

Abstract
Background
Management of pediatric post‐transplantation lymphoproliferative disorder (PTLD) after hematopoietic stem cell (HSCT) and solid organ transplantation (SOT) is challenging.
Aim
This study of 34 PTLD patients up to 19‐years old diagnosed in Austria from 2000 to 2018 aimed at assessing initial characteristics, therapy, response, and outcome as well as prognostic markers of this rare pediatric disease.
Methods and results
A retrospective data analysis was performed. Types of allografts were kidney (n = 12), liver (n = 7), heart (n = 5), hematopoietic stem cells (n = 4), lungs (n = 2), multi‐visceral (n = 2), small intestine (n = 1), and vessels (n = 1). Eighteen/34 were classified as monomorphic PTLD, with DLBCL accounting for 15 cases. Polymorphic disease occurred in nine, and non‐destructive lesions in six cases. One patient had a non‐classifiable PTLD. Thirteen/34 patients are surviving event‐free in first remission (non‐destructive, n = 4/6; polymorphic, n = 4/9; monomorphic, n = 6/18). Fourteen/34 patients lacked complete response to first‐line therapy, of whom seven died. Four/34 patients relapsed, of whom two died. In 3/34 patients, death occurred as a first event. The 5‐year overall and event‐free survival rates were 64% ± 9% and 35% ± 9% for the whole cohort. Among all parameters analyzed, only malignant disease as the indication for transplantation had a significantly poor influence on survival.
Conclusions
This study shows PTLD still to be a major cause of mortality following SOT or HSCT in children. A continued understanding of the molecular biology of the disease shall allow to decrease treatment intensity for lower risk patients and to identify patients who may benefit from newer therapy approaches to improve outcome and decrease morbidity.
Keywords: hematopoietic stem cell transplantation, outcome, post‐transplant lymphoproliferative disease, solid organ transplantation, treatment
Abbreviations
- BM
bone marrow
- B‐NHL
B‐cell non‐Hodgkin lymphoma
- CCR
continuous complete remission
- CHOP
cyclophosphamide, doxorubicin, vincristine, and prednisolone
- CNS
central nervous system
- CR
complete remission
- CSF
cerebrospinal fluid
- DLBCL
diffuse large B‐cell lymphoma
- EBER
EBV‐encoded RNA
- EBV
Epstein‐Barr virus
- EFS
event‐free survival
- EPOCH
etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin
- GIT
gastrointestinal tract
- HLA
human leukocyte antigen
- HSCT
hematopoietic stem cell transplantation
- IPNHLSS
International Pediatric Non‐Hodgkin Lymphoma Staging System
- LDH
lactate dehydrogenase
- LPD
lymphoproliferative disease
- m‐COMP
modified ‐ cyclophosphamide, vincristine, methotrexate, and prednisolone
- NK‐cells
natural killer cells
- OS
overall survival
- PB
peripheral blood
- PCR
polymerase chain reaction
- PD
progressive disease
- PH
plasmacytic hyperplasia
- PR
partial remission
- PTLD
post‐transplant lymphoproliferative disease
- R‐CHOP
rituximab‐cyclophosphamide, doxorubicin, vincristine, and prednisolone
- RIS
reduction of immunosuppression
- SD
stable disease
- SOT
solid organ transplantation
- WHO
World Health Organization
1. INTRODUCTION
In transplantation medicine prevention and treatment of graft rejection is the primary aim of immunosuppressive medication. Besides this, iatrogenic immunosuppression is accompanied by serious side effects, such as an increased infection susceptibility and a higher risk of developing malignancies due to reduced tumor surveillance. 1 The latter can result in post‐transplant lymphoproliferative disease (PTLD), which is a heterogeneous disorder ranging from benign hyperplasia to malignant lymphomas. 2 , 3 , 4
In pediatric patients, PTLD represents the most frequent type of malignant diseases secondary to solid organ transplantation (SOT) or hematopoietic stem cell transplantation (HSCT), with the overall risk of developing malignancies being 45‐fold higher than in healthy individuals. 5 , 6 , 7 Epstein‐Barr virus (EBV) infection is detected in most cases of pediatric PTLD. 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 Whereas >90% of the world's adult population already harbor EBV, the infestation rate is comparatively low in children. 16 , 17 Transplantation of grafts from EBV‐seropositive adults results in primary EBV infection of children, thereby explaining a 2‐ to 4‐fold higher risk of PTLD in children compared to adults. 5 , 15 , 18 , 19 , 20 Reflecting the age‐dependent seroprevalence rates, EBV‐positivity occurs in only 50% of adult as compared to 85%‐90% of pediatric PTLD. 15 , 16 , 19 , 21 , 22 , 23 Notably, pediatric EBV‐negative PTLD typically develops late, that is, >1 year after transplantation. 4 , 8 , 11 , 16 , 24
The World Health Organization (WHO) classification system differentiates between (i) non‐destructive, benign forms, showing polyclonal cell populations only, (ii) polymorphic subtypes, which can present with either polyclonal or monoclonal proliferation patterns, and (iii) monomorphic, monoclonal subtypes, which are genuine malignant lymphomas, mostly of B‐ and, less frequently, of T‐ or NK‐cell origin. 8 , 9
The incidence of lymphoproliferative diseases (LPD) following transplantations varies between 1% and 30%, depending on the presence of risk factors, 25 , 26 the most important of which are a mismatched EBV serostatus between recipient and donor, 8 , 10 , 11 , 27 , 28 a high‐intensity immunosuppressive regimen, 3 , 11 , 12 , 19 , 29 and the type of allograft. The highest risk for PTLD after SOT results from having an EBV‐positive organ donor and an EBV‐naïve recipient. For PTLD following HSCT, a converse serological EBV constellation of donor and recipient is an established risk factor. Transplantation of intestine or lungs poses the highest risk for PTLD (20%), 12 , 26 , 30 , 31 , 32 followed by cardiac or liver (2%‐10%), 29 , 33 , 34 and kidney transplantations (1%‐4%). 5 , 19 , 29 , 35 , 36 HSCT shows an incidence of 1%‐8%, likewise depending on a range of risk factors, such as transplantation of T‐cell depleted bone marrow (BM) or HLA‐mismatched grafts from unrelated donors, as well as the use of anti‐thymocyte globulin. 37 , 38 , 39 , 40 Concerning the latency period after transplantation, LPDs can be grouped into early‐ and late‐onset diseases, referring to cases within and later than the first year after transplantation. 25
Herein, we describe 34 SOT and HSCT patients with PTLD diagnosed in Austria within a period of nearly 20 years.
2. PATIENTS AND METHODS
2.1. Patients
Patients had to fulfill three inclusion criteria to be enrolled into this retrospective study: (i) diagnosis of PTLD as established by a reference pathologist according to the WHO classification valid at the time of diagnosis, (ii) diagnosis and treatment of PTLD between 2000 and 2018, and (iii) treatment of PTLD carried out at one of the four participating Austrian centers offering performance and aftercare of SOT and/or HSCT. Thirty‐four patients aged <19 years meeting the inclusion criteria were identified and, hence, included in our study.
3. METHODS
Classification of PTLD was based on histopathology according to the contemporary WHO classification system. 41 , 42 EBV‐association was defined by a positive in‐situ hybridization test of EBV‐encoded RNA (EBER) in the tissue(s) analyzed. Patient data included demographics and information on disease, treatment, response, and outcome. Serum analysis was performed to detect EBV in peripheral blood. In dependence of the laboratory, to which the samples were submitted, the threshold for EBV positivity was either ≥5 or ≥ 100 copies/ml. Tumor stage was retrospectively defined according to the International Pediatric Non‐Hodgkin Lymphoma Staging System (IPNHLSS) whenever possible. 43 , 44 Evaluation of response was documented at the end of each therapy line (complete remission, CR; partial remission, PR; stable disease, SD; progressive disease, PD). Accordingly, two subgroups were defined: (i) patients with complete response, including patients with CR at the end of a respective therapy line, and (ii) patients who lacked complete response, including all patients with PR, SD, and PD. Due to the lack of standardized imaging procedures used throughout the therapy and the retrospective nature of our study, our data on response were based on the results documented by the local treating physicians. Fatalities were classified as either (i) PTLD‐related, (ii) therapy of PTLD‐related, or (iii) PTLD‐unrelated deaths.
