

Abstract
Background/Aims:
Binding of histones to molecular pattern recognition receptors on endothelial cells and leukocytes provokes proinflammatory responses and promotes activation of coagulation. Histones also bind therapeutic heparins, thereby neutralizing their anticoagulant functions. The aim of this study was to test the hypothesis that histones can interact with the antithrombin (AT)-binding vascular glycosaminoglycans (GAGs) to induce inflammation and inhibit the anti-inflammatory function of AT.
Materials:
We evaluated the heparin-binding function of histones by an AT-dependent protease-inhibition assay. Furthermore, we treated endothelial cells with histones in the absence and presence of AT and monitored cellular phenotypes employing established signaling assays.
Results:
Histones neutralized AT-dependent anticoagulant function of heparin in both purified protease-inhibition and plasma-based assays. Histones also disrupted endothelial cell barrier-permeability function by a GAG-dependent mechanism as evidenced by the GAG-antagonist, surfen, abrogating their disruptive effects. Further studies revealed histones and AT compete for overlapping binding-sites on GAGs, thus increasing concentrations of one protein abrogated effects of the other. Histones elicited proapoptotic effects by inducing nuclear localization of PKC-δ in endothelial cells and barrier-disruptive effects by destabilizing VE-cadherin, which were inhibited by AT, but not by a D-helix mutant of AT incapable of interacting with GAGs. Finally, histones induced release of Weibel-Palade body contents, VWF and angiopoietin-2, and promoted expression of cell adhesion molecules on endothelial cells, which were all downregulated by AT but not by D-helix mutant of AT.
Conclusion:
We conclude that histones and AT compete for overlapping binding sites on vascular GAGs to modulate coagulation and inflammation.
Keywords: Histones, antithrombin, glycosaminoglycans, signaling
Introduction
Histones are chromatin-associated nuclear proteins that may be released into extracellular spaces by activated innate immune cells, damaged tissues and necrotic cells in response to bacterial endotoxin and/or trauma [1–4]. Activated neutrophils and macrophages can release their nuclear contents including DNA and histones as extracellular traps to bind and neutralize invading microorganisms [5–8]. The interaction of histones with damage-associated molecular pattern (DAMP) recognition receptors, including the receptor for advanced glycation end-products (RAGE) and toll-like receptors (TLRs), initiates potent cytotoxic and proinflammatory signaling responses in target cells [9, 10]. Binding of histones to DAMP receptors on endothelial cells is known to induce expression of adhesion molecules and stimulate the production of an array of proinflammatory cytokines that are involved in mediating leukocyte adherence, increased vascular permeability, coagulation activation and microvascular thrombosis [10–13]. Extracellular histone levels in plasma are elevated in animals and patients with infection, inflammation and cancer, suggesting pathogenic roles for these nuclear proteins in human diseases when they are released from the nucleus to the circulation [2–4]. A high concentration of histones in the plasma of patients with severe sepsis correlates with a poor prognosis and high mortality [2–4, 14]. Consistent with a pathogenic role for histones in severe sepsis, their pharmacological inhibition is associated with improved survival in experimental models of endotoxemia, while their infusion into mice can cause highly cytotoxic effects and death due to multiple organ failure [2–4].
In addition to their proinflammatory roles, recent results have indicated that histones can also promote activation of coagulation by inducing exocytosis of von Willebrand Factor (VWF) from Weibel-Palade bodies (WPB) by unknown mechanisms [15]. A recent study further showed that histones can promote thrombin generation by a platelet-dependent mechanism that involves their interaction with TLR2 and TLR4 on platelet surfaces [16]. Because of their basic nature, histones can also interact with heparin and neutralize the anticoagulant effects of therapeutic heparins [17]. However, the heparin-binding property of histones can have beneficial effect in eliminating the cytotoxicity of circulating nuclear proteins in inflammatory disorders [18]. Therapeutic heparins bind to D-helix of AT and function as cofactors to promote inhibition of coagulation proteases by the serpin [19, 20]. In addition to this anticoagulant function, binding of AT to glycosaminoglycans (GAGs) on vascular endothelial cells can elicit anti-inflammatory signaling in response to proinflammatory cytokines [21, 22]. The protective effect of AT through its D-helix-dependent interaction with vascular GAGs culminates in inhibition of NF-κB activation, downregulation of expression of vascular cell adhesion molecules and inhibition of barrier-disruptive effects of proinflammatory stimuli [21, 22]. Whether histones can interact with vascular GAGs to modulate the D-helix-dependent anti-inflammatory function of AT is not known. We addressed this question in this study by analyzing the signaling effect of histones in endothelial cells in the absence and presence of AT and a D-helix mutant of AT (AT-4Mut) that cannot interact with GAGs [23]. Results showed that histones compete with AT for binding on vascular GAGs, thereby eliciting a barrier-disruptive effect and inhibiting the anti-inflammatory signaling function of AT in endothelial cells. Because of the competitive nature of interaction, increasing concentrations of AT but not AT-4Mut inhibited the GAG-dependent proinflammatory effects of histones. The GAG antagonist, surfen, which is known to inhibit the anti-inflammatory function of AT, also inhibited the proinflammatory function of histones, supporting the hypothesis that AT and histones compete for overlapping binding sites on vascular GAGs. The GAG-dependent interaction of histones also induced exocytosis of WPB contents in endothelial cells which were counteracted by AT but not by AT-4Mut. In addition to GAGs, interaction with RAGE was also required for the signaling mechanism of histones.
Materials and Methods
Reagents
Human plasma-derived antithrombin (AT) was purchased from Enzyme Research Laboratories (South Bend, IN, USA). Histone H3 (#11034758001), H4 (#M2504S) and calf thymus histone (CTH#10223565001) were purchased from Roche (Indianapolis, IN, USA) and New England Biolabs (Ipswich, MA, USA). The plasma protein factor Xa (FXa) was purchased from Hematologic Technologies (Essex Junction, VT, USA). Expression and purification of soluble RAGE and the D-helix mutant of AT (AT-4Mut) have been described [13, 23]. Therapeutic unfractionated heparin (average MW ~15 kDa) and the AT-binding pentasaccharide, fondaparinux sodium (MW=1.728 kD), were purchased from Quintiles Clinical Supplies (Mt. Laurel, NJ, USA). The chromogenic substrates Spectrozymes FXa (SpFXa) was purchased from American Diagnostica Inc. (Stamford, CT, USA) and S2238 from Instrumentation Laboratory Company (Lexington, MA, USA). Normal pooled plasma was purchased from George King Bio-Medical, Inc. (Overland Park, KS, USA). The aPTT reagent (KONTACT) was from Thermo Scientific (Middletown, VA, USA). Human umbilical vein endothelial cells (HUVECs) and HUVEC immortalized with hTERT (immortalized HUVEC) were obtained from ATCC (Manassas, VA, USA). Anti-VE-cadherin (#2500S) and anti-β-actin (#4967) antibodies were from cell signaling technology (Beverly, MA, USA). Anti-phospho-VE-cadherin antibody (#441144), Alexa Flour 488-conjugated goat anti-rabbit IgG (#A11008), protease and phosphatase inhibitor cocktail were from Invitrogen (Carlsbad, CA, USA). Fluorescein isothiocyanate (FITC)-conjugated intercellular adhesion molecule-1 (ICAM-1) antibody (#353108) was purchased from Bio-Legend (San Diego, CA, USA). VWF (#DY2764) and angiopoietin-2 (Ang-2) (#DY623) ELISA kits, and anti-RAGE antibody (#AF1145) were from R&D systems (Minneapolis, MN, USA). Anti-PKC-δ antibody (#MA5–32482), Alexa Fluor 555-conjugated goat anti-rabbit IgG (#11011), and nuclear and cytoplasmic extraction (NE-PER #78833) kit were purchased from ThermoFisher (Waltham, MA, USA). TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay kit (#11684795910) was from Roche (Mannheim, Germany). Proliferating cell nuclear antigen (PCNA) (#sc-56) antibody was obtained from Santa Cruz Biotechnology (Dallas, Texas, USA). Endothelial (EA.hy926) cells stably overexpressing Protein kinase C delta (PKC-δ-OE) or its dominant negative form (PKC-δ-DN) were established as described previously [24].
