

Abstract

Weakening the triplet–triplet annihilation (TTA) self-quenching effect induced by sensitizers remains a tremendous challenge due to the very few investigations carried out on them. Herein, benzo-21-crown-7 (B21C7)-functionalized 2,6-diiodo-1,3,5,7-tetramethyl-8-phenyl-4,4-difluoroboradiazaindacene (DIBDP) was synthesized to investigate the influences of huge bulks and electron-rich cavities of B21C7 moieties on the fluorescence emission and triplet-state lifetimes of DIBDP moieties. Density functional theory (DFT)/time-dependent DFT (TDDFT) computable results preliminarily predicted that B21C7 moieties had influences on the fluorescence emissions of DIBDP moieties but not on their localization of triplet states of B21C7-functionalized DIBDP (B21C7-DIBDP). The UV–vis absorption spectra, fluorescence emission spectra, and cyclic voltammograms verified that there was an electron-transfer process from the B21C7 moiety to the DIBDP moiety in B21C7-DIBDP. However, the calculated results of ΔGCS and ECS values and nanosecond time-resolved transient absorption spectra demonstrated that the electron-transfer process from the B21C7 moiety to the DIBDP moiety in B21C7-DIBDP had direct influences on the fluorescence emission of DIBDP moieties but not on the triplet states of DIBDP moieties. The experimental values of triplet-state lifetimes of B21C7-DIBDP were obviously longer than those of DIBDP at a high concentration (1.0 × 10–5 M); however, the fitted values of intrinsic triplet-state lifetimes of B21C7-DIBDP were slightly greater than those of DIBDP in the same solvent. These results demonstrated that the steric hindrance of B21C7 moieties could weaken the TTA self-quenching effect of DIBDP moieties at a high concentration and the a-PET effect induced a proportion of the produced singlet states of DIBDP moieties and could not emit fluorescence in the form of radiation transition but they could be transformed into triplet states through intersystem crossing (ISC) processes due to the iodine atoms in the DIBDP moiety. The stronger a-PET effects in polar solvents induced smaller fluorescence quantum yields so that more singlet states of DIBDP moieties were transformed into triplet states to weaken the TTA self-quenching effects.
1. Introduction
Triplet photosensitizers have demonstrated a strong potential for a broad range of applications in numerous areas, such as photocatalysis,1−7 photodynamic therapy (PDT),8−19 photoelectric devices,20−22 organic room-temperature phosphorescence (RTP) materials,23−30 and triplet–triplet annihilation (TTA) upconversion.31−41 Significantly, to complete the triplet–triplet energy-transfer (TTET) processes from photosensitizers to acceptors following the Dexter energy-transfer mechanism in most of these applications, the photosensitizers and acceptors must diffuse within a distance smaller than the Dexter radius (usually 0–1 nm) before the total triplet states of the sensitizers decaying;31,35,36 therefore, a long-enough triplet excited-state is pivotal for a perfect photosensitizer besides the abilities of strong absorption of visible light and efficient intersystem crossing (ISC).1,6,7,14,31,37,38,40 The triplet-state lifetimes of sensitizers can be influenced by many factors, and previous efforts have been devoted to prolonging their triplet-state lifetimes.1,6,7,40,42−50 However, limited by the repulsive aggregation-caused quenching (ACQ) effect of the π-conjugated-system-containing and planar rigid sensitizers (e.g., 2,6-diiodo-1,3,5,7-tetramethyl-8-phenyl-4,4-difluoroboradiazaindacene (DIBDP)),51,52 the TTA self-quenching effect induced by these photosensitizers will cause additional lifetime quenching in the decaying of their transient absorption accompanied by their concentration increase, which will result in the experimental values of the triplet-state lifetimes to be much shorter than their intrinsic values in most cases due to the TTA self-quenching effects.47,49−51 Although we have reported that huge bulks of pillar[5]arene moieties in DIBDP-functionalized pillar[5]arene could weaken the TTA self-quenching effect of DIBDP moieties,51 owing to only one report, weakening the photosensitizers’ TTA self-quenching effect is still a great challenge for their further applications.
Crown ethers have been widely exploited and applied in numerous areas, such as supramolecular polymers,53−57 fluorescence probes and sensors,58−61 molecular machines,62−67 and ionic channels,68 since the 1987 Nobel Prize for Chemistry was awarded to Pedersen,69 Cram,70 and Lehn.71 Among them, fluorophore-functionalized crown ethers utilized in constructing supramolecular fluorescence systems that were usually based on photoinduced electron-transfer (PET) mechanisms between the hosts and guests were widely applied in fluorescence probes and sensors;58−61 however, fabricating triplet-photosensitizer-functionalized crown ether systems remains an enormous challenge due to very few investigations on them.72 Previously, we prepared a DIBDP-functionalized ethoxypillar[5]arene (EtP5) and confirmed that the steric hindrance of EtP5 could reduce collision and further weaken the TTA self-quenching effects of DIBDP moieties.51 However, to the best of our knowledge, the electron-transfer mechanism in triplet-photosensitizer-functionalized crown ethers has not been investigated so far, and the influences of electron-rich cavities of crown ethers on the triplet-state lifetimes of photosensitizers in photosensitizer-functionalized crown ether are still not clear. All of these are major obstacles and challenges to designing and assembling supramolecular smart nanostructures, including excellent PDT materials, photocatalysis, and photovoltaic devices based on photosensitizer-functionalized crown ethers.
Herein, we designed and synthesized a benzo-21-crown-7 (B21C7)-functionalized DIBDP (B21C7-DIBDP) (Scheme 1). Systematic investigations of calculations of density functional theory (DFT), photophysical properties, electrochemical properties, and nanosecond time-resolved transient absorption spectra were performed to work out the electron-transfer mechanisms between crown ethers and DIBDP moieties, as well as the strategies of weakening the TTA self-quenching effect of DIBDP moieties.
Scheme 1. Synthetic Routes of B21C7-DIBDP.

The molecular structure of the reference compound DIBDP is also shown in this scheme.
2. Results and Discussion
2.1. Design and Synthesis of the Target Molecule
As stated in Section 1, the TTA self-quenching effect induced by sensitizers is an obstacle for applications of triplet sensitizers,47,49−51 and how to weaken the TTA self-quenching effect of triplet sensitizers is a critical issue that needs to be resolved. Inspired by our previous work, owing to the huge bulks, pillar[5]arenes connected by methylene bridges at the para-positions of five 2,5-dialkoxybenzene rings could weaken the TTA self-quenching effect induced by the photosensitizers.51 Herein, we attempted to introduce another supramolecular macrocycle (B21C7) connected by seven ethylene glycol units that contain electron-rich cavities and huge bulks as well.57 Moreover, due to the excellent photophysical properties of DIBDP,42,47,48,50−52 as well as both DIBDP and B21C7 being readily functionalized,42,47,48,50−52,57B21C7-substituted BODIPY (B21C7-BDP) was prepared according to a literature report,73 followed by the iodization of B21C7-BDP to obtain B21C7-DIBDP. Both B21C7-BDP and B21C7-DIBDP could be prepared in satisfactory yields (Scheme 1). The molecular structure of B21C7-DIBDP was systematically verified by melt points, 1H nuclear magnetic resonance (NMR), 13C NMR, and high-resolution mass spectra (HRMS) (Figures S1–S3). Furthermore, similar to DIBDP-functionalized pillar[5]arene in our previous work,51 the electron-transfer effect of electron-rich B21C7 moieties on both singlet and triplet states of DIBDP moieties was also expected to be systematically investigated in B21C7-functionalized DIBDP.
