

Abstract
Lipoprotein(a) (Lp(a)) is a prothrombotic and anti-fibrinolytic lipoprotein, whose role has not been clearly defined in the pathogenesis of coronavirus disease 2019 (COVID-19). In this prospective observational study, serum Lp(a) as well as outcomes were measured in 50 COVID-19 patients and 30 matched sick controls. Lp(a) was also assessed for correlation with a wide panel of biomarkers. Serum Lp(a) did not significantly differ between COVID-19 patients and sick controls, though its concentration was found to be significantly associated with severity of COVID-19 illness, including acute kidney failure stage (r = 0.380, p = 0.007), admission disease severity (r = 0.355, p = 0.013), and peak severity (r = 0.314; p = 0.03). Lp(a) was also positively correlated with interleukin (IL)-8 (r = 0.308; p = 0.037), fibrinogen (r = 0.344; p = 0.032) and creatinine (r = 0.327; p = 0.027), and negatively correlated with ADAMTS13 activity/VWF:Ag (r = − 0.335; p = 0.021); but not with IL-6 (r = 0.241; p = 0.106). These results would hence suggest that adverse outcomes in patients with COVID-19 may be aggravated by a genetically determined hyper-Lp(a) state rather than any inflammation induced elevations.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11239-021-02597-y.
Keywords: Lipoprotein(a), Coronavirus disease 2019, Acute kidney injury, Coagulopathy, Thrombosis
Highlights
Serum Lp(a) elevation is correlated with illness severity at admission and at peak, as well as acute kidney injury stage in COVID-19 patients.
Given no correlation with IL-6, an inherited hyper-Lp(a) state rather than an inflammation-driven increase may contribute to enhanced risk of micro- and macro-thrombosis in COVID-19 patients.
Secondary thrombotic microangiopathy-induced severe AKI may also be mediated by elevated Lp(a) and IL-8.
Further research should focus on examining for associations between apo(a) gene polymorphisms and COVID-19 outcomes, as well as elucidating the precise mechanism of Lp(a) and IL-8 in the pathophysiology of COVID-19.
Introduction
Multiple organ failure (MOF) in coronavirus disease 2019 (COVID-19) patients has been associated with a dysregulated immune response and an eventual vasculopathy-mediated hypercoagulable state called COVID-19 associated coagulopathy (CAC), often culminating in clinically apparent thrombosis [1–3]. Klok et al. reported that nearly one-third of ICU COVID-19 patients developed thrombotic complications [4], whilst Middeldorp and colleagues found an 59% incidence of venous thromboembolisms (VTE) in ICU COVID-19 patients [5]. These findings have been attributed to a virus-mediated impairment of endothelial integrity and platelet function, followed by hemostasis activation and decreased fibrinolysis [6]. No evidence of a direct effect of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) on atherosclerotic plaque progression has been found [6]. Previous research has largely focused on the interplay between SARS-CoV-2 and the most representative subsets of lipid and lipoprotein (e.g., total cholesterol, triglycerides, high- and low-density lipoproteins), linking hypolipidemia with disease severity and length of hospitalization [7–9], however overlooking other lipids and lipoproteins which may play an equally important role in this clinical setting.
Given its highly atherogenic and anti-fibrinolytic properties [10], and capacity to be activated by interleukin(IL)-6 [11], lipoprotein(a) (Lp(a)) might be an important etiological factor in COVID-19 complications, bridging a dysregulated immune response with thrombosis. Besides being a well-established risk factor for cardiovascular disease, particularly through accumulation in arterial intima, and binding of proinflammatory oxidized phospholipids [12, 13], Lp(a) has also been found to increase plasminogen activator inhibitor-1 (PAI-1) expression and antagonize tissue factor pathway inhibitor. While apolipoprotein(a) (apo(a)), a unique protein moiety characterizing this lipoprotein, can directly inhibit the conversion of plasminogen into plasmin, and the binding of plasminogen to fibrin clots, thus triggering a highly pathogenic state of concomitant hypercoagulation and hypofibrinolysis [14, 15]. A case control study by von Depka et al. found 3.2-fold increased odds of elevated Lp(a) levels in patients with VTE compared to healthy controls [16], while a meta-analysis by Nave et al. discovered 41% increased odds of Lp(a) elevations in patients with ischemic stroke [17]. Research on whether Lp(a) levels increase during inflammatory states is mixed. Lp(a) levels have been correlated with CRP in rheumatoid arthritis and dialysis patients [18, 19]. However, a larger study on a total of 34,829 individuals found that elevated CRP had only a minimal influence on Lp(a) levels, and did not alter the ability of Lp(a) to predict cardiovascular risk [20]. Nonetheless, in an attempt to help elucidate this matter, a more recent study found not only that Lp(a) levels correlated with IL-6, but through transcriptomic analysis and interleukin blockade by tocilizumab uncovered that the gene encoding for apo(a), LPA, contains an IL-6 responsive element, which when activated during pro-inflammatory states (e.g., infections), may boost mRNA expression and liver synthesis of Lp(a) [11]. It is hence conceivable that the hyperinflammatory state of COVID-19, involving significant increases in IL-6, may elevate serum Lp(a), and increase thrombotic risk.
