

Abstract
Background and Objectives
To determine the genetic cause of the disease in the previously reported family with adult-onset autosomal dominant distal myopathy (myopathy, distal, 3; MPD3).
Methods
Continued clinical evaluation including muscle MRI and muscle pathology. A linkage analysis with single nucleotide polymorphism arrays and genome sequencing were used to identify the genetic defect, which was verified by Sanger sequencing. RNA sequencing was used to investigate the transcriptional effects of the identified genetic defect.
Results
Small hand muscles (intrinsic, thenar, and hypothenar) were first involved with spread to the lower legs and later proximal muscles. Dystrophic changes with rimmed vacuoles and cytoplasmic inclusions were observed in muscle biopsies at advanced stage. A single nucleotide polymorphism array confirmed the previous microsatellite-based linkage to 8p22-q11 and 12q13-q22. Genome sequencing of three affected family members combined with structural variant calling revealed a small heterozygous deletion of 160 base pairs spanning the second last exon 10 of the heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1) gene, which is in the linked region on chromosome 12. Segregation of the mutation with the disease was confirmed by Sanger sequencing. RNA sequencing showed that the mutant allele produces a shorter mutant mRNA transcript compared with the wild-type allele. Immunofluorescence studies on muscle biopsies revealed small p62 and larger TDP-43 inclusions.
Discussion
A small exon 10 deletion in the gene HNRNPA1 was identified as the cause of MPD3 in this family. The new HNRNPA1-related phenotype, upper limb presenting distal myopathy, was thus confirmed, and the family displays the complexities of gene identification.
Distal myopathies are hereditary disorders with progressive muscle weakness in the hands and/or ankles as the first symptoms, and they consist of dozens of diseases.1-3 Finland and Sweden are known for a high prevalence of some dominant distal myopathies such as Welander distal myopathy4 and tibial muscular dystrophy.5 In East Asia and Middle East, founder mutations cause the higher prevalence of recessive GNE myopathy,3 whereas early-onset Laing distal myopathy occurs in all populations with many different mutations, some of which are de novo and independently reoccurring.6
We have previously described an adult-onset distal myopathy (MPD3, OMIM: 610099) in a large Finnish family with a dominant mode of inheritance7. The disease onset ranged from 32 to 45 years and was generally earlier in the men of the family. Hand weakness and stumbling on the feet were the first symptoms. Muscle atrophy started in the small hand muscles and continued to both anterior and posterior compartments of the lower legs, the forearm muscles, and later to proximal muscles. Asymmetry of muscle involvement was common. Muscle imaging showed fatty degeneration of the affected muscles, and muscle biopsy revealed frequent rimmed vacuoles and some cytoplasmic eosinophilic inclusions, besides general dystrophic changes with the loss of fibers and increase of connective tissue. In 2004, a genome-wide scan of microsatellites was performed on DNA samples from the family. Two different chromosomal regions, 8p22-q11 and 12q13-q22, showed linkage with equal maximum multipoint logarithm of odds (LOD) score 3.01, but Sanger sequencing revealed no rare variants in the protein coding genes in the linked regions.8 Recently, we renewed the analysis with a dense genome-wide genotyping array, and the linkage was confirmed for both chromosomal regions. Genome sequencing was then performed on 3 DNA samples from affected members, and a small heterozygous deletion within the linked region on chromosome 12 encompassing the second last exon 10 of heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1) (NM_031157.4, exon 9 in the shorter transcript NM_002136.4) encoding the HNRNPA1 was identified. The deletion was confirmed by Sanger sequencing and shown to be expressed as a transcript lacking exon 10 on the RNA level. This deletion segregated with the disease in the large family.
Methods
Patients
The patients provided written informed consent. The study, approved by the ethics committee of the Helsinki University Hospital, was performed in accordance with the Declaration of Helsinki. Patients and healthy family members were available in three consecutive generations (Figure 1). Clinical examination and laboratory investigations were performed earlier and described in detail in 6 patients.7,8 During this study, the material was expanded with the assessment of 4 additional family members, II-9, II-10, IV-10, and IV-11. Two of these 4 individuals, II-10 and IV-11, were affected. Patient IV-11 was diagnosed after clinical examination, showing early atrophy with thin muscle bulks of thenar and hypothenar muscles. Previous CT scans showed early minor lesions in lower leg muscles in this male patient at the age of 28 years. His older brother IV-10 was unaffected. Patient II-10, a younger sister of the proband’s mother, was not available for further clinical examinations, but according to her medical records, she had clear walking difficulties at the age of 50 and used a cane for walking.
