

Abstract
Tumor hypoxia is viewed as one of the major factors responsible for tumor progression, metastasis, invasion, and treatment resistance, leading to low local tumor control and recurrence after radiotherapy in different types of cancers. Novel hypoxia agents are therefore in great need for accurate detection and monitoring of tumor hypoxia. Herein, we report the development of 18F-PET (positron emission tomography) probes for visualizing viable hypoxic cells in biopsies. Pimonidazole derivatives and nitroimidazole based agents bearing sulfonyl linker were evaluated. Small animal PET study showed the tumor uptake of [18F]-23 (PEG-sulfonyl-linker with tumor uptake of 3.36 ± 0.29%ID/g) was significantly higher (P < 0.01) than that of [18F]-20 (piperazine-linker tracer with tumor uptake of 2.55 ± 0.49 %ID/g) at 2 h post-injection in UPPL tumor. The tumor-to-muscle uptake ratio of [18F]-23 (2.46 ± 0.48 at 2 h p.i.) is well improved compared with the positive control ([18F]-FMISO, 1.25 ±0.14 at 2 h p.i.). Comparable distribution pattern was observed between ex vivo autoradiography of [18F]-23 and pimonidazole staining of neighboring slice, indicating [18F]-23 is a promising PET agent for hypoxia imaging and worth further evaluation in future studies.
Keywords: hypoxia, imidazole, positron emission tomography (PET), fluorine-18
Graphical abstract
Introduction
Cancer is the second leading cause of death globally, claiming over 9.5 million lives worldwide. In addition to conventional cancer therapies, such as surgery, chemotherapy, and radiotherapy, significant amount of effort has been devoted to the development of new anticancer agents, including small molecular compounds,1,2 metal complexes,3–5 and antibody therapy.6,7 Consequently, providing patients with suitable therapy becomes especially important. Hypoxia, a condition with low oxygen levels in tissues, plays an important role in promoting tumor progression, angiogenesis and resistance to radiotherapy and chemotherapy.8,9
Hypoxia is a common phenomenon in solid malignant tumors, characterized by higher resistance to therapy and an indicator for poor prognosis.10 For example, the degree of hypoxia in prostate cancer was shown to be a strong predictor for treatment failure in patients with localized prostate cancer after brachytherapy.11 Various methods have been developed for hypoxia detection, such as Eppendorf O2 polarographic needle electrode,12 exogenous markers,13 immunohistochemical (IHC) analysis of endogenous tumor hypoxia markers,14 magnetic resonance imaging15 and hypoxia positron emission tomography (PET).16 As a non-invasive, quantitative and highly sensitive imaging method,17 PET has been used to delineate the behavior of tumor hypoxia during therapy, thus offering improved treatment plan and prognosis for patients.18
A number of hypoxia PET probes have been developed in the last three decades, including F-18, Ga-68, Cu-64 or I-123 labeled hypoxia PET agents.19–21 Among them, the F-18 labeled nitroimidazole derivatives, such as 18F-FMISO and 18F-FAZA,22,23 have been applied in basic and clinical research because of their suitable half-life and contrast (Figure 1). The nitroimidazole analogues have been widely used as a motif targeting hypoxic environments, due to the hypoxia-selective mechanism of action. Normally, the nitro group in nitroimidazole compounds are enzymatically reduced in all tissues. The intermediates are immediately oxidized back to the starting material in the presence of normal levels of oxygen. In hypoxic tissues, however, these electrophilic intermediates react further and form adducts with DNA and proteins.
Figure 1.
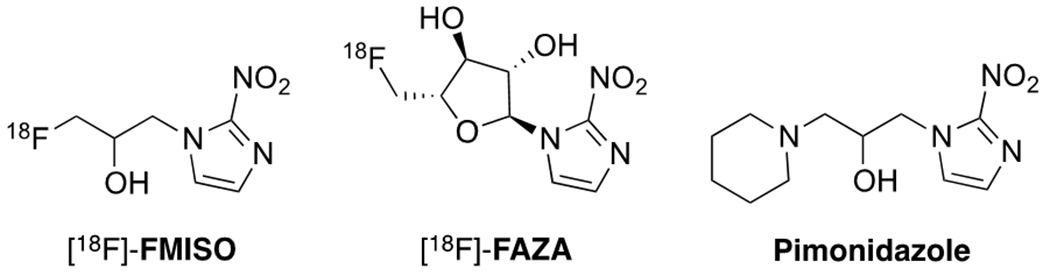
Nitroimidazole based hypoxia PET tracers and staining agent pimonidazole.
Despite the advances in nitroimidazole-based probes, there remains a great need to develop new hypoxia PET agents. Pimonidazole is widely used in hypoxia staining due to its relatively short plasma half-life and low background. Thus, PET agents derived from the pimonidazole skeleton may seamlessly integrate PET images with current pimonidazole based staining to provide favorable tumor-to-reference tissue contrast.24 Unfortunately, investigations of pimonidazole-based PET agents have been limited due to challenges to efficiently introduce 18F into the parent pimonidazole compound. Herein, we report our initial attempts to develop PET agents based on the pimonidazole core structure. Moreover, neutral PEG-sulfonyl linkers were introduced between 18F and imidazole motif, which were then evaluated in vivo in order to study the effects of side chains on nitroimidazole-based hypoxia probes. The goal of this study was to design and evaluate novel hypoxia PET agents centering on the modifications of the side chains.
Results and Discussion
Chemical and Radiochemical Syntheses.
Our initial approach focused on synthesizing pimonidazole-based PET tracers by a NIS-mediated cyclization, a novel radiofluorination method that we have recently reported.25 The synthetic attempts are outlined in Scheme 1. We started with the fluorination of compound 1, a readily accessible starting material. Yet fluorinated product 2 was obtained in extremely low yield, potentially due to the steric hindrance of the methyl group. Alternatively, compound 3 was synthesized and subjected to the NIS-mediated fluorination reaction to afford standard 4 smoothly (34% yield, detail in supporting information). However, the radiofluorination of 3 failed to form [18F]-4, possibly owing to the presence of a free hydroxyl group. While a THP-protected precursor 5 successfully led to the formation of [18F]-6, the deprotection of THP group of [18F]-6 with hydrochloric acid failed to yield [18F]-4. Finally, in an attempt to introduce 18F to the piperidine ring using the compound 7, the radiolabeled [18F]-8 was not detected, but rather undesired elimination reaction was a dominant pathway.
Scheme 1.

Initial trials of pimonidazole-based PET tracer synthesis. i) NIS, RT, 10 min, followed by incubation with [19F]/[18F]-TBAF at RT for 10 min; ii) 1 N HCl, 80 °C, 5 min; iii) [18F]-TBAF, 110 °C, 10 min.
Compounds with PEG linkers.
In light of these unexpected problems, we modified our probe design and focused on labeling the imidazole with diamine-linked polyethylene glycols (PEG) (Scheme 2). A series of diamine-linked polyethylene glycol hypoxia tracers were then synthesized. Starting from di-tosylate-PEG1 (9) and di-tosylate-PEG2 (12) respectively, the reactions with 18F-TBAF at 110 °C26 followed by purification with semi-HPLC gave the intermediate [18F]-10 and [18F]-13. Substitution reactions of [18F]-10 and [18F]-13 with diamines produced [18F]-11, [18F]-14 or [18F]-15, which then reacted with 1-(2,3-epoxypropyl)-2-nitroimidazole to provide final products [18F]-20, [18F]-21 and [18F]-22, in moderate radiochemical yield (RCY).
Scheme 2.

Synthesis of PET agents [18F]-20 to [18F]-25. i) [18F]-TBAF, 110 °C, 15 min; ii) piperazine, 110 °C, 10 min; iii) homopiperazine, 110 °C, 10 min; iv) potassium carbonate, 1-(2,3-epoxypropyl)-2-nitroimidazole, 90 °C, 20 min; v) [18F]-TBAF, 110 °C, 20 min; vi) 2-nitroimidazole, borate buffer, pH 8.5, DMSO; vii) 4-nitroimidazole, borate buffer, pH 8.5, DMSO.
Compounds with sulfonyl linkers.