Coagulation protease inhibition assay
The competitive effect of histones on heparin-mediated anticoagulant effect of AT (promotion of inhibition of FXa) was evaluated by a discontinuous inhibition assay method as described [25]. Briefly, FXa (1 nM) was incubated with 100 nM AT in the presence of 0–5 nM therapeutic heparin at room temperature in 0.1 M NaCl, 0.02 mM Tris-HCl, pH 7.4, and 5 mM Ca2+ (TBS/Ca2+) containing 0.1 mg/mL BSA and 0.1% PEG-8000. The cofactor function of heparin in accelerating the inhibition of FXa by AT was monitored in the absence and presence of increasing concentrations of histones (0–50 nM). The reactions were carried out in 50 μL volumes in 96-well plates for 1 to 2 min following which 50 μL chromogenic substrate for FXa (SpFXa, 200 μM) in TBS was added to each reaction and the remaining activity of FXa was measured at 405 nm using a Vmax Kinetic Plate Reader (Molecular Devices, Menlo Park, CA, USA). The experimental conditions were set up such that approximately 70–80% of the FXa activity in the absence of histones was inhibited.
Clotting assay
The competitive effect of histones on heparin-mediated prolongation of activated partial thromboplastin time (aPTT) in plasma was measured in the clotting assay using STart 4 fibrinometer (Diagnostica/Stago, Asnieres, France). Briefly, 50 µL citrated human plasma was mixed with 30 nM heparin with or without increasing concentrations of histones (H3 and CTH), followed by addition of 50 µL of the aPTT reagent for 5 min before initiating clotting by the addition of 50 µL CaCl2 (25 mM) at 37 °C as described [26].
Cell permeability assay
Cell permeability in response to histones in the presence and absence of AT was monitored by spectrophotometric analysis of the leakage of Evans blue-bound albumin across endothelial cell monolayer in a modified two-compartment chamber model. Briefly, hTERT-HUVECs (2 × 105 cells/well) were seeded on trans-well culture inserts (3.0 μm pore size) in complete growth medium and allowed to become confluent. Cells were then simultaneously treated with different concentrations of histones and AT (2.5 µM, AT-WT or the D-helix mutant AT-4Mut). In some experiments, cells were treated with histones in the presence of the GAG-antagonist, surfen (10 µM). After treatments, cells were washed with TBS to remove AT and permeability was induced by thrombin. The activity of added thrombin remained 100% as determined by an enzymatic assay in a control well. The permeability by histones alone were also examined by incubating cells with histones for 4h. In all experiments, cells were washed and Evans blue dye (0.67 mg/mL) and BSA (4%) was added to the upper chamber. Permeability was measured by collecting media from the lower chamber and measuring the absorbance of the leaked Evans blue dye at 650 nm as described [25]. Final values were plotted as fold change over untreated controls.
Western-blotting
Treated cells were lysed with the lysis buffer (Tris-pH-7.4, NaCl 150 mM) containing 1% Triton X-100, 5 mM EDTA and protease inhibitor cocktail. Lysates were boiled in the loading buffer with 5% β-mercaptoethanol and resolved on 8–10% SDS-PAGE. The resolved proteins were then electro-blotted onto a polyvinylidene difluoride (PVDF) membrane, blocked with 5% skim milk and incubated with primary antibodies including anti-phospho VE-cadherin, anti-total VE-cadherin and anti-β-actin. The immunoblotted PVDF membranes were then incubated with the respective horseradish peroxidase-conjugated secondary antibodies for 1h and bands were detected using enhanced chemiluminescence reagent. Protein bands were quantified using NIH Image J software.
Flow cytometry analysis
Endothelial cells were treated with histones (1 µM) in the presence and absence of AT (2.5 µM) and incubated at 37° C for 4h. Cells were washed and detached using HBSS containing 10 mM EDTA, washed and resuspended in HBSS containing 2 mM EDTA and 0.1% HSA. Cells were stained using FITC-conjugated anti-ICAM-1 antibody and surface expression of ICAM-1 was detected using FACS Celesta and data was analyzed by FlowJo software (BD Biosciences).
Immunofluorescence
Endothelial cells were treated with histones (1 µM) for 1h or 4h both in the presence and absence of AT-WT and its D-helix variant AT-4Mut (2.5 µM each). Following treatments, cells were fixed in 4% paraformaldehyde and permeabilized in 0.2% Triton X-100/PBS, followed by blocking for 1h with normal goat serum. Cells were then incubated with anti-VE-cadherin or anti-PKC-δ antibodies overnight at 4° C, followed by Alexa Fluor 488-conjugated goat anti-rabbit IgG (for VE-Cadherin) or Alexa Fluor 555-conjugated goat anti-rabbit IgG (for PKC-δ). Cells were then washed and the nucleus was stained with DAPI. Photomicrographs were captured using a Nikon C2 Confocal Microscope.
Nuclear Translocation and confocal microscopy
HTERT-HUVECs were cultured on glass coverslips. Cells were treated with histone H3, H4 and CTH for 1h and TNFα was used as a positive control. After treatment, cells were washed with PBS, fixed with 2% paraformaldehyde, permeabilized with 1% Triton-X and blocked with 2% BSA in PBS. To analyze the PKC-δ localization, cells were incubated with anti-PKC-δ rabbit monoclonal antibody in 4°C for overnight followed by staining with goat anti-rabbit Alexa Fluor 568-conjugated secondary antibody and counterstained with DAPI. Images were obtained with a Nikon C2 Confocal Microscope (Melville, NY, USA
Nuclear fraction
Nuclear fractions were prepared according to the protocol described in the NE-PER nuclear and cytoplasmic extraction kit. Briefly, cells were treated with histones (1 µM) for 1h in the absence or presence of RAGE blocking antibody. Following treatments, cells were washed with PBS, nuclear extracts were prepared and analyzed by Western-blot.
TUNEL assay
Cell death were analyzed with TUNEL assay kit according to the protocol provided by the manufacturer. Briefly, wild type, PKC-δ-OE and PKC-δ-DN cells were treated with histones for 16h under serum free conditions. After treatment, cells were washed and processed for TUNEL assay.
Statistical analysis
Data are presented as mean ± SD from ≥3 independent experiments. Data were analyzed by the Student t-test, and group data were analyzed using ANOVA followed by Bonferroni post hoc test using GraphPad Prism 7 (GraphPad, San Diego, CA). A p value of <0.05 was considered statistically significant.
Results
Histones inhibit heparin-dependent anticoagulant and GAG-dependent anti-inflammatory functions of AT
Previous results have indicated that histones can bind and neutralize the anticoagulant function of therapeutic heparins [17, 18]. Here, we tested the hypothesis that upon interaction with heparin, histones can inhibit the cofactor function of heparin in promotion of coagulation proteases by AT. The coagulation protease factor Xa (FXa) was chosen as prototype since it is sensitive to inhibition by both heparin-mediated conformational activation of AT and its template mechanism [27]. Results showed that histone H3 (Fig. 1A), histone H4 (Fig. 1B) and calf thymus histone (CTH) (Fig. 1C) effectively compete with AT for interaction with heparin, as evidenced by their ability to inhibit the cofactor function of heparin in promoting the AT-mediated inhibition of FXa. The apparent IC-50 for the histone-mediated neutralization of the heparin cofactor function was ~5–10 nM. Histones did not have any effect on the chromogenic substrate activity of FXa in this assay. The ability of histones to inhibit the cofactor function of heparin was also examined in an aPTT assay. Results showed that histones dose-dependently inhibit the cofactor function of heparin in prolonging plasma-clotting time in the aPTT assay (Fig. 1D, E). These results suggest that histones bind to heparin and blocks its interaction with D-helix of AT. Histones alone had no effect on AT function.
Fig. 1-.