Figure 1. Pedigree Chart of the Family.

The proband (III-3) is indicated with an arrow. Available DNA samples indicated with asterisk (*). All available samples were used in the single nucleotide polymorphism array.
Genome-wide Genotyping
A genome-wide single nucleotide polymorphism microarray analysis (Illumina GSAMD-24 v2) with 730,000 markers was performed on 13 available samples from three generations of the family. The results were pooled. The arrays were run according to the manufacturer's instructions at the Institute for Molecular Medicine Finland (FIMM, Helsinki, Finland)9 and analysed with the GenomeStudio Software using the genomic build hg19. The data were filtered using PLINK (version 1.9) (zzz.bwh.harvard.edu/plink/) before linkage analysis and then filtered for minor allele frequency < 0.01. The LOD score was calculated using single point analysis with Merlin (version 1.1.2) (csg.sph.umich.edu/abecasis/merlin/index.html). The internal Merlin error detection algorithm was used for filtering and removing of erroneous genotypes. A parametric linkage analysis was performed on the data for complete penetrance model. Haplotypes were constructed, allowing a minimum number of recombination events. The SLINK program was used to estimate the maximum possible LOD score for this pedigree.10
Genome Sequencing
DNA extracted from the blood of 3 affected family members (II-7, IV-6, IV-11) was genome sequenced (GS), with an average sequencing depth of 30 × (90 Gb) for two of the samples (IV-6 and IV-11) and 50 × (150 Gb) for one of them (II-7).11,12 The library construction and sequencing on a DNBSEQ platform with paired end reads of 150 base pair (bp) were performed at BGI, Tech Solutions, Hong Kong. Standard bioinformatics and variant calling of the data were performed by BGI.
CNV Detection Methods
The programs DELLY (v0.8.3)13 and LUMPY (v0.3.1)14 were both used with their default recommended settings. The programs use information on paired end reads, split reads, and read depth to delineate genomic rearrangements and copy number variants (deletions and duplications), inversions and translocations. Structural variant calling was performed separately for each of the three genome-sequenced family members, and the results were evaluated separately for each program. An in-house version of the program cnvScan15 was used for further filtering of the results for rare structural variants against both internal and public (gnomAD v2.1)16 databases of unrelated control samples. The comparison was performed with a strict requirement of <1% frequency and different overlap degrees (1 bp overlap, and 50% and 90% reciprocal overlap). Both DELLY and LUMPY enable the examination of possible translocations between the linked regions by analysis of hybrid sequences.
Sanger Sequencing
PCR was performed on genomic DNA of the available samples from the family to confirm the 160 bp deletion in the HNRNPA1 gene (hg19: chr12:54677979-54678138). The primers were designed using the Primer3 program (bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi).17 The forward primer was: tgggacctctttaccacctcc and the reverse primer was actatgttgcactgctcagct. PCR products obtained from the proband were cloned, and several clones were Sanger sequenced to map the breakpoints.
RNA Extraction and Sequencing
RNA was extracted from the tibialis anterior muscle biopsy of patient III-5 with Qiagen RNeasy Plus Universal Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The strand-specific RNAseq library was prepared using the NEBNext Ultra II Directional RNA Library Prep kit (E7760) for Illumina (NEB, Beverly, MA) at the Oxford Genomics Center, Wellcome Trust Institute, Oxford, United Kingdom. Whole transcriptome sequencing (RNAseq) was performed on HiSeq4000 (Illumina), generating 59 million paired end reads with 75 bp read length. Trimming was performed using fastq:s, and the trimmed sequences were mapped with STAR 2.7.0d using STAR 2-pass method.18 Gene level read summarization and quantification was performed by featureCounts, and fragment counts were converted into TPM counts.19 RNAseq data were analyzed as described before.20,21
Immunofluorescence
Snap-frozen muscle biopsies of patients III-6, IV-7, and IV-11 were used for the preparation of 8 µm sections for hematoxylin & eosin staining and immunofluorescent analysis using the standard methodology. Primary antibodies used were monoclonal mouse (mAb) anti-HNRNPA1 (Abcam ab5832), pAb anti-p62 (Sigma-Aldrich P0067), and mAb anti-TDP-43 (Sigma-Aldrich, clone 2E2-D3). Alexa fluor-488 and -546 conjugated secondary antirabbit and antimouse antibodies were used for detection, and the nuclei were counterstained using Hoechst. The slides were analyzed using the Zeiss Axio Imager M2 system using a 20× NA 0.80 objective (Carl Zeiss AG) or with a Leica TCS SP8 confocal microscope using a 63× NA 1.12 objective (Leica Microsystems).