It has been shown that the overall charge and electronic properties of molecules can significantly alter the compounds’ biodistribution potency.27 The diamine linker in 20–22 is generally considered to be positively charged candidates, as they are likely to be protonated in aqueous media. In contrast, sulfone moieties (–SO2R) have been widely used in medicinal chemistry as a neutral linker.28 The sulfone group can act as a less polar variant of diphosphate and as a pro-drug by releasing sulfonic acid after hydrolysis to mimic some bioactive agents including antibiotics analogue.29 The Michael addition between vinyl sulfone and amine or thiol groups in biomolecules has demonstrated its great potential in developing imaging tracers or treatment agents.30–32 We therefore designed compound 23–25 bearing a sulfone linker as neutrally charged candidates. In brief, [18F]-17 and [18F]-19 were synthesized from 16 and 18 through SN2 reaction with [18F]-TBAF. The reactions of [18F]-17 with 2-nitroimidazole were performed under pH 8.5 in borate buffer and DMSO mixture, which lead to [18F]-23 with a separation RCY of 58.7%. [18F]-24 was synthesized from [18F]-17 using 4-nitroimidazole. [18F]-25 was analogous to [18F]-23, but with an increased length of PEG chain. RCY results of the above PET tracers are summarized in Table 1. Generally, the labeling yields of sulfone-based hypoxia tracers (53.1–66.8%) were higher than those of piperazine-based hypoxia tracers (22.8–29.0%). The identities of these agents ([18F]-20–[18F]-25) were confirmed by co-injection with the non-radiolabeled standards ([19F]-20–[19F]-25) (HPLC spectra presented in the supporting information).
Table 1.
Radiosynthetic yields for [18F]-20, [18F]-21, [18F]-22, [18F]-23, [18F]-24 and [18F]-25.
We also attempted to develop a more concise synthesis of [18F]-23 through precursor 26 (Scheme 3). Although the process was greatly simplified, one-step radiofluorination of 26 only led to [18F]-23 in 1.4% RCY. Further elevated reaction temperature or switching to K18F/Kriptofix failed to improve the labeling yield due to increased precursor decomposition. We therefore used the synthetic method shown in Scheme 2 to prepare [18F]-23 for its evaluation in tumor imaging profile.
Scheme 3.
Alternative synthetic protocol of [18F]-23. i) 2-nitroimidazole, borate buffer, pH 8.5, DMSO; ii) [18F]-TBAF, 110 °C, acetonitrile, 20 min.
Small animal PET imaging.
Screening of PET agents [18F]-20–[18F]-23 was first performed on nude mice bearing Fadu tumors (a well-established model for hypoxia imaging).33 PET scanning was performed at 0.5 h and 2 h post tracer injection. Representative images of [18F]-20 to [18F]-23 on Fadu tumor-bearing mice are shown in Figure 2A. The tumor uptakes of [18F]-20 to [18F]-23, which were presented as mean ± SD, were 2.21 ± 0.31%ID/g, 1.43 ± 0.25%ID/g, 1.52 ± 0.30%ID/g and 2.82% ± 0.66%ID/g at 0.5 h p.i., and 1.80 ± 0.27%ID/g, 0.95 ± 0.34%ID/g, 0.77 ± 0.31%ID/g and 2.27 ± 0.64%ID/g at 2 h p.i., respectively. Compared with [18F]-20 (tumor uptake value of 2.21 ± 0.31%ID/g and tumor-to-muscle ratio of 1.37 ± 0.18 at 2 h p.i.), the lower tumor uptake of [18F]-21 (1.43 ± 0.25%ID/g) and low tumor-to-muscle ratio of [18F]-22 (0.74 ± 0.16) did not warrant them to be investigated further as hypoxia imaging agents. Replacing the piperazine motif with homopiperazine ([18F]-22) also lead to decreased tumor uptake in vivo. [18F]-23, the agent with a sulfone linker, demonstrated highest tumor uptake among these four agents. These preliminary studies suggested [18F]-20 and [18F]-23 are promising hypoxia tracers, although relatively high background was still observed. This also indicated that the neutral PEG-sulfonyl linker may be beneficial in the biodistribution of the tracer.
Figure 2.

Evaluation of [18F]-20 to [18F]-23 in tumor bearing mice. (A) PET image of FaDu tumor bearing mice at 2 h post injection. (B) Major organ and tumor uptakes of [18F]-20, [18F]-21, [18F]-22, and [18F]-23 in nude mice bearing FaDu tumor at 0.5 h post injection (three mice per group). (C) Major organ and tumor uptakes of [18F]-20, [18F]-21, [18F]-22, and [18F]-23 in nude mice bearing FaDu tumor at 2 h post injection (three mice per group).
In addition to FaDu tumor, hypoxia is also common in the microenvironment of various solid tumors, including bladder cancer. Recent studies have shown that hypoxic bladder cancer cells remodel tumor microenvironment to facilitate tumor growth 34 and treatments targeting hypoxic cells have gained promising results in bladder cancer therapy.35 A hypoxia imaging agent would work as a companion diagnostic and prognosis biomarker in the study of hypoxic bladder cancers. Therefore, we evaluated our newly developed agents in bladder cancers using UPPL tumors (Luminal-like bladder cancer cell line (named UPPL)).36
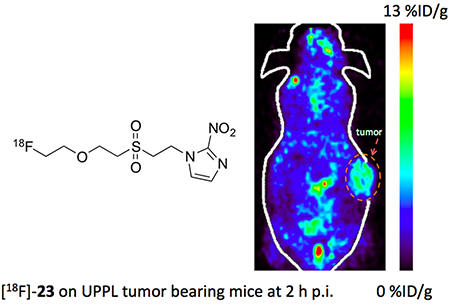
As shown in Figure 3A, [18F]-23 demonstrated higher uptake (3.36 ± 0.29%ID/g at 2 h p.i.) than [18F]-20 (2.55 ± 0.50%ID/g at 2 h p.i.) in UPPL tumor. Both agents showed increased uptake in UPPL tumor (P < 0.05) compared with that of FaDu tumor (P > 0.05). The tumor uptake of [18F]-23 in UPPL model is 3.80 ± 0.35%ID/g, 4.02 ± 0.48%ID/g and 3.36 ± 0.29%ID/g at 30, 60 and 120 min p.i. respectively. The comparison with well-established hypoxia agent [18F]-FMISO was also performed in UPPL model (Figure S36). [18F]-23 demonstrated higher tumor uptake but comparable contrast compared with [18F]-FMISO at 0.5 h p. i. (P < 0.01). The contrast of [18F]-23 became significantly higher than [18F]-FMISO at 2 h p. i. (P < 0.01, Figure 3B). The tumor/muscle uptake ratios of [18F]-23 is 0.93 ± 0.08 and 2.46 ± 0.48 at 0.5, 2 h p.i., respectively, compared with 0.84 ± 0.20 and 1.25 ± 0.14 at 0.5, 2 h p.i., respectively for [18F]-FMISO. The representative images of tracer [18F]-23 in UPPL tumor bearing mice show the contrast was improved at 2 h timepoint post the tracer injection compared to the images at 0.5 h and 1 h p.i. (Figure 3C).
Figure 3.

(A) Tumor uptake of [18F]-20 and [18F]-23 in nude mice bearing FaDu tumor or black mice bearing UPPL tumor at 2 h post injection (three mice per group). (B) Major organ uptakes of [18F]-23 and [18F]-FMISO in black mice bearing UPPL tumor at 2 h post injection (three mice per group). (C) Representative PET images of UPPL tumor bearing mouse at 30 min, 60 min and 120 min post injection of [18F]-23.
In addition to [18F]-23, [18F]-24 and [18F]-25 were also evaluated in mice bearing UPPL tumors and the results were summarized in Figure 4A. The replacement of 2-nitroimidazole ([18F]-23, 3.36 ± 0.29 %ID/g, n = 3/group) to 4-nitroimidazole (Figure 4B, [18F]-24) resulted in the significant decrease in tumor uptake (1.18 ± 0.04 %ID/g, n = 3/group, P < 0.01). Extending the PEG linker led to comparable tumor uptake ([18F]-25, 3.30 ± 0.47 %ID/g, n = 3/group, P > 0.05). Overall, PEG1-sulfonyl-2-nitro-imidazole tracer [18F]-23 was the most promising candidate among the six PET tracers with various linkers. We observed bone uptake when the mice were scanned ([18F]-25, Figure S37) at 4 h post injection, suggesting defluorination of this kind of tracer at a later time point.
Figure 4.
