

After central nervous system (CNS) injury, severed axons fail to regenerate and their disconnections to the original targets result in permanent functional deficits in patients (Mahar and Cavalli, 2018). Both the diminished intrinsic regenerative capacity of mature neurons and the inhibitory CNS milieu contribute to the regenerative failure following CNS injury. Glial cells have important physiological functions, including maintaining homeostasis, supporting and protecting neurons, regulating neuronal activities, and forming myelin (Gaudet and Fonken, 2018). In response to CNS injury, reactive glial cells shift their phenotype and activities and contribute to scar formation. Gliosis is a defense response of the CNS to diminish primary damage and to repair injured tissues and has numerous beneficial effects, such as preventing the spread of damage from injury site. Various glia present around the lesion, including astrocytes, may express multiple positive molecules that promote axon regrowth, such as laminins, syndecans, glypicans, and decorin. Accordingly, preventing scar formation after injury or removing chronic astrocytic scars failed to promote axon regeneration (Anderson et al., 2016). However, reactive glial cells and scar tissues ultimately produce detrimental effects by upregulating numerous molecules that suppress neuronal elongation and form potent barriers to axon regeneration. Shortly after CNS injury, chondroitin sulfate proteoglycans (CSPGs) are upregulated dramatically and form part of the extracellular matrix components. CSPGs remain around the lesion epicenter for at least months, form an inhibitory milieu around the lesion, and suppress regrowth of injured axons into and beyond the lesion area (Hara et al., 2017). Two transmembrane protein tyrosine phosphatases (LAR and PTP σ) are important for mediating inhibition by CSPGs.
The CNS environment is nonpermissive to regeneration in contrast to the conducive milieu of peripheral nervous system, but several studies have demonstrated to convert the inhibitory CNS environment into a permissive one for axon growth by altering the status of glial cells. Changing CNS glial environment by peripheral nerve transplants allowed the central neurons to regenerate into the grafts for several millimeters in adult rodents. Immature CNS environment is conducive for axon regeneration because transplantation of immature astrocytes into mature CNS stimulated axon regeneration in acallosal animals. In neonate mice with spinal cord injury (SCI), microglia are essential for orchestrating the injury response, scar-free healing, and axon regeneration (Li et al., 2020b). Transplanted neonatal or adult microglia combined with proteinase inhibitors into lesioned spinal cord of adult mice promoted significant tissue healing and axon regrowth.
Recently, an interesting study demonstrated the novel role of glia in regulating CNS regeneration. Reprogrammed glia by targeting their intracellular pathways, specifically PI3K and EGFR activation, stimulated significant axon regeneration after CNS injury. Enhanced production of glycolytic metabolites, including L-lactate and L-2-hydroxyglutarate (L-2HG), in reprogrammed glia, promotes dramatic axon regeneration by acting on neuronal GABAB receptors (Figure 1). This study reported the novel approach to promote neuronal regeneration and synaptogenesis by glial reprogramming and revealed innovative glia-neuron interactions coupled with glycolytic metabolites (Li et al., 2020a). Consistently, glia programming has been shown to improve regeneration of the retina neurons (Jorstad et al., 2020). Activation of the proneural transcription factor Ascl1 and its target genes (e.g., Apobec2a and Apobec2b) in Müller glia is required for axon regeneration after retinal damage in zebrafish, and overexpressing Ascl1 specifically in Müller glia combined with histone deacetylase inhibition enables Müller glia to generate retinal neurons after retinal injury in adult mice (Jorstad et al., 2017).
Figure 1.