All patients were treated after informed consent from the patient, patient's parents or legal guardians had been obtained.
3.1. Statistical analysis
Event‐free survival (EFS) was calculated from the date of diagnosis to the date of first event. Events considered were lacking complete response at the end of a therapy line, relapse, or death, whichever occurred first. Overall survival (OS) was defined as the time from diagnosis to death from any cause or the date of last follow‐up. EFS and OS were estimated according to the Kaplan‐Meier method; differences between groups were evaluated with the log‐rank test. P‐values ≤0.05 were considered statistically significant.
4. RESULTS
4.1. Initial characteristics
Demographics, histopathological results and EBV‐status are shown in Tables 1 and 2. Among the 34 PTLD patients analyzed, median age was 8.78 years and the male‐to‐female ratio 1:1. Types of allografts were kidney (n = 12), liver (n = 7), heart (n = 5), hematopoietic stem cells (n = 4), lung (n = 2), multi‐visceral (n = 2), small intestine (n = 1), and vessels (n = 1). All patients received one or more immunosuppressive drugs at the time of PTLD onset with 24 receiving ≥2 immunosuppressive drugs.
TABLE 1.
Clinical and laboratory characteristics of the 34 patients with PTLD according to the histological subtype
| Non‐destructive PTLD | Polymorphic PTLD | Monomorphic PTLD | Not classifiable PTLD | ∑ | |
|---|---|---|---|---|---|
| No. of patients | 6 (18%) | 9 (26%) | 18 (53%) | 1 (3%) | 34 |
| Gender | |||||
| female | 2 (33%) | 5 (56%) | 10 (56%) | 0 | 17 (50%) |
| male | 4 (67%) | 4 (44%) | 8 (44%) | 1 | 17 (50%) |
| Condition leading to Tx | |||||
| malignant | 0 | 0 | 3 (17%) | 0 | 3 (9%) |
| non‐malignant, congenital | 4 (67%) | 5 (56%) | 13 (72%) | 0 | 22 (64%) |
| non‐malignant, acquired | 2 (33%) | 4 (44%) | 2 (11%) | 1 | 9 (27%) |
| Organ Tx | |||||
| liver a | 3 (50%) | 1 (11%) | 3 (17%) | 0 | 7 (21%) |
| heart | 0 | 3 (33%) | 2 (11%) | 0 | 5 (15%) |
| lung | 0 | 0 | 2 (11%) | 0 | 2 (6%) |
| kidney | 2 (33%) | 3 (33%) | 7 (39%) | 0 | 12 (35%) |
| HSCT | 0 | 0 | 3 (17%) | 1 | 4 (12%) |
| other | 1 (17%) | 2 (22%) | 1 (5%) | 0 | 4 (12%) |
| Time from Tx to PTLD | |||||
| median (years) | 1.23 | 1.04 | 0.63 | / | 0.65 |
| range (years) | 0.35‐2.56 | 0.19‐12.56 | 0.06‐11.68 | / | 0.06‐12.56 |
| <1 year | 3 (50%) | 4 (44%) | 11 (61%) | 1 | 19 (56%) |
| ≥1 year | 3 (50%) | 5 (56%) | 7 (39%) | 0 | 15 (44%) |
| Age at PTLD onset | |||||
| median | 3.04 | 5.1 | 11.69 | / | 8.78 |
| range | 0.88‐16.63 | 2.52‐16.45 | 3.07‐21.81 | / | 0.88‐21.81 |
| <10 years | 4 (67%) | 6 (67%) | 9 (50%) | 0 | 19 (56%) |
| ≥10 years | 2 (33%) | 3 (33%) | 9 (50%) | 1 | 15 (44%) |
| Immunsuppression at onset of PTLD | |||||
| tacrolimus | 6 (100%) | 8 (89%) | 12 (67%) | 0 | 26 (77%) |
| prednisolone | 3 (50%) | 4 (44%) | 11 (61%) | 0 | 18 (53%) |
| mycophenolate mofetil | 2 (33%) | 5 (56%) | 9 (50%) | 0 | 16 (47%) |
| cyclosporin A | 0 | 1 (11%) | 5 (28%) | 1 | 7 (21%) |
| other | 1 (17%) | 2 (22%) | 2 (11%) | 0 | 5 (15%) |
| 1 drug | 3 (50%) | 1 (11%) | 5 (28%) | 1 | 10 (29%) |
| ≥2 drugs | 3 (50%) | 8 (89%) | 13 (72%) | 0 | 24 (71%) |
| B‐Symptoms | 3 (50%) | 5 (56%) | 4 (22%) | 1 | 13 (38%) |
| Pre‐therapeutic LDH level | |||||
| ≥500 U/L | 4 (67%) | 7 (78%) | 13 (72%) | 1 | 25 (74%) |
| <500 U/L | 2 (33%) | 2 (22%) | 5 (28%) | 0 | 9 (26%) |
Abbreviations: No., number; Tx, transplantation; LDH; lactate dehydrogenase; PTLD, post‐transplant lymphoproliferative disease.
There was one patient included who developed the PTLD after liver transplantation, but had also undergone a previous kidney transplantation.
TABLE 2.
Immunohistochemistry, EBV‐status, and site/stage of disease of the 34 patients with PTLD according to the histological subtype
| Non‐destructive PTLD | Polymorphic PTLD | Monomorphic PTLD | Not classifiable PTLD | ∑ | |
|---|---|---|---|---|---|
| No. of patients | 6 (18%) | 9 (27%) | 18 (53%) | 1 (3%) | 34 |
| CD20‐expression | |||||
| positive | 4 (67%) | 8 (89%) | 15 (83%) | 1 | 28 (82%) |
| negative | 2 (33%) | 1 (11%) | 3 (17%) | 0 | 6 (18%) |
| EBER status of samples | |||||
| EBER‐positive | 4 (67%) | 9 (100%) | 16 (89%) | 1 | 30 (88%) |
| EBER‐negative | 2 (33%) | 0 | 2 (11%) | 0 | 4 (12%) |
| EBV‐PCR results of PB | |||||
| positive | 5 (83%) | 9 (100%) | 15 (83%) | 1 | 30 (88%) |
| negative | 1 (17%) | 0 | 1 (6%) | 0 | 2 (6%) |
| not available | 0 | 0 | 2 (11%) | 0 | 2 (6%) |
| <10.000 copies/ml | 4 (66%) | 4 (44%) | 8 (44%) | 0 | 16 (47%) |
| ≥10.000 copies/ml | 1 (17%) | 5 (56%) | 7 (39%) | 1 | 14 (41%) |
| not available | 1 (17%) | 0 | 3 (17%) | 0 | 4 (12%) |
| <100.000 copies/ml | 4 (66%) | 6 (67%) | 11 (61%) | 0 | 21 (62%) |
| ≥100.000 copies/ml | 1 (17%) | 3 (33%) | 4 (22%) | 1 | 9 (26%) |
| not available | 1 (17%) | 0 | 3 (17%) | 0 | 4 (12%) |
| Stage of disease | |||||
| I | 0 | 0 | 0 | 0 | 0 |
| II | 1 (17%) | 0 | 1 (5%) | 0 | 2 (6%) |
| III | 1 (17%) | 2 (22%) | 5 (28%) | 0 | 8 (24%) |
| IV/Burkitt leukemia | 0 | 1 (11%) | 5 (28%) | 0 | 6 (18%) |
| not available | 4 (66%) | 6 (67%) | 7 (39%) | 1 | 18 (53%) |
| Site of involvement | |||||
| single site | 0 | 0 | 1 (6%) | 0 | 1 (3%) |
| graft | 0 | 2 (22%) | 2 (11%) | 0 | 4 (12%) |
| lungs | 0 | 1 (11%) | 3 (17%) | 0 | 4 (12%) |
| lymph nodes regarless of other sites | 5 (83%) | 9 (100%) | 16 (89%) | 1 | 31 (91%) |
| lymph nodes only | 1 (17%) | 3 (33%) | 4 (22%) | 0 | 8 (24%) |
| gastrointestinal tract | 2 (33%) | 2 (22%) | 3 (17%) | 0 | 7 (21%) |
| upper airway | 3 (50%) | 1 (11%) | 6 (33%) | 0 | 10 (29%) |
| central nervous system | 0 | 0 | 2 (11%) | 0 | 2 (6%) |
| bone marrow | 0 | 1 (11%) | 4 (22%) | 0 | 5 (15%) |
| others | 0 | 2 (22%) | 6 (33%) | 1 | 9 (27%) |
Abbreviations: No., number; EBER, EBV‐encoded RNA; EBV, Epstein–Barr virus; PCR, polymerase chain reaction; PB, peripheral blood; PTLD, post‐transplant lymphoproliferative disease.
