

Abstract
Common variable immunodeficiency (CVID) is one of the most common primary immunodeficiency disorders characterized by hypogammaglobulinemia and inadequate antibody response to immunizations. The impaired antibody response occurs due to the failure of B cells to differentiate into plasma cells resulting in low immunoglobulins levels and increased frequency of infections. Granulomatous and Lymphocytic Interstitial Lung Disease (GLILD) is a non-infectious complication of CVID that is seen in 10-30% of cases. GLILD is a multisystem inflammatory disease involving the lungs, lymph node, liver, spleen and gastrointestinal tract that mimics sarcoidosis. This report describes a series of cases who presented with dyspnea, recurrent respiratory infections or autoimmunity and on further evaluation revealed features suggestive of GLILD. There is very limited understanding of GLILD in terms of clinical presentation, the histo-pathological logical findings, and the diagnostic criteria by itself are limited. A diagnosis of GLILD is established in cases of CVID when there is evidence of lymphoproliferation, cytopenia, autoimmune processes and a lung biopsy demonstrating lymphocytic interstitial pneumonia, follicular bronchiolitis, lymphoid hyperplasia, and/or non-necrotizing granulomas. We review the treatment strategies, including replacement of immunoglobulin and agents targeting B and T lymphocytes. Systematic characterization of GLILD cases and long term follow up studies are sorely needed to understand the natural history of GLILD.
Keywords: Common variable immunodeficiency, CVID, granulomatous lymphocytic interstitial lung disease, GLILD, hypogammaglobulinemia
Introduction
Primary humoral immunodeficiencies are a group of immunodeficiency disorders with impaired antibody formation due to intrinsic B cell abnormalities resulting in decreased numbers or function of B lymphocytes. Common variable immunodeficiency (CVID) is one of the most common primary humoral immunodeficiency disorders with heterogenous manifestations that affects nearly 1 in 250,000 individuals (1). CVID is associated with polymorphisms in costimulatory molecules, (CD18, CD19, CD20, CD21) transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), and B cell-activating factor of the tumor necrosis family (TNF) (2). Despite numerically normal B cells, their function is impaired, which in part is related to defective peripheral maturation of B cells. The diagnosis of CVID requires reduced age-adjusted serum immunoglobulin G (IgG) levels along with abnormally low levels of at least one of the isotypes immunoglobulin M (IgM) or immunoglobulin A (IgA), or both IgA and IgM, and an inability to mount a specific antibody response to exogenous antigens such as vaccines or infections (3). Other causes of humoral immunodeficiency states such as malnutrition or medications, e.g., corticosteroids, anti-inflammatory drugs (hydroxychloroquine, sulfasalazine), immunosuppressive agents (cyclosporine, tacrolimus, mycophenolate mofetil), and biologic agents (Rituximab) must be excluded. CVID has a bimodal peak in presentation, usually in late teens/early adulthood and another peak in mid adult life. Patients commonly present with recurrent sinopulmonary tract and mucosal infections especially caused by encapsulated bacteria, similar to many other primary humoral immunodeficiency syndromes. However, in contrast to other humoral immunodeficiency disorders like Bruton’s agammaglobulinemia, a select subgroup of CVID patients may develop non-infectious complications (4).
Non-infectious complications of CVID include autoimmunity as well as lymphoproliferative disorders mimicking malignancy, inflammatory and lymphoid malignancies. Mutations in TACI, reduction of isotype-switched memory B cells, increase in CD21low B cells and an interferon gene signature are associated with both autoimmunity and lymphoproliferation in CVID (5). Autoimmunity occurs in 25-30% of patients with CVID (1) and can be a presenting manifestation in up to 12% of patients (6). Autoimmune manifestation may pre-date recurrent infections in many patients. Severe cytopenia may be the first manifestation of autoimmunity (7). Hematologic abnormalities such as autoimmune hemolytic anemia (AIHA) and immune thrombocytopenia (ITP) are the most common autoimmune manifestations of CVID. Patients with hematologic autoimmune disorders may be less likely to have recurrent infections (8). Other autoimmune diseases that can occur in patients with CVID include inflammatory bowel disease, enteropathy, seronegative arthritis, pernicious anemia, Sjogren syndrome, uveitis, vasculitis, thyroiditis, alopecia, vitiligo, hepatitis, primary biliary cirrhosis, sicca syndrome, and systemic lupus erythematosus (9). Lymphocytic infiltration and granuloma formation in lymph nodes, spleen, liver, and lungs may occur. Up to 20% of patients with CVID have lymphoid hyperplasia (10). Cervical, mediastinal, and intestinal lymph nodes are commonly involved. The common biopsy findings are atypical lymphoid hyperplasia, reactive lymphoid hyperplasia, or granulomatous inflammation (3). Lymph nodes may lack plasma cells and have ill-defined germinal centers (11). Nodular lymphoid hyperplasia of the gastrointestinal tract is seen in the small intestine (12), and when present, is associated with an increased risk of intestinal and extraintestinal lymphoma (12). Splenomegaly may be present in 26% of patients with CVID (1). The histological changes seen in the spleen are granulomatous lesions, congestive red pulp, follicular hyperplasia, and atrophic germinal centers/white pulp are (3). Ten percent of the patients with CVID have liver dysfunction with noncirrhotic portal hypertension, primary biliary cholangitis, or granulomatous inflammation and nodular regenerative hyperplasia on biopsy (12-14). CVID patients with liver disease have a higher rate of autoimmune disease and nonceliac enteropathy. Non-Hodgkin lymphomas (NHL) is the most common malignancy seen in CVID followed by gastric cancer. NHLs in CVID are mostly extra-nodal B cell origin, well-differentiated, EBV negative and secrete immunoglobulin. Unlike lymphomas in other congenital immune defects, CVID associated lymphomas typically present in the fourth to seventh decade (15). The increased prevalence of gastric cancer is likely related to increased prevalence of pernicious anemia or Helicobacter pylori infection, which has been reported in 41% of CVID patients with dyspepsia (14).
