

ABSTRACT
Cell-cell adhesion between oral bacteria plays a key role in the development of polymicrobial communities such as dental plaque. Oral streptococci such as Streptococcus gordonii and Streptococcus oralis are important early colonizers of dental plaque and bind to a wide range of different oral microorganisms, forming multispecies clumps or “coaggregates.” S. gordonii actively responds to coaggregation by regulating gene expression. To further understand these responses, we assessed gene regulation in S. gordonii and S. oralis following coaggregation in 25% human saliva. Coaggregates were formed by mixing, and after 30 min, RNA was extracted for dual transcriptome sequencing (RNA-Seq) analysis. In S. oralis, 18 genes (6 upregulated and 12 downregulated) were regulated by coaggregation. Significantly downregulated genes encoded functions such as amino acid and antibiotic biosynthesis, ribosome, and central carbon metabolism. In total, 28 genes were differentially regulated in Streptococcus gordonii (25 upregulated and 3 downregulated). Many genes associated with transporters and a two-component (NisK/SpaK) regulatory system were upregulated following coaggregation. Our comparative analyses of S. gordonii-S. oralis with different previously published S. gordonii pairings (S. gordonii-Fusobacterium nucleatum and S. gordonii-Veillonella parvula) suggest that the gene regulation is specific to each pairing, and responses do not appear to be conserved. This ability to distinguish between neighboring bacteria may be important for S. gordonii to adapt appropriately during the development of complex biofilms such as dental plaque.
IMPORTANCE Dental plaque is responsible for two of the most prevalent diseases in humans, dental caries and periodontitis. Controlling the formation of dental plaque and preventing the transition from oral health to disease requires a detailed understanding of microbial colonization and biofilm development. Streptococci are among the most common colonizers of dental plaque. This study identifies key genes that are regulated when oral streptococci bind to one another, as they do in the early stages of dental plaque formation. We show that specific genes are regulated in two different oral streptococci following the formation of mixed-species aggregates. The specific responses of S. gordonii to coaggregation with S. oralis are different from those to coaggregation with other oral bacteria. Targeting the key genes that are upregulated during interspecies interactions may be a powerful approach to control the development of biofilm and maintain oral health.
KEYWORDS: bioinformatics, coaggregation, oral streptococci, Streptococcus gordonii, Streptococcus oralis, transcriptome, biofilms
INTRODUCTION
Oral streptococci, including Streptococcus gordonii and Streptococcus oralis, are among the most common bacteria in biofilms on the hard and soft tissues in the mouth (1). While S. gordonii predominantly colonizes tooth surfaces, S. oralis is frequently found both in dental plaque and in biofilms on soft tissues in the oral cavity (2, 3). Like many other oral streptococci, S. gordonii and S. oralis are able to adhere to cells of different species through specific adhesin-receptor interactions (4). Adhesion specificity is not fully conserved between different strains of a species due to differences in the key adhesins or receptors. For example, S. gordonii SK120 coaggregates with different strains of Actinomyces species compared with S. gordonii DL1 (Challis), M5, and SK184 (5). Genomic sequence analysis has revealed marked differences in the structure of a genetic locus encoding the coaggregation receptor polysaccharide (RPS) in S. gordonii SK120 compared with those of S. gordonii DL1, M5, and SK184, which likely underpins the differences in coaggregation specificity (6).
Many different coaggregation interactions can be detected between different strains of bacteria isolated from the mouth of an individual (7). It is thought that these interactions are critical for the colonization of surfaces in the mouth by microorganisms. For example, in a mouse model, the introduction of Candida albicans to the oral cavity in the absence of sucrose enhances mucosal biofilm formation by S. oralis (8, 9). S. gordonii expresses a range of cell surface adhesins that mediate adhesion with components of the acquired enamel pellicle, a layer of proteins and glycoproteins that coats the tooth surface (10). S. gordonii also adheres to a range of bacteria and is thought to be important for recruiting the periodontal keystone pathogen Porphyromonas gingivalis to dental plaque biofilms (11).
Coaggregation interactions will bring cells into close proximity with one another in oral microbial communities. It has been proposed that this enables cells to sense different species and to adapt in order to optimize their growth and survival within polymicrobial biofilms (12). Recently, a number of studies have investigated the impact of coaggregation or mixed-species biofilm formation on gene expression. S. gordonii has become a model organism for such studies due to its multifarious interactions with different partners. Thus, studies have explored interactions between S. gordonii and Aggregatibacter actinomycetemcomitans (13), C. albicans (14), P. gingivalis (15), or Actinomyces oris (16). However, each of these studies has used different models for bringing the cells together, and it is therefore difficult to identify genes that are regulated by cell-cell binding independently of the adhesion partner. In an attempt to standardize this approach, we have developed a simple method for studying coaggregation-mediated gene regulation by mixing suspensions of different bacteria in 25% human saliva to form coaggregates, incubating for 30 min, extracting RNA, and assessing gene expression by dual transcriptome sequencing (RNA-Seq). Using this approach, we have identified a number of genes that are regulated in S. gordonii in response to coaggregation with Fusobacterium nucleatum or Veillonella parvula (17, 18).
So far, studies on gene regulation responses to coaggregation have focused on intergeneric or interkingdom interactions. However, intrageneric coaggregation interactions have also been demonstrated. For example, a protein adhesin of S. gordonii DL1 recognizes RPS on the cell surface of S. oralis 34 that results in coaggregation (5, 19). Using antibodies against S. gordonii DL1 and the type of RPS expressed by S. oralis 34, interactions were also shown to occur between these bacteria in dental plaque developed in situ in the mouth of a volunteer (20). However, it is not yet clear whether the coaggregation between cells of the same genus results in cell-cell sensing and gene regulation in the partner organisms.
