

Abstract
Breast cancer causes the most cancer fatalities in women worldwide. Approximately one‐third of breast cancers metastasize, or spread from primary tumors to other tissues, and have a 70% 5‐year mortality rate. Current breast cancer treatments like doxorubicin and paclitaxel become ineffective when breast cancer cells develop multi‐drug resistance and overexpress ATP‐binding cassette transporters, as the transporters cause a substantial efflux of the chemotherapies. Jadomycins, a group of molecules isolated from Streptomyces venezuelae ISP5230, are shown to be cytotoxic against a variety of cancers, especially breast cancer. Furthermore, jadomycins retain their cytotoxic properties in multi‐drug resistant breast cancer cells, as they are not expelled through ATP‐binding cassette transporters. Here, we describe the research that supports the potential use of jadomycins as a novel chemotherapy in the treatment of multi‐drug resistant, metastatic breast cancer. We present the supportive findings, as well as the mechanisms of action investigated thus far. These include copper‐mediated reactive oxygen species generation, aurora B kinase inhibition, and topoisomerase IIα and IIβ inhibition. We also suggest future directions of jadomycin research, which will help to determine if jadomycins can be used as a breast cancer chemotherapy in clinical practice.
Keywords: chemotherapy, jadomycins, metastatic breast cancer, multi‐drug resistance
Jadomcyins are isolated from the soil bacterium Streptomyces venezuelae and are cytotoxic against a variety of human cancer cells, including those that express multi‐drug resistance transporters. Here we describe the research that supports the potential use of jadomycins, especially against human breast cancer cells, and the mechanisms of action investigated so far. We also suggest novel future directions to develop jadomycin and breast cancer research.

Abbreviations
- 3AMBA
3‐(aminomethyl)benzoic acid
- 4AMBA
4‐(aminomethyl)benzoic acid
- ABC
ATP‐binding cassette
- ABK
aurora B kinase
- DDC
sodium diethyldithiocarbamate
- DOX
doxorubicin
- dsDNA
double‐strand DNA
- EA
ellagic acid
- ER
estrogen receptor
- ETP
etoposide
- H2O2
hydrogen peroxide
- HER2
human epidermal growth factor receptor 2
- HMEC
human microvascular epithelial cells
- IC50
half maximal inhibitory concentration
- MDR
multi‐drug resistant
- MITX
mitoxantrone
- MTS
phenazine methosulfate
- MTT
thiazolyl blue methyltetrazolium bromide
- NAC
N‐acetyl cysteine
- NSCLC
non‐small cell lung carcinoma
- pHis3
phosphorylated H3
- PR
progesterone receptor
- ROS
reactive oxygen species
- SRB
sulforhodamine B
- TrxR
thioredoxin reductase
- TXL
paclitaxel
- WaterLOGSY
Water‐Ligand Observed via Gradient SpectroscopY
- γH2AX
phosphorylated histone H2AX
1. INTRODUCTION
Cancer universally claims the lives of over 9.5 million individuals each year. 1 , 2 Breast cancer is of particular concern as it is the most common cause of cancer fatalities in women worldwide. 1 Approximately 30% of breast cancers metastasize, a process in which the cancer spreads to other body regions. Metastatic breast cancers respond poorly to current treatment options and the prognosis for patients is dire, with a 5‐year mortality rate of over 70%. 3 Metastatic breast cancers are especially difficult to treat as they are often multi‐drug resistant (MDR) and have little to no expression of estrogen receptors (ER) and progesterone receptors (PR), and lack overexpression of human epidermal growth factor receptor 2 (HER2). These receptors are key breast cancer drug targets and serve as diagnostics to identify specific types and stages of breast cancer. 4 When breast cancers lack all three of these receptors, they are known as triple‐negative breast cancer. There are currently few effective treatments for MDR metastatic breast cancers, and despite treatment breast cancer cells often continue to proliferate and gain resistance to most available chemotherapies, which become ineffective. 5 Thus, research for new MDR metastatic breast cancer therapies is imperative.
Jadomycins, a group of molecules first isolated in 1991 from the soil bacteria Streptomyces venezuelae ISP5230, have demonstrated promising cytotoxicity against triple‐negative and hormone receptor positive MDR metastatic breast cancer cells, and may therefore be useful as a novel chemotherapy. 6 , 7 , 8 , 9 , 10 Here, we review the current evidence pertaining to jadomycin cytotoxicity in various cancer cell lines, with a specific focus on MDR breast cancer. Our search strategy utilized Google Scholar and the PubMed and Novanet databases, and included the key terms “jadomycin,” “copper dependency,” “reactive oxygen species,” “aurora B kinase,” and “topoisomerase,”. We explain the present knowledge of jadomycin cytotoxic mechanisms in MDR breast cancer cells, including their copper‐dependent reactive oxygen species (ROS) generation and aurora B kinase (ABK), and topoisomerase II interactions. 7 , 8 , 11 , 12 We also discuss future directions for the study of jadomycins, including the need to investigate jadomycin anticancer effects in animal models, and to further define jadomycin mechanism of action to predict possible adverse drug effects and drug–drug interactions.
2. JADOMYCIN BIOSYNTHESIS
Jadomycins belong to the angucycline, type‐II polyketide‐derived family of molecules, and are produced by S. venezuelae under stress conditions of ethanol treatment, phage infection, or heat shock (Figure 1A). 10 , 12 S. venezuelae is known for its production of the antibiotic chloramphenicol, and jadomycins exhibit similar antimicrobial activity against both Gram positive and negative bacteria (Figure 1B). 13 , 14 Additionally, jadomycins demonstrate anticancer properties against hepatic, lung, multiple myeloma, cervical, colon, and breast cancer cell lines. 7 , 8 , 9 , 11 , 15 , 16 , 17 For a more complete description of jadomycin biosynthesis we refer the interested reader to the recent review article by de Koning et al. 18
FIGURE 1.
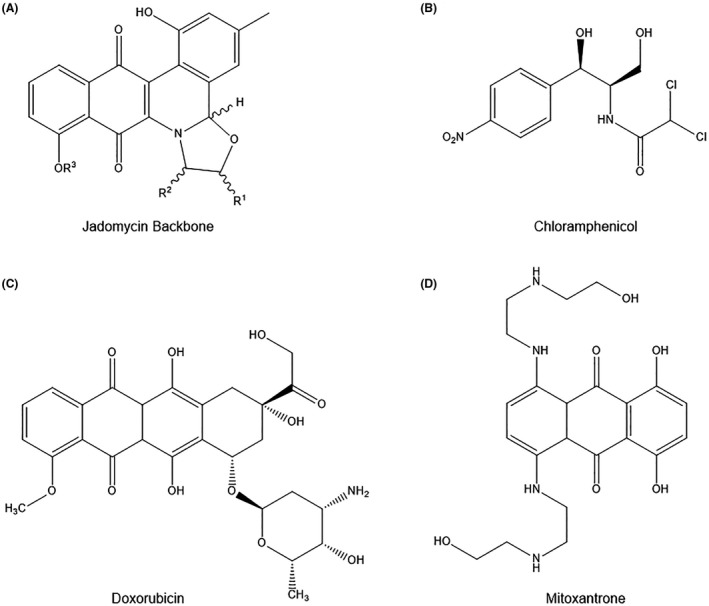
(A) Jadomycin backbone structure compared to (B) antibiotic chloramphenicol and triple‐negative breast cancer chemotherapies (C) doxorubicin and (D) mitoxantrone. Jadomycin backbone structure; “R1”‐group indicates location of =O or ‐H attachment, creating an oxazolone or oxazolidine ring, respectively, “R2”‐group indicates attachment of amino acid analog, and “R3”‐group indicates ‐H or deoxy sugar attachment
While most jadomycins maintain the same polyaromatic core structure, usually consisting of an A‐, B‐, C‐, D‐, and E‐ring, their various amino acid side groups make them unique (Figure 2A). 10 S. venezuelae grown in media containing a single amino acid as the nitrogen source selectively produces diverse analogs of the jadomycin molecule. 10 Interestingly, S. venezuelae incorporates amino acids into the jadomycin polyaromatic core through chemical biosynthesis in a non‐enzymatic manner. 10 , 19 This is an important feature, as over 70 jadomycins are conveniently synthesized in vitro through chemical methods, which facilitates structural diversity around the E‐ring and produces jadomycins with unique pharmacological properties (Figure 2A). 10 However, the ability to synthesize jadomycin derivatives through the use of selective media is not always achieved in the presence of a single amino acid. 20 For example, media containing 3‐ or 4‐(aminomethyl)benzoic acid (3AMBA and 4AMBA) normally inhibits S. venezuelae proliferation, but the addition of D‐serine to the media allows the bacteria to add 3AMBA or 4AMBA acid to the jadomycin backbone. 21 This suggests that D‐serine promotes the incorporation of other products to create novel jadomycins, which could further enhance jadomycin pharmacological diversity. 21
FIGURE 2.