2.2. DFT/TDDFT Computations
DFT calculations using Gaussian 09W were effective auxiliary tools for predicting the electron-transfer process from the B21C7 moiety to the DIBDP moiety in B21C7-DIBDP prior to the experimental investigations.51 From the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) distributions of the low-lying singlet state (S1) of B21C7-DIBDP (Figure 1), no overlaps were observed between the initial and the destination molecular orbital (MO) involved in the transitions of B21C7-DIBDP, which indicates that the forbidden S0 → S1 transition and the emission intensity of B21C7-DIBDP may be weaker than DIBDP. It could be speculated from the DFT/TDDFT calculated results that there may be an electron-transfer process from the B21C7 moiety to the DIBDP moiety in B21C7-DIBDP.
Figure 1.

Selected frontier molecular orbitals of B21C7-DIBDP. The calculations are based on the optimized ground-state geometry (S0 state) without any solvent at the B3LYP/6-31G(d) level using Gaussian 09W.
The isosurfaces of spin densities of DIBDP and B21C7-DIBDP were exported from their optimized lowest-lying triplet-state (T1) geometries based on their optimized geometries of the S0 state, respectively (Figure 2), at the DFT//B3LYP/6-31G(d) level using Gaussian 09W.74−76 However, unlike the HOMO–LUMO distributions of the S1 state of B21C7-DIBDP, the isosurface of the spin density of B21C7-DIBDP was not localized on the B21C7 moiety but on the DIBDP moiety, which indicated that the electron-rich cavity of B21C7 had no direct effect on the T1 state of the DIBDP moiety. As seen from Figures 1 and 2, the possible existing electron-transfer process in B21C7-DIBDP from the B21C7 moiety to the DIBDP moiety could only affect the S0 → S1 transition of the DIBDP moiety but not the ISC of S1 states. Next, these computational results will be further verified using experimental strategies.
Figure 2.
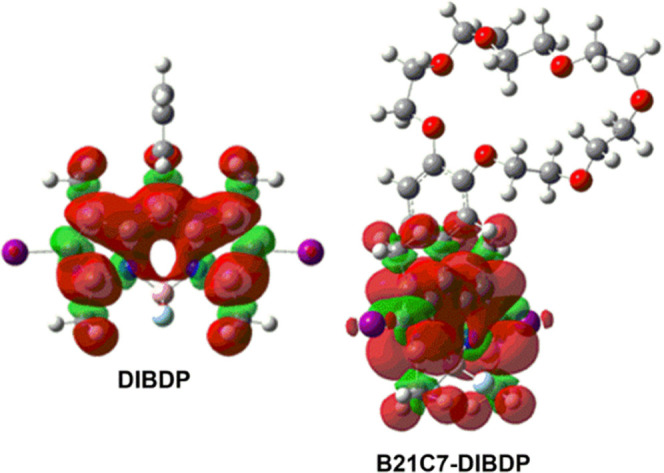
Isosurfaces of the spin densities of DIBDP and B21C7-DIBDP based on the optimized triplet-state geometries. The calculations were performed at the B3LYP/6-31G(d) level using Gaussian 09W.
2.3. Spectroscopic and Photophysical Properties
The basic spectroscopic (Figures 3 and 5) and photophysical properties (Table 1) were investigated first. In comparison to the characteristic absorptions of DIBDP in Figure 3, the strong absorption bands of B21C7-DIBDP at ∼530 nm in toluene, CHCl3, acetone, and CH3CN are all absorptions of DIBDP chromophores in B21C7-DIBDP (Figure 3). Both maximum absorption wavelengths of DIBDP and B21C7-DIBDP exhibited a hypsochromic shift to shorter wavelengths, and the decreasing intensities at maximum absorption wavelengths with the polarity of solvents increasing (Table 1) indicated that there were also n−π transitions besides π–π transitions in these two molecules because both contained nonbonding electrons in the atoms of nitrogen, iodine, and fluorine. However, the absorption intensities of B21C7-DIBDP at maximum absorption wavelengths were weaker than those of DIBDP in the same solvent (Figure 3 and Table 1), which could be attributed to the electron transfer from B21C7 moieties to DIBDP moieties. Furthermore, the maximum absorption intensities of B21C7-DIBDP became weaker when 1.0 equiv of KPF6 was added in the above-mentioned four solvents (Figure 3 and Table 1). Thus, it could be seen that the hyperchromic effect of electron-rich B21C7 on the DIBDP moiety became weaker after the K+ entered the B21C7’s cavity because a proportion of electrons of B21C7 was transferred to K+, as well as the increase in the solvents’ polarities after adding KPF6 could have weakened the maximum absorption intensities of B21C7-DIBDP due to the n−π transitions in B21C7-DIBDP. The UV–vis absorption spectra in Figure 3 preliminarily verified that there were electron-transfer processes from the B21C7 moiety to the DIBDP moiety in B21C7-DIBDP in toluene, CHCl3, acetone, and CH3CN.
Figure 3.

UV–vis absorption spectra of DIBDP and B21C7-DIBDP (1.0 × 10–5 M, 25 °C) in toluene (a), CHCl3 (b), acetone (c), and CH3CN (d).
Figure 5.

Fluorescence emission spectra of DIBDP and B21C7-DIBDP (1.0 × 10–5 M, 25 °C) in toluene (a), CHCl3 (b), acetone (c), and CH3CN (d).
Table 1. Photophysical Parameters of DIBDP and B21C7-DIBDP in Toluene (TOL), CHCl3 (CHL), Acetone (ACE), and CH3CN (ACN).
| solvent | compd | λabs (nm)a | εb | λem (nm)c | ΦF (%)d | τF (ns)e | τT1 (μs)f | τT (μs)g |
|---|---|---|---|---|---|---|---|---|
| TOL | DIBDP | 537 | 0.87 | 556 | 1.9i | 0.27 | 45 | 126 |
| B21C7-DIBDP | 536 | 0.83 | 553 | 1.7 | 0.28 | 59 | 125 | |
| B21C7-DIBDP⊃K+h | 536 | 0.73 | 554 | 1.3 | 0.18 | 60 | 136 | |
| CHL | DIBDP | 536 | 0.88 | 553 | 2.0i | 0.29 | 66 | 142 |
| B21C7-DIBDP | 536 | 0.81 | 552 | 1.7 | 0.32 | 99 | 145 | |
| B21C7-DIBDP⊃K+h | 537 | 0.72 | 555 | 1.4 | 0.16 | 104 | 144 | |
| ACE | DIBDP | 531 | 0.83 | 550 | 1.5i | 0.12 | 43 | 130 |
| B21C7-DIBDP | 529 | 0.78 | 548 | 1.1 | 0.07 | 66 | 136 | |
| B21C7-DIBDP⊃K+h | 530 | 0.72 | 548 | 1.3 | 0.15 | 63 | 139 | |
| ACN | DIBDP | 529 | 0.81 | 550 | 1.5i | 0.17 | 41 | 114 |
| B21C7-DIBDP | 529 | 0.76 | 548 | 0.8 | 0.04 | 78 | 117 | |
| B21C7-DIBDP⊃K+h | 529 | 0.69 | 548 | 1.1 | 0.10 | 75 | 118 |
Maximum absorption wavelength (c = 1.0 × 10–5 M).
Molar extinction coefficient, in 105 M–1 cm–1.
Maximum emission wavelength (c = 1.0 × 10–5 M).
Absolute fluorescence quantum yields.
Fluorescence lifetimes.
Triplet-state lifetimes (c = 1.0 × 10–5 M).
Intrinsic triplet-state lifetimes obtained by fitting experimental curves using a kinetic model supposing the TTA self-quenching effect eliminated.77
Molar ratio of 1:1.
Literature-reported value.