Here, we performed transcriptome profiling using a dual RNA-Seq approach to concurrently identify global changes in gene expression in S. gordonii DL1 and S. oralis 34 following coaggregation. This work builds on and improves our understanding of the interactions between S. gordonii and S. oralis and provides insights into their potential roles during the formation of mixed-species biofilms. We also compared these genes with the sets of genes that we identified in the interactions between S. gordonii and other bacterial species (e.g., F. nucleatum and V. parvula) in order to examine whether there are any mechanisms that are common among these S. gordonii-related bacterial pairings.
RESULTS
Generation of reference genome for S. oralis.
Due to the lack of a reference genome of S oralis 34, which was required for this transcriptomic study, we sequenced the genome of S. oralis 34 using Illumina HiSeq sequencing technology. The sequencing yielded 175,190 reads representing approximately 37-fold mean genome coverage. The assembly of these reads yielded six contigs with a GC content of 41.2% (see Table S1 in the supplemental material). The total assembly size was 1,904,876 bp with an N50 of 1,534,347 bp, suggesting that the assembly is suitable to be used as a reference genome for downstream transcriptomic analysis.
Coaggregation of Streptococcus gordonii and Streptococcus oralis.
To assess the formation of coaggregates between S. gordonii and S. oralis, coaggregation was assessed semiquantitatively by vigorously mixing concentrated suspensions of cells in coaggregation buffer. Substantial aggregates were observed with a clear background and were scored “4+” on the visual coaggregation scale (21). Coaggregation was also monitored in freshly collected 25% human saliva; again, strong coaggregation was observed within seconds and was designated 4+ in reference to the standard visual scoring system.
To more closely assess the interactions between S. gordonii and S. oralis, prestained cells of each species were mixed in 25% human saliva to induce coaggregation and visualized by confocal laser scanning microscopy (Fig. 1). Large coaggregates were observed that contained at least 100 cells of each species. S. gordonii and S. oralis cells were interspersed throughout these structures, indicating that there was significant potential for cell-cell sensing and responses, as would occur in surface-associated biofilms. Therefore, to explore gene regulation in each species in response to coaggregation, monocultures and equivalent cultures containing coaggregated bacteria were set up in 25% human saliva, incubated for 30 min, harvested, and subjected to RNA extraction. The quality of each RNA preparation was assessed by NanoDrop spectroscopy, Bioanalyzer, and agarose gel electrophoresis, prior to sequencing on the Illumina HiSeq platform.
FIG 1.
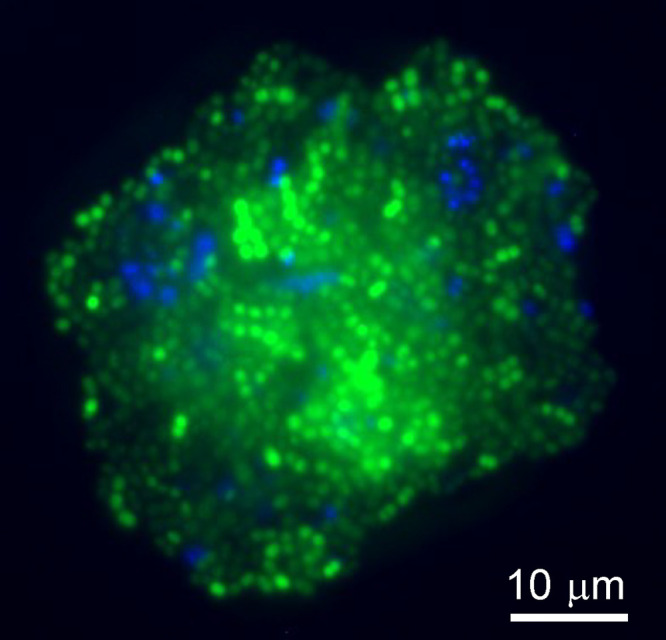
Visualization of coaggregation between Streptococcus gordonii and Streptococcus oralis. Example of a coaggregate formed between S. gordonii (Syto 9; green) and S. oralis (4′-6-diamidino-2-phenylindole [DAPI]; blue) in 25% human saliva. Cells were prestained before mixing and were visualized by fluorescence microscopy. The image shows a large aggregate.
Dual RNA-Seq data analysis.
Dual transcriptome sequencing was carried out with 5 or 6 independent biological repeats for monocultures or mixed cultures. A total of 16 sequencing libraries, comprising 5 mixed (S. gordonii-S. oralis), 6 monoculture S. gordonii, and 5 monoculture S. oralis biological replicates were sequenced, yielding approximately 339 million paired-end raw reads with read length of 100 bp (Table 1). After the removal of low-quality reads and adapter content by Trimmomatic v. 0.36, a total of approximately 314 million clean reads (clean ratio = 93%) were obtained.
TABLE 1.