The aim of this study was to measure Lp(a) levels in a cohort of COVID-19 patients presenting to the emergency department (ED), and explore its role in the pathophysiology of the disease by looking for relationships with biomarkers of inflammation and hemostasis, disease severity, as well as development of severe acute kidney injury (AKI) given our previous observations of secondary thrombotic microangiopathy (TMA) involving the kidneys [21].
Serum Lp(a) levels were measured in blood samples collected from patients with reverse transcriptase-polymerase chain reaction (RT-PCR) confirmed SARS-CoV-2 infection, as well as in matched sick controls (respiratory symptoms with a non-COVID-19 discharge diagnosis and confirmed RT-PCR negative), at ED presentation to the University of Cincinnati Medical Center. Serum Lp(a) was quantified with an immunonephelometric assay on a Behring Nephelometer II System (BN II; Siemens Medical Solutions USA, Inc.). The primary outcome was development of severe AKI over the course of illness, defined by Kidney Disease Improving Global Outcomes (KDIGO) Stage 2 and 3 based on serum creatinine (SCr) criteria [22]. Secondary outcomes were need for renal replacement therapy (RRT) within 30 days of index ED visit, and initial ED and peak COVID-19 severity, defined as mild (ambulatory), moderate (hospitalized, no ICU), or severe (ICU or death). Continuous data was reported as median and interquartile range (IQR). Comparison of serum Lp(a) levels between groups was performed using Mann–Whitney U test. Spearman’s correlation coefficient was used to assess for correlation between Lp(a) and admission disease severity, peak disease severity, and KDIGO AKI stage. Additionally, correlations between Lp(a) and inflammatory, hemostatic, and fibrinolytic biomarkers such as C-reactive protein (CRP), IL-6, IL-8, IL-10, lactate dehydrogenase (LDH), fibrinogen, plasminogen, plasminogen activator inhibitor-1 (PAI-1), a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 activity to von Willebrand factor antigen ratio (ADAMTS13 activity/VWF:Ag), D-dimer, platelets, prothrombin time (PT), as well as kidney function tests, including ED creatinine, and serum creatinine (SCr) increase ratio were examined using Spearman’s correlation coefficient. Statistical analysis was performed using R software (version 4.0.2, R Foundation for Statistical Computing, Vienna, Austria), with p < 0.05 considered significant. This study was approved by the Institutional Review Board (IRB) of the University of Cincinnati and received a waiver of informed consent due to minimal risk to participants. This study was conducted in accordance with the Declaration of Helsinki, under the terms of relevant local and national legislation.
A total of 80 patients were enrolled. The COVID-19 cohort included 50 patients, 30 (60%) of whom were males, with median age of 50.5 (IQR 40.5–66.0) years. Of the 30 sick controls, 20 (66.7%) were males, with median age of 55.5 (IQR 33.2–63.7) years. There were no significant differences with regard to sex (p = 0.721) or age (p = 0.591). Serum Lp(a) levels didn’t differ significantly between COVID-19 patients and sick controls (0.1 [IQR 0.03–0.26] vs. 0.2 [IQR 0.08–0.47] g/L; p = 0.098).
