0286 |
| Gentamicin | Bio Basic | Cat#105-41-0 |
| Tetracycline | Sigma-Aldrich | 87128-25G |
| Bluo-Gal | Thermo Fisher Scientific | Cat#15519-028 |
| IPTG | UBPBio | P1010-100 |
| Phenylmethylsulfonyl fluoride (PMSF) | Bio Basic | PB0425 |
| DL-Dithiothreitol (DTT) | Soltec Ventures | M112_100g |
| Ethylenediaminetetraacetic acid (EDTA) | Sigma-Aldrich | E9884-1KG |
| cOmplete™, Mini, EDTA-free Protease inhibitor Cocktail (PIC) | Roche | Cat#4693159001 |
| CellfectinTM II Reagent | Thermo Fisher Scientific | Cat#10362100 |
| HyQ CCM®3 | Cytiva | SH30061 |
| S.O.C. medium | Thermo Fisher Scientific | Cat#15544-034 |
| 2-Mercaptoethanol (BME) | Sigma-Aldrich | M6250-250ML |
| Anti-FLAG M2 affinity resin | Sigma-Aldrich | A2220-25ML |
| FLAG-peptide | Apeptide | APG1474 |
| Nickel-nitrilotriacetic acid resin (Ni-NTA) | QIAGEN | Cat#30230 |
| Glutathione SepharoseTM 4B | Cytiva | #17075605 |
| Recombinant TEV protease | This paper | N/A |
| Other | ||
| Pyrex® 2,800 mL Fernbach culture flask | Pyrex | 4425-2XL |
| MF-Millipore Membrane Filter, 0.22 μm pore size | Millipore | GSWP04700 |
| 6-Well plate | SPL Solutions | 30066 |
| 75 cm2 Culture flask | SPL Solutions | Cat#70075 |
| 150 cm2 Cell culture dish | SPL Solutions | Cat#20151 |
| Minisart® Syringe Filter | Sartorius | S6534-FMOSK |
| Steriflip-GP Sterile Centrifuge Tube Top Filter Unit | Millipore | SCGP00525 |
| Corning® 500 mL Bottle Top Vacuum Filter, 0.22 μm Pore | Corning | 431118 |
| Superdex 200 HiLoad 26/60 Column | GE Healthcare | Cat#28–9893-36 |
| Spectra/Por® 1 Dialysis Membrane | Spectrum Labs | Cat#132660 |
| Amicon Ultra 100-KDa filter | Millipore | ACS510024 |
| INFORS HT shaking incubator | INFORS HT | Multitron |
| Econo-Column® Chromatography Columns, 2.5 × 10 cm | Bio-Rad | #7372512 |
Materials and equipment
Preparation of DH10Bac LB agar plates and medium
-
•
DH10Bac LB agar plates (1% Tryptone, 0.5% Yeast Extract, 1.0% NaCl, and 15 g/L agar) contain 50 μg/mL kanamycin, 7 μg/mL gentamycin, 10 μg/mL tetracycline, 100 μg/mL Bluo-Gal and 40 μg/mL IPTG.
| Reagent | Final concentration | Amount |
|---|---|---|
| Tryptone | 1% (w/v) | 5 g |
| Yeast extract | 0.5% (w/v) | 2.5 g |
| NaCl | 1% (w/v) | 5 g |
| Agar | 1.5% (w/v) | 7.5 g |
| Sterile Water | N/A | Up to 500 mL |
| Total | N/A | 500 mL |
| Kanamycin (50 mg/mL) | 50 μg/mL | 500 μL |
| Gentamycin (50 mg/mL) | 7 μg/mL | 70 μL |
| Tetracycline (5 mg/mL) | 10 μg/mL | 1 mL |
| Bluo-Gal (200 mg/mL) | 100 μg/mL | 250 μL |
| IPTG (119 mg/mL) | 40 μg/mL | 168 μL |
-
•
DH10Bac LB medium contains 50 μg/mL kanamycin, 7 μg/mL gentamycin and 10 μg/mL tetracycline.
| Reagent | Final concentration | Amount |
|---|---|---|
| Tryptone | 1% (w/v) | 0.5 g |
| Yeast extract | 0.5% (w/v) | 0.25 g |
| NaCl | 1% (w/v) | 0.5 g |
| Agar | 1.5% (w/v) | 0.75 g |
| Sterile Water | N/A | Up to 50 mL |
| Total | N/A | 50 mL |
| Kanamycin (50 mg/mL) | 50 μg/mL | 50 μL |
| Gentamycin (50 mg/mL) | 7 μg/mL | 7 μL |
| Tetracycline (5 mg/mL) | 10 μg/mL | 100 μL |
Note: The DH10Bac LB agar plates containing Bluo-Gal should be stored at 4°C in the dark place for less than one month. DH10Bac LB medium is freshly made from autoclaved LB medium by adding the antibiotics.
Maintenance of insect cells
-
•
The optimal growth conditions of Sf9 cells with HyQ-CCM®3 medium (Cytiva) are 27°C and 100 rpm in an orbital shaker incubator.