Mapping statistics of mixed and monoculture transcriptomes of S. gordonii and S. oralis in the coaggregation experiment
| Sample name | No. of raw reads | No. of preprocessed reads | No. of mapped reads (%) |
|---|---|---|---|
| Monocultures | |||
| S. gordonii | |||
| Sg1 | 13,702,956 | 13,629,680 | 13,104,542 |
| Sg2 | 15,266,848 | 15,185,702 | 14,640,981 |
| Sg3 | 12,783,626 | 12,715,336 | 12,435,361 |
| Sg4 | 34,884,736 | 32,450,626 | 31,924,924 |
| Sg5 | 26,850,598 | 24,852,990 | 24,448,922 |
| Sg6 | 30,372,844 | 28,134,060 | 21,986,544 |
| All reads | 133,861,608 | 127,187,392 | 118,194,982 (92.9%) |
| S. oralis | |||
| So1 | 14,451,066 | 14,451,066 | 13,926,290 |
| So2 | 15,621,300 | 15,619,918 | 14,879,371 |
| So3 | 12,780,306 | 12,779,208 | 12,164,752 |
| So4 | 32,061,362 | 29,339,116 | 28,867,869 |
| So5 | 31,849,734 | 29,208,704 | 28,709,530 |
| All reads | 106,763,768 | 101,398,012 | 98,547,812 (97.1%) |
| Coaggregates | |||
| SgSo1 | 13,414,958 | 4,269,588 | 1,095,399 (S. gordonii); 2,984,283 (S. oralis) |
| SgSo2 | 15,148,212 | 15,146,904 | 5,229,367 (S. gordonii); 9,382,155 (S. oralis) |
| SgSo3 | 12,743,546 | 12,742,346 | 5,885,024 (S. gordonii); 6,499,028 (S. oralis) |
| SgSo4 | 33,322,784 | 30,982,486 | 15,833,006 (S. gordonii); 15,370,738 (S. oralis) |
| SgSo5 | 24,359,624 | 22,479,16 | 15,833,006 (S. gordonii); 12,065,953 (S. oralis) |
| Mixed cultures | 98,989,124 | 85,620,486 | 38,563,039 (45.04%; S. gordonii); 46,302,157 (54.08%; S. oralis) |
| All reads (monoculture and coaggregate) | 339,614,500 | 314,205,890 | 301,607,990 (96%) |
Read mapping and transcript abundance estimation in mixed and monocultures.
For S. gordonii monoculture samples, approximately 92.9% of preprocessed reads were mapped to the reference genome of S. gordonii DL1 (NCBI accession number NC_009785.1), whereas for the S. oralis monoculture samples, 97.1% of the reads were successfully mapped to the assembled genome of S. oralis 34 (Table 1). The high mapping rate indicated that our sequencing data were high quality and suitable for downstream analyses. For mixed culture samples, we bioinformatically separated the read sequences of the two different transcriptomes by mapping the reads to the reference genomes of the two bacterial species. On average, 45% of the reads of the mixed cultures were mapped to the reference genome of S. gordonii (data set SgSo_Sg), and 54% of the reads were mapped to the reference genome of S. oralis (data set SgSo_So) (Table 1). Read counts of the mixed and monoculture samples were normalized using the trimmed mean of M values (TMM). The normalized distributions of data were comparable between the mixed and coaggregate and monoculture samples, and no apparent batch effects were observed (Fig. S1).
Differential expression analysis in mixed and monocultures.
To investigate the impact of coaggregation on gene expression, differential gene expression analysis was performed using DESeq2 (22). Comparing the SgSo_So coaggregate and S. oralis monoculture identified 18 differentially expressed genes (6 upregulated and 12 downregulated) in S. oralis using a significance cutoff of a P value of <0.05 and a fold change of at least 2 (Table 2). After comparison between SgSo_Sg and S. gordonii monoculture, we identified 28 significant differentially expressed genes (25 upregulated and 3 downregulated genes) in S. gordonii (Table 3). The differentially expressed genes were visualized using volcano plots (Fig. S2).
TABLE 2.
Full list of differentially expressed genes found in S. oralis
| Identifier | Gene name | Protein name | Regulation | Fold change | P |
|---|---|---|---|---|---|
| 13396_Soralis34_01263a | rpsR | 30S ribosomal protein S18 | Upregulated | 4.11 | 6.12E−03 |
| 13396_Soralis34_01748a | rpmGA | 50S ribosomal protein L33 1 | Upregulated | 2.67 | 1.90E−02 |
| 13396_Soralis34_00180a | rpsS | 30S ribosomal protein S19 | Upregulated | 2.59 | 1.38E−03 |
| 13396_Soralis34_01130a | rplL | 50S ribosomal protein L7/L12 | Upregulated | 2.42 | 4.55E−02 |
| 13396_Soralis34_00682 | Upregulated | 2.30 | 2.04E−02 | ||
| 13396_Soralis34_01310 | gpmA_3 | 2,3-Bisphosphoglycerate-dependent phosphoglycerate mutase | Upregulated | 2.25 | 2.78E−02 |
| 13396_Soralis34_01404b | trpF | N-(5′-phosphoribosyl)anthranilate isomerase | Downregulated | 2.76 | 1.06E−02 |
| 13396_Soralis34_01406b | trpD | Anthranilate phosphoribosyltransferase | Downregulated | 2.41 | 2.24E−04 |
| 13396_Soralis34_01408b | trpE | Anthranilate synthase component 1 | Downregulated | 2.39 | 3.52E−06 |
| 13396_Soralis34_01407 | folP | Dihydropteroate synthase | Downregulated | 2.38 | 1.92E−05 |
| 13396_Soralis34_01405b | trpC | Indole-3-glycerol phosphate synthase | Downregulated | 2.27 | 9.91E−03 |
| 13396_Soralis34_00768 | opuBA_1 | Choline transport ATP-binding protein OpuBA | Downregulated | 2.26 | 9.84E−03 |
| 13396_Soralis34_00766 | Downregulated | 2.24 | 2.39E−02 | ||
| 13396_Soralis34_00578 | Downregulated | 2.24 | 4.89E−02 | ||
| 13396_Soralis34_01403b | trpB | Tryptophan synthase beta chain | Downregulated | 2.24 | 2.25E−02 |
| 13396_Soralis34_01314 | Downregulated | 2.15 | 3.81E−02 | ||
| 13396_Soralis34_01402b | trpA | Tryptophan synthase alpha chain | Downregulated | 2.14 | 3.88E−02 |
| 13396_Soralis34_00767 | Downregulated | 2.09 | 4.86E−02 |
Ribosomal proteins that were significantly upregulated in S. oralis in response to coaggregation with S. gordonii.