Twelve (24%) COVID-19 patients developed severe AKI, with 8 (67%) needing RRT. At ED presentation, 19 (38%) patients had mild severity, 25 (50%) moderate severity, and 6 (12%) severe COVID-19. At peak, 15 (30%) developed severe disease. Full demographics and comorbidities are presented in Supplementary Table 1. Lp(a) levels according to AKI status are presented in Fig. 1. COVID-19 patients who developed severe AKI had significantly higher Lp(a) (0.24 [IQR 0.13–0.29] vs 0.07 [0.02–0.21] g/L; p = 0.008) and levels were positively correlated with KDIGO AKI stage (r = 0.380, p = 0.007). Patients needing RRT had elevated Lp(a) levels; however, the difference did not reach statistical significance (0.21 [IQR 0.13–0.26] vs. 0.08 [IQR 0.03–0.25] g/L, p = 0.059). Serum Lp(a) levels were significantly correlated with admission disease severity (r = 0.355, p = 0.013), and peak severity (r = 0.314; p = 0.03). Lp(a) was also positively correlated with IL-8 (r = 0.308; p = 0.037), fibrinogen (r = 0.344; p = 0.032) and ED creatinine (r = 0.327; p = 0.027), and negatively with ADAMTS13 activity/VWF:Ag (r = − 0.335; p = 0.021) (Supplemental Table 2). Notably, Lp(a) and IL-6 levels were not correlated (r = 0.241; p = 0.106).
Fig. 1.
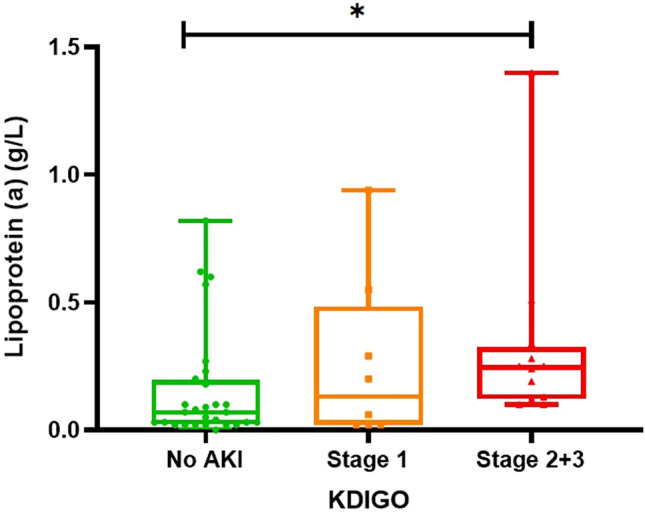
Lipoprotein(a) levels measured at emergency department presentation according to AKI severity defined by KDIGO. *p = 0.007 for Spearman’s correlation coefficient
To the best of our knowledge, this study is the first to report Lp(a) measurements in COVID-19, exploring its relationship with disease severity. Taken together, our results demonstrate that elevated Lp(a) in COVID-19 patients is significantly correlated with higher admission and peak disease severity, as well as development of severe AKI. COVID-19 patients with severe AKI are predisposed to significantly worse outcomes, including prolonged ICU stays and high rates of mortality [23, 24]. Therefore, findings of elevated Lp(a) offer valuable insights into therapeutic potential of Lp(a)-lowering interventions.
Our study also investigated whether Lp(a) could associate with poor outcomes in COVID-19 patients via aggravating thrombosis. Moriarty et al. speculated that the hyperinflammatory state of COVID-19, particularly with high IL-6 levels, would boost circulating Lp(a), and lead to thrombosis by disturbing hemostatic balance [14, 15]. We observed that not only was there an absence of correlations with plasminogen and PAI-1, but also no correlation with IL-6, which was hypothesized to bridge the gap between hyperinflammation and thrombosis via IL-6 responsive elements in the LPA gene [11]. Notably, the IL-6 values found in our cohort, as well as those found in other studies, are not extraordinarily high compared to other pathologies accompanied by a “cytokine storm” (trauma, bacterial sepsis, and so forth) [25], so that it may be conceivable that this IL-6 boost was actually too low to significantly raise Lp(a) concentration. Given the strong genetic basis of circulating Lp(a) (i.e., up to 80–90% of apo(a) concentration is genetically determined) [26], an inherited hyper-Lp(a) state rather than an inflammation-driven increase may be a more important contributor to adverse outcomes in COVID-19.