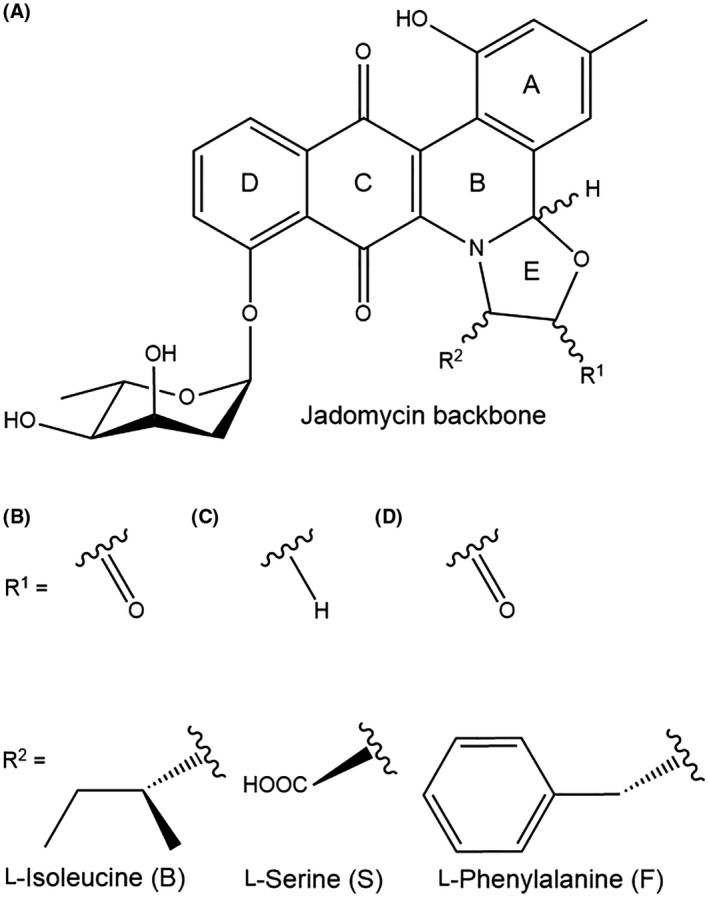
Jadomycin chemical structure and key analogs. (A) Jadomycin backbone structure with A‐, B‐, C‐, D‐, and E‐rings, and deoxy sugar attached to D‐ring. “R1”‐group indicates location of =O or ‐H attachment, creating an oxazolone or oxazolidine ring, respectively, while “R2”‐group indicates attachment of amino acid analog (B) jadomycin B, L‐Isoleucine, (C) jadomycin S, L‐Serine, or (D) jadomycin F, L‐Phenylalanine
Jadomycin A was the first to be discovered and is structurally identical to jadomycin B, but contains a hydroxyl group on the D‐ring instead of a deoxy sugar (Figure 2A). 10 Jadomycin A and B were named in order of discovery, but subsequent jadomycins were named based on their R2 side groups (Table 1). Most jadomycins exhibit some pharmacological significance, but jadomycin A and W in particular have little cytotoxic activity against bacteria and cancer cells, attributed to the lack of a glycone ring and a large aromatic structure, respectively. 9 , 15 , 16 Large amino acids on the E‐ring generally prevent jadomycins from interacting precisely with cellular targets. However, there is additional versatility in the E‐ring, as the R2 attachment is not strictly limited to amino acids. Jadomycin N, for example, incorporates a carboxyl group in this location, yet still possesses anticancer properties. 16 Notably, various enzymes can also catalyze prejadomycin, from which jadomycin A is derived, into alternative natural products that may possess additional benefits. 22
TABLE 1.
Jadomycin analogs and corresponding E‐ring attachment
| Jadomycin name | E‐ring |
|---|---|
| 3AMBA | N/A |
| 4AMBA | N/A |
| βala | N/A |
| A | L‐Isoleucine |
| Ala | L‐Alanine |
| Abu | L‐2‐Aminobutanoic Acid |
| B | L‐Isoleucine |
| DM | D‐Methionine |
| DNV | D‐Norvaline |
| DS | D‐Serine |
| DT | D‐Threonine |
| DV (D‐Val) | D‐Valine |
| F | L‐Phenylalanine |
| G | Glycine |
| H | L‐Histidine |
| Hse | L‐Homoserine |
| L | L‐Leucine |
| LN | L‐Asparagine |
| M | L‐Methionine |
| N | Carboxyl Group |
| Nle | L‐Norleucine |
| Orn | L‐Ornithine |
| R‐Phe | R‐Phenylglycine |
| S | L‐Serine |
| SPhG (S‐Phe) | S‐Phenylglycine |
| T | L‐Threonine |
| V | L‐Valine |
| W | L‐Tryptophan |
| Y | L‐Tyrosine |
Jadomycin analog names as discussed in this article are listed in correspondence to their respective nitrogen sources used to incorporate amino acids or alternative side chains into jadomycin E‐rings through a chemical process.
Jadomycins are believed to maintain their cytotoxic properties in MDR breast cancer cells because unlike standard metastatic breast cancer chemotherapies including taxanes, anthracyclines, and epothilones, many of the most potent jadomycins exhibit an oxazolone or oxazolidine ring side chain which may result in unique interactions with cancer cells (Figure 2B–D). 9 , 16 Nonetheless, researchers have noticed that jadomycins with different chemical structures can also exhibit anticancer properties. For instance, jadomycins with a 3AMBA side group lack an E‐ring, but are still cytotoxic against breast cancer cells. 21 Jadomycins with a triazole group attached to the E‐ring are also cytotoxic against various cancer cells, while those with a non‐triazole group attachment demonstrate little to no cytotoxic activity. 23 This argues that while the A, B, C, and D‐rings are involved in the anticancer effect of jadomycins, the glycone ring attachment on the D‐ring and structural diversity around the E‐ring are most involved in changing the potency and efficacy of jadomycins.
Several experiments have shown that jadomycins have the unique ability to avoid ATP‐binding cassette (ABC) drug efflux transporters. ABC transporters are pumps that expel intracellular drugs and toxins, and are often overexpressed in MDR breast cancer cells, which may contribute to the avoidance of cell death. 7 , 24 , 25 Jadomycins have demonstrated the ability to maintain their potency and efficacy in both drug‐sensitive and drug‐resistant breast cancer cells, unlike the structurally similar doxorubicin (DOX) and mitoxantrone (MITX) chemotherapies used to treat triple‐negative breast cancer (Figure 1C and D). Thus, jadomycins are a potentially attractive chemotherapeutic alternative, and may contribute to an increasingly important area of cancer research. 7
3. JADOMYCINS ARE CYTOTOXIC TO BREAST CANCER CELL LINES
Several studies have verified that jadomycins exhibit anticancer properties in multiple human breast cancer cell lines. 7 , 8 , 9 , 16 The evidence supporting jadomycin cytotoxicity against each breast cancer type is highlighted in the following sections.
3.1. Hormone‐receptor positive breast cancer
3.1.1. T‐47D cells
Borissow et al. were among the first to explore jadomycin cytotoxicity in breast cancer cells. 16 They assessed cytotoxicity profiles of jadomycin βala, B, DM, DT, DV, F, G, H, M, N, R‐Phe, S, SPhG, T, V, W, and Y against the ER positive (+), PR+, and HER2 negative (−) human breast cancer cell line T‐47D. 16 , 26 The results indicated that jadomycin DT and S were the most potent overall, while jadomycin βala, H, and Y were among the least potent (Table 2). 16 Jadomycin DT and S both have polar carboxy side chains, which may be responsible for their ability to induce cancer cell death. 10 , 16 Polarity of the side chain is perhaps more important than size, as jadomycin G has one of the smallest side chains but is non‐polar and exhibits little pharmacological activity. 16 Aromatic amino acids also appear to sterically hinder jadomycin interaction with cancer cells, as jadomycin H and Y with histidine and tyrosine groups, respectively, were less efficacious than jadomycins with nonaromatic groups. 16
TABLE 2.
