

Summary
Cytotoxic natural killer cells kill tumors and infected cells. We carried out CRISPR-based gene editing and transcriptional regulation in hard-to-manipulate NK-92 cells. NK-92-based therapies were found to be safe and efficacious in preclinical studies of cancers. Here, we have pioneered the generation and validation of NK-92 cells constitutively expressing Cas9 or dCas9 for knockout (CRISPRko), transcriptional activation (CRISPRa), or transcriptional repression (CRISPRi) of genes. Our CRISPR-engineered NK-92 cell platforms can be modified for research and off-the-shelf therapeutic applications.
Subject areas: Cell Biology, Cancer, Immunology, Molecular Biology, CRISPR
Graphical abstract
Highlights
-
•
Constitutively repressing or activating genes in human NK cells is difficult
-
•
CRISPR-engineered natural killer cells are attractive for therapeutic applications
-
•
Our approach generates stable CRISPRko, CRISPRi, and CRISPRa human NK cell lines
Cytotoxic natural killer cells kill tumors and infected cells. We carried out CRISPR-based gene editing and transcriptional regulation in hard-to-manipulate NK-92 cells. NK-92-based therapies were found to be safe and efficacious in preclinical studies of cancers. Here, we have pioneered the generation and validation of NK-92 cells constitutively expressing Cas9 or dCas9 for knockout (CRISPRko), transcriptional activation (CRISPRa), or transcriptional repression (CRISPRi) of genes. Our CRISPR-engineered NK-92 cell platforms can be modified for research and off-the-shelf therapeutic applications.
Before you begin
Cell culture
Timing: ∼ 10–15 days for NK-92 cells; ∼ 3 days for K-562 cells; ∼ 3–6 days for HEK cells
-
1.
Thaw NK-92 natural killer (NK), chronic myeloid leukemia K-562, and Lenti-X293T human embryonic kidney (HEK) cell lines at 37°C in a metallic bead bath. Refer to materials and equipment section for details of NK-92, K-562, and HEK cell culture growth media.
-
2.
Transfer thawed NK-92 and K-562 cells into two separate 15 mL tubes containing 10 mL each of complete RPMI media. Transfer HEK cells into a single 15 mL tube containing 10 mL complete DMEM medium.
-
3.
Pellet down the cells by centrifugation at 300 × g for 5 min at room temperature (RT; 18°C–22°C) and aspirate the supernatant.
-
4.
Wash the NK-92, K-562, and HEK cells again with 10 mL of their respective media for completely removing DMSO (300 × g, 5 min at RT).
-
5.Resuspend NK-92 cells in 1 mL of complete RPMI media, count the cells, and seed 0.5–1 × 105 cells/200 μL/well in a 96-well plate (flat bottom) in NK cell media (complete RPMI + rhIL-2) at 37°C.
-
a.When cells become confluent (media color changes to light yellow), spin down the cells (300 × g at RT for 5 min) and aspirate the media.
-
b.Resuspend the cells in 500 μL of fresh NK cell media and culture them in a 48-well plate at 37°C.
-
c.Wait until cells become confluent, then spin down, aspirate the media and transfer these cells to a T-25 flask containing 5 mL of fresh NK cell media.
-
d.After expansion in a T-25 flask for 3 days, cells will be ready to be used in transduction experiments.
-
a.
CRITICAL: For successful NK-92 cell transduction, use cells that have been cultured for at least 7 days after thawing. Once thawed, we do not recommend culturing NK-92 cells beyond 3 months.
-
6.
Resuspend K-562 cells in 1 mL of complete RPMI media, count the cells, and seed ∼ 1 × 106 cells in 5 mL complete RPMI media in a T-25 culture flask. After 3 days of culture in T-25 flask, cells will be ready to be used as targets in NK cell cytotoxicity experiments.
-
7.
Resuspend HEK cells in 1 mL of complete DMEM media, count the cells, and seed ∼5 × 105 cells in a 150 mm culture dish in 15 mL complete DMEM.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC anti-human CD132 (IL2Rγ); working dilution 1:100 | BioLegend | Cat# 338607, RRID: AB_2123585 |
| Bacterial and virus strains | ||
| NEB 5-alpha competent E. coli | New England Biolabs | Cat# C2987U |
| Critical commercial assays | ||
| GeneJET Gel Extraction Kit | Thermo Fisher Scientific | Cat# K0691 |
| GeneJET Plasmid Miniprep Kit | Thermo Fisher Scientific | Cat# K0502 |
| CellTrace Violet Cell Proliferation Kit | Thermo Fisher Scientific | Cat# C34557 |
| NucleoSpin RNA Purification Kit | MACHEREY-NAGEL | Cat# 740955.250 |
| NucleoBond Xtra Maxi Plus EF | MACHEREY-NAGEL | Cat# 740426.50 |
| Lenti-X Concentrator | Takara Bio | Cat# 631231 |
| SuperScript IV First Strand Synthesis System | Thermo Fisher Scientific | Cat# 18091050 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s phosphate-buffered saline (DPBS), pH 7.4 without Ca2+ and Mg2+ | Thermo Fisher Scientific | Cat# 14190144 |
| Recombinant human interleukin-2 (rhIL-2) | PeproTech | Cat# 200-02 |
| Poly-L-lysine | R&D Systems | Cat# 3438-100-01 |
| Polybrene | Millipore Sigma | Cat# TR-1003-G |
| Sodium butyrate | Fisher Scientific | Cat# AAA1107922 |
| Sodium azide | Sigma-Aldrich | Cat# S2002-100G |
| Puromycin | Thermo Fisher Scientific | Cat# A1113803 |
| Trypsin/EDTA Solution (TE) | Thermo Fisher Scientific | Cat# R001100 |
| LB Agar, Miller (Pre-Buffered Capsules) | Fisher Scientific | Cat# BP9734-500 |
| Ethanol 70% mol. Grade | Fisher Scientific | Cat# BP8201500 |
| Glycerol | Fisher Scientific | Cat# BP2291 |
| Ampicillin | Fisher Scientific | Cat# BP1760-5 |
| Certified Agarose Powder | Bio-Rad | Cat# 1613100 |
| 4,6-Diamidino-2-phenylindole (DAPI); working dilution 1:5000 | BioLegend | Cat# 422801 |
| 7-AAD Viability Staining Solution (200 tests) | BioLegend | Cat# 420403 |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15140122 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | Cat# 21985023 |
| MEM Non-essential Amino Acids (100X) | Thermo Fisher Scientific | Cat# 11140050 |
| GlutaMAX (100X) | Thermo Fisher Scientific | Cat# 35050061 |
| Sodium pyruvate | Thermo Fisher Scientific | Cat# 11360070 |
| Fetal Bovine Serum (FBS) | Thermo Fisher Scientific | Cat# 26140079 |
| DMEM (Dulbecco's Modified Eagle Medium) | Thermo Fisher Scientific | Cat# 11965126 |
| RPMI (Roswell Park Memorial Institute)-1640 | Thermo Fisher Scientific | Cat# 11875119 |
| SOS medium | New England Biolabs | Cat# B9020S |
| Terrific Broth (granulated) | Fisher Scientific | Cat# BP9728-500 |
| PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | Cat# A25742 |
| BsmBI-v2 | New England Biolabs | Cat# R0739S |
| CutSmart buffer (10X) | New England Biolabs | Cat# B7204S |
| T4 DNA ligase | New England Biolabs | Cat# M0202S |
| 50X TAE Buffer | Bio-Rad | Cat# 1610743 |
| 1 kb Plus DNA Ladder | New England Biolabs | Cat# N3232S |
| Quick-Load Purple 1 kb DNA Ladder | New England Biolabs | Cat# N0552S |
| SYBR Green Nucleic Acid Gel Stain (10000X) | Thermo Fisher Scientific | Cat# S7563 |
| Gel Loading Dye, Purple (6X), no SDS | New England Biolabs | Cat# B7025S |
| HyPure Molecular-Grade Water | Cytiva Lifesciences | Cat# SH30538.03 |
| Experimental models: cell lines | ||
| NK-92 cell line | ATCC | Cat# CRL-2407 |
| Lenti-X 293T cell line | Takara Bio | Cat# 632180 |
| K-562 cell line | ATCC | Cat# CCL-243 |
| Recombinant DNA | ||
| Lenti Cas9-EGFP | Addgene | Cat# 63592, RRID: Addgene_63592 |
| pCAG dCas9-KRAB-2A-EGFP | Addgene | Cat# 92396, RRID: Addgene_92396 |
| dCAS9-VP64-GFP | Addgene | Cat# 61422, RRID: Addgene_61422 |
| ipUSEPR | From Chun-Wei Chen | House-made plasmid (Yang et al., 2021) |
| pMD.2G | Addgene | Cat# 12259, RRID: Addgene_12259 |
| psPAX2 | Addgene | Cat# 12260, RRID: Addgene_12260 |
| Oligonucleotides (5’→3′) | ||
| Control (Scramble) sgRNA (forward); CRISPRko/i/a | Derived from (Wang et al., 2014); ordered form Integrated DNA Technologies (IDT) | GTAGCGAACGTGTCCGGCGT |
| Control (Scramble) sgRNA (reverse) | Reverse complement of control sgRNA (forward); CRISPRko/i/a, (https://www.bioinformatics.org/sms/rev_comp.html) | ACGCCGGACACGTTCGCTAC |
| Red fluorescent protein (RFP) sgRNA (forward); CRISPRko | From Chun-Wei Chen Lab | GTCACCACATACGAAGACGG |
| RFP sgRNA (reverse) | Reverse complement of RFP sgRNA (forward); CRISPRko, (https://www.bioinformatics.org/sms/rev_comp.html) | CCGTCTTCGTATGTGGTGAC |
| IL2RG sgRNA (1) (forward); CRISPRko | https://portals.broadinstitute.org/gppx/crispick/public | GGTGCAGTACCGGACTGACT |
