

Abstract
Right aortic arch (RAA) is an abnormal embryologic development of the aorta characterized by its descendance on the right side of the trachea. This anomaly is accompanied often by other intracardiac and extracardiac anomalies and it is also known for potential association with genetic aberrations, most common being 22q11.2 deletion. The aim of the study was to evaluate the incidence of chromosomal anomalies and in particular 22q11.2 deletion in RAA. Moreover, we assessed the prognosis of fetuses with isolated RAA. Our second objective was to evaluate the prevalence of hypoplastic or absent thymus in RAA fetuses diagnosed with 22q11.2 deletion. We conducted a retrospective study of all fetuses with RAA over a period of 10 years diagnosed prenatally in a tertiary referral center in Romania. A detailed ultrasound was obtained in each case. We extracted the cases that were investigated genetically and selected the cases positive for 22q11.2 deletion. These fetuses were followed up until pregnancy termination or birth to confirm the ultrasound findings. Deletion 22q11.2 was present in 23.52% (4/17) cases. The incidence was particularly high when the fetuses presented a small thymus. In conclusion, we believe that all cases of RAA, including when isolated, should be referred for genetic testing and especially 22q11.2 deletion exclusion. Also, we suggest considering hypoplastic thymus to be a soft marker for this deletion.
Keywords: Right aortic arch, DiGeorge Syndrome, 22q11.2, prenatal diagnosis, prenatal ultrasound, congenital heart disease
Introduction
Right aortic arch (RAA) is a congenital heart defect (CHD), a structural anomaly of the aortic arch, where the arch crosses the trachea and descends to its right. This abnormal laterality can be diagnosed sonographically on the three vessel and trachea view (3VT) during fetal anomaly scan, as early as the first trimester. The origin of this anomaly lays in the abnormal regression of the embryonic aortic arches [1,2].
The incidence of RAA is reported around 1/1000 low risk pregnancies [1,3,4]. The rationale for RAA prenatal diagnosis is to recognize the cases at risk for postpartum mediastinal compressive complications (tracheal and/or esophageal compression) [5] and more important, cases at risk for cardiac, extracardiac and genetic anomalies, given the frequent associations [5,6,7,8,9].
RAA represents 5% of the total cases of CHD and the number of cases is increasing gradually due to the modern fetal echocardiography methods [9,10]. In one large cohort study, RAA was present in 3.6% of the individuals diagnosed postpartum with 22q11.2DS that also associated a CHD [11].
Other studies suggest the prevalence of iRAA to be around 9%-12% [12] in 22q11.2DS postpartum/adulthood symptomatic cases and a prevalence of 3.8% when diagnosed prenatally [13]. The difference between the two prevalence values signals a need to increase testing rates for 22q11.2 deletion antepartum, especially when a CHD is suspected.
This study focuses on the association between RAA and chromosomal abnormalities, particularly 22q11.2 deletion syndrome (22q11.2DS). Another point of interest is the incidence of these genetic diseases in isolated RAA (iRAA) and non-isolated cases of RAA in our center, in comparison with other tertiary institutes. We define RAA as isolated if it does not associate other cardiac (C) or extracardiac (EC) abnormalities.
DiGeorge syndrome, defined by the 22q11.2 deletion, is associated with a development anomaly of the 3rd and 4th aortic arches. It represents a serious health problem because it causes a wide range of anatomical defects: frequent congenital heart defects, hypoplastic or absent thymus and parathyroids, face defects (eye anomalies, micrognathia, cleft palate), musculoskeletal anomalies, psychiatric and neurological complications (seizures, development delay), cerebral, respiratory tract and renal anomalies [14,15,16].
Even if our study group is narrow, the thoroughness with which each case was investigated is a strong asset of our study and provides important information for clinicians.
Matherials and Methods
We conducted a retrospective study over a period of 10 years (2012-2021) in the Obstetrics and Gynecology Clinics of the Emergency County Hospital, Craiova, Romania. Approximately 11 500 pregnant women were examined in this period of time in this tertiary center. The indications for the fetal examinations were: first trimester screening, first or second trimester morphological scans, other routine fetal scans and second opinion scans for referred pregnancies suspected with a fetal pathology.