We aimed at staging the 34 patients according to the IPNHLSS. In 18 patients staging was incomplete, as pre‐therapeutic bone marrow (BM) and cerebrospinal fluid (CSF) were not assessed. Of the 16 patients evaluable for staging, two showed stage II disease, eight stage III disease, and six stage IV disease or Burkitt's leukemia. The patient with Burkitt's leukemia showed lesions in the CNS, too.
The most frequently involved sites of disease were the lymph nodes (n = 31/34), however, only in 8/34 patients the disease was solely restricted to the lymph nodes. Other sites of involvement were the gastrointestinal tract (GIT; n = 7/34), upper airways including the Waldeyer's ring (n = 10/34), lungs (n = 4/34), BM (n = 5/34), CNS (n = 2/34), and others (n = 9/34). Graft involvement was diagnosed in four cases (lungs, n = 1; kidney, n = 1; GIT, n = 2). Only 1/34 patients presented with uni‐locular disease (lingula).
Eighteen/34 cases were classified as monomorphic PTLD, all of which were of B‐cell origin. Diffuse large B‐cell lymphoma (DLBCL) accounted for the vast majority of this group (n = 15) occurring after SOT in 12/15 and after HSCT in 3/15 cases. The other three patients had a plasmacytoma‐like PTLD, plasmablastic lymphoma, and Burkitt's leukemia, respectively, all occurring after SOT. Polymorphic disease occurred in 9/34 cases, and non‐destructive lesions in 6/34 cases, all occurring after SOT. The latter group included five patients with plasmacytic hyperplasia (PH) and one with mononucleosis‐like PTLD. In 1/34 patients (after HSCT), the PTLD could not be subclassified. In two patients, synchronous detection of different subtypes was seen: one case of polymorphic disease showed elements of mononucleosis‐like PTLD, and one case with PH showed aspects of florid follicular hyperplasia. Twenty‐eight/34 demonstrated CD20‐expression. The CD20‐negative cases included plasmacytic hyperplasia (n = 2), polymorphic PTLD (n = 1), plasmablastic lymphoma (n = 1), and DLBCL (n = 2), respectively.
Depending on the latency period between transplantation and occurrence of PTLD, 19 occurred within the first year after transplantation. The median interval between transplantation and PTLD diagnosis was 0.65 years for all patients. All four cases of PTLD following HSCT occurred within the first year.
In 32/34 patients, serum PCR analysis was performed for EBV detection at the time of diagnosis with 30/32 being positive. EBV‐positive disease was defined by histological examination of the tumor, showing that 30 were EBER‐positive. Notably, two patients presented with EBER‐positivity of the samples, but had negative EBV‐PCR in PB.
All nine cases of polymorphic PTLD presented with EBV‐positive PTLD, while this was seen in 4/6 and 16/18 of non‐destructive and monomorphic PTLD, respectively. The patient with non‐classifiable PTLD was EBV‐positive. Of all cases occurring within 1 year after transplantation, all but one patient showed EBER‐positivity (n = 18/19). The remaining patient had a negative EBER‐status of the tumor, but positive EBV‐PCR in PB. Of the four EBV‐negative cases, two had non‐destructive and two monomorphic PTLD, and, interestingly, all were CD20‐negative. In three of the four cases with negative EBER‐reactions, EBV was detectable in PB.
4.2. Treatment and response
A detailed description of the therapy, response and outcome of the 34 patients is given in Figure 1 and Table 3. One/34 patients died from disease before any therapy. Reduction of immunosuppression (RIS) was the first therapeutic step taken in 32 of the remaining 33 individuals. The patient without RIS directly received rituximab followed by chemotherapy, which led to a CR. Three/32 patients with RIS required no further therapy. Two of them achieved a CR, the remaining patient's lesions switched from polymorphic to non‐destructive PTLD (classified as PR). Two/32 patients with RIS, including one with localized disease of the lingula and one with a supraglottic tumor and cervical lymphadenopathy, had a surgical approach of the disease only. Thereby both achieved a CR.
FIGURE 1.
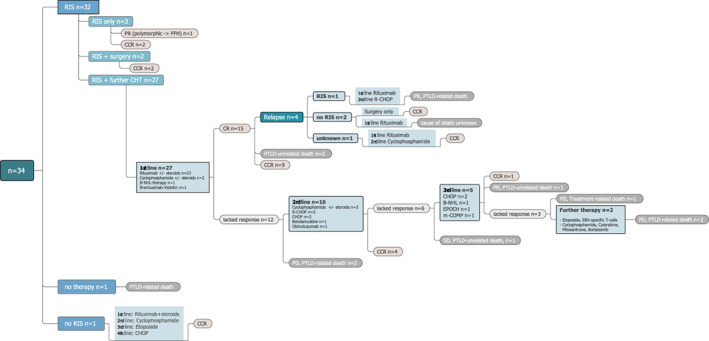
Flow chart of the therapy and outcome of the 34 PTLD patients. RIS, reduction of immunosuppression; PR, partial remission; FFH, florid follicular hyperplasia; CCR, continuous complete remission; B‐NHL, B‐cell non‐Hodgkin lymphoma; PTLD, post‐transplant lymphoproliferative disease; CHOP, cyclophosphamide, doxorubicin, vincristine and prednisolone; CR, complete response; R‐CHOP, rituximab‐CHOP; PD, progressive disease; EPOCH, etoposide, prednisolone, vincristine, cyclophosphamide and doxorubicin; m‐COMP, modified ‐ cyclophosphamide, vincristine, methotrexate and prednisone; SD, stable disease; EBV, Epstein–Barr virus
TABLE 3.
Response and outcome of the 34 patients with PTLD according to the histological subtype
| Non‐destructive PTLD | Polymorphic PTLD | Monomorphic PTLD | Not classifiable PTLD | ∑ | |
|---|---|---|---|---|---|
| No. of patients | 6 (18%) | 9 (27%) | 18 (53%) | 1 (3%) | 34 |
| Response of initial disease | |||||
| complete response | 6 (100%) | 5 (56%) | 14 (78%) | 0 | 25 (74%) |
| partial response | 0 | 1 (11%) | 1 (6%) | 0 | 2 (6%) |
| stable disease | 0 | 1 (11%) | 0 | 0 | 1 (3%) |
| progressive disease | 0 | 1 (11%) | 3 (17%) | 1 | 5 (15%) |
| not evaluable | 0 | 1 a (11%) | 0 | 0 | 1 (3%) |
| First Event | |||||
| no event | 4 (67%) | 3 (33%) | 6 (33%) | 0 | 13 (38%) |
| lack of complete response | 2 (33%) | 3 (33%) | 8 (44%) | 1 | 14 (41%) |
| relapse | 0 | 2 (22%) | 2 (11%) | 0 | 4 (12%) |
| death | 0 | 1 | 2 | 0 | 3 (9%) |
| Response of first relapse | 0 | 2 | 2 | 0 | 4 |
| complete response | 0 | 1 (50%) | 1 (50%) | 0 | 2 (50%) |
| partial response | 0 | 1 (50%) | 0 | 0 | 1 (25%) |
| stable disease | 0 | 0 | 0 | 0 | 0 |
| progressive disease | 0 | 0 | 0 | 0 | 0 |
| unknown | 0 | 0 | 1 (50%) | 0 | 1 (25%) |
| Overall outcome | |||||
| median follow‐up (years) | 1.27 | 7.81 | 6.48 | / | 5.68 |
| range of follow‐up (years) | 0.37‐2.54 | 1.45‐16.05 | 0.14‐19.56 | / | 0.14‐19.56 |
| CCR | 6 (100%) | 4 (44%) | 11 (61%) | 0 | 21 (62%) |
| First CCR | 6 (100%) | 3 (33%) | 10 (56%) | 0 | 19 (56%) |
| Second CCR | 0 | 1 (11%) | 1 (6%) | 0 | 2 (6%) |
| death | 0 | 4 (44%) | 7 (39%) | 1 | 12 (35%) |
| PTLD‐related | 0 | 3 (33%) | 3 (17%) | 0 | 6 (18%) |
| therapy of PTLD‐related | 0 | 0 | 0 | 1 | 1 (3%) |
| not‐PTLD‐related | 0 | 1 (11%) | 3 (17%) | 0 | 4 (12%) |
| cause of death unknown | 0 | 0 | 1 (6%) | 0 | 1 (3%) |
Abbreviations: No., number; CCR, continuous complete remission; PTLD, post‐transplant lymphoproliferative disease.