Jadomycin IC50 values in various cancer cell lines
| Study | Cell type | Cell line | Exposure time (h) | Assay used | IC50 (µM) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| βala | Abu | Ala | B | DM | DNV | DT | DV | F | G | H | Hse | |||||
| Borissow 2007 16 | Breast | T‐47D | 48 | MTT | 23.11 | — | — | 9.92 | 7.80 | — | 2.57 | 6.42 | 8.84 | 8.27 | 9.82 | — |
| Fu 2008 11 | Breast | MCF7‐CON | 48 | MTT | — | — | — | 27.2 | — | — | — | — | — | — | — | — |
| Fan 2012 17 | Breast | MCF7‐CON | 72 | SRB | — | 2.7 | — | 2.8 | — | — | — | — | — | — | — | 66.8 |
| Issa 2014 9 | Breast | MCF7‐CON | 72 | MTT | — | — | — | 4.4 | — | 1.3 | — | — | 0.9 | — | — | — |
| Hall 2015 8 | Breast | MCF7‐CON | 72 | MTT | — | — | — | 2.6 | — | — | — | — | 3.6 | — | — | — |
| Issa 2014 9 | Breast | MCF7‐TXL | 72 | MTT | — | — | — | 5.8 | — | 2.0 | — | — | 3.1 | — | — | — |
| Issa 2014 9 | Breast | MCF7‐ETP | 72 | MTT | — | — | — | 8.0 | — | 2.2 | — | — | 3.2 | — | — | — |
| Issa 2014 9 | Breast | MCF7‐MITX | 72 | MTT | — | — | — | 9.9 | — | 4.2 | — | — | 3.6 | — | — | — |
| Hall 2015 8 | Breast | BT474 | 72 | MTT | — | — | — | 4.2 | — | — | — | — | 5.1 | — | — | — |
| Hall 2015 8 | Breast | SKBR3 | 72 | MTT | — | — | — | 3.8 | — | — | — | — | 4.7 | — | — | — |
| Borissow 2007 16 | Breast | MDA‐MB‐435 | 48 | MTT | 10.66 | — | — | 6.89 | 2.87 | — | 1.15 | 3.24 | 6.11 | 3.35 | 20.99 | — |
| Hall 2015 8 | Breast | 231‐CON | 72 | MTT | — | — | — | 1.8 | — | — | — | — | 3.3 | — | — | — |
| Hall 2017 7 | Breast | 231‐CON | 72 | MTT | — | — | — | 2.8 | — | — | — | — | 3.0 | — | — | — |
| Hall 2017 7 | Breast | 231‐TXL | 72 | MTT | — | — | — | 2.5 | — | — | — | — | 3.2 | — | — | — |
| Zheng 2005 15 | Hepatic | HepG2 | 24 | MTT | — | — | 100.0 | 10.8 | — | — | — | — | 49.0 | — | — | — |
| Zheng 2005 15 | Myeloma | IM9 | 24 | MTS | — | — | 40.0 | 8.5 | — | — | — | — | 29.0 | — | — | — |
| Zheng 2005 15 | Lung | H460 | 24 | MTT | — | — | 30.7 | 21.8 | — | — | — | — | 12.4 | — | — | — |
| Fu 2008 11 | Lung | A549 | 48 | MTT | — | — | — | 11.3 | — | — | — | — | — | — | — | — |
| Fu 2008 11 | Cervix | HeLa | 48 | MTT | — | — | — | 18.2 | — | — | — | — | — | — | — | — |
| Fan 2012 17 | Colon | HCT116 | 72 | SRB | — | 8.3 | — | 3.8 | — | — | — | — | — | — | — | 56.8 |
| Fan 2012 17 | Epithelium | HMEC | 72 | SRB | — | 3.0 | — | 2.3 | — | — | — | — | — | — | — | 62.9 |
| Study | Cell type | Cell line | Exposure time (h) | Assay used | IC50 (µM) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L | M | N | Nle | Orn | R‐Phe | S | SPhG | T | V | W | Y | |||||
| Borissow 2007 16 | Breast | T‐47D | 48 | MTT | — | 6.70 | 8.16 | — | — | 5.16 | 2.90 | 7.57 | 4.04 | 7.44 | 9.03 | 31.07 |
| Fu 2008 11 | Breast | MCF7‐CON | 48 | MTT | — | — | — | — | — | — | 23.3 | — | 29.4 | — | — | — |
| Fan 2012 17 | Breast | MCF7‐CON | 72 | SRB | 1.4 | — | — | 8.5 | 4.5 | — | 4.4 | — | 0.97 | 1.9 | — | — |
| Issa 2014 9 | Breast | MCF7‐CON | 72 | MTT | 3.7 | — | — | — | — | — | 1.9 | 2.8 | 2.5 | — | 19.3 | — |
| Hall 2015 8 | Breast | MCF7‐CON | 72 | MTT | — | — | — | — | — | — | 3.4 | — | — | — | — | — |
| Issa 2014 9 | Breast | MCF7‐TXL | 72 | MTT | 5.5 | — | — | — | — | — | 3.6 | 5.2 | 6.1 | — | 73.3 | — |
| Issa 2014 9 | Breast | MCF7‐ETP | 72 | MTT | 6.8 | — | — | — | — | — | 3.6 | 3.5 | 4.8 | — | 50.2 | — |
| Issa 2014 9 | Breast | MCF7‐MITX | 72 | MTT | 8.3 | — | — | — | — | — | 5.2 | 6.5 | 5.6 | — | 75.1 | — |
| Hall 2015 8 | Breast | BT474 | 72 | MTT | — | — | — | — | — | — | 3.1 | — | — | — | — | — |
| Hall 2015 8 | Breast | SKBR3 | 72 | MTT | — | — | — | — | — | — | 3.1 | — | — | — | — | — |
| Borissow 2007 16 | Breast | MDA‐MB‐435 | 48 | MTT | — | 3.57 | 4.01 | — | — | 1.57 | 1.06 | 3.50 | 2.82 | 3.76 | 7.60 | 10.65 |
| Hall 2015 8 | Breast | 231‐CON | 72 | MTT | — | — | — | — | — | — | 2.8 | — | — | — | — | — |
| Hall 2017 7 | Breast | 231‐CON | 72 | MTT | — | — | — | — | — | — | 2.6 | — | — | — | — | — |
| Hall 2017 7 | Breast | 231‐TXL | 72 | MTT | — | — | — | — | — | — | 2.8 | — | — | — | — | — |
| Zheng 2005 15 | Hepatic | HepG2 | 24 | MTT | — | — | — | — | — | — | 9.8 | — | 27.0 | 27.0 | — | — |
| Zheng 2005 15 | Myeloma | IM9 | 24 | MTS | — | — | — | — | — | — | 6.3 | — | 9.1 | 8.2 | — | — |
| Zheng 2005 15 | Lung | H460 | 24 | MTT | — | — | — | — | — | — | 19.2 | — | 19.6 | — | — | — |
| Fu 2008 11 | Lung | A549 | 48 | MTT | — | — | — | — | — | — | 38.4 | — | 47.8 | — | — | — |
| Fu 2008 11 | Cervix | HeLa | 48 | MTT | — | — | — | — | — | — | 24.5 | — | 26.7 | — | — | — |
| Fan 2012 17 | Colon | HCT116 | 72 | SRB | 3.8 | — | — | 14.5 | 5.4 | — | 4.6 | — | 1.3 | 4.4 | — | — |
| Fan 2012 17 | Epithelium | HMEC | 72 | SRB | 2.1 | — | — | 9.2 | 10.2 | — | 4.3 | — | 1.4 | 2.0 | — | — |
Jadomycin IC50 values (µM) determined by MTS, MTT, or SRB assays are compared across studies by cell line. Highlighted values are the lowest jadomycin IC50 values and generally most potent jadomycin derivative for the indicated cell line, as determined by the specified study. IC50, half maximal inhibitory concentration, MTS, phenazine methosulfate, MTT, thiazolyl blue methyltetrazolium bromide, SRB, sulforhodamine B.
3.1.2. MCF7 cells
Most studies on jadomycin cytotoxicity against breast cancer have used the human metastatic, ER+/PR+/HER2‐ breast cancer cell line MCF7, as it is an ideal model for the study of anticancer drug mechanisms and is widely accessible. 27 Fu et al. used thiazolyl blue methyltetrazolium bromide (MTT) colorimetric assays to quantify jadomycin cytotoxicity. 11 The MTTs determined that jadomycin B, S, and T half maximal inhibitory concentration (IC50) values, the concentration of drug that inhibits 50% of cell growth, ranged from 23.3 to 29.4 µM and produced cytotoxic effects in MCF7 cells (Table 2). In comparison, Fan et al. assessed the cytotoxicity of jadomycin Abu, B, Hse, L, Nle, Orn, S, T, and V in MCF7 cells using sulforhodamine B (SRB) assays, which measure cell density. Their results revealed that while jadomycin T had the lowest IC50, jadomycin Abu, B, L, and V all shared similar values (Table 2). 17 , 28
Issa et al. revisited the interactions between jadomycins and MCF7 cells using jadomycin B, DNV, F, L, S, SPhG, T, and W in MCF7 control (MCF7‐CON), paclitaxel‐resistant (MCF7‐TXL), etoposide‐resistant (MCF7‐ETP), and MITX‐resistant (MCF7‐MITX) cells. 9 A corresponding increase of mRNA verified that the MCF7‐TXL, MCF7‐ETP, and MCF7‐MITX cells overexpressed ABCB1, ABCC1, and ABCG2 transporters, respectively. 9 With IC50s ranging from 1.3 to 4.4 µM, jadomycin B, DNV, F, L, S, SPhG, and T were all able to reduce MCF7‐CON cell viability to approximately 0%, while jadomycin W had a higher IC50 of 19 µM and only reduced cell viability to approximately 20% (Table 2). 9 These findings indicate that jadomycin W, which contains a large aromatic side group, has stereochemical hinderance and limited interaction in cancer cells. All jadomycins tested also reduced cell viability in MCF7‐TXL, MCF7‐ETP, and MCF7‐MITX cells with a small decrease in potency, suggesting that their anticancer properties are largely retained in ABC‐overexpressing MDR breast cancer. 9
3.1.3. BT474 and SKBR3 cells
Jadomycin cytotoxicity was tested in the human ER+/PR+/HER2+ BT474 and ER‐/PR‐/HER2+ SKBR3 breast cancer cells lines, and the results were compared to MCF7‐CON and triple‐negative ER‐/PR‐/HER2‐ MDA‐MB‐231 breast cancer cells. 7 , 8 , 26 Jadomycin B, F, and S had similar cytotoxicity in BT474, SKBR3, MDA‐MB‐231, and MCF7‐CON cell lines, with slightly varying potencies (Table 2). 7 , 8 These findings indicate that jadomycin cytotoxicity is likely independent of hormone receptor status.
3.2. Triple‐negative breast cancer
3.2.1. MDA‐MB‐435 cells
Borissow et al. tested jadomycin βala, B, DM, DT, DV, F, G, H, M, N, R‐Phe, S, SPhG, T, V, W, and Y against ER‐/PR‐/HER2‐ MDA‐MB‐435 cancer cells, and the results were very similar to those observed in T‐47D cells (Table 2). 16 Jadomycin DT and S were most potent against MDA‐MB‐435, while jadomycin βala, H, and Y were least potent. Most jadomycins demonstrated greater potency against MDA‐MB‐435 than T‐47D, but jadomycin H was two‐fold more potent in T‐47D cells, suggesting it may interact differently in hormone receptor positive breast cancer. 16 However, some doubt surrounds the authenticity of the MDA‐MB‐435 cell line. At the time of publication, it was believed that MDA‐MB‐435 was a triple‐negative breast cancer cell line, but it has since been proposed that MDA‐MB‐435 cell samples derive from a melanoma cell line. 29 , 30 , 31 As such, these results may give insight into jadomycin cytotoxicity against melanoma, but it is necessary to repeat the experiments in authentic melanoma cells to verify this presumption.