| IL2RG sgRNA (1) (reverse); CRISPRko | Reverse complement of IL2RG sgRNA (1) (forward); CRISPRko, (https://www.bioinformatics.org/sms/rev_comp.html) | AGTCAGTCCGGTACTGCACC |
| IL2RG sgRNA (2) (forward); CRISPRko | https://portals.broadinstitute.org/gppx/crispick/public | AGCCGCTTTAACCCACTCTG |
| IL2RG sgRNA (2) (reverse); CRISPRko | Reverse complement of IL2RG sgRNA (2) (forward); CRISPRko, (https://www.bioinformatics.org/sms/rev_comp.html) | CAGAGTGGGTTAAAGCGGCT |
| IL2RG sgRNA (1) (forward); CRISPRi | https://portals.broadinstitute.org/gppx/crispick/public | TGGTAATGATGGCTTCAACA |
| IL2RG sgRNA (1) (reverse); CRISPRi | Reverse complement of IL2RG sgRNA (1) (forward); CRISPRi, (https://www.bioinformatics.org/sms/rev_comp.html) | TGTTGAAGCCATCATTACCA |
| IL2RG sgRNA (2) (forward); CRISPRi | https://portals.broadinstitute.org/gppx/crispick/public | AGGGATGTGAATGGTAATGA |
| IL2RG sgRNA (2) (reverse); CRISPRi | Reverse complement of IL2RG sgRNA (2) (forward); CRISPRi, (https://www.bioinformatics.org/sms/rev_comp.html) | TCATTACCATTCACATCCCT |
| IL-15 sgRNA (forward); CRISPRa | https://portals.broadinstitute.org/gppx/crispick/public | ACACCTCCCGCGGAGACTGG |
| IL-15 sgRNA (reverse); CRISPRa | Reverse complement of IL-15 sgRNA (forward); CRISPRa, (https://www.bioinformatics.org/sms/rev_comp.html) | CCAGTCTCCGCGGGAGGTGT |
| GAPDH (forward); qPCR | Derived from (Kooreman et al., 2017) | GTCTCCTCTGACTTCAACAGCG |
| GAPDH (reverse); qPCR | Derived from (Kooreman et al., 2017) | ACCACCCTGTTGCTGTAGCCAA |
| IL-15 (forward); qPCR | Derived from (Kooreman et al., 2017) | AACAGAAGCCAACTGGGTGAATG |
| IL-15 (reverse); qPCR | Derived from (Kooreman et al., 2017) | CTCCAAGAGAAAGCACTTCATTGC |
| Software and algorithms | ||
| FlowJo 10.7.1 software | FlowJo (Becton Dickinson) | https://www.flowjo.com/ |
| sgRNA Designer, CRISPick | Broad Institute | https://portals.broadinstitute.org/gppx/crispick/public |
| Other | ||
| 150 mm Surface-treated tissue culture dishes | Fisher Scientific | Cat# FB012925 |
| Steriflip-HV Sterile Centrifuge Tube Top Filter Unit | Millipore Sigma | Cat# SE1M003M00 |
| Fisherbrand Disposable PES filter Units (500 mL) | Fisher Scientific | Cat# FB12566504 |
| Olympus 384-well PCR plate | Genesee Scientific | Cat# 24-305 |
| 96-Well Cell Culture Plates (flat bottom) | Genesee Scientific | Cat# 25-109 |
| 96-Well Cell Culture Plates (U bottom) | Genesee Scientific | Cat# 25-221 |
| 48-Well Cell Culture Plates | Genesee Scientific | Cat# 25-108 |
| 24-Well Cell Culture Plates | Genesee Scientific | Cat# 25-107 |
| TC Treated Flasks, 25 mL, Vent (T-25) | Genesee Scientific | Cat# 25-205 |
| 0.2 mL 8-Strip PCR Tubes, Natural | Genesee Scientific | Cat# 27-125 |
| 50 mL Conical centrifuge tubes | Genesee Scientific | Cat# 28-106 |
| 15 mL Conical centrifuge tubes | Genesee Scientific | Cat# 28-103 |
| FACS tubes (Falcon round-bottom polypropylene tubes) | Fisher Scientific | Cat# 14-959AA |
| 14 mL Polypropylene round-bottom tubes with caps | Corning Life Sciences | Cat# 352059 |
| 1.7 mL Microtubes | Genesee Scientific | Cat# 22-281 |
| 0.6 mL Microtubes | Genesee Scientific | Cat# 24-272 |
| 1000 μL Pipette tips | Genesee Scientific | Cat# 24-830 |
| 200 μL Pipette tips | Genesee Scientific | Cat# 24-812 |
| 20 μL Pipette tips | Genesee Scientific | Cat# 24-804 |
| 10 μL Pipette tips | Genesee Scientific | Cat# 24-801 |
| 200 μL Round gel loading tips, 0.57 mm | Genesee Scientific | Cat# 24-113 |
| FACSymphonyTM A5 flow cytometer | Becton Dickinson (BD) | n/a |
| ProFlex PCR System | Thermo Fisher Scientific | n/a |
| NanoDropTM One Spectrophotometer | Thermo Fisher Scientific | n/a |
Materials and equipment
Growth media for NK-92 cell line
| Reagent | Final concentration |
|---|---|
| RPMI-1640 | N/A |
| FBS | 10% (v/v) |
| Penicillin-Streptomycin (100X) | 1% (v/v) |
| Sodium pyruvate (100 mM) | 1% (v/v) |
| Minimum Essential Media (MEM) non-essential amino acid (100X) | 1% (v/v) |
| GlutaMAX (100X) | 1% (v/v) |
| 2-Mercaptoethanol (50 mM) | 0.01% (v/v) |
Note: Mix all components and filter the media using sterile disposable vacuum filter with polyethersulfone (PES) membrane. This is complete RPMI media, to grow NK-92 cells, supplement complete media with recombinant human (rh) IL-2 (100 U/mL). Store the media at 4°C for up to 1 month.
Growth media for Lenti-X 293T cell line
| Reagent | Final concentration |
|---|---|
| DMEM | N/A |
| FBS | 10% (v/v) |
| Penicillin-Streptomycin (100X) | 1% (v/v) |
| Sodium pyruvate (100 mM) | 1% (v/v) |
| MEM non-essential amino acid (100X) | 1% (v/v) |
| GlutaMAX (100X) | 1% (v/v) |
Note: Mix all these components and filter the media using sterile disposable vacuum filter with PES membrane. This is complete DMEM media. Store the media at 4°C for up to 1 month.
Growth media for K-562 cell line
| Reagent | Final concentration |
|---|---|
| RPMI-1640 | N/A |
| FBS | 10% (v/v) |
| Penicillin-Streptomycin (100X) | 1% (v/v) |
| Sodium pyruvate (100 mM) | 1% (v/v) |
| Minimum Essential Media (MEM) non-essential amino acid (100X) | 1% (v/v) |
| GlutaMAX (100X) | 1% (v/v) |
| 2-Mercaptoethanol (50 mM) | 0.01% (v/v) |
Note: K-562 cell line is grown in complete RPMI media described above. Mix all above components and filter the media using sterile disposable vacuum filter with PES membrane. Store the media at 4°C for up to 1 month.
Terrific Broth (TB) media (1 L)
| Component | Amount |
|---|---|
| Terrific Broth powder | 47.6 g |
| Glycerol | 4 mL |
| Double distilled (dd) H2O | Up to 1000 mL |
| Ampicillin antibiotic | 100 mg |
Note: Dissolve 47.6 g TB powder in 500 mL double distilled (dd) H2O and add 4 mL glycerol. Mix well and makeup the final media volume to 1 L with ddH2O and autoclave for 15 min at 121°C. Cool down to RT and add ampicillin antibiotic (working concentration is 100 μg/mL). Store the media at 4°C for up to 1 month.
Flow cytometry
Flow cytometry analysis was performed on BD FACSymphonyTM A5 flow cytometer and data were analyzed using FlowJo 10.7.1 software.
| Reagent | Final concentration |
|---|---|
| Anti-human IL2Rγ (APC) | 1:100 |
| 4,6-diamidino-2-phenylindole (DAPI) (Stock 5 mg/mL) | 1:5000 |
| Fluorescence activated cell sorting (FACS) Buffer (Ca2+ and Mg2+ free DPBS + 2% FBS + 0.09% sodium azide) | N/A |
Fluorescence activated cell sorting (FACS) Buffer
| Component | Final concentration | Amount |
|---|---|---|
| Ca2+ and Mg2+ free DPBS | N/A | 490 mL |
| FBS | 2% (v/v) | 10 mL |
| Sodium azide | 0.09% (w/v) | 450 mg |
Note: Store the buffer at 4°C for up to 6 months.
Primers used in qPCR
| Name | Forward sequence | Reverse sequence |
|---|---|---|
| GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA |
| IL-15 | AACAGAAGCCAACTGGGTGAATG | CTCCAAGAGAAAGCACTTCATTGC |
Step-by-step method details
Lentivirus production
-
1.
On day 1, coat a 150 mm culture dish with ∼3–5 mL of poly-L lysine solution (0.01%) by gently swirling to ensure even coating of the plate surface. Aspirate the solution and let the coated plate dry for 15 min at RT.
-
2.
Aspirate the cell culture media from Lenti-X 293T HEK cells grown in a 150 mm culture dish, wash these cells with PBS, add 2 mL of 1X Trypsin solution to the plate. Gently swirl the plate to evenly cover the whole surface area and incubate the plate for 1 min at 37°C. Detach the cells by gently tapping the plate and inactivate the Trypsin enzyme by adding 10 mL of complete DMEM media. Spin them down at 350 × g for 5 min.
-
3.
Resuspend the cells in 10 mL of complete DMEM media, count the cells, and seed 7–10 × 106 Lenti-X 293T HEK cells on the poly-L lysine-coated 150 mm plate. Make up the total cell culture media volume to 20 mL by adding complete DMEM media, gently swirl the plate so that cells are uniformly seeded and incubate them at 37°C.
-
4.On day 2, take two 15 mL tubes, add 4 mL of plain DMEM media (media without FBS and PS and containing only 1X GlutaMAX (glutamine), non-essential amino acids and sodium pyruvate) in each tube. In the first tube, add 20 μg of the plasmid containing the gene of interest (here, Lenti-Cas9-EGFP or dCas9-VP64-GFP or dCas9-KRAB-GFP), 20 μg of packaging plasmid (psPAX2), and 5 μg of envelope plasmid (pMD.2G). In the second tube, add 80 μL of lipofectamine 2000. Incubate both tubes at RT for 5 min.CRITICAL: Use plain DMEM i.e. serum-free DMEM, as serum inhibits transfection.