The aortic arch and ductus arteriosus were imaged in relation to the trachea in the standard three vessels and trachea (3VT) view, in line with the recommendations of the International Society of Ultrasound in Obstetrics and Gynecology [17]. The thymic-thoracic ratio was obtained after measuring the anteroposterior thymic and the intrathoracic mediastinal diameters in the 3VT view.
When fetal RAA was diagnosed, the couple was counseled for extensive sonographic and genetic investigations. All parents agreed to participate in further scientific studying and written consent was obtained in the medical files. The parents were offered amniocentesis followed by karyotyping and 22q11.2 evaluation using fluorescence in situ hybridization (FISH) analysis of metaphase chromosome with specific 22q11.2 probes. Some parents chose extended chromosomal microarray analysis (CMA) on their own cost, as well. The latter investigation was not covered by standard medical services in Romania, thus having important economic limitations.
Right aortic arch cases were divided in 3 categories, according to the genetic test results: normal genetic result, 22q11.2DS positive and other genetic anomalies. The fetuses were also divided according to the type of RAA: type I (RAA with right ductus arteriosus-DA), type II (RAA with left DA) and DAA and also in isolated and non-isolated cases. All the cases were followed until delivery or termination of pregnancy for confirmation of the anatomic status by neonatal evaluation or pathology results.
Results
During this period of time, we diagnosed 19 cases of RAA. The study group included double aortic arch (DAA) cases as well and consisted of 17 singleton pregnancies and two fetuses from twin pregnancies. The incidence of RAA in this study group was 0.165%. One patient was lost to follow-up after first trimester diagnosis (RAA associated with complex CHD and alobar holoprosencephaly), the couple chose pregnancy termination by elective curettage in another center. Another couple refused genetic testing and gave birth to a living fetus with a favorable postnatal evolution. The remaining 17 couples accepted further sonographic evaluations, fetal echocardiography and prenatal invasive testing.
Figure 1 presents the distribution of the fetuses with RAA based on the genetic results. Out of the 19 cases, 17 were tested for chromosomal anomalies.
Figure 1.
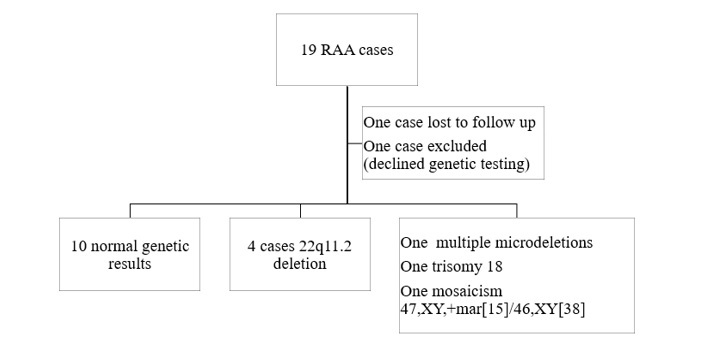
Genetic results of fetuses diagnosed with RAA
In ten cases the results came back normal, one case had multiple microdeletions (1p33 microdeletion, 4q32.1 microdeletion, 10q26.3 microduplication, 12p.11.21 microduplication, 14q.2 microduplication and 17p11.2 microdeletion), one case involved trisomy 18, one case presented mosaicism 47,XY/46,XY and 4 cases were diagnosed with 22q11.2 deletion. The incidence of 22q11.2DS in our study group was 23.52%.