Western Blotting
For western blotting analysis, the muscle biopsy from IV to 11 was homogenized in the Laemmli sample buffer and heated at 95°C for 5 minutes. The soluble fraction after centrifugation (13,000 × g, 10 minutes) was collected and used for SDS-PAGE using 4%–20% TGX gradient gels and Bio-Rad Mini-PROTEAN system (Bio-Rad Laboratories, CA). After separation, the proteins were transferred onto PVDF membranes using the Bio-Rad TransBlot Turbo device. The membranes were incubated with anti-HNRNPA1 (Abcam ab5832) antibody at 1:10,000, diluted in 5% nonfat milk powder/TBST, overnight at +8°C. After HRP-conjugated secondary antibody incubation, ECL reaction (SuperSignal West Femto, Thermo Fisher Scientific) and Bio-Rad ChemiDoc reader were used for detection of bands. The gels were recovered after transfer and stained with Coomassie, and the myosin heavy chain bands were used as loading control.
Standard Protocol Approvals, Registrations, and Patient Consents
Informed consent was collected from the participants, and the local ethics committee approved this study.
Data Availability
Pseudonymized data not published in this article are available in RD-connect, as genomic data and clinical HPOs, and will also be shared upon request to any qualified investigator.
Results
Clinical Investigations
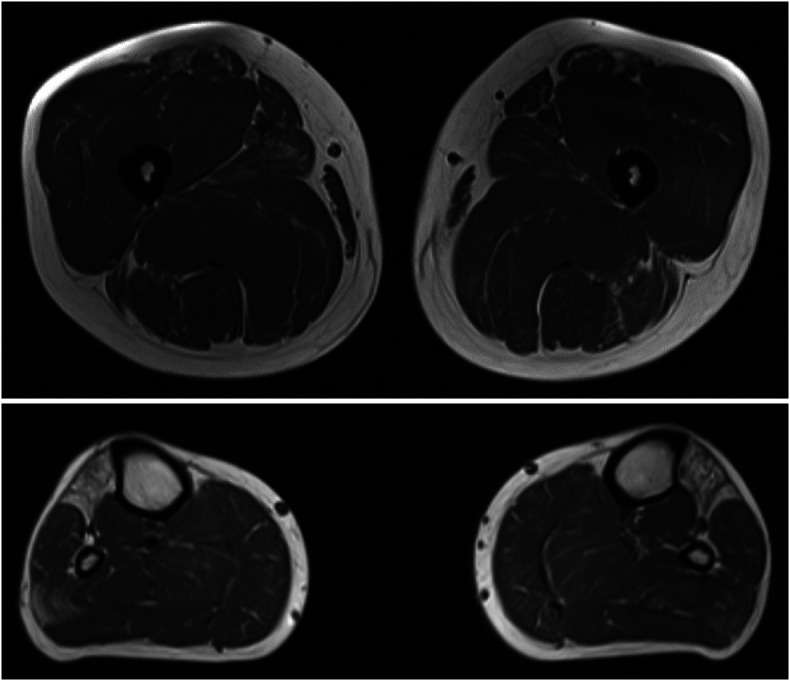
Follow-up studies on the patients 15–20 years after initial examinations showed a relatively benign evolution of the disease. Patient III-3 (Figure 1) was still living by her own at the age of 76 years and walked short distance without a walker using dynamic ankle foot orthoses (DAFOs). She had severe atrophies of hand muscles, weakness in the fingers, making writing and similar hand skills very difficult despite aids to improve hand grip. Patient III-5 had a similar disease evolution and was still walking without aids at the age of 70 years using DAFOs and could even rise from the squat without hand help. He was single living at home and could drive his car. Reduced fine finger movements and hand weakness impaired daily life activities despite wrist orthoses. No cardiac abnormalities were noted. Patient III-6 died of unrelated cause at the age of 66 years. His son IV–11 was re-evaluated at age of 42 years and now presented a clear phenotype with foot drop, atrophy of small hand muscles, and preserved proximal muscle strength. Muscle MRI showed fatty replacement of the anterior compartment muscles of the lower legs (Figure 2).
Figure 2. Muscle MRI of Patient IV-11 Show Selective Fatty Replacement of Tibialis Anterior and Long Toe Extensor Muscles and Mild Early Degenerative Change in the Lateral Part of the Right Soleus Muscle and Both Rectus Femoris Muscles.