-
a.Mix lipofectamine-containing media with media containing the lentiviral vectors by gentle swirling or inverting and incubate the mix for 20 min at RT.
-
b.After incubation, aspirate the culture media from Lenti-X 293T HEK cells and add the lipofectamine and plasmid mix to these cells in a dropwise fashion over the entire plate. Make up the total media volume to 15 mL by including additional 7 mL of plain DMEM to Lenti-X293T HEK cells. Gently swirl the plate and incubate at 37°C for 3 h.
-
c.After incubation, add 15 mL complete FBS-containing DMEM media to the plate, making the total media volume to 30 mL. Incubate overnight (12–16 h) at 37°C.
-
a.
-
5.
On day 3, aspirate the 30 mL of media from culture dish and add 20 mL of fresh complete DMEM media containing 400 μL of 500 mM sodium butyrate. Then, incubate for 5 h at 37°C to enhance the lentiviral production. After sodium butyrate induction, discard the media, replenish the cells with 16 mL fresh complete DMEM media, and incubate at 37°C for 24 h.
-
6.On day 4, filter the media containing lentivirus using a sterile disposable vacuum filter unit (0.45 μm).Alternatives: Clarification of media can also be done with centrifugation (400 × g for 5 min).
-
a.Concentrate lentivirus by adding 4 mL of lenti-X concentrator to 16 mL of viral supernatant, mixing well by pipetting, and incubating overnight (12–16 h) at 4°C.
-
a.
-
7.
On day 5, centrifuge the mix at 1500 × g for 45 min, 4°C. After centrifugation, an off-white pellet will form at the bottom of centrifugation tube, without disturbing the pellet, aspirate the supernatant by suction using a glass Pasteur pipette. Finally, resuspend the off-white pellet in 1 mL of complete RPMI media.
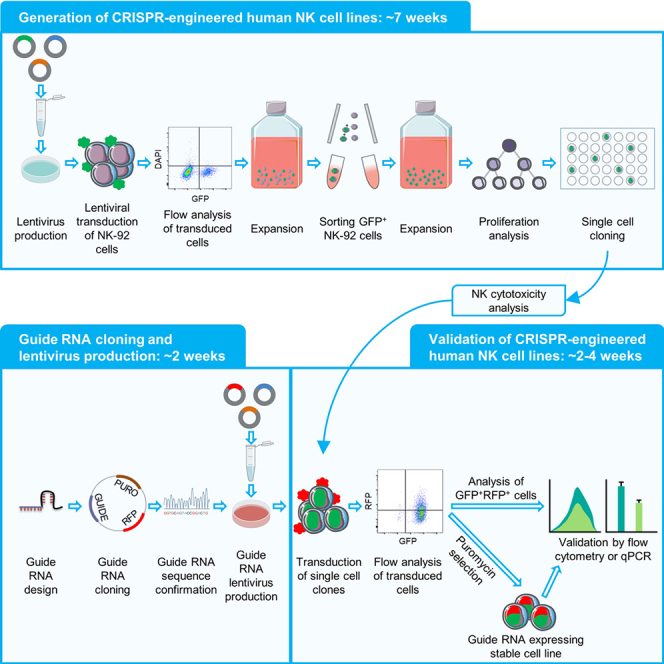
Designing and cloning single guide RNAs (sgRNAs) for CRISPR-mediated gene editing and transcriptional modulation
-
8.
Oligo design for sgRNA production. Go to https://portals.broadinstitute.org/gppx/crispick/public Broad institute web portal (Doench et al., 2016) .
-
9.
Select the reference genome (here, Human GRCh37) and then select the mechanism of CRISPR editing (CRISPRko or CRISPRa or CRISPRi). Enzyme will be SpyoCas9.
-
10.
Select quick look up in the target section and enter the target gene I.D. (e.g., IL2RG for IL2Rγ).
-
11.
Enter the value in CRISPICK Quota box (this value represents how many sgRNA per gene will be designed) and submit.
-
12.
Download the results by selecting picking result file (tab-limited text file).
-
13.
Open the file in excel, this file contains the summary statistics for on- and off-target scores by pick order. Choose the guide RNA sequence-based on pick order rank (choose minimum 2–5 sgRNAs per gene). Also check the orientation of the sgRNA (sense or antisense).
-
14.
Design the reverse complement of the sgRNA sequence.
-
15.Add the following overhang to the sgRNA sequence which will facilitate its ligation to linearized ipUSEPR plasmid
-
a.Forward oligo: 5′ CACCG…guide RNA 3′
-
b.Reverse oligo: 5′ AAAC…guide RNA…C 3′
-
a.
-
16.
Order the oligos from Integrated DNA Technologies (IDT) Inc.
-
17.
Annealing. Thaw and spin down the oligo vial at 300 × g for 2 min at RT and reconstitute the oligos in nuclease-free water to make a 100 μM stock (the amount of water added into the oligo vial to make the 100 μM stock = 10X number of nanomoles of oligo).
-
18.
Mix 10 μL of the forward oligo (100 μM) + 10 μL of the reverse oligo in a PCR tube and spin down at 300 × g for 2 min.
Use a thermocycler to anneal forward and reverse oligos:-
a.95°C 10 min × 1 cycle
-
b.Ramp down temperature from 95°C to 25°C, decrease rate = 0.5°C /1min × 140 cycles (total time = 140 min)
-
c.Hold at 4°C.
-
a.
-
19.
Take 1 μL of annealed oligo from step 18, add into 2 mL of ddH2O to reach a final dilution of 1/2000. These diluted oligos will be used as the insert for ligation.
Linearizing ipUSEPR plasmid for ligation
-
20.
Digestion of ipUSEPR plasmid. Prepare the digestion mixture in 0.6 mL microtubes as follows:
| Reagent | Volume/Conc |
|---|---|
| ipUSEPR plasmid | 40 μg |
| Cutsmart 10X (Cat# B7204S) | 40 μL |
| BsmBI-v2 (Cat# R0739S) | 16 μL |
| Rnase free water | Fill up to 400 μL |
-
21.
Incubate the mixture overnight (12–16 h) at 55°C in a heat block.
-
22.
Isolate the linearized plasmid by electrophoresis (0.8% agarose gel, 120 V for 2 h) followed by gel extraction.
-
23.
Add 80 μL of 6X loading dye into 400 μL BsmbI digestion and load 50 μL in each well (Marker: NEB# N0552S, Gel loading dye: NEB# B7025S).
-
24.Gel extraction (GeneJET Gel Extraction kit # K0691):
-
a.Weigh empty Eppendorf tubes and clip the expected band from gel into Eppendorf tubes.
-
b.Purify the linearized ipUSEPR plasmid as per manufacturer’s instructions and after elution, measure DNA concentration using a NanoDropTM One spectrophotometer and store the linearized plasmid at −20°C.
-
a.
Ligation
-
25.
Prepare ligation mixture in 0.2 mL PCR tubes as follows:
| Reagents | Volume |
|---|---|
| Linearized ipUSEPR plasmid (60–100 ng/μL) | 1.5 μL |
| 10X T4 ligase buffer (supplied with T4 DNA ligase) | 0.6 μL |
| T4 DNA ligase (NEB # M0202S) | 0.3 μL |
| Master Mix (all above three products) | 2.4 μL |
| 1/2000 diluted oligo (remember adding H2O control) | 3.6 μL |
| Total | 6 μL |
-
26.Ligate using a thermocycler using following conditions:
-
a.25°C 1 h × 1 cycle
-
b.16°C 12 h × 1 cycle
-
c.4°C hold
-
a.
-
27.
Store ligation product at −20°C until transformation
Transformation (NEB-5 alpha competent E. coli# C2987U)
-
28.
Thaw NEB 5-alpha competent E. coli cells on ice (50 μL/tube), and bring Ampicillin plates to RT.
-
29.
Take 10 μL of competent cells and mix it with 6 μL of the ligated product in a 0.6 mL microtube and then, incubate on ice for 30 min.
-
30.
Heat shock at 42°C for 30s in a heat block and put back on ice for 5 min.
-
31.
Add 180 μL of SOC medium and grow the bacteria at 37°C in a shaker incubator for 1 h.
-
32.
Spread all the medium onto the Ampicillin plate evenly and incubate the plates upside down at 37°C.
-
33.
After 12–16 h, store the plates at 4°C until miniprep.
Miniprep (GeneJET Plasmid Miniprep Kit # K0502)
-
34.
Prepare 4 mL TB with Ampicillin in a 14 mL tube, pick a single colony with the help of a pipette tip from the bacterial plate into each tube, and shake overnight (12–16 h) at 37°C.
-
35.
Take 1.8 mL bacteria into a 2 mL tube, and centrifuge at 11000 × g for 30s at RT to pellet down the bacteria.
-
36.
Prepare the miniprep of plasmid DNA as per manufacturer’s instructions.
-
37.
In the final elution step, use 50 μL of elution buffer to elute the plasmid DNA from the column and measure concentration of the plasmid and store at −20°C.
-
38.
Send 8 μL of 100–200 ng/μL DNA and 5 μL of 5 μM hU6 primer: “GAGGGCCTATTTCCCATGAT” per sample to Eton Biosciences for sequencing to confirm the sgRNA sequence.
Maxiprep (NucleoBond Xtra Maxi Plus EF # 740426.50)
-
39.
Take 4 mL of TB medium in a 14 mL tube and inoculate with a single colony picked from ampicillin plate. Incubate at 37°C in a shaker incubator at 250 RPM for 5–6 h.
-
40.
Add ∼1 mL of the inoculated media to 250 mL of TB media and grow the culture overnight (12–16 h) at 37°C in a shaker incubator at 250 RPM.
-
41.
Harvest bacterial cells by centrifuging at 6000 × g for 15 min at 4°C. Then discard the supernatant.
-
42.
Isolate the plasmid DNA as per manufacturer’s instructions. In the final elution step, use 400 μL of elution buffer to elute plasmid DNA from NucleoBond finalizer (provided in the kit).