Tryptophan metabolism genes that were significantly downregulated in S. oralis in response to coaggregation with S. gordonii.
TABLE 3.
Full list of differentially expressed genes found in S. gordonii
| Locus tag | Gene name | Protein name | Regulation | Fold change | P |
|---|---|---|---|---|---|
| SGO_RS09355a | SGO_1911 | ABC-type transporter, ATPase component | Upregulated | 18.15 | 1.08E−07 |
| SGO_RS04515 | Upregulated | 14.94 | 1.68E−05 | ||
| SGO_RS09350a | SGO_1910 | Membrane protein, putative | Upregulated | 12.15 | 1.32E−12 |
| SGO_RS04530 | Upregulated | 10.52 | 3.86E−07 | ||
| SGO_RS04520 | Upregulated | 5.50 | 1.01E−03 | ||
| SGO_RS04535 | Upregulated | 4.10 | 3.50E−06 | ||
| SGO_RS04525 | Upregulated | 3.98 | 2.98E−02 | ||
| SGO_RS04510a | SGO_0920 | Cobalt ABC transporter, ATP-binding protein | Upregulated | 3.96 | 2.83E−23 |
| SGO_RS04505a | SGO_0919 | ABC transporter, ATP-binding protein | Upregulated | 3.74 | 6.21E−17 |
| SGO_RS05260 | SGO_1071 | Uncharacterized protein | Upregulated | 3.07 | 1.87E−02 |
| SGO_RS01830 | Upregulated | 3.06 | 8.47E−03 | ||
| SGO_RS04500a | ABC-type transporter permease | Upregulated | 3.02 | 1.25E−08 | |
| SGO_RS01720 | SGO_0348 | Reductase (EC 1.5.1.3) | Upregulated | 2.90 | 2.19E−03 |
| SGO_RS06370 | SGO_1298 | Uncharacterized protein | Upregulated | 2.79 | 2.84E−02 |
| SGO_RS08955 | SGO_1825 | Acetyltransferase, GNAT family | Upregulated | 2.63 | 4.13E−02 |
| SGO_RS03395 | SGO_0689 | Uncharacterized protein | Upregulated | 2.62 | 3.96E−02 |
| SGO_RS04495a | SGO_0917 | Membrane protein, putative | Upregulated | 2.61 | 6.98E−09 |
| SGO_RS09340a | SGO_1908 | DNA response regulator | Upregulated | 2.53 | 1.04E−03 |
| SGO_RS04115 | SGO_0839 | TfoX N-terminal domain superfamily | Upregulated | 2.43 | 2.27E−02 |
| SGO_RS01965 | SGO_0394 | Membrane protein, putative | Upregulated | 2.37 | 3.54E−02 |
| SGO_RS08490 | SGO_1732 | Histidine kinase (EC 2.7.3.–) | Upregulated | 2.36 | 1.52E−07 |
| SGO_RS04130 | Upregulated | 2.20 | 3.64E−02 | ||
| SGO_RS02465 | dsg (SGO_0498) | Putative permease | Upregulated | 2.19 | 2.15E−03 |
| SGO_RS01835 | Upregulated | 2.16 | 3.37E−02 | ||
| SGO_RS04940 | SGO_1008 | Phosphohydrolase (MutT/nudix family protein) (EC 3.6.1.–) | Upregulated | 2.02 | 1.47E−02 |
| SGO_RS01520 | Downregulated | 4.85 | 2.69E−02 | ||
| SGO_RS07630 | SGO_1557 | NrdH-redoxin | Downregulated | 2.37 | 1.87E−03 |
| SGO_RS01275 | Downregulated | 2.05 | 2.02E−04 |
Transporter and two-component system genes that were upregulated in S. gordonii in response to coaggregation with S. oralis.
Gene regulation in S. oralis in response to coaggregation with S. gordonii.
To get better insights into the interactome of genes regulated in S. oralis, we performed a network analysis using STRING. The downregulated genes were mostly interacting and formed two prominent clusters. Cluster 1, the largest cluster, was comprised of downregulated genes involved in tryptophan biosynthesis (Fig. 2). Cluster 2 was comprised of the upregulated genes rpsS (S19 protein), rpsR (S18 protein), rpmGA (L33 protein), and rpsL (L7/L12 protein), encoding ribosomal proteins that were upregulated from 2.42- to 4.11-fold (Table 2). In each of these clusters, gene interactions were based on multiple lines of evidence indicating that they are likely to be functionally related. The STRING functional enrichment analysis revealed three major functions: phenylalanine, tyrosine, and tryptophan biosynthesis (false-discovery rate [FDR] = 1.08E−06), biosynthesis of amino acids (FDR = 0.00024), and ribosome (FDR = 0.00076) (Fig. 2). Cluster 1 genes involved in phenylalanine, tyrosine, and tryptophan biosynthesis were downregulated between 2.14 and 2.76-fold in S. oralis following coaggregation (Table 2). The genes in this pathway are all involved in the tryptophan biosynthesis pathway.