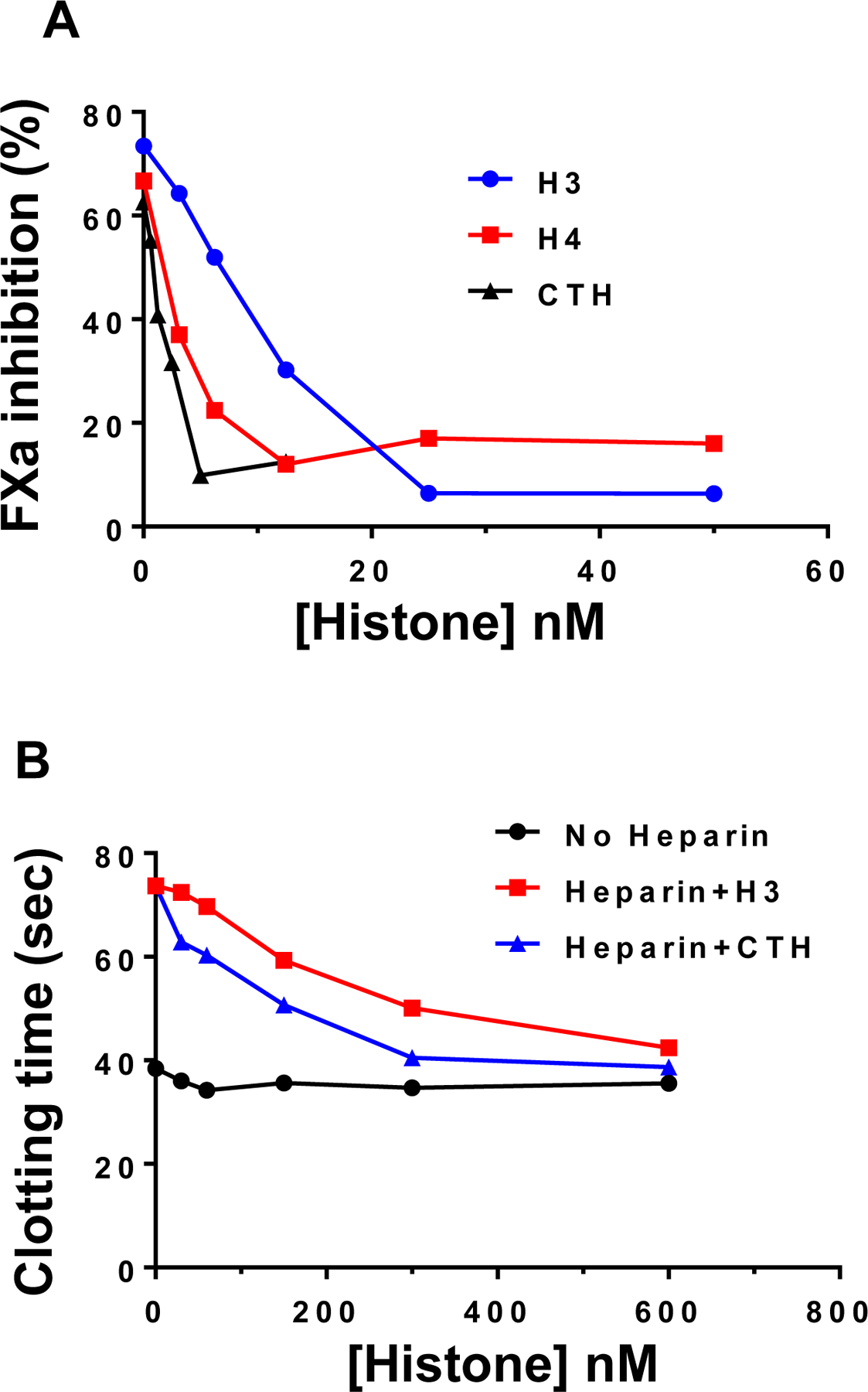
Competitive effect of histones on inhibiting the cofactor function of heparin. (A) Competitive effect of increasing concentrations of histones on inhibiting the cofactor function of heparin in accelerating the AT inhibition of factor Xa (FXa) was monitored as described in methods. (B) The competitive effect of increasing concentrations of histone H3 and CTH on neutralization of the anticoagulant effect of heparin was monitored in an aPTT assay using citrated normal plasma as described in methods.
The ability of histones to neutralize the cofactor function of heparin in promoting the AT inhibition of FXa suggests that histones compete with the basic D-helix of the AT for interaction with heparin. D-helix of AT also interacts with endothelial cell surface GAGs to elicit cytoprotective signaling responses [21]. We used an endothelial cell permeability assay to determine whether histones can interact with the AT-binding vascular GAGs to inhibit the intracellular signaling function of AT. Results indicated that increasing concentrations of histone H3 inhibit the barrier-protective function of AT in response to thrombin in endothelial cells (Fig. 2A). Histone H3 induced a barrier-disruptive effect in endothelial cells, and as expected from the nature of competitive binding interactions, increasing concentrations of AT inhibited the barrier-disruptive effect of histone H3 in this assay (Fig. 2B). These results suggest that histones and AT utilize overlapping binding sites on GAGs to elicit barrier-disruptive and barrier-protective signaling responses, respectively. To confirm that the proinflammatory effect of histone is mediated through interaction with GAGs, the cell permeability assay with histone H3 was conducted in the presence of the GAG antagonist surfen. Results indicated that surfen effectively inhibits histone-mediated hyper-permeability in endothelial cells (Fig. 2C). Further support for the hypothesis that AT and histones bind to overlapping sites on GAGs is provided by the observation that, unlike wild-type AT (WT-AT), the D-helix mutant of AT (AT-4Mut) which exhibits no affinity for GAGs [23], exhibited no protective effect against barrier-disruptive function of histones in endothelial cells (Fig. 2D-F).
Fig. 2-.
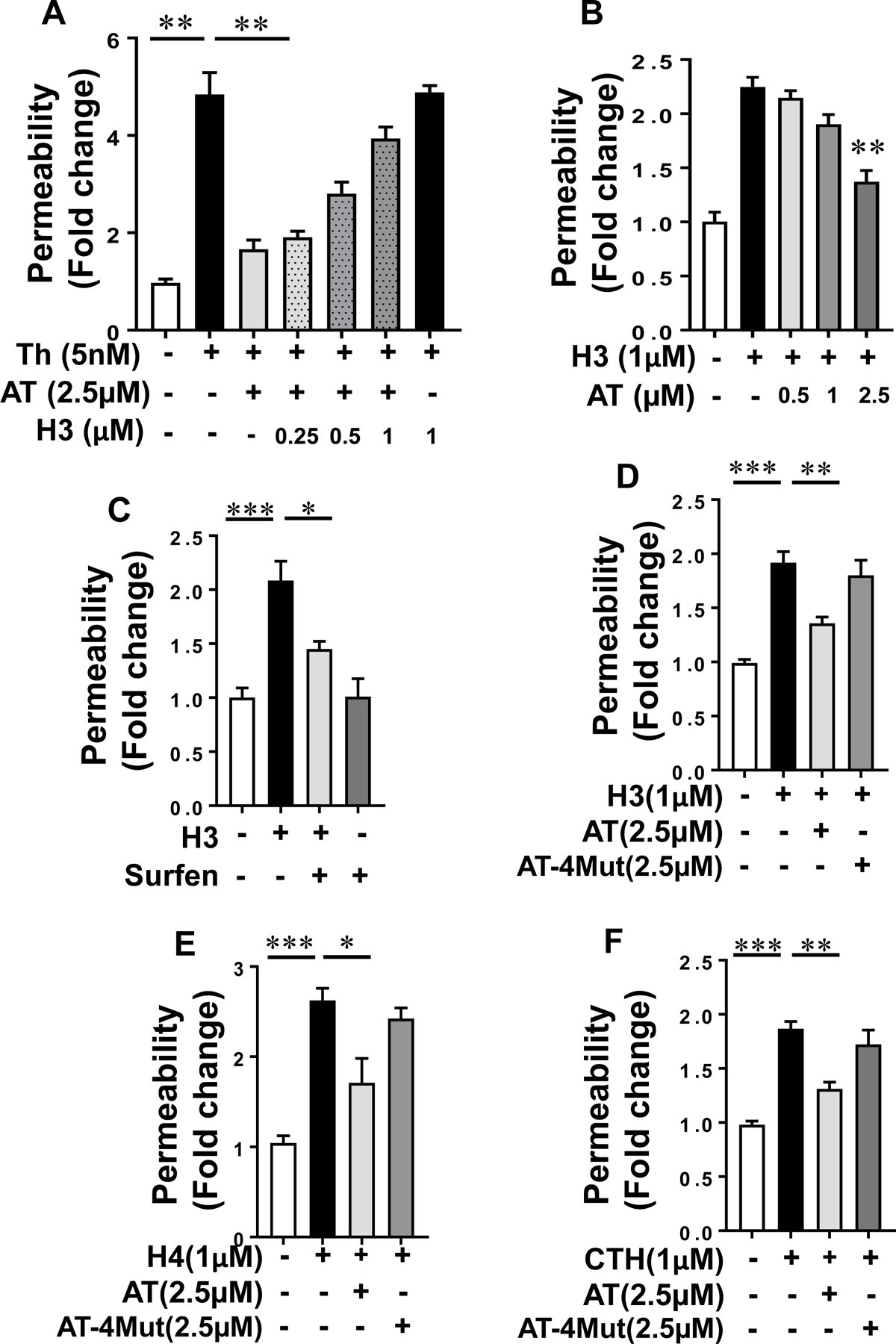
Histone H3 inhibits the endothelial barrier protective function of AT. (A) Confluent HTERT-HUVECs were simultaneously incubated with a fixed concentration of AT (2.5 µM) and increasing concentration of histone H3 for 4h followed by measuring cell permeability in response to thrombin by spectrophotometric measurement of the flux of Evans blue-bound albumin across functional endothelial cell monolayer as described in methods. (B) The same as (A) except that the fold change in cell permeability was measured by simultaneous incubation of a fixed concentration of histone H3 with increasing concentration of AT. (C) The cell permeability induced by histone H3 (1 µM for 4h) was monitored in the absence and presence of the GAG-antagonist surfen (10 µM). (D) Protective effect of WT-AT and its D-helix mutant (AT-4Mut) on histone H3-mediated endothelial cell permeability was measured by influx of albumin-bound Evans blue across functional endothelial cell monolayer. (E) The same as (D) except that the effect of WT-AT and AT-4Mut on histone H4-mediated permeability was monitored. (F) The same as (D) except that the effect of WT-AT and AT-4Mut on calf thymus histone (CTH)-mediated cell permeability was monitored. Neither AT-WT nor AT-4Mut had an effect on cell permeability in the absence of histones (not shown). All results are shown as means ± SD of three different experiments. * p < 0.05, ** p < 0.01, *** p < 0.001.