The PET processes, which included a-PET (the photoinduced electron-transfer process from the electron donor to the fluorophore that is an electron acceptor) and d-PET processes (the photoinduced electron-transfer process from the fluorophore (donor) to the electron acceptor), are illustrated in Figure 4. We have verified the a-PET (Figure 4a) effect between EtP5 and DIBDP moieties in DIBDP-functionalized pillar[5]arene in our previous work.51 To investigate the electron-transfer process between B21C7 and DIBDP moieties in B21C7-DIBDP, the fluorescence emissions of DIBDP, B21C7-DIBDP, and B21C7-DIBDP⊃K+ were investigated in a variety of solvents with different polarities (Figures 5 and S4–S9). First, the emission intensities of B21C7-DIBDP were slightly weaker than those of DIBDP in toluene and CHCl3; however, the emission intensities of B21C7-DIBDP were quenched more seriously when K+ was added (Figure 5a,b), which indicated that there were more obvious electron-transfer processes when the polarities of the solvents became higher after K+ was added. Second, interestingly, the opposite experimental results were obtained in acetone and CH3CN (Figure 5c,d). The emission intensities of B21C7-DIBDP were quenched more seriously in acetone and CH3CN than those in toluene and CHCl3, which verified again that the electron-transfer processes could easily occur in higher polar solvents. When K+ was added, the emission intensities of B21C7-DIBDP were boosted in acetone and CH3CN because the complexations of B21C7 moiety with K+ made the electron-transfer effects from the B21C7 moiety to the DIBDP moiety weaker. Upon investigating the fluorescence emission experiments of DIBDP, B21C7-DIBDP, and B21C7-DIBDP⊃K+ in a variety of solvents with different polarities, the electron-transfer effect between B21C7 and DIBDP moieties in B21C7-DIBDP (a-PET) was further confirmed. In addition, the other photophysical properties of DIBDP and B21C7-DIBDP are listed in Table 1. According to the a-PET process in Figure 4a, the electron transfer from the B21C7 moiety to the DIBDP moiety resulted in the HOMO orbitals of DIBDP moieties being occupied by the electrons of B21C7 moieties, and a proportion of the produced singlet states of DIBDP moieties could not emit fluorescence in the form of radiation transition. Therefore, the fluorescence quantum yields of B21C7-DIBDP were smaller than those of DIBDP in the same solvents (Table 1).
Figure 4.

Frontier molecular orbitals of fluorescecet molecules with PET processes. (a) a-PET process: photoinduced electron-transfer process from the electron donor to the fluorophore, which is an electron acceptor; (b) d-PET process: photoinduced electron-transfer process from the fluorophore (donor) to the electron acceptor.
2.4. Electrochemical Investigations
To verify the a-PET process from the B21C7 moiety to the DIBDP moiety in B21C7-DIBDP qualitatively and quantitatively, the electrochemical properties of B21C7-DIBDP and reference compounds DIBDP and B21C7 were investigated using cyclic voltammetry (Figure 6). B21C7 showed a reversible oxidation potential at +0.98 V (vs Fc/Fc+), and no reduction potential was observed from 0 to −2.0 V, indicating that the B21C7 moiety was suitable for being an electron donor (Figure 6 and Tables 2 and 3).51 However, interestingly, after the B21C7 moiety modified to DIBDP, the oxidation potential of B21C7-DIBDP was at +0.87 V (vs Fc/Fc+) and the initial oxidation potential of B21C7 at +0.98 V disappeared due to the electron transfer from the B21C7 moiety to the DIBDP moiety. Moreover, both oxidation and reduction potentials of B21C7-DIBDP were similar to those of DIBDP (Figure 6 and Tables 2 and 3). Cyclic voltammograms in Figure 6 qualitatively demonstrate the existence of the electron-transfer process from the B21C7 moiety to the DIBDP moiety in B21C7-DIBDP.
Figure 6.

Cyclic voltammograms of B21C7, DIBDP, and B21C7-DIBDP in deaerated CH2Cl2 containing 1.0 mM compound to be measured and ferrocene (Fc), 0.10 M Bu4NPF6 as the supporting electrolyte, and Ag/AgNO3 as the reference electrode; scan rate was 0.1 V/s, 25 °C. Fc was used as the internal reference [E1/2 = +0.64 V (Fc+/Fc) vs the standard hydrogen electrode].
Table 2. Electrochemical Redox Potentialsa, Gibbs Free Energy Changes (ΔGCS), and Charge Separation Energy States (ECS)b for B21C7, DIBDP, and B21C7-DIBDP in Toluene (TOL), CHCl3 (CHL), Acetone (ACE), and CH3CN (ACN).
| ΔGCS (eV) |
ECS (eV) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | EOX (V) | ERED (V) | TOL | CHL | ACE | ACN | TOL | CHL | ACE | ACN |
| B21C7 | +0.98 | c | d | d | d | d | d | d | d | d |
| DIBDP | +0.89 | –1.47 | d | d | d | d | d | d | d | d |
| B21C7-DIBDP | +0.87 | –1.48 | 0.01 | –0.10 | –0.19 | –0.21 | 2.25 | 2.14 | 2.06 | 2.05 |
The values of redox potentials were obtained by setting the potential of Fc+/Fc as 0 V, measured in deaerated CH2Cl2. The counter electrode was a Pt electrode, and the working electrode was a glassy carbon electrode, with a Ag/AgNO3 couple as the reference electrode.
E00 is the energy level of the fluorescence emission.
Not been observed.
Unable to be determined.
Table 3. Electrochemical Redox Potentialsa, Gibbs Free Energy Changes (ΔGCS), and Charge Separation Energy States (ECS)b for B21C7, DIBDP, and B21C7-DIBDP in Toluene (TOL), CHCl3 (CHL), Acetone (ACE), and CH3CN (ACN).
| ΔGCS (eV) |
ECS (eV) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | EOX (V) | ERED (V) | TOL | CHL | ACE | ACN | TOL | CHL | ACE | ACN |
| EtP5 | +0.98 | c | d | d | d | d | d | d | d | d |
| DIBDP | +0.89 | –1.47 | d | d | d | d | d | d | d | d |
| EtP5-DIBDP | +0.87 | –1.48 | 0.59 | 0.49 | 0.41 | 0.38 | 2.25 | 2.14 | 2.06 | 2.05 |
The values of redox potentials were obtained by setting the potential of Fc+/Fc as 0 V, measured in deaerated CH2Cl2. The counter electrode was a Pt electrode, and the working electrode was a glassy carbon electrode, with a Ag/AgNO3 couple as the reference electrode.
E00 is the energy level of the T1 state.
Not been observed.
Unable to be determined.
To clarify the direction and limitation of the influences of electron-transfer processes in B21C7-DIBDP on the fluorescence emission and the T1 state of the DIBDP moiety quantitatively, Gibbs free energy changes (ΔGCS) of the electron transfer as well as the energy levels of charge separation states (ECS) of B21C7-DIBDP were determined using Weller equations (eqs 1–3).78
| 1 |
| 2 |
| 3 |
where ΔGS is the static Coulombic energy, which was determined by eq 1. e is the elementary charge, EOX is the half-wave potential for one electron oxidation in the electron-donor unit, ERED is the half-wave potential for one electron reduction of the electron-acceptor unit, and E00 is the energy level calculated with the fluorescence emission wavelength (S1) or the phosphorescent emission wavelength (T1). εS is the static dielectric constant of the solvent. RCC is the center-to-center separation distance between the electron donor and the electron acceptor, determined by the optimized geometry of DFT calculations. RD is the radius of the electron donor; RA is the radius of the electron acceptor. εREF is the static dielectric constant of the solvent used for the electrochemical studies. ε0 is the permittivity of free space. ΔGCS is the Gibbs free energy changes, which was determined by eq 2. ECS is energy of the charge-separated states, which was determined by eq 3. The calculated results of ΔGCS and ECS for the influence on fluorescence emission (E00 is the energy level approximated with the fluorescence emission wavelength) of the DIBDP moiety in B21C7-DIBDP are listed in Table 2; however, the results for the influence on the T1-state lifetime (E00 is the energy level approximated with phosphorescent emission wavelength) of the DIBDP moiety in B21C7-DIBDP are listed in Table 3.