Genetic Studies
The genome-wide linkage analysis was performed on DNA of 13 members of the family, of which 8 were affected and the rest were healthy family members (Figure 1). Two potentially linked areas on chromosomes 8 (hg19 chr8:18745476-41425848) and 12 (hg19 chr12:48808721-92048193) were identified, confirming the two previously linked genomic areas.8 The highest LOD scores were 2.6 for both linked areas and comparable with the expected maximum LOD score calculated for this pedigree data set by the SLINK program, which gave maximum scores between 2.3 and 2.7 (Figure 3). Both loci are large. The 22.6 Mb region on chromosome 8 includes 435 known or predicted genes, of which 141 are protein coding. The chromosome 12 region is approximately 43.2 Mb long including 986 known or predicted genes, of which 387 are protein coding.
Figure 3. Genetic Analyses.

(A) LOD results for the single nucleotide polymorphism array for chromosome 8 and 12. Highest LOD scores 2. (B) A drop in read depth indicates the deletion surrounding HNRNPA1 exon 10 in genomic sequencing of III-5. (C) The exact breakpoint was mapped by Sanger sequencing of a cloned PCR product. (D) Effects on RNA and protein levels as seen in Sashimi plots of RNA sequencing data from a control sample and patient III-5. (Compare with the exon diagram in E.) The patient sample shows reduced read count for HNRNPA1 exon 10, splicing of exon 9 to exon 11, and increased splicing to the alternative exons in the far end of the 3'-UTR. (E) A schematic view of the 3’end of the HNRNPA1 gene (wild-type and deleted alleles) and theoretical protein products of the observed transcripts. Splicing of exon 9 to exon 11 is predicted to result in the major mutant protein p.Gly356Asnfs*4, where the C-terminal amino acids are replaced by a short tail encoded by exon 11. The possible minor protein product would contain a longer C-terminal tail encoded by the alternative far-3'-UTR exons. HNRNPA1 = heterogeneous nuclear ribonucleoprotein A1; LOD = logarithm of odds.
None of the SNV or indel variants identified by GS in these linked regions by standard bioinformatic analysis seemed potentially interesting. No translocations between the regions on chromosomes 8 and 12 were evident. After filtering for structural variant detections in the three affected samples, some structural variants shared by the affected were found in the linked regions, but most of these had frequencies of 0.15–0.5 in the gnomAD structural variant database and could hence be excluded. However, a small heterozygous deletion encompassed the second last exon of the HNRNPA1 gene (exon 10 of the long A1b isoform NM_031157.4) (Figure 3). The breakpoints were in the intronic regions surrounding the exon. This variant was not present in the gnomAD database with any of the overlap evaluations. Sanger sequencing (Figure 3) of cloned PCR products mapped the 160 bp deletion at chr12:54677979–54678138 and confirmed the segregation of the variant with the disease.
RNAseq was performed on RNA from a biopsy of patient III-5. The RNAseq data were analyzed for the expression of transcripts of the HNRNPA1 gene20-22 and identified the expression of an abnormal transcript without exon 10 in addition to the normal transcript (Figure 3). A transcript skipping exon 10 of HNRNPA1 was not observed in any of our internal polyA RNAseq samples (n = 95), confirming it is a direct consequence of the observed deletion (Figure 3). The observed splicing of exon 9 to exon 11 would result in the predicted protein product p.Gly356Asnfs*4 (NP_112420.1) partially encoded by exon 11. Some reported HNRNPA1 transcripts skip much of the 3’-UTR and use two alternative exons (exons 10–11 in the transcript NR_135167.2). Our RNAseq results suggested increased inclusion of exons in the far 3'-UTR, with 24% of mutant transcripts using the far 3'-UTR vs 5% for the wild-type transcripts. The predicted protein sequences are shown in Figure 3.
Immunofluorescence and Western Blotting
Muscle biopsies from patients III-6, IV-7, and IV-11 were available for further analyses. The biopsies of III-6 (obtained at age 48) and IV-7 (at age 37) showed severe myopathology including a high number of very atrophic fibers, with increased endomysial connective tissue and fatty degeneration (Figure 4). Frequent rimmed vacuoles were present, together with some eosinophilic protein inclusions, and immunofluorescent staining showed several fibers with smaller p62 inclusions in rimmed vacuolated fibers and a few larger TDP-43 positive protein accumulations (Figure 4). In a subset of p62-positive fibers, separate modest HNRNPA1 positivity was observed (Figure 4). In addition, HNRNPA1 showed diffuse sarcoplasmic labeling in several fibers.