-
•
We maintain the cell density of Sf9 between 1×106 to 8×106 cells/mL and split the cells at every 3 or 4 days. Once Sf9 cells are thawed from frozen cell stocks, the cells can be maintained over 6 months.
Note: The density of cells should be maintained above 0.5×106 cells/mL.
Note: Healthy Sf9 cells should double every 24 h.
Note: We recommend using a PYREX® 2,800 mL Fernbach culture flask with a triple baffle and a membrane filtered cap for cell culture. The recommended cell culture volume in a Fernbach culture flask is 500 - 1,000 mL.
Prepare buffers
Note: All the buffers should be kept at 4°C during the purification.
TE buffer
TE buffer contains 1 mM EDTA (pH8.0) and 10 mM Tris-HCl (pH8.0).
| Reagent | Final concentration | Amount |
|---|---|---|
| 0.5 M EDTA (pH8.0) | 1 mM | 0.1 mL |
| 1 M Tris-HCl (pH8.0) | 10 mM | 0.5 mL |
| Sterile Water | N/A | 49.4 mL |
| Total | N/A | 50 mL |
Note: TE buffer must be filtered through a 0.22 μm MCE membrane filter (Millipore™) as this buffer will be treated directly with Sf9 cells for transfection. Filtered TE buffer can be stored at room temperature (RT; 20°C–24°C) for 1 yr.
Cell harvest buffer
Cell harvest buffer contains 100 mM NaCl, 50 mM Tris-HCl (pH8.0), 5% (v/v) glycerol, 1 mM PMSF and 1×Protease Inhibitor Cocktail (PIC) (Roche).
| Reagent | Final concentration | Amount |
|---|---|---|
| 5 M NaCl | 100 mM | 1 mL |
| 1 M Tris-HCl (pH8.0) | 50 mM | 2.5 mL |
| 100% Glycerol | 5% (v/v) | 2.5 mL |
| Sterile Water | N/A | 43 mL |
| Total | N/A | 50 mL |
| 1 M PMSF | 1 mM | 50 μL |
| PIC (Roche) | N/A | 1 tablet |
Note: We recommend adding PMSF and PIC immediately before harvesting the cells. The cell harvest buffer can be stored at 4°C for a month.
Wash buffers
Three different buffers are used to remove nonspecifically bound contaminants from the anti-FLAG M2 resin. The wash buffers can be stored at 4°C for a month.
Wash buffer A contains 100 mM NaCl, 50 mM Tris-HCl (pH8.0) and 5% (v/v) glycerol.
| Reagent | Final concentration | Amount |
|---|---|---|
| 5 M NaCl | 100 mM | 20 mL |
| 1 M Tris-HCl (pH8.0) | 50 mM | 50 mL |
| 100% Glycerol | 5% (v/v) | 50 mL |
| Sterile Water | N/A | 880 mL |
| Total | N/A | 1,000 mL |
Wash buffer B contains 300 mM NaCl, 50 mM Tris-HCl (pH8.0) and 5% (v/v) glycerol.
| Reagent | Final concentration | Amount |
|---|---|---|
| 5 M NaCl | 300 mM | 60 mL |
| 1 M Tris-HCl (pH8.0) | 50 mM | 50 mL |
| 100% Glycerol | 5% (v/v) | 50 mL |
| Sterile Water | N/A | 840 mL |
| Total | N/A | 1,000 mL |
Wash buffer C contains 500 mM NaCl, 50 mM Tris-HCl (pH8.0) and 5% (v/v) glycerol.
| Reagent | Final concentration | Amount |
|---|---|---|
| 5 M NaCl | 500 mM | 100 mL |
| 1 M Tris-HCl (pH8.0) | 50 mM | 50 mL |
| 100% Glycerol | 5% (v/v) | 50 mL |
| Sterile Water | N/A | 800 mL |
| Total | N/A | 1,000 mL |
Anti-FLAG M2 resin elution buffer (freshly made)
Anti-FLAG M2 elution buffer contains 150 mM NaCl, 50 mM Tris-HCl (pH8.0), 5% (v/v) glycerol, and 0.4 mg/mL FLAG peptides (sequence: DYKDDDDK).
| Reagent | Final concentration | Amount |
|---|---|---|
| 5 M NaCl | 150 mM | 1.5 mL |
| 1 M Tris-HCl (pH8.0) | 50 mM | 2.5 mL |
| 100% Glycerol | 5% (v/v) | 2.5 mL |
| Sterile Water | N/A | 43.5 mL |
| Total | N/A | 50 mL |
| FLAG peptides | 0.4 mg/mL | N/A |
Note: FLAG peptides (Apeptide) in powder were dissolved immediately before use.