FIG 2.

Network of genes regulated in S. oralis in coaggregation with S. gordonii visualized using the STRING database. Nodes were clustered by Markov cluster (MCL) clustering into three groups, represented by a single gene (gpmA, encoding 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase) and two gene clusters indicated in circles of different colors. Interactions between nodes are depicted by colored solid lines. Different colors represent evidence from different sources, such as gene neighborhood (green), gene cooccurrence (dark blue), text mining (yellow), curated databases (cyan), experimentally determined (magenta), coexpression (black), protein homology (light blue), and gene fusions (red). Genes involved in phenylalanine, tyrosine, and tryptophan biosynthesis (red nodes), biosynthesis of amino acids (blue nodes), and ribosome (green nodes). Following coaggregation, all genes in cluster 1 were downregulated, whereas the genes in cluster 2 were upregulated in S. oralis.
Gene regulation in S. gordonii in response to coaggregation with S. oralis.
Genes regulated in S. gordonii were dominated by transporter genes and particularly ATP-binding cassette (ABC)-type transporters (Table 3). Two clusters encoding transporters, which included a two-component (NisK/SpaK) regulatory system, were upregulated in response to coaggregation (Fig. 3). Two-component systems consist of a transmembrane sensor and response regulator that induce or repress transcription of target genes in response to an external stimulus (23, 24). A tblastn homology analysis of our two-component system showed 33% to 35% similarity with Lactococcus lactis and Streptococcus suis NisK/NisR systems, which are involved in sensing lantibiotics. Mature lantibiotics in streptococci can be sensed by two-component systems, leading to an autoinduction process that results in the production and activation of lantibiotics in neighboring cells (25).
FIG 3.

Network of genes regulated in S. gordonii in coaggregation with S. oralis. The largest connected group of genes encodes components of transporters (Table 2). Nodes were clustered by MCL clustering into two groups (cluster 1 and cluster 2). Interactions between nodes are depicted by colored lines. Different colors represent evidence from different sources such as gene neighborhood (green), gene cooccurrence (dark blue), text mining (yellow), curated databases (cyan), experimentally determined (magenta), coexpression (black), protein homology (light blue), and gene fusions (red). All genes in cluster 1 were upregulated in S. gordonii following coaggregation. Solid lines indicate interactions between genes in the same gene cluster, whereas dotted lines indicate interactions between genes in different gene clusters.
Comparative analysis between different S. gordonii pairings.
We assessed the impact of coaggregation between S. gordonii and two key initial colonizers of dental plaque, Fusobacterium nucleatum and Veillonella parvula, on gene expression in each partner using the same approach described in this study (17, 18). We hypothesized that there are common mechanisms or pathways that are regulated in S. gordonii in response to coaggregation, independently of partner species. To examine this, we compared the differentially expressed genes of S. gordonii in each pairing (S. gordonii-S. oralis [SgSo], S. gordonii-F. nucleatum [SgFn], and S. gordonii-V. parvula [SgVp]) (Fig. 4). None of the genes were regulated commonly by coaggregation in all three bacterial pairings (Table S2).
FIG 4.

Venn diagram showing overlaps between three S. gordonii pairings. In total, six genes were common between S. gordonii-Fusobacterium nucleatum (SgFn) and S. gordonii-Veillonella parvula (SgVp), whereas only one gene was found in common between SgVp and S. gordonii-S. oralis (SgSo), and there were no genes in common between the SgSo versus SgFn pairings (see Table S2 in the supplemental material). Among the six genes that were regulated in SgFn and SgVp pairings, four genes were regulated in the same direction in two different bacterial pairings. These genes encode the tagatose-6-phosphate kinase (fold change = −3.2 in SgFn and −2.72 in SgVp), truncated hypothetical protein (fold change = 2.5 in SgFn and 2.62 in SgVp), short-chain dehydrogenase (fold change = −2.32 in SgFn and −2.75 in SgVp), and thiamine biosynthesis protein (fold change = −2.46 in SgFn and −2.04 in SgVp). Interestingly, two genes were regulated in the reverse direction in two different bacterial pairings. These genes encode the recombination regulator SGO_RS03085 (fold change = 2.84 in SgFn and −2.88 in SgVp) and Fur family transcriptional regulator (fold change = 2.42 in SgFn and −2.68 in SgVp). The one gene (pf08796 family protein) that was common between the SgSo and SgVp pairings was regulated in the same direction in both pairings.
It was noteworthy that the pairing with the evolutionarily most distant species, F. nucleatum, led to the highest number of S. gordonii genes regulated (119 genes). The pairing with V. parvula, another member of the phylum Firmicutes, led to regulation of 69 genes, whereas only 27 genes were regulated in S. gordonii following interactions with S. oralis. F. nucleatum cells are long and may bind multiple S. gordonii cells, further enhancing the potential to trigger gene regulation.
DISCUSSION
Coaggregation has been suggested to play a key role in promoting interactions between different bacteria that lead to profound phenotypic changes in the partner cells that enable them to proliferate in biofilm formation. Previous studies have shown that cell-cell interactions during coaggregation or biofilm formation lead to changes in gene expression in the partner organisms that may be important for adaptation to a community lifestyle (14, 15, 26, 27). Here, we studied the interaction between S. gordonii and S. oralis. Both were shown to form 3-dimensional coaggregate structures with cells of different species that were relatively evenly spread throughout. This is similar to the arrangements of cells that we previously observed in S. gordonii-F. nucleatum and S. gordonii-V. parvula coaggregates (17, 18). The close proximity of different cell types in these structures facilitates the exchange of signals or cues that modulate cell-cell sensing and gene regulation.