When E00 was the energy level of the fluorescence emission (S1), the ΔGCS values were 0.01, −0.10, −0.19, and −0.21 eV for the electron-transfer processes from the B21C7 moiety to the DIBDP moiety in toluene, CHCl3, acetone, and CH3CN, respectively (Table 2). The corresponding ECS values in CHCl3, acetone, and CH3CN were 2.14, 2.06, and 2.05, respectively (Table 2), and were all lower than the corresponding S1-state energies of DIBDP moieties (2.25 eV). However, in toluene, the ΔGCS value for the electron-transfer process in B21C7-DIBDP was greater than 0 and the ECS value of B21C7-DIBDP was higher than the S1-state energy of DIBDP moieties (2.25 eV). These calculation results demonstrated that the electron-transfer processes from B21C7 moieties to DIBDP moieties influenced the fluorescence emission of DIBDP moieties in CHCl3, acetone, and CH3CN except for that in toluene. Simultaneously, the fluorescence emissions of DIBDP moieties were easier to be affected by the electron-transfer processes in higher polar solvents, which were in agreement with the fluorescence emission spectra in Figure 5.
When E00 was the energy level of the T1 state of DIBDP (1.61 eV), the ΔGCS values were 0.59, 0.49, 0.41, and 0.38 eV for the electron-transfer processes from the B21C7 moiety to the DIBDP moiety in toluene, CHCl3, acetone, and CH3CN, respectively (Table 3). The ΔGCS values in these four solvents were all greater than 0, and the corresponding ECS values were all higher than the T1-state energy of DIBDP moieties (Table 3). These calculated results demonstrated that the electron-transfer processes from B21C7 moieties to DIBDP moieties in B21C7-DIBDP could not directly affect the T1 states of DIBDP moieties in all of the above four solvents due to the exceedingly low T1 energy state of DIBDP (1.61 eV).
2.5. Nanosecond Time-Resolved Transient Absorption Spectroscopy
DFT calculations and electrochemical investigations demonstrated that there were no direct influences on the electron-transfer process on the T1 states of DIBDP moieties in B21C7-DIBDP. Herein, nanosecond time-resolved transient absorption spectra were used to verify the results of DFT calculations and electrochemical investigations further (Figures S26–S34). Simultaneously, we fitted the intrinsic triplet-state lifetimes of DIBDP and B21C7-DIBDP in these four solvents according to eqs 4–6.47,50,77 The differential equation in the decaying process of the triplet state is expressed in eq 4.
| 4 |
where cT is the concentration of the triplet state, t is the decaying time, k1 is the spontaneous decaying rate constant without the TTA self-quenching effect, and k2 is the bimolecular decaying rate constant induced by the TTA self-quenching effect. Moreover, the corresponding integral equation is expressed in eq 5.
| 5 |
where c0 is the initial concentration of triplet states and cT (t) is the concentration of triplet states at time t. The transient absorption intensity at time t is expressed in eq 6.
| 6 |
where A(t) is the transient absorption intensity at time t. A0 is the initial transient absorption intensity prior to decaying. τ1 is the unimolecular lifetime of the triplet state without the TTA self-quenching effect induced by the photosensitizers. τ2 = 1/c0k2, which is the reciprocal of the initial concentration of the triplet state multiply the bimolecular decaying rate constant induced by the TTA self-quenching effect of the photosensitizers. The triplet-state lifetime values of the photosensitizers were fitted using eq 6 with three variables (A0, τ1, τ2) supposing that the TTA self-quenching effect was eliminated.47,50,77
First, the maps of transient absorption for DIBDP and B21C7-DIBDP in various solvents were very similar (Figures S26–S33), verifying that B21C7 moieties could not affect the T1 states of DIBDP moieties in B21C7-DIBDP. Second, the lifetimes of B21C7-DIBDP in toluene, CHCl3, acetone, and CH3CN were obviously longer than those of DIBDP at a high concentration (1.0 × 10–5 M); moreover, the lifetimes of B21C7-DIBDP in these four solvents almost remained unchanged when K+ was added (Figure S34). Third, the intrinsic triplet-state lifetimes values of B21C7-DIBDP were slightly greater than DIBDP in the same solvents, for instance, in Figure S34b,f,j. The above experimental results demonstrated that there were no direct effects of the electron transfer from the B21C7 moiety to the DIBDP moiety in B21C7-DIBDP on the intrinsic triplet-state lifetimes. Moreover, the huge bulks of B21C7 moieties resulting in steric hindrance (Figure 7) could reduce the collision of DIBDP moieties and further weaken the TTA self-quenching effect (the TTA process needs collision of sensitizers in the range of 0–1 nm31,35,36) of DIBDP moieties in B21C7-DIBDP at a high concentration (1.0 × 10–5 M). The fluorescence quantum yields of B21C7-DIBDP were observed to be greater in nonpolar solvents compared to those in polar solvents; this was because the stronger a-PET effects induced smaller fluorescence quantum yields in polar solvents, while the TTA self-quenching effects were weakened more efficiently in polar solvents (Table 1). As illustrated in Figure 4a, when the outer-layer valence electron of DIBDP moieties in B21C7-DIBDP was transited from the HOMO orbital to the LUMO orbital, as a result of the a-PET effect, the HOMO orbitals of DIBDP moieties were occupied by the electrons in B21C7 moieties so that singlet states of DIBDP moieties could not emit fluorescence in the form of radiation transition but could be transformed to triplet states through the ISC process due to the iodine atoms in the DIBDP moiety. Therefore, the stronger a-PET effects in polar solvents induced smaller fluorescence quantum yields but weaker TTA self-quenching effects (Table 1).
Figure 7.

Optimized ground-state geometries of DIBDP and B21C7-DIBDP at the DFT//B3LYP/6-31G(d) level using Gaussian 09W.
3. Conclusions
In summary, we designed and synthesized a B21C7-functionalized DIBDP (B21C7-DIBDP) to investigate the influences of huge bulks and electron-rich cavities of B21C7 moieties on the fluorescence emission and triplet-state lifetimes of DIBDP moieties. DFT/TDDFT computable results preliminarily predicted that B21C7 moieties had direct influences on the fluorescence emissions of DIBDP moieties but not on their localization of triplet states of B21C7-DIBDP. The UV–vis absorption spectra, fluorescence emission spectra, and cyclic voltammograms verified that there was an electron-transfer process from the B21C7 moiety to the DIBDP moiety (a-PET) in B21C7-DIBDP. However, the calculated results of ΔGCS and ECS values, nanosecond time-resolved transient absorption spectra, and intrinsic triplet-state lifetimes demonstrated that the electron-transfer processes from the B21C7 moiety to the DIBDP moiety in B21C7-DIBDP had direct influences on the fluorescence emission of DIBDP moieties but not on the triplet states of DIBDP moieties. The triplet-state lifetimes of B21C7-DIBDP were obviously longer than DIBDP at a high concentration (1.0 × 10–5 M); however, the fitted values of intrinsic triplet-state lifetimes of B21C7-DIBDP were only slightly greater than those of DIBDP in the same solvent. Besides the steric hindrance of B21C7 moieties, which could weaken the TTA self-quenching effect of DIBDP moieties at a high concentration, the a-PET effect resulted in a proportion of the produced singlet states of DIBDP moieties not being able to emit fluorescence in the form of radiation transition, but they could be transformed to triplet states through ISC processes due to the iodine atoms in the DIBDP moiety. The stronger a-PET effects in polar solvents induced smaller fluorescence quantum yields so that more singlet states of DIBDP moieties were transformed to triplet states to weaken the TTA self-quenching effects. These experimental results provided another strategy of weakening the TTA self-quenching effect induced by the sensitizers; thus, they will be significant for designing and assembling supramolecular smart nanostructures, including excellent PDT materials, photocatalysis, and photovoltaic devices based on photosensitizer-functionalized crown ethers.