In support of this hypothesis, Sticchi et al. explored the potential association between the risk of VTE and quantitative (number of KIV-2 repeats) and qualitative (single nucleotide polymorphisms) LPA gene [27]. A significant association was found between LPA size polymorphism, whereby patients in the lower quintile of LPA KIV-2 repeats (i.e., ≤ 7) had 3.8-fold higher risk of developing VTE than those in the higher quintile (i.e., ≥ 23). Moreover, two specific LPA haplotypes (i.e., TAAC and TAGT) had borderline moderate impact on VTE risk (p = 0.014 and p = 0.033, respectively). This observation reinforces earlier evidence of a well-established association between LPA gene size polymorphisms and arterial thrombosis, whereby patients with smaller apo(a) isoforms have increased risk of developing myocardial infarction [28].
We also found a correlation between Lp(a) and IL-8, a proinflammatory, prothrombotic, and proatherogenic cytokine, which attracts leukocytes, triggers tissue factor production, and promotes adhesion of monocytes to early atherosclerotic plaques [29, 30]. In a study by Scipione et al., Lp(a) directly induced IL-8 expression [29], illustrating an important mechanism by which this lipoprotein may enhance plaque instability, fostering an already elevated cardiovascular risk at baseline in COVID-19. Moreover, IL-8 is a potent activator of neutrophil extracellular trap (NET) release from granulocytes [31]. These chromatin based extracellular fibers have been strongly implicated in coronary atherosclerosis, venous thrombosis, as well as TMA [32–34]. Given our findings of negative correlation between Lp(a) and ADAMTS13 activity/VWF:Ag, which suggests an underlying secondary TMA driving progression to severe AKI in COVID-19 as observed in our prior research [21], it is tempting to propose that Lp(a) may be synergistic with secondary TMA in COVID-19 via activation of IL-8 and subsequent NETosis. However, we cannot conclude a cause-and-effect relationship based on our study design, and additional studies should be conducted to investigate these hypotheses. Nonetheless, we find support for this in studies demonstrating evidence of lipoprotein glomerulopathy leading to TMA, and linking Lp(a) to elevations in VWF [35, 36].
In conclusion, we observed a significant correlation between higher serum Lp(a) and worse outcomes in COVID-19 patients, suggesting a potential role in the pathogenesis of SARS-CoV-2 infection, such as enhancing the risk of micro- and macro-thrombosis. This association may be unrelated to hyperinflammation, but rather genetically determined. We also found that Lp(a) was positively correlated with IL-8 and negatively with ADAMTS13 activity/VWF:Ag, thus underpinning that secondary TMA-induced severe AKI may also be mediated by elevated Lp(a) and IL-8.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contributions
GL and BMH conceived and designed the study. SWB, JLB, and BMH collected samples and ran experiments. JLB and MHSO performed data acquisition and collection. GL, IS, MHSO, and BMH did data analysis. GL, IS, SWB, JLB, EJF, and BMH interpreted the data. IS prepared the first draft. GL, MHSO, SWB, JLB, EJF, and BMH critically revised the manuscript for important intellectual content. All authors have approved the final article.
Funding
This study was funded by the University of Cincinnati College of Medicine Special Coronavirus (COVID-19) Research Pilot Grant Program.
Data availability
Data available on reasonable request from the authors.
Declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Ethical approval
This study was approved by the Institutional Review Board (IRB) of the University of Cincinnati. This study was performed in line with the principles of the Declaration of Helsinki.