The characteristics of the fetuses diagnosed with 22q11.2DS are presented in Table 1. The four cases diagnosed with 22q11.2DS were as following: one fetus with RAA type I, two fetuses with RAA type II and one fetus with DAA. All fetuses were diagnosed in the second trimester at a mean gestational age of 21 weeks and 5 days. Two fetuses (50%) associated Tetralogy of Fallot (TOF) and the other 2 fetuses had an isolated RAA (no other C anomalies). In Figure 2 we present a case of RAA type I with TOF. Regarding EC anomalies, hypoplastic thymus was the most common, present in three cases (75%). In Figure 3 we present a case of thymic hypoplasia diagnosed antepartum. One fetus presented hypoplastic nasal bone. No other EC anomalies were discovered in the 22q11.2DS cases. Three couples decided upon termination of pregnancy and the autopsies confirmed RAA, hypoplastic thymuses and in the case with a hypoplastic nasal bone, cranio-cerebral dysmorphism was noted as well. The fetus that was birthed alive had a good postnatal evolution. It was the only 22q11.2DS positive fetus with a normal thymus that underwent surgery for correction of TOF and had a good outcome.
Table 1.
Characteristics of the fetuses diagnosed with 22q11.2 deletion. (RAA: right aortic arch; TOP: termination of pregnancy; TOF: Tetralogy of Fallot; CHD: congenital heart disease; DAA: double aortic arch)
|
|
Type of RAA |
Associated intracardiac anomalies |
Associated extracardiac anomalies |
Gestational age at diagnosis (weeks) |
Outcome of pregnancy |
|
1 |
II |
- |
Hypoplastic nasal bone Thymus hypoplasia |
20 |
TOP The autopsy confirmed RAA, cranio-cerebral dysmorphism and a small thymus. |
|
2 |
I |
TOF |
Thymus hypoplasia |
23 |
TOP The autopsy confirmed the CHD association and a small thymus. |
|
3 |
DAA |
- |
Thymus hypoplasia |
22 |
TOP The autopsy confirmed the DAA and the small thymus. |
|
4 |
II |
TOF |
- |
22 |
Live birth Postnatal confirmation of RAA and TOF. Surgical treatment for TOF. Favorable postnatal evolution. |
Figure 2.
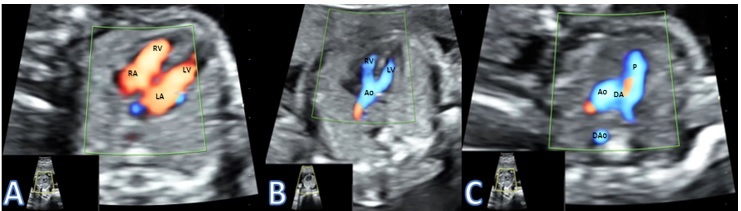
Presenting a case of RAA type I with TOF. A) Normal 4 chamber view; B) Overriding Aorta with Ventricular Septal Defect; C) Right aorta with right ductus arteriosus. RAA-right aortic arch; TOF-Tetralogy of Fallot; RA-Right Atrium; LA-Left Atrium; RV-Right Atrium; LV-Left Atrium; Ao-Aorta; P-Pulmonary artery; DA-Ductus Arteriosus; DAo- Descending Aorta
Figure 3.
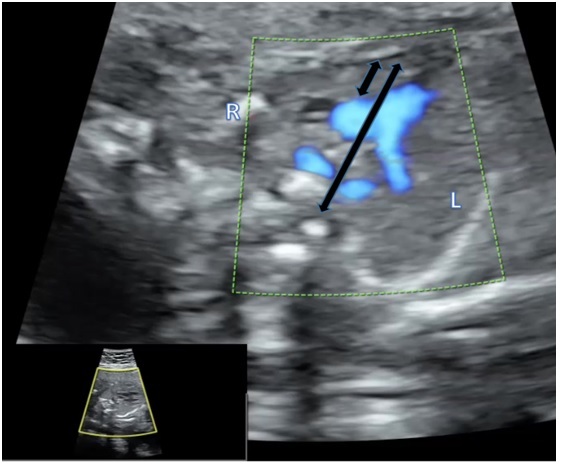
Presenting a hypoplastic thymus diagnosed using thymus-thoracic measurement in a fetus with double aortic arch
Discussions
Our study group is not to be considered a low-risk pregnancy population, since the research has been conducted in a tertiary referral institution. The incidence of RAA in our study was found to be 0.165%, a value similar to those reported in other studies, ranging from 0.05% to 0.18% [1,8,18,19,20,21,22] in the general population and as high as 0.35% to 0.6% [9,23,24] in high-risk population. Thus, the incidence in our group can be considered at the high end of the prevalence interval of low-risk groups or of medium-risk.