-
43.
Measure plasmid DNA concentration and store at −20°C.
Significance of genetically manipulating natural killer cells using CRISPR
Natural killer (NK) cells are innate immune cells that protect against viral infections (Strauss-Albee and Blish, 2016) and development of cancers (Cerwenka and Lanier, 2001). NK cell-mediated immune surveillance is defective in infections and cancers for various reasons including NK cell exhaustion, defective homeostasis, and impaired NK cytotoxicity (Duault et al., 2021; Wilk et al., 2021). Our overall goal is to develop therapeutic NK cells which are functional and evade exhaustion through CRISPR-based genetic manipulation. To this end, we manipulated NK-92 cells , one of the platforms for NK therapies in clinical trials (Klingemann et al., 2016), using CRISPRko/Cas9 for knockout (Jinek et al., 2012), CRISPRi/dCas9-KRAB for transcriptional repression (Gilbert et al., 2013), and CRISPRa/dCas9-VP64 for transcriptional activation (Chavez et al., 2015) of genes that promote NK cytotoxicity and reduce NK exhaustion.
Below, we detail the protocols for generation of each of these multifunctional CRISPR-based NK cell platforms.
Generating Cas9+ NK-92 cells (CRISPRko NK-92 platform)
-
44.
Count NK-92 cells, resuspend ∼0.5–1.5 × 106 cells in 500 μL of complete RPMI media, and add 500 μL of concentrated Cas9-EGFP lentivirus (concentrated from 16 mL of lentiviral supernatant to 1 mL using lenti-X concentrator). Now the total volume will be 1 mL; to this, add polybrene (10 μg/mL) and rhIL-2 (100 U/mL). Seed these cells in a 24-well plate (1.5 × 106/mL/well) and incubate at 37°C for 48 h.
-
45.
On day 2, wash the cells with complete RPMI media, and replenish the NK-92 cells with NK cell media (RPMI media + rhIL-2). Pipette a small aliquot of the transduced cells (∼100 μL) into a 1.7 mL tube to measure percentages of EGFP+ Cas9-transduced NK-92 cells by flow cytometry (Figure 1A upper panel).
-
46.
Expand the Cas9-transduced NK-92 cells for the next 8–10 days by changing the NK cell media and re-plating the cells on every third day. Then, pipette a small aliquot of cells (∼0.5–1 × 106) and stain them with DAPI and check the percentages of DAPI−Cas9-EGFP+ viable cells (Figure 1A lower panel). This step is to ensure that transduction with Cas9 does not reduce the viability of NK-92 cells. We observed that NK cell viability was similar in untransduced and Cas9-transduced NK-92 cells (Figure 1A).
-
47.After expansion, stain the cells with DAPI and sort DAPI−EGFP+ cells. Check the post-sort purity by flow cytometry (Figure 1B, Day 0). Expand the sorted cells as described below:
-
a.Count the sorted cells and seed ∼ 0.5–1 × 105 cells/well in a 96-well flat bottom plate.
-
b.Expand these cells in NK media at 37°C. When cells become confluent (media color changes to light yellow), spin down the cells (300 × g at RT for 5 min) and aspirate the media.
-
c.Resuspend the cells in fresh 500 μL NK cell media and culture them in a 48 well plate at 37°C.
-
d.Wait until the cells become confluent, spin down, aspirate the media and transfer these cells to T-25 flask containing 5 mL of fresh NK cell media.
-
e.After expansion of NK-92 cells in a T-25 flask for 3 days, once again conduct flow cytometry to ensure that only EGFP+ cells are expanding (Figure 1B, Day 10). Please note that cells from a single well of 96-well plate will be transferred to single well of 48-well plate and then to a T-25 flask.
-
a.
-
48.Compare the proliferation potential of transduced NK-92 cells with that of untransduced NK-92 cells to ensure that transduction with Cas9 does not reduce NK-92 cell proliferation:
-
a.Reconstitute a vial of CFSE-violet dye (Thermo Scientific# C34557) in DMSO and add 20 μL of DMSO in the vial to make a 5 mM stock.
-
b.Spin down the untransduced and Cas9-transduced NK-92 cells (300 × g for 5 min at RT). Aspirate the culture media and resuspend the cells in PBS. Adjust the cell count to 1 × 106 NK-92 cells/mL in a 15 mL tube. Add 0.5 μL/mL of CFSE-violet dye and incubate these cells for 20 min at 37°C in the dark.
-
c.After incubation, add at least 5 times the original staining volume of complete RPMI media and incubate for 5 min at 37°C.
-
d.Centrifuge the cells (300 × g for 5 min), discard the media, and wash the cells again with complete RPMI media.
-
e.Adjust the cell count to 1 × 106 NK-92 cells/mL in NK cell media and seed 200 μL in 96-well flat-bottom plate (0.2 × 106 cells/well).
-
f.Incubate the cells for 96 h at 37°C and then analyze using flow cytometry for CFSE dilution (Figure 1C).
-
g.Analyze the data using FlowJo software and note down the median fluorescence intensity (MFI) values for CFSE and calculate the proliferation score (PS) using formula PS = Log2 (MFICFSE day 0/MFICFSE day 4) (Ahlen et al., 2009). We found a less than 2-fold difference in the PS of untransduced NK-92 cells (PS=4.4) and Cas9+ NK-92 cells (PS=3.5), suggesting that transduction with Cas9 did not abrogate or markedly impair NK-92 cell proliferation.
-
a.
-
49.Single cell isolation using limiting dilution technique:
-
a.Count Cas9-EGFP+ NK-92 cells using a hemocytometer and adjust the cell count to 5 × 103 cells/mL.
-
b.The final cell counts to seed a 96-well plate will be 5 cells/mL, and to achieve this count, mix 50 μL of 5 × 103 cells/mL solution with 49.95 mL of NK cell media.
-
c.Transfer 100 μL of 5 cells/mL solution to each well of 96-well round-bottom plates and incubate at 37°C. Carefully monitor these cells over a period of two weeks. When single-cell clumps have grown in size, remove these cells from 96-well plates using a 1 mL pipette, wash, and transfer to 48-well culture plate in NK cell media (500 μL/well).
-
d.After three days of culture in 48-well plate, analyze percentages of Cas9-EGFP+ NK-92 cells in each clone using flow cytometry (Figure 1D) and culture the cells in a T-25 flask in NK cell media.
-
a.
Figure 1.

Generation and validation of Cas9+ NK-92 cell line
(A and B) Flow cytometry dot plots showing (A) frequencies of Cas9-EGFP+ NK-92 cells on days 2 (upper panel) and 11 (lower panel) after transduction with CAS9-EGFP lentivirus, and (B) purity of Cas9-EGFP-transduced NK-92 cells immediately after sorting (day 0) and after expanding the sorted cells for 10 days.
(C) Histogram overlays comparing untransduced and Cas9-EGFP-transduced NK-92 cells for changes in CFSE-Violet fluorescence with respect to the day the cells were labeled with CFSE (day 0).
(D) Flow cytometry dot plots of Cas9-EGFP+ NK-92 single cell clones expanded by limiting dilution method.
(E) Representative flow cytometry dot plots showing frequencies of sgRNA-RFP+Cas9-EGFP+ NK-92 cells after transduction of clone-5 of Cas9-EGFP+ NK-92 cells with either control sgRNA-RFP or RFP sgRNA-RFP to knockout RFP.
(F) Specific cytotoxicities of untransduced NK-92, polyclonal, and monoclonal (clone-5) dCas9-EGFP+ NK-92 cells. Error bars represent mean ± SEM of three technical replicates.
(G and H) Graph showing fold change in frequencies of sgRNA-RFP+Cas9-EGFP+ NK-92 cells with respect to day 1 after transduction with control sgRNA-RFP or IL-2RG sgRNA1-RFP or IL-2RG sgRNA2-RFP targeting IL2Rγ chain.
Alternative method for single cell isolation: Flow cytometry-based sorting of single cell clones. However, sorting is not preferred because it is stressful for sensitive cells such as, human NK cells.
Selecting cytotoxic Cas9+ NK-92 single-cell clones for CRISPRko editing
Cas9+ NK-92 cells single cell clones were first evaluated for CRISPR editing efficiency by using a puromycin-tagged sgRNA that targets and knocks out the RFP present in the ipUSEPR sgRNA cloning plasmid, as described below:
-
50.To transduce single cell clones:
-
a.Resuspend each single cell clone (here, clone-1 to -5, Figure 1D) in 1000 μL complete RPMI media (1 × 106 cells/clone) in 1.7 mL microtubes.
-
b.Divide single cell suspension from each clone into two parts and transfer into 1.7 mL microtubes.
-
c.To one portion, add 500 μL control sgRNA lentivirus and to other half add 500 μL lentivirus harboring sgRNA targeting RFP of ipUSEPR plasmid.
-
d.Now the total volume in each tube will be 1 mL, to this, add polybrene (10 μg/mL) and rhIL-2 (100 U/mL). Seed these cells in 24-well plate (0.5 × 106/well) and incubate at 37°C for 48 h.
-
a.
-
51.
On day 2, analyze frequencies of sgRNA-transduced RFP+ cells by flow cytometry. Resuspend the remaining cells in 2 mL complete RPMI media containing rhIL-2 (100 U/mL) and puromycin (1 μg/mL) (Figure 1E, left panels).
-
52.
After selecting transduced cells using puromycin for ∼10 days, we found that clone-5 showed the best CRISPR editing efficiency with percentages of RFP+ cells reducing from 11.9% at day 2 to 0.46% at day 10 after puromycin selection (Figure 1E). Note that procedures for design and cloning of sgRNAs and production of sgRNA lentiviruses have been described previously (steps 1–43).