One patient did not receive any therapy at all.
Of the 27/32 patients with RIS who received further treatment, 15 achieved a CR, while 12 lacked complete response including two progressing during first‐line therapy and dying from the PTLD. Four/15 patients with CR following first‐line therapy relapsed, 2/15 died in first CR due to PTLD‐unrelated causes and 9/15 remained in first continuous CR (CCR). Two of the four relapsed patients achieved a second CCR, the other two died (unknown cause, n = 1; PTLD‐related, n = 1).
Out of the 12 patients who lacked complete response to first‐line therapy, four achieved a first CCR with second‐line therapy, one with stable disease died from a PTLD‐unrelated cause, and five lacked complete response to second‐line therapy. One of the latter achieved a first CCR following third‐line therapy, one attained a PR and died (PTLD‐unrelated), and another patient progressed and died from third‐line therapy‐associated toxicity. The remaining two patients lacking complete response to third‐line therapy proceeded to further chemotherapy, but subsequently succumbed to PTLD.
4.3. Outcome according to therapy
Twenty‐three/27 patients with RIS and further therapy were treated with rituximab as part of their first‐line therapy with or without additional steroids. First‐line therapy in the remaining four consisted of cyclophosphamide and steroids in two, and brentuximab‐vedotin and B‐cell non‐Hodgkin lymphoma (B‐NHL)‐based chemotherapy in each one patient. Fifteen of the 27 patients achieved a CR.
Ten/12 patients with RIS and additional treatment, who lacked complete response to first‐line therapy, proceeded to polychemotherapy in nine (R‐CHOP, n = 3; CHOP, n = 2; cyclophosphamide±steroids, n = 3; bendamustine, n = 1) and obinutuzumab in one patient. Five/six patients without complete response to second‐line therapy proceeded to chemotherapy (CHOP, n = 2; B‐NHL therapy, n = 1; EPOCH, n = 1; m‐COMP, n = 1), and two of three patients, who lacked complete response to third‐line therapy, subsequently received other therapies such as etoposide and EBV‐specific T‐cells in one, and cyclophosphamide followed by cytarabine, mitoxantrone, and bortezomib in the other patient.
Of all 14 patients treated with rituximab only (in addition to RIS), 13 achieved a CR, one showed disease progression at the end of therapy, and three patients relapsed. Classical B‐NHL therapies were applied in one case of Burkitt's leukemia as first‐line and in another case of DLBCL as third‐line therapy. The latter achieved a PR and died from a PTLD‐unrelated cause; the former achieved a CCR.
4.4. Outcome according to histology
Of all six patients with non‐destructive disease, four received rituximab±steroids, and two lacked complete response and received second‐line therapy (Table 3). All patients finally achieved a CCR.
Among the nine patients with polymorphic disease, eight received RIS upfront, and one patient died before having received any therapy. Two/eight patients received RIS only, by which one achieved a CCR. The other patient's lesions regressed from polymorphic to non‐destructive PTLD (classified as PR). Six/8 patients received rituximab±steroids, with four achieving a CR and two who did not (both died). Two of the four patients relapsed, of whom one died from the PTLD, and the other one achieved a second CCR. Of all eight patients with polymorphic disease having received treatment, three achieved a first CCR, and one a second CCR, while one showed a PR and three patients died (PTLD‐related, n = 2; PTLD‐unrelated, n = 1) (Table 3).
Monomorphic PTLD was documented in 18 cases, including the only patient without RIS upfront. One/18 achieved a CCR by RIS and surgery, the other 17 received additional therapy (rituximab±steroids, n = 13; cyclophosphamide±steroids, n = 2; B‐NHL therapy, n = 1; brentuximab‐vedotin, n = 1). Eleven/17 patients received first‐line therapy only, whereby nine achieved a CR, and the other two died of progressive disease. Of the nine patients in CR after first‐line treatment, five remained in CCR, two relapsed (death, n = 1; second CCR, n = 1), and two died from a PTLD‐unrelated cause.
Six/17 patients received second‐line therapy (cyclophosphamide, n = 2; R‐CHOP, n = 2; CHOP, n = 1; bendamustine, n = 1), by which two achieved a CR, whereas four proceeded to third‐line therapy due to lack of complete response (B‐NHL therapy, n = 1; etoposide, n = 1; m‐COMP, n = 1; EPOCH, n = 1). Thereby, one achieved a CCR, and another one died from a PTLD‐unrelated cause. The remaining two patients subsequently received further therapy, by which one patient achieved a CCR, and the other one died from PTLD progression. Overall, 11/18 patients with monomorphic disease achieved a CCR, while 7/18 patients died (PTLD‐related, n = 3, PTLD‐unrelated, n = 3, unknown cause, n = 1).
The one/34 patients with unclassifiable PTLD received RIS and rituximab+steroids first, to which he lacked response and proceeded to cyclophosphamide and polychemotherapy (CHOP). He died from a treatment‐related complication.
Notably, two/24 patients having received rituximab within first‐line therapy showed CD20‐negative disease. One of them presented with plasmacytic hyperplasia and was treated with RIS + rituximab, and the other one had a DLBCL and received rituximab followed by chemotherapy (cyclophosphamide, etoposide, and CHOP). Both achieved a CR.
4.5. Outcome according to type of transplantation
Response and outcome of the 4 HSCT as compared to the 30 SOT patients are shown in Supplemental Table 1. Although numbers are small, results suggest a worse response and outcome for the HSCT patients.
4.6. Events
Thirteen/34 patients experienced no events and are alive in first CCR. Fourteen/34 patients lacked complete response to first‐line therapy. Out of them, seven died (PTLD‐related, n = 4, PTLD‐unrelated, n = 2, treatment‐related, n = 1). Four/34 patients relapsed, of whom 2 died (PTLD‐related, n = 1; unknown cause, n = 1). Among the remaining 3/34 patients, death occurred as a first event, of which one was PTLD‐related, and the other two were PTLD‐unrelated.
Six/12 deaths were considered PTLD‐related, with five occurring during first‐line treatment (progression, n = 4; EBV‐associated hemophagocytic lymphohistiocytosis, n = 1). The remaining one died from PTLD‐related gastrointestinal perforation during relapse therapy. One patient died amidst relapse therapy due to an unknown cause. One case of candida septicemia was considered a PTLD‐treatment‐related death. The group of 4/12 PTLD‐unrelated deaths consisted of a septicemia, an acute respiratory distress syndrome combined with secondary graft failure following HSCT, a diffuse alveolar hemorrhage syndrome, and a cardiac arrest, respectively.
4.7. Survival rates
Median follow‐up for surviving patients was 5.68 years as calculated from the time of diagnosis of primary PTLD diagnosis. On the date of last follow‐up 22 of all 34 patients were alive in CCR. The 5‐year OS and EFS rates were 64% ± 9% and 35% ± 9% for the whole study cohort, respectively (Figure 2). When evaluating the 5‐year OS according to the PTLD subtypes, it was 100% for non‐destructive, 53% ± 17% for polymorphic, and 65% ± 12% for monomorphic PTLD. Five‐year EFS according to the PTLD subtypes was 53% ± 25% for non‐destructive, 33% ± 16% for polymorphic, and 36% ± 12% for monomorphic PTLD.