4. Methods
4.1. Synthesis and Characterizations
All of the raw materials were analytically pure and used without further purification. The organic solvents were dried or distilled before use. Thin-layer chromatography (TLC) and column chromatography were carried out using silica gel (200–300 mesh) as the stationary phase. Melting points were measured on an SHPSIC WRS-2 automatic melting point apparatus (China). 1H NMR and 13C NMR spectra were obtained on a Bruker AVANCE 400 spectrophotometer (Germany) with CDCl3 as the solvent and Si(CH3)4 as the internal standard (0.00 ppm). High-resolution mass spectra (HRMS) were recorded in an ion trap ESI-HRMS spectrometer. Detailed synthetic routes for B21C7-DIBDP are shown in Scheme 1; please see the Supporting Information for the structural characterizations.
4.2. Instruments for Spectral Measurement
The UV–vis absorption spectra were recorded on an HP8453 UV–vis spectrophotometer. The fluorescence emission spectra were measured with an FS5 spectrofluorometer (Edinburgh Instrument Ltd., U.K.). The absolute fluorescence quantum yields were determined by a C13534 UV–NIR spectrometer, equipped with an optical integration sphere (Hamamatsu Photonics K.K., Japan). Fluorescence lifetimes were recorded on an OB920 luminescent lifetime spectrometer (Edinburgh Instruments Ltd., U.K.) equipped with a 510 nm EPL picosecond pulsed laser for excitation, and the decay traces were measured with the time-correlated single photon counting (TCSPC) technique. Cyclic voltammograms were performed using a CHI610D electrochemical workstation (Shanghai, China). The nanosecond time-resolved transient absorption spectra were measured with an LP980 laser flash photolysis spectrometer (Edinburgh Instruments Ltd., U.K.). Opolette HE 355 UV tunable laser systems (OPOTEK Inc.) were used for excitation (210–2400 nm). Typical laser power was 5 mJ per pulse. The signal was digitized on a Tektronix TDS 3012B oscilloscope.
4.3. Details of Theoretical Computations
The ground-state (S0) geometries of DIBDP and B21C7-DIBDP were optimized based on DFT with the B3LYP functional74 and the 6-31G(d) basis set.75 The isosurfaces of spin density and excited-state energies of DIBDP and B21C7-DIBDP were calculated using DFT and time-dependent DFT (TDDFT), respectively, based on the optimized geometries of the S0 state. All of these calculations were performed using the Gaussian 09W program package suite in a Dell server.76
4.4. Methods of Electrochemical Studies
Cyclic voltammogram curves were recorded at a scan rate of 0.1 V/s. The electrolytic cell was a three-electrode-containing cell. Electrochemical measurements were performed in CH2Cl2 with ferrocene (Fc) as the internal reference and 0.1 M Bu4NPF6 as the supporting electrolyte after deaerated by high-purity N2. The working electrode was a glassy carbon electrode, and the counter electrode was a platinum electrode. In addition, a nonaqueous Ag/AgNO3 (0.1 M in CH3CN) reference electrode was contained in a separate compartment connected to the solution via a semipermeable membrane.78
4.5. Measuring Method for Nanosecond Time-Resolved Transient Absorption Spectroscopy
All of the samples of DIBDP and B21C7-DIBDP were deaerated by high-purity N2 for 15 min and well-sealed by a parafilm prior to measurement. The fitted values of triplet-state lifetimes and the maps of transient absorption of the samples were obtained in L900 software. The intrinsic triplet-state lifetimes were obtained by fitting experimental curves using a kinetic model, supposing that the TTA self-quenching effect was eliminated.77
4.6. Synthetic Procedures of B21C7-DIBDP
Compounds 1 and B21C7-BDP were prepared according to a literature report,73 and the synthetic procedures of B21C7-DIBDP have been described as follows.
B21C7-BDP (1.2 g, 2.0 mmol) was dissolved in anhydrous CH2Cl2 (250 mL), and then excess N-iodosuccinimide (1.8 g, 8.0 mmol) was added. The mixture was stirred at room temperature overnight. The reaction mixture was then washed using brine three times and concentrated under a vacuum. The crude product was purified by flash column chromatography (CH2Cl2/EtOAc, 5:1, v/v). The red band was collected, and the solvent was removed under reduced pressure to give the product as a red solid. Yield: 1.5 g, 85.0%. MP: 168.8–169.5 °C. The 1H NMR spectrum of B21C7-DIBDP is shown in Figure S1. 1H NMR (400 MHz, CDCl3): δ 6.99–6.97 (d, 1H, J = 8.0 Hz), 6.78–6.74 (m, 2H), 4.24–4.22 (t, 2H, J = 4.0 Hz), 4.12–4.10 (t, 2H, J = 4.0 Hz), 3.99–3.97 (t, 2H, J = 4.0 Hz), 3.93–3.91 (t, 2H, J = 4.0 Hz), 3.86–3.84 (t, 2H, J = 4.0 Hz), 3.81–3.75 (m, 6H), 3.70 (s, 8H), 2.64 (s, 6H), 1.48 (s, 6H). The 13C NMR spectrum of B21C7-DIBDP is shown in Figure S2. 13C NMR (100 MHz, CDCl3): δ 156.6, 149.8, 144.8, 141.2, 132.9, 131.6, 129.8, 128.0, 127.1, 120.8, 114.0, 113.3, 85.6, 70.5, 68.6, 63.6, 31.9, 29.6, 21.6, 17.1, 16.0. The ESI-HRMS spectrum of B21C7-DIBDP is shown in Figure S3. ESI-HRMS: calcd [C31H39BF2I2N2O7 + NH4+] m/z = 872.1246, found m/z = 872.1253, error: 0.0007; calcd [C31H39BF2I2N2O7 + Na+] m/z = 877.0800, found m/z = 877.0805, error: 0.0005.
Acknowledgments
J.S. is thankful to the Jingying Plan of Shandong University of Science and Technology (0104060541012) for the financial support. J.Z. is grateful to the National Natural Science Foundation of China (U2001222) and the State Key Laboratory of Fine Chemicals for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c04540.