The pulmonary involvement occurs due to infections by atypical and encapsulated bacteria and non-infectious involvement with diverse manifestations causing interstitial lung disease (16). Most patients are susceptible to Hemophilus influenzae, pneumococcus and mycoplasma infections (17). Opportunistic infections are rare and should raise suspicion of a combined T and a B cell immunodeficiency disorders. The non-infectious pulmonary complications that are commonly encountered include bronchiectasis due to recurrent infections, organizing pneumonia and diffuse non-necrotizing granulomatous disease with lymphoid interstitial pneumonia or follicular bronchitis (3, 18). In this case series, we describe three cases with non-infectious pulmonary involvement with granulomatous inflammation interstitial lung disease (GLILD), a clinical manifestation which is a sarcoidosis mimic.
Clinical Cases
Case 1
A 44-year-old male presented to the pulmonary clinic for evaluation of abnormal chest computed tomography (CT). He presented with eight months history of cough, fatigue, and decreased exercise capacity. He had recurrent pulmonary infections with one hospitalization due to severe pneumonia in his 20s and possible sinusitis. He smoked briefly in the past, about 4 years, having quit in his 20s. Chest X-Ray (CXR) and chest CT (Figure 1) showed upper lobe predominant peri-bronchial consolidation with micronodular changes, calcified and noncalcified mediastinal adenopathy, and axillary lymphadenopathy and splenomegaly.
Figure 1.
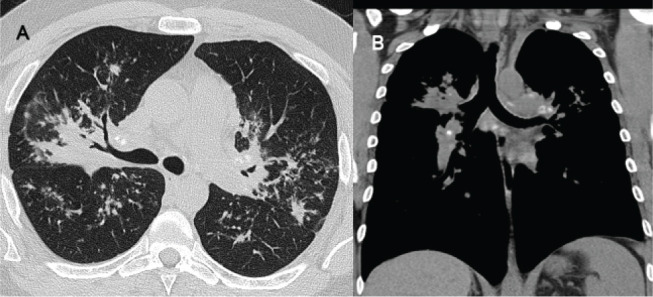
(A) HRCT of chest demonstrating upper lope predominant peribronchial micronodular infiltrates with consolidation and fibrosis with some central airway narrowing. Spiculated and flame shaped nodules in a perilymphatic distribution were also noted, along with bilateral hilar and mediastinal calcifies and non-calcified lymph nodes. (B) Splenomegaly was seen on the coronal images. This was interpreted as compatible with sarcoidosis.
Pulmonary function tests (PFT) showed mild obstruction without a bronchodilator response, with normal lung volumes and diffusing capacity for carbon monoxide (DLCO). The bronchoalveolar lavage (BAL) had 126 WBCs / ml (basophils 1%, eosinophils 1%, monocytes / macrophages 83% and lymphocytes 15% with a CD4/CD8 ratio of 3.8). The BAL fluid culture grew Hemophilus influenza, for which a course of antibiotic was prescribed. Endobronchial biopsies (Figure 2) demonstrated non-necrotizing granulomas. CVID was diagnoses based on serum IgG levels of 478 mg/d (normal- 695-1620) and low serum IgA levels of 55 mg/dL(normal: 70-380 mg/dl), IgM levels 57 mg/dL (normal: 60-265 mg/dl) and low H influenzae titers suggesting impaired immune response. Immunophenotyping and B cell phenotyping showed normal B lymphocyte and NK cell counts with mild T cell lymphopenia. Quantitative B cell subset immunophenotyping was normal for age, with the only exception a mild decreases in class-switched memory B cells (CD27+M-D-) driven by the proportionate decreases in both marginal and class-switched memory B cells at 45% of total memory B cells with marginal zone B cells at 37%. Due to persistent low immunoglobulin levels, IV immunoglobulin (IVIG) therapy was initiated which stabilized his pulmonary symptoms.
Figure 2.
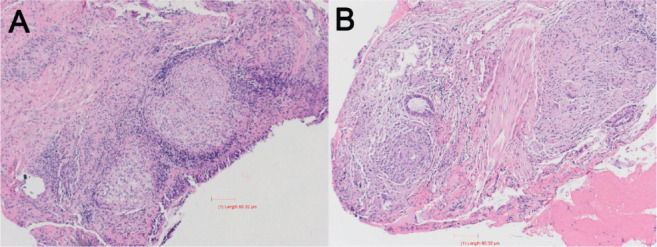
(A Low Power) Right lung endobronchial biopsy shows multiple large non-necrotizing granulomas present in the respiratory submucosa. (B High Power) Shows presence of scattered multinucleated giant cells and minimal, if any, associated lymphocytic infiltrate, giving the appearance of so-called “naked granulomas”, characteristic for sarcoidosis.
Case 2
A 38 y/o male was evaluated for management of granulomatous inflammation diagnosed fourteen years back by axillary lymph node biopsy (Figure 3).
Figure 3.
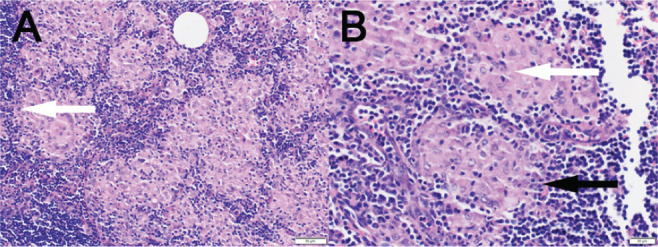
(A Low power) Excisional biopsy of right axillary lymph node shows multiple non-necrotizing granulomas completely effacing the lymph node architecture. Rare multinucleated giant cells are observed (white arrow). (B High Power) The granulomas are formed of tightly packed histiocytes (white arrow) in a background of lymphoid tissue representing the lymph node (black arrow).