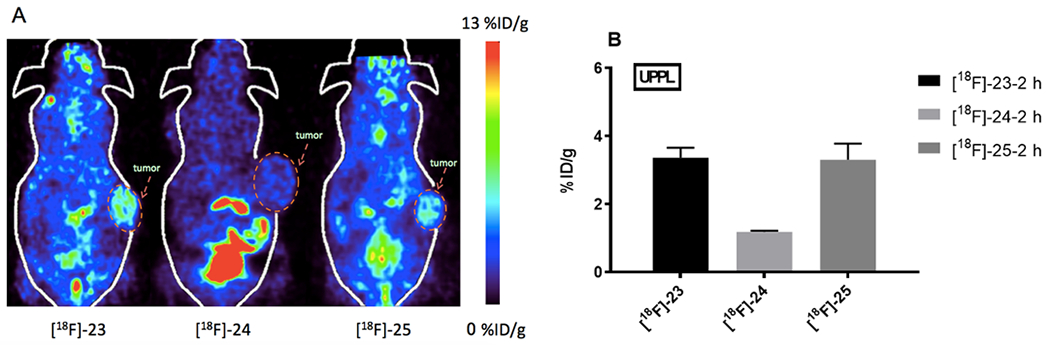
(A) PET image on UPPL tumor bearing mice at 2 h post injection. (B) Tumor uptake quantification of [18F]-23, [18F]-24 and [18F]-25 in black mice bearing UPPL tumor at 2 h post injection (three mice per group).
In vitro and in vivo stability.
The in vitro and in vivo stability of [18F]-23 was evaluated (Figure S29–S34). As shown in Figure S29–S31, [18F]-23 was stable in plasma with no apparent decomposition after 2 h incubation. After injecting into an animal, the agent decomposed quickly when the bloods were analyzed (Figure S32–S34). Only 41.76% of [18F]-23 remained unchanged at 30 min post-injection, which was further decreased to baseline level at the 1 and 2 h p.i.. This may be caused by the fast clearance of the agent from blood and the complexity of in vivo enzymatic environment. In one aspect, the hypoxia agents need be retained in tumor cell due to the hypoxic environment; yet, the agent also needs to be cleared from normal tissues to improve contrast. Although we observed improved contrast of [18F]-23 in hypoxia tumor, detailed mechanism studies are still needed in the future to better design the agent.
Autoradiography.
To investigate the distribution of our PET agent within tumor, UPPL tumor was collected, followed by ex vivo autoradiography and IHC of pimonidazole staining using neighboring slices (Figure 5). Pimonidazole is a hypoxia marker widely applied in the study of tumor hypoxia and cell proliferation in tumor, so comparison of pimonidazole IHC and ex vivo autoradiography would confirm the tracer distribution.37–39 As shown in Figure 5, the pimonidazole-positive hypoxia regions were observed in staining (right image). Similarly, in the neighboring slice, [18F]-23 was also heterogeneously distributed within the tumor (left image). The intra-tumor distribution of [18F]-23 on ex vivo autoradiography was visually similar to the histopathological distribution of pimonidazole. The distribution of [18F]-23 shown at the lower area of tumor on autoradiography but not on pimonidazole staining was most likely caused by either a cutting and section mounting error, or using neighboring slices instead of the same slice. Nonetheless, it is notable that the staining experiment provided promising preliminary result. Additional study is needed in the future to further confirm the hypoxia specificity of the [18F]-23.
Figure 5.

Autoradiography (left) and pimonidazole staining (right) of tumor tissues with [18F]-23
Conclusion
Six potential hypoxia agents were synthesized and evaluated in our studies. PEG-Sulfonyl based hypoxia agent [18F]-23 has demonstrated prominent uptake (3.36 ± 0.29%ID/g at 2 h p. i. in UPPL tumor model) with relatively low muscle uptake. Autoradiography and pimonidazole staining of neighboring slides showed a similar distribution pattern, indicating promising potential of [18F]-23 to image hypoxia tumor tissue. Our findings warrant the use of neutral sulfonyl group as a suitable scaffold to further expand the library of novel PET hypoxia tracers and/or hypoxia treatment agents.
Experimental Section
General.
All chemicals were used as received from their manufacturer (Sigma-Aldrich, Fisher Scientific, and Alfa Aesar) unless otherwise noted. Nuclear magnetic resonance spectra were obtained on a Bruker AVANCE 400 MHz spectrometer. HPLC was carried out with either a Shimadzu Nexera XR LC-20AD equipped with a PDA detector or a Shimadzu LC-20AT Prominence equipped with UV detection. Purity of all final compounds was ≥95% as determined by HPLC analysis.
Synthesis of compound 1, 3-7.
1-((2,2-dimethylpent-4-en-1-yl)amino)-3-(2-nitro-1H-imidazol-1-yl)propan-2-ol (1) :
A 25-mL round bottom flask (RBF) was charged with 4-methylpent-4-en-1-amine (338 mg, 2.0 mmol, 1.0 equiv), 2-nitro-1-(oxiran-2-ylmethyl)-1H-imidazole (396 mg, 4.0 mmol, 2.0 equiv) and EtOH (5 mL). The resulting mixture was heated to 50 °C and allowed to stir overnight. Solvent was then removed in vacuo and the crude reaction mixture was subjected to flash column chromatography (50% EtOAc/Hexanes–5% MeOH/DCM) to afford product 1 as a yellow oil (54%). Rf = 0.16 (5% NEt3, 50% EtOAc/Hexane); 1H NMR (500 MHz, CD3OD): δ 7.45 (s, 1H), 7.12 (s, 1H), 4.47 (m, 2H), 4.66 (dd, J = 13.8, 3.6 Hz, 1H), 4.33 (dd, J = 13.8, 8.6 Hz, 1H), 4.07 (tt, J = 8.6, 3.6 Hz, 1H), 2.75 (dd, J = 12.2, 3.6 Hz, 1H), 2.64–2.61 (m, 3H), 2.07 (t, J = 7.5 Hz, 2H), 1.73 (s, 3H), 1.67 (p, J = 7.5 Hz, 2H); 13C NMR (126 MHz, CD3OD): δ 146.3, 129.2, 128.1, 113.2, 110.8, 69.5, 54.8, 53.4, 50.1, 36.3, 28.1, 22.4; FTIR (thin film): 2937, 1647, 1361, 1265 cm−1; HRMS-ESI (m/z) calcd. for C12H21N4O3 ([M+H]+): 269.1608; found: 269.1612.
1-((2,2-dimethylpent-4-en-1-yl)amino)-3-(2-nitro-1H-imidazol-1-yl)propan-2-ol (3) :
A 50-mL RBF was charged with 2,2-dimethylpent-4-en-1-amine (676 mg, 4.0 mmol, 1.0 equiv), 2-nitro-1-(oxiran-2-ylmethyl)-1H-imidazole (904 mg, 8.0 mmol, 2.0 equiv.) and EtOH (10 mL). The resulting mixture was heated to 60 °C and stirred overnight. Solvent was then removed in vacuo, affording 3 as an orange oil (98%). Rf = 0.14 (5% NEt3, 50% EtOAc/Hexane); 1H NMR (500 MHz, CDCl3): δ 7.16 (s, 1H), 6.87 (s, 1H), 5.65 (ddt, J = 16.1, 10.8, 7.5 Hz, 1H), 4.87 (d, J = 10.8 Hz, 1H), 4.86 (d, J = 16.1 Hz, 1H), 4.56 (dd, J = 13.8, 3.0 Hz, 1H), 4.15 (d, J = 13.8, 8.0 Hz, 1H), 3.94–3.91 (m, 1H), 2.64 (d, J = 12.2, 4.2 Hz, 1H), 2.43 (d, J = 12.2, 7.7 Hz, 1H), 2.27 (d, J = 11.5 Hz, 1H), 2.20 (d, J = 11.5 Hz, 1H), 1.86 (d, J = 7.5 Hz, 2H), 0.74 (s, 6H); 13C NMR (126 MHz, CDCl3): δ 144.4, 134.9, 127.7, 127.3, 116.8, 67.8, 59.8, 53.5, 52.7, 44.3, 34.1, 25.2; FTIR (thin film): 3334, 2958, 1639, 1488, 1361, 1162 cm−1; HRMS-ESI (m/z) calcd. for C13H23N4O3 ([M+H]+): 283.1765; found: 238.1768.