Characterization data, spectroscopic data, and DFT/TDDFT calculation details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chen K. K.; Guo S.; Liu H.; Li X.; Zhang Z. M.; Lu T. B. Strong Visible-Light-Absorbing Cuprous Sensitizers for Dramatically Boosting Photocatalysis. Angew. Chem., Int. Ed. 2020, 59, 12951–12957. 10.1002/anie.202003251. [DOI] [PubMed] [Google Scholar]
- Hari D. P.; Konig B. The Photocatalyzed Meerwein Arylation: Classic Reaction of Aryl Diazonium Salts in a New Light. Angew. Chem., Int. Ed. 2013, 52, 4734–4743. 10.1002/anie.201210276. [DOI] [PubMed] [Google Scholar]
- Linic S.; Christopher P.; Ingram D. B. Plasmonic-Metal Nanostructures for Efficient Conversion of Solar to Chemical Energy. Nat. Mater. 2011, 10, 911–921. 10.1038/nmat3151. [DOI] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J.; Xu Z. Y.; Zhang D. W.; Wang H.; Xie S. H.; Xu D. W.; Ren Y. H.; Wang H.; Liu Y.; Li Z. T. Supramolecular Metal-Organic Frameworks That Display High Homogeneous and Heterogeneous Photocatalytic Activity for H2 Production. Nat. Commun. 2016, 7, 11580 10.1038/ncomms11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Guo S.; Wang H. J.; Chen K. K.; Zhang N.; Zhang Z. M.; Lu T. B. A Broadband and Strong Visible-Light-Absorbing Photosensitizer Boosts Hydrogen Evolution. Nat. Commun. 2019, 10, 3155 10.1038/s41467-019-11099-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Wu W.; Sun J.; Guo S. Triplet Photosensitizers: From Molecular Design to Applications. Chem. Soc. Rev. 2013, 42, 5323–5351. 10.1039/c3cs35531d. [DOI] [PubMed] [Google Scholar]
- Chen H.; Zeng X.; Tham H. P.; Phua S. Z. F.; Cheng W.; Zeng W.; Shi H.; Mei L.; Zhao Y. NIR-Light-Activated Combination Therapy with a Precise Ratio of Photosensitizer and Prodrug Using a Host–Guest Strategy. Angew. Chem., Int. Ed. 2019, 58, 7641–7646. 10.1002/anie.201900886. [DOI] [PubMed] [Google Scholar]
- Gao J.; Li J.; Geng W. C.; Chen F. Y.; Duan X.; Zheng Z.; Ding D.; Guo D. S. Biomarker Displacement Activation: A General Host–Guest Strategy for Targeted Phototheranostics in Vivo. J. Am. Chem. Soc. 2018, 140, 4945–4953. 10.1021/jacs.8b02331. [DOI] [PubMed] [Google Scholar]
- Kwon N.; Kim H.; Li X.; Yoon J. Supramolecular Agents for Combination of Photodynamic Therapy and Other Treatments. Chem. Sci. 2021, 12, 7248–7268. 10.1039/D1SC01125A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Shao Y.; Kim J. H.; Pu Z.; Zhao X.; Huang H.; Xiong T.; Kang Y.; Li G.; Shao K.; Fan J.; Foley J. W.; Kim J. S.; Peng X. Unimolecular Photodynamic O2-Economizer to Overcome Hypoxia Resistance in Phototherapeutics. J. Am. Chem. Soc. 2020, 142, 5380–5388. 10.1021/jacs.0c00734. [DOI] [PubMed] [Google Scholar]
- Liu B.; Jiao J.; Xu W.; Zhang M.; Cui P.; Guo Z.; Deng Y.; Chen H.; Sun W. Highly Efficient Far-Red/NIR-Absorbing Neutral Ir(III) Complex Micelles for Potent Photodynamic/Photothermal Therapy. Adv. Mater. 2021, 33, 2100795 10.1002/adma.202100795. [DOI] [PubMed] [Google Scholar]
- Liu S.; Li Y.; Kwok R. T. K.; Lam J. W. Y.; Tang B. Z. Structural and Process Controls of Aiegens for NIR-II Theranostics. Chem. Sci. 2021, 12, 3427–3436. 10.1039/D0SC02911D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V. N.; Yan Y.; Zhao J.; Yoon J. Heavy-Atom-Free Photosensitizers: From Molecular Design to Applications in the Photodynamic Therapy of Cancer. Acc. Chem. Res. 2021, 54, 207–220. 10.1021/acs.accounts.0c00606. [DOI] [PubMed] [Google Scholar]
- Sun C.; Zhang H.; Yue L.; Li S.; Cheng Q.; Wang R. Facile Preparation of Cucurbit[6]Uril-Based Polymer Nanocapsules for Targeted Photodynamic Therapy. ACS Appl. Mater. Interfaces 2019, 11, 22925–22931. 10.1021/acsami.9b04403. [DOI] [PubMed] [Google Scholar]
- Xue L.; Shen Q.; Zhang T.; Fan Y.; Xu X.; Shao J.; Yang D.; Zhao W.; Dong X.; Mou X. Fluorescence Resonance Energy Transfer Enhanced Photothermal and Photodynamic Antibacterial Therapy Post a Single Injection. Mater. Chem. Front. 2021, 5, 6061–6070. 10.1039/D1QM00631B. [DOI] [Google Scholar]
- Yuan B.; Wu H.; Wang H.; Tang B.; Xu J. F.; Zhang X. A Self-Degradable Supramolecular Photosensitizer with High Photodynamic Therapeutic Efficiency and Improved Safety. Angew. Chem., Int. Ed. 2021, 60, 706–710. 10.1002/anie.202012477. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Ma S.; Liu X.; Xu Y.; Zhao J.; Si X.; Li H.; Huang Z.; Wang Z.; Tang Z.; Song W.; Chen X. Supramolecular Assembled Programmable Nanomedicine as in Situ Cancer Vaccine for Cancer Immunotherapy. Adv. Mater. 2021, 33, 2007293 10.1002/adma.202007293. [DOI] [PubMed] [Google Scholar]
- Zhu H.; Wang H.; Shi B.; Shangguan L.; Tong W.; Yu G.; Mao Z.; Huang F. Supramolecular Peptide Constructed by Molecular Lego Allowing Programmable Self-Assembly for Photodynamic Therapy. Nat. Commun. 2019, 10, 2412 10.1038/s41467-019-10385-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittner E. R.; Lankevich V.; Gelinas S.; Rao A.; Ginger D. A.; Friend R. H. How Disorder Controls the Kinetics of Triplet Charge Recombination in Semiconducting Organic Polymer Photovoltaics. Phys. Chem. Chem. Phys. 2014, 16, 20321–20328. 10.1039/C4CP01776E. [DOI] [PubMed] [Google Scholar]
- Qin L.; Liu X.; Zhang X.; Yu J.; Yang L.; Zhao F.; Huang M.; Wang K.; Wu X.; Li Y.; Chen H.; Wang K.; Xia J.; Lu X.; Gao F.; Yi Y.; Huang H. Triplet Acceptors with a D-A Structure and Twisted Conformation for Efficient Organic Solar Cells. Angew. Chem. 2020, 132, 15153–15159. 10.1002/ange.202006081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Gu W.; Lv L.; Chen Y.; Yang Y.; Ye P.; Wu J.; Hong L.; Peng A.; Huang H. Triplet Tellurophene-Based Acceptors for Organic Solar Cells. Angew. Chem., Int. Ed. 2018, 57, 1096–1102. 10.1002/anie.201712011. [DOI] [PubMed] [Google Scholar]
- Bhatia H.; Dey S.; Ray D. Effect of π–π Interactions of Donor Rings on Persistent Room-Temperature Phosphorescence in D4–A Conjugates and Data Security Application. ACS Omega 2021, 6, 3858–3865. 10.1021/acsomega.0c05666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. Y.; Hu J. W.; Huang C. H.; Huang T. H.; Chen D. G.; Ho S. Y.; Chen K. Y.; Li E. Y.; Chou P. T. Sulfur-Based Intramolecular Hydrogen-Bond: Excited-State Hydrogen-Bond on/off Switch with Dual Room-Temperature Phosphorescence. J. Am. Chem. Soc. 2019, 141, 9885–9894. 10.1021/jacs.9b02765. [DOI] [PubMed] [Google Scholar]
- Ma L.; Sun S.; Ding B.; Ma X.; Tian H. Highly Efficient Room–Temperature Phosphorescence Based on Single–Benzene Structure Molecules and Photoactivated Luminescence with Afterglow. Adv. Funct. Mater. 2021, 31, 2010659 10.1002/adfm.202010659. [DOI] [Google Scholar]