He was treated with anti-inflammatory medication for a diagnosis of sarcoidosis. The patient provided a history of recurrent sinus symptoms and exertional dyspnea. Over the previous year, the patient needed hospitalization for recurrent pneumonias. There was a concern for asbestos exposure during employment with the naval services. He had no history of cigarette smoking, significant drug exposure, or significant family history. Chest CT scan showed perilymphatic nodular infiltrates, airway centered fibrotic changes with traction bronchiectasis, axillary adenopathy, and splenomegaly (Figure 4A). CT findings also suggested pulmonary hypertension based on increased diameter of the pulmonary artery and bronchial artery collaterals detected on coronal CT reconstructions (Figure 4B). Pulmonary function testing demonstrated moderate restriction with a total lung capacity (TLC) of 65% predicted, moderately decreased DLCO, and exertional hypoxia for which oxygen supplementation was recommended. A right heart catheterization revealed pulmonary hypertension with pulmonary artery pressure of 42/20 mm Hg (mean 33 mmHg), pulmonary capillary wedge pressure of 8 mm Hg and normal cardiac index of 2.9 L/min/m2. Further workup revealed extremely low total IgG levels at 90 mg/dL, IgA levels <7 mg/dL, IgM levels <4mg/dL, Diphtheria antibody titers were < 0.01 IU/mL and H influenza were 0.6 µgm/mL suggesting impaired response to vaccination. He had been tapered to 5mg daily of prednisone at that time which was not felt to be enough to suppress his IgG levels significantly. He has subsequently been tapered off prednisone and is currently on IgG replacement therapy, which he is tolerating well.
Figure 4.
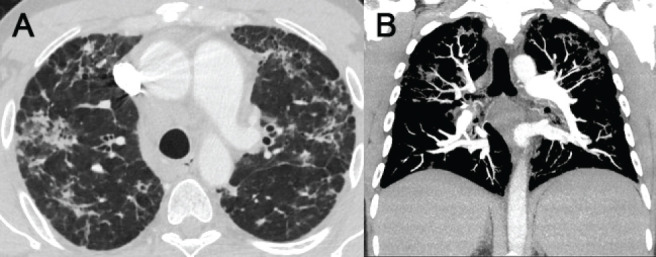
(A) CT of chest demonstrating peri lymphatic nodular infiltrates, airway centered fibrotic changes with traction bronchiectasis. (B) Coronal images also suggested pulmonary hypertension based on increased diameter of the pulmonary artery and bronchial artery collaterals. Splenomegaly was also noted.
Case 3
49 y/o female with a diagnosis of CVID established at age 38 presented to the pulmonary clinic with a history of recurrent pulmonary infections for the past decade and a 12 month history of dyspnea on exertion. She had a history of cigarette smoking in the past. She reported prior hospital admission for influenza A. Her PFTs and six-minute walk test were within normal limits. Chest CT scans demonstrated bilateral perilymphatic nodules without significant mediastinal or axillary lymphadenopathy (Figure 5A). No splenomegaly was present (Figure 5B).
Figure 5.
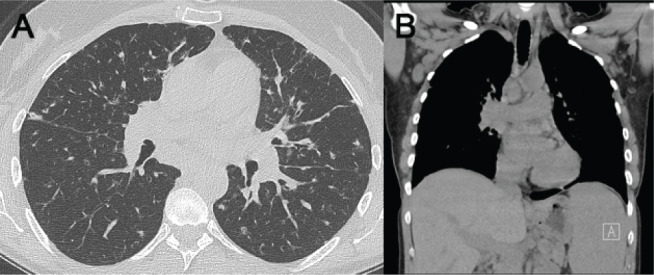
(A) Chest CT scan demonstrated bilateral perilymphatic nodules without significant mediastinal lymphadenopathy. (B) No splenomegaly or axillary adenopathy was present on coronal images.
Transbronchial lung biopsy demonstrated peribronchiolar lymphoid aggregates and interstitial lymphocytic infiltrates with non-necrotizing granulomatous inflammation (Figure 6). Her total IgG level was <109 mg/dl and both IgM and IgA were severely decreased at <5 mg/dL. Intravenous IgG replacement therapy was initiated but was tolerated poorly die to anaphylactoid-like reaction, pruritus, and nausea and hence she was transitioned to subcutaneous IgG replacement which she is tolerating well.
Figure 6.
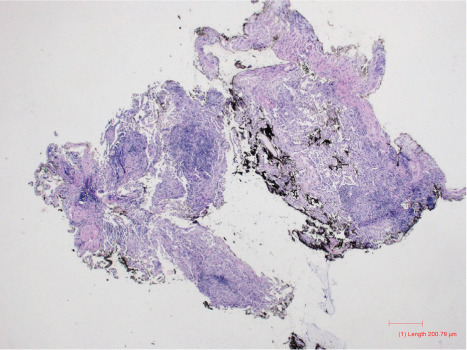
Transbronchial biopsies of right middle and lower lobes show large peribronchiolar lymphoid aggregates associated with diffuse interstitial lymphocytic infiltrate with granulomatous inflammation, consistent with granulomatous-lymphocytic interstitial lung disease (GLILD). GMS and AFB special stains were performed to exclude fungal and acid-fast infections, respectively, and were negative.
Discussion
In this report we describe variable clinical presentations of GLILD including recurrent pulmonary infections, dyspnea, abnormal imaging studies and histology demonstrating granulomatous inflammation, mimicking sarcoidosis. While one of the three patients had a working diagnosis of CVID, the other two patients were initially managed with a diagnosis of sarcoidosis. Evaluation for alternate causes of granulomatous inflammation revealed hypogammaglobulinemia, with reduced isotype-switched memory B cells and an impaired immune response. Based on history, physical examination, and laboratory findings, GLILD was diagnosed. These cases emphasize that sarcoidosis is a diagnosis of exclusion (19) and the workup for sarcoidosis should routinely include testing of immunoglobulin levels.
GLILD is non-infectious pulmonary complication seen in 10-30% of patients with CVID (20,21). The frequency is approximately equivalent in both males and females (21-23) with a diagnosis made usually in the third and fourth decade of life. Unlike sarcoidosis, GLILD is less common in Blacks (21-23). Environmental triggers remain unknown for both sarcoidosis and GLILD. Past or current cigarette smoking was reported in 65% of GLILD patients in one study (23), whereas twelve of thirteen cases were non-smokers in another report (21). At present, neither genetic predisposition nor specific major histocompatibility complex (MHC) is linked to GLILD (24). GLILD and sarcoidosis patients commonly have multisystem disease. Clinical features that help in differentiating GLILD and sarcoidosis are outlined in Table 1.