Dialysis buffer (freshly made)
This dialysis buffer is used when incubation with TEV protease to remove tags. The dialysis buffer contains 150 mM NaCl, 50 mM Tris-HCl (pH8.0), 5% (v/v) glycerol, 1 mM DTT, 0.5 mM EDTA (pH8.0) and 20 mM Imidazole.
| Reagent | Final concentration | Amount |
|---|---|---|
| 5 M NaCl | 150 mM | 60 mL |
| 1 M Tris-HCl (pH8.0) | 50 mM | 100 mL |
| 100% Glycerol | 5% (v/v) | 100 mL |
| 1 M DTT | 1 mM | 2 mL |
| 0.5 M EDTA (pH8.0) | 0.5 mM | 2 mL |
| 1 M Imidazole (pH8.0) | 20 mM | 40 mL |
| Sterile Water | N/A | 1,696 mL |
| Total | N/A | 2,000 mL |
Gel filtration buffer
Gel filtration buffer contains 150 mM NaCl and 20 mM HEPES (pH7.5).
| Reagent | Final concentration | Amount |
|---|---|---|
| 5 M NaCl | 150 mM | 30 mL |
| 1 M HEPES (pH7.5) | 20 mM | 20 mL |
| Sterile Water | N/A | 950 mL |
| Total | N/A | 1,000 mL |
Note: The buffer should be degassed and filtered through a 0.22 μm MCE membrane filter (Millipore™). The gel filtration buffer can be stored at 4°C for a month.
Step-by-step method details
Note: The purification scheme was visualized with a flow diagram (Figure 2).
Figure 2.

A workflow of Huntingtin protein expression and purification
DH10Bac transformation
Generate DH10Bac strains containing the bacmid inserted with the HTT cDNA.
-
1.
Add 100 ng of pFastBac1 plasmid containing the full-length HTT cDNA to 100 μL of DH10Bac™ (Thermo Fisher Scientific) competent cells.
Note: Mix gently. Do not pipet.
-
2.
Incubate the cells on ice for 30 min.
-
3.
Heat-shock at 42°C for 45 s.
-
4.
Incubate the cells on ice again for 2 min.
-
5.
Add 900 μL of S.O.C. medium to the cells and incubate at 37°C for 4 h in a shaking incubator.
-
6.
Prepare a series of 10×, 100×, and 1000× diluted cells with S.O.C. medium.
-
7.
Plate 100 μL of each diluted cell sample on DH10Bac LB agar plates.
-
8.
Incubate the plates for 2 days at 37°C in an incubator.
-
9.
Pick 3–4 white colonies and restreak them onto new DH10Bac LB agar plates.
Note: This step is essential to distinguish true white colonies from blue colonies.
Note: If it is difficult to distinguish between blue and white colonies due to the light blue color of some colonies, refer to the troubleshooting problem 1.
-
10.
Incubate the restreaked plates for 2 days at 37°C.
Note: The resteaked plates can be stored at 4°C for 2–3 days.
-
11.
Inoculate a single white colony into 5 mL of DH10Bac LB medium.
Note: We usually grow 3–5 cultures from several single white colonies.
-
12.
Incubate for 2 days at 37°C in a shaking incubator.
Bacmid isolation and analysis
Isolate bacmid from the culture of the selected white colony and confirm the HTT cDNA transposition into the bacmid using Polymerase Chain Reaction (PCR)
Note: We use Sol I, Sol II and Sol III buffers from the Plasmid DNA Purification Kit Mini (Labopass)
-
13.
Harvest the cells by centrifugation at 3,000×g for 20 min at 4°C.
-
14.
Remove the supernatant and resuspend the cell pellet in 300 μL of Sol I buffer.
-
15.
Add 300 μL of Sol II buffer and mix gently by inverting. Incubate the sample at room temperature (RT; 20°C–24°C) for 5 min.
-
16.
Add 300 μL of Sol III buffer to the mixture and gently mix by inverting. Incubate the sample on ice for 10 min.
-
17.
Centrifuge the sample at 17,000×g for 20 min at 4°C.
-
18.
Transfer 750 μL of the supernatant to 800 μL of 100% isopropanol and mix gently the sample by inverting. Incubate the sample on ice for 10 min.
-
19.
Centrifuge the incubated sample at 17,000×g for 20 min at RT.
Note: After step #19, a white DNA pellet should be observed at the bottom of the tube.
-
20.
Decant the supernatant and add 1 mL of 70% ethanol on the top of the white pellet. Wash the pellet by inverting the tube.
-
21.
Centrifuge the sample at 17,000×g for 5 min at RT.
-
22.
After centrifugation, remove the supernatant with a pipet without disturbing the pellet. Dry the white DNA pellet with airdry while the tubes are upside-down on a Kimwipe until the white pellet becomes transparent.
Note: If the white pellet does not become transparent and stays white, it is an indication of impure bacmid. Refer to the troubleshooting problem 2.
-
23.
Dissolve the transparent DNA pellet by adding 40 μL of TE buffer. Resuspend the DNA pellet gently by tapping. Do not use a pipette.
Note: Store the bacmid at 4°C, and use the bacmid within a day.
Note: In general, the concentration of DNA (bacmid) is ∼4 μg/μL in 40 μl TE (total 160 μg).
Note: The purity of the bacmid can be monitored by the A260/A280 ratio. The A260/A280 ratio of the bacmid should be approximately 2.