FIGURE 2.
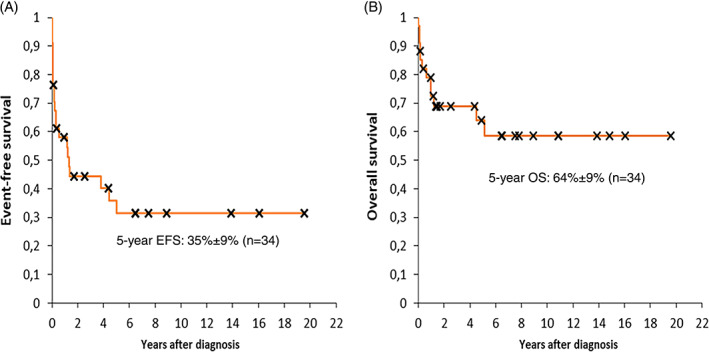
5‐year event‐free survival (A) and overall survival (B) of the 34 patients with PTLD. EFS, event‐free survival; OS, overall survival. [Correction added on 12 April 2021, after first online publication: The text “5‐year EFS: 35%±9% (n=34)” has been added in figure 2 A image in this version]
A variety of factors were evaluated concerning their impact on OS and EFS (Supplemental Table 2): Diagnosis indicating transplantation was the only factor having an effect on OS with a significantly poorer survival in cases with malignant diseases (P = 0.002), though numbers were very small. None of the factors analyzed had a significant influence on EFS.
5. DISCUSSION
PTLD represents a heterogenous group of excessive lymphoid proliferation, generally B‐lymphocytes, that occurs in the setting of suppressed T‐cell function, usually after SOT. In HSCT patients, it often represents a fatal risk occurring relatively early after transplantation. EBV infection has long been implicated as a causative factor in the development of PTLD. 8 , 11 , 21 , 45 , 46 , 47 , 48 In our study, 30 of 34 cases showed a positive EBER‐reaction of the tumor, thereby confirming the high rates of association seen in previous pediatric studies. Our data matched previous reports on monomorphic and polymorphic PTLD with 16/18 and 9/9 of the patients being EBER‐positive, while only 4/6 patients with non‐destructive PTLD were EBER‐positive. Interestingly, among the four EBV‐negative cases, median time from transplantation until PTLD was 6.0 years as compared to 2.3 years among the EBV‐positive patients. Accordingly, all four EBER‐negative patients had a CD20‐negative tumor, but no reduced survival.
The incidence of LPD following transplantations shows a biphasic distribution, with a first peak occurring within 12 months, and another either at 3 to 5 or at 7 to 10 years after transplantation. 11 , 25 , 49 , 50 , 51 , 52 Our study showed that half of the patients presented with early‐onset disease at a median time of 0.44 years and half with late‐onset disease at a median time of 5.61 years after transplantation. In accordance with the literature, all four patients with PTLD after HSCT were diagnosed within the first year after transplantation. 9 , 37 , 53
The majority of PTLDs present as multi‐locular disease, affecting both nodal and extranodal sites. Virtually every organ can be involved, the graft itself being a common site, whereas the most frequently affected extranodal site is the intestine. 15 , 54 , 55 , 56 We found that the vast majority (n = 33) involved multiple sites and the most frequently affected site were the lymph nodes (n = 31) followed by the upper airways (n = 10) and intestinal tract (n = 7). The graft itself was affected in four of our 34 cases.
Since experience with PTLD in childhood is limited to small case series and studies, treatment is not fully standardized and frequently adjusted to the affected individual, thereby addressing the multitude of clinical and histological presentations. Restoration of the patients' EBV‐specific T‐cell response achieved by RIS is widely accepted as the treatment of first choice independent of the organ graft. However, there is no consensus about the impact of a single immunosuppressive agent and the risk of acute graft‐rejection is assessed differently by the respective organ specialist. 11 , 35 , 57 , 58 Remarkably, none of the 32 patients included in our study who received RIS suffered from graft‐failure or ‐loss due to PTLD‐therapy. Non‐destructive PTLD and low‐risk patients show high response rates to RIS alone, but it is not recommended as a sole therapy in cases of monomorphic PTLD, aggressive disease or patients with a high tumor burden. 11 , 35 Among our cohort, three underwent RIS only (non‐destructive, n = 1; polymorphic, n = 2) and thereby achieved a CCR (n = 2) or PR (n = 1), and another 2 patients achieved CCR by RIS and surgery only (non‐destructive, n = 1; monomorphic, n = 1).
Another substantial part of PTLD‐therapy is B‐cell depletion due to the use of rituximab. It is recommended as first‐line treatment for all CD20‐positive subtypes sequential or in parallel to RIS. Altogether, treatment with rituximab has led to better overall survival in children and adolescents with PTLD. 22 , 59 , 60 , 61 Eighty‐five percent of our patients having received first‐line treatment required further therapy, with 86% of them having received rituximab±steroids as first‐line therapy. Forty‐six percent of patients lacked response to rituximab, including one patient who died from progressive disease and all others proceeding to chemotherapy. Of the 54% of patients having achieved CR after rituximab‐containing therapy, 23% relapsed.
In cases of DLBCL non‐responding to RIS and rituximab, as well as “non‐DLBCL” monomorphic PTLD and primary CNS‐lymphoma, polychemotherapy is suggested, mostly consisting of R‐CHOP, CHOP, and COP regimens. 35 , 62 Overall, 56% of our patients with monomorphic disease underwent polychemotherapy at some time, 40% of whom already received it as first‐line therapy, whereas 60% proceeded to chemotherapy after lacking response to rituximab±steroids or brentuximab‐vedotin.
Novel therapeutic approaches include immunotherapies, such as EBV‐specific CTLs and chimeric antigen receptor T‐cells, small molecule inhibitors, such as the mTOR inhibitor everolimus or tyrosine kinase inhibitor ibrutinib, as well as risk‐adapted chemotherapy regimens. Aside from rituximab, targeted therapies such as the anti‐CD30 antibody brentuximab‐vedotin yield promising results. 11 , 35 , 63 Within our cohort, one patient with plasmablastic lymphoma received brentuximab‐vedotin as first‐line therapy, but proceeded to further chemotherapy due to refractory disease.
Several factors reportedly influence the prognosis of pediatric patients with PTLD. 13 , 36 , 60 , 64 , 65 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 Parameters associated with a poor prognosis are a history of malignant disease indicating transplantation, advanced disease, multifocal and extranodal disease, CNS, BM and graft involvement, female gender, B‐symptoms and elevated LDH levels at PTLD onset, high EBV load in PB at the time of diagnosis, CD20‐ and EBV‐negative as well as monomorphic and late‐onset PTLD. Considering the type of allograft, lung, liver, and hematopoietic stem cells were associated with poorer prognosis. None of the factors referred to could be identified as relevant prognostic factors affecting EFS or OS in our patient cohort, which may at least be partially owed to the relatively small number of patients included in our study.
Children diagnosed with PTLD tend to have a better prognosis compared to adults, which seems to be due to more favorable PTLD‐subtypes (EBV‐positive) and less treatment‐related complications. 15 , 20 The prospective PTLD‐1 trial implemented sequential treatment with rituximab followed by a CHOP‐regimen in adult CD20‐positive patients with mostly monomorphic PTLD, resulting in a median OS of 6.6 years. Considering that response to rituximab predicted favorable OS, another prospective study subsequently treated the patients with CR to 4 weeks of rituximab with further rituximab consolidation, whereas non‐responders switched to R‐CHOP. Thereby, a 3‐year OS of 78% (compared to 69% in PTLD‐1) was achieved. 59 , 61 The prospective Ped‐PTLD 2005 trial treated pediatric patients with CD20‐positive PTLD following SOT with 3 weeks of rituximab followed by either rituximab or chemotherapy (mCOMP) depending on initial response, achieving a 2‐year OS of 86%, and 67% surviving event‐free. 64 According to the literature available, 5‐year survival rates for pediatric PTLD are around 53‐80%. 25 , 61 , 65 , 74 , 75 The 3‐ and 5‐year OS rates in our patient cohort were 69% ± 8% and 64% ± 9%, respectively, fitting well to the hitherto reports.