Informed consent
This study received a waiver of informed consent due to no greater than minimal risk to participants.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Henry BM, Vikse J, Benoit S, Favaloro EJ, Lippi G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: a novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin Chim Acta Int J Clin Chem. 2020;507:167–173. doi: 10.1016/j.cca.2020.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becker RC. COVID-19 update: Covid-19-associated coagulopathy. J Thromb Thrombolysis. 2020;50(1):54–67. doi: 10.1007/s11239-020-02134-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lippi G, Sanchis-Gomar F, Favaloro EJ, Lavie CJ, Henry BM. Coronavirus disease 2019–associated coagulopathy. Mayo Clin Proc. 2021;96(1):203–217. doi: 10.1016/j.mayocp.2020.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Müller MCA, et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost JTH. 2020;18(8):1995–2002. doi: 10.1111/jth.14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grzegorowska O, Lorkowski J. Possible correlations between atherosclerosis, acute coronary syndromes and COVID-19. J Clin Med. 2020;9:3746. doi: 10.3390/jcm9113746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan J, Wang H, Ye G, Cao X, Xu X, Tan W, et al. Letter to the Editor: Low-density lipoprotein is a potential predictor of poor prognosis in patients with coronavirus disease 2019. Metabolism. 2020;107:154243. doi: 10.1016/j.metabol.2020.154243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang G, Zhang Q, Zhao X, Dong H, Wu C, Wu F, et al. Low high-density lipoprotein level is correlated with the severity of COVID-19 patients: an observational study. Lipids Health Dis. 2020;19(1):204. doi: 10.1186/s12944-020-01382-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei X, Zeng W, Su J, Wan H, Yu X, Cao X, et al. Hypolipidemia is associated with the severity of COVID-19. J Clin Lipidol. 2020;14(3):297–304. doi: 10.1016/j.jacl.2020.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lippi G, Guidi G. Lipoprotein(a): an emerging cardiovascular risk factor. Crit Rev Clin Lab Sci. 2003;40(1):1–42. doi: 10.1080/713609328. [DOI] [PubMed] [Google Scholar]
- 11.Müller N, Schulte DM, Türk K, Freitag-Wolf S, Hampe J, Zeuner R, et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans. J Lipid Res. 2015;56(5):1034–1042. doi: 10.1194/jlr.P052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353(1):46–57. doi: 10.1056/NEJMoa043175. [DOI] [PubMed] [Google Scholar]
- 13.Missala I, Kassner U, Steinhagen-Thiessen E. A systematic literature review of the association of Lipoprotein(a) and autoimmune diseases and atherosclerosis. Int J Rheumatol. 2012;2012:4807084. doi: 10.1155/2012/480784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moriarty PM, Gorby LK, Stroes ES, Kastelein JP, Davidson M, Tsimikas S. Lipoprotein(a) and its potential association with thrombosis and inflammation in COVID-19: a testable hypothesis. Curr Atheroscler Rep. 2020;22(9):48. doi: 10.1007/s11883-020-00867-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caplice NM, Panetta C, Peterson TE, Kleppe LS, Mueske CS, Kostner GM, et al. Lipoprotein (a) binds and inactivates tissue factor pathway inhibitor: a novel link between lipoproteins and thrombosis. Blood. 2001;98(10):2980–2987. doi: 10.1182/blood.V98.10.2980. [DOI] [PubMed] [Google Scholar]
- 16.von Depka M, Nowak-Göttl U, Eisert R, Dieterich C, Barthels M, Scharrer I, et al. Increased lipoprotein (a) levels as an independent risk factor for venous thromboembolism. Blood. 2000;96(10):3364–3368. doi: 10.1182/blood.V96.10.3364. [DOI] [PubMed] [Google Scholar]
- 17.Nave AH, Lange KS, Leonards CO, Siegerink B, Doehner W, Landmesser U, et al. Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis. 2015;242(2):496–503. doi: 10.1016/j.atherosclerosis.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 18.Dursunoğlu D, Evrengül H, Polat B, Tanrıverdi H, Çobankara V, Kaftan A, et al. Lp(a) lipoprotein and lipids in patients with rheumatoid arthritis: serum levels and relationship to inflammation. Rheumatol Int. 2005;25(4):241–245. doi: 10.1007/s00296-004-0438-0. [DOI] [PubMed] [Google Scholar]