-
24.
Perform PCR using the M13 forward and hHTT R146 primers (the sequences of each primer are described in the key resources table). The PCR product should appear at approximately 2.9 kbp when the HTT c cDNA is properly integrated into bacmids.
Table 1.
The detailed PCR composition
| Component | Amount (μl) |
|---|---|
| Bacmid DNA (final 100 ng) | 1 |
| 2×TOPsimple™ PreMIX-Tenuto | 25 |
| 5×Band Doctor™ | 10 |
| M13 forward primer (final 10 pmol) | 1 |
| hHTT R146 reverse primer (final 10 pmol) | 1 |
| Sterile Water | 12 |
| Total | 50 |
Table 2.
The reaction conditions of the PCR reaction for the bacmid plasmid containing HTT gene
| PCR cycling conditions | |||
|---|---|---|---|
| Steps | Temperature | Time | Cycles |
| Initial Denaturation | 94°C | 3 min | 1 |
| Denaturation | 94°C | 45 s | 30 cycles |
| Annealing | 55°C | 45 s | |
| Extension | 72°C | 3+α min a | |
| Final extension | 72°C | 7 min | 1 |
| Hold | 4°C | - | |
Depending on the number of CAG repeats, add extra min for extension (α=0.18 s per one CAG repeat).
Generating and amplify baculovirus
Transfect the bacmid into Sf9 cells to generate baculovirus and amplify the baculovirus for large scale infection to produce HTT proteins.
-
25.
Plate 1.0×106 Sf9 cells in 2 mL CCM3 media (0.5×106 cells/mL) into each well in a 6-well plate.
Note: Sf9 cells were used in the log phase of the growth where the cell density was between 2–4×106.
-
26.
Incubate the cells to attach to the bottom of the plate at 27°C for 1 h.
-
27.
While the cells attach to the plate, prepare 1 μg of bacmid in 100 μL of CCM3 media and 6 μL of Cellfectin II (Thermo Fisher Scientific) in 100 μL of CCM3 media separately.
-
28.
Combine the bacmid in CCM3 and the Cellfectin II in CCM3 from step #27, and mix gently by inverting. Incubate the mixture for 45 min at RT.
-
29.
Remove the medium from each well from step #26 and add 2 mL of fresh CCM medium to each well.
-
30.
After incubating the mixture of bacmid and Cellfectin II at step #28, add 800 μL of fresh CCM medium to the mixture and mix gently by inverting.
-
31.
Remove the medium from each well again from step #29 and add the bacmid-Cellfectin II mixture (∼1 mL) to the cells. Incubate the plate at 27°C for 5 h.
Note: Ensure that one well remains as a negative control without bacmid at step #27–28.
-
32.
After 5 h, remove the medium and add 2 mL of fresh CCM media to each well, and incubate the plate at 27°C for 3 days.
Note: The media loss by evaporation during incubation can be prevented by incubating the cell plate in a sealed chamber such as a Ziploc container with wet Kimwipes.
-
33.
After 3 days, collect the medium containing baculovirus from the wells and filter the medium by using a 0.22 μm syringe filter (Sartorius). This is a P1 virus stock.
Note: The volume of P1 virus stock is approximately around 1.5 mL.
Note: We do not observe clear indication of the infection such as enlarged cells and detached cells, at this stage.
-
34.
Plate 5×106 Sf9 cells in 10 mL into a 75 cm2 cell culture T-flask (or 10 cm2 cell culture dish). Wait for 30 min to let the cells attach to the bottom of the plate. Add 50 μL of the harvested P1 virus from step #33 to the cells. Shake gently the flask to mix the virus in the cell media.
Note: As a control, leave at least one plate of cells without baculovirus.
-
35.
Incubate the flask at 27°C for 3–5 days
Figure 3.

Representative images of baculovirus-infected Sf9 cells
(A and B) Images of Sf9 cells were taken under a light-microscopy after 5 days of post-infection of P2 baculovirus. The infected cells (B) are enlarged compared with the control cells (A).
-
36.
Collect and filter using Steriflip filter unit (Merck Millipore). This is a P2 virus stock.
-
37.
Plate 15×106 Sf9 cells in 25 mL (final 0.6×106 cells/mL) into 150 cm2 cell culture dish. Wait for 30 min to let the cells attach to the bottom of the dish. In addition, add 50 μL of the P2 virus to cells.
Note: As a control, leave at least one culture dish of cells without baculovirus.
-
38.
Incubation at 27°C for 5–6 days.
-
39.
As there are a lot of cells in the media, centrifuge the media at 4,000×g for 10 min and collect the media.
-
40.
Filter the media using 0.22 μm bottle top filters (PES, Corning). This is a P3 virus stock.
Note: We usually make 50 mL of P2 and 500 mL of P3.
Note: Store the virus at 4°C and protect it from light.
Note: The virus can be stored for one month without affecting the level of protein expression.
Large-scale protein expression
Infect baculovirus into Sf9 cells to express HTT proteins.