We therefore compared the cytotoxic function of untransduced, bulk (polyclonal), and single cell Cas9-transduced NK-92 cells using a flow cytometry-based NK cytotoxicity assay. In this assay, we measured the ability of untransduced and Cas9+ NK-92 cells to kill the standard NK-sensitive target human erythroleukemia cell line, K-562. Among the Cas9+ NK-92 single cell clones, only clone-5 was utilized for the NK cytotoxicity/functionality assay because it was found to have the highest gene editing efficiency (Figure 1E). Below, we describe the procedure to conduct the NK cytotoxicity assay:
-
53.To distinguish NK-92 effector cells from target K-562 cells, label K-562 cells with CFSE using the procedure below:
-
a.Reconstitute a vial of CFSE-violet dye (Thermo Scientific# C34557) by adding 20 μL DMSO in the vial to make a 5 mM stock.
-
b.Spin down K-562 cells (300 × g for 5 min at RT), resuspend the cell pellet in PBS in a 15 mL tube and adjust the cell count to 1 × 106 K-562 cells/mL. Add 0.5 μL/mL of CFSE-violet dye and incubate these cells for 20 min at 37°C in dark.
-
c.After incubation, add at least 5 times the original staining volume of complete RPMI-1640 media (RPMI-1640 + 10% FBS) and incubate for 5 min at 37°C.
-
d.Centrifuge the cells at 300 × g for 5 min at RT, discard the media, and wash the cells again with 5 mL of complete RPMI media before using them in cytotoxicity assays.
-
a.
-
54.
Count NK-92 effector and K-562 target cells. Pipette mix these cells at different effector to target ratios in a U bottom 96-well plate in 200 μL complete RPMI media. Centrifuge the plate at 300 × g for 5 min at RT and incubate for 5 h at 37°C.
-
55.
After incubation, add 3 μL of 7-AAD (50 μg/mL) in each well and mix by pipetting. Incubate the plate for 10 min at RT followed by transferring of cells into FACS tubes and analyze the cells by flow cytometry (Figure 1F). We calculate the specific cytotoxicity using the formula: [(7-AAD+ target-cell frequency in coculture with effector cells − 7-AAD+ target-cell frequency alone) / (100 − 7-AAD+ target-cell frequency alone)] × 100.
Validating gene knockout in Cas9+ NK-92 cells
As NK-92 cells are dependent on the cytokines IL-2 and IL-15 for their survival, knockout of the IL2R, to which these cytokines bind, should lead to the death of NK-92 cells (Carson et al., 1997; Törnroos et al., 2019). Therefore, we knocked out a required signaling component of the IL2R, the IL2Rγ chain (Waickman et al., 2016) to validate the successful generation of Cas9+ NK-92 cells.
The procedures for designing and cloning IL2Rγ sgRNA, and that for producing IL2Rγ sgRNA lentiviruses have been described earlier.
-
56.To validate IL2Rγ gene knockout in Cas9+ NK-92 cells:
-
a.Count Cas9-EGFP+ NK-92 cells, resuspend 1 × 106 cells in 1000 μL of complete RPMI media.
-
b.Divide the cell suspension into two equal parts and transfer to 1.7 mL microtubes.
-
c.Add 500 μL of concentrated lentivirus for control sgRNA or sgRNA targeting the IL2Rγ chain.
-
d.Now the total volume in each tube will be 1 mL. To these tubes, add polybrene (10 μg/mL) and rhIL-2 (100 U/mL).
-
e.Seed these cells in a 24-well plate (0.5 × 106 cells/well) and incubate at 37°C for 48 h. Refer to “Single guide RNA (sg) target sequences table” in materials and equipment section for sequences of control or IL-2RG sgRNA used for CRISPRko.
-
a.
-
57.
On day 2, wash the cells with complete RPMI media, resuspend the cells in 2 mL of media containing rhIL-2 (100 U/mL), and seed them in 24-well plate. Before seeding, take a small aliquot of cells to determine the frequencies of transduced cells by flow cytometry.
-
58.
On day 2 after sgRNA transduction, monitor the levels of IL2Rγ chain on transduced NK-92 cells by flow cytometry (Figures 1G and 1H). Successful IL2R knockout is confirmed by the reduction in percentages of IL2Rγ sgRNA-RFP+ Cas9-EGFP+ cells over time as these cells fail to respond to IL-2 present in the culture medium. However, frequencies of control sgRNA-RFP+ Cas9-EGFP+ NK-92 cells do not reduce with time (Figures 1G and 1H), confirming that the transduction procedure itself doesn’t kill NK cells.
Single guide RNA (sg) target sequences
| Name | Forward sequence (5’→3′) | Reverse sequence (5’→3′) | Application |
|---|---|---|---|
| Control (Scramble) sgRNA | GTAGCGAACGTGTCCGGCGT | ACGCCGGACACGTTCGCTAC | CRISPRko/i/a |
| RFP sgRNA | GTCACCACATACGAAGACGG | CCGTCTTCGTATGTGGTGAC | CRISPRko |
| IL2RG sgRNA (1) | GGTGCAGTACCGGACTGACT | AGTCAGTCCGGTACTGCACC | CRISPRko |
| IL2RG sgRNA (2) | AGCCGCTTTAACCCACTCTG | CAGAGTGGGTTAAAGCGGCT | CRISPRko |
| IL2RG sgRNA (1) | TGGTAATGATGGCTTCAACA | TGTTGAAGCCATCATTACCA | CRISPRi |
| IL2RG sgRNA (2) | AGGGATGTGAATGGTAATGA | TCATTACCATTCACATCCCT | CRISPRi |
| IL-15 sgRNA | ACACCTCCCGCGGAGACTGG | CCAGTCTCCGCGGGAGGTGT | CRISPRa |
Forward sgRNAs each with a 5′ overhang “CACCG” and reverse sgRNAs each with 5’ “AAAC” and 3’ “C” overhangs were ordered form integrated DNA technologies (IDT). Refer to the section titled “oligo design for sgRNA production” for sgRNA design details.
Generating dCas9-KRAB+ NK-92 cells (CRISPRi NK-92 platform)
The CRISPRi system employs a catalytically dead Cas9 (dCas9) without endonuclease activity fused to the KRAB transcriptional repressor (Qi et al., 2013). Specific sgRNAs allow the recruitment and binding of the dCas9-KRAB transcriptional repressor to target genes (Gilbert et al., 2013). The dCas9-KRAB-mediated transcriptional repression of genes is an alternative approach to the Cas9-mediated CRISPRko system. The dCas9-KRAB system mitigates the risk of off-target effects and exogenous DNA integration caused by the active Cas9 nuclease in the CRISPRko system (Tycko et al., 2019; Hanlon et al., 2019; Saayman et al., 2016).
Below, we detail the protocol for generating and validating NK-92 cells that constitutively express the dCas9-KRAB for CRISPRi-mediated transcriptional repression.
-
59.
As with the generation of Cas9+ NK-92 cells, resuspend ∼0.5–1.5 × 106 cells in 500 μL complete RPMI media + 500 μL concentrated dCas9-KRAB-GFP lentivirus + polybrene (10 μg/mL) + rhIL-2 (100 U/mL). Seed these cells in 24-well plate and incubate 37°C for 48 h.
-
60.
After incubation, wash and resuspend in complete RPMI media supplemented with rhIL-2. Analyze percentages of dCas9-KRAB-GFP-transduced NK-92 cells using flow cytometry. Expand these cells to get enough cells for sorting with intermittent flow cytometry analysis of cells for GFP positivity (Figure 2A).
-
61.
Stain the cells with DAPI and sort DAPI−GFP+ cells, check post sort purity (Figure 2B).
-
62.
Analyze proliferation potential (Figure 2C) and calculate the proliferation score (PS) as described previously (step 48) for Cas9+ NK-92 cells. We found PS of dCas9-KRAB+ NK-92 cells to be 5.4 which is less than 2-fold different from that of untransduced NK-92 cells (PS = 4.4). The comparable PS of untransduced and dCas9-KRAB+ NK-92 cells suggest that constitutive expression of the dCas9-KRAB protein in NK-92 cells for CRISPRi does not abolish or markedly impair their natural proliferation.
-
63.
Isolate single cell clones (Figure 2D) using the limiting dilution technique as described previously (step 49).
Figure 2.

Generation and validation of dCas9-KRAB+ NK-92 cell line
(A and B) Flow cytometry dot plots showing (A) frequencies of dCas9-KRAB-GFP+ NK-92 cells on days 2, 3, 8, and 22 after transduction with dCas9-KRAB-GFP lentivirus, and (B) purity of dCas9-KRAB-GFP-transduced NK-92 cells immediately after sorting (day 0) and after expanding the sorted cells for 10 days.
(C) Histogram overlays comparing untransduced and dCas9-KRAB-GFP-transduced NK-92 cells for changes in CFSE-Violet fluorescence with respect to the day the cells were labeled with CFSE (day 0).
(D) Flow cytometry dot plots of dCas9-KRAB-GFP+ NK-92 single cell clones expanded by limiting dilution method.
(E) Specific cytotoxicities of untransduced NK-92, polyclonal, and monoclonal (clones 1 to 5) dCas9-KRAB-GFP+ NK-92 cells. Error bars represent mean ± SEM of three technical replicates.
(F−G) Graphs showing fold change in frequencies of sgRNA-RFP+dCas9-KRAB-GFP+ NK-92 cells with respect to day 1 after transduction with control sgRNA-RFP or IL-2RG sgRNA1-RFP or IL-2RG sgRNA2-RFP targeting IL2Rγ chain.
Selecting cytotoxic dCas9-KRAB+ NK-92 single-cell clones for CRISPRi editing
Because the dCas9-KRAB system uses catalytically dead Cas9 without endonuclease activity, we did not use the sgRNA targeting RFP that we previously used to measure Cas9 editing efficiency. Instead, we screened all dCas9-KRAB+ NK-92 single cell clones that had expanded in culture for NK cytotoxicity by flow cytometry as described below.
-
64.
Using a flow cytometry-based NK cytotoxicity assay (as described in steps 53–55), we measured the ability of untransduced and dCas9-KRAB+ NK-92 cells to kill the standard NK-sensitive target human erythroleukemia cell line, K-562, as described earlier.
-
65.
Among the Cas9-KRAB+ NK-92 single cell clones, only clone-1 showed the highest cytotoxic activity against K-562 cells (Figure 2E). Therefore, only this clone was utilized for validation of the dCas9-KRAB system.