Histone binding to vascular GAGs disrupts VE-cadherin
The mechanism by which histones exert a GAG-dependent barrier-disruptive effect was investigated by analysis of the phosphorylation state of the endothelial cells junctional protein, VE-cadherin. Results indicated that histones induce VE-cadherin phosphorylation in endothelial cells and a physiological concentration of WT-AT, but not AT-4Mut, inhibits this effect (Fig. 3A, B). The phosphorylation of VE-cadherin by histone H3 required its interaction with GAGs since surfen inhibited phosphorylation of VE-cadherin by histone H3 (Fig. 3C, D). Immunofluorescence analysis also suggested that histone-mediated phosphorylation of VE-cadherin is associated with disruption of the junctional integrity of endothelial cells (Fig. 4A). The disruptive effect of histone H3 was inhibited by simultaneous treatment with WT-AT but not AT-4Mut, suggesting that D-helix of AT and histone H3 are binding to overlapping sites on endothelial cell surface GAGs (Fig. 4A). Histone-mediated destabilization of VE-cadherin was also attenuated by surfen (Fig. 4B), providing further support for the hypothesis that interaction with GAGs plays a key role in the barrier-disruptive function of histones in endothelial cells.
Fig. 3-.
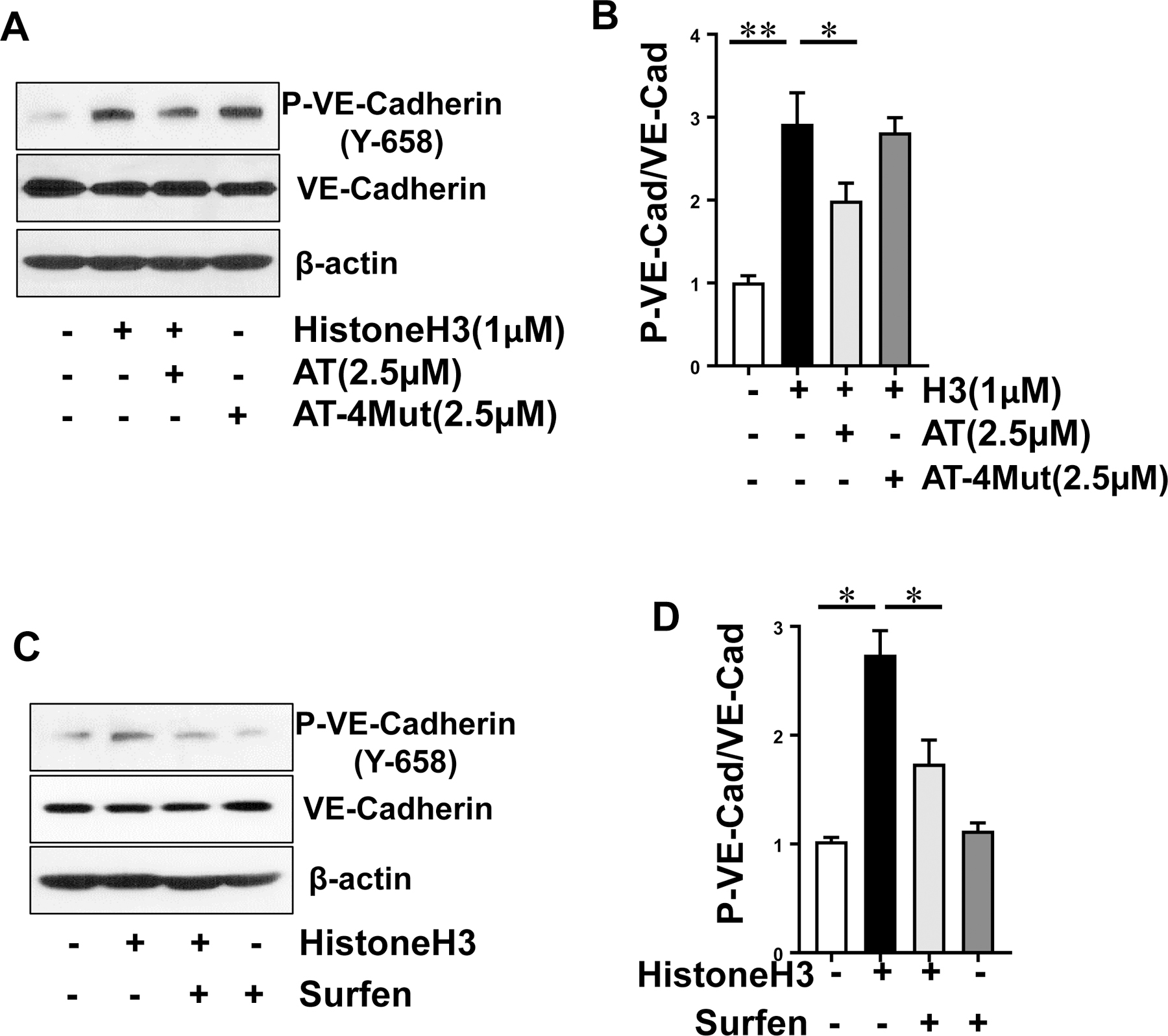
Analysis of VE-cadherin phosphorylation by histone H3 in the absence and presence of AT. (A) Confluent HTERT-HUVECs were incubated with histone H3 for 15 min in the absence or presence of WT-AT and AT-4Mut and phosphorylated (Y-658) VE-cadherin, total VE-cadherin and β-actin levels in cell lysates were measured by Western blot. Data is representative of three independent repeats. (B) Densitometric analysis of the panel A Western blot. (C) The same as (A) except that histone H3-mediated phosphorylation of VE-cadherin was monitored in the absence or presence of surfen (10 µM). Data is representative of three independent repeats. (D) Densitometric analysis of the panel C Western blot. All results are shown as means ± SD of three different experiments. * p < 0.05, ** p < 0.01.
Fig. 4-.