The primary aim of this study was to investigate the characteristics and outcome of PTLD in Austrian patients over a period of 20 years. Special attention was paid to factors influencing the patient's prognosis. In childhood and adolescence, PTLD is the largest group of secondary malignant diseases, making it essential to define coherent diagnostic and therapeutic guidelines addressing the heterogeneity of this disease. Considering the absolute number of affected people, it is still defined as an Orphan Disease, and both, a transnational cooperation and a compilation of comprehensive scientific studies, including series like the present one, are essential to optimize the management of PTLD, as it is still a major cause of mortality following transplantation.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHORS' CONTRIBUTIONS
A.A., A.F., and R.C. designed and planned the study; A.A., A.F., G.K., and R.C. wrote the manuscript; A.A., A.F., and G.K. collected and analyzed the data; I.S.K. was in charge of the histopathological analyses; M.B., M.D., S.G., W.D.H., H.H., A.L., G.M., I.M.B., T.M.S., H.P., W.S., Z.S., A.A., G.K., and R.C. recruited, treated, and identified the patients. A.F., G.K., M.B., M.D., S.G., W.D.H., H.H., A.L., G.M., I.M.B., T.M.S., H.P., I.S.K., W.S., Z.S., R.C., and A.A. read and approved the final version of the manuscript.
ETHICAL STATEMENT
Ethical approval for this study (No. 1919/2018) was granted by the Ethics Committee of the Medical University of Vienna on 11th November 2018.
PATIENT CONSENT STATEMENT
This was a retrospective study involving access to existing medical records, with the research personnel reviewing these records normally having access to these records. Therefore, patient consent was waivered.
Supporting information
Supplemental Table 1 Response and outcome of the 4 HSCT patients as compared to the 30 SOT patients with PTLD
Supplemental Table 2 Event‐free and overall survival of the 34 patients with PTLD according to prognostic factors analyzed
ACKNOWLEDGEMENTS
We thank all participating institutions and their physicians, nurses, pathologists, and biologists for their support of the study.
Füreder A, Kropshofer G, Benesch M, et al. Characteristics, management, and outcome of pediatric patients with post‐transplant lymphoproliferative disease—A 20 years' experience from Austria. Cancer Reports. 2021;4:e1375. 10.1002/cnr2.1375
Anna Füreder and Gabriele Kropshofer are co‐first authors.
Contributor Information
Roman Crazzolara, Email: roman.crazzolara@i-med.ac.at.
Andishe Attarbaschi, Email: andishe.attarbaschi@stanna.at.
DATA AVAILABILITY STATEMENT
The data that support the findings of the study are available on request from the corresponding authors.
REFERENCES
- 1. Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715‐2729. 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 2. JK‐C C. Post‐transplant lymphoproliferative disorders: a simplified overview. Hong Kong J Nephrol. 2001;3(2):57‐66. 10.1016/s1561-5413(09)60060-0. [DOI] [Google Scholar]
- 3. Penn I. Cancers complicating organ transplantation. N Engl J Med. 1990;323(25):1767‐1769. 10.1056/NEJM199012203232510. [DOI] [PubMed] [Google Scholar]
- 4. Harris NL, Ferry JA, Swerdlow SH. Posttransplant lymphoproliferative disorders: summary of society for hematopathology workshop. Semin Diagn Pathol. 1997;14:8‐14. [PubMed] [Google Scholar]
- 5. Mynarek M, Schober T, Behrends U, Maecker‐Kolhoff B. Posttransplant lymphoproliferative disease after pediatric solid organ transplantation. Clin Dev Immunol. 2013;10:814973. 10.1155/2013/814973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guerra‐García P, Hirsch S, Levine DS, Taj MM. Preliminary experience on the use of PET/CT in the management of pediatric post‐transplant lymphoproliferative disorder. Pediatr Blood Cancer. 2017;64(12):e26685. 10.1002/pbc.26685. [DOI] [PubMed] [Google Scholar]
- 7. Mynarek M, Hussein K, Kreipe HH, Maecker‐Kolhoff B. Malignancies after pediatric kidney transplantation: more than PTLD? Pediatr Nephrol. 2014;29(9):1517‐1528. 10.1007/s00467-013-2622-5. [DOI] [PubMed] [Google Scholar]
- 8. Crombie JL, LaCasce AS. Epstein Barr virus associated B‐cell lymphomas and iatrogenic lymphoproliferative disorders. Front Oncol. 2019;9:109. 10.3389/fonc.2019.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Loren AW, Porter DL, Stadtmauer EA, Tsai DE. Post‐transplant lymphoproliferative disorder: a review. Bone Marrow Transplant. 2003;31(3):145‐155. 10.1038/sj.bmt.1703806. [DOI] [PubMed] [Google Scholar]
- 10. Walker RC, Paya CV, Marshall WF, et al. Pretransplantation seronegative Epstein‐Barr virus status is the primary risk factor for posttransplantation lymphoproliferative disorder in adult heart, lung, and other solid organ transplantations. J Heart Lung Transplant. 1995;14:214. [PubMed] [Google Scholar]
- 11. Evens AM, Roy R, Sterrenberg D, Moll MZ, Chadburn A, Gordon LI. Post‐transplantation lymphoproliferative disorders: diagnosis, prognosis, and current approaches to therapy. Curr Oncol Rep. 2010;12:383‐394. 10.1007/s11912-010-0132-1. [DOI] [PubMed] [Google Scholar]
- 12. Opelz G, Döhler B. Lymphomas after solid organ transplantation: a collaborative transplant study report. Am J Transplant. 2004;4:222‐230. 10.1046/j.1600-6143.2003.00325.x. [DOI] [PubMed] [Google Scholar]
- 13. Ghobrial IM, Habermann TM, Maurer MJ, et al. Prognostic analysis for survival in adult solid organ transplant recipients with post‐transplantation lymphoproliferative disorders. J Clin Oncol. 2005;46(2):191‐196. 10.1200/JCO.2005.01.0934. [DOI] [PubMed] [Google Scholar]
- 14. Uhlin M, Wikell H, Sundin M, et al. Risk factors for Epstein‐Barr virus‐related post‐transplant lymphoproliferative disease after allogeneic hematopoietic stem cell transplantation. Haematologica. 2014;99(2):346‐352. 10.3324/haematol.2013.087338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hussein K, Tiede C, Maecker‐Kolhoff B, Kreipe H. Posttransplant lymphoproliferative disorder in pediatric patients. Pathobiology. 2013;80(6):289‐296. 10.1159/000350331. [DOI] [PubMed] [Google Scholar]
- 16. Everly MJ, Bloom RD, Tsai DE, Trofe J. Posttransplant lymphoproliferative disorder. Ann Pharmacother. 2007;41(11):1850‐1858. 10.1345/aph.1G706. [DOI] [PubMed] [Google Scholar]
- 17. Cohen JI. Epstein–Barr virus infection. N Engl J Med. 2000;343:481‐492. [DOI] [PubMed] [Google Scholar]
- 18. Hussein K, Maecker‐Kolhoff B, Klein C, Kreipe H. Transplantations‐assoziierte Lymphoproliferationen. Pathologe. 2011;32(2):152‐158. 10.1007/s00292-010-1407-x. [DOI] [PubMed] [Google Scholar]
- 19. Shroff R, Rees L. The post‐transplant lymphoproliferative disorder ‐ a literature review. Pediatr Nephrol. 2004;19:369‐377. 10.1007/s00467-003-1392-x. [DOI] [PubMed] [Google Scholar]