Note: This protocol is based on a purification procedure from 8 L of Sf9 cell culture.
-
41.
Prepare cells (final density of cells in 1,000 mL media: 1.0–1.5×106 cells/mL) in 1,000 mL of CCM3 media in a 2,800 mL Fernbach flask a day before the large-scale infection.
Note: We dilute Sf9 cells on a large scale from Sf9 cells at 6–8×106 cells/mL.
-
42.
Incubate the cell until 2.0–2.5×106 cells/mL density at 27°C with 100 rpm in an orbital shaker incubator.
Note: Typically, it will take 24 h to reach 2.0×10 6 cells/mL from 1.0×106 cells/mL at 27°C at 100 rpm in an INFORS HT shaking incubator
-
43.
When the proper cell density is reached, add 50 mL of the P3 virus to 1,000 mL of the cells in a 2,800 mL Fernbach flask.
-
44.
Incubate at 27°C for 42–48 h.
-
45.
Harvest the cells by centrifugation at 4,600×g for 20 min at 4°C.
-
46.
Decant the medium and resuspend the cell pellet with 30 mL of the cell harvest buffer for the cells from 1,000 mL of cell culture.
Note: Ensure that 1 mM PMSF and final 1× Protease inhibitor Cocktail are added to the cell harvest buffer before use.
-
47.
Freeze the resuspended cells in liquid nitrogen and store them at −80°C.
FLAG affinity chromatography
Isolate the recombinant FLAG tag-HTT with anti-FLAG M2 affinity gel from cell lysate.
-
48.
The resuspended cells were lysed by repeatedly freezing and thawing in liquid nitrogen and in a water bath at 25°C for 3 times.
Note: No other reagent such as DNase is necessary for cell lysis.
-
49.
Centrifuge the cell lysate at 27,000×g for 2 h using 50 mL centrifuge tubes.
Note: If the supernatant is still turbid after this step, centrifuge again with the same condition for another 1 hr.
Note: There should be clear supernatant and soft sponge like pellet. We don’t expect see solid pellet.
-
50.
Collect the supernatant by pouring out without disturbing the soft pellet.
Note: We usually obtain 30 mL of the supernatant from 1 L of Sf9 cell culture. Therefore, the volume of the supernatant for 8 L culture is approximately 240 mL.
Note: Equally distribute the supernatant to six 50 mL conical tubes.
-
51.
Equilibrate the anti-FLAG M2 affinity resin (Sigma) with the wash buffer A.
Note: We use 10 mL (packed bead volume) of M2 resin for 8 L of Sf9 cell culture.
-
52.
Equally distribute 10 mL of the M2 resin to the supernatants from step #50.
-
53.
Incubate the mixture of the supernatant and the M2 resin for 3 h at 4°C in a tube rotator or a tumbler at 10 turns per min.
-
54.
Collect the M2 resin by centrifugation at 1,500×g for 5 min.
-
55.
After slowly pouring out the supernatant from the centrifuged M2 resin, collect all the resin in a single 50 mL tube with the wash buffer A and bring the solution to 50 mL by adding the wash buffer A.
Note: After centrifugation, the M2 resin sits at the bottom of the 50 mL tubes. Slowly decant the supernatant by pouring out.
-
56.
Collect the M2 resin by centrifugation at 1,500×g for 5 min and decant the supernatant by pouring out.
-
57.
Add 40 mL of the wash buffer A to the M2 resin and resuspend by inverting the tube.
-
58.
Repeat step #56–57 for 3 times with the wash buffer A.
-
59.
Repeat step #56–57 for 3 times with the wash buffer B.
-
60.
Repeat step #56–57 for 3 times with the wash buffer C.
-
61.
Repeat step #56–57 for 3 times with the wash buffer B.
-
62.
Repeat step #56–57 for 3 times with the wash buffer A.
-
63.
After washing, load the M2 resin to an Econo-Column® Chromatography Columns, 2.5 × 10 cm (Bio-Rad).
-
64.
Wash the resin with the wash buffer A until the absorbance of the flow-through becomes lower than 0.01 at the OD280 reading.
-
65.
Decant the wash buffer A to the surface of the M2 resin.
-
66.
Add 1 column volume (CV, 10 mL) of the M2 elution buffer slowly on the top of the resin without disturbing the M2 resin and incubate for 10 min.
-
67.
Collect the incubated elution buffer.
-
68.
Repeat step #66–67 until the OD280 reading of the eluted sample is less than 0.01.
Note: We usually collect HTT proteins with 5–6 elutions (total 50–60 mL).
Note: The expected amount of the proteins at this stage is approximately 15–20 mg from 8 L of the Sf9 cell culture. The amounts of proteins were estimated by OD280 with Abs 0.1% (1 g/L) of 0.78 (the molar extinction coefficient of Q23 HTT is 272,755 cm−1M−1 at 280 nm) and the purity of the samples is higher than 80% based on estimation by SDS-PAGE.
TEV protease treatment
Remove the N-terminal tags of HTT with TEV protease.