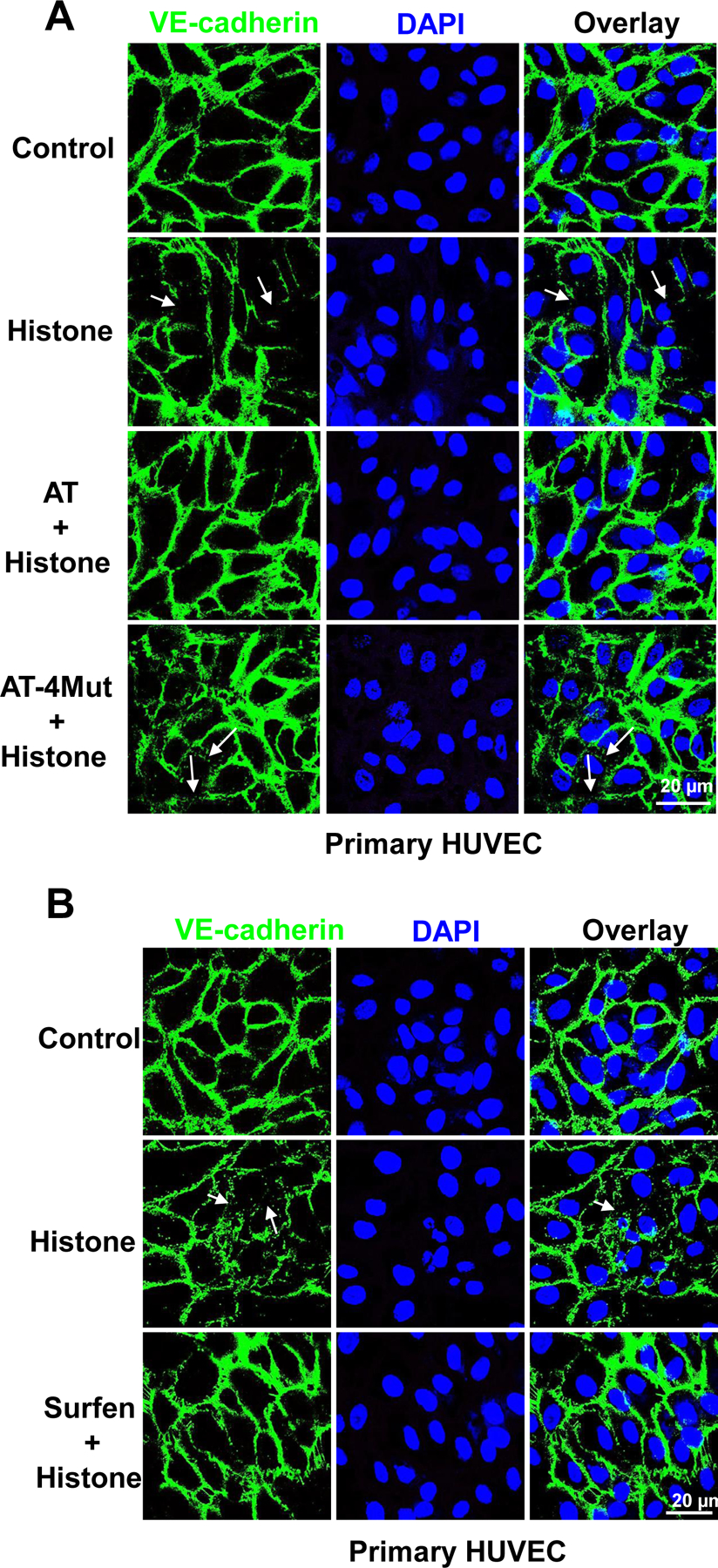
Immunofluorescence analysis of histone-mediated VE-cadherin disruption in endothelial cells. (A) Primary HUVECs were incubated with Histone H3 (1 µM) for 1h in the absence or presence of WT-AT and AT-4Mut (2.5 µM each). Cells were then fixed, permeabilized and incubated with rabbit anti-VE-cadherin antibody and Alexa Fluor 488-conjugated goat anti-rabbit IgG. The nucleus was stained with DAPI. Immunofluorescence images were obtained with confocal microscopy. (B) The same as (A) except that primary HUVECs were incubated with histone H3 for 1h in the absence or presence of surfen (10 µM). Arrows indicate loss of VE-cadherin at junctions.
Histone-mediated endothelial cell dysfunction is inhibited by AT
In addition to their proinflammatory functions, it has been demonstrated that extracellular histones induce exocytosis of VWF from Weibel-Palade bodies (WPB), thereby promoting the activation of coagulation and interaction of platelets with vascular endothelial cells [15]. Histone-mediated VWF release in endothelial cell supernatants was measured for both CTH and histone H3 by ELISA. Results showed that histone concentrations of as low as 1 µM can induce VWF release from endothelial cells (Fig. 5A). Simultaneous treatment of cells with WT-AT, but not with the D-helix mutant of AT, was associated with significant inhibition of histone-mediated VWF release (Fig. 5B, C). The VWF releasing effect of histone was mediated through interaction with GAGs since it was significantly inhibited by surfen (Fig. 5D). Ang-2 is another signaling molecule that is known to be stored in WPBs. To provide further support for the hypothesis that AT attenuates histone-mediated WPB exocytosis, Ang-2 release was also measured in supernatants of cells treated with both histone H3 and AT. Results showed that H3-mediated Ang-2 release was significantly inhibited by AT (Fig. 5E). Flow cytometry analysis indicated that histones also induce the expression of ICAM-1 on endothelial cells, which is significantly downregulated by AT (Fig. 5F, G).
Fig. 5-.
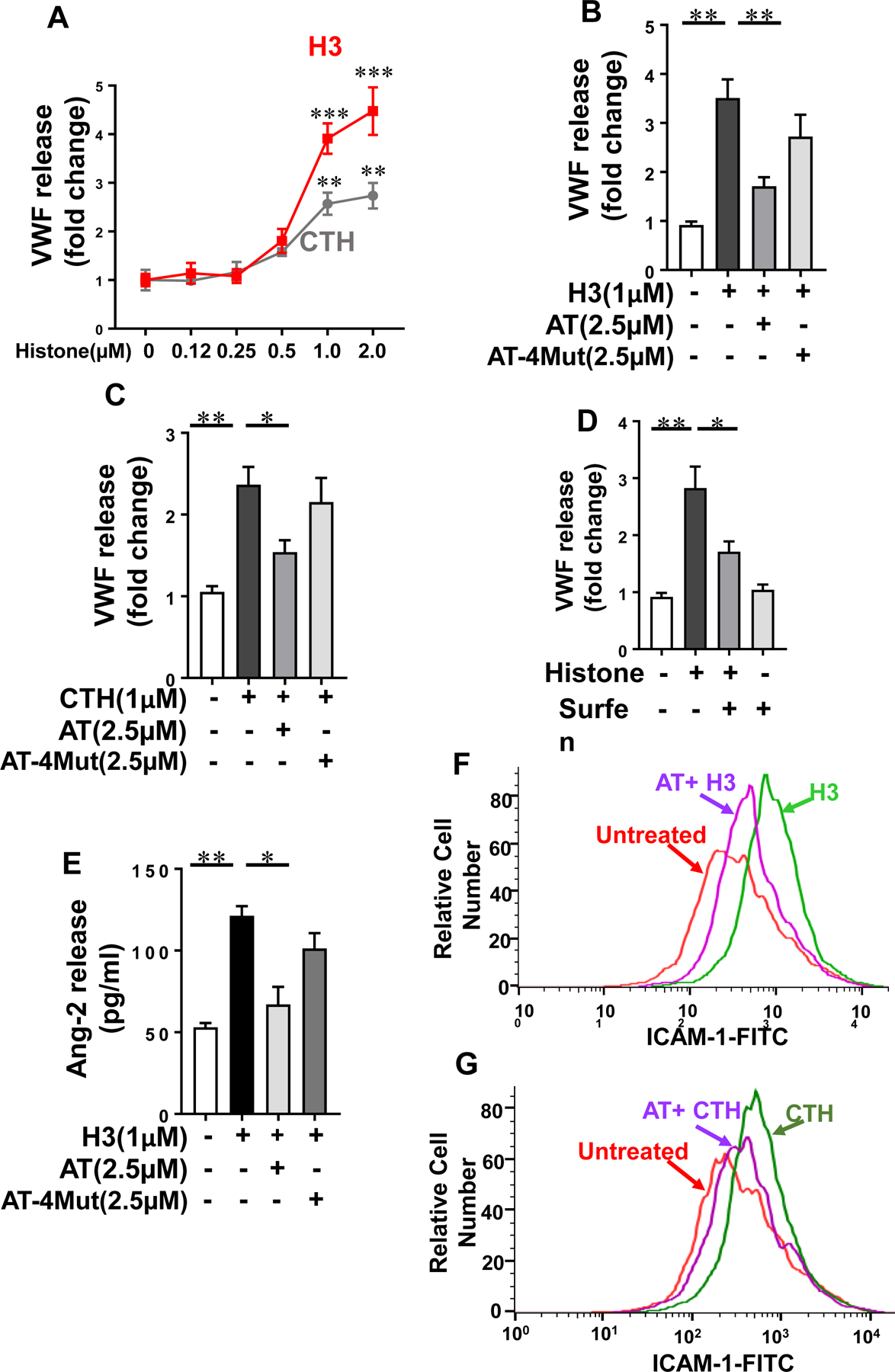
Histone-mediated WPB exocytosis and ICAM-1 expression in endothelial cells. (A) HTERT-HUVECs were incubated with increasing concentrations of histone H3 (red) or calf thymus histone (CTH, grey) for 4h and VWF release was measured by a sandwich ELISA. (B) The same as (A) except that cells were incubated with histone H3 for 4h in the absence or presence of WT-AT or AT-4Mut and VWF release was measured by a sandwich ELISA. (C) The same as (B) except that cells were incubated with calf thymus histone (CTH). (D) The same as (B) except that cells were incubated with histone H3 in the absence or presence of surfen and VWF release was measured by a sandwich ELISA. (E) The same as (B) except that cells were incubated with histone H3 in the absence or presence of WT-AT or AT-4Mut and Ang-2 release was measured by a sandwich ELISA. (F) HTERT-HUVECs were incubated with histone H3 (1 µM) for 4h in the absence or presence of WT-AT (2.5 µM) followed by measuring expression of surface level of ICAM-1 by flow cytometry. The data is representative of three independent experiments. (G) The same as (F) except that CTH-mediated expression of ICAM-1 in the presence of WT-AT was measured by flow cytometry.