Validating transcriptional repression in dCas9-KRAB+ NK-92 cells
We validated transcriptional repression in dCas9-KRAB in NK-92 cells using the same validation strategy as that described in Cas9+ NK-92 cells, i.e., blocking the expression of the IL2Rγ chain required for NK-92 cell proliferation and survival, and examining the effects of this blockade on NK-92 cell turnover. The procedures for designing and cloning IL2Rγ sgRNAs, and that for producing IL2Rγ sgRNA lentiviruses have been described earlier (steps 1–43). Also, the procedure for sgRNA lentiviral transduction of dCas9-KRAB+ NK-92 cells is identical to that outlined for Cas9+ NK-92 cells (step 44). Starting on day 2 after transduction, we serially monitored the levels of IL2Rγ chain on transduced NK-92 cells by flow cytometry (Figures 2F and 2G). Successful IL2R knockout is confirmed by the reduction in percentages of IL2Rγ sgRNA-RFP+ dCas9-KRAB-GFP+ cells over time as these cells fail to respond to IL-2 present in the culture medium.
Generating dCas9-VP64+ NK-92 cells (CRISPRa NK-92 platform)
The CRISPRa system employs a catalytically dead Cas9 (dCas9) without endonuclease activity fused to the VP64 transcriptional activator (Qi et al., 2013). Specific sgRNAs allow the recruitment and binding of the dCas9-VP64 transcriptional activator to target genes (Gilbert et al., 2013). Like the dCas9-KRAB system, the dCas9-VP64 system has reduced risk of off-target effects and exogenous DNA integration unlike the CRISPRko system (Tycko et al., 2019; Hanlon et al., 2019; Saayman et al., 2016). The CRISPRa approach to transcriptionally activate genes in human NK cells is superior to conventional lentiviral overexpression for two reasons: First, it is easier to introduce ∼20 nt single guide (sgRNAs) to simultaneously transcriptionally activate multiple genes by CRISPRa as opposed to sequentially transducing NK cells with large lentiviral overexpression vectors encoding the large open reading frame of each gene. Second, because transcript levels increase by ∼3-5-fold in CRISPRa as opposed to >20-fold in conventional overexpression systems, cellular toxicity caused by excessive production of proteins can be avoided in CRISPRa.
Below, we detail the protocol for generating and validating NK-92 cells that constitutively express the dCas9-VP64 for CRISPRa-mediated transcriptional activation.
-
66.
As with the generation of Cas9+ and dCAS9-KRAB+ NK-92 cells, count NK-92 cells, resuspend ∼0.5–1.5 × 106 cells in 500 μL of complete RPMI media in a 1.7 mL microtube, and add 500 μL of concentrated dCas9-VP64-GFP lentivirus. Now the total volume will be 1 mL. To this, add polybrene (10 μg/mL) and rhIL-2 (100 U/mL). Seed these cells in a 24-well plate and incubate at 37°C for 48 h.
-
67.
On day 2, wash and then replenish the cells with complete RPMI media + rhIL2 (100 U/mL). Take a small aliquot of cells to measure the percentages of GFP+ dCas9-VP64-transduced NK-92 cells using flow cytometry. We found that the percentages of dCas9-VP64-transduced NK-92 cells were 14.8% at day 2 and 14.5% at day 11 after expansion (Figure 3A).
-
68.
Stain the cells with DAPI and sort DAPI−GFP+ NK-92 cells by FACS. Check the post-sort purity by flow cytometry. We found that post-sort purities of dCas9-VP64-GFP+ NK-92 cells were 99.8% at day 0 and 96.9% at day 10 after expansion (Figure 3B).
-
69.
Compare the proliferation of dCas9-VP64+ NK-92 cells and that of untransduced NK-92 cells (Figure 3C) by measuring the proliferation score (PS) using the formula previously described (step 48) for Cas9+ and dCAS9-KRAB+ NK-92 cells. We found PS of dCas9-VP64+ NK-92 cells to be 3.4 which is less than 2-fold different from that of untransduced NK-92 cells (PS=4.4). The comparable PS of untransduced and dCas9-VP64+ NK-92 cells suggest that constitutive expression of the dCas9-VP64 protein in NK-92 cells for CRISPRa does not abolish or markedly impair their natural proliferation.
-
70.
Isolate the single cell clones of dCas9-VP64+ NK-92 cells (Figure 3D) using the limiting dilution procedure previously described (step 49) for Cas9+ and dCas9-KRAB+ NK-92 cells in Figures 1D and 2D.
Figure 3.

Generation and validation of dCas9-VP64+ NK-92 cell line
(A and B) Flow cytometry dot plots showing (A) frequencies of dCas9-VP64-GFP+ NK-92 cells on days 2 (upper panel) and 11 (lower panel) after transduction with dCas9-VP64-GFP lentivirus, and (B) purity of dCas9-VP64-GFP-transduced NK-92 cells immediately after sorting (day 0) and after expanding the sorted cells for 10 days.
(C) Histogram overlays comparing untransduced and dCas9-VP64-GFP-transduced NK-92 cells for changes in CFSE-Violet fluorescence with respect to the day the cells were labeled with CFSE (day 0).
(D) Flow cytometry of dCas9-VP64-GFP+ NK-92 single cell clones expanded by limiting dilution method.
(E) Specific cytotoxicities of untransduced NK-92, polyclonal, and monoclonal (clone-3) dCas9-VP64-GFP+ NK-92 cells. Error bars represent mean ± SEM of three technical replicates.
(F) Flow cytometry dot plots showing the percentages of dCas9-VP64-GFP+ NK-92 cells transduced with control sgRNA-RFP or IL-15 sgRNA-RFP on days 2 (upper panel) and 15 (lower panel) after transduction and puromycin selection.
(G) Fold change in IL-15 mRNA expression levels by qPCR in sgRNA-RFP+ dCas9-VP64-GFP+ NK-92 cells on day 15 after being transduced with control sgRNA-RFP or IL-15 sgRNA-RFP. Error bars represent mean ± SEM of three technical replicates, Exact p-value was calculated by Student's t-test.
Selecting cytotoxic dCas9-VP64+ NK-92 single-cell clones for CRISPRa editing
Because the dCas9-VP64 system uses catalytically dead Cas9 without endonuclease activity, we did not use the sgRNA targeting RFP that we previously used to measure Cas9 editing efficiency. Instead, we directly screened dCas9-VP64+ NK-92 single cell clones that had expanded in culture for NK cytotoxicity by flow cytometry. Of note, all dCas9-VP64+ NK-92 cells except clone-3 had contamination from GFP negative (dCas9-VP64−) NK-92 cells (Figure 3D). Therefore, the cytotoxicity assay was performed only using clone-3 of dCas9-VP64+ NK-92 cells (Figure 3E). The procedure for performing cytotoxicity assays is similar to that described earlier (steps 53–55) for Cas9+ and dCas9-KRAB+ NK-92 cells.
Validating transcriptional activation in dCas9-VP64+ NK-92 cells
To validate dCas9-VP64+ NK-92 cells, we transcriptionally activated the IL-15 gene which is required for not just survival and proliferation but also the cytotoxic function of NK cells (Cooper et al., 2002; Zhang et al., 2018). Alternately, one could transcriptionally activate IL-2.
The procedures for designing and cloning IL-15 sgRNA, and that for producing IL2Rγ sgRNA lentiviruses have been described earlier.
-
71.To validate transcriptional activation of IL-15 gene in dCas9-VP64+ NK-92 cells:
-
a.Resuspend 1 × 106 dCas9-VP64-GFP+ NK-92 cells in 1000 μL of complete RPMI media in 1.7 mL microtube.
-
b.Divide the cell suspension into two parts and transfer to 1.7 mL microtubes.
-
c.Add 500 μL of concentrated lentivirus for control sgRNA-RFP or IL-15 sgRNA-RFP targeting the IL-15 gene.
-
d.Now the total volume in each tube will be 1 mL. To these tubes, add polybrene (10 μg/mL) and rhIL-2 (100 U/mL).
-
e.Seed these cells in a 24-well plate (0.5 × 106 cells/well) and incubate at 37°C for 48 h. See “Single guide RNA (sg) target sequences” table in materials and equipment section for sequences of control sgRNA or IL-15 sgRNA used for CRISPRa.
-
a.
-
72.
On day 2, before puromycin selection, take a small aliquot of transduced cells and analyze the frequencies of dCas9-VP64-GFP+ sgRNA-RFP+ transduced cells by flow cytometry (Figure 3F upper panel).
-
73.
Resuspend the remaining cells in NK cell media containing puromycin (1 μg/mL) and select these cells in puromycin for ∼ 2–4 weeks until frequencies of dCas9-VP64-GFP+ sgRNA-RFP+ cells in the control sgRNA and IL-15 sgRNA groups reaches >95% (Figure 3F lower panel).
Alternative Method: The time period of puromycin selection may be prolonged and/or selection may be difficult if the gene modulated negatively impacts NK cell turnover. For such sgRNAs, we recommend flow-based sorting of dCas9-VP64-GFP+ sgRNA-RFP+ NK cells.
-
74.
Proceed with RNA isolation (NucleoSpin RNA purification kit # 740955.250), cDNA synthesis (SuperScript IV First Strand Synthesis System # 18091050) and quantitate the mRNA expression of IL-15 in cells transduced with control sgRNA or IL-15 sgRNA by quantitative real-time PCR (Figure 3G).
Expected outcomes
This protocol sequentially details the steps for genetically manipulating NK-92 cells using Cas9 mediated gene knockout as well as dCas9-mediated transcriptional repression and activation of genes. We also provide optimized and working sgRNA sequences which can be used by others to functionally validate the successful establishment of the Cas9+ and dCas9+ NK-92 cell lines. The effects of targeting specific genes in human NK cells on homeostasis, function, and therapeutic potential of NK cells can be studied using our Cas9+ and dCas9+ NK-92 platforms. We anticipate that our protocols can be extended to modify human NK cells derived from cord blood or peripheral blood of healthy donors using CRISPR for developing off-the-shelf NK cell-based therapies (Suck et al., 2016).