- 19.Kario K, Matsuo T, Kobayashi H, Matsuo M, Asada R, Koide M. High lipoprotein (a) levels in chronic hemodialysis patients are closely related to the acute phase reaction. Thromb Haemost. 1995;74(4):1020–1024. doi: 10.1055/s-0038-1649872. [DOI] [PubMed] [Google Scholar]
- 20.Langsted A, Kamstrup PR, Nordestgaard BG. Lipoprotein(a): fasting and nonfasting levels, inflammation, and cardiovascular risk. Atherosclerosis. 2014;234(1):95–101. doi: 10.1016/j.atherosclerosis.2014.01.049. [DOI] [PubMed] [Google Scholar]
- 21.Henry BM, Benoit SW, de Oliveira MHS, Lippi G, Favaloro EJ, Benoit JL. ADAMTS13 activity to von Willebrand factor antigen ratio predicts acute kidney injury in patients with COVID-19: evidence of SARS-CoV-2 induced secondary thrombotic microangiopathy. Int J Lab Hematol. 2020;00:1–8. doi: 10.1111/ijlh.13415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.KDIGO AKI Working Group KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;2(1):1. doi: 10.1038/kisup.2012.1. [DOI] [Google Scholar]
- 23.Chan L, Chaudhary K, Saha A, Chauhan K, Vaid A, Zhao S, et al. AKI in hospitalized patients with COVID-19. J Am Soc Nephrol. 2021;32(1):151–160. doi: 10.1681/ASN.2020050615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gupta S, Coca SG, Chan L, Melamed ML, Brenner SK, Hayek SS, et al. AKI treated with renal replacement therapy in critically Ill patients with COVID-19. J Am Soc Nephrol. 2021;32(1):161–176. doi: 10.1681/ASN.2020060897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kox M, Waalders NJB, Kooistra EJ, Gerretsen J, Pickkers P. Cytokine levels in critically Ill patients with COVID-19 and other conditions. JAMA. 2020;324(15):1565. doi: 10.1001/jama.2020.17052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kotani K, Serban M-C, Penson P, Lippi G, Banach M. Evidence-based assessment of lipoprotein(a) as a risk biomarker for cardiovascular diseases—some answers and still many questions. Crit Rev Clin Lab Sci. 2016;53(6):370–378. doi: 10.1080/10408363.2016.1188055. [DOI] [PubMed] [Google Scholar]
- 27.Sticchi E, Magi A, Kamstrup PR, Marcucci R, Prisco D, Martinelli I, et al. Apolipoprotein(a) Kringle-IV Type 2 copy number variation is associated with venous thromboembolism. PLoS ONE. 2016;11(2):e0149427. doi: 10.1371/journal.pone.0149427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmer SR, Hengstenberg C, Kraft H-G, Mayer B, Pöll M, Kürzinger S, et al. Association of polymorphisms of the Apolipoprotein(a) gene with Lipoprotein(a) levels and myocardial infarction. Circulation. 2003;107(5):696–701. doi: 10.1161/01.CIR.0000048125.79640.77. [DOI] [PubMed] [Google Scholar]
- 29.Scipione CA, Sayegh SE, Romagnuolo R, Tsimikas S, Marcovina SM, Boffa MB, et al. Mechanistic insights into Lp(a)-induced IL-8 expression: a role for oxidized phospholipid modification of apo(a) J Lipid Res. 2015;56(12):2273–2285. doi: 10.1194/jlr.M060210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Aken BE, Reitsma PH, Rosendaal FR. Interleukin 8 and venous thrombosis: evidence for a role of inflammation in thrombosis. Br J Haematol. 2002;116(1):173–177. doi: 10.1046/j.1365-2141.2002.03245.x. [DOI] [PubMed] [Google Scholar]
- 31.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 32.Fuchs TA, Brill A, Wagner DD. NET impact on deep vein thrombosis. Arterioscler Thromb Vasc Biol. 2012;32(8):1777–1783. doi: 10.1161/ATVBAHA.111.242859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol. 2013;33(8):2032–2040. doi: 10.1161/ATVBAHA.113.301627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lämmle B. Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood. 2012;120(6):1157–1164. doi: 10.1182/blood-2012-02-412197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Y, Chen X, Yang Y, Wang B, Liu X, Tao Y, et al. A case of lipoprotein glomerulopathy with thrombotic microangiopathy due to malignant hypertension. BMC Nephrol. 2013;14(1):53. doi: 10.1186/1471-2369-14-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin-Lazaro JF, Fabregate R, Bajo-Martinez A, Bernal E, Ugalde A, Calbacho M, et al. Selective elevation of von willebrand factor in patients with high lipoprotein (a) levels: an unknown prothrombotic risk factor in patients with hypertension and other risk factors. Am J Hypertens. 2003;16(S1):71A–71A. doi: 10.1016/S0895-7061(03)00255-3. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data available on reasonable request from the authors.