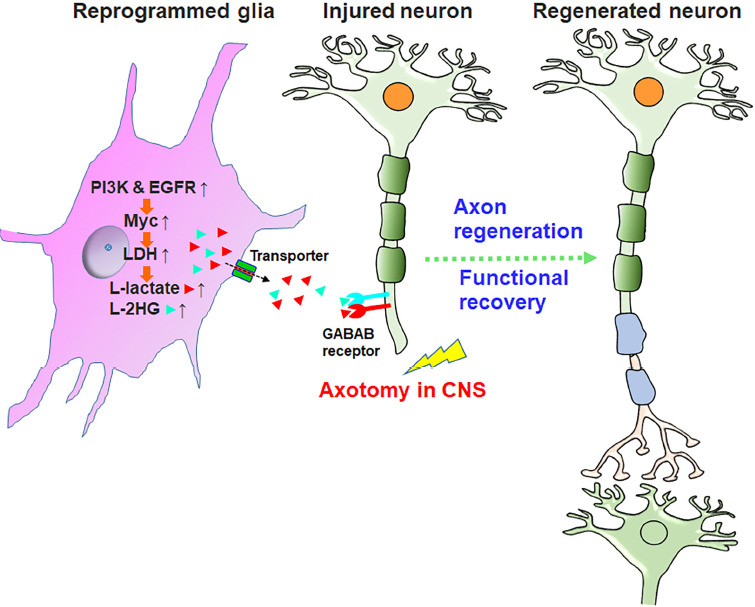
Glial reprogramming by PI3K and EGFR activation promotes functional regeneration after CNS injury.
Glial reprogramming by activating PI3K and EGFR pathways activates transcriptional factor Myc and increases LDH-associated metabolites, including L-lactate (red triangles) and L-2HG (light blue triangles). After transporting out of glia through monocarboxylate transporters, these metabolites bind and inactivate neuronal GABAB-R and enhance the cAMP levels, thus promoting functional axon regeneration in the CNS. GABAB receptors are expressed in the CNS axons of adult mice, and local lactate application enhanced the levels of their expression around the lesion. It requires to determine whether these metabolites bind the same site as GABA and compete with GABA ligand. CNS: Central nervous system; EGFR: epidermal growth factor receptor; L-2HG: L-2-hydroxyglutarate; LDH: lactate dehydrogenase; PI3K: phosphoinositide 3-kinase.
Glial reprogramming by targeting intracellular pathways promotes functional CNS regeneration: Activating intracellular PI3K and EGFR signals in glia promotes substantial neuronal regeneration and functional recovery after axotomy in Drosophila. A glial overexpression screen of candidate genes for intracellular signals in flies indicated that activating P13K, EGFR, or cell-cycle regulator in glia significantly enhanced axon regeneration. Simultaneously activating both P13K and EGFR enables axons to regrow more extensively than targeting each of them alone. Moreover, evaluation of larval escaping behaviors triggered by thermal stimuli revealed that regenerated axons induced by glial reprogramming significantly improved functional recovery after CNS injury (53% vs. 6% of recovery in control; Li et al., 2020a). Transcription factor c-Myc is essential for mediating glial actions of EGFR-PI3K activation by regulating glycolytic genes. Myc is essential signaling downstream of EGFR-PI3K activation in reprogrammed glia (Li et al., 2020a) and glioma cells in Drosophila. Myc suppression by loss-of-function or glial-specific knockdown with RNAi reduced axon regeneration in flies (Li et al., 2020a).
Glial upregulation of glycolysis stimulates CNS axon regeneration in flies: EGFR-PI3K activation upregulated several major genes in glycolytic pathway, including lactate dehydrogenase (LDH; Li et al., 2020a). Mutants of LDH and glial-specific knockdown of LDH, hexokinase A, and phosphofructokinase prevented axon regeneration by glial EGFR-PI3K-Myc activation. Previously, c-Myc has been linked to regulation of glycolysis in cancer cells. Glial LDH deletion prohibited axon regeneration induced by EGFR-PI3K activation while LDH upregulation enhanced regeneration. Upregulating glycolysis by targeting other enzymes, including knocking down pyruvate dehydrogenase complex or mitochondrial pyruvate carrier, is also sufficient to promote axon regeneration (Li et al., 2020a). These enzymes decrease lactate levels by converting pyruvate into acetyl-CoA or transporting pyruvate into mitochondria for decarboxylation.
Glycolytic metabolites mediate CNS axon regeneration induced by glial reprogramming: Glial reprogramming increased levels of LDH-associated metabolites, including L-lactate and L-2GH, which are essential for stimulating axon regeneration. Elevated levels of L-lactate and L-2GH were detected in the hemolymph of glia-reprogrammed larval, suggesting that direct application of these metabolites may effectively promote regeneration. Indeed, injecting a blood-brain-barrier-permeable solution of L-lactate or L-2GH into Drosophila larvae resulted in considerable CNS axon regeneration. Consistently, decreasing L-2GH by upregulating its dehydrogenase, which breaks down L-2GH, significantly reduced axon regeneration induced by glial reprogramming, and increasing L-2GH by downregulating this dehydrogenase enhanced regeneration. D-2HG, the enantiomer of L-2HG, also promoted regeneration when produced in glia, not in neurons.