Interestingly, our analysis showed the downregulation of a cluster of tryptophan biosynthesis pathway-related genes in S. oralis. This cluster of genes was recently identified in S. oralis subsp. tigurinus (formerly Streptococcus tigurinus [27]) and S. gordonii DL1 and was suggested to represent a novel pathway for production of indole. In some Gram-negative bacteria, tryptophan and indole play important roles in cell-cell communication and biofilm formation (28). For example, the production of indole by Escherichia coli interferes with cell-cell communication pathways of Pseudomonas aeruginosa and promotes the growth of E. coli in mixed cultures (29). On the other hand, tryptophan inhibits biofilm formation by both E. coli and P. aeruginosa (30, 31). Recently, indole has been shown to enhance biofilm formation by the cariogenic oral bacterium Streptococcus mutans (32). It is possible that the exchange of tryptophan and/or indole between S. gordonii and S. oralis may modulate cell-cell sensing and biofilm formation.
The downregulation of ribosomal protein expression has previously been shown to be associated with growth rate (33). A similar effect of downregulation on S. oralis ribosomal proteins by Anaeroglobus geminatus has been demonstrated in proteomic analysis of polymicrobial biofilm model (34). It is possible that competition from S. gordonii led to a decrease in the rate of S. oralis growth in coaggregates, although the short timescale of the experiments here did not allow the measurement of growth rate. It is noteworthy that a decreased protein synthesis rate has been shown to be linked to expression of tryptophan biosynthesis genes. Thus, it was shown that trp genes were downregulated when protein synthesis was reduced in Escherichia coli (35). Therefore, the coaggregation-mediated downregulation of the trp operon in S. oralis may be linked to a more general decrease in growth rate.
It can be hypothesized that proximity of S. gordonii and S. oralis in coaggregates may enhance the interbacterial competition between them, resulting in upregulation of sensing systems that detect competitive molecules such as lantibiotics. However, at present there is no experimental evidence regarding the role of this two-component system in S. gordonii, and further work is needed to confirm a function in sensing antimicrobial peptides.
Our data suggest that the gene regulation is very specific to each pairing and that responses do not appear to be conserved. This indicates that the process of aggregation and the resultant increase in cell density is not the main driver behind gene regulation, even though autoaggregation has been shown to lead to changes in gene expression in other bacteria, such as F. nucleatum (36). This ability to distinguish between neighboring bacteria may be important for S. gordonii to adapt appropriately during the development of complex biofilms such as dental plaque. It is interesting that stronger gene regulation was observed in the pairing with the most distantly related microorganism (F. nucleatum), and the lowest regulation was observed with the intrageneric interaction (S. oralis). It is important to note that the absolute number of genes regulated is highly dependent on the thresholds applied and can be influenced by batch effects. A more rigorous comparison of gene regulation during interactions with a wider range of different oral microorganisms in experiments performed alongside one another is required to show whether the extent of gene regulation following cell-cell interactions is associated with evolutionary distance between the partner strains.
This study and our previous two S. gordonii pairing studies described a range of genes and pathways in S. gordonii-F. nucleatum, S. gordonii-V. parvula, and S. gordonii-S. oralis in response to coaggregation with each other (17, 18). Coaggregation was successfully employed as a model to interpret transcriptional changes involved in biofilm formation. Oral streptococci may have hundreds of different coaggregation partners in the oral cavity (37, 38). Our work indicates that the transcriptional responses of streptococci such as S. gordonii will be highly dependent upon their cell-cell interactions as oral biofilms develop. Consequently, it may be difficult to identify genes that are critical for biofilm development under all conditions and that may be targeted for biofilm control approaches. Nevertheless, more detailed analyses of transcriptomic and metatranscriptomic changes during the formation of dental plaque will continue to provide insights into how different species of oral bacteria adapt to the formation of polymicrobial communities.
MATERIALS AND METHODS
Routine culture of bacteria.
S. gordonii DL1 (Challis; ATCC 35105) and S. oralis 34 (formerly S. sanguis 34) (39) were routinely cultured statically at 37°C in THYE medium consisting of Todd Hewitt Broth (30 g · liter−1; Difco, Becton, Dickinson and Company, Oxford, UK) and yeast extract (5 g · liter−1; Melford Laboratories, Ipswich, UK) or on solidified THYE medium containing Bacto agar (15 g · liter−1; Difco, Becton, Dickinson). Alternatively, bacteria were cultured in BHYG medium containing (per liter) 37 g brain heart infusion (Becton, Dickinson), 5 g yeast extract, 2.5 g sodium glutamate (Sigma-Aldrich, Dorset, UK). All media were sterilized by autoclaving at 121°C for 15 min before use. For long-term storage, stocks of bacteria were maintained at −80°C in THYE medium supplemented with 50% glycerol. The purity of cultures was checked frequently by phase-contrast microscopy and by plating aliquots on agar plates.
DNA extraction and whole-genome sequencing.
Genomic DNA was purified from a 20-ml culture of S. oralis 34 using the MasterPure complete DNA and RNA purification kit (Epicentre Biotechnologies, Madison, WI) as instructed by the manufacturer. The extracted DNA was checked by agarose gel electrophoresis and NanoDrop spectrophotometry prior to being sent to the sequencing service group, MicrobesNG, at the University of Birmingham for sequencing. The sequencing was done using the Illumina HiSeq 2500 platform with a paired-end strategy with 100-bp reads. The de novo genome assembly was done using SPAdes v. Dec-2017 (40).