-
69.
Collect the eluted samples and add TEV protease.
Note: We use home-made TEV protease, which has an N-terminal 6×His-tag. Add 40 μg of TEV protease per 1 mg of the eluted samples (800 μg of TEV protease for 20 mg of HTT protein).
Note: TEV proteases are commercially available (Thermo Fisher, Cat# 1257023)
-
70.
Transfer the sample with TEV protease to Spectra/Por® 1 Dialysis membrane (30 Kd MWCO).
-
71.
Dialyze the sample for 12–16 h against 2 L of the dialysis buffer.
Nickel affinity chromatography
Remove the uncleaved protein and the remaining TEV protease with Ni affinity chromatography.
-
72.
Load the Ni-nitrilotriacetic acid resin (Ni-NTA, Qiagen) to a Econo column (2.5×10 cm).
Note: Use 10 mL of Ni-NTA.
Note: Ensure that Ni-NTA is resistant to 1 mM DTT and 0.5 mM EDTA in the dialysis buffer. We found that Ni-NTA from Qiagen is suitable for this step.
-
73.
Equilibrate the Ni-NTA resin with the dialysis buffer.
-
74.
Load the sample from step #71 to pass through the equilibrated Ni-NTA column and collect the flow-through.
-
75.
Reload the collected flow-through to the Ni-NTA again.
-
76.
Repeat step #74–75 for 3 times.
-
77.
Concentrate the sample using an Amicon Ultra 100-KDa filter up to 10 mL (less than 2 mg/mL concentration) by repeated centrifugation at 1,500×g for 5 min each time.
Size exclusion chromatography
Note: Elution profiles of the size exclusion chromatography and protein quality of final concentrated samples are shown in Figure 4.
Figure 4.

Size exclusion chromatography and PAGE analysis of recombinant HTT
(A) The elution profile of HTT purification from size exclusion chromatography with 10% SDS-PAGE analysis of the fractions. Asterisk indicates the position of the fraction in the gel.
(B) The purity of purified HTT was analyzed by 10% SDS-PAGE (lefts) and 5% native-PAGE (right). The proteins were visualized by Coomassie staining.
-
78.
Equilibrate a Superdex 200 HiLoad 26/60 column (Cytiva) with the gel filtration buffer.
-
79.
Load the concentrated sample from step #77 to the equilibrated column and run the column with a 2.0 mL/min flow rate and 5 mL fraction size.
-
80.
Evaluate the purity of each fraction by SDS-PAGE.
-
81.
Collect the fractions containing HTT proteins and concentrate the sample using an Amicon Ultra 100-KDa filter up to 5 mg/mL concentration.
-
82.
Aliquot the sample into ∼20 μL per tube and snap freeze with liquid nitrogen. Store the samples at −80°C.
Note: The sample can remain stable at −80°C up to 1 year.
Note: With this protocol, we obtained 8 mg of recombinant HTT proteins from 8 L of Sf9 cell culture.
Note: The expected purity of samples at this step is over 95%.
Expected outcomes
SDS-PAGE and western blotting assay on HTT proteins
To confirm the quality of the purified recombinant HTT proteins from this protocol, SDS-PAGE and western blotting assay were performed with HTT proteins. The purity of the purified proteins is more than 95%, as assessed by SDS- and native-PAGE gels stained with Coomassie blue (Figure 4B), showing that HTT proteins form multimers.
Using this protocol, we generated HTT proteins with allelic series of polyQ lengths (Q2, Q23, Q46, Q67 and Q78). The size differences of the samples with different polyQ lengths were distinguished by 5% SDS-PAGE (Figure 5A). The presence of expanded polyQ was examined with mAb2166 (Millipore) and 1F8 (Wheeler et al., 2000) monoclonal antibodies, which recognize N-HEAT (181–810 a.a) and expanded polyQ respectively (Figure 5B)
Figure 5.

SDS-PAGE and western blot analysis on the purified HTT proteins with allelic series of polyQ lengths
(A) The purity of recombinant full-length HTT with various polyQ (Q2, Q23, Q46, Q67, Q78) was analyzed by 5% SDS-PAGE and Coomassie staining.
(B) Purified recombinant full-length HTT were analyzed via western blotting with mAb2166 (above) and 1F8 (below) recognizing N-HEAT (181–810 a.a.) and expanded polyQ respectively.