The prevalence of 22q11.2 deletion in the general population is considered around 9.6-37.5 per 100,000 live births [25]. This deletion is present in 11.9% of fetuses with conotruncal heart defects, the most frequent being: TOF, DORV (double outlet right ventricle) and RAA [16]. It is also important to mention that out of all 22q11.2 deletion cases, 48.5% to 74% cases associate some form of CHD [11]. Another study reports that 92% of the fetuses diagnosed with DiGeorge syndrome associates a conotruncal heart defect, 86% thymus hypoplasia and 34% urinary tract anomalies [14]. Therefore, when a CHD is diagnosed antepartum, including RAA, the pregnant woman must be offered genetic counseling and testing using fetal karyotyping and FISH technique for the exclusion of 22q11.2 deletion.
In our study group, the incidence of chromosomal anomalies and 22q11.2DS in fetuses diagnosed with RAA was 41.17% (7/17) and 23.52% (4/17), respectively. Half of the fetuses positive for 22q11.2DS associated TOF.
Literature data suggests that 15,3 to 30% of RAA cases associate chromosomal abnormalities [6,9] and up to 30% of iRAA cases are 22q11.2DS positive [6]. Another author published a very different incidence for chromosomal anomalies in iRAA and non-iRAA: 3,7% of iRAA associate a genetic anomaly, compared to 19,4% of non-iRAA that associate a genetic anomaly [9]. The same study highlights the increased incidence of extracardiac anomalies in non-iRAA compared to iRAA (39,4% vs. 11,1%). The incidence of extracardiac anomalies associated with 22q11.2 deletion was reported lower in iRAA compared to non-iRAA [26,27]. In our study group, we had two cases of iRAA that associated extracardiac anomalies and two cases of non-iRAA (both with TOF) of which one associated extracardiac malformations. Our results do not confirm the literature findings, but our group is too small to be statistically important on its own. However, the awareness of these genetic and structural cardiac associations is important because it provides important information for genetic counseling and prenatal ultrasound scan.
When detecting an iRAA, counseling parents towards genetic testing can be difficult. However, despite the high cost, CMA should be suggested in these cases. This method represents the only way to detect microdeletions and should be the first-tier test, rather than 22q11.2 deletion specific FISH and karyotyping. The relative risk for pathologic CMA results in iRAA cases varies between 4.56% (95% Cl 2.05-10.13, p<0.001)[28] and 6.32% (95% Cl 2.83-14.10, p<0.001) [29].
One limitation of this small study group is the fact that we cannot establish if there is a higher risk for 22q11.2 incidence in the three different types of RAA (with right DA, left DA, or DAA). Two cases presented type II RAA, one case type I RAA and one case DAA. The incidence was higher for type II RAA (2/4 cases), but the small number of cases does not allow for statistical interpretation.
In other studies the incidence of 22q11.2 deletion in DAA was 0% [30] and therefore, these authors do not consider an isolated DAA at risk for chromosomal abnormalities. In RAA type I and II the data is conflicting, but the general belief is that type I has a higher risk for abnormal genetic results [19,31].
However, we strongly recommend, based on our experience and on the data extracted from the literature, genetic testing, especially CMA, in all cases of RAA, including.
One important finding of our work was the association of 22q11.2DS in RAA cases with a small thymus. Out of the four cases included into this study, three had hypoplastic thymus (75%). All three cases were terminated and the anomalies were confirmed at autopsy. Searching the literature, we highlighted that a small thymus is highly correlated with 22q11.2 deletion. One study had a 100% rate of association between 22q11.2 deletion and thymic hypoplasia, all cases of deletion presented this extracardiac anomaly and all cases of RAA that were 22q11.2 negative had normal thymuses [7].