Figure 4. Histopathology.

(A–B) Hematoxylin and eosin staining of patients IV-7 and III-6 showed advanced myopathology with rimmed vacuoles (arrow) and eosinophilic protein inclusions (white arrowheads). (C) Immunofluorescent staining showed predominantly nuclear localization of HNRNPA1(magenta in all images) in patient IV-11 with minor myopathology. (D) In patient III-6 HNRNPA1 shows increased sarcoplasmic labeling in several highly atrophic fibers. (E) In patient IV-7, there is moderate HNRNPA1 accumulation pathology in one fiber (arrow), together with but apart from prominent p62 (green) inclusions. (F) TDP-43 (green) inclusions in patient III-6. Scale bars 100 µm. (G) Confocal microscopy shows nuclear localization of HNRNPA1 in healthy control muscle. Nuclear localization was less prominent in patient IV-7 and with some myonuclei being negative (arrowheads). Sarcoplasmic accumulation of p62 (green) and moderate accumulation of HNRNPA1 (small arrows) was observed. Scale bar 20 µm. HNRNPA1 = heterogeneous nuclear ribonucleoprotein A1
Biopsy of IV-11, obtained at the age of 39, represented early-stage pathology, with minor fiber size variation and internal nuclei, and very few rimmed vacuoles. No focal accumulation pathology was observed, but a few atrophic fibers showed diffuse HNRNPA1 signal (not shown). In the immunofluorescence analysis, HNRNPA1 showed the anticipated nuclear localization in healthy control muscle. In biopsies from III-6 and IV-7, a proportion of myonuclei were devoid of HNRNPA1 and TDP-43 (Figure 4).
In western blotting of patient IV-11 with early disease-stage biopsy, the HNRNPA1 isoforms A1a and A1b both showed approximately 50% reduced expression in the patient muscle (Figure 5). Additional bands of higher molecular weight, possibly representing HNRNPA1 oligomers, were observed, showing similarly reduced intensity in the patient. The epitope of the used HNRNPA1 antibody is just upstream of the mutation; hence, the antibody should recognize both wild-type and mutant protein isoforms.23 Muscle samples from other patients were not available for western blotting.
Figure 5. Western Blotting.
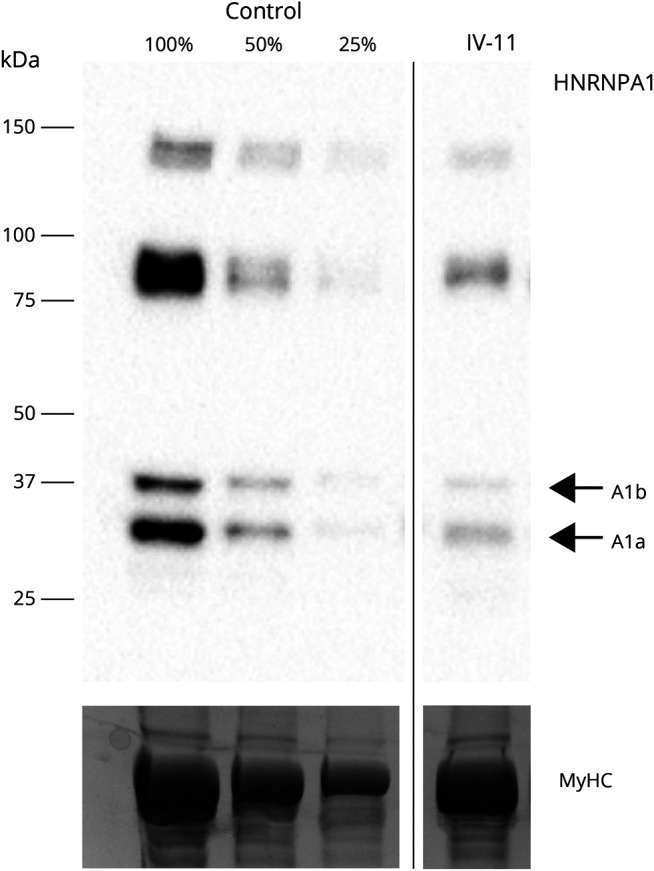
Both heterogeneous nuclear ribonucleoprotein A1 protein isoforms A1a (34 kDa) and A1b (39 kDa) were decreased in the biopsy from patient IV-11, compared with a pooled control sample. Myosin heavy chain (MyHC) is shown as loading control.