3.2.2. MDA‐MB‐231 cells
Hall et al. continued to explore jadomycin avoidance of ABC transporters in triple‐negative MDA‐MB‐231 control (231‐CON) and MDA‐MB‐231 paclitaxel‐resistant (231‐TXL) breast cancer cells. 7 As expected, triple‐negative breast cancer chemotherapies DOX and MITX were effective against 231‐CON cells (Figure 3A) but were significantly less potent in 231‐TXL cells (Figure 3B). 7 Jadomycins, however, retained their cytotoxic potency in 231‐TXL cells, despite the overexpression of ABCB1 transporters (Table 2; Figure 3B). 7 These findings indicate that jadomycins may be useful as a chemotherapy for MDR breast cancer regardless of ER, PR, or HER2 status. Additionally, this supports that jadomycins should be explored as an alternative or additive to current breast cancer chemotherapies as they retain their efficacy in ABC‐overexpressing breast cancer cells. 7 , 9
FIGURE 3.
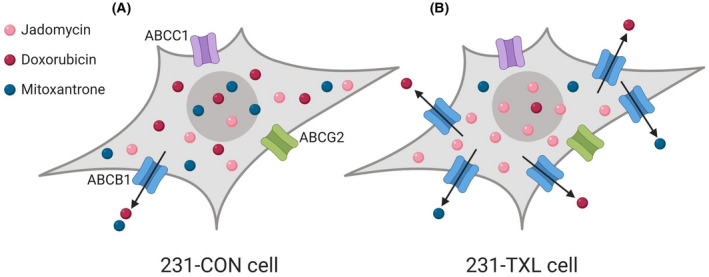
The effects of jadomycins on MDR triple‐negative MDA‐MB‐231 drug sensitive control (231‐CON) and paclitaxel‐resistant (231‐TXL) cells compared to triple‐negative breast cancer doxorubicin (DOX) and mitoxantrone (MITX) chemotherapies. (A) In 231‐CON cells, jadomycins, DOX, and MITX are all effective and cytotoxic against breast cancer cells. Small amounts of DOX and MITX are expelled from the cell through ABC drug efflux transporter ABCB1, but most is retained, and both drugs maintain maximum efficacy. (B) In 231‐TXL cells, there is an upregulation of ABCB1, causing the cell to expel larger amounts of DOX and MITX relative to 231‐CON cells, and the drugs become largely ineffective. However, it has been observed that jadomycins retain their efficacy in 231‐TXL cells as they are not removed through ABC drug efflux transporters. 7 Image created with BioRender.com
4. JADOMYCIN MECHANISMS OF ACTION
Understanding the mechanism(s) of jadomycins is necessary to determine their clinical usefulness against specific types of breast cancer and to predict adverse drug effects and drug–drug interactions. The following sections summarize previously proposed jadomycin mechanisms of action, and the current evidence supporting jadomycin polypharmacology.
4.1. APOPTOSIS
Apoptosis is a process that controls cell death to maintain homeostasis in body tissues and is defective in many cancers through up‐ or downregulation of various genes that result in the unchecked proliferation of cancer cells. 32 , 33 , 34 Evidence from earlier studies of HepG2 hepatic cancer cells, A549 non‐small cell lung carcinoma (NSCLC) cells, and IM‐9 myeloma cells supports that jadomycins activate apoptosis as a mechanism of cancer cell death. 11 , 15 Jadomycin cytotoxicity was also tested in the Bcl‐2 overexpressing myeloma sub‐line IM‐9/Bcl‐2. Bcl‐2 is a protein that regulates apoptosis, and overexpression of Bcl‐2 leads to resistance to standard chemotherapies. Jadomycins were able to overcome Bcl‐2 overexpression, albeit at exceptionally high doses as the cells demonstrated more than two‐fold resistance. 15
Conclusively, apoptosis is also indicated as the main jadomycin‐induced cell death process in breast cancer cells. One study showed that jadomycins B, S, and T caused apoptosis in MCF7 cells. 11 Annexin V/PI staining indicated that jadomycin B, F, and S also caused apoptosis in 231‐CON and 231‐TXL cells. 7 While jadomycins are also cytotoxic against NSCLC H460, cervical HeLa, and colon HCT116 cancer cells, it is unclear if they induce cell death through apoptosis in these cells. Hence, the most basic mechanistic details of jadomycin activity in lung, cervical, and colon cancers remains largely unknown and requires further investigation. Finally, while jadomycins are repeatedly shown to induce cell death through apoptosis, other non‐apoptotic mechanisms of cell death are possible, including mitotic catastrophe or senescence, and may provide more insight into jadomycin cytotoxicity. 35
4.2. CU(II)‐DEPENDANT ROS
Once jadomycin‐induced apoptosis was observed in several cancer cell lines, investigation of the events leading to apoptosis, such as ROS induction, became a primary focus. ROS are highly reactive molecules that contain oxygen and often accumulate in cancer cells, leading to cancer progression and metastasis. 36 , 37 However, chemotherapeutic drugs often induce additional ROS production in cancer cells and cause an accumulation of ROS beyond the tolerable threshold, resulting in cell toxicity, and death. 8 , 37 Thus, depending on the intracellular level, ROS can be either carcinogenic or anticarcinogenic. 36 , 37 In the presence of copper, jadomycins have been shown to increase ROS production and the rate of cell death. 8 Breast cancer cells often have increased intracellular levels of copper compared to normal breast tissue, which may facilitate the higher potency of jadomycins observed in breast cancer cells compared to other cell lines. 38
Researchers have explored the jadomycin‐copper interaction that may propagate chemotherapeutic benefits. In extracellular bacterial supercoiled plasmid models, jadomycin B reduced Cu(II) to Cu(I) and resulted in DNA strand scission through the production of hydrogen peroxide (H2O2) and hydroxyl radicals. 12 When 100 mM of copper chelator EDTA was added to the jadomycin B treatment no DNA damage was observed, suggesting that the presence of copper ions is necessary for jadomycin‐induced DNA cleavage. 12 H2O2 is also a necessity as treatment with catalase, a catalytic enzyme that reduces H2O2, resulted in total inhibition of DNA scission. 12 Still, it is important to note that these plasmids contained high guanine‐cytosine content and the results of this study may be slightly exaggerated since guanine is more susceptible to oxidative stress, which can cause DNA strand breaks. 12 , 39
Though the studies Monro et al. performed were impressive, they were limited to the observation of ROS effects in non‐cellular assays. 12 Therefore, it was important to determine if jadomycins would also increase ROS in breast cancer cells and lead to cytotoxicity. CM‐DCFH2‐DA assays, used to measure ROS production in cells, revealed that CuSO4 cotreated with jadomycin B, F, S, or SPhG in MCF7 cells increased ROS production and decreased cell viability. 8 CuSO4 treatment alone increased ROS but produced no cytotoxic effects, confirming an interaction between Cu and jadomycins is required for the observed increase in cell death. 8 Furthermore, the Cu(II)‐chelator D‐penicillamine cotreated with jadomycin B, F, S, or SPhG in MCF7 cells reduced ROS production and jadomycin cytotoxicity, supporting the hypothesis that copper is either required for or supports the enhancement of jadomycin cytotoxicity. 8
To further investigate jadomycin ROS production and dependency, Hall et al. cotreated cells with antioxidants or inhibitors of cellular antioxidant pathways. 8 When MCF7 cells were cotreated with antioxidant N‐acetyl cysteine (NAC) and jadomycin B, F, S, or SPhG, decreased ROS production and cytotoxic potency was observed for each jadomycin. 8 , 40 NAC treatment affected the cytotoxicity of jadomycin B, F, and S more than jadomycin SPhG, suggesting that ROS‐mediated toxicity depends on the specific jadomycin. Overall, these results indicated that jadomycin cytotoxicity is reduced when ROS production is hindered. However, increased doses of jadomycin B, F, S, or SPhG were still able to reduce the number of viable cells to less than 5%, even in the presence of NAC. 8 It is possible that higher doses of jadomycin exceed the protective effect of NAC or stimulate a ROS‐independent cytotoxic mechanism.