Histones promote nuclear localization of PKC-δ
Extracellular histones exert their proinflammatory function largely through interaction with pattern recognition receptors (PRR) that culminates in coordinated activation of intracellular signaling pathways that promote inflammatory responses. RAGE has been identified as a critical PRR involved in transmitting signaling effects of histones. However, in light of the observation that AT and histones bind to overlapping sites on GAGs and AT can directly signal through interaction with GAGs by subcellular localization of PKC-δ [24], the possibility that histones can also signal via GAGs was investigated. Immunofluorescence analysis indicated that histones promote the nuclear localization of PKC-δ in endothelial cells (Fig. 6A). In support of the immunofluorescence data, Western-blot analysis of nuclear fractions derived from cells treated with histones also indicated a higher amount of PKC-δ localization in the nucleus (Fig. 6B). TNFα is known to induce a proapoptotic effect by nuclear localization of PKC-δ in endothelial cells [24, 28], thus it was used as a positive control in these experiments (Fig. 6B). The nuclear localization of PKC-δ by histones appears to play an important role in their cytotoxic and proapoptotic effects since in the TUNEL assay, histones (H3 and H4) caused cell death in endothelial cells and cells overexpressing PKC-δ (δ-OE), but the effect was significantly downregulated in PKC-δ dominant negative (δ-DN) cells (Fig. 6C, shown for histone H3 only). To determine whether nuclear localization of PKC-δ is a direct effect of histones interacting with GAGs, endothelial cells were treated with histone H3 in the absence and presence of an anti-RAGE antibody followed by analysis of the nuclear fraction of PKC-δ by Western-blotting. The anti-RAGE antibody significantly inhibited nuclear localization of PKC-δ in endothelial cells (Fig. 6D). Moreover, the ability of soluble RAGE (sRAGE) to inhibit the proinflammatory signaling effects of histone H3 was also investigated as described [13]. Results showed that a 2-fold molar excess of sRAGE significantly inhibits histone-mediated barrier-disruption (Fig. 6E) and VWF release (Fig. 6F) in endothelial cells, suggesting that in addition to GAGs, interaction with the RAGE receptor is required for the signaling mechanism of histones.
Fig. 6-.
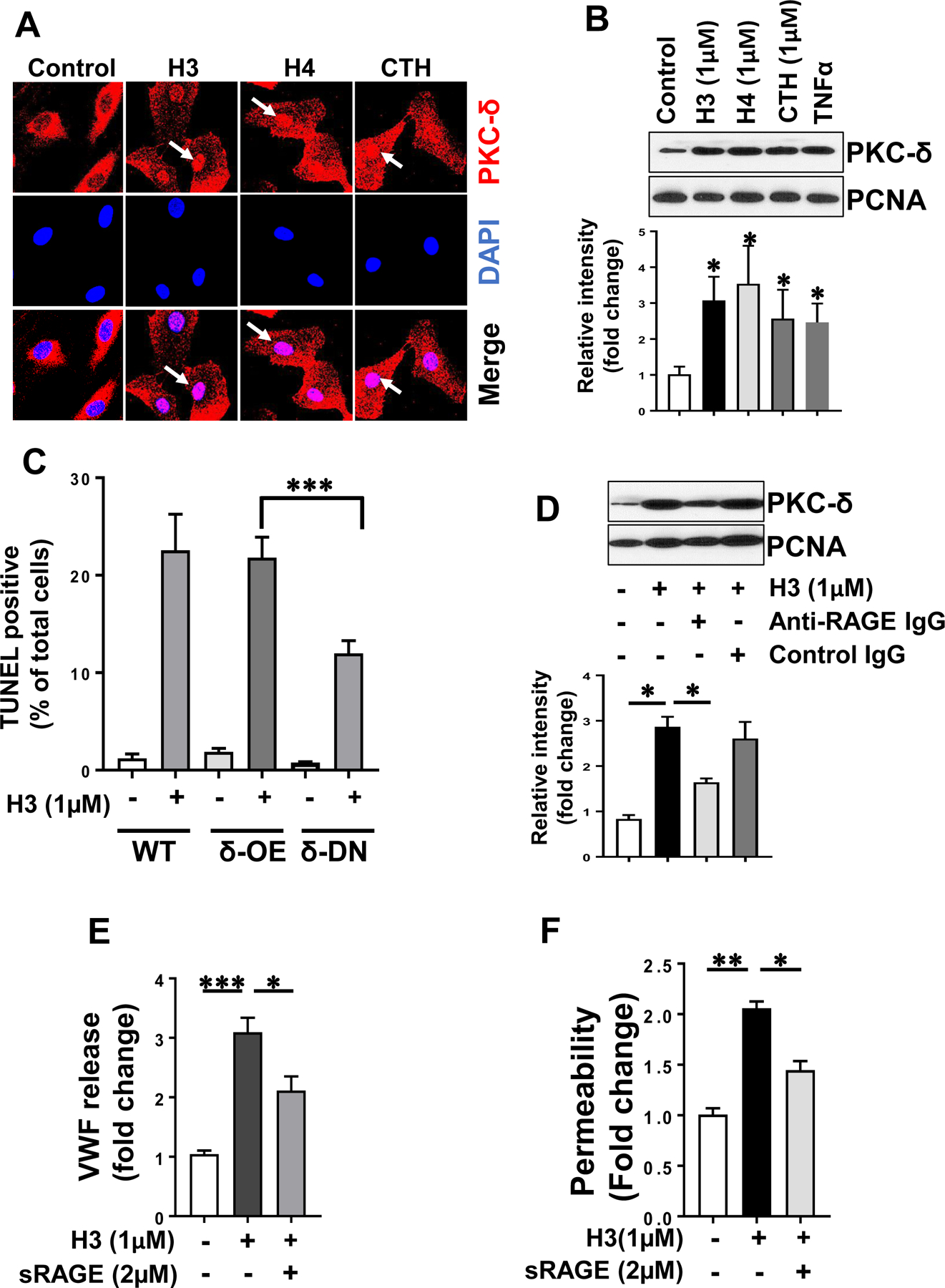
RAGE signaling is required for histone-mediated nuclear localization of PKC-δ in endothelial cells. (A) HTERT-HUVECs were treated with histones (H3, H4, CTH) for 1h and PKC-δ nuclear localization was analyzed by confocal microscopy. PKC-δ was stained with rabbit monoclonal antibody followed by Alexa Fluor 555-conjugated goat anti-rabbit IgG. DAPI was used to stain the nucleus. (B) The same as (A) except that nuclear PKC-δ in histone-treated cells was monitored through by Western-blotting. TNFα was used as a positive control and PCNA was used as a loading control. Densitometric analysis of the data in the Western-blot is shown below the panel. (C) Wild-type (WT), PKC-δ overexpressing (PKC-δ-OE) and PKC-δ dominant negative (PKC-δ-DN) endothelial (EA.hy926) cells were treated with histones for 16h and cell death was analyzed by a TUNEL assay. TUNEL-positive cells were counted using Nikon C2 Confocal Microscope for at least 10 randomly selected fields from 3 independent samples. (D) HTERT-HUVECs were incubated with histone H3 (1 µM) for 1h in the absence and presence of anti-RAGE antibody (20µg/ml) followed by the analysis of PKC-δ in the nuclear fractions by Western-blotting. Densitometric analysis of the Western-blotting data is shown below the panel. (E) Confluent HTERT-HUVECs were incubated with histone H3 (1 µM) for 4h in the absence and presence of sRAGE (2 µM) followed by measuring the cell permeability by spectrophotometric measurement of the flux of Evans blue-bound albumin across functional endothelial cell monolayer. (F) The same as (E) except that histone H3-mediated VWF release from endothelial cells was measured by an ELISA. Results in all panels are shown as means ± SD of three different experiments. * p < 0.05, ** p < 0.01, *** p < 0.001.