Discussion
HNRNPs are evolutionarily conserved RNA-binding proteins that assemble with nuclear and cytoplasmic RNA to form ribonucleoproteins. They are associated with the processing of RNAs into mature mRNAs by splicing, activation of gene expression, and modulation of protein translation.22,24 The chromosomal deletion identified in our patients contains an exon encoding part of a distinctive prion-like domain (PrLD), which is essential for the assembly of ribonucleoprotein granules. At least 240 human RNA-binding proteins contain PrLDs, low-complexity domains with similar composition to prions.25 HNRNPA1 variations have been associated with congenital heart defects,26 multiple sclerosis,27 and different phenotypes of the multisystem proteinopathy (MSP) spectrum including myopathy.28-30 Most mutations in the PrLD have been reported to accelerate the polymerization of HNRNPA1 and to interfere with stress granule dynamics.27,28,30,31 Abnormal stress granule dynamics leading to protein aggregation underlies also other degenerative diseases, such as distal myopathies caused by mutations in the PrLD of TIA1.4,32 Thus, HNRNPA1 was a good candidate gene for causing the observed phenotype.
No potentially interesting missense variants nor copy number variants (CNVs) were detected in the linked region on chromosome 8, and we assume that the linkage to chromosome 8 seems just by chance without having any impact on the disease. The Finnish population represents a genetic isolate with several lines of remote common ancestries increasing the likelihood of shared haplotypes.33
The small 160 bp deletion spanning exon 10 of the HNRNPA1 gene in our family escaped several genetic attempts over the years. Generally, small CNVs (<1000 bp) are challenging to detect from short-read sequencing data, with the evaluation of only-read depth differences. Therefore, the choice of structural variant analysis programs used on the genomic sequencing data was essential for the initial detection of this variant, and the RNAseq results showed convincingly that the deletion results in a transcript skipping exon 10. This aberrant transcript is predicted to result in the mutant protein p.Gly356Asnfs*4 (corresponding to p.Gly304Asnfs*4 in HNRNPA1a, NP_002127.1), where the 17 most C-terminal amino acids, encoded by the second last exon, are replaced by three amino acids encoded by the last exon, which normally is in the 3'-UTR (Figure 3). It is impossible to unequivocally predict the consequences at the protein level because splicing of exon 9 to the far 3'-UTR is another possible event, which apparently is increased in the mutant transcript according to our RNAseq data. If translated, this would lead to a novel 55 amino acid tail with no homology to any known proteins.
While this article was under review, Beijer et al.30 reported several novel pathogenic HNRNPA1 variants. Among these, 2 de novo mutations—a splice site mutation seen in a motor neuropathy patient and a 500-bp deletion in a myopathy patient—lead to the same p.Gly356Asnfs*4 protein change as seen in our family, adding strong support to the pathogenicity of these variants. In the functional studies of Beijer et al.,30 the mutant HNRNPA1a protein showed reduced phase separation and fibrillization propensity, and accelerated stress granule clearance, suggesting a pathomechanism distinct from most other PrLD mutations. Of note, their RNAseq data also indicated increased inclusion of the far 3’-UTR exons in the patient harboring the 500-bp deletion, potentially giving rise to the long minor mutant protein.
The fate of the mutant protein product(s) in patient muscle is, however, unclear. In an IF analysis of patient IV-11 muscle biopsy displaying minor pathology, HNRNPA1 showed, as expected, primarily nuclear localization and no sarcoplasmic expression. WB of the same biopsy indicated approximately 50% reduced expression of both HNRNPA1 isoforms (Figure 5). We did not detect any additional bands compatible with the predicted major or the possible minor mutant protein products. However, the major mutant proteins are only 1.4 kDa smaller than the respective wild-type forms and might not separate well in SDS-PAGE. The lower HNRNPA1 level in early-stage muscle could suggest reduced stability of the mutant protein. The major predicted mutant protein has a C-terminal alanine residue and could show increased turnover because of this C-degron,34 although the normal expression level of the mutant protein in transfection studies30 argues against this possibility.