Table 1.
Differences in Granulomatous lymphocytic interstitial lung disease and sarcoidosis
| Feature | GLILD | Sarcoidosis |
| Organ system involvement (22, 55) | ||
| Pulmonary | 51% | 95% |
| Spleen | 46% | 6.7 |
| Lymph node | 15.2 | |
| Liver | 41% | 11.5 |
| Skin | 7% | 15.9 |
| Bone marrow | 8% | 3,9% |
| CNS | 5% | 4.6% |
| GI tract | 15% | Rare |
| Recurrent infections | Common | May occur if architectural distortion of lung |
| Autoimmunity | Frequently report | Not seen |
| Immunoglobulin levels | Low | Normal or high. May be low in patients on long term steroids |
| Chest CT | ||
| Distribution | Could have lower lobe disease | Upper lobe predominant disease |
| Common finding | Larger nodule with random or perilymphatic distribution | Perilymphatic micronodular infiltrate in bronchovascular distribution |
| Flame shape hemorrhage, ‘halo*’ sign (16) | More common than sarcoidosis | Could be seen |
| Bronchiectasis | Common due to recurrent infections | Cicatricial bronchiectasis in setting of architectural distortion |
| Mediastinal / hilar adenopathy | Present | More prominent |
| Bronchoalveolar fluid findings | ||
| Cultures | Rules out infection in cases of CVID (42) | Usually negative. Must rule our etiologies that may mimic clinical or radiologic manifestations of sarcoidosis such as histoplasma or tuberculosis |
| CD4: CD8 ratio | Usually, normal | High (> 3.5) |
* Halo sign: nodules surrounded by ground glass opacities
Ocular disease, erythema nodosum and myocardial disease has not been reported in GLILD (24). Immunoglobulin assessment is usually a reliable differentiator of the two entities, with GLILD presenting with hypogammaglobinemia, while immunoglobulin levels may be high in sarcoidosis (25). However, long-term steroid therapy for sarcoidosis may result in abnormally low immunoglobulin levels. Thus, in sarcoidosis low immunoglobulin levels should be interpreted in context of anti-inflammatory therapy. While spontaneous remissions are seen in sarcoidosis these are speculated to be rare in GLILD, however long-term data is limited.
An aberrant humoral response as well as immune dysregulation, causing lymphoid infiltration and systemic immune dysfunction contribute to the development of GLILD (9). While several molecular defects result in the failure of B cells to differentiate into plasma cells, the defect in the majority of the patients is unknown. CVID patients have a lower number of isotype switched memory and CD27+IgM- B cells (26, 27), with transitional expansion CD21low associated with splenomegaly (27, 28) and autoimmunity (28). Heterozygosity of the C104R sequence variant of the TACI alleles is linked to lower number of switched B cells, benign lymphoproliferation and autoimmune manifestations in CVID patients (29). Immunoregulatory cytokines like lymphotoxin-α and TNF-α may also contribute to the granulomatous disease. High TNF levels likely due to TNF +488A allele, or alleles in linkage disequilibrium with +488A, could drive the granulomatous inflammation in some CVID cases (30). Besides genetic association, infections such as Epstein Barr Virus (EBV) that promote B cell proliferation (31), human immunodeficiency virus (HIV) (32), and HHV-8 (33) are linked to the development of GLILD. As a result of immune dysregulation, lymphoid interstitial pneumonitis is seen on histology in GLILD (34,35), which responds to corticosteroid therapy (35). There is an increased risk of B cell lymphoma (36) and gastric malignancy.
The clinical presentation of GLILD is nonspecific with symptoms like dyspnea, arthralgia and swelling of more than four joints, and recurrent respiratory infections (24). Physical exam findings that indicate GLILD include splenomegaly (69%) (23,37), hepatomegaly (50%) (37), lymphadenopathy (75%) (24) and cytopenias (38). Laboratory studies to establish a diagnosis of CVID (3) and granulomatous inflammation are needed to confirm GLILD. Hypogammaglobulinemia as well as decreased levels of either isotype IgM or IgA and an impaired response to antigenic stimulus are necessary to consider a diagnosis of CVID. Ninety percent of patients have low serum IgA levels (39). Serum ACE levels can be elevated in patients with GLILD (40). CD4:CD8 ratio in bronchoalveolar lavage (BAL) was reported to be lower than in sarcoidosis, and thus can be a useful diagnostic tool (16). A restrictive ventilatory pattern on spirometry with reduced diffusion capacity for carbon monoxide (DLCO) is typically seen (24).
Recurrent infections and autoimmunity, especially cytopenia, should prompt evaluation for CVID. A combination of splenomegaly, history of immune thrombocytopenic purpura (ITP) or autoimmune hemolytic anemia (AIHA), low IgA levels, and a relative increase in the percentage of CD21low B cells should increase the clinical suspicion of GLILD and prompt evaluation by High Resolution CT (HRCT). Randomly distributed nodules (24), nodules surrounded by groundglass abnormality and the halo sign (16) indicates GLILD in cases with CVID. Other findings are bronchiectasis, groundglass abnormalities, areas of consolidation, and hilar/mediastinal/axillary lymphadenopathy (41). Abnormality on HRCT should prompt bronchoscopy with immunocompromised host protocol and consideration of lung biopsy (23). The primary role of bronchoscopy in evaluation of CVID patients with possibility of GLILD is excluding pulmonary infections. Surgical lung biopsy should also strongly be considered in CVID patients with HRCT abnormalities as transbronchial forceps biopsy may not sample adequate tissue (42, 43). Transbronchial cryoprobe biopsy may present a less invasive alternative with better tissue sampling, but further research is needed to validate it as a diagnostic tool in GLILD (44). In addition to non-necrotizing granulomatous inflammation, other findings such as lymphocytic interstitial pneumonia, follicular bronchiolitis and lymphoid hyperplasia, organizing pneumonia, can occur in GLILD (21, 24). Lymph node biopsy may demonstrate reactive follicular hyperplasia with prominent germinal centers and paracortical hyperplasia (45). CD4+ T cells and B cells are predominantly seen on immunohistochemistry (16).