When MCF7‐CON cells were cotreated with jadomycin B, F, S, or SPhG and auranofin, a prooxidant thioredoxin reductase (TrxR) inhibitor, ROS activity and jadomycin cytotoxicity significantly increased. 8 , 41 Similar results were observed when the cells were cotreated with sodium diethyldithiocarbamate (DDC), a superoxide dismutase I inhibitor that blocks conversion of superoxide to H2O2. 8 However, when MCF7 cells were cotreated with jadomycin and MitoTEMPO, a superoxide dismutase II inhibitor, there was no change in ROS activity or cell viability. This may indicate that jadomycins increase ROS concentrations in the cytosol of MCF7 cells rather than in the mitochondria, the usual location of superoxide dismutase II. Therefore, jadomycins might have an advantage over DOX, which has an affinity for mitochondria‐rich heart cells and causes cardiotoxicity. 8 , 36 Interestingly, the glutathione S‐transferase inhibitor ellagic acid (EA) did not affect ROS activity but still enhanced the cytotoxicity of jadomycin S and SPhG, again suggesting that some aspects of jadomycin cytotoxicity may be ROS‐independent. 8 Also, in the presence of jadomycin B, MCF7‐CON cells significantly increased expression of the antioxidant gene TrxR1. 8 This may be a cellular response to increased ROS, and lead to the activation of the Prx/Trx pathway to remove ROS and enhance survival. These findings are consistent with the earlier discussed experiment involving increased ROS activity during cotreatment with the TrxR inhibitor auranofin and jadomycin B. 8
When CM‐DCFH2‐DA assays confirmed that jadomycins increase intracellular ROS activity in both 231‐CON and 231‐TXL cells, Hall et al. investigated if accumulation of double‐strand DNA (dsDNA) breaks occurred as a downstream result of ROS activation. 7 , 42 To do this Hall et al. measured nucleus‐contained phosphorylated histone H2AX (γH2AX) protein using western blots to identify dsDNA breaks in both 231‐CON and 231‐TXL cells. 7 Jadomycin B, F, S, and MITX all increased γH2AX protein expression in 231‐CON cells. As a representative jadomycin, jadomycin S significantly increased γH2AX protein expression in 231‐TXL cells while MITX lost its effect, a finding consistent with MITX being an ABCB1 substrate and jadomycins avoiding efflux transporters. 7 Initially, most of these results appeared to indicate that jadomycins primarily cause dsDNA breaks and apoptosis through ROS‐dependent pathways. However, when jadomycin F or S was cotreated with NAC, ROS decreased significantly in 231‐CON cells, but there was no change in dsDNA damage. 7 Likewise, when jadomycin F or S was cotreated with auranofin, ROS increased significantly but did not affect dsDNA damage. 7 These results suggest that while copper‐dependent ROS production augments jadomycin cytotoxicity, it is not the primary driving factor. 7 , 8 It is also possible that such results are specific to certain breast cancer cell types.
4.3. ABK INHIBITION
While jadomycin‐induction of ROS was being explored, jadomycins were also found to inhibit genes and proteins like ABK, which participates in chromatid separation and cytokinesis in eukaryotic mitosis. 43 Inhibition of ABK causes cells to cease rapid proliferation. Furthermore, overexpression of ABK is common in hepatic, lung, and breast cancers, leading to increased proliferation. 44 , 45 , 46 Hence, ABK may be an ideal anticancer target.
In 2008, Fu et al. investigated jadomycin B cytotoxic mechanisms in budding yeast cells containing the IPL1 gene that encodes for Ipl1 aurora kinase, an analog of human ABK. 11 Since the amino acid residues surrounding the ATP‐binding site of Ipl1 kinase and ABK are nearly identical, the binding pocket where the ABK‐competitive inhibitor hesperadin interaction was modeled and used as a virtual screening tool, which identified jadomycin B as a potential ABK inhibitor. 11 Supporting the molecular docking prediction, jadomycin B was shown to competitively inhibit purified ABK in the presence of various concentrations of ATP. Two yeast strains were then used to compare the activity of potential ABK inhibitors: wild‐type yeast, and ipl1‐321 temperature‐sensitive yeast, the latter of which expresses compromised Ipl1 kinase activity and was expected to have increased sensitivity to ABK inhibitors. 11 Both strains were treated with 10 μg/ml jadomycin B, which decreased the percentage of ipl1‐321 mutant growth to approximately 20% but had little to no effect on wild‐type growth. 11 Interestingly, jadomycin S and T demonstrated no growth inhibition against either yeast strain, and did not inhibit purified ABK, reinforcing the idea that jadomycin structure dictates its pharmacological interactions with ABK. 11
Although these findings initially supported the idea that jadomycin B acts as an ABK inhibitor, there were several outstanding questions. First, much of the research was conducted based on the assumption that Ipl1 kinase and ABK are homologues. While this is true, the researchers reached this conclusion based on the small ATP‐binding site amino acid sequence rather than protein homology analysis, as the crystal structure of Ipl1 kinase was unavailable. 11 , 47 Whereas it was acknowledged that jadomycin B may interact with other aurora kinases, the possibility of jadomycin B interacting with other non‐mitotic proteins that could lead to downstream cascades and inhibit ABK was not considered. The anticancer effects of jadomycin S and T also appeared to occur through a pathway outside of ABK inhibition and suggested that other cytotoxic mechanisms of action must exist. Nonetheless, the overall results of jadomycin B as a potential ABK inhibitor were impressive and became a viable area of research.
Since ABK is also known to phosphorylate histone H3 within cells, Fu et al. used western blots to compare the amount of phosphorylated H3 (pHis3) in untreated versus jadomycin B‐treated NSCLC A549 cells. 11 While the untreated cells showed a clear gel band indicating pHis3, pHis3 was undetectable in cells treated with 10 μg/ml of jadomycin B, indirectly demonstrating that jadomycin B blocks ABK activity. A dose‐dependent decrease in pHis3 was also observed in jadomycin B‐treated hepatic HepG2 and cervical HeLa cells. 11 Later, Issa et al. revealed that jadomycin B similarly inhibited pHis3 in both MCF7‐CON and MCF7‐TXL cells in a dose‐dependent manner, with a slightly reduced effect in MCF7‐MITX cells. 9 This finding suggested that the overexpression of ABCB1 and ABCG2 transporters does not interfere with ABK inhibition. 9 Further research demonstrated that ABK inhibition proceeds through a ROS‐independent mechanism, as jadomycin B‐mediated reduction in pHis3 and cytotoxicity was retained when ROS were inhibited, though with decreased potency. 8 , 9 While there is evidence supporting direct jadomycin B inhibition of ABK, this may not be the primary mechanism that causes dsDNA damage. DNA damage leads to poly(ADP‐ribosyl)ation of ABK, and subsequently inhibits ABK activity. 7 , 48 Thus, ABK inhibition is no longer viewed as a direct mechanism of action, but rather as a consequence of other disrupted cellular processes.
Our previously unpublished data further supports the idea that ABK inhibition is not a direct mechanism of action. A direct, extracellular kinase activity assay of ABK in the presence of jadomycin B, F, S, or vehicle control was conducted using the ADP‐Glo™ Kinase Assay (Promega) and ABK Enzyme System (Promega), as per the manufacturer's instructions. Only 25–30% inhibitory effect was observed for 50 µM jadomycin B, F, and S in these in vitro kinase assays (Figure 4). The concentration of jadomycin B, F, and S was 10 to 50‐fold higher than the IC50s in breast cancer cells (Table 2). Thus, jadomycin‐mediated reductions in pHis3 likely occur as a consequence of the dsDNA damage response rather than through direct ABK inhibition and new jadomycin mechanisms have been proposed to induce DNA damage, namely, topoisomerase II inhibition.
FIGURE 4.
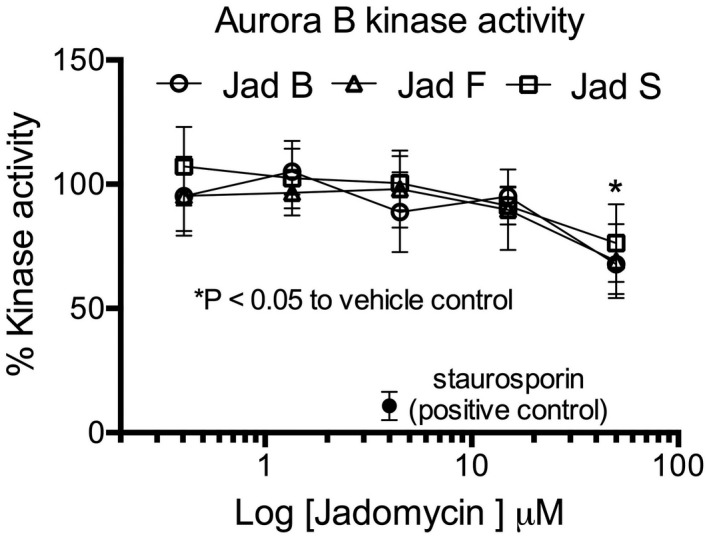
Jadomycin B, F, and S inhibit aurora B kinase (ABK) activity in vitro. Staurosporin is included as a positive control of ABK activity inhibition. Data presented are mean ± SEM with an n = 3. *p < .05, where indicated concentration of each jadomycin inhibited the % kinase activity significantly compared to vehicle control as determined by a one‐way analysis of variance with Tukey's post hoc test
4.4. TOPOISOMERASE II INHIBITION
Inhibition of topoisomerases was originally proposed as a potential mechanism based on jadomycin structural similarity to the chemotherapy DOX and the antibiotic family of fluoroquinolones, as these drugs inhibit topoisomerases to hinder cancer and bacteria growth, respectively. 12 , 19 , 49 Topoisomerases assist in DNA replication and transcription through cleavage and realignment of positive and negative supercoiled DNA. Inhibition of topoisomerase activity can lead to DNA damage and apoptosis. 7 , 50 There are two isoforms of topoisomerase II, topoisomerase IIα, and topoisomerase IIβ, which aid in the replication of cells and neural development. 51
Martinez‐Farina et al. performed experiments that provided initial support for topoisomerase II as a jadomycin target. 19 Using Water‐Ligand Observed via Gradient SpectroscopY (WaterLOGSY), a technique used to analyze the binding affinity between enzymes and substrates, jadomycin DS was found to loosely bind human topoisomerase IIβ while jadomycin LN exhibited no binding interaction. 19 These findings suggested that jadomycins have the ability to bind to topoisomerase IIβ, but the binding relies on jadomycin structure. 19 Although this study provided the first insight into jadomycin‐topoisomerase interactions, the results were somewhat limited since jadomycin DS interaction with topoisomerase IIα and other jadomycins with more established cytotoxicity profiles were not investigated.