1-(5-fluoro-3,3-dimethylpiperidin-1-yl)-3-(2-nitro-1H-imidazol-1-yl)propan-2-ol (4):
To a solution of 3 (141 mg, 0.5 mmol, 1.0 equiv) in t-BuOH (7.5 mL) was added NIS (113 mg, 0.5 mmol, 1.0 equiv.) dissolved in t-BuOH (2.5 mL). The reaction was allowed to stir at room temp for 1.5 h. To the mixture was then added AgOTf (386 mg, 1.5 mmol, 3.0 equiv.) and TBAF (1 M in THF, 1.5 mL, 1.5 mmol, 3.0 equiv.). The resulting mixture was heated to 70 °C and allowed to stir for 15 min. The reaction was cooled to room temperature, filtered through a plug of Celite, and the filtrate was concentrated in vacuo. The crude reaction mixture was purified by flash column chromatography (50% EtOAc/Hexane–100% EtOAc) to afford 4 as a yellow solid (34%); Rf = 0.27 (5% NEt3, 50% EtOAc/Hex); 1H NMR (500 MHz, CDCl3): δ 7.26 (s, 1H), 7.11 (s, 1H), 4.78–4.63 (m, 2H), 4.22 (ddd, J = 13.8, 10.1, 2.5 Hz, 1H), 4.12–4.04 (m, 1H), 2.86–2.72 (m, 1H), 2.53 (ddd, J = 12.4, 5.5, 4.0 Hz, 1H), 2.46–2.37 (m, 1H), 2.33–2.26 (m, 2H), 2.12–2.01 (m, 1H), 1.72–1.59 (m, 1H), 1.53–1.41 (m, 1H), 1.03 (s, 3H), 0.96 (s, 3H); 13C NMR (126 MHz, CDCl3): δ 144.9, 128.2, 127.7, 87.8 (d, J = 44.4 Hz), 86.4 (d, J = 43.6 Hz) 66.2, 60.3 (d, J = 15.4 Hz), 58.0 (d, J = 23.7 Hz), 53.5 (d, J = 11.8 Hz), 43.4 (d, J = 17.5 Hz), 31.8 (d, J = 34.3 Hz), 28.3 (d, J = 41.8 Hz), 27.3 (d, J = 60.3 Hz); FTIR (thin film): 3283, 3107, 2941, 1486, 1365 cm−1; HRMS-ESI (m/z) calcd. for C13H22FN4O3 ([M+H]+): 301.1671; found: 301.1671.
2,2-dimethyl-N-(3-(2-nitro-1H-imidazol-1-yl)-2-((tetrahydro-2H-pyran-2-yl)oxy)propyl)pent-4-en-1-amine (5):
To a solution of 1-(3-azido-2-((tetrahydro-2H-pyran-2-yl)oxy)propyl)-2-nitro-1H-imidazole (148.1 mg, 0.5 mmol, 1.0 equiv.) in DCM was added solution of trimethylphosphine (1.0 M in toluene, 0.6 mL, 0.6 mmol, 1.2 equiv.) under N2 atmosphere. Reaction was stirred at room temp for 1.5 h, then 2,2-dimethylpent-4-enal (112 mg, 1.0 mmol, 2.0 equiv.) was added and stirred for 16 h. Then, NaBH(OAc)3 (211.9 mg, 1.0 mmol, 2.0 equiv.) was added at room temp and stirred for 10 h. Reaction was quenched with saturated aqueous NaHCO3 and extracted with DCM. Organic layers were concentrated in vacuo, then subjected to flash column chromatography (100% EtOAc) to afford the product (55%). 1H NMR (400 MHz, CDCl3): δ 7.30 (s, 1H), 7.10 (s, 1H), 5.84–5.73 (m, 1H), 5.07–5.03 (m, 2H), 4.73–4.67 (m, 2 H), 4.36 (s, 1H), 3.44–3.35 (m, 2H), 3.02 (m, 1H), 2.68 (dd, J =12.6, 5.7 Hz, 1H), 2.54 (dd, J =41.2, 11.7 Hz, 2H), 2.05 (d, J = 7.4 Hz, 2H), 1.72–1.70 (m, 1H), 1.55–1.39 (m, 6H), 0.96 (s, 6H). HRMS-ESI (m/z) calcd. for C18H31N4O4 ([M+H]+): 367.2345; found: 267.2340.
5-fluoro-3,3-dimethyl-1-(3-(2-nitro-1H-imidazol-1-yl)-2-((tetrahydro-2H-pyran-2-yl)oxy)propyl)piperidine (6):
To a solution of 5 (137 mg, 0.1 mmol, 1.0 equiv.) in t-BuOH (0.5 mL) was added NIS (22.5 mg, 0.1 mmol, 1.0 equiv.) dissolved in t-BuOH (0.5 mL). The reaction was allowed to stir at room temp for 1 h. To the mixture was then added AgOTf (76.8 mg, 0.3 mmol, 3.0 equiv.) and TBAF (1 M in THF, 0.3 mL, 0.3 mmol, 3.0 equiv.). The resulting mixture was heated to 70 °C and allowed to stir for 1 h. The reaction was cooled to room temperature, filtered through a plug of Celite, and the filtrate was concentrated in vacuo. The crude reaction mixture was purified by flash column chromatography (50% EtOAc/Hexane–100% EtOAc) to afford 6 as a yellow solid (15%); Rf = 0.20 (10% MeOH/DCM); HRMS-ESI (m/z) calcd. for C18H30FN4O4 ([M+H]+): 385.2251; found: 385.2249.
1-(3-(2-nitro-1H-imidazol-1-yl)-2-((tetrahydro-2H-pyran-2-yl)oxy)propyl)piperidin-4-yl 4-methylbenzenesulfonate (7):
1H NMR (400 MHz, CDCl3): δ 7.78 (dd, J = 8.3, 2.2 Hz, 2H), 7.22 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 1.1 Hz, 1H), 7.08 (d, J = 1.1 Hz, 1H), 4.79 (dd, J = 14.0, 3.2 Hz, 1H), 4.62 (dd, J = 4.8, 2.5 Hz, 1H), 4.53 (tt, J = 3.4, 11.1 Hz, 1H), 4.40 (dd, J = 14.0, 7.8 Hz, 1H), 4.09–4.18 (m, 1H), 3.25 (dd, J = 6.1, 4.0 Hz, 1H), 3.18 (dd, J = 7.3, 3.5 Hz, 1H), 2.55-2.63 (m, 3H), 2.44 (s, 3H), 2.26–2.32 (m, 5H), 1.37–1.47 (m, 8H).
Radio chemistry.
Typical radiolabeling protocol for [18F]-20 to [18F]-22:
To the solution of ethane-1,2-diyl bis(4-methylbenzenesulfonate) (2 mg) in anhydrous acetonitrile (50 μL) within cap sealed v-vial, the [18F]-TBAF (216 mCi) in acetonitrile was added in. The sealed reactor allows the reaction to be performed under pressure at 110 °C for 15 min. After diluted with 1.5 mL of solvent (water/acetonitrile 2/3 v/v), the mixture was load to HPLC for purification. HPLC method: Solvent A: 0.1% TFA water; solvent B: 0.1% TFA acetonitrile; 0 to 2 min: isocratic elution of 40% solvent B, 2 to 22 min, 40−95% of solvent B. Flow rate: 3 mL/min. HPLC column: Gemini 5 u C18 110 A, 250 × 10.00 mm. 77 mCi of [18F]-2-fluoroethyl 4-methylbenzenesulfonate was collected at 15 min. The collected [18F]-2-fluoroethyl 4-methylbenzenesulfonate was diluted with 10 mL of water and then passed through a Sep-Pak tC18 cartridge. The loaded activity was washed out with 1 mL of acetonitrile. The elution was blow dried in a v-vial, and then 20 μL piperazine solution (0.33 μmol/mL) and 60 μL acetonitrile were added in and heated at 110 °C for further 10 min. Then K2CO3 (2 mg), 1-(2,3-epoxypropyl)-2-nitroimidazole (3.8 mg) and 60 μL acetonitrile were added and heated at 90 °C for 20 min. The resulting tracer was purified by HPLC with the method described as: solvent A: 0.1% Et3N water; solvent B: 0.1% Et3N acetonitrile, 0 to 45 min: 5–30% of solvent B. Flow rate: 1 mL/min, collected at 17.6 min. RCY: 29.0%. [18F]-21 (0 to 35 min: 5–60% of solvent B. Flow rate: 1 mL/min, collected at 14.0 min. 22.8%) and [18F]-22 (0 to 35 min: 5–60% of solvent B. Flow rate: 1 mL/min, collected at 15.4 min, 28.8%) were synthesized accordingly by using oxybis(ethane-2,1-diyl) bis(4-methylbenzenesulfonate) and homopiperazine as starting material.