- Nara M.; Orita R.; Ishige R.; Ando S. White-Light Emission and Tunable Luminescence Colors of Polyimide Copolymers Based on FRET and Room-Temperature Phosphorescence. ACS Omega 2020, 5, 14831–14841. 10.1021/acsomega.0c01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas H.; Fries F.; Gmelch M.; Barschneider T.; Kroll M.; Vavaleskou T.; Reineke S. Purely Organic Microparticles Showing Ultralong Room Temperature Phosphorescence. ACS Omega 2021, 6, 13087–13093. 10.1021/acsomega.1c00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T.; Ma X.; Wu H.; Zhu L.; Zhao Y.; Tian H. Molecular Engineering for Metal-Free Amorphous Materials with Room-Temperature Phosphorescence. Angew. Chem., Int. Ed. 2020, 59, 11206–11216. 10.1002/anie.201915433. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Gao L.; Zheng X.; Wang Z.; Yang C.; Tang H.; Qu L.; Li Y.; Zhao Y. Ultraviolet Irradiation-Responsive Dynamic Ultralong Organic Phosphorescence in Polymeric Systems. Nat. Commun. 2021, 12, 2297 10.1038/s41467-021-22609-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.; He Z.; Tang B. Z. Room-Temperature Phosphorescence from Organic Aggregates. Nat. Rev. Mater. 2020, 5, 869–885. 10.1038/s41578-020-0223-z. [DOI] [Google Scholar]
- Fan C.; Wu W.; Chruma J. J.; Zhao J.; Yang C. Enhanced Triplet–Triplet Energy Transfer and Upconversion Fluorescence through Host–Guest Complexation. J. Am. Chem. Soc. 2016, 138, 15405–15412. 10.1021/jacs.6b07946. [DOI] [PubMed] [Google Scholar]
- Kouno H.; Ogawa T.; Amemori S.; Mahato P.; Yanai N.; Kimizuka N. Triplet Energy Migration-Based Photon Upconversion by Amphiphilic Molecular Assemblies in Aerated Water. Chem. Sci. 2016, 7, 5224–5229. 10.1039/C6SC01047D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonat A.; Chan C. F.; Liu T.; Platas-Iglesias C.; Liu Z.; Wong W. T.; Wong W. K.; Wong K. L.; Charbonniere L. J. Room Temperature Molecular Upconversion in Solution. Nat. Commun. 2016, 7, 11978 10.1038/ncomms11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravetz B. D.; Pun A. B.; Churchill E. M.; Congreve D. N.; Rovis T.; Campos L. M. Photoredox Catalysis Using Infrared Light via Triplet Fusion Upconversion. Nature 2019, 565, 343–346. 10.1038/s41586-018-0835-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon Y. C.; Weder C. Low-Power Photon Upconversion through Triplet–Triplet Annihilation in Polymers. J. Mater. Chem. 2012, 22, 20817–20830. 10.1039/c2jm33654e. [DOI] [Google Scholar]
- Singh-Rachford T. N.; Castellano F. N. Photon Upconversion Based on Sensitized Triplet–Triplet Annihilation. Coord. Chem. Rev. 2010, 254, 2560–2573. 10.1016/j.ccr.2010.01.003. [DOI] [Google Scholar]
- Wang Z.; Hou Y.; Huo Z.; Liu Q.; Xu W.; Zhao J. Spatially Confined Photoexcitation with Triplet–Triplet Annihilation Upconversion. Chem. Commun. 2021, 57, 9044–9047. 10.1039/D1CC03309C. [DOI] [PubMed] [Google Scholar]
- Xiao X.; Tian W.; Imran M.; Cao H.; Zhao J. Controlling the Triplet States and Their Application in External Stimuli-Responsive Triplet–Triplet–Annihilation Photon Upconversion: From the Perspective of Excited State Photochemistry. Chem. Soc. Rev. 2021, 50, 9686–9714. 10.1039/D1CS00162K. [DOI] [PubMed] [Google Scholar]
- Yang D.; Han J.; Liu M.; Duan P. Photon Upconverted Circularly Polarized Luminescence via Triplet–Triplet Annihilation. Adv. Mater. 2019, 31, 1805683 10.1002/adma.201805683. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Zhao J.; Wu S.; Wang Z.; Wu W.; Ma J.; Guo S.; Huang L. Intramolecular RET Enhanced Visible Light-Absorbing Bodipy Organic Triplet Photosensitizers and Application in Photooxidation and Triplet–Triplet Annihilation Upconversion. J. Am. Chem. Soc. 2013, 135, 10566–10578. 10.1021/ja405170j. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Liu Z.; Li F. Upconversion Nanophosphors for Small-Animal Imaging. Chem. Soc. Rev. 2012, 41, 1323–1349. 10.1039/C1CS15187H. [DOI] [PubMed] [Google Scholar]
- Sun J.; Zhong F.; Yi X.; Zhao J. Efficient Enhancement of the Visible-Light Absorption of Cyclometalated Ir(III) Complexes Triplet Photosensitizers with Bodipy and Applications in Photooxidation and Triplet–Triplet Annihilation Upconversion. Inorg. Chem. 2013, 52, 6299–6310. 10.1021/ic302210b. [DOI] [PubMed] [Google Scholar]
- Ji S.; Guo H.; Wu W.; Wu W.; Zhao J. Ruthenium(II) Polyimine-Coumarin Dyad with Non-Emissive 3IL Excited State as Sensitizer for Triplet–Triplet Annihilation Based Upconversion. Angew. Chem., Int. Ed. 2011, 50, 8283–8286. 10.1002/anie.201008134. [DOI] [PubMed] [Google Scholar]
- Ji S.; Wu W.; Wu W.; Guo H.; Zhao J. Ruthenium(II) Polyimine Complexes with a Long-Lived 3IL Excited State or a 3MLCT/3IL Equilibrium: Efficient Triplet Sensitizers for Low-Power Upconversion. Angew. Chem., Int. Ed. 2011, 50, 1626–1629. 10.1002/anie.201006192. [DOI] [PubMed] [Google Scholar]
- Cui X.; Zhao J.; Lou Z.; Li S.; Wu H.; Han K. L. Switching of the Triplet Excited State of Rhodamine/Naphthaleneimide Dyads: An Experimental and Theoretical Study. J. Org. Chem. 2015, 80, 568–581. 10.1021/jo502554y. [DOI] [PubMed] [Google Scholar]
- Huang L.; Li Z.; Zhao Y.; Yang J.; Yang Y.; Pendharkar A. I.; Zhang Y.; Kelmar S.; Chen L.; Wu W.; Zhao J.; Han G. Enhancing Photodynamic Therapy through Resonance Energy Transfer Constructed Near-Infrared Photosensitized Nanoparticles. Adv. Mater. 2017, 29, 1604789 10.1002/adma.201604789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y.; Dick B.; Zhao J. Twisted Bodipy Derivative as a Heavy-Atom-Free Triplet Photosensitizer Showing Strong Absorption of Yellow Light, Intersystem Crossing, and a High-Energy Long-Lived Triplet State. Org. Lett. 2020, 22, 5535–5539. 10.1021/acs.orglett.0c01903. [DOI] [PubMed] [Google Scholar]
- Hou Y.; Liu Q.; Zhao J. An Exceptionally Long-Lived Triplet State of Red Light-Absorbing Compact Phenothiazine-StyrylBodipy Electron Donor/Acceptor Dyads: A Better Alternative to the Heavy Atom-Effect?. Chem. Commun. 2020, 56, 1721–1724. 10.1039/C9CC09058D. [DOI] [PubMed] [Google Scholar]
- Liu D.; El-Zohry A. M.; Taddei M.; Matt C.; Bussotti L.; Wang Z.; Zhao J.; Mohammed O. F.; Di Donato M.; Weber S. Long-Lived Charge-Transfer State Induced by Spin-Orbit Charge Transfer Intersystem Crossing (SOCT-ISC) in a Compact Spiro Electron Donor/Acceptor Dyad. Angew. Chem., Int. Ed. 2020, 59, 11591–11599. 10.1002/anie.202003560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Huang L.; Yan Y.; El-Zohry A. M.; Toffoletti A.; Zhao J.; Barbon A.; Dick B.; Mohammed O. F.; Han G. Elucidation of the Intersystem Crossing Mechanism in a Helical Bodipy for Low-Dose Photodynamic Therapy. Angew. Chem., Int. Ed. 2020, 59, 16114–16121. 10.1002/anie.202005269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Dai Y.; Hou Y.; Wu Q.; Ma L.; Zhao J.; Wang B. Weakened Triplet–Triplet Annihilation of Diiodo-Bodipy Moieties without Influence on Their Intrinsic Triplet Lifetimes in Diiodo-Bodipy-Functionalized Pillar[5]Arenes. J. Phys. Chem. A 2021, 125, 2344–2355. 10.1021/acs.jpca.1c01088. [DOI] [PubMed] [Google Scholar]