Discussion
Extracellular histones bind pattern recognition receptors (PRR) to promote inflammation and coagulation. In this study, we demonstrate that interaction of histones with GAGs plays a key role in the signaling mechanism of histones in endothelial cells. We discovered that histones bind to overlapping AT-binding sites on GAGs not only to competitively inhibit the anti-inflammatory signaling function of AT, but also induce barrier-disruptive and proapoptotic effects and exocytosis of WPB contents in cultured endothelial cells. The GAG antagonist, surfen, effectively inhibited both barrier-disruptive and VWF-releasing effects of histones, supporting the GAG-dependent proinflammatory signaling function of histones. Histones induced phosphorylation of VE-cadherin in endothelial cells, thereby destabilizing intercellular junctions. Simultaneous incubation of histones with WT-AT, but not with AT-4Mut, inhibited the proinflammatory signaling function of histones, supporting the hypothesis that histones and AT interact with overlapping binding sites on endothelial cell GAGs to transmit their signaling effects.
We recently demonstrated that AT transduces its anti-inflammatory signaling effect by binding GAGs on syndecans 4, thereby recruiting PKC-δ to the plasma membrane and catalyzing phosphorylation of the cytoplasmic domain of the receptor at Ser179, which culminates in induction of prostacyclin synthesis by endothelial cells [24]. However, unlike WT-AT, protease-cleaved and latent conformers of AT, which have much lower affinity for GAGs, exhibited potent proapoptotic activities by also binding to GAGs, but instead of recruiting PKC-δ to the cytoplasmic membrane, they enhanced the perinuclear/nuclear localization of PKC-δ, thereby activating a proapoptotic pathway [24]. Thus, the possibility that, similar to cleaved and latent AT, histones through interaction with GAGs directly transduce signals by subcellular localization of PKC-δ was investigated. Interestingly, analysis of the nuclear extracts of endothelial cells suggested that histone H3 markedly increases the nuclear localization of PKC-δ in endothelial cells. However, further analysis in the presence of an anti-RAGE antibody revealed that the histone-mediated nuclear localization of PKC-δ might not be directly induced by the binding of the histone to GAGs but rather through interaction and signaling via RAGE since a function-blocking anti-RAGE antibody significantly inhibited histone-mediated nuclear localization of PKC-δ. TNFα also induces a proapoptotic effect through nuclear localization of PKC-δ [24, 28]. Histones appear to share this property of TNFα and exert a cytotoxic effect by a similar mechanism through interaction with GAGs and RAGE.
In addition to their proinflammatory signaling functions, extracellular histones promote thrombosis by activation of coagulation and release of VWF from WPBs of endothelial cells [4, 15]. The observation of the current study that surfen inhibited histone-mediated VWF release from endothelial cells suggests that interaction with vascular GAGs plays a key role in the procoagulant function of extracellular histones. AT binds to distinct vascular GAGs via its D-helix to exert its anti-inflammatory signaling effects in endothelial cells [21]. Further support for the hypothesis that histones induce proinflammatory and procoagulant responses through interaction with vascular GAGs was provided by the findings that WT-AT, but not AT-4Mut with no affinity for GAGs, inhibited both barrier-disruptive and VWF-releasing functions of histones by a competitive binding mechanism. Histones promote inflammation and coagulation by primarily binding to pattern recognition receptors TLR-2, TLR-4 and RAGE on endothelial cells and platelets [9, 13, 16]. However, a previous study proposing an immunothrombosis role for histones [15], did not find a role for either TLR2 or TLR4 in promoting the procoagulant function of histones since the inhibitory antibodies against either one of these receptors had no effect on histone-mediated exocytosis of the WPB contents. On the other hand, in this study we discovered that, similar to the GAG antagonist surfen, sRAGE significantly downregulated both the barrier-disruptive and VWF-releasing functions of histones in endothelial cells, suggesting that interactions with both GAG and RAGE are essential for the proinflammatory and procoagulant signaling mechanism of histones. A recent study investigating the mechanism of RAGE signaling demonstrated that heparan sulfate-mediated oligomerization of RAGE is required for the receptor to transduce signals across the cell membrane [29]. Thus, this previous study discovered that binding of several RAGE ligands including HMGB1, S100A8/A9 and S100A12 to heparan sulfates is essential for RAGE signaling in endothelial cells [29]. These ligands and heparan sulfate were found to bind on separate surfaces on RAGE to exert their signaling functions [29]. In the context of these previous results and our findings in this study, we envision a hypothetical model for the histone signaling in which histone-loaded vascular GAGs by binding to specific sites on RAGE not only facilitate activation and oligomerization of the receptor, but also concentrate and present histones to the receptor, thereby amplifying the signaling function of the nuclear protein (Fig. 7). The binding of AT to GAGs not only exerts a cytoprotective effect against the proinflammatory functions of histones, but also competitively inhibits the interaction of histones with vascular GAGs.
Fig. 7-.
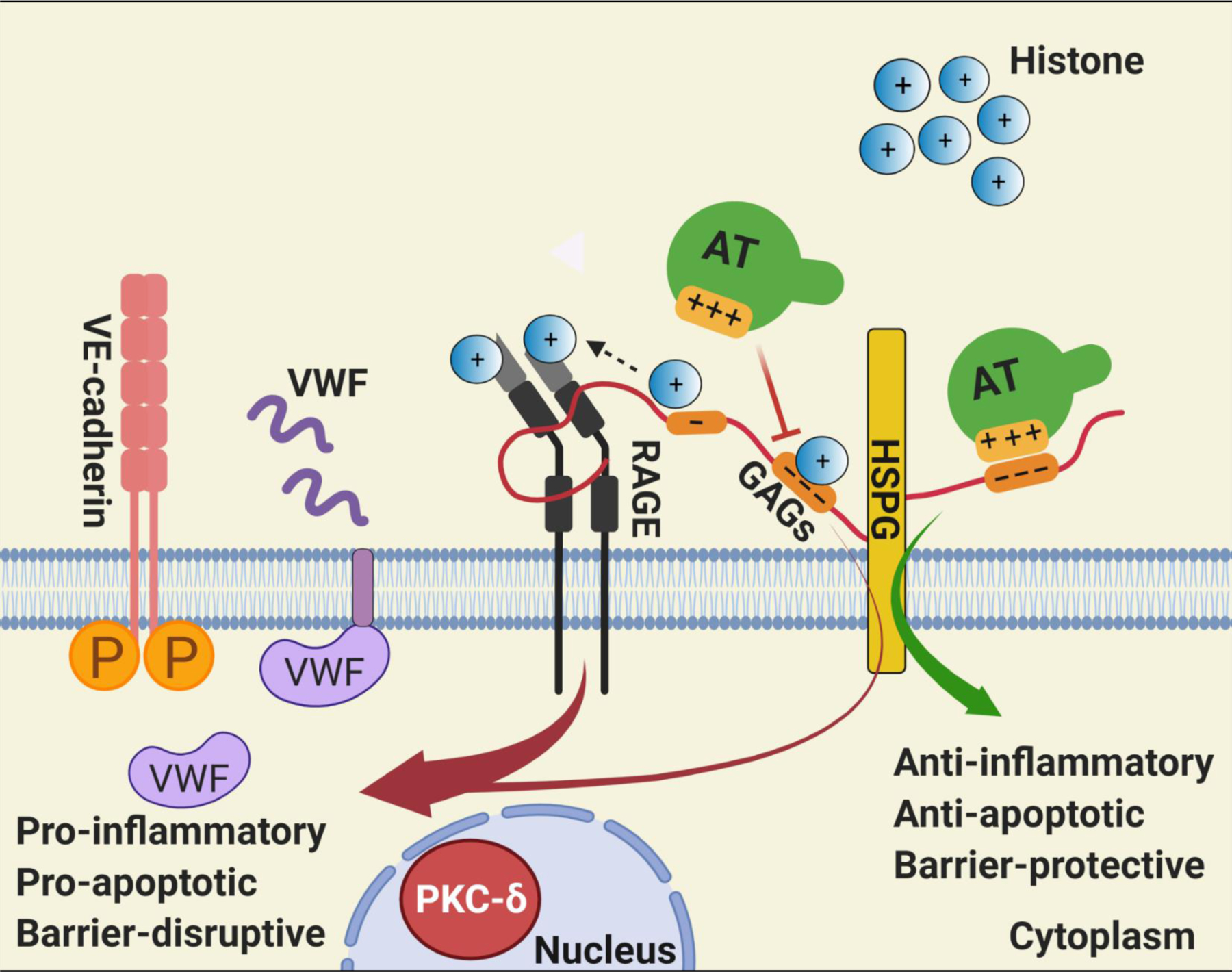
Hypothetical model of GAG-dependent AT and histone signaling in endothelial cells. Binding of positively charged residues of D-helix of AT on distinct GAGs covalently linked to heparan sulfate proteoglycans (HSPG) culminates in anti-inflammatory, anti-apoptotic and barrier-protective effects. Histones can bind to the same and/or overlapping AT-binding sites on GAGs, thereby excluding AT from binding GAGs. GAGs are known to bind RAGE, activate the receptor by oligomerization and also present histones to RAGE [29], thereby promoting the pro-inflammatory, pro-apoptotic and barrier-disruptive signaling functions of nuclear proteins. The GAG-dependent RAGE signaling by histones induces VWF release from endothelial cells. The pro-apoptotic activity of histones is associated with RAGE-dependent nuclear localization of PKC-δ in endothelial cells. The GAG-dependent histone signaling through RAGE also leads to phosphorylation of VE-cadherin and destabilization of endothelial cell junctions. See the text for further details. Figure was prepared by software provided by Biorender.com.