Typical radiolabeling protocol for [18F]-23 to [18F]-25:
The precursor 2-(2-(vinylsulfonyl)ethoxy)ethyl 4-methylbenzenesulfonate (2 mg) was added into a v-vial and then the v-vial was sealed. Then the [18F]-TBAF (200 mCi) in acetonitrile was added in and the vial was heated at 110 °C for 20 min. After diluted with 1 mL of water, the mixture was load to HPLC for purification. HPLC method: Solvent A: 0.1%TFA water; solvent B: 0.1%TFA acetonitrile; 0 to 2 min: isocratic elution of 5% solvent B, 2 to 22 min, 5–95% of solvent B. Flow rate: 3 mL/min. HPLC column: Gemini 5u C18 110A, 250 × 10.00 mm. 79 mCi of [18F]-((2-(2-fluoroethoxy)ethyl)sulfonyl)ethene was collected at 13.6 min. Then 2-nitroimidazole (2 mg) was mixed with the collected [18F]-((2-(2-fluoroethoxy)ethyl)sulfonyl)ethene (5 mCi) in a solution of 50 μL DMSO and 200 μL 20× borate buffer (pH 8.5) and then heated at 80 °C for 20 min. The resulting tracer [18F]-23 was purified by HPLC with the method described as: 0 to 2 min: isocratic elution of 10% solvent B, 2 to 22 min, 10–55% of solvent B. Flow rate: 1 mL/min, collected at 12.4 min. RCY: 58.7%. [18F]-24 (12.4 min, 66.8%) and [18F]-25 (13.8 min, 53.1%) were synthesized accordingly by using 4-nitroimidazole/[18F]-((2-(2-fluoroethoxy)ethyl)sulfonyl)ethene and 2-nitroimidazole /[18F]-((2-(2-(2-fluoroethoxy)ethoxy)ethyl)sulfonyl)ethene as starting material.
Chemistry.
Synthesis of precursor 2-(2-(2-(vinylsulfonyl)ethoxy)ethoxy)ethyl 4-methylbenzenesulfonate (18):
The 2-(2-hydroxyethoxy)ethyl 4-methylbenzenesulfonate (384.9 mg, 1.0 equiv.), which was synthesized according to the literature,40was stirred at room temperature with divinyl sulfone (262.0 mg, 1.5 equiv.) and triphenylphosphine (39.3 mg, 0.1 equiv.) in anhydrous dichloromethane (200 μL) for 2 hours.41 Then the mixture was directly loaded on silica gel column chromatography for purification (ethyl acetate/hexane 1/15-1/1). 1 H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 8.3 Hz, 2 H), 7.33 (d, J = 8.1 Hz, 2 H), 6.79-6.72 (m, 1 H), 6.36 (d, J = 16.6 Hz, 1 H), 6.08 (d, J = 9.9 Hz, 1 H), 4.13–4.11 (m, 2 H), 3.84 (t, J = 5.6 Hz, 2 H), 3.65 (t, J = 4.8 Hz, 2 H), 3.55 (s, 4 H), 3.21 (t, J = 5.7 Hz, 2 H), 2.42 (s, 3 H). 13 C NMR (101 MHz, CDCl3) δ145.0, 137.9, 132.8, 129.9, 128.8, 127.9, 70.3, 69.2, 68.6, 64.6, 55.0. The precursor 2-(2-(vinylsulfonyl)ethoxy)ethyl 4-methylbenzenesulfonate (16) was synthesized accordingly with ethane-1,2-diol as starting material. 1 H NMR (400 MHz, CDCl3) δ 7.79 (d, J = 8.4 Hz, 2 H), 7.36 (d, J = 8.1 Hz, 2 H), 6.69 (m, 1 H), 6.38 (d, J = 16.6 Hz, 1 H), 6.07 (d, J = 9.9 Hz, 1 H), 4.14 (m, 2 H), 3.83 (t, J = 6.0 Hz, 2 H), 3.65 (m, 2 H), 3.19 (t, J = 5.6 Hz, 2 H), 2.44 (s, 3 H).13 C NMR (101 MHz, CDCl3) δ 145.1, 137.7, 132.8, 129.9, 129.0, 127.9, 68.7, 68.7, 64.8, 54.8, 21.6.
Typical procedure for the synthesis of [19F]-20:
The tert-butyl 4-(2-fluoroethyl)piperazine-1-carboxylate (880 mg), which was prepared according the literature method,42 was dissolved in 2 mL 50% v/v trifluoroacetic acid in dichloromethane. The reaction mixture was stirred at room temperature overnight and then evaporated under vacuum to remove excess trifluoroacetic acid. Further purification was achieved by silica gel chromatography with ethyl acetate/methanol (10/1–2/1 v/v) as eluent. 1-(2-fluoroethyl)piperazine: 1 H NMR (400 MHz, CDCl3) δ 9.49 (s, 1H), 4.62 (t, J = 4.6 Hz, 1 H), 4.50 (t, J = 4.7 Hz, 1 H), 3.21 (t, J = 5.0 Hz, 4 H), 2.83-2.79 (m, 5 H), 2.73 (t, J = 4.7 Hz, 1 H).
The mixture of 1-(2-fluoroethyl)piperazine (96.7 mg, 1.0 equiv.), 2-nitro-1-(oxiran-2-ylmethyl)-1H-imidazole (85.8 mg, 1.3 equiv.), and potassium carbonate (70.5 mg, 1.3 equiv.) was mixed in 1 mL of acetonitrile and refluxed for 30 min, them the mixture was cooled down to room temperature and stirred overnight. After purification from the column chromatography with an elution of ethyl acetate/methanol (10/1–2/1 v/v), the product was collected as off-white solid.
1-(4-(2-fluoroethyl)piperazin-1-yl)-3-(2-nitro-1H-imidazol-1-yl)propan-2-ol ([19F]-20):
Off-white solid. 1 H NMR (400 MHz, CDCl3) δ 7.26 (d, J = 0.9 Hz, 1 H), 7.12 (d, J = 0.9 Hz, 1 H), 4.70 (dd, J = 14.0, 2.5 Hz, 1 H), 4.61 (t, J = 4.8 Hz, 1 H), 4.49 (t, J = 4.8 Hz, 1 H), 4.25 (dd, J = 14.0, 7.4 Hz, 1 H), 4.05 (m, 1 H), 2.73-2.45 (m, 12 H), 2.28 (dd, J = 12.3, 10.2 Hz, 1 H).13 C NMR (101 MHz, CDCl3) δ 144.9, 128.0, 127.4, 82.7, 81.0, 65.7, 60.3, 58.1, 57.9, 53.4, 53.4, 53.3, 53.0. 19 F NMR (376 MHz, CDCl3) δ -218.32. HRMS calcd for C12H21FN5O3(M+H), 302.1628; found, 302.1630.
The synthesis of standard [19F]-21 and [19F]-22 were according to the typical procedure.
1-(4-(2-(2-fluoroethoxy)ethyl)piperazin-1-yl)-3-(2-nitro-1H-imidazol-1-yl)propan-2-ol (21):
Off-white solid. 1 H NMR (400 MHz, CDCl3) δ7.25 (d, J = 0.9 Hz, 1 H), 7.11 (d, J = 0.9 Hz, 1 H), 4.68 (dd, J = 14.0 Hz, 1 H), 4.58 (t, J = 4.1 Hz, 1 H), 4.47 (t, J = 4.1 Hz, 1 H), 4.23 (dd, J = 14.0, 7.4 Hz, 1 H), 4.02 (m, 1 H), 3.71 (t, J = 4.2 Hz, 1 H), 3.64-3.60 (m, 3 H), 2.65-2.63 (m, 2 H), 2.58 (t, J = 5.8 Hz, 2 H), 2.51-2.42 (m, 7 H), 2.25 (dd, J = 12.2, 10.2 Hz, 1 H). 13 C NMR (101 MHz, CDCl3) δ 144. 9, 128.0, 127.4, 83.9, 82.3, 70.3, 70.1, 69.1, 65.6, 60.3, 57.6, 53.5, 53.3. 19 F NMR (376 MHz, CDCl3) δ -222.72. HRMS calcd for C14H25FN5O4 (M+H), 346.1891; found, 346.1892.