Recombinant HTT forms a complex with HAP40
HTT protein forms a complex with Huntingtin-associated protein 40 (HAP40) (Guo et al., 2018). To examine whether the HAP40 and HTT proteins form a complex using this protocol, we coinfected Sf9 cells with baculoviruses harboring either HAP40 or HTT expressing to induce HTT_HAP40 complex (50 mL of each virus to 1 L of Sf9 cells). After harvesting the expressed cells, the HTT_HAP40 complex was purified with the same protocol as HTT protein alone. The elution profile from Superdex S200 chromatography followed by SDS-PAGE analysis (Figure 6B) show that HTT forms a stoichiometric complex with HAP40. The quality of the complex was examined by SDS- and native-PAGE (Figure 6B). We also noticed that HTT_HAP40 complex forms a multimer, which is contrast to the previous works (Guo et al., 2018; Harding et al., 2021; Huang et al., 2021). However, it is possible that the multimer of HTT_HAP40 complex may be induced in high concentration, which is in our case. Furthermore, we analyzed whether isolated HTT can interact with isolated HAP40. We performed an in-vitro pulldown assay with recombinant GST-HAP40 (Figure 6C). GST or GST-HAP40 were expressed in Sf9 cells and the cells were harvested in a buffer A containing 150 mM NaCl, 20 mM HEPES (pH7.5). The cells were lysed by repeated freezing and thawing 3 times and cell debris was cleared with centrifugation at 17,000×g for 1 h at 4°C. The proteins were purified with glutathione-SepharoseTM 4B resins (Cytiva). The GST resins bound with GST or GST-HAP40 were incubated with purified recombinant HTT (300 μg). After incubation the resins were washed with a buffer A to remove the unbound HTT proteins. The proteins were then eluted from the resins with a buffer B containing150 mM NaCl, 20 mM HEPES (pH7.5) and 20 mM reduced glutathione. The eluted proteins were analyzed by SDS-PAGE with Coomassie blue staining.
Figure 6.

HTT proteins form a complex with HAP40
(A) The elution profile of size exclusion chromatography and 10% SDS-PAGE analysis of the purified HTT-HAP40, which were co-expressed. Asterisk indicates the position of the fraction.
(B) The purity of the HTT and HTT_HAP40 complex was confirmed by SDS-PAGE (left) and Native-PAGE (right).
(C) GST pull-down assay with GST-HAP40 and isolated HTT protein.
Although the purified HTT protein can be pulled down with GST-HAP40, HTT_HAP40 does not form a stable complex when compared with the complex coexpressed, which is consistent with previous works (Guo et al., 2018; Harding et al., 2021; Huang et al., 2021). These data demonstrated that the HTT complexes can be generated through this protocol.
Recombinant HTT proteins form a complex with HAP1
Huntingtin-associated protein 1 (HAP1) is the first identified protein as a HTT interacting protein using yeast two-hybrid screen (Y2H) (Li et al., 1995). The HAP1 (278–599 a.a.) and HTT (1–253 a.a.) was shown to be an essential region for binding to each other (Li et al., 1998). To examine the binding of the recombinant HTT proteins purified from this protocol to HAP1, we performed a GST pulldown assay using GST-HAP1 (300–381 a.a.) (Figure 7A). Purified GST or GST-HAP1 were incubated with glutathione-SepharoseTM 4B resins (Cytiva) in a total volume of 500 μL of buffer A containting 150 mM NaCl, 20 mM HEPES (pH7.5) for 1 h at 4°C. The resins were washed with the buffer A followed by incubation with purified recombinant HTT (300 μg). After incubation, the resins were washed with the buffer A and the proteins were eluted with the buffer B and analyzed by SDS-PAGE with Coomassie blue staining. As shown in the results, the purified recombinant HTT can directly bind to the HAP1 fragments. These results indicate that the HTT protein generated through this protocol retains its HAP1 binding activity.
Figure 7.

Invitro binding assay using GST-HAP1 and recombinant HTT protein
The GST pull-down assay was used to test the binding between purified HTT and GST-HAP1 (300–381 a.a.). The result of the GST pull-down assay was analyzed by SDS-PAGE and Coomassie staining.
Limitations
Since the normal function of HTT is still unclear, experiments to directly confirm the activity of recombinant HTT were not conducted in this paper. However, we have shown that the recombinant HTT purified by using this protocol activates the histone H3K27 tri-methylase activity of Polycomb Repressive Complex 2 (PRC2) in vitro (Seong et al., 2010). In addition, our cryo-EM analysis (EMDB: EMD-4944) (Jung et al., 2020) on full-length HTT proteins obtained using this method shows that they have similar structures to those previously resolved high-resolution cryo-EM structure of HTT (Guo et al., 2018; Harding et al., 2021; Huang et al., 2021) and maintains the similar phosphorylation pattern to the previously identified phosphorylation sites (Harding et al., 2019; Jung et al., 2020; Saudou and Humbert, 2016). These results suggest that this protocol to generate recombinant full-length HTT proteins with allelic series of polyQ lengths will provide key reagents for various experiments to investigate the functions of HTT and the molecular mechanism of HD pathology. It should be noted that other protocols have been used to generate recombinant full-length HTT proteins (Harding et al., 2019; Huang et al., 2015).
Troubleshooting
Problem 1
The blue color of colonies is too weak in step #9.
Potential solution
When Bluo-Gal is added to the autoclaved LB agar when it is too hot, Bluo-Gal goes bad and does not show clear blue color. Bluo-Gal was added when the temperature of the autoclaved LB agar was below 50°C.
Problem 2
The pellet was still turbid after dry in step #22.
Potential solution
This is an indication that bacmid is not pure. Pick new white colonies (step #11) and purify the bacmid again.