Other glycolytic metabolites, such as L-2-hydroxybutyrate and L-tartrate, share similar structures to L-lactate and L-2HG (i.e., a carboxylic group with a hydroxyl group on the adjacent carbon) and display comparable efficacy for improving CNS regeneration. Notably, monocarboxylate transporters, especially Silnoon, actively transport these metabolites out of glia (Li et al., 2020a). Knocking down glial Silnoon efficiently impeded axon regeneration induced by glial reprogramming. It will be interesting to determine whether other metabolites transported by Silnoon, such as butyrate and pyruvate, have similar effects as L-lactate. The metabolic coupling between glia and neurons, including the glia-neuron lactate shuttle, has been studied frequently, but this study uniquely links the glial glycolysis to neuronal regeneration by molecular signaling. Besides metabolites, numerous other components, including proteins, lipids, nucleic acids, and the extracellular vesicle carriers, can be transported from glia to neurons and mediate the glia-neuron interactions (Pistono et al., 2020).
The interactions between glycolytic metabolites and neuronal GABAB receptors are essential for axon regeneration mediated by glial reprogramming: By binding and inactivating neuronal transmembrane GABAB receptors, glycolytic metabolites L-lactate and L-2HG act as signaling molecules to increase the intracellular cAMP levels, thus promoting axon regeneration. A biochemical assay showed that relatively high concentrations of L-lactate and L-2HG inversely activated GABAB receptors (Li et al., 2020a). Either deleting GABAB-R1 or knocking down GABAB-R2 in fly sensory C4da neurons efficiently prohibited axon regeneration induced by glial reprogramming. Consistently, functional GABAB receptors require assembling transmembrane heterodimers of both GABAB-R1 and GABAB-R2 subunits (Jones et al., 1998). Intracellularly elevated cAMP signaling is crucial for enhancing neuronal growth by glial reprogramming (Li et al., 2020a) and other mechanisms. Applying L-lactate or L-2HG increased neuronal cAMP levels in wildtype larvae but not GABAB-R1-deleted larvae.
CNS axon regeneration mediated by glycolytic metabolites is conserved in mammals: Importantly, local treatment with glycolytic metabolite L-lactate stimulated dramatic regeneration of descending motor fibers and significant recovery of locomotor function in adult mice with mid-thoracic SCI (Li et al., 2020a). Application of lactate to the injured spinal cord in adult mice stimulated substantial regeneration of tracer-labeled corticospinal tracts, which are important for controlling voluntary movements but are particularly refractory to regeneration after injury. Lactate-treated mice displayed extensive regeneration of injured motor axons into the lesion and caudal spinal cord, in contrast to their termination at the lesion site in SCI controls. Notably, lactate treatment improved the recovery of locomotor function, including increased locomotion scores and successful grasping rate, as well as reduced grid walk errors of the hindlimbs. Therefore, CNS regeneration mediated by glycolytic metabolites is conserved in adult mammals.
Perspective: Given complicated functions of CNS environment on neuronal regeneration, it is very interesting to couple the metabolic status of glial cells with CNS regeneration and to covert the inhibition of regeneration by glia into permission through glial reprogramming. It is very exciting to identify the novel glia-neuron interactions mediated by glycolytic metabolites and to promote substantial CNS axon regeneration with the specific metabolites recognized. Because CNS regeneration mediated by glycolytic metabolites is conserved in mammalians, it will be extremely important to further dissect the molecular targets of glial reprogramming-based regeneration and to translate these promising findings into treating human disorders, including SCI. Because the reported glycolytic metabolites partially mediate the actions of glial PI3K-EGFR activation, it is vital to study additional genes and molecules that mediate glial reprogramming. It is also interesting to reprogram glia by targeting other signaling pathways (e.g., Hedgehog and hypoxia-inducible factor) and to combine glial reprogramming with other mechanisms, including enhancing intrinsic growth capability of mature neurons (Nathan et al., 2020) and overcoming potent suppression by scar-sourced inhibitors (Lang et al., 2015). Although flies and mammals respond differently to CNS injury, fly CNS lesions induce a glial-scar-like structure and upregulate the homolog of CSPG phosphacan (called Ptp99A) around the lesion (Li et al., 2020a). Beyond the reported glycolysis-related biomolecules, such as L-lactate and L-2GH, other structurally similar metabolites and their related genes may also become attractive targets for CSN regeneration. Also, future translational studies are required to validate the major findings and promising regenerative strategies in other translation models, such as cervical or chronic SCI, stroke, traumatic brain injury, and axonal injuries in large animals. Together, the interesting study (Li et al., 2020a) opens new avenues for CNS regeneration study and neural repair of injured CNS.