Hall et al. conducted a series of experiments in 231‐CON breast cancer cells to further explore the topoisomerase mechanism. 7 The expression of TOP2A and TOP2B genes, which encode for topoisomerase IIα and topoisomerase IIβ, respectively, were significantly diminished in 231‐CON cells treated with jadomycin B, F, and S versus the vehicle control. 7 Jadomycin S also significantly decreased expression of the TOP1 gene encoding for topoisomerase I, but jadomycin B and F did not. 7 Further investigation through western blots revealed that the topoisomerase II inhibitor MITX, and jadomycin B, F, and S, all significantly lowered the expression of topoisomerase IIα protein compared to the vehicle control in 231‐CON cells. 7 In 231‐TXL cells, jadomycin S significantly lowered expression of topoisomerase IIα protein but MITX did not. 7 Again, this verified that jadomycin S pharmacological activity was retained within the ABC‐overexpressing 231‐TXL cells, and was not affected by the drug efflux mechanism through which MITX was removed. 7 These data suggest that jadomycins could reduce topoisomerase II function through transcription and translational mechanisms.
DNA decatenation assays demonstrated that jadomycins directly inhibited topoisomerase IIα and IIβ isoforms in a dose‐dependent manner. 7 The next step was to determine whether jadomycins were topoisomerase II poisons or catalytic inhibitors. Topoisomerase II poisons complex with the enzyme to directly cause dsDNA breaks, while catalytic inhibitors reduce topoisomerase activity to break dsDNA indirectly. 52 Jadomycins B, S, and F similarly inhibited topoisomerase IIα and IIβ plasmid decatenation. 7 Jadomycin F was the most potent and Jadomycin B was the least potent. 7 All three jadomycins were significantly less potent than DOX, suggesting they are weak and non‐selective catalytic inhibitors of topoisomerase IIα and IIβ. In comparison jadomycin B and F but not S increased topoisomerase IIβ‐mediated cleavage of supercoiled plasmid DNA in a manner consistent with the activity of a topoisomerase poison, albeit at low potency. 7 , 53 Jadomycin B, F, and S did not display activity consistent with topoisomerase IIα poisoning. The nature of the selective interaction of jadomycin B and F with topoisomerase IIβ should be further explored as it could pose some concern for cardiotoxicity at higher concentrations that will need to be assessed through in vivo studies.
In summary, these findings show that jadomycins induce DNA cleavage, apoptosis, and cytotoxicity in MDR breast cancer cells through the reduction of Cu(II), production of ROS, and/or the inhibition of topoisomerase II. 7 , 8 , 12 These events are hypothesized to inhibit ABK downstream, leading to cessation of mitosis and cell proliferation, and ultimately result in breast cancer cell death (Figure 5). 7 , 9 , 11
FIGURE 5.
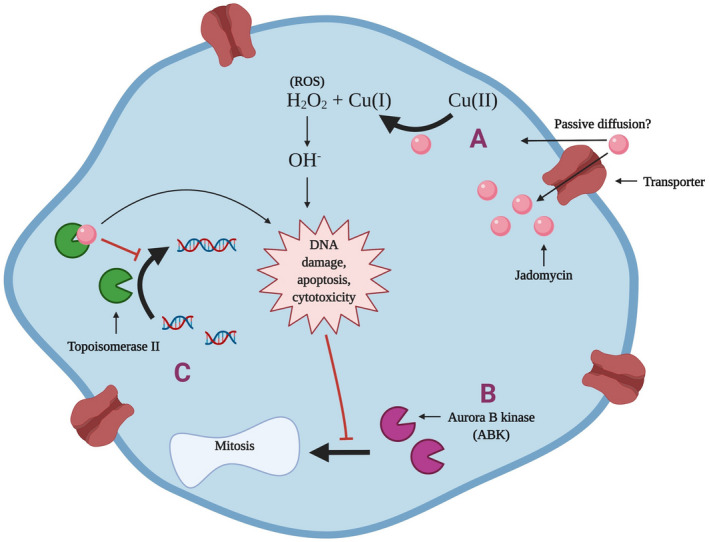
Jadomycin proposed mechanisms of action in breast cancer cells. (A) Some jadomycins are copper‐dependent and convert Cu(II) to Cu(I), leading to the production of ROS and subsequent DNA damage. 8 , 12 (B) The DNA damage induced by jadomycins inhibits ABK and prevents mitosis and proliferation of breast cancer cells. 7 (C) Jadomycins B, S, and F have also demonstrated the ability to inhibit or poison topoisomerase II, which leads to further DNA damage and apoptosis. 7 Image created with BioRender.com
5. LIMITATIONS AND FUTURE DIRECTIONS FOR JADOMYCIN BREAST CANCER CELL CYTOTOXICITY STUDIES
In cancer research, the study of chemotherapeutic effects in noncancerous cells and benign tumors is often excluded. This is partially due to the additional costs that such experiments would involve as well as possible redundancy, since animal models usually reveal the level of toxicity to noncancerous body cells and organ systems. It is important to note that although studies conducted thus far have verified jadomycin cytotoxicity and efficacy in MDR breast cancer cells in vitro, the effects of jadomycins in noncancerous cells remains largely unknown. Only a single study has investigated a noncancerous cell line and determined the IC50s of several jadomycins in human microvascular epithelial cells (HMEC) in vitro. 17 Interestingly, jadomycin Orn exhibited an appreciably higher IC50 in noncancerous cells as compared to MCF7 and HCT116 cells (Table 2). This difference suggests that jadomycins can possess selectivity against different cellular targets, but it will be important for future research to establish the distribution of jadomycins to tissues and measure biomarkers of toxicity in in vivo models.
There is also some variability in the reported jadomycin IC50 values from different studies (Table 2). There could be a number of explanations for this, including methodological differences, such as the exposure time and type of assay used, or jadomycin purity and stability. Overall, however, IC50 values in the literature suggest that jadomycin B, F, S, and T have very similar cytotoxic profiles against cancer cells. Therefore, while different amino acids may alter jadomycin functionality, they do not always cause a significant change in potency and efficacy. As jadomycin B, F, S, and T appear to be the most cytotoxic, future development of new jadomycins should be based on these structures, with slight alterations to reduce adverse effects or improve pharmacological parameters if needed.
Moreover, while numerous studies have provided insight into jadomycin structure, chemical synthesis, and cytotoxicity, the exact cascade of events and specific cytotoxic mechanisms have yet to be determined. Research to date has focused mainly on intracellular jadomycin mechanisms and their ability to evade drug efflux transporters. Jadomycin cellular entry is an important area to explore as it will broaden the understanding of jadomycin mechanisms and allow researchers to investigate jadomycin metabolism and possible pharmacogenetic variations.
Although drug efflux is a common mechanism of MDR, the reduced uptake of transport‐mediated anticancer drugs is also a major threat. Decreased uptake limits chemotherapy‐induced cytotoxic DNA damage, apoptosis, and alteration of the cell cycle. 54 Specifically, the role of the solute carrier transporter family should be investigated as these transporters mediate the uptake of several anticancer drugs. 55 Within the family of solute carrier transporters, the organic anion transporter polypeptides OATP1B3, OATP3A1, and OATP4A1 are highly expressed in breast cancers and represents possible mechanisms for jadomycin uptake. 56 , 57 , 58 OATP1B3 is of particular interest as it is responsible for the uptake of chemotherapies TXL, docetaxel, SN‐38, methotrexate, and imatinib. 56 MDR breast cancers can express different transporter phenotypes and are essential to explore as patients may have diverse pharmacokinetic responses to jadomycins. 56
Ideally, we should also broaden our knowledge of less‐studied jadomycins, and explore other pathways that involve jadomycins. For example, a recent study indicated that jadomycin Orn may induce more ROS production and be more efficacious than jadomycin B. 59 Additional mechanisms such as the possibility of the Prx/Trx and GST/GPx antioxidant systems primarily reducing jadomycin‐induced H2O2 have been proposed, but not yet explored. 8 While in vitro cell experiments are ideal for identifying initial evidence of cytotoxicity and mechanistic details, in vivo animal models are needed to observe and predict additional medical benefits, toxicities, and adverse effects. For example, as DOX is a known topoisomerase II poison and can pose serious cardiotoxic effects, jadomycins may be a desired chemotherapy alternative if they are shown to be less cardiotoxic. 60 , 61
While the potential role of jadomycins in the treatment of MDR breast cancers is being explored, the question of how cancer cells may develop resistance to jadomycins remains. An important addition to the literature would be the development of a resistance model, to establish what changes confer a selective advantage against jadomycin treatment as this may further inform on mechanism of action.
Jadomycin pharmacogenomics is another area of interest. The MCF7, BT474, SKBR3, and MDA‐MB‐231 breast cancer cell lines are all derived from the breast tissues of Caucasian women, with the exception of T‐47D cells for which the ethnic origins are unknown. 62 , 63 , 64 , 65 As Gleason et al. have observed, certain breast cancer phenotypes determined through the presence and/or absence of ER and PR are more prevalent in certain ethnicities. 66 Black women, regardless of age, tend to have higher rates of ER‐ and PR‐ breast cancers than White women, which reduces their therapeutic options. 66 Evidence suggests that women of Chinese ethnic origins may also have a number of genetic differences in breast cancer phenotype, compared to other ethnicities, that could influence clinical care. 67
Furthermore, jadomycins may exhibit anticancer activity through other factors related to ethnicity, such as the inhibition of p53 or Ki‐67 protein expression, which are responsible for initiating DNA‐damaged cell death and can be used to measure cell growth rate, respectively. 68 Notably, the expression of Ki‐67 and p53 proteins is often upregulated in triple‐negative breast cancer, and the P53 gene encoding for p53 has increased expression in 30% of breast cancers. 68 Significantly higher expression of both Ki‐67 and p53 proteins are seen in African‐American women versus Caucasian women. 69 Therefore, it would be ideal to investigate the potency of jadomycins in breast cancer cell lines of various ethnic origins to compare results. This could help determine if jadomycins have an unrecognized benefit in the treatment of patients with different ethnic backgrounds.