Problem 3
The infected Sf9 cells are not enlarged nor detached from the plates in step #35.
Potential solution
You should see that the infected Sf9 cells become enlarged and detached from the plates during P2 and P3 virus generation, but not necessary during P1 virus generation. This is an indication that the quality of the virus is poor. We recommend returning to step #1.
Problem 4
The A260/A280 ratio of the eluted samples is over 1.0 in step #68.
Potential solution
This indicates the presence of the genetic material contamination, which results in significant loss of the proteins during the gel-filtration chromatography step. This problem can be solved by washing with additional wash buffer C in step #60.
Problem 5
The yield of final product is less than suggested in step #82.
Potential solution
Return to step #37–39 with the harvested P3 virus. This amplified virus is a P4 virus stock and is used in infection for expression. If that does not work, consider making a new virus from a fresh bacmid. Please refer to the critical points of this protocol in “before you begin”.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ji-Joon Song (songj@kaist.ac.kr).
Materials availability
Htt plasmids with allelic series of CAG are available at Song Lab, KAIST. As it is very tricky to maintain the stable number of CAG, the plasmid will be directly distributed from the lead contact upon request.
Acknowledgments
We thank the members of the Song Lab at KAIST. This work is partially supported by a grant (NRF-2016K1A1A2912057 to J.-J.S. and I.S.S.) from the National Research Foundation of Korea.
Author contributions
H.K. established a protocol for HTT protein purification. H.K., A.L., and I.S.S. established a protocol for plasmid amplification. H.K., K.-g.H., and J.-J.S. wrote the manuscript. All authors reviewed the data.
Declaration of interests
J.S. is a co-founder and CTO of PCG Biotech.
Data and code availability
This study did not generate/analyze datasets/code.
References
- Guo Q., Bin H., Cheng J., Seefelder M., Engler T., Pfeifer G., Oeckl P., Otto M., Moser F., Maurer M. The cryo-electron microscopy structure of huntingtin. Nature. 2018;555:117–120. doi: 10.1038/nature25502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding R.J., Deme J.C., Hevler J.F., Tamara S., Lemak A., Cantle J.P., Szewczyk M.M., Zuo X., Loppnau P., Seitova A. HAP40 orchestrates huntingtin structure for differential interaction with polyglutamine expanded exon 1. bioRxiv. 2021 doi: 10.1101/2021.04.02.438217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding R.J., Loppnau P., Ackloo S., Lemak A., Hutchinson A., Hunt B., Holehouse A.S., Ho J.C., Fan L., Toledo-Sherman L. Design and characterization of mutant and wildtype huntingtin proteins produced from a toolkit of scalable eukaryotic expression systems. J. Biol. Chem. 2019;294:6986–7001. doi: 10.1074/jbc.RA118.007204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B., Guo Q., Niedermeier M.L., Cheng J., Engler T., Maurer M., Pautsch A., Baumeister W., Stengel F., Kochanek S., Fernandez-Busnadiego R. Pathological polyQ expansion does not alter the conformation of the Huntingtin-HAP40 complex. Structure. 2021;29:804–809 e805. doi: 10.1016/j.str.2021.04.003. [DOI] [PubMed] [Google Scholar]
- Huang B., Lucas T., Kueppers C., Dong X., Krause M., Bepperling A., Buchner J., Voshol H., Weiss A., Gerrits B., Kochanek S. Scalable production in human cells and biochemical characterization of full-length normal and mutant huntingtin. PLoS One. 2015;10:e0121055. doi: 10.1371/journal.pone.0121055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung T., Shin B., Tamo G., Kim H., Vijayvargia R., Leitner A., Marcaida M.J., Astorga-Wells J., Jung R., Aebersold R. The polyglutamine expansion at the N-terminal of huntingtin protein modulates the dynamic configuration and phosphorylation of the C-terminal HEAT domain. Structure. 2020;28:1035–1050 e1038. doi: 10.1016/j.str.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.H., Gutekunst C.A., Hersch S.M., Li X.J. Interaction of huntingtin-associated protein with dynactin P150Glued. J. Neurosci. 1998;18:1261–1269. doi: 10.1523/jneurosci.18-04-01261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.J., Li S.H., Sharp A.H., Nucifora F.C., Jr., Schilling G., Lanahan A., Worley P., Snyder S.H., Ross C.A. A huntingtin-associated protein enriched in brain with implications for pathology. Nature. 1995;378:398–402. doi: 10.1038/378398a0. [DOI] [PubMed] [Google Scholar]
- Saudou F., Humbert S. The biology of huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- Seong I.S., Woda J.M., Song J.J., Lloret A., Abeyrathne P.D., Woo C.J., Gregory G., Lee J.M., Wheeler V.C., Walz T. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010;19:573–583. doi: 10.1093/hmg/ddp524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler V.C., White J.K., Gutekunst C.A., Vrbanac V., Weaver M., Li X.J., Li S.H., Yi H., Vonsattel J.P., Gusella J.F. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze datasets/code.