Muscle biopsies from the patients III-6 and IV-7 with more advanced muscle pathology were not available for WB. As reported in previous publications on this family,7,8 the biopsies from these patients showed extensive rimmed vacuolation and large cytoplasmic protein inclusions. TDP-43 and SQSTM1/p62 showed clear accumulation pathology, similar to previously described HNRNPA1-related myopathies.29,30 HNRNPA1 seemed upregulated in a proportion of atrophic fibers in the III-6 and IV-7 biopsies showing advanced myopathology. However, no strong focal cytoplasmic accumulation, similar to biopsies with point mutations affecting HNRNPA1 aa 314,28,29 was observed in these samples. This difference in this family could be due to a more C-terminal mutation, and it is in line with reduced fibrillization propensity and accelerated stress granule clearance observed by Beijer et al.30
Although identification of HNRNPA1 as the gene underlying MPD3 resolves the genetic background of this disease, the relationship of HNRNPA1 variants and the associated clinical outcomes remains enigmatic. Besides myopathy, the MPD3 patients of our Finnish family did not show any symptoms of the MSP spectrum such as Paget disease of the bone, frontotemporal dementia, or ALS. Pure myopathy was also reported in families with the p.Asp314Asn mutation,29 originally identified in patients with ALS.28 On the other hand, variants resulting in the same p.Gly356Asnfs*4 change as in our family were reported in both myopathy and motor neuropathy patients (families D and A in ref. 30 respectively). The effects of p.Asp314Asn and p.Gly356Asnfs*4 on HNRNPA1 fibrillization propensity are essentially opposite.28,30 There is hence no obvious correlation between the primary molecular pathomechanism and the clinical phenotype, and this needs to be clarified in further studies.
Altogether, HNRNPA1 is a gene to consider in all unsolved distal myopathies especially in dominant families.
Acknowledgment
The authors wish to thank the family for participating in this study and CSC – IT Center for Science, Finland, for computational resources. Confocal microscopy was performed at the Biomedicum Imaging Unit, University of Helsinki. Sanger sequencing was performed at the sequencing unit of Institute for Molecular Medicine Finland FIMM Technology Centre, University of Helsinki. The sequencing unit is supported by Biocenter Finland. The study was supported by the Jane and Aatos Erkko Foundation, the Sigrid Jusélius Foundation, Solve-RD, and Finska läkaresällskapet.
Glossary
- bp
base pair
- CNV
copy number variant
- DAFO
dynamic ankle foot orthosis
- GS
genome sequencing
- HNRNPA1
heterogeneous nuclear ribonucleoprotein A1
- LOD
logarithm of odds
- MPD3
myopathy, distal, 3
- MSP
multisystem proteinopathy
- PrLD
prion-like domain
Appendix. Authors
Contributor Information
Salla M. Rusanen, Email: salla.valipakka@helsinki.fi.
Mridul Johari, Email: mridul.johari@helsinki.fi.
Anna Vihola, Email: anna.vihola@helsinki.fi.
Per Harald Jonson, Email: per-harald.jonson@helsinki.fi.
Jaakko Sarparanta, Email: jaakko.sarparanta@helsinki.fi.
Kati Donner, Email: kati.donner@helsinki.fi.
Päivi Lahermo, Email: paivi.lahermo@helsinki.fi.
Sampo Koivunen, Email: sampo.koivunen@alumni.helsinki.fi.
Helena Luque, Email: helena.luque@folkhalsan.fi.
Merja Soininen, Email: merja.x.soininen@helsinki.fi.
Ibrahim Mahjneh, Email: ibrahim.mahjneh@jakobstad.fi.
Mari Auranen, Email: mari.auranen@hus.fi.
Meharji Arumilli, Email: meharji.arumilli@helsinki.fi.
Marco Savarese, Email: marco.savarese@helsinki.fi.
Bjarne Udd, Email: bjarne.udd@netikka.fi.
Study Funding
Jane and Aatos Erkko Foundation, the Sigrid Jusélius Foundation, Solve-RD, and Finska läkaresällskapet.