The same study cites a higher incidence of 22q11.2DS when extracardiac anomalies are present and considers the small thymus as a soft marker for this deletion. When we reevaluated our initial study group, the 19 RAA cases, the three cases that associated 22q11.2DS with thymus hypoplasia were the only cases with abnormal thymuses. Therefore, we support the affirmation that a hypoplastic or absent thymus represents an important marker for 22q11.2DS in RAA cases.
DiGeorge syndrome associates CHD and small thymus in approximately 90% of fetuses and infants, the TT ratio defining a small thymus when it is less than minus two standard deviations [7,32,33,34].
This calculation has a 96% sensitivity, 95% Cl 0.79-0.99 and if normal, reduces the risk of 22q11.2 by 90% [7,32].
Assuming that hypoplastic or absent thymus is an indicator for 22q11.2DS testing, the diagnosis criteria involves a lower thymus-thoracic (TT) ratio, less than 0.25 [16,32].
Albeit limited, the available experience concerning the TT ratio in DiGeorge syndrome is highly important and can guide the practitioner towards a fair prognosis and correct management, especially in low-income countries or populations, where a limited genetic testing,-karyotyping and FISH for the exclusion of 22q11.2 deletion, is not always available for RAA cases detected antepartum.
RAA has also a high risk of dysphonia and dysphagia due to tracheal and esophageal compression, especially in cases of DAA and RAA with a left DA [5,9,35].
Some authors highlight the concomitance of 22q11.2DS with superior airway tract anomalies in 70% of children diagnosed with this deletion, most important being the enlargement of the trachea and larynx and subglottic stenosis [36].
In our limited study group, we did not discover cases that associated RAA, 22q11.2DS and superior airway tract anomalies. The neonate who associated RAA, TOF and 22q11.2DS, did not present tracheal or esophageal compression symptoms after birth. However, these manifestations might occur in the following years, during childhood. The fetus was born in 2018 and until present days we did not receive data from the parents that might suspect these complications. It is important to mention that an extended follow-up, over a period of at least 10 years postpartum would be beneficial for a thoroughly investigation and accurate antenatal prognosis of the fetuses detected with this chronic illness.
Conclusions
We believe that this study demonstrates the need for invasive testing in all cases of RAA, including iRAA, because of the significant incidence of 22q11.2 deletion associated with these cases. Karyotyping and exclusion of this deletion should always be offered. CMA is also recommended, but the costs involved may limit the addressability, especially in low-resources settings.
A small or absent thymus represents a reliable marker for DiGeorge syndrome in fetuses with RAA and when detected 22q11.2DS testing should be recommended. The TT ratio can be easily obtained during fetal anomaly scan and is considered pathological when it is less than minus two standard deviations or less than 0.25.
Conflict of interests
The authors declare that they have no conflict of interests.