- 20. Jain A, Nalesnik M, Reyes J, et al. Posttransplant lymphoproliferative disorders in liver transplantation: a 20‐year experience. Ann Surg. 2002;236(4):429‐436. 10.1097/00000658-200210000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luskin MR, Heil DS, Tan KS, et al. The impact of EBV status on characteristics and outcomes of posttransplantation lymphoproliferative disorder. Am J Transplant. 2015;15:2665‐2673. 10.1111/ajt.13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abbas F, El Kossi M, Shaheen IS, Sharma A, Halawa A. Post‐transplantation lymphoproliferative disorders: current concepts and future therapeutic approaches. World J Transplant. 2020;10(2):29‐46. 10.5500/wjt.v10.i2.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Choquet S, Varnous S, Deback C, Golmard JL, Leblond V. Adapted treatment of epstein‐barr virus infection to prevent posttransplant lymphoproliferative disorder after heart transplantation. Am J Transplant. 2014;14:857‐866. 10.1111/ajt.12640. [DOI] [PubMed] [Google Scholar]
- 24. Chadburn A, Frizzera G, Pan L, Shulman H, Knowles D. Posttransplantation lymphoproliferative disorders in bone marrow transplant and solid organ transplant recipients [abstract]. Mod Pathol. 2000;13:144. [Google Scholar]
- 25. Schober T, Framke T, Kreipe H, et al. Characteristics of early and late PTLD development in pediatric solid organ transplant recipients. Transplantation. 2013;95(1):240‐246. 10.1097/TP.0b013e318277e344. [DOI] [PubMed] [Google Scholar]
- 26. Ibrahim HAH, Naresh KN. Posttransplant lymphoproliferative disorders. Adv Hematol. 2012;11:230173. 10.1155/2012/230173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Walker RC, Marshall WF, Strickler JG, et al. Pretransplantation assessment of the risk of lymphoproliferative disorder. Clin Infect Dis. 1995;20:1346‐1353. 10.1093/clinids/20.5.1346. [DOI] [PubMed] [Google Scholar]
- 28. Newell KA, Alonso EM, Whitington PF, et al. Posttransplant lymphoproliferative disease in pediatric liver transplantation: interplay between primary Epstein‐Barr virus infection and immunosuppression. Transplantation. 1996;62(3):370‐375. 10.1097/00007890-199608150-00012. [DOI] [PubMed] [Google Scholar]
- 29. Lewin KJ. Post‐transplant lymphoproliferative disorders. Pathol Oncol Res. 1997;3(3):177‐182. [DOI] [PubMed] [Google Scholar]
- 30. Reyes J, Bueno J, Kocoshis S, et al. Current status of intestinal transplantation in children. J Pediatr Surg. 1998;33(2):243‐254. 10.1016/S0022-3468(98)90440-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quintini C, Kato T, Gaynor JJ, et al. Analysis of risk factors for the development of posttransplant lymphoprolipherative disorder among 119 children who received primary intestinal transplants at a single center. Transplant Proc. 2006;38(6):1755‐1758. 10.1016/j.transproceed.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 32. Elidemir O, Kancherla BS, Schecter MG, et al. Post‐transplant lymphoproliferative disease in pediatric lung transplant recipients: recent advances in monitoring. Pediatr Transplant. 2009;13(5):606‐610. 10.1111/j.1399-3046.2008.01085.x. [DOI] [PubMed] [Google Scholar]
- 33. Raymond E, Tricottet V, Samuel D, Reynes M, Bismuth H, Misset J. Epstein Barr virus related hepatic lymphoproliferative disorder after liver transplantation. Cancer. 1999;76:1344‐1349. [DOI] [PubMed] [Google Scholar]
- 34. Chen JM, Barr ML, Chadburn A, et al. Management of lymphoproliferative disorders after cardiac transplantation. Ann Thorac Surg. 1993;56(3):527‐531. 10.1016/0003-4975(93)90893-M. [DOI] [PubMed] [Google Scholar]
- 35. Dierickx D, Habermann TM. Post‐transplantation lymphoproliferative disorders in adults. N Engl J Med. 2018;378:549‐562. 10.1056/NEJMra1702693. [DOI] [PubMed] [Google Scholar]
- 36. Dierickx D, Tousseyn T, Sagaert X, et al. Single‐center analysis of biopsy‐confirmed posttransplant lymphoproliferative disorder: incidence, clinicopathological characteristics and prognostic factors. Leuk Lymphoma. 2013;5(11):2433‐2440. 10.3109/10428194.2013.780655. [DOI] [PubMed] [Google Scholar]
- 37. Landgren O, Gilbert ES, Rizzo JD, et al. Risk factors for lymphoproliferative disorders after allogeneic hematopoietic cell transplantation. Blood. 2009;113(20):4992‐5001. 10.1182/blood-2008-09-178046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shapiro RS, McCLain K, Frizzera G, et al. Epstein‐Barr virus associated B cell lymphoproliferative disorders following bone marrow transplantation. Blood. 1988;71(5):1234‐1243. 10.1182/blood.v71.5.1234.bloodjournal7151234. [DOI] [PubMed] [Google Scholar]
- 39. Jagadeesh D, Woda BA, Draper J, Evens AM. Post transplant lymphoproliferative disorders: risk, classification, and therapeutic recommendations. Curr Treat Options in Oncol. 2012;13(1):122‐136. 10.1007/s11864-011-0177-x. [DOI] [PubMed] [Google Scholar]
- 40. Chiang KY, Hazlett LJ, Godder KT, et al. Epstein‐Barr virus‐associated B cell lymphoproliferative disorder following mismatched related T cell‐depleted bone marrow transplantation. Bone Marrow Transplant. 2001;28:1117‐1123. 10.1038/sj.bmt.1703311. [DOI] [PubMed] [Google Scholar]
- 41. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. In: Swerdlow S, Campo E, Harris N, eds. World Health Organization Calssification of Tumours of Haematopoietic and Lymphoid Tissue. 4th ed. Lyon, France: IARC Press; 2017:453‐462. 10.1017/CBO9781107415324.004. [DOI] [Google Scholar]
- 42. Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375‐2390. 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murphy SB. Classification, staging and end results of treatment of childhood non‐Hodgkin's lymphomas: dissimilarities from lymphomas in adults. Semin Oncol. 1980;7:332‐339. 10.5555/uri:pii:0093775480900408. [DOI] [PubMed] [Google Scholar]
- 44. Rosolen A, Perkins SL, Pinkerton CR, et al. Revised international pediatric non‐Hodgkin lymphoma staging system. J Clin Oncol. 2015;33(18):2112‐2118. 10.1200/JCO.2014.59.7203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carbone A, Gloghini A, Dotti G. EBV‐associated lymphoproliferative disorders: classification and treatment. Oncologist. 2008;13(5):577‐585. 10.1634/theoncologist.2008-0036. [DOI] [PubMed] [Google Scholar]
- 46. Herreman A, Dierickx D, Morscio J, et al. Clinicopathological characteristics of posttransplant lymphoproliferative disorders of T‐cell origin: single‐center series of nine cases and meta‐analysis of 147 reported cases. Leuk Lymphoma. 2013;54(10):2190‐2199. 10.3109/10428194.2013.775436. [DOI] [PubMed] [Google Scholar]
- 47. Hussein K, Maecker‐Kolhoff B, Klein C, Kreipe H. Transplantations‐assoziierte Lymphoproliferationen. Pathologe. 2011;32:152‐158. https://www.springermedizin.de/transplantations-assoziierte-lymphoproliferationen/8300250. [DOI] [PubMed] [Google Scholar]
- 48. Nagle SJ, Reshef R, Tsai DE. Posttransplant lymphoproliferative disorder in solid organ and hematopoietic stem cell transplantation. Clin Chest Med. 2017;38(4):771‐783. 10.1016/J.CCM.2017.08.001. [DOI] [PubMed] [Google Scholar]
- 49. Bishnoi R, Minish J, Franke AJ, et al. Single‐institution retrospective analysis of prognostic factors influencing very late‐onset post‐transplant lymphoproliferative disorder. Cureus. 2020;12(2):e6912. 10.7759/cureus.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Caillard S, Lamy FX, Quelen C, et al. Epidemiology of posttransplant lymphoproliferative disorders in adult kidney and kidney pancreas recipients: report of the French registry and analysis of subgroups of lymphomas. Am J Transplant. 2012;12:682‐693. 10.1111/j.1600-6143.2011.03896.x. [DOI] [PubMed] [Google Scholar]