Disclosure
The authors report no disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Udd B. Distal Myopathies. Neuromusc Disord. 2012;22(1):5-12. [DOI] [PubMed] [Google Scholar]
- 2.Udd B, Hackman P. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. University of Washington, Seattle; 1993–2020. [Google Scholar]
- 3.Savarese M, Sarparanta J, Vihola A, et al. Panorama of the distal myopathies. Acta Myol. 2020;39(4):245-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hackman P, Sarparanta J, Lehtinen S, et al. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Annal Neurol. 2013;73(4):500-509. [DOI] [PubMed] [Google Scholar]
- 5.Hackman P, Vihola A, Haravuori H, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamont P, Wallefeld W, Hilton-Jones Det al. New mutations widen the phenotypic spectrum of slow skeletal/ β-cardiac myosin (MYH7) distal myopathy. Hum Mutation. 2014;35(7):868-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahjneh I, Haravuori H, Paetau A, et al. A distinct phenotype of distal myopathy in a large Finnish family. Neurology. 2003;61:87-92. [DOI] [PubMed] [Google Scholar]
- 8.Haravuori H, Siitonen HA, Mahjneh I, et al. Linkage to two separate loci in a family with a novel distal myopathy phenotype (MPD3). Neuromuscul Disord. 2004;14:183-187. [DOI] [PubMed] [Google Scholar]
- 9.Siggberg L, Ala-Mello S, Linnankivi T, et al. High-resolution SNP array analysis of patients with developmental disorder and normal array CGH results. BMC Med Genet. 2012;13:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weeks DE, Ott J, Lathrop GM. SLINK: a general simulation program for linkage analysis. Am J Hum Genet. 1990;47:A203. [Google Scholar]
- 11.Di Resta C, Pipitone GB, Carrera P, et al. Current scenario of the genetic testing for rare neurological disorders exploiting next generation sequencing. Neural Regen Res. 2021;3:475-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, Kamatani Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019;20(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333-i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014;15(6):R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Samarakoon PS, Sorte HS, Stray-Pedersen A, Rødningen OK, Rognes T, Lyle R. cnvScan: a CNV screening and annotation tool to improve the clinical utility of computational CNV prediction from exome sequencing data. BMC Genomics. 2016;17:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins R, Brand H, Karczewski K, et al. A structural variation reference for medical and population genetics. Nature. 2020;581(7809):444-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007;35:W71-W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobin A, Davis CA, Schlesinger F, et al. Ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923-30. [DOI] [PubMed] [Google Scholar]
- 20.Savarese M, Jonson PH, Huovinen S, Paulin L, Auvinen P, Udd B, Hackman P. The complexity of titin splicing pattern in human adult skeletal muscles. Skelet Muscle. 2018;8(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saeidian AH, Youssefian L, Vahidnezhad H, Uitto J. J research techniques made simple: whole-transcriptome sequencing by RNA-seq for diagnosis of monogenic disorders. Invest Dermatol. 2020(6):1117-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy R, Huang Y, Seckl MJ, Pardo OE. Emerging roles of hnRNPA1 in modulating malignant transformation. Wiley Interdiscip Rev RNA. 2017;8:1-18. [DOI] [PubMed] [Google Scholar]
- 23.Kim JH, Lee JH, Kim J, Kim JK. Identification of phage-displayed peptides which bind to the human HnRNPA1 protein-specific monoclonal antibody. Mol Cells. 1999;9(4):452-454. [PubMed] [Google Scholar]
- 24.Geuens T, Bouhy D, Timmerman V. The hnRNP family: insights into their role in health and disease. Hum Genet. 2016;135(8):851-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fomicheva A, Ross E. From prions to stress granules: defining the compositional features of prion-like domains that promote different types of assemblies. J Mol Sci. 2021;22(3):1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu Z, Tang PL, Wang J, et al. Mutations in HNRNPA1 cause congenital heart defects. JCI Insight. 2018;3(2):1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke JWE, Thibault PA, Salapa HE, Kim DE, Hutchinson C, Levin MC. Multiple sclerosis-associated hnRNPA1 mutations alter HNRNPA1 dynamics and influence stress granule formation. Int J Mol Sci. 2021;22(6):2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HJ, Kim NC, Wang YD, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495(7442):467-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Izumi R, Warita H, Niihori T, et al. Isolated inclusion body myopathy caused by a multisystem proteinopathy–linked hnRNPA1 mutation. Neurol Genet. 2015;1(3):e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beijer D, Kim HJ, Guo L, et al. Characterization of HNRNPA1 mutations defines diversity in pathogenic mechanisms and clinical presentation. JCI Insight. 2021;6(14):e148363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison AF, Shorter J. RNA-binding proteins with prion-like domains in health and disease. Biochem J. 2017;474(8):1417-1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee Y, Jonson PH, Sarparanta J, et al. TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J Clin Invest. 2018;128(3):1164-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peltonen L, Jalanko A, Varilo T. Molecular genetics of the Finnish disease heritage. Hum Mol Genet. 1999;8:1913-1923. [DOI] [PubMed] [Google Scholar]
- 34.Timms RT, Koren I. Tying up loose ends: the N-degron and C-degron pathways of protein degradation. Biochem Soc Trans. 2020;48(4):1557-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Pseudonymized data not published in this article are available in RD-connect, as genomic data and clinical HPOs, and will also be shared upon request to any qualified investigator.