- Wu W.; Guo H.; Wu W.; Ji S.; Zhao J. Organic Triplet Sensitizer Library Derived from a Single Chromophore (Bodipy) with Long-Lived Triplet Excited State for Triplet–Triplet Annihilation Based Upconversion. J. Org. Chem. 2011, 76, 7056–7064. 10.1021/jo200990y. [DOI] [PubMed] [Google Scholar]
- Wang F.; Han C.; He C.; Zhou Q.; Zhang J.; Wang C.; Li N.; Huang F. Self-Sorting Organization of Two Heteroditopic Monomers to Supramolecular Alternating Copolymers. J. Am. Chem. Soc. 2008, 130, 11254–11255. 10.1021/ja8035465. [DOI] [PubMed] [Google Scholar]
- Ashton P. R.; Fyfe M. C. T.; Glink P. T.; Menzer S.; Stoddart J. F.; White A. J. P.; Williams D. J. Multiply Stranded and Multiply Encircled Pseudorotaxanes. J. Am. Chem. Soc. 1997, 119, 12514–12524. 10.1021/ja9714806. [DOI] [Google Scholar]
- Dong S.; Zheng B.; Xu D.; Yan X.; Zhang M.; Huang F. A Crown Ether Appended Super Gelator with Multiple Stimulus Responsiveness. Adv. Mater. 2012, 24, 3191–3195. 10.1002/adma.201200837. [DOI] [PubMed] [Google Scholar]
- Huang F.; Scherman O. A. Supramolecular Polymers. Chem. Soc. Rev. 2012, 41, 5879–5880. 10.1039/c2cs90071h. [DOI] [PubMed] [Google Scholar]
- Yan X.; Xu D.; Chi X.; Chen J.; Dong S.; Ding X.; Yu Y.; Huang F. A Multiresponsive, Shape-Persistent, and Elastic Supramolecular Polymer Network Gel Constructed by Orthogonal Self-Assembly. Adv. Mater. 2012, 24, 362–369. 10.1002/adma.201103220. [DOI] [PubMed] [Google Scholar]
- Li J.; Yim D.; Jang W. D.; Yoon J. Recent Progress in the Design and Applications of Fluorescence Probes Containing Crown Ethers. Chem. Soc. Rev. 2017, 46, 2437–2458. 10.1039/C6CS00619A. [DOI] [PubMed] [Google Scholar]
- Wu D.; Sedgwick A. C.; Gunnlaugsson T.; Akkaya E. U.; Yoon J.; James T. D. Fluorescent Chemosensors: The Past, Present and Future. Chem. Soc. Rev. 2017, 46, 7105–7123. 10.1039/C7CS00240H. [DOI] [PubMed] [Google Scholar]
- Weißenstein A.; Saha-Moller C. R.; Wurthner F. Optical Sensing of Aromatic Amino Acids and Dipeptides by a Crown-Ether-Functionalized Perylene Bisimide Fluorophore. Chem. – Eur. J. 2018, 24, 8009–8016. 10.1002/chem.201800870. [DOI] [PubMed] [Google Scholar]
- Erbas-Cakmak S.; Kolemen S.; Sedgwick A. C.; Gunnlaugsson T.; James T. D.; Yoon J.; Akkaya E. U. Molecular Logic Gates: The Past, Present and Future. Chem. Soc. Rev. 2018, 47, 2228–2248. 10.1039/C7CS00491E. [DOI] [PubMed] [Google Scholar]
- Zhu X.; Chen C. A Highly Efficient Approach to [4]Pseudocatenanes by Threefold Metathesis Reactions of a Triptycene-Based Tris[2]pseudorotaxane. J. Am. Chem. Soc. 2005, 127, 13158–13159. 10.1021/ja0546020. [DOI] [PubMed] [Google Scholar]
- Amano S.; Fielden S. D. P.; Leigh D. A. A Catalysis-Driven Artificial Molecular Pump. Nature 2021, 594, 529–534. 10.1038/s41586-021-03575-3. [DOI] [PubMed] [Google Scholar]
- Ma X.; Zhang J.; Cao J.; Yao X.; Cao T.; Gong Y.; Zhao C.; Tian H. A Room Temperature Phosphorescence Encoding [2]Rotaxane Molecular Shuttle. Chem. Sci. 2016, 7, 4582–4588. 10.1039/C6SC00769D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao M.; Zhang X. K.; Xu T. Y.; Wang X. D.; Rao S. J.; Liu Y.; Qu D. H.; Tian H. Towards a Hexa-Branched [7]Rotaxane from a [3]Rotaxane Via a [2+2+2] Alkyne Cyclotrimerization Process. Chem. Commun. 2019, 55, 3525–3528. 10.1039/C9CC00363K. [DOI] [PubMed] [Google Scholar]
- Rao S. J.; Zhang Q.; Mei J.; Ye X. H.; Gao C.; Wang Q. C.; Qu D. H.; Tian H. One-Pot Synthesis of Hetero[6]Rotaxane Bearing Three Different Kinds of Macrocycle through a Self-Sorting Process. Chem. Sci. 2017, 8, 6777–6783. 10.1039/C7SC03232C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C.; Fielden S. D. P.; Whitehead G. F. S.; Vitorica-Yrezabal I. J.; Leigh D. A. Weak Functional Group Interactions Revealed through Metal-Free Active Template Rotaxane Synthesis. Nat. Commun. 2020, 11, 744 10.1038/s41467-020-14576-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Wang Y.; Nie T.; Bao C.; Wang C.; Xu T.; Lin Q.; Qu D.; Gong X.; Yang Y.; Zhu L.; Tian H. An Artificial Molecular Shuttle Operates in Lipid Bilayers for Ion Transport. J. Am. Chem. Soc. 2018, 140, 17992–17998. 10.1021/jacs.8b09580. [DOI] [PubMed] [Google Scholar]
- Pedersen C. J. The Discovery of Crown Ethers (Noble Lecture). Angew. Chem., Int. Ed. 1988, 27, 1021–1027. 10.1002/anie.198810211. [DOI] [Google Scholar]
- Cram D. J. The Design of Molecular Hosts, Guests, and Their Complexes (Nobel Lecture). Angew. Chem., Int. Ed. 1988, 27, 1009–1112. 10.1002/anie.198810093. [DOI] [PubMed] [Google Scholar]
- Lehn J.-M. Supramolecular Chemistry–Scope and Perspectives Molecules, Supermolecules, and Molecular Devices (Nobel Lecture). Angew. Chem., Int. Ed. 1988, 27, 89–112. 10.1002/anie.198800891. [DOI] [Google Scholar]
- Ozlem S.; Akkaya E. U. Thinking Outside the Silicon Box Molecular and Logic as an Additional Layer of Selectivity in Singlet Oxygen Generation for Photodynamic Therapy. J. Am. Chem. Soc. 2009, 131, 48–49. 10.1021/ja808389t. [DOI] [PubMed] [Google Scholar]
- Kim H. J.; Kim S. H.; Kim J. H.; Lee E.-H.; Kim K.-W.; Kim J. S. BODIPY Appended Crown Ethers: Selective Fluorescence Changes for Hg2+ Binding. Bull. Korean Chem. Soc. 2008, 29, 1831–1834. 10.5012/bkcs.2008.29.9.1831. [DOI] [Google Scholar]
- Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. A 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Hariharan P. C.; Pople J. A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/BF00533485. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.. et al. Gaussian 09, revision D.01; Gaussian Inc.: Wallingford CT, 2013.
- Wang Z.; Sukhanov A. A.; Toffoletti A.; Sadiq F.; Zhao J.; Barbon A.; Voronkova V. K.; Dick B. Insights into the Efficient Intersystem Crossing of Bodipy-Anthracene Compact Dyads with Steady-State and Time-Resolved Optical/Magnetic Spectroscopies and Observation of the Delayed Fluorescence. J. Phys. Chem. C 2019, 123, 265–274. 10.1021/acs.jpcc.8b10835. [DOI] [Google Scholar]
- El-Khouly M. E.; Amin A. N.; Zandler M. E.; Fukuzumi S.; D’Souza F. Near-IR Excitation Transfer and Electron Transfer in a BF2-Chelated Dipyrromethane-Azadipyrromethane Dyad and Triad. Chem. – Eur. J. 2012, 18, 5239–5247. 10.1002/chem.201103074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.