1-(4-(2-fluoroethyl)-1,4-diazepan-1-yl)-3-(2-nitro-1H-imidazol-1-yl)propan-2-ol ([19F]-22):
Off-white solid. 1 H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 0.9 Hz, 1 H), 7.12 (d, J = 0.9 Hz, 1 H), 4.69 (dd, J = 13.9, 2.4 Hz, 1 H), 4.57 (t, J = 5.0 Hz, 1 H), 4.45 (t, J = 5.2 Hz, 1 H), 4.22 (dd, J = 13.9, 7.6 Hz, 1 H), 3.96 (m, 1 H), 2.88-2.67 (m, 11 H), 2.29 (dd, J = 12.4, 10.4 Hz, 1 H), 1.80 (m, 2 H). 13 C NMR (101 MHz, CDCl3) δ 142.8, 128.1, 127.4, 83.2, 81.6, 66.4, 60.4, 57.9, 57.7, 55.6, 55.6, 55.4, 54.5, 54.3, 54.3, 53.3, 28.0. 19 F NMR (376 MHz, CDCl3) δ -219.37. HRMS calcd for C13H23FN5O3(M+H), 316.1785; found, 316.1791.
Typical procedure for the synthesis of [19F]-23:
To the solution of 2-fluoroethanol (64.1 mg, 1.0 equiv.) and divinyl sulfone (177.2 mg, 1.5 equiv.) in anhydrous dichloromethane (200 μL), triphenylphosphine (26.2 mg, 0.1 equiv.) was added as portion.28The mixture was stirred at room temperature for 2 hours and then directly loaded on silica gel column chromatography for purification (ethyl acetate/hexane 1/20−1/5). The ((2-(2-fluoroethoxy)ethyl)sulfonyl)ethane (51.2 mg, 1.0 equiv.) and 2-nitroimidazole (63.5 mg, 2.0 equiv.) was mixed in a solution of 200 μL DMSO and 500 μL 20×Borate buffer (pH 8.5) and then heated at 80 °C for 2 hour. The 5 mL of water and 5 mL ethyl acetate were added to the reaction mixture. After extracted the aqueous phase for three times, the organic solutions were combined and dried over anhydrous MgSO4. Evaporated the solvent under reduced pressure, and the residue was purified with an elution of ethyl acetate/hexane (1/5-2/1) to get the product as off-white solid. 1-(2-((2-(2-fluoroethoxy)ethyl)sulfonyl)ethyl)-2-nitro-1H-imidazole ([19F]-23): Off-white solid. 1 H NMR (400 MHz, acetone-d6) δ 7.58 (d, J = 0.9 Hz, 1 H), 7.14 (d, J = 0.9 Hz, 1 H), 4.98 (t, J = 6.7 Hz, 2 H), 4.62 (t, J = 4.0 Hz, 1 H), 4.50 (t, J = 4.0 Hz, 1 H), 3.93 (t, J = 5.3 Hz, 2 H), 3.82 (t, J = 6.7 Hz, 2 H), 3.79 (d, J = 4.0 Hz, 1 H), 3.71 (t, J = 4.0 Hz, 1 H), 3.31 (t, J = 5.4 Hz, 2 H). 13 C NMR (101 MHz, acetone-d6) δ 128.6, 128.5, 84.4, 82.8, 71.2, 71.0, 65. 5, 54.9, 54.8, 44.1. 19 F NMR (376 MHz, acetone-d6) δ −223.39. HRMS calcd for C9H15FN3O5S (M+H), 296.0716; found, 296.0712.
The synthesis of standard for [19F]-24 and [19F]-25 were according to the typical procedure.
1-(2-((2-(2-fluoroethoxy)ethyl)sulfonyl)ethyl)-4-nitro-1H-imidazole ([19F]-24):
Off-white solid. 1 H NMR (400 MHz, acetone-d6) δ 8.26 (d, J = 1.5 Hz, 1 H), 7.80 (d, J = 1.4 Hz, 1 H), 4.72 (t, J = 6.8 Hz, 2 H), 4.65 (t, J = 4.0 Hz, 1 H), 5.53 (t, J = 4.0 Hz, 1 H), 3.95 (t, J = 5.2 Hz, 2 H), 3.83 (t, J = 6.8 Hz, 2 H), 3.80 (d, J = 8.0 Hz, 1 H), 3.74 (t, J = 4.0 Hz, 1 H), 3.38 (t, J = 5.4 Hz, 1 H). 13 C NMR (101 MHz, acetone-d6) δ 137.2, 137.2, 120.7, 83.6, 81.9, 70.3, 70.1, 64.7, 54.3, 54.0, 41.2. 19 F NMR (376 MHz, acetone-d6) δ -223.35. HRMS calcd for C9H15FN3O5S (M+H), 296.0716; found, 296.0708.
1-(2-((2-(2-(2-fluoroethoxy)ethoxy)ethyl)sulfonyl)ethyl)-2-nitro-1H-imidazole ([19F]-25):
White solid. 1 H NMR (400 MHz, acetone-d6) δ 7.59 (s, 1H), 7.13 (s, 1H), 4.97 (t, J = 6.6 Hz, 2H), 4.56 (t, J = 4.0 Hz, 1H), 4.44 (t, J = 4.0 Hz, 1H), 3.89 (t, J = 5.2 Hz, 2H), 3.84 (t, J = 6.6 Hz, 2H), 3.72 (t, J = 4.1 Hz, 1H), 3.66-3.64 (m, 5H), 3.28 (t, J = 5.3 Hz, 2H). 13 C NMR (101 MHz, acetone-d6) δ 127.7, 127.6, 83.8, 82.2, 70.2, 70.1, 70.0, 69.9, 64.6, 54.0, 53.9, 43.4. 19 F NMR (376 MHz, acetone-d6) δ −223.35. HRMS calcd for C11H19FN3O6S (M+H), 340.0979; found, 340.0966.
The synthesis of [18F]-FMISO was modified from the literature.43 1-(2’-nitro-1’-imidazolyl)-2-O-tetra-hydropyranyl-3-O-toluenesulfonyl propanediol (NITTP, 2 mg) precursor was heated with 247 mCi of [18F]-TBAF in acetonitrile at 110 °C for 10 min. After removing the solvent by blow dry, 1 mL 1N HCl was added to the reaction mixture and kept on heating at 105 °C for 5 min. Cooled down the mixture to room temperature, and 0.5 mL 2 N NaOH was added in to neutralize the reaction mixture. After passing the mixture through a Sep-Pak alumina N cartridge, the activity was loaded on HPLC (isocratic ethanol/water 5/95). 145 mCi of [18F]-FMISO was collected at 13.5 min after separation.
The 2-(2-((2-(2-nitro-1H-imidazol-1-yl)ethyl)sulfonyl)ethoxy)ethyl 4-methyl benzenesulfonate (26) was synthesized with 2-(2-(vinylsulfonyl)ethoxy)ethyl 4-methylbenzenesulfonate and 2-nitroimidazole as starting material in borate buffer. 1 H NMR (400 MHz, acetone-d6) δ 7.81 (d, J = 8.3 Hz, 2 H), 7.58 (d, J = 1.0 Hz, 1 H), 7.49 (d, J = 8.0 Hz, 2 H), 7.13 (d, J = 1.0 Hz, 1 H), 4.94 (t, J = 6.7 Hz, 2 H), 4.23 (t, J = 4.4 Hz, 2 H), 3.86 (t, J = 5.2 Hz, 2 H), 3.78-3.72 (m, 4 H), 3.30 (t, J = 5.4 Hz, 2 H), 2.45 (s, 3 H). 13 C NMR (101 MHz, acetone-d6) δ 145.1, 133.3, 130.0, 127.8, 127.8, 127.8, 127.6, 69.3, 68.6, 64.6, 53.9, 53.8, 43.1, 20.6. HRMS calcd for C16H22N3O8S2(M+H), 448.0848; found, 448.0820.
Mouse models.
All animal studies were reviewed and approved by The University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee. The FaDu tumor cell was obtained from the LCCC tissue culture facility (University of North Carolina at Chapel Hill, Chapel Hill, NC). The UPPL tumor cell was received from Prof. Kim’s group as a gift. The FaDu tumor bearing nude mice and UPPL tumor bearing C57BL/6 mice were prepared with the literature method. 44,45
Small-animal PET.