References
- 1.Achiron R, Rotstein Z, Heggesh J, Bronshtein M, Zimand S, Lipitz S, Yagel S. Anomalies of the fetal aortic arch: a novel sonographic approach to in-utero diagnosis. Ultrasound Obstet Gynecol. 2002;20(6):553–557. doi: 10.1046/j.1469-0705.2002.00850.x. [DOI] [PubMed] [Google Scholar]
- 2.Schleich JM. Images in cardiology. Development of the human heart: days 15-21. Heart. 2002;87(5):487–487. doi: 10.1136/heart.87.5.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavoretto PI, Sotiriadis A, Girardelli S, Spinillo S, Candiani M, Amodeo S, Farina A, Fesslova V. Postnatal Outcome and Associated Anomalies of Prenatally Diagnosed Right Aortic Arch with Concomitant Right Ductal Arch: A Systematic Review and Meta-Analysis. Diagnostics (Basel) 2020;10(10):831–831. doi: 10.3390/diagnostics10100831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prabhu S, Mehra S, Kasturi S, Tiwari R, Joshi A, John C, Karl TR. Anatomic classification of the right aortic arch. Cardiology in the Young. 2020;30(11):1694–1701. doi: 10.1017/S1047951120003601. [DOI] [PubMed] [Google Scholar]
- 5.Campanale CM, Pasquini L, Santangelo TP, Iorio FS, Bagolan P, Sanders SP, Toscano A. Prenatal echocardiographic assessment of right aortic arch. Ultrasound in Obstetrics & Gynecology. 2019;54(1):96–102. doi: 10.1002/uog.20098. [DOI] [PubMed] [Google Scholar]
- 6.Db M, Bj 3rd C, Pm W, Ml K, D M-M, Da D, Eh Z, E G. Association of chromosome 22q11 deletion with isolated anomalies of aortic arch laterality and branching. J Am Coll Cardiol. 2001;37(8):2114–2119. doi: 10.1016/s0735-1097(01)01286-4. [DOI] [PubMed] [Google Scholar]
- 7.Perolo A, De Robertis V, Cataneo I, Volpe N, Campobasso G, Frusca T, Ghi T, Prandstraller D, Pilu G, Volpe P. Risk of 22q11.2 deletion in fetuses with right aortic arch and without intracardiac anomalies. Ultrasound Obstet Gynecol. 2016;48(2):200–203. doi: 10.1002/uog.15766. [DOI] [PubMed] [Google Scholar]
- 8.Velipasaoglu M, Sentürk M, Ayaz R, Atesli B, Tanir HM. Characteristics of prenatally detected right aortic arch cases in a single institution. Journal of Obstetrics and Gynaecology. 2018;38(7):895–898. doi: 10.1080/01443615.2018.1430126. [DOI] [PubMed] [Google Scholar]
- 9.Miranda JO, Callaghan N, Miller O, Simpson J, Sharland G. Right aortic arch diagnosed antenatally: associations and outcome in 98 fetuses. Heart. 2014;100(1):54–59. doi: 10.1136/heartjnl-2013-304860. [DOI] [PubMed] [Google Scholar]
- 10.Zidere V, Tsapakis EG, Huggon IC, Allan LD. Right aortic arch in the fetus. Ultrasound in Obstetrics & Gynecology. 2006;28(7):876–881. doi: 10.1002/uog.3841. [DOI] [PubMed] [Google Scholar]
- 11.Poirsier C, Besseau-Ayasse J, Schluth-Bolard C, Toutain J, Missirian C, Le Caignec C, Bazin A, de Blois MC, Kuentz P, Catty M, Choiset A, Plessis G, Basinko A, Letard P, Flori E, Jimenez M, Valduga M, Landais E, Lallaoui H, Cartault F, Lespinasse J, Martin-Coignard D, Callier P, Pebrel-Richard C, Portnoi M-F, Busa T, Receveur A, Amblard F, Yardin C, Harbuz R, Prieur F, Le Meur N, Pipiras E, Kleinfinger P, Vialard F, Doco-Fenzy M. A French multicenter study of over 700 patients with 22q11 deletions diagnosed using FISH or aCGH. Eur J Hum Genet. 2016;24(6):844–851. doi: 10.1038/ejhg.2015.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindsay EA, Goldberg R, Jurecic V, Morrow B, Carlson C, Kucherlapati RS, Shprintzen RJ, Baldini A. Velo-cardio-facial syndrome: frequency and extent of 22q11 deletions. Am J Med Genet. 1995;57(3):514–522. doi: 10.1002/ajmg.1320570339. [DOI] [PubMed] [Google Scholar]