Our data suggest that the five major steps of the protocol, namely, transduction of NK cells with Cas9/dCas9, sorting of Cas9/dCas9+ NK cells, single cell cloning of sorted Cas9/dCas9+ NK cells, confirmation of cytotoxic function, and sgRNA-based validation of the CRISPR-engineered NK cells are feasible. However, at each of these steps, it is likely that the desired outcome may not be obtained due to technical difficulties. Therefore, in the troubleshooting section, we outline potential pitfalls and provide solutions to ensure that each of the steps leading to generation and validation of CRISPR-engineered NK cells is successful.
Quantification and statistical analysis
Before proceeding with CRIPSR-mediated target gene modification in Cas9+ and dCas9+ NK-92 cells, we ensured that transduction with Cas9 or dCas9 did not impair NK cell viability, proliferation, and cytotoxicity. Only single cell Cas9+ and dCas9+ NK-92 clones without disruptions in normal NK homeostasis were selected for functional validation and will be used by us in future experiments. For manipulating a given gene in Cas9+ and dCas9+ NK-92 cells during functional validation of the NK cell platforms, we selected two top-scoring sgRNAs with low off-target effects using the Broad Institute Genetic Perturbation Platform (GPP) web portal (Doench et al., 2016). We tested the abilities of each of these sgRNAs to knockout (Cas9), repress transcription (dCas9-KRAB), and activate transcription (dCas9-VP64) of genes in NK-92 cells. We found that for the Cas9 and dCas9-KRAB systems, both top scoring sgRNAs had identical effects. However, in the dCas9-VP64 system, only one sgRNA was successful in transcriptionally activating the target gene, and is shown. For experiments aimed at modulating specific genes of interest in these platforms, we recommend testing at least 2 sgRNAs per gene, as we demonstrate. Cytotoxicity assays and assays for functional validation of the Cas9 and dCas9 systems were conducted in three technical replicates per sample. P-values have been calculated using the Student’s t-test in GraphPad software. The level of statistical significance for all experiments has been set at P<0.05.
Limitations
As innate immune cells, NK cells are generally refractory to viral transductions (Schmidt et al., 2021). For this reason, other groups have previously electroporated the Cas9 ribonucleoprotein complex into primary human NK cells (Pomeroy et al., 2020; Rautela et al., 2020; Berrien-Elliott et al., 2020). While electroporation is suitable for knocking out genes where transient expression of Cas9 protein is sufficient, dCas9-KRAB-mediated transcriptional repression and dCas9-VP64-mediated transcriptional activation require constitutive expression of dCas9-KRAB and dCas9-VP64, respectively. Therefore, NK-92 lymphoblasts, which are easier to manipulate using lentiviruses than normal NK cells, were used. Furthermore, because NK-92 cell-based therapies are currently in clinical trials for both hematological malignancies and solid tumors ((Li et al., 2015; Tang et al., 2018; Chu et al., 2014; Rezvani and Rouce, 2015; Klingemann et al., 2016), https://immunitybio.com/pipeline/), we anticipate that our protocols for CRISPR-based editing of NK-92 cells will have clinical applications.
Although our protocol details the steps for engineering NK-92 cells using CRISPR, it can be extended to other malignant NK cell lines including NKL (Robertson et al., 1996), and NK101 (Yang et al., 2019) which are also being used in research and clinical applications. In the future, we intend to genetically manipulate primary human NK cells derived from cord blood or from peripheral blood of healthy donors using the above-described CRISPR-based methods. For modifying healthy human NK cells, we acknowledge that our current protocol may have to be modified because normal NK cells are more difficult to transduce than NK cell lines (Schmidt et al., 2021) . At present, it is difficult to predict the extent of modification necessary to constitutively edit healthy NK cells for CRISPR-based applications. However, we expect the sequence of steps to constitutively modify healthy NK cells for CRISPR-based applications to be identical to that described here for NK-92.
Because we used fresh lentiviral supernatant for transducing NK-92 without determining the lentiviral titer, frequencies of transduced cells vary in each experiment. Unfortunately, determining lentivirus titer using permissive cell lines including HEK cells does not directly translate to innate immune cells such as NK cells. Also, we do not recommend using MOI for estimating efficiencies of Cas9- and dCas9- transductions in NK-92 cells for two reasons:
First, increasing the MOI does not significantly improve transduction efficiencies of human NK cells (Su et al., 2011; Rautela et al., 2020), suggesting that MOI is not a reliable measure of estimating NK transduction efficiencies.
Second, a more recent study suggested that transduction efficiencies of cells in culture cannot be accurately estimated using MOI because cell cultures are heterogenous with some cells being more resistant than others to transduction and because virions may not contact each cell with the same efficiency. Furthermore, when a virus particle interacts with and infects the cell, it is a discrete event. Even at MOI=1, the same cell may become infected many times. Therefore, MOI can only be applied to a homogeneous cell population with each cell having identical transduction efficiency (Shabram and Aguilar-Cordova, 2000), which is not the case with NK cells cultures. In our experiments, this situation is further complicated by the general resistance of all human NK cells to viral transduction (Schmidt et al., 2021).
Instead of using MOI, we therefore recommend users of our protocol to apply 1% of transduced cells (on day 2 post infection), as the minimum cut off in Cas9 and dCas9 transduction experiments to facilitate expansion and sorting of these cells for next steps in CRISPR engineering.
The constitutive expression of the active Cas9 nuclease in NK-92 cells in the CRISPRko system may lead to off-target and deleterious genomic alterations in the NK cells. There are several methods to mitigate this potential problem. One method is to use electroporation of the Cas9 ribonucleoprotein (RNP) which will lower the time frame for which active Cas9 is present in the NK cell, thereby reducing the risk of off-target genetic alterations (Hildreth et al., 2020).
A better solution which avoids the use of Cas9 but achieves the same effect is the dCas9-KRAB system (Gilbert et al., 2013). In this system, as Cas9 is catalytically dead and the specificity of the gene targeted for transcriptional repression is decided by the sgRNA sequence (Qi et al., 2013), the off-target toxicity of Cas9 is drastically reduced. Furthermore, unlike the Cas9 system which knocks out the entire protein, the sgRNAs in the dCas9-KRAB system can be tailored to modulate the level of transcriptional repression of a target gene. Such downmodulation is especially important while engineering NK cells for therapeutic applications where complete knockout of a given gene using Cas9 may be detrimental to the NK cell.
Troubleshooting
Problem 1
No transduction or low frequencies of Cas9- and dCas9- transduced NK-92 cells (step 44)
Potential solutions
Because NK cells are innate immune cells, they are difficult to transduce with lentiviruses (Schmidt et al., 2021). We found that low levels of transduction (<1%) are mostly attributed to the lentivirus quality/titer. Potential solutions for this problem include:
-
a.
making a fresh lot of virus, and on day 4 of the virus production, avoid filtering the lentiviral supernatant but rather centrifuge (300 × g for 5 min) it to remove debris before concentrating the virus.
-
b.
at the final step of virus concentration on day 5, using 500 μL of fresh viral supernatant to transduce the NK-92 cells as freeze thawing of viral supernatant can affect the lentivirus quality and titer.
-
c.
tracking the cells with low transduction efficiencies for at least a week. In some cases, although the efficiency of transduction is initially low, after a week the frequencies of GFP+ transduced cells were found to be increased (Figure 2A).
If none of the above potential solutions work, the only option is to restart the experiment from the first stage of virus production.
Problem 2
Transduction with Cas9 or dCas9 slows down or blocks the proliferation of NK cells (step 48)
Potential solutions
We suggest the following solutions to overcome the potential pitfall 2.
-
a.
One must ensure that the NK-92 cell growth media is being supplemented with rhIL-2 (100 U/mL).
-
b.
Because NK-92 cell proliferation is affected by the seeding density at the time of culture initiation, we suggest growing the post-sort Cas9+ or dCas9+ NK-92 cells in 96-well plates at high densities (∼0.5 × 106/well) and transferring cells to 48-well plates and finally into T-25 flasks when these cells begin proliferating. If this method doesn’t work, one would infer that the reduced proliferation of transduced NK-92 cells may be caused by lentiviral integrations that disrupt genes involved in NK-92 cell proliferation. The only recourse in this case would be to restart the experiment.
Depending on the type of application, we recommend the users to either proceed further after checking NK-92 proliferation or troubleshoot. For example, proliferation may be less critical for clinical applications where NK cytotoxicity is the most important and NK cells are irradiated before use (Klingemann et al., 2016; Tang et al., 2018).
Problem 3
Contamination of sorted and expanding Cas9+ and dCas9+ single cell clones with untransduced GFP- cells (step 49)
Potential solutions
FACS-based purification protocols do not lead to the isolation of the desired populations at 100% purity. For example, we observed an outgrowth of DAPI−GFP− cells after expanding some of the sorted dCas9-VP64+ single cell clones (Figure 3E). We propose two potential solutions for this problem:
-
a.
Sorting polyclonal dCas9-VP64+ to remove DAPI-GFP- cells and then performing single cell cloning.
-
b.
Seeding multiple 96 well plates with single cell clones to get at least ∼5–10 pure single cell colonies of dCas9-VP64+ NK-92 cells.
Problem 4
None of the selected clones show cytotoxicity at levels comparable to that of unmodified NK-92 cells (steps 53–55)
Potential solutions
We suggest seeding multiple (∼5–10) single cells clones to increase the chances of getting at least one Cas9/dCas9+ NK cell clone with cytotoxicity comparable to that of untransduced NK-92 cells. We have observed that cytotoxicity varies at a clonal level, i.e., a Cas9/dCas9-transduced polyclonal NK population with cytotoxicity comparable to that of untransduced cells does not guarantee that single cells clones will be equally cytotoxic and vice-versa. For example, in Figure 2E we observe that polyclonal dCas9-KRAB+ NK-92 cells have lower cytotoxicity whereas clone-1 of same population showed cytotoxicity at levels similar to untransduced NK-92 cells.
Problem 5
Transduction with the sgRNA does not yield the desired effect, i.e., knockout or transcriptional activation or transcriptional repression (steps 58,74)
Potential solutions
Selection of ∼2–5 sgRNAs per gene using the Broad Institute Genetic Perturbation Platform (Doench et al., 2016) increases the likelihood of finding the best working sgRNA which yields the desired gene regulation.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Srividya Swaminathan (sswaminathan@coh.org).