FaDu or UPPL tumor bearing mice was intravenously injected via the tail vein with [18F]-tracer (three mice in a group). The animals were fresh and only injected once for PET image. For FaDu tumor bearing mice (Injection dose: 6.0 μCi/g–8.9 μCi/g), a 10-min static emission scan was acquired with a SuperArgus small-animal PET/CT scanner at 30 min and 120 min post injection. For UPPL tumor bearing mice (Injection dose: 5.0 μCi/g–9.1 μCi/g), a 10-min static emission scan was acquired at 30 min, 1, 2 and 4 hour p.i.. The regions of interests (ROIs) were drawn over the tumor and other organs and calculated as %ID/g. The mean uptake and standard deviation (mean ± SD) were calculated for each group, and the number of organs we tested in each group was calculated as n value.
In vitro stability.
The in vitro stability study was carried out on Agilent HPLC. In brief, [18F]-23 (238 μCi) was incubated with 400 μL plasm at 37 °C for 0.5, 1 and 2 hours. After the serum protein was precipitated and centrifuged, the liquid supernatant was then analyzed by radio-HPLC.
In vivo stability.
The in vivo stability study was performed using normal mice. In brief, [18F]-23 (207–213 μCi) was injected into the animal and blood samples were collected at 0.5, 1 and 2 h p.i.. After the blood protein was precipitated and centrifuged, the liquid supernatant was then analyzed by radio-HPLC or gamma counting.
Pimonidazole staining and autoradiography study.
Pimonidazole was injected i. v. around 120 min before euthanasia followed by [18F]-23 injection. After PET imaging with [18F]-23, the tumors were collected and 10 μm thin tissue sections were obtained from the frozen tumor blocks using a cryostat with an optimal cutting temperature compound. For cutting, four slices were obtained at each location with two sections mount on each slice. Odd number slices were used for autoradiography and even number ones were used for pimonidazole fluorescence staining. Then about 15–20 sections were skipped and the tumor sections were collected as describe above until the whole tumor was cut. Thereafter, these fresh frozen sections were dried out. Autoradiography studies were performed using the Cyclone Plus storage phosphor system (Perkin Elmer) and all phosphor images in these studies had a 25-μm pixel resolution. For pimonidazole staining of the neighboring slices, the sections were fixed with acetone for 10 minutes, washed twice in PBS (5 minutes each) and immersed in 0.3% Tween-20 for 20 min at RT. Then the slices were blocked in primary AB dilution buffer (Biomeda, Foster City) for 30 min to block nonspecific binding, and incubated with mouse anti-pimonidazole Mab HP7-DyLight™549 (1:500 in PBS) in dark for 1 hour at RT. After washed with PBS 3 times (5 minutes each), the slices were covered with EverBrite Mounting Medium containing 4, 6-diamino-2-phenylindole (DAPI, Biotium, CA, USA) and observed under Zeiss LSM 710 laser scanning microscope (Zeiss, Jena, German).
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to Prof. William Kim for providing UPPL cancer cells and Dr. Gerald T. Bida for excellent technical support on cyclotron operation.
Funding Sources
The research was partially supported by the UNC Department of Radiology, Biomedical Research Imaging Center, and UNC Lineberger Comprehensive Cancer Center (start-up fund to Z.W.), the National Institutes of Health (5R01CA233904, 1R01EB029451, 1S10OD023611) (Z.L.), and the Camille and Henry Dreyfus Foundation (Q.W.).
ABBREVIATIONS
- NIS
N-iodosuccinimide
- THP
2-tetrahydropyranyl
- TLC
thin layer chromatography
- RT
room temperature
- TBAF
tetra-n-butylammonium fluoride
- PEG
polyethylene glycol
- RCY
radio chemistry yield
Footnotes
Supporting Information. Additional figures illustrating HPLC data, experimental details, Molecular Formula Strings, and NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Ali I; Lone MN; Aboul-Enein HY Imidazoles as potential anticancer agents. Med. Chem. Commun 2017, 8, 1742–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ren Y; de Blanco EJC; Fuchs JR; Soejarto DD; Burdette JE; Swanson SM; Kinghorn AD Potential anticancer agents characterized from selected tropical plants. J. Nat. Prod 2019, 82, 657–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Saleem K; Wani WA; Haque A; Lone MN; Hsieh M; Jairajpuri MA; Ali I Synthesis, DNA binding, hemolysis assays and anticancer studies of copper (II), nickel (II) and iron (III) complexes of a pyrazoline-based ligand. Future Med. Chem 2013, 5, 135–146. [DOI] [PubMed] [Google Scholar]
- (4).Ali I; Saleem K; Wesselinova D; Haque A Synthesis, DNA binding, hemolytic, and anti-cancer assays of curcumin I-based ligands and their ruthenium (III) complexes. Med. Chem. Res 2013, 22, 1386–1398. [Google Scholar]
- (5).Ali I; Wani WA; Saleem K; Hseih M Design and synthesis of thalidomide based dithiocarbamate Cu (II), Ni (II) and Ru (III) complexes as anticancer agents. Polyhedron 2013, 56, 134–143. [Google Scholar]
- (6).Scott AM; Wolchok JD; Old LJ Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287 [DOI] [PubMed] [Google Scholar]
- (7).Park S; Nam J The force awakens: metastatic dormant cancer cells. Exp. Mol. Med 2020, 52, 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Wilson WR; Hay MP Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [DOI] [PubMed] [Google Scholar]
- (9).Horsman MR; Mortensen LS; Petersen JB; Busk M; Overgaard J Imaging hypoxia to improve radiotherapy outcome. Nat. Rev. Clin. Oncol 2012, 9, 674–687. [DOI] [PubMed] [Google Scholar]
- (10).Harris AL Hypoxia--a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [DOI] [PubMed] [Google Scholar]
- (11).Milosevic M; Warde P; Ménard C; Chung P; Toi A; Ishkanian A; McLean M; Pintilie M; Sykes J; Gospodarowicz M; Catton C; Hill RP; Bristow R Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin. Cancer Res 2012, 18, 2108–2114. [DOI] [PubMed] [Google Scholar]
- (12).Nordsmark M; Loncaster J; Aquino-Parsons C; Chou SC; Ladekarl M; Havsteen H; Lindegaard JC; Davidson SE; Varia M; West C; Hunter R; Overgaard J; Raleigh JA Measurements of hypoxia using pimonidazole and polarographic oxygen-sensitive electrodes in human cervix carcinomas. Radiother Oncol. 2003, 67, 35–44. [DOI] [PubMed] [Google Scholar]
- (13).Bussink J; Kaanders JHAM; van der Kogel AJ Tumor hypoxia at the micro-regional level: clinical relevance and predictive value of exogenous and endogenous hypoxic cell markers. Radiother Oncol, 2003, 67, 3–15. [DOI] [PubMed] [Google Scholar]
- (14).Brinkhuizen T; Weijzen CA; Eben J; Thissen MR; van Marion AM; Lohman BG; Winnepenninckx VJ; Nelemans PJ; van Steensel MA Immunohistochemical analysis of the mechanistic target of rapamycin and hypoxia signalingpathways in basal cell carcinoma and trichoepithelioma. PLoS One. 2014, 9, e106427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fan Q; Tang CY; Gu D; Zhu J; Li G; Wu Y; Tao X Investigation of hypoxia conditions using oxygen-enhanced magnetic resonance imaging measurements in glioma models. Oncotarget. 2017, 8, 31864–31875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Verwer EE; Boellaard R; van der Veldt AA Positron emission tomography to assess hypoxia and perfusion in lung cancer. World J. Clin. Oncol 2014, 5, 824–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lopci E; Grassi I; Chiti A; Nanni C; Cicoria G; Toschi L; Fonti C; Lodi F; Mattioli S; Fanti S PET radiopharmaceuticals for imaging of tumor hypoxia: a review of the evidence. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 365–384. [PMC free article] [PubMed] [Google Scholar]
- (18).Stieb S; Eleftheriou A; Warnock G; Guckenberger M; Riesterer O Longitudinal PET imaging of tumor hypoxia during the course of radiotherapy. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2201–2217. [DOI] [PubMed] [Google Scholar]
- (19).Tong X; Srivatsan A; Jacobson O; Wang Y; Wang Z; Yang X; Niu G; Kiesewetter DO; Zheng H; Chen X Monitoring tumor hypoxia using (18)F-FMISO PET and pharmacokinetics modeling after photodynamic therapy. Sci. Rep 2016, 6, 31551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Verwer EE; Bahce I; van Velden FH; Yaqub M; Schuit RC; Windhorst AD; Raijmakers P; Hoekstra OS; Lammertsma AA; Smit EF; Boellaard R Parametric methods for quantification of 18F-FAZA kinetics in non-small cell lung cancer patients. J. Nucl. Med 2014, 55, 1772–1777. [DOI] [PubMed] [Google Scholar]
- (21).Hoigebazar L; Jeong JM; Hong MK; Kim YJ; Lee JY; Shetty D; Lee YS; Lee DS; Chung JK; Lee MC Synthesis of 68Ga-labeled DOTA-nitroimidazole derivatives and their feasibilities as hypoxia imaging PET tracers. Bioorg. Med. Chem 2011, 19, 2176–2181. [DOI] [PubMed] [Google Scholar]
- (22).ClinicalTrials.gov Identifier: NCT03573986.