- 13.Lee M-Y, Won H-S, Baek JW, Cho J-H, Shim J-Y, Lee P-R, Kim A. Variety of prenatally diagnosed congenital heart disease in 22q11.2 deletion syndrome. Obstet Gynecol Sci. 2014;57(1):11–16. doi: 10.5468/ogs.2014.57.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noël A-C, Pelluard F, Delezoide A-L, Devisme L, Loeuillet L, Leroy B, Martin A, Bouvier R, Laquerriere A, Jeanne-Pasquier C, Bessieres-Grattagliano B, Mechler C, Alanio E, Leroy C, Gaillard D. Fetal phenotype associated with the 22q11 deletion. Am J Med Genet A. 2014;164(11):2724–2731. doi: 10.1002/ajmg.a.36720. [DOI] [PubMed] [Google Scholar]
- 15.Schindewolf E, Khalek N, Johnson MP, Gebb J, Coleman B, Crowley TB, Zackai EH, McDonald-McGinn DM, Moldenhauer JS. Expanding the fetal phenotype: Prenatal sonographic findings and perinatal outcomes in a cohort of patients with a confirmed 22q11.2 deletion syndrome. Am J Med Genet A. 2018;176(8):1735–1741. doi: 10.1002/ajmg.a.38665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Traisrisilp K, Tongprasert F, Srisupundit K, Luewan S, Tongsong T. Prenatal screening of DiGeorge (22q11.2 deletion) syndrome by abnormalities of the great arteries among Thai pregnant women. Obstet Gynecol Sci. 2020;63(3):330–336. doi: 10.5468/ogs.2020.63.3.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carvalho J, Allan L, Chaoui R, Copel J, DeVore G, Hecher K, Lee W, Munoz H, Paladini D, Tutschek B, Yagel S. ISUOG Practice Guidelines (updated): sonographic screening examination of the fetal heart: ISUOG Guidelines. Ultrasound in Obstetrics & Gynecology. 2013;41(3):348–359. doi: 10.1002/uog.12403. [DOI] [PubMed] [Google Scholar]
- 18.Galindo A, Nieto O, Nieto MT, Rodríguez‐Martín MO, Herraiz I, Escribano D, Granados MA. Prenatal diagnosis of right aortic arch: associated findings, pregnancy outcome, and clinical significance of vascular rings. Prenatal Diagnosis. 2009;29(10):975–981. doi: 10.1002/pd.2327. [DOI] [PubMed] [Google Scholar]
- 19.Bravo C, Gámez F, Pérez R, Álvarez T, De León-Luis J. Fetal Aortic Arch Anomalies: Key Sonographic Views for Their Differential Diagnosis and Clinical Implications Using the Cardiovascular System Sonographic Evaluation Protocol. J Ultrasound Med. 2016;35(2):237–251. doi: 10.7863/ultra.15.02063. [DOI] [PubMed] [Google Scholar]
- 20.Mogra R, Zidere V, Allan LD. Prenatally detectable congenital heart defects in fetuses with Down syndrome. Ultrasound Obstet Gynecol. 2011;38(3):320–324. doi: 10.1002/uog.8977. [DOI] [PubMed] [Google Scholar]
- 21.Gül A, Güngördük K, Yıldırım G. Perinatal outcomes and anomalies associated with fetal right aortic arch. J Turk Ger Gynecol Assoc. 2012;13(3):184–186. doi: 10.5152/jtgga.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans WN, Acherman RJ, Ciccolo ML, Carrillo SA, Mayman GA, Luna CF, Rollins RC, Castillo WJ, Galindo A, Rothman A, Alexander JA, Kwan TW, Restrepo H. Right aortic arch with situs solitus frequently heralds a vascular ring. Congenital Heart Disease. 2017;12(5):583–587. doi: 10.1111/chd.12487. [DOI] [PubMed] [Google Scholar]
- 23.Wójtowicz A, Respondek-Liberska M, Słodki M, Kordjalik P, Płużańska J, Knafel A, Huras H. The significance of a prenatal diagnosis of right aortic arch. Prenatal Diagnosis. 2017;37(4):365–374. doi: 10.1002/pd.5020. [DOI] [PubMed] [Google Scholar]