Materials availability
All plasmids described in this study have been deposited in Addgene. The NK-92, K-562 and Lenti-X 293T cell lines were obtained from American Type Culture Collection and Takara Bio respectively. The research described in this manuscript is covered by a pending US patent application.
Acknowledgments
This study was supported by the following awards to S.S.: Special Fellow Award from the Leukemia and Society (LLS 3366-17), American Society of Hematology Scholar Award, PhRMA Foundation Research Starter Grant in Drug Discovery (2021 RSGDS 730217), Toni Stephenson Lymphoma Center Pilot Award, P50 CA107399-12 National Cancer Institute Lymphoma SPORE Career Enhancement Pilot Award (PIs: Stephen J. Forman, Larry W. Kwak), and Research Start-Up from City of Hope. C.-W.C is funded by American Society of Hematology Scholar Award, Alex’s Lemonade Stand Foundation Innovation Award, Stand Up To Cancer & Cancer Research UK Pediatric Cancer New Discoveries Challenge Award, and National Institutes of Health Grants CA197498, CA233691, CA236626, CA233922, and CA243124.
Author contributions
A.K. performed most of the experiments, designed all experiments, and wrote the manuscript. S.J.L. assisted A.K. with experiments. Q.L. A.K.N.C., and S.P.P. provided reagents and technical expertise on CRISPR. J.Y. provided scientific expertise on NK cell biology. C.-W.C provided scientific expertise in CRISPR. S.S. conceived and supervised the study and edited the manuscript.
Declaration of interests
The research described in this manuscript is covered by a pending US patent application.
Contributor Information
Anil Kumar, Email: anikumar@coh.org.
Srividya Swaminathan, Email: sswaminathan@coh.org.
Data and code availability
This study did not generate or analyze any code or datasets.
References
- Ahlen M.T., Husebekk A., Killie M.K., Skogen B., Stuge T.B. T-cell responses associated with neonatal alloimmune thrombocytopenia: isolation of HPA-1a–specific, HLA-DRB3∗ 0101–restricted CD4+ T cells. J. Am. Soc. Hematol. 2009;113:3838–3844. doi: 10.1182/blood-2008-09-178475. [DOI] [PubMed] [Google Scholar]
- Berrien-Elliott M.M., Cashen A.F., Cubitt C.C., Neal C.C., Wong P., Wagner J.A., Foster M., Schappe T., Desai S., McClain E.J. Multidimensional analyses of donor memory-like NK cells reveal new associations with response after adoptive immunotherapy for leukemia. Cancer Discov. 2020;10:1854–1871. doi: 10.1158/2159-8290.CD-20-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson W.E., Fehniger T.A., Haldar S., Eckhert K., Lindemann M.J., Lai C.-F., Croce C.M., Baumann H., Caligiuri M.A. A potential role for interleukin-15 in the regulation of human natural killer cell survival. J. Clin. Invest. 1997;99:937–943. doi: 10.1172/JCI119258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerwenka A., Lanier L.L. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 2001;1:41–49. doi: 10.1038/35095564. [DOI] [PubMed] [Google Scholar]
- Chavez A., Scheiman J., Vora S., Pruitt B.W., Tuttle M., Iyer E.P., Lin S., Kiani S., Guzman C.D., Wiegand D.J. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J., Deng Y., Benson D.M., He S., Hughes T., Zhang J., Peng Y., Mao H., Yi L., Ghoshal K. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–927. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper M.A., Bush J.E., Fehniger T.A., Vandeusen J.B., Waite R.E., Liu Y., Aguila H.L., Caligiuri M.A. In vivo evidence for a dependence on interleukin 15 for survival of natural killer cells. J. Am. Soc. Hematol. 2002;100:3633–3638. doi: 10.1182/blood-2001-12-0293. [DOI] [PubMed] [Google Scholar]
- Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duault C., Kumar A., Khani A.T., Lee S.J., Yang L., Huang M., Hurtz C., Manning B., Ghoda L., McDonald T.J. Activated natural killer cells predict poor clinical prognosis in high-risk B-and T-cell acute lymphoblastic leukemia. Blood. 2021 doi: 10.1182/blood.2020009871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:1–11. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildreth A.D., Riggan L., O’Sullivan T.E. CRISPR-Cas9 ribonucleoprotein-mediated genomic editing in primary innate immune cells. STAR Protoc. 2020;1:100113. doi: 10.1016/j.xpro.2020.100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingemann H., Boissel L., Toneguzzo F. Natural killer cells for immunotherapy–advantages of the NK-92 cell line over blood NK cells. Front. Immunol. 2016;7:91. doi: 10.3389/fimmu.2016.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooreman N.G., De Almeida P.E., Stack J.P., Nelakanti R.V., Diecke S., Shao N.-Y., Swijnenburg R.-J., Sanchez-Freire V., Matsa E., Liu C. Alloimmune responses of humanized mice to human pluripotent stem cell therapeutics. Cell Rep. 2017;20:1978–1990. doi: 10.1016/j.celrep.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Yin J., Li T., Huang S., Yan H., Leavenworth J., Wang X. NK cell-based cancer immunotherapy: from basic biology to clinical application. Sci. China Life Sci. 2015;58:1233–1245. doi: 10.1007/s11427-015-4970-9. [DOI] [PubMed] [Google Scholar]
- Pomeroy E.J., Hunzeker J.T., Kluesner M.G., Lahr W.S., Smeester B.A., Crosby M.R., Lonetree C.-L., Yamamoto K., Bendzick L., Miller J.S. A genetically engineered primary human natural killer cell platform for cancer immunotherapy. Mol. Ther. 2020;28:52–63. doi: 10.1016/j.ymthe.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautela J., Surgenor E., Huntington N.D. Drug target validation in primary human natural killer cells using CRISPR RNP. J. Leukoc. Biol. 2020;108:1397–1408. doi: 10.1002/JLB.2MA0620-074R. [DOI] [PubMed] [Google Scholar]
- Rezvani K., Rouce R.H. The application of natural killer cell immunotherapy for the treatment of cancer. Front. Immunol. 2015;6:578. doi: 10.3389/fimmu.2015.00578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson M., Cochran K., Cameron C., Le J., Tantravahi R., Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp. Hematol. 1996;24:406–415. [PubMed] [Google Scholar]
- Saayman S.M., Lazar D.C., Scott T.A., Hart J.R., Takahashi M., Burnett J.C., Planelles V., Morris K.V., Weinberg M.S. Potent and targeted activation of latent HIV-1 using the CRISPR/dCas9 activator complex. Mol. Ther. 2016;24:488–498. doi: 10.1038/mt.2015.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt P., Raftery M.J., Pecher G. Engineering NK cells for CAR therapy—recent advances in gene transfer methodology. Front. Immunol. 2021;11:3404. doi: 10.3389/fimmu.2020.611163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabram P., Aguilar-Cordova E.J. Multiplicity of infection/multiplicity of confusion. Mol. Ther. 2000;2:420–421. doi: 10.1006/mthe.2000.0212. [DOI] [PubMed] [Google Scholar]
- Strauss-Albee D.M., Blish C.A. Human NK cell diversity in viral infection: ramifications of ramification. Front. Immunol. 2016;7:66. doi: 10.3389/fimmu.2016.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S., Betters D.M., Ramanathan M., Keyvanfar K., Smith A., Feng X., Furutani E., Carlsten M., Lundqvist A., Childs R. Optimizing lentiviral transduction of human natural killer cells. Am. Soc. Hematol. 2011;118:4714. [Google Scholar]
- Suck G., Odendahl M., Nowakowska P., Seidl C., Wels W.S., Klingemann H.G., Tonn T. NK-92: an ‘off-the-shelf therapeutic’for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol. Immunother. 2016;65:485–492. doi: 10.1007/s00262-015-1761-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Yang L., Li Z., Nalin A.P., Dai H., Xu T., Yin J., You F., Zhu M., Shen W. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018;8:1083. [PMC free article] [PubMed] [Google Scholar]
- Törnroos H., Hägerstrand H., Lindqvist C. Culturing the human natural killer cell line NK-92 in interleukin-2 and interleukin-15–Implications for clinical trials. Anticancer Res. 2019;39:107–112. doi: 10.21873/anticanres.13085. [DOI] [PubMed] [Google Scholar]
- Tycko J., Wainberg M., Marinov G.K., Ursu O., Hess G.T., Ego B.K., Li A., Truong A., Trevino A.E., Spees K. Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements. Nat. Commun. 2019;10:1–14. doi: 10.1038/s41467-019-11955-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waickman A.T., Park J.-Y., Park J.-H. The common γ-chain cytokine receptor: tricks-and-treats for T cells. Cell Mol Life Sci. 2016;73:253–269. doi: 10.1007/s00018-015-2062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk A.J., Lee M.J., Wei B., Parks B., Pi R., Martínez-Colón G.J., Ranganath T., Zhao N.Q., Taylor S., Becker W. Multi-omic profiling reveals widespread dysregulation of innate immunity and hematopoiesis in COVID-19. J. Exp. Med. 2021;218:e20210582. doi: 10.1084/jem.20210582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.G., Kang M.C., Kim T.Y., Hwang I., Jin H.T., Sung Y.C., Eom K.-S., Kim S.W. Discovery of a novel natural killer cell line with distinct immunostimulatory and proliferative potential as an alternative platform for cancer immunotherapy. J. Immunother. Cancer. 2019;7:1–17. doi: 10.1186/s40425-019-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Chan A., Miyashita K., Delaney C., Wang X., Li H., Pokharel S., Li S., Li M., Xu X. High-resolution characterization of gene function using single-cell CRISPR tiling screen. Nat. Commun. 2021;12:4063. doi: 10.1038/s41467-021-24324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Wen B., Anton O.M., Yao Z., Dubois S., Ju W., Sato N., Dilillo D.J., Bamford R.N., Ravetch J.V. IL-15 enhanced antibody-dependent cellular cytotoxicity mediated by NK cells and macrophages. Proc. Natl. Acad. Sci. U S A. 2018;115:E10915–E10924. doi: 10.1073/pnas.1811615115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate or analyze any code or datasets.