Saliva preparation.
Ethical approval for the collection of saliva from healthy volunteers was obtained from the Newcastle University Research Ethics Committee (reference 14898/2018). All saliva donors were given a participant information sheet and gave written informed consent to participate in the study. Saliva was collected and pooled from five healthy individuals who had not eaten for at least 2 h prior to collection. Saliva was stimulated by chewing on Parafilm and was placed on ice immediately after collection. The reducing agent dithiothreitol (DTT) was added to a final concentration of 2.5 mM, and saliva samples were gently stirred on ice for 10 min. Aggregated particles were removed by centrifugation at 15,000 × g and 4°C for 30 min. Three volumes of H2O were added to 1 volume of saliva and sterilized by filtration through a 0.22-μm-pore membrane. Aliquots were stored at −20°C. The 25% saliva was thawed at 37°C immediately before use and any precipitate that had formed was removed by centrifugation at 1,400 × g and 20°C for 10 min.
Coaggregation assays.
S. gordonii DL1 and S. oralis 34 were cultured at 37°C in THYE medium for 18 h, harvested by centrifugation at 3,800 × g for 10 min, and washed three times with one volume of phosphate-buffered saline (PBS; pH 7.3). Cells were resuspended in one volume of PBS and adjusted to an optical density at 600 nm (OD600) of 1.0 to give a final concentration of approximately 1 × 109 CFU/ml. To visualize S. gordonii cells, Syto 9 (Life Technologies Ltd., Paisley, UK) was added to cells to achieve 7.5 μM. S. oralis 34 was stained by addition of 4′-6-diamidino-2-phenylindole (DAPI) (2.5 μg/ml final concentration; Thermo Scientific) in 1 ml of PBS solution containing bacterial cells. Cells were incubated at 37°C in the dark for 10 min. Fluorescently stained bacteria were washed twice with PBS and resuspended in 1 ml of 25% cell-free saliva. To induce coaggregation in dual-species cultures, 500 μl of each species were mixed by vortex for 10 s in glass test tubes and gently rocked by hand until coaggregation was visible. Samples were visualized using a 60× lens objective on an Olympus BX61 microscope (Olympus Corporation, Tokyo, Japan), equipped with a dichroic mirror to split the excitation and emission wavelengths. Images were captured using an Olympus XM10 monochrome camera.
To assess gene regulation responses to coaggregation, S. gordonii and S. oralis were cultured for 18 h at 37°C in BHYG medium, subcultured into fresh medium, and grown at 37°C to the mid-exponential phase (OD600 = 0.4 to 0.6). Cells were harvested at 3,800 × g and 20°C in a swing-out rotor for 10 min and adjusted to an OD600 of 1.0 ± 0.2. A 5-ml aliquot of each culture was harvested at 3,800 × g and 20°C for 5 min and resuspended in 0.5 ml of 25% saliva. Samples were divided into two equal portions. One was used for monoculture controls, while the other samples of each species were mixed together. Samples were mixed vigorously using a vortex mixer for 10 s. All samples were made up to 5 ml by the addition of 25% saliva and were incubated at 37°C for 30 min. RNAlater (5 ml; Invitrogen) was added, and the tubes were vortex mixed for 5 s and incubated at 20°C for 5 min. Cells were harvested at 3,000 × g for 15 min at 20°C, and the pellets were frozen at −80°C for subsequent RNA extraction.
RNA-Seq data sets.
Six biological replicates of S. gordonii monoculture, five replicates of S. oralis monoculture, and five replicates for the S. gordonii-S. oralis mixed culture were used. In total, 16 samples were used in this study.
RNA extraction.
To disrupt cells for RNA extraction, samples were thawed at 20°C and resuspended in 100 μl spheroplasting buffer containing 0.1 mg/ml spectinomycin (41). Mutanolysin was added to 500 U/ml, and cells were incubated at 37°C for 5 min. Total RNA was extracted using the Ambion RiboPure bacteria RNA purification kit (Life Technologies) according to the manufacturer’s instructions. RNA concentrations were determined using a NanoDrop ND-1000 spectrophotometer (Thermo Scientific). To ensure that RNA had not degraded during extraction, an aliquot of each sample was analyzed by gel electrophoresis.
Library preparation and whole-transcriptome sequencing.
Library preparation and sequencing were performed by the established and internationally recognized sequencing provider BGI Tech Solutions (Hong Kong). Following an initial rRNA depletion, first-strand cDNA synthesis was performed using random hexamer primers. The second-strand cDNA was synthesized using buffer, deoxynucleoside triphosphates (dNTPs), RNase H, and DNA polymerase I, respectively, after removing dNTPs. Short fragments were purified with the QIAquick PCR extraction kit and resuspended in elution buffer for end repair and addition of poly(A) tails. The short fragments were ligated to sequencing adapters. Uracil N-glycosylase enzyme was used to degrade the second-strand cDNA, and products were purified by MinElute PCR purification kit before PCR amplification. All libraries were sequenced using the Illumina HiSeq 2500 platform with a paired-end sequencing strategy.
Read mapping and preprocessing.