There are no established criteria for the treatment of GLILD. Immunoglobulin replacement, either by subcutaneous (SCIG) (46) or intravenous route (IVIG), decreases the frequency of infections in CVID patients (47). Most guideline recommend a starting dose of 0.4 -0.6 gm/kg/month for SCIG and 0.4-0.5 gm/kg/month for IVIG (3). In a Delphi processes there was good consensus for either SCIG or IVIG treatment in GLILD cases who have symptoms with abnormal and deteriorating lung function, symptoms with normal but deteriorating lung function, and asymptomatic patients who have abnormal or deteriorating lung function (42). However, treatment with SCIG or IVIG may not change the trajectory of GLILD and may not be needed in asymptomatic cases with normal and stable lung function (42,48). Although the evidence is limited, when specific treatment is needed for GLILD, corticosteroids are believed to be initial treatment (42) in doses varying between 30-70 mg/day. Other immunosuppressive agents such as cyclosporine, methotrexate, azathioprine, and chlorambucil are used when there is intolerance or side effects due to corticosteroids (49), with strongest support for use of azathioprine or mycophenolate mofetil combined with rituximab (45,50,51). Rituximab administered intravenously in a dose of 375 mg/m2 weekly for four weeks plus oral azathioprine (1-2 mg/kg/day) for 18 months improved spleen size, FVC, FEV1, and HRCT score in patients with GLILD (45). Intravenous infusion of Rituximab in doses 375 mg/m2 weekly for four doses and repeated at six months for three to four courses plus antimetabolites, either azathioprine (1-2 mg/kg per day) or mycophenolate mofetil (250-1000 mg twice daily), also improve HRCT score, FVC, FEV1 and TLC but not DLCO (50). Once symptoms and lung function are stabilized, if close monitoring is possible, immunosuppression can be weaned in selected patients with GLILD (52). A recent report described successful treatment of CVID-associated GLILD with Rituximab monotherapy (53). TNF blockade with infliximab could be used when there is no response to immunosuppression and immunoglobulin replacement (54). There are many unanswered questions about the natural history of the disease, as there are no longitudinal or prospective studies published. The common causes of death in GLILD are infection, lymphoma, liver disease, bronchiectasis and progressive pulmonary insufficiency (24).
References
- 1.Gathmann B, Mahlaoui N, Ceredih, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134(1):116–26. doi: 10.1016/j.jaci.2013.12.1077. Epub 2014/03/04. doi: 10.1016/j.jaci.2013.12.1077. PubMed PMID: 24582312. [DOI] [PubMed] [Google Scholar]
- 2.van de Ven AA, Compeer EB, van Montfrans JM, Boes M. B-cell defects in common variable immunodeficiency: BCR signaling, protein clustering and hardwired gene mutations. Crit Rev Immunol. 2011;31(2):85–98. doi: 10.1615/critrevimmunol.v31.i2.10. Epub 2011/05/06. doi: 10.1615/critrevimmunol.v31.i2.10. PubMed PMID: 21542788. [DOI] [PubMed] [Google Scholar]
- 3.Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59. doi: 10.1016/j.jaip.2015.07.025. Epub 2015/11/14. doi: 10.1016/j.jaip.2015.07.025. PubMed PMID: 26563668; PubMed Central PMCID: PMCPMC4869529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chun JK, Lee TJ, Song JW, Linton JA, Kim DS. Analysis of clinical presentations of Bruton disease: a review of 20 years of accumulated data from pediatric patients at Severance Hospital. Yonsei Med J. 2008;49(1):28–36. doi: 10.3349/ymj.2008.49.1.28. Epub 2008/02/29. doi: 10.3349/ymj.2008.49.1.28. PubMed PMID: 18306466; PubMed Central PMCID: PMCPMC2615253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maglione PJ. Autoimmune and Lymphoproliferative Complications of Common Variable Immunodeficiency. Curr Allergy Asthma Rep. 2016;16(3):19. doi: 10.1007/s11882-016-0597-6. Epub 2016/02/10. doi: 10.1007/s11882-016-0597-6. PubMed PMID: 26857017. [DOI] [PubMed] [Google Scholar]
- 6.Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27(3):308–16. doi: 10.1007/s10875-007-9075-1. Epub 2007/05/19. doi: 10.1007/s10875-007-9075-1. PubMed PMID: 17510807. [DOI] [PubMed] [Google Scholar]
- 7.Chapel H, Lucas M, Patel S, et al. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J Allergy Clin Immunol. 2012;130(5):1197–8. doi: 10.1016/j.jaci.2012.05.046. e9. Epub 2012/07/24. doi: 10.1016/j.jaci.2012.05.046. PubMed PMID: 22819511. [DOI] [PubMed] [Google Scholar]
- 8.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650–7. doi: 10.1182/blood-2011-09-377945. Epub 2011/12/20. doi: 10.1182/blood-2011-09-377945. PubMed PMID: 22180439; PubMed Central PMCID: PMCPMC3286343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agarwal S, Cunningham-Rundles C. Autoimmunity in common variable immunodeficiency. Ann Allergy Asthma Immunol. 2019;123(5):454–60. doi: 10.1016/j.anai.2019.07.014. Epub 2019/07/28. doi: 10.1016/j.anai.2019.07.014. PubMed PMID: 31349011; PubMed Central PMCID: PMCPMC7310570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gompels MM, Hodges E, Lock RJ, et al. Lymphoproliferative disease in antibody deficiency: a multi-centre study. Clin Exp Immunol. 2003;134(2):314–20. doi: 10.1046/j.1365-2249.2003.02253.x. Epub 2003/11/18. doi: 10.1046/j.1365-2249.2003.02253.x. PubMed PMID: 14616793; PubMed Central PMCID: PMCPMC1808874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Unger S, Seidl M, Schmitt-Graeff A, et al. Ill-defined germinal centers and severely reduced plasma cells are histological hallmarks of lymphadenopathy in patients with common variable immunodeficiency. J Clin Immunol. 2014;34(6):615–26. doi: 10.1007/s10875-014-0052-1. Epub 2014/05/03. doi: 10.1007/s10875-014-0052-1. PubMed PMID: 24789743. [DOI] [PubMed] [Google Scholar]