- 24.Razon Y, Berant M, Fogelman R, Amir G, Birk E. Prenatal diagnosis and outcome of right aortic arch without significant intracardiac anomaly. J Am Soc Echocardiogr. 2014;27(12):1352–1358. doi: 10.1016/j.echo.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 25.Orphanet Report Series, Rare Diseases collection, 2021, Prevalence of rare diseases: Bibliographic data[online] Available at: https://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf[ Accessed 02.07.2021]
- 26.O'Mahony EF, Hutchinson DP, McGillivray G, Nisbet DL, Palma-Dias R. Right-sided aortic arch in the age of microarray. Prenatal Diagnosis. 2017;37(5):440–445. doi: 10.1002/pd.5029. [DOI] [PubMed] [Google Scholar]
- 27.Luo Q, Chen J, Zhang Y, Li J, Su X, Wang Q, Ding Y. Incidence of chromosomal anomalies in fetuses with isolated right aortic arch: A meta-analysis. Prenatal Diagnosis. 2020;40(3):294–300. doi: 10.1002/pd.5606. [DOI] [PubMed] [Google Scholar]
- 28.Maya I, Singer A, Baris HN, Goldberg Y, Shalata A, Khayat M, Ben-Shachar S, Sagi-Dain L. Prenatal microarray analysis in right aortic arch-a retrospective cohort study and review of the literature. J Perinatol. 2018;38(5):468–473. doi: 10.1038/s41372-018-0062-6. [DOI] [PubMed] [Google Scholar]
- 29.Callaway JLA, Shaffer LG, Chitty LS, Rosenfeld JA, Crolla JA. The clinical utility of microarray technologies applied to prenatal cytogenetics in the presence of a normal conventional karyotype: a review of the literature. Prenatal Diagnosis. 2013;33(12):1119–1123. doi: 10.1002/pd.4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu X, Li Y, Su L, Xie X, Cai M, Lin N, Huang H, Lin Y, Xu L. Chromosomal Microarray Analysis for the Fetuses with Aortic Arch Abnormalities and Normal Karyotype. Molecular Diagnosis & Therapy. 2020;24(5):611–619. doi: 10.1007/s40291-020-00474-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berg C, Bender F, Soukup M, Geipel A, Axt-Fliedner R, Breuer J, Herberg U, Gembruch U. Right aortic arch detected in fetal life. Ultrasound Obstet Gynecol. 2006;28(7):882–889. doi: 10.1002/uog.3883. [DOI] [PubMed] [Google Scholar]
- 32.Chaoui R, Heling KS, Lopez AS, Thiel G, Karl K. The thymic-thoracic ratio in fetal heart defects: a simple way to identify fetuses at high risk for microdeletion 22q11. Ultrasound Obstet Gynecol. 2011;37(4):397–403. doi: 10.1002/uog.8952. [DOI] [PubMed] [Google Scholar]
- 33.Chaoui R, Kalache KD, Heling KS, Tennstedt C, Bommer C, Körner H. Absent or hypoplastic thymus on ultrasound: a marker for deletion 22q11.2 in fetal cardiac defects. Ultrasound in Obstetrics & Gynecology. 2002;20(6):546–552. doi: 10.1046/j.1469-0705.2002.00864.x. [DOI] [PubMed] [Google Scholar]
- 34.Oskarsdóttir S, Persson C, Eriksson BO, Fasth A. Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr. 2005;164(3):146–153. doi: 10.1007/s00431-004-1577-8. [DOI] [PubMed] [Google Scholar]
- 35.Shah RK, Mora BN, Bacha E, Sena LM, Buonomo C, Del Nido P, Rahbar R. The presentation and management of vascular rings: an otolaryngology perspective. Int J Pediatr Otorhinolaryngol. 2007;71(1):57–62. doi: 10.1016/j.ijporl.2006.08.025. [DOI] [PubMed] [Google Scholar]
- 36.Sacca R, Zur KB, Crowley TB, Zackai EH, Valverde KD, McDonald-McGinn DM. Association of airway abnormalities with 22q11.2 deletion syndrome. Int J Pediatr Otorhinolaryngol. 2017;96:11–14. doi: 10.1016/j.ijporl.2017.02.012. [DOI] [PubMed] [Google Scholar]