- 51. Faull RJ, Hollett P, McDonald SP. Lymphoproliferative disease after renal transplantation in Australia and New Zealand. Transplantation. 2005;80:193‐197. 10.1097/01.TP.0000165098.49658.F3. [DOI] [PubMed] [Google Scholar]
- 52. Maecker‐Kolhoff B, Klein C. Das pädiatrische PTLD‐Register (Ped‐PTLD‐Register). Nephrol Ther. 2009;4(5):339‐344. 10.1007/s11560-009-0306-8. [DOI] [Google Scholar]
- 53. Curtis RE, Travis LB, Rowlings PA, et al. Risk of lymphoproliferative disorders after bone marrow transplantation: a multi‐institutional study. Blood. 1999;94(7):2208‐2216. 10.1182/blood.V94.7.2208.419k21_2208_2216. [DOI] [PubMed] [Google Scholar]
- 54. Bakker NA, Van Imhoff GW, Verschuuren EAM, Van Son WJ. Presentation and early detection of post‐transplant lymphoproliferative disorder after solid organ transplantation. Transpl Int. 2007;20(3):207‐218. 10.1111/j.1432-2277.2006.00416.x. [DOI] [PubMed] [Google Scholar]
- 55. Oton A, Wang H, Leleu X, et al. Clinical and pathological prognostic markers for survival in adult patients with post‐transplant lymphoproliferative disorders in solid transplant. Leuk Lymphoma. 2008;49:1738‐1744. 10.1080/10428190802239162. [DOI] [PubMed] [Google Scholar]
- 56. Tsai DE, Hardy CL, Tomaszewski JE, et al. Reduction in immunosuppression as initial therapy for posttransplant lymphoproliferative disorder: analysis of prognostic variables and long‐term follow‐up of 42 adult patients. Transplantation. 2001;71(8):1076‐1088. 10.1097/00007890-200104270-00012. [DOI] [PubMed] [Google Scholar]
- 57. Allen UD, Preiksaitis JK. Post‐transplant lymphoproliferative disorders, Epstein‐Barr virus infection, and disease in solid organ transplantation: guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin Transpl. 2019;33(9):1‐22. 10.1111/ctr.13652. [DOI] [PubMed] [Google Scholar]
- 58. Reshef R, Vardhanabhuti S, Luskin MR, et al. Reduction of immunosuppression as initial therapy for posttransplantation lymphoproliferative disorder. Am J Transplant. 2011;11(2):336‐347. 10.1111/j.1600-6143.2010.03387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Trappe RU, Dierickx D, Zimmermann H, et al. Response to rituximab induction is a predictive marker in B‐cell post‐transplant lymphoproliferative disorder and allows successful stratification into rituximab or r‐chop consolidation in an international, prospective, multicenter phase II trial. J Clin Oncol. 2017;35(5):536‐543. 10.1200/JCO.2016.69.3564. [DOI] [PubMed] [Google Scholar]
- 60. Choquet S, Oertel S, LeBlond V, et al. Rituximab in the management of post‐transplantation lymphoproliferative disorder after solid organ transplantation: proceed with caution. Ann Hematol. 2007;86:599‐607. 10.1007/s00277-007-0298-2. [DOI] [PubMed] [Google Scholar]
- 61. Trappe R, Oertel S, Leblond V, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B‐cell post‐transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD‐1 trial. Lancet Oncol. 2012;13(2):196‐206. 10.1016/S1470-2045(11)70300-X. [DOI] [PubMed] [Google Scholar]
- 62. Major A, Kamdar M. Management of non‐diffuse large B cell lymphoma post‐transplant lymphoproliferative disorder. Curr Treat Options in Oncol. 2018;19:33. 10.1007/s11864-018-0549-6. [DOI] [PubMed] [Google Scholar]
- 63. DeStefano CB, Desai SH, Shenoy AG, Catlett JP. Management of post‐transplant lymphoproliferative disorders. Br J Haematol. 2018;182(3):330‐343. 10.1111/bjh.15263. [DOI] [PubMed] [Google Scholar]
- 64. Maecker‐Kolhoff B, Beier R, Zimmermann M, et al. Response‐adapted sequential Immuno‐chemotherapy of post‐transplant lymphoproliferative disorders in pediatric solid organ transplant recipients: results from the prospective Ped‐PTLD 2005 trial. Blood. 2014;124(21):4468‐4468. 10.1182/blood.v124.21.4468.4468. [DOI] [Google Scholar]
- 65. Caillard S, Porcher R, Provot F, et al. Post‐transplantation lymphoproliferative disorder after kidney transplantation: report of a nationwide French registry and the development of a new prognostic score. J Clin Oncol. 2012;31(10):1302‐1309. 10.1200/JCO.2012.43.2344. [DOI] [PubMed] [Google Scholar]
- 66. Kumarasinghe G, Lavee O, Parker A, et al. Post‐transplant lymphoproliferative disease in heart and lung transplantation: defining risk and prognostic factors. J Heart Lung Transplant. 2015;34(11):1406‐1414. 10.1016/j.healun.2015.05.021. [DOI] [PubMed] [Google Scholar]
- 67. García‐Cadenas I, Yáñez L, Jarque I, et al. Frequency, characteristics, and outcome of PTLD after Allo‐SCT: a multicenter study from the Spanish group of blood and marrow transplantation (GETH). Eur J Haematol. 2019;102(6):465‐471. 10.1111/ejh.13226. [DOI] [PubMed] [Google Scholar]
- 68. Knight JS, Tsodikov A, Cibrik DM, Ross CW, Kaminski MS, Blayney DW. Lymphoma after solid organ transplantation: risk, response to therapy, and survival at a transplantation center. J Clin Oncol. 2009;27(20):3354‐3362. 10.1200/JCO.2008.20.0857. [DOI] [PubMed] [Google Scholar]
- 69. Bishnoi R, Bajwa R, Franke AJ, et al. PTLD: survival and analysis of prognostic factors in a cohort of 138 patients from a single institution. J Clin Oncol. 2017;35(15):348‐349. 10.1200/jco.2017.35.15_suppl.e19048. [DOI] [Google Scholar]
- 70. Montanari F, Radeski D, Seshan V, Alobeid B, Bhagat G, O'Connor OA. Recursive partitioning analysis of prognostic factors in post‐transplant lymphoproliferative disorders (PTLD): a 120 case single institution series. Br J Haematol. 2015;171(4):491‐500. 10.1111/bjh.13621. [DOI] [PubMed] [Google Scholar]
- 71. Ligeti K, Müller LP, Müller‐Tidow C, Weber T. Risk factors, diagnosis, and management of posttransplant lymphoproliferative disorder: improving patient outcomes with a multidisciplinary treatment approach. Transpl Res Risk Manag. 2017;9:1‐14. 10.2147/TRRM.S84744. [DOI] [Google Scholar]
- 72. Shipp M, Harrington D, Anderson J, Armitage JA. Predictive model for aggressive non‐Hodgkin's lymphoma. N Engl J Med. 1993;329(14):987‐994. 10.1056/NEJM199309303291402. [DOI] [PubMed] [Google Scholar]
- 73. Trappe RU, Choquet S, Dierickx D, et al. International prognostic index, type of transplant and response to rituximab are key parameters to tailor treatment in adults with cd20‐positive b cell ptld: clues from the ptld‐1 trial. Am J Transplant. 2015;15:1091‐1100. 10.1111/ajt.13086. [DOI] [PubMed] [Google Scholar]
- 74. Maecker B, Jack T, Zimmermann M, et al. CNS or bone marrow involvement as risk factors for poor survival in post‐transplantation lymphoproliferative disorders in children after solid organ transplantation. J Clin Oncol. 2007;25:4902‐4908. 10.1200/JCO.2006.10.2392. [DOI] [PubMed] [Google Scholar]
- 75. Gross TG, Orjuela MA, Perkins SL, et al. Low‐dose chemotherapy and rituximab for posttransplant lymphoproliferative disease (PTLD): a children's oncology group report. Am J Transplant. 2012;12(11):3069‐3075. 10.1111/j.1600-6143.2012.04206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 Response and outcome of the 4 HSCT patients as compared to the 30 SOT patients with PTLD
Supplemental Table 2 Event‐free and overall survival of the 34 patients with PTLD according to prognostic factors analyzed
Data Availability Statement
The data that support the findings of the study are available on request from the corresponding authors.