- 12.Albuquerque A. Nodular lymphoid hyperplasia in the gastrointestinal tract in adult patients: A review. World J Gastrointest Endosc. 2014;6(11):534–40. doi: 10.4253/wjge.v6.i11.534. Epub 2014/11/18. doi: 10.4253/wjge.v6.i11.534. PubMed PMID: 25400867; PubMed Central PMCID: PMCPMC4231492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ward C, Lucas M, Piris J, Collier J, Chapel H. Abnormal liver function in common variable immunodeficiency disorders due to nodular regenerative hyperplasia. Clin Exp Immunol. 2008;153(3):331–7. doi: 10.1111/j.1365-2249.2008.03711.x. Epub 2008/07/24. doi: 10.1111/j.1365-2249.2008.03711.x. PubMed PMID: 18647320; PubMed Central PMCID: PMCPMC2527366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zullo A, Romiti A, Rinaldi V, et al. Gastric pathology in patients with common variable immunodeficiency. Gut. 1999;45(1):77–81. doi: 10.1136/gut.45.1.77. Epub 1999/06/16. doi: 10.1136/gut.45.1.77. PubMed PMID: 10369708; PubMed Central PMCID: PMCPMC1727591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham-Rundles C. Common variable immunodeficiency. Curr Allergy Asthma Rep. 2001;1(5):421–9. doi: 10.1007/s11882-001-0027-1. Epub 2002/03/15. doi: 10.1007/s11882-001-0027-1. PubMed PMID: 11892068. [DOI] [PubMed] [Google Scholar]
- 16.Bouvry D, Mouthon L, Brillet PY, et al. Granulomatosis-associated common variable immunodeficiency disorder: a case-control study versus sarcoidosis. Eur Respir J. 2013;41(1):115–22. doi: 10.1183/09031936.00189011. Epub 2012/08/21. doi: 10.1183/09031936.00189011. PubMed PMID: 22903958. [DOI] [PubMed] [Google Scholar]
- 17.Roifman CM, Rao CP, Lederman HM, Lavi S, Quinn P, Gelfand EW. Increased susceptibility to Mycoplasma infection in patients with hypogammaglobulinemia. Am J Med. 1986;80(4):590–4. doi: 10.1016/0002-9343(86)90812-0. Epub 1986/04/01. doi: 10.1016/0002-9343(86)90812-0. PubMed PMID: 3963038. [DOI] [PubMed] [Google Scholar]
- 18.Vieira AL, Vale A, Melo N, et al. Organizing pneumonia revisited: insights and uncertainties from a series of 67 patients. Sarcoidosis Vasc Diffuse Lung Dis. 2018;35(2):129–38. doi: 10.36141/svdld.v35i2.6860. Epub 2018/01/01. doi: 10.36141/svdld.v35i2.6860. PubMed PMID: 32476892; PubMed Central PMCID: PMCPMC7170093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crouser ED, Maier LA, Wilson KC, et al. Diagnosis and Detection of Sarcoidosis. An Official American Thoracic Society Clinical Practice Guideline. Am J Respir Crit Care Med. 2020;201(8):e26–e51. doi: 10.1164/rccm.202002-0251ST. Epub 2020/04/16. doi: 10.1164/rccm.202002-0251ST. PubMed PMID: 32293205; PubMed Central PMCID: PMCPMC7159433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vultaggio A, Matucci A, Parronchi P, et al. Association between “sarcoidosis-like” disease and common variable immunodeficiency (CVI): a new CVI variant showing an activation of the immune system. Sarcoidosis Vasc Diffuse Lung Dis. 2007;24(2):127–33. Epub 2008/05/24. PubMed PMID: 18496983. [PubMed] [Google Scholar]
- 21.Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol. 2004;114(2):415–21. doi: 10.1016/j.jaci.2004.05.057. Epub 2004/08/19. doi: 10.1016/j.jaci.2004.05.057. PubMed PMID: 15316526. [DOI] [PubMed] [Google Scholar]
- 22.Boursiquot JN, Gerard L, Malphettes M, et al. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol. 2013;33(1):84–95. doi: 10.1007/s10875-012-9778-9. Epub 2012/09/19. doi: 10.1007/s10875-012-9778-9. PubMed PMID: 22986767. [DOI] [PubMed] [Google Scholar]
- 23.Hartono S, Motosue MS, Khan S, et al. Predictors of granulomatous lymphocytic interstitial lung disease in common variable immunodeficiency. Ann Allergy Asthma Immunol. 2017;118(5):614–20. doi: 10.1016/j.anai.2017.01.004. Epub 2017/03/04. doi: 10.1016/j.anai.2017.01.004. PubMed PMID: 28254202. [DOI] [PubMed] [Google Scholar]
- 24.Verbsky JW, Routes JM. Sarcoidosis and common variable immunodeficiency: similarities and differences. Semin Respir Crit Care Med. 2014;35(3):330–5. doi: 10.1055/s-0034-1376862. Epub 2014/07/10. doi: 10.1055/s-0034-1376862. PubMed PMID: 25007085. [DOI] [PubMed] [Google Scholar]