- (23).ClinicalTrials.gov Identifier: NCT01542177.
- (24).Busk M; Jakobsen S; Horsman MR; Mortensen LS; Iversen AB; Overgaard J; Nordsmark M; Ji X; Lee DY; Raleigh JR PET imaging of tumor hypoxia using 18F-labeled pimonidazole. Acta Oncologica 2013, 52, 1300–1307. [DOI] [PubMed] [Google Scholar]
- (25).Ortiz GX Jr.; Chansaenpak K; Wang M; Ma X; Wang H; Li Z; Wang Q A novel 18F-labeling method for the synthesis of [18F]-piperidine-containing ligands as potential PET radiotracers for σ receptors. Synlett 2018, 29, 410–414. [Google Scholar]
- (26).Erlandsson M; Karimi F; Takahashi K; Långström B 18F-Labelled vorozole analogues as PET tracer for aromatase. J. Labelled Compd. Rad 2008, 51, 207–212. [Google Scholar]
- (27).Perruchon J; Ortmann R; Altenkämper M; Silber K; Wiesner J; Jomaa H; Klebe G; Schlitzer M Studies addressing the importance of charge in the binding of fosmidomycin-like molecules to deoxyxylulosephosphate reducto isomerase. ChemMedChem. 2008, 3, 1232–1241. [DOI] [PubMed] [Google Scholar]
- (28).Kapadnis PB; Hall E; Ramstedt M; Galloway WR; Welch M; Spring DR Towards quorum-quenching catalytic antibodies. Chem. Commun 2009, 7, 538–540. [DOI] [PubMed] [Google Scholar]
- (29).Fairlamb IJ; Dickinson JM; O’Connor; Cohen LH; van Thiel CF. Synthesis and antimicrobial evaluation of farnesyl diphosphate mimetics. Bioorg. Chem 2003, 31, 80–97. [DOI] [PubMed] [Google Scholar]
- (30).Morales-Sanfrutos J; Lopez-Jaramillo J; Ortega-Muñoz M; Megia-Fernandez A; Perez-Balderas F; Hernandez-Mateo F; Santoyo-Gonzalez F Vinyl sulfone: a versatile function for simple bioconjugation and immobilization. Org. Biomol. Chem 2010, 8, 667–675. [DOI] [PubMed] [Google Scholar]
- (31).Wu Z; Li L; Liu S; Yakushijin F; Yakushijin K; Horne D; Conti PS; Li Z; Kandeel F; Shively JE Facile preparation of a thiol-reactive 18F-labeling agent and synthesis of 18F-DEG-VS-NT for PET imaging of a neurotensin receptor–positive tumor. J. Nucl. Med 2014, 55, 1178–1184 [DOI] [PubMed] [Google Scholar]
- (32).Li L; Tsai SW; Anderson AL; Keire DA; Raubitschek AA; Shively JE Vinyl sulfone bifunctional derivatives of DOTA allow sulfhydryl- or amino-directed coupling to antibodies. Conjugates retain immunoreactivity and have similar biodistributions. Bioconjug. Chem 2002, 13, 110–115. [DOI] [PubMed] [Google Scholar]
- (33).McCall KC; Humm JL; Bartlett R; Reese M; Carlin S Copper-64-diacetyl-bis(N(4)-methylthiosemicarbazone) pharmacokinetics in FaDu xenograft tumors and correlation with microscopic markers of hypoxia. Int. J. Radiat. Oncol. Biol. Phys 2012, 84, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Xue M; Chen W; Xiang A; Wang R; Chen H; Pan J; Pang H; An H; Wang X; Hou H; Li X Hypoxic exosomes facilitate bladder tumor growth and development through transferring longnon-coding RNA-UCA1. Mol. Cancer 2017, 16, 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ord JJ; Streeter EH; Roberts IS; Cranston D; Harris AL Comparison of hypoxia transcriptome in vitro with in vivo gene expression in human bladder cancer. Br. J. Cancer 2005, 93, 346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Saito R; Smith CC; Utsumi T; Bixby LM; Kardos J; Wobker SE; Stewart KG; Chai S; Manocha U; Byrd KM; Damrauer JS; Williams SE; Vincent BG; Kim WY Molecular subtype-specific immunocompetent models of high-grade urothelial carcinoma reveal differential neoantigen expression and response to immunotherapy. Cancer Res. 2018, 78, 3954–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Dubois L; Landuyt W; Haustermans K; Dupont P; Bormans G; Vermaelen P; Flamen P; Verbeken E; Mortelmans L Evaluation of hypoxia in an experimental rat tumor model by [(18)F]fluoromisonidazole PET and immunohistochemistry. Br. J. Cancer 2004, 91, 1947–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Troost EG; Laverman P; Philippens ME; Lok J; van der Kogel AJ; Oyen WJ; Boerman OC; Kaanders JH; Bussink J Correlation of [18F]FMISO autoradiography and pimonidazole [corrected] immunohistochemistry in human head and neck carcinoma xenografts. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1803–1811. [DOI] [PubMed] [Google Scholar]
- (39).Busk M; Mortensen LS; Nordsmark M; Overgaard J; Jakobsen S; Hansen KV; Theil J; Kallehauge JF; D’Andrea FP; Steiniche T; Horsman MR PET hypoxia imaging with FAZA: reproducibility at baseline and during fractionated radiotherapy in tumor-bearing mice. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 186–197. [DOI] [PubMed] [Google Scholar]
- (40).Davis OA; Bull JA Synthesis of di-, tri-, and tetrasubstituted oxetanes by rhodium-catalyzed O-H insertion and C-C bond-forming cyclization. Angew. Chem. Int. Ed. Engl 2014, 53, 14230–14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Strassera S; Slugovc C Nucleophile-mediated oxa-Michael addition reactions of divinylsulfone–a thiol-free option for step-growth polymerizations. Catal. Sci. Technol 2015, 5, 5091–5094. [Google Scholar]
- (42).Breyholz HJ; Wagner S; Faust A; Riemann B; Höltke C; Hermann S; Schober O; Schäfers M; Kopka K Radiofluorinated pyrimidine-2,4,6-triones as molecular probes for noninvasive MMP-targetedimaging. ChemMedChem. 2010, 5, 777–789. [DOI] [PubMed] [Google Scholar]
- (43).Tang G; Wang M; Tang X; Gan M; Luo L Fully automated one-pot synthesis of [18F]fluoromisonidazole. Nucl. Med. Biol 2005, 32, 553–558. [DOI] [PubMed] [Google Scholar]
- (44).Li F; Jørgensen JT; Forman J; Hansen AE; Kjaer A 64Cu-ATSM reflects pO2 levels in human head and neck cancer xenografts but not in colorectal cancer xenografts: Comparison with 64CuCl2. J. Nucl. Med 2016, 57, 437–443. [DOI] [PubMed] [Google Scholar]
- (45).Jiang W; Yin L; Chen H; Paschall AV; Zhang L; Fu W; Zhang W; Todd T; Yu KS; Zhou S; Zhen Z; Butler M; Yao L; Zhang F; Shen Y; Li Z; Yin A; Yin H; Wang X; Avci FY; Yu X; Xie J NaCl nanoparticles as a cancer therapeutic. Adv. Mater 2019, 31, e1904058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.