Plasma levels of histones are elevated in acute and chronic inflammatory disorders like sepsis, trauma and cancer. In inflammatory disorders, the activation of coagulation can decrease the plasma level of AT due to consumption and/or its proteolytic cleavage by the neutrophil elastase. Based on findings of this study, it is expected that acquired or genetic deficiency of AT will have a negative impact on the course of the inflammatory diseases in which histones play a pathogenic role. Thus, the beneficial effect of AT supplementation, in particular AT variants with higher affinity for GAGs, in inflammatory disorders warrants further investigation.
Acknowledgements
We would like to thank Cindy Carter for technical assistance and Audrey Rezaie for editorial work on the manuscript.
Funding
This study was supported by the National Heart, Lung, and Blood Institute of the National Institutes of Health HL101917 and HL062565 to ARR.
Footnotes
Statement of Ethics
The authors have no ethical conflicts to disclose.
Disclosure Statement
The authors declare no conflict of interests.
References
- 1.Campos EI, Reinberg D. Histones: annotating chromatin. Annu Rev Genet 2009;43:559–599. [DOI] [PubMed] [Google Scholar]
- 2.Chen R, Kang R, Fan XG, Tang D. Release and activity of histone in diseases. Cell Death Dis 2014;5:e1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT. Extracellular histones are major mediators of death in sepsis. Nat Med 2009;15:1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gould TJ, Lysov Z, Liaw PC. Extracellular DNA and histones: double-edged swords in immunothrombosis. J Thromb Haemost 2015;Suppl 1:S82–91. [DOI] [PubMed]
- 5.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science 2004;303:1532–5153. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 2010;107:15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okubo K, Kurosawa M, Kamiya M, Urano Y, Suzuki A, Yamamoto K, Hase K, Homma K, Sasaki J, Miyauchi H, Hoshino T, Hayashi M, Mayadas TN, Hirahashi J. Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat Med 2018;24:232–238. [DOI] [PubMed] [Google Scholar]
- 8.Cai X, Panicker SR, Biswas I, Giri H, Rezaie AR. Protective Role of Activated Protein C against Viral Mimetic Poly(I:C)-Induced Inflammation. Thromb Haemost [DOI] [PMC free article] [PubMed]
- 9.Fritz G RAGE: a single receptor fits multiple ligands. Trends Biochem Sci 2011;36: 625–632. [DOI] [PubMed] [Google Scholar]
- 10.Ibrahim ZA, Armour CL, Phipps S, Sukkar MB. RAGE and TLRs: relatives, friends or neighbours? Mol Immunol 2013;56:739–744. [DOI] [PubMed] [Google Scholar]
- 11.Esmon CT. Molecular circuits in thrombosis and inflammation. Thromb Haemost 2013;109:416–420. [DOI] [PubMed] [Google Scholar]
- 12.Cohen J. The immunopathogenesis of sepsis. Nature 2004;420:885–891. [DOI] [PubMed] [Google Scholar]
- 13.Dinarvand P, Hassanian SM, Qureshi SH, Manithody C, Eissenberg JC, Yang L, Rezaie AR. Polyphosphate amplifies proinflammatory responses of nuclear proteins through interaction with receptor for advanced glycation end products and P2Y1 purinergic receptor. Blood 2014;123:935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalbitz M, Grailer JJ, Fattahi F, Jajou L, Herron TJ, Campbell KF, Zetoune FS, Bosmann M, Sarma JV, Huber-Lang M, Gebhard F, Loaiza R, Valdivia HH, Jalife J, Russell MW, Ward PA. Role of extracellular histones in the cardiomyopathy of sepsis. FASEB J 2015;29:2185–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michels A, Albánez S, Mewburn J, Nesbitt K, Gould TJ, Liaw PC, James PD, Swystun LL, Lillicrap D. Histones link inflammation and thrombosis through the induction of Weibel-Palade body exocytosis. J Thromb Haemost 2016;14:2274–2286. [DOI] [PubMed] [Google Scholar]
- 16.Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 2011;118:1952–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longstaff C, Hogwood J, Gray E, Komorowicz E, Varjú I, Varga Z, Kolev K. Neutralisation of the anti-coagulant effects of heparin by histones in blood plasma and purified systems. Thromb Haemost 2016;115:591–599. [DOI] [PubMed] [Google Scholar]
- 18.Hogwood J, Pitchford S, Mulloy B, Page C, Gray E. Heparin and non-anticoagulant heparin attenuate histone-induced inflammatory responses in whole blood. PLoS One 2020;15:e0233644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olson ST, Björk I, Sheffer R, Craig PA, Shore JD, Choay J. Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J Biol Chem 1992;267:12528–12538. [PubMed] [Google Scholar]
- 20.Belzar KJ, Zhou A, Carrell RW, Gettins PG, Huntington JA. Helix D elongation and allosteric activation of antithrombin. J Biol Chem 2002;277:8551–8558. [DOI] [PubMed] [Google Scholar]
- 21.Bae JS, Rezaie AR. Mutagenesis studies toward understanding the intracellular signaling mechanism of antithrombin. J Thromb Haemost 2009;7:803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rezaie AR, Giri H. Anticoagulant and signaling functions of antithrombin. J Thromb Haemost 2020;18: 3142–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang L, Manithody C, Qureshi SH, Rezaie AR. Contribution of exosite occupancy by heparin to the regulation of coagulation proteases by antithrombin. Thromb Haemost 2010;103:277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Panicker SR, Biswas I, Giri H, Cai X, Rezaie AR. PKC (Protein Kinase C)-δ Modulates AT (Antithrombin) Signaling in Vascular Endothelial Cells. Arterioscler Thromb Vasc Biol 2020;40:748–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dinarvand P, Yang L, Biswas I, Giri H, Rezaie AR. Plasmodium falciparum histidine rich protein HRPII inhibits the anti-inflammatory function of antithrombin. J Thromb Haemost 2020;18:1473–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rezaie AR. Heparin chain-length dependence of factor Xa inhibition by antithrombin in plasma. Thromb Res 2007;119:481–488. [DOI] [PubMed] [Google Scholar]
- 27.Rezaie AR. Heparin-binding exosite of factor Xa. Trends Cardiovasc Med 2000;10: 333–338. [DOI] [PubMed] [Google Scholar]
- 28.DeVries TA, Neville MC, Reyland ME. Nuclear import of PKCdelta is required for apoptosis: identification of a novel nuclear import sequence. EMBO J 2002;21:6050–6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu D, Young JH, Krahn JM, Song D, Corbett KD, Chazin WJ, Pedersen LC, Esko JD. Stable RAGE-heparan sulfate complexes are essential for signal transduction. ACS Chem Biol 2013;8:1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]