- 25.Te HS, Perlman DM, Shenoy C, et al. Clinical characteristics and organ system involvement in sarcoidosis: comparison of the University of Minnesota Cohort with other cohorts. BMC Pulm Med. 2020;20(1):155. doi: 10.1186/s12890-020-01191-x. Epub 2020/06/04. doi: 10.1186/s12890-020-01191-x. PubMed PMID: 32487134; PubMed Central PMCID: PMCPMC7268634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez-Ramon S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. 2008;128(3):314–21. doi: 10.1016/j.clim.2008.02.013. Epub 2008/07/16. doi: 10.1016/j.clim.2008.02.013. PubMed PMID: 18620909; PubMed Central PMCID: PMCPMC2692232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wehr C, Kivioja T, Schmitt C, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111(1):77–85. doi: 10.1182/blood-2007-06-091744. Epub 2007/09/28. doi: 10.1182/blood-2007-06-091744. PubMed PMID: 17898316. [DOI] [PubMed] [Google Scholar]
- 28.Piqueras B, Lavenu-Bombled C, Galicier L, et al. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol. 2003;23(5):385–400. doi: 10.1023/a:1025373601374. Epub 2003/11/07. doi: 10.1023/a:1025373601374. PubMed PMID: 14601647. [DOI] [PubMed] [Google Scholar]
- 29.Salzer U, Bacchelli C, Buckridge S, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113(9):1967–76. doi: 10.1182/blood-2008-02-141937. Epub 2008/11/05. doi: 10.1182/blood-2008-02-141937. PubMed PMID: 18981294; PubMed Central PMCID: PMCPMC2651012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mullighan CG, Fanning GC, Chapel HM, Welsh KI. TNF and lymphotoxin-alpha polymorphisms associated with common variable immunodeficiency: role in the pathogenesis of granulomatous disease. J Immunol. 1997;159(12):6236–41. Epub 1998/04/29. PubMed PMID: 9550427. [PubMed] [Google Scholar]
- 31.Barbera JA, Hayashi S, Hegele RG, Hogg JC. Detection of Epstein-Barr virus in lymphocytic interstitial pneumonia by in situ hybridization. Am Rev Respir Dis. 1992;145(4 Pt 1):940–6. doi: 10.1164/ajrccm/145.4_Pt_1.940. Epub 1992/04/01. doi: 10.1164/ajrccm/145.4_Pt_1.940. PubMed PMID: 1313215. [DOI] [PubMed] [Google Scholar]
- 32.Travis WD, Fox CH, Devaney KO, et al. Lymphoid pneumonitis in 50 adult patients infected with the human immunodeficiency virus: lymphocytic interstitial pneumonitis versus nonspecific interstitial pneumonitis. Hum Pathol. 1992;23(5):529–41. doi: 10.1016/0046-8177(92)90130-u. Epub 1992/05/01. doi: 10.1016/0046-8177(92)90130-u. PubMed PMID: 1314778. [DOI] [PubMed] [Google Scholar]
- 33.Wheat WH, Cool CD, Morimoto Y, et al. Possible role of human herpesvirus 8 in the lymphoproliferative disorders in common variable immunodeficiency. J Exp Med. 2005;202(4):479–84. doi: 10.1084/jem.20050381. Epub 2005/08/17. doi: 10.1084/jem.20050381. PubMed PMID: 16103407; PubMed Central PMCID: PMCPMC2212861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koss MN, Hochholzer L, Langloss JM, Wehunt WD, Lazarus AA. Lymphoid interstitial pneumonia: clinicopathological and immunopathological findings in 18 cases. Pathology. 1987;19(2):178–85. doi: 10.3109/00313028709077131. Epub 1987/04/01. doi: 10.3109/00313028709077131. PubMed PMID: 3453998. [DOI] [PubMed] [Google Scholar]
- 35.Levinson AI, Hopewell PC, Stites DP, Spitler LE, Fudenberg HH. Coexistent lymphoid interstitial pneumonia, pernicious anemia, and agammaglobulinemia. Arch Intern Med. 1976;136(2):213–6. Epub 1976/02/01. PubMed PMID: 1082325. [PubMed] [Google Scholar]
- 36.Chua I, Quinti I, Grimbacher B. Lymphoma in common variable immunodeficiency: interplay between immune dysregulation, infection and genetics. Curr Opin Hematol. 2008;15(4):368–74. doi: 10.1097/MOH.0b013e328302c7b6. Epub 2008/06/10. doi: 10.1097/MOH.0b013e328302c7b6. PubMed PMID: 18536576. [DOI] [PubMed] [Google Scholar]
- 37.Mannina A, Chung JH, Swigris JJ, et al. Clinical Predictors of a Diagnosis of Common Variable Immunodeficiency-related Granulomatous-Lymphocytic Interstitial Lung Disease. Ann Am Thorac Soc. 2016;13(7):1042–9. doi: 10.1513/AnnalsATS.201511-728OC. Epub 2016/04/12. doi: 10.1513/AnnalsATS.201511-728OC. PubMed PMID: 27064856. [DOI] [PubMed] [Google Scholar]
- 38.Ramyar A, Aghamohammadi A, Moazzami K, et al. Presence of Idiopathic Thrombocytopenic Purpura and autoimmune hemolytic anemia in the patients with common variable immunodeficiency. Iran J Allergy Asthma Immunol. 2008;7(3):169–75. Epub 2008/09/11. doi: 07.03/ijaai.169175. PubMed PMID: 18780952. [PubMed] [Google Scholar]
- 39.Cunningham-Rundles C. How I treat common variable immune deficiency. Blood. 2010;116(1):7–15. doi: 10.1182/blood-2010-01-254417. Epub 2010/03/25. doi: 10.1182/blood-2010-01-254417. PubMed PMID: 20332369; PubMed Central PMCID: PMCPMC2904582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fasano MB, Sullivan KE, Sarpong SB, et al. Sarcoidosis and common variable immunodeficiency. Report of 8 cases and review of the literature. Medicine (Baltimore) 1996;75(5):251–61. doi: 10.1097/00005792-199609000-00002. Epub 1996/09/01. doi: 10.1097/00005792-199609000-00002. PubMed PMID: 8862347. [DOI] [PubMed] [Google Scholar]
- 41.Tanaka N, Kim JS, Bates CA, et al. Lung diseases in patients with common variable immunodeficiency: chest radiographic, and computed tomographic findings. J Comput Assist Tomogr. 2006;30(5):828–38. doi: 10.1097/01.rct.0000228163.08968.26. Epub 2006/09/07. doi: 10.1097/01.rct.0000228163.08968.26. PubMed PMID: 16954938. [DOI] [PubMed] [Google Scholar]