6. CONCLUSIONS
In this review, we have presented the key findings that support jadomycins as a potential chemotherapy for MDR metastatic breast cancer. Jadomycins are repeatedly shown to avoid efflux from the overexpression of ABC‐transporters and are equally cytotoxic against a variety of breast cancer cell lines regardless of hormone receptor status, which may give them an additional advantage to hormone‐restricted treatment options. 7 Conclusively, jadomycin B, S, and F are demonstrated to be the most potent jadomycins, and often mediate cancer cell death through the induction of apoptosis and DNA strand breaks. 7 , 11 , 15 There are several jadomycin mechanisms of action that may mediate cancer cell death, including copper‐dependant ROS generation, ABK inhibition, and topoisomerase II inhibition. 7 , 8 , 11 , 12 Other mechanisms that remain to be explored include jadomycin interactions with cellular uptake transporters, jadomycin resistance, and jadomycin effects on the Prx/Trx and GST/GPx antioxidant systems and on the expression of Ki‐67 and p53 proteins, which may provide additional pharmacogenomic benefits for certain patients. 8 , 68 , 69 The summarized findings present jadomycins as a potential candidate for MDR breast cancer chemotherapy, and we strongly encourage further preclinical research in this area which may identify new and effective breast cancer treatments options.
7. NOMENCLATURE OF TARGETS AND LIGANDS
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019). 70
DISCLOSURE
There are no conflicts of interest to report by any author.
AUTHOR CONTRIBUTIONS
Esther Bonitto developed the literature search strategy, completed the literature search, drafted the review article, and finalized the revisions. Brendan McKeown assisted with the literature search and drafting the review article, and reviewed and edited the final version. Kerry Goralski chose the topic for the review, provided supervisory support during the literature search and manuscript drafting phases, and edited and revised the drafted review article.
ACKNOWLEDGEMENTS
This work was supported by a research grant from the Dalhousie Pharmacy Endowment. E.P.B. is supported by funds from the Natural Sciences and Engineering Research Council of Canada Undergraduate Student Research Award (NSERC USRA), and the Pharmacology Summer Research Award, Department of Pharmacology, Dalhousie University. B.T.M. is a trainee in the Cancer Research Training Program of the Beatrice Hunter Cancer Research Institute, with funds provided by the Terry Fox Research Institute. B.T.M. is supported by funds from the Natural Sciences and Engineering Research Council of Canada (Create grant number 510963). The authors would like to thank Dr. Jaime Wertman for critical reading and editing of the manuscript and Matthew Ladda for technical contributions to the ABK activity assay.
Bonitto EP, McKeown BT, Goralski KB. Jadomycins: a potential chemotherapy for multi‐drug resistant metastatic breast cancer. Pharmacol Res Perspect. 2021;9:e00886. doi: 10.1002/prp2.886
Contributor Information
Esther P. Bonitto, Email: esther.bonitto@dal.ca.
Kerry B. Goralski, Email: kerry.goralski@dal.ca.
DATA AVAILABILITY STATEMENT
The new data that support the findings of this study (Figure 4) are available from the corresponding author Dr. Kerry Goralski upon reasonable request (kerry.goralski@dal.ca).
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941‐1953. [DOI] [PubMed] [Google Scholar]
- 3. Ha R, Chow D, Mango V, Friedlander L, Desperito E, Wynn R. Have we given up on breast cancer metastasis? Global trends in breast cancer metastasis research productivity. Breast J. 2015;21(4):442‐444. [DOI] [PubMed] [Google Scholar]
- 4. Mouttet D, Laé M, Caly M, et al. Estrogen‐receptor, progesterone‐receptor and HER2 status determination in invasive breast cancer. Concordance between immuno‐histochemistry and MapQuant™ microarray based assay. PLoS One. 2016;11(2):e0146474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marquette C, Nabell L. Chemotherapy‐resistant metastatic breast cancer. Curr Treat Options Oncol. 2012;13(2):263‐275. [DOI] [PubMed] [Google Scholar]
- 6. Ayer SW, McInnes AG, Thibault P, et al. Jadomycin, a novel 8H‐benz[b]oxazolo[3,2‐f]phenanthridine antibiotic from from streptomyces venezuelae ISP5230. Tetrahedron Lett. 1991;32(44):6301‐6304. [Google Scholar]
- 7. Hall SR, Toulany J, Bennett LG, et al. Jadomycins inhibit type II topoisomerases and promote DNA damage and apoptosis in multidrug‐resistant triple‐negative breast cancer cells. J Pharmacol Exp Ther. 2017;363(2):196‐210. [DOI] [PubMed] [Google Scholar]
- 8. Hall SR, Blundon HL, Ladda MA, et al. Jadomycin breast cancer cytotoxicity is mediated by a copper‐dependent, reactive oxygen species‐inducing mechanism. Pharmacol Res Perspect. 2015;3(2):e00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Issa ME, Hall SR, Dupuis SN, Graham CL, Jakeman DL, Goralski KB. Jadomycins are cytotoxic to ABCB1‐, ABCC1‐, and ABCG2‐overexpressing MCF7 breast cancer cells. Anticancer Drugs. 2014;25(3):255‐269. [DOI] [PubMed] [Google Scholar]
- 10. MacLeod JM, Forget SM, Jakeman DL. The expansive library of jadomycins. Can J Chem. 2018;96(6):495‐501. [Google Scholar]
- 11. Fu D‐H, Jiang W, Zheng J‐T, et al. Jadomycin B, an Aurora‐B kinase inhibitor discovered through virtual screening. Mol Cancer Ther. 2008;7(8):2386‐2393. [DOI] [PubMed] [Google Scholar]
- 12. Monro SMA, Cottreau KM, Spencer C, et al. Copper‐mediated nuclease activity of jadomycin B. Bioorg Med Chem. 2011;19(11):3357‐3360. [DOI] [PubMed] [Google Scholar]
- 13. Chater KF. Recent advances in understanding Streptomyces. F1000Res. 2016;5:2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jakeman DL, Bandi S, Graham CL, Reid TR, Wentzell JR, Douglas SE. Antimicrobial activities of jadomycin B and structurally related analogues. Antimicrob Agents Chemother. 2009;53(3):1245‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zheng J‐T, Rix U, Zhao L, et al. Cytotoxic activities of new jadomycin derivatives. J Antibiot. 2005;58(6):405‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Borissow CN, Graham CL, Syvitski RT, Reid TR, Blay J, Jakeman DL. Stereochemical integrity of oxazolone ring‐containing jadomycins. ChemBioChem. 2007;8(10):1198‐1203. [DOI] [PubMed] [Google Scholar]
- 17. Fan K, Zhang X, Liu H, et al. Evaluation of the cytotoxic activity of new jadomycin derivatives reveals the potential to improve its selectivity against tumor cells. J Antibiot. 2012;65:449‐452. [DOI] [PubMed] [Google Scholar]
- 18. de Koning CB, Ngwira KJ, Rousseau AL. Biosynthesis, synthetic studies, and biological activities of the jadomycin alkaloids and related analogues. Alkaloids Chem Biol. 2020;84:125‐199. [DOI] [PubMed] [Google Scholar]
- 19. Martinez‐Farina CF, McCormick N, Robertson AW, et al. Investigations into the binding of jadomycin DS to human topoisomerase IIβ by WaterLOGSY NMR spectroscopy. Org Biomol Chem. 2015;13(41):10324‐10327. [DOI] [PubMed] [Google Scholar]
- 20. Dupuis SN, Veinot T, Monro SMA, et al. Jadomycins derived from the assimilation and incorporation of norvaline and norleucine. J Nat Prod. 2011;74(11):2420‐2424. [DOI] [PubMed] [Google Scholar]
- 21. Robertson AW, MacLeod JM, MacIntyre LW, et al. Post polyketide synthase carbon‐carbon bond formation in type‐II PKS‐derived natural products from Streptomyces venezuelae . J Org Chem. 2018;83(4):1876‐1890. [DOI] [PubMed] [Google Scholar]
- 22. Fewer DP, Metsä‐Ketelä M. A pharmaceutical model for the molecular evolution of microbial natural products. Febs J. 2020;287(7):1429‐1449. [DOI] [PubMed] [Google Scholar]
- 23. Dupuis SN, Robertson AW, Veinot T, et al. Synthetic diversification of natural products: semi‐synthesis and evaluation of triazole jadomycins. Chem Sci. 2012;3(5):1640‐1644. [Google Scholar]
- 24. Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. Revisiting the role of ABC transporters in multidrug‐resistant cancer. Nat Rev Cancer. 2018;18(7):452‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szakács G, Paterson JK, Ludwig JA, Booth‐Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5(3):219‐234. [DOI] [PubMed] [Google Scholar]
- 26. Dai X, Cheng H, Bai Z, Li J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J Cancer. 2017;8(16):3131‐3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Comşa Ş, Cîmpean AM, Raica M. The story of MCF‐7 breast cancer cell line: 40 years of experience in research. Anticancer Res. 2015;35(6):3147‐3154. [PubMed] [Google Scholar]
- 28. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1(3):1112‐1116. [DOI] [PubMed] [Google Scholar]
- 29. Rae JM, Creighton CJ, Meck JM, Haddad BR, Johnson MD. MDA‐MB‐435 cells are derived from M14 melanoma cells–a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res Treat. 2007;104(1):13‐19. [DOI] [PubMed] [Google Scholar]