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Zhao LJ, Li JY; T-Editor: Jia Y
References
- 1.Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV. Astrocyte scar formation aids central nervous system axon regeneration. Nature. 2016;532:195–200. doi: 10.1038/nature17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gaudet AD, Fonken LK. Glial cells shape pathology and repair after spinal cord injury. Neurotherapeutics. 2018;15:554–577. doi: 10.1007/s13311-018-0630-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hara M, Kobayakawa K, Ohkawa Y, Kumamaru H, Yokota K, Saito T, Kijima K, Yoshizaki S, Harimaya K, Nakashima Y, Okada S. Interaction of reactive astrocytes with type I collagen induces astrocytic scar formation through the integrin-N-cadherin pathway after spinal cord injury. Nat Med. 2017;23:818–828. doi: 10.1038/nm.4354. [DOI] [PubMed] [Google Scholar]
- 4.Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, Tang C, Shen Q, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- 5.Jorstad NL, Wilken MS, Grimes WN, Wohl SG, VandenBosch LS, Yoshimatsu T, Wong RO, Rieke F, Reh TA. Stimulation of functional neuronal regeneration from Muller glia in adult mice. Nature. 2017;548:103–107. doi: 10.1038/nature23283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jorstad NL, Wilken MS, Todd L, Finkbeiner C, Nakamura P, Radulovich N, Hooper MJ, Chitsazan A, Wilkerson BA, Rieke F, Reh TA. STAT signaling modifies ascl1 chromatin binding and limits neural regeneration from Muller glia in adult mouse retina. Cell Rep. 2020;30:2195–2208. doi: 10.1016/j.celrep.2020.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lang BT, Cregg JM, DePaul MA, Tran AP, Xu K, Dyck SM, Madalena KM, Brown BP, Weng YL, Li S, Karimi-Abdolrezaee S, Busch SA, Shen Y, Silver J. Modulation of the proteoglycan receptor PTPsigma promotes recovery after spinal cord injury. Nature. 2015;518:404–408. doi: 10.1038/nature13974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li F, Sami A, Noristani HN, Slattery K, Qiu J, Groves T, Wang S, Veerasammy K, Chen YX, Morales J, Haynes P, Sehgal A, He Y, Li S, Song Y. Glial metabolic rewiring promotes axon regeneration and functional recovery in the central nervous system. Cell Metab. 2020a;32:767–785. doi: 10.1016/j.cmet.2020.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, He X, Kawaguchi R, Zhang Y, Wang Q, Monavarfeshani A, Yang Z, Chen B, Shi Z, Meng H, Zhou S, Zhu J, Jacobi A, Swarup V, Popovich PG, Geschwind DH, He Z. Microglia-organized scar-free spinal cord repair in neonatal mice. Nature. 2020b;587:613–618. doi: 10.1038/s41586-020-2795-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahar M, Cavalli V. Intrinsic mechanisms of neuronal axon regeneration. Nat Rev Neurosci. 2018;19:323–337. doi: 10.1038/s41583-018-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nathan FM, Ohtake Y, Wang S, Jiang X, Sami A, Guo H, Zhou FQ, Li S. Upregulating lin28a promotes axon regeneration in adult mice with optic nerve and spinal cord injury. Mol Ther. 2020;28:1902–1917. doi: 10.1016/j.ymthe.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pistono C, Bister N, Stanova I, Malm T. Glia-derived extracellular vesicles: role in central nervous system communication in health and disease. Front Cell Dev Biol. 2020;8:623771. doi: 10.3389/fcell.2020.623771. [DOI] [PMC free article] [PubMed] [Google Scholar]