- 42.Hurst JR, Verma N, Lowe D, et al. British Lung Foundation/United Kingdom Primary Immunodeficiency Network Consensus Statement on the Definition, Diagnosis, and Management of Granulomatous-Lymphocytic Interstitial Lung Disease in Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract. 2017;5(4):938–45. doi: 10.1016/j.jaip.2017.01.021. Epub 2017/03/30. doi: 10.1016/j.jaip.2017.01.021. PubMed PMID: 28351785. [DOI] [PubMed] [Google Scholar]
- 43.Rao N, Mackinnon AC, Routes JM. Granulomatous and lymphocytic interstitial lung disease: a spectrum of pulmonary histopathologic lesions in common variable immunodeficiency--histologic and immunohistochemical analyses of 16 cases. Hum Pathol. 2015;46(9):1306–14. doi: 10.1016/j.humpath.2015.05.011. Epub 2015/07/04. doi: 10.1016/j.humpath.2015.05.011. PubMed PMID: 26138782; PubMed Central PMCID: PMCPMC4554947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravaglia C, Wells AU, Tomassetti S, et al. Transbronchial Lung Cryobiopsy in Diffuse Parenchymal Lung Disease: Comparison between Biopsy from 1 Segment and Biopsy from 2 Segments - Diagnostic Yield and Complications. Respiration. 2017;93(4):285–92. doi: 10.1159/000456671. Epub 2017/03/01. doi: 10.1159/000456671. PubMed PMID: 28245447. [DOI] [PubMed] [Google Scholar]
- 45.Chase NM, Verbsky JW, Hintermeyer MK, et al. Use of combination chemotherapy for treatment of granulomatous and lymphocytic interstitial lung disease (GLILD) in patients with common variable immunodeficiency (CVID) J Clin Immunol. 2013;33(1):30–9. doi: 10.1007/s10875-012-9755-3. Epub 2012/08/30. doi: 10.1007/s10875-012-9755-3. PubMed PMID: 22930256; PubMed Central PMCID: PMCPMC3557581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kobrynski L. Subcutaneous immunoglobulin therapy: a new option for patients with primary immunodeficiency diseases. Biologics. 2012;6:277–87. doi: 10.2147/BTT.S25188. Epub 2012/09/08. doi: 10.2147/BTT.S25188. PubMed PMID: 22956859; PubMed Central PMCID: PMCPMC3430092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Busse PJ, Razvi S, Cunningham-Rundles C. Efficacy of intravenous immunoglobulin in the prevention of pneumonia in patients with common variable immunodeficiency. J Allergy Clin Immunol. 2002;109(6):1001–4. doi: 10.1067/mai.2002.124999. Epub 2002/06/14. doi: 10.1067/mai.2002.124999. PubMed PMID: 12063531. [DOI] [PubMed] [Google Scholar]
- 48.Kohler PF, Cook RD, Brown WR, Manguso RL. Common variable hypogammaglobulinemia with T-cell nodular lymphoid interstitial pneumonitis and B-cell nodular lymphoid hyperplasia: different lymphocyte populations with a similar response to prednisone therapy. J Allergy Clin Immunol. 1982;70(4):299–305. doi: 10.1016/0091-6749(82)90066-5. Epub 1982/10/01. doi: 10.1016/0091-6749(82)90066-5. PubMed PMID: 6981662. [DOI] [PubMed] [Google Scholar]
- 49.Davies CW, Juniper MC, Gray W, Gleeson FV, Chapel HM, Davies RJ. Lymphoid interstitial pneumonitis associated with common variable hypogammaglobulinaemia treated with cyclosporin A. Thorax. 2000;55(1):88–90. doi: 10.1136/thorax.55.1.88. Epub 1999/12/23. doi: 10.1136/thorax.55.1.88. PubMed PMID: 10607809; PubMed Central PMCID: PMCPMC1745598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verbsky JW, Hintermeyer MK, Simpson PM, et al. Rituximab and antimetabolite treatment of granulomatous and lymphocytic interstitial lung disease in common variable immunodeficiency. J Allergy Clin Immunol. 2020 doi: 10.1016/j.jaci.2020.07.021. Epub 2020/08/04. doi: 10.1016/j.jaci.2020.07.021. PubMed PMID: 32745555. [DOI] [PubMed] [Google Scholar]
- 51.Tashtoush B, Memarpour R, Ramirez J, Bejarano P, Mehta J. Granulomatous-lymphocytic interstitial lung disease as the first manifestation of common variable immunodeficiency. Clin Respir J. 2018;12(1):337–43. doi: 10.1111/crj.12511. Epub 2016/06/01. doi: 10.1111/crj.12511. PubMed PMID: 27243233. [DOI] [PubMed] [Google Scholar]
- 52.Beaton TJ, Gillis D, Morwood K, Bint M. Granulomatous lymphocytic interstitial lung disease: limiting immunosuppressive therapy-a single-centre experience. Respirol Case Rep. 2020;8(5):e00565. doi: 10.1002/rcr2.565. Epub 2020/05/08. doi: 10.1002/rcr2.565. PubMed PMID: 32377343; PubMed Central PMCID: PMCPMC7199072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ng J, Wright K, Alvarez M, Hunninghake GM, Wesemann DR. Rituximab Monotherapy for Common Variable Immune Deficiency-Associated Granulomatous-Lymphocytic Interstitial Lung Disease. Chest. 2019;155(5):e117–e21. doi: 10.1016/j.chest.2019.01.034. Epub 2019/05/08. doi: 10.1016/j.chest.2019.01.034. PubMed PMID: 31060706; PubMed Central PMCID: PMCPMC6689079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hatab AZ, Ballas ZK. Caseating granulomatous disease in common variable immunodeficiency treated with infliximab. J Allergy Clin Immunol. 2005;116(5):1161–2. doi: 10.1016/j.jaci.2005.08.041. Epub 2005/11/09. doi: 10.1016/j.jaci.2005.08.041. PubMed PMID: 16275393. [DOI] [PubMed] [Google Scholar]
- 55.Baughman RP, Teirstein AS, Judson MA, et al. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001;164(10 Pt 1):1885–9. doi: 10.1164/ajrccm.164.10.2104046. Epub 2001/12/06. doi: 10.1164/ajrccm.164.10.2104046. PubMed PMID: 11734441. [DOI] [PubMed] [Google Scholar]