- 30. Chambers AF. MDA‐MB‐435 and M14 cell lines: identical but not M14 melanoma? Can Res. 2009;69(13):5292–5293. [DOI] [PubMed] [Google Scholar]
- 31. Korch C, Hall EM, Dirks WG, et al. Authentication of M14 melanoma cell line proves misidentification of MDA‐MB‐435 breast cancer cell line. Int J Cancer. 2018;142(3):561‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mourtada‐Maarabouni M, Pickard MR, Hedge VL, Farzaneh F, Williams GT. GAS5, a non‐protein‐coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene. 2009;28(2):195‐208. [DOI] [PubMed] [Google Scholar]
- 33. Oskouian B, Sooriyakumaran P, Borowsky AD, et al. Sphingosine‐1‐phosphate lyase potentiates apoptosis via p53‐ and p38‐dependent pathways and is down‐regulated in colon cancer. Proc Natl Acad Sci. 2006;103(46):17384‐17389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu L, Yu X, Guo X, et al. miR‐143 is downregulated in cervical cancer and promotes apoptosis and inhibits tumor formation by targeting Bcl‐2. Mol Med Rep. 2012;5(3):753‐760. [DOI] [PubMed] [Google Scholar]
- 35. Ricci MS, Zong WX. Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 2006;11(4):342‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Deavall DG, Martin EA, Horner JM, Roberts R. Drug‐induced oxidative stress and toxicity. J Toxicol. 2012;2012:645460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther. 2008;7(12):1875‐1884. [DOI] [PubMed] [Google Scholar]
- 38. Karginova O, Weekley CM, Raoul A, et al. Inhibition of copper transport induces apoptosis in triple‐negative breast cancer cells and suppresses tumor angiogenesis. Mol Cancer Ther. 2019;18(5):873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Joshi PC, Li HH, Merchant M, Keane TC. Total inhibition of 1O2‐induced oxidative damage to guanine bases of DNA/RNA by turmeric extracts. Biochem Biophys Res Comm. 2014;452(3):515‐519. [DOI] [PubMed] [Google Scholar]
- 40. Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N‐acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther. 2008;8(12):1955‐1962. [DOI] [PubMed] [Google Scholar]
- 41. Liu C, Liu Z, Li M, et al. Enhancement of auranofin‐induced apoptosis in MCF‐7 human breast cells by selenocystine, a synergistic inhibitor of thioredoxin reductase. PLoS One. 2013;8(1):e53945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khanna KK, Jackson SP. DNA double‐strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27(3):247‐254. [DOI] [PubMed] [Google Scholar]
- 43. Lee M, Kim IS, Park KC, Kim J‐S, Baek SH, Kim KI. Mitosis‐specific phosphorylation of Mis18α by Aurora B kinase enhances kinetochore recruitment of polo‐like kinase 1. Oncotarget. 2018;9(2):1563‐1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin Z‐Z, Jeng Y‐M, Hu F‐C, et al. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC Cancer. 2010;10(1):461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vischioni B, Oudejans JJ, Vos W, Rodriguez JA, Giaccone G. Frequent overexpression of aurora B kinase, a novel drug target, in non–small cell lung carcinoma patients. Mol Cancer Ther. 2006;5(11):2905. [DOI] [PubMed] [Google Scholar]
- 46. Zhang Y, Jiang C, Li H, et al. Elevated Aurora B expression contributes to chemoresistance and poor prognosis in breast cancer. Int J Clin Exp Pathol. 2015;8(1):751‐757. [PMC free article] [PubMed] [Google Scholar]
- 47. Vieillemard A, Prouzet‐Mauléon V, Hugues M, et al. The Saccharomyces cerevisiae RhoGAP Rgd1 is phosphorylated by the Aurora B like kinase Ipl1. Biochem Biophys Res Commun. 2013;433(1):1‐5. [DOI] [PubMed] [Google Scholar]
- 48. Monaco L, Kolthur‐Seetharam U, Loury R, Murcia J M‐D, de Murcia G, Sassone‐Corsi P. Inhibition of Aurora‐B kinase activity by poly(ADP‐ribosyl)ation in response to DNA damage. Proc Natl Acad Sci. 2005;102(40):14244‐14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ehrmann E, Jolivet‐Gougeon A, Bonnaure‐Mallet M, Fosse T. Role of DNA gyrase and topoisomerase IV mutations in fluoroquinolone resistance of Capnocytophaga spp. clinical isolates and laboratory mutants. J Antimicrob Chemother. 2017;72(8):2208‐2212. [DOI] [PubMed] [Google Scholar]
- 50. Liu Z, Deibler RW, Chan HS, Zechiedrich L. The why and how of DNA unlinking. Nucleic Acids Res. 2009;37(3):661‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jadhav AK, Karuppayil SM. Molecular docking studies on thirteen fluoroquinolines with human topoisomerase II a and b. In Silico Pharmacol. 2017;5(4):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee JH, Wendorff TJ, Berger JM. Resveratrol: a novel type of topoisomerase II inhibitor. J Biol Chem. 2017;292(51):21011‐21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gao R, Schellenberg MJ, Huang S‐Y, et al. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2·DNA and Top2·RNA covalent complexes by tyrosyl‐DNA‐phosphodiesterase 2 (TDP2). J Biol Chem. 2014;289(26):17960‐17969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Okabe M, Szakacs G, Reimers MA, et al. Profiling SLCO and SLC22 genes in the NCI‐60 cancer cell lines to identify drug uptake transporters. Mol Cancer Ther. 2008;7(9):3081‐3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hagenbuch B, Stieger B. The SLCO (former SLC21) superfamily of transporters. Mol Aspects Med. 2013;34(2‐3):396‐412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nakanishi T, Tamai I. Solute carrier transporters as targets for drug delivery and pharmacological intervention for chemotherapy. J Pharm Sci. 2011;100(9):3731‐3750. [DOI] [PubMed] [Google Scholar]
- 57. Nozawa T, Suzuki M, Takahashi K, et al. Involvement of estrone‐3‐sulfate transporters in proliferation of hormone‐dependent breast cancer cells. J Pharmacol Exp Ther. 2004;311(3):1032‐1037. [DOI] [PubMed] [Google Scholar]
- 58. Nozawa T, Suzuki M, Yabuuchi H, Irokawa M, Tsuji A, Tamai I. Suppression of cell proliferation by inhibition of estrone‐3‐sulfate transporter in estrogen‐dependent breast cancer cells. Pharm Res. 2005;22(10):1634‐1641. [DOI] [PubMed] [Google Scholar]
- 59. Forget SM, Robertson AW, Hall SR, et al. Isolation of a jadomycin incorporating l‐ornithine, analysis of antimicrobial activity and jadomycin reactive oxygen species (ROS) generation in MDA‐MB‐231 breast cancer cells. J Antibiot. 2018;71(8):722‐730. [DOI] [PubMed] [Google Scholar]
- 60. Ashley N, Poulton J. Anticancer DNA intercalators cause p53‐dependent mitochondrial DNA nucleoid re‐modelling. Oncogene. 2009;28(44):3880‐3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Prathumsap N, Shinlapawittayatorn K, Chattipakorn SC, Chattipakorn N. Effects of doxorubicin on the heart: from molecular mechanisms to intervention strategies. Eur J Pharmacol. 2020;866:172818. [DOI] [PubMed] [Google Scholar]
- 62. Cailleau R, Olivé M, Cruciger QVJ. Long‐term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro. 1978;14(11):911‐915. [DOI] [PubMed] [Google Scholar]
- 63. Kaushik V, Azad N, Yakisich JS, Iyer AKV. Antitumor effects of naturally occurring cardiac glycosides convallatoxin and peruvoside on human ER+ and triple‐negative breast cancers. Cell Death Discov. 2017;3:17009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee AV, Oesterreich S, Davidson NE. MCF‐7 cells–changing the course of breast cancer research and care for 45 years. J Natl Cancer Inst. 2015;107(7):1‐4. [DOI] [PubMed] [Google Scholar]
- 65. Kaplan MH, Contreras‐Galindo R, Jiagge E, et al. Is the HERV‐K HML‐2 Xq21.33, an endogenous retrovirus mutated by gene conversion of chromosome X in a subset of African populations, associated with human breast cancer? Infect Agent Cancer. 2020;15:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gleason MX, Mdzinarishvili T, Sherman S. Breast cancer incidence in black and white women stratified by estrogen and progesterone receptor statuses. PLoS One. 2012;7(11):e49359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen M, Xu R, Turner JW, Warhol M, August P, Lee P. Race and the molecular origins of breast cancer in Chinese women: breast cancer in Chinese women. Ann Surg Oncol. 2012;19(13):4085‐4093. [DOI] [PubMed] [Google Scholar]
- 68. Pan Y, Yuan Y, Liu G, Wei Y. P53 and Ki‐67 as prognostic markers in triple‐negative breast cancer patients. PLoS One. 2017;12(2):e0172324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Morris GJ, Naidu S, Topham AK, et al. Differences in breast carcinoma characteristics in newly diagnosed African‐American and Caucasian patients: a single‐institution compilation compared with the National Cancer Institute's Surveillance, Epidemiology, and End Results database. Cancer. 2007;110(4):876‐884. [DOI] [PubMed] [Google Scholar]
- 70. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2019: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46:D1091‐D1106. doi: 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The new data that support the findings of this study (Figure 4) are available from the corresponding author Dr. Kerry Goralski upon reasonable request (kerry.goralski@dal.ca).