All raw reads generated from Illumina sequencing platform were preprocessed before mapping to reference genomes. Illumina adapters and low-quality reads (Q < 20) were removed with Trimmomatic v. 0.36. FastQC was used to verify removal of low-quality reads and adapters. Reads from S. gordonii monoculture were mapped to the NCBI reference genome (accession number NC_009785.1), whereas the reads from S. oralis were mapped to the assembled genome of S. oralis 34 that we sequenced in this study, using TopHat v1.0.14 with default parameters. Five replicates of mixed samples were mapped separately in two rounds to the reference genomes of S. gordonii and S. oralis and designated “SgSo_Sg” (reads of coaggregate culture mapped to S. gordonii reference genome from NCBI) and “SgSo_So” (reads of coaggregate culture mapped to S. oralis). After read mapping, SAMtools (42) was employed to calculate mapping statistics.
Gene expression quantification, normalization, and differential expression analysis.
All mapped reads were used for quantifying gene expression using HTseq-count. HTseq (43) required a gene feature format (GFF) annotation file (mode = union, -t = gene, -i = locus_tag), and the standard gene annotations provided with reference genomes were used. Box plots were generated using in-house scripts to evaluate whether the normalization works well for all samples before downstream analyses. Comparisons were made between monoculture (S. gordonii or S. oralis) and coaggregate samples (SgSo_Sg and SgSo_So). Differential expression analyses between monoculture (S. gordonii or S. oralis) and coaggregate samples (SgSo_Sg and SgSo_So) were performed using the Bioconductor package DESeq v. 3.854 in the R statistical software program. DESeq-normalized gene count data were based on “size factors” to account for RNA-Seq library size differences, and dispersion estimates were calculated. Pairwise comparisons of expression were made between the monoculture and mixed-sample group for every replicate based on a negative binomial model. Fold changes were obtained along with their associated P values. A gene was defined as significantly expressed if it had a P value of <0.05 and a fold change of at least 2.
STRING interaction network analysis.
The STRING v. 11.0 database was used to predict if there were any functional associations of differentially regulated significant genes (44). The search tool for retrieval of interacting genes/proteins (STRING) was used to identify known and predicted interactions based on evidence from different sources such as experiments, databases, neighborhood, text mining, cooccurrence, coexpression, gene fusion, and databases) using default settings. Nodes represent differentially expressed genes, and edges indicate the level of confidence in the association, with thicker lines indicating greater confidence. The network was clustered using the Markov cluster (MCL) clustering method with a specified “MCL inflation parameter” of 3. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed using STRING.
Comparative analysis of S. gordonii in response to coaggregation with F. nucleatum, V. parvula, or S. oralis.
Using a Venn diagram, the S. gordonii genes that were common to three pairs of comparisons and the genes that were shared between any two pairs were identified. The genes commonly expressed from the three pairings were further investigated with STRING database analysis to explain possible common genes and pathways.
Data availability.
Raw sequence reads were deposited in the NCBI Sequence Read Archive (SRA) database under accession numbers SRR12650300, SRR12650301, SRR12650302, SRR12650303, SRR12650304, SRR12650305, SRR12650306, SRR12650307, SRR12650308, SRR12650309, SRR12650310, SRR12650311, SRR12650312, SRR12650313, SRR12650314, and SRR12650315. The genome sequence of S. oralis 34 can be accessed in the GenBank database under accession number JAHKGX000000000.
ACKNOWLEDGMENTS
We gratefully acknowledge Genliang Zhu for his bioinformatics support and for assisting in data analyses. We thank Ekaterina Kozhevnikova and Nicola Griffiths for expert technical assistance.
W.K.M. was supported by a PhD studentship from the Ministry of Higher Education and Scientific Research (Iraq). N.K. was supported from EPSRC Grants EP/J004111/2 and EP/N031962/1. This publication was supported by NSFC International Young Scientists Fund (project no. 31750110452) and by the high-level talent recruitment program for academic and research platform construction (reference no. 5000105) from Wenzhou-Kean University.
S.W.C., G.Y.A.T., and N.S.J. conceived the project; N.V.R.M., W.K.M., N.R., and H.A. conducted experimental work and data analysis; N.K., G.Y.A.T., Y.L., S.W.C., and N.S.J. advised on the analysis of data; S.W.C., N.V.R.M., W.K.M., and N.S.J. wrote the manuscript; all authors critically reviewed and approved the final version.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Siew Woh Choo, Email: cwoh@wku.edu.cn.
Nicholas S. Jakubovics, Email: nick.jakubovics@ncl.ac.uk.
Andrew J. McBain, University of Manchester
REFERENCES
- 1.Zaura E, Keijser BJ, Huse SM, Crielaard W. 2009. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol 9:259. 10.1186/1471-2180-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eren AM, Borisy GG, Huse SM, Mark Welch JL. 2014. Oligotyping analysis of the human oral microbiome. Proc Natl Acad Sci USA 111:E2875–E2884. 10.1073/pnas.1409644111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mark Welch JL, Dewhirst FE, Borisy GG. 2019. Biogeography of the oral microbiome: the site-specialist hypothesis. Annu Rev Microbiol 73:335–358. 10.1146/annurev-micro-090817-062503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jakubovics NS, Yassin SA, Rickard AH. 2014. Community interactions of oral streptococci. Adv Appl Microbiol 87:43–110. 10.1016/B978-0-12-800261-2.00002-5. [DOI] [PubMed] [Google Scholar]
- 5.Hsu S, Cisar JO, Sandberg AL, Kilian M. 1994. Adhesive properties of viridans streptoccocal species. Microb Ecol Health Dis 7:125–137. [Google Scholar]
- 6.Zheng W, Tan MF, Old LA, Paterson IC, Jakubovics NS, Choo SW. 2017. Distinct biological potential of Streptococcus gordonii and Streptococcus sanguinis revealed by comparative genome analysis. Sci Rep 7:2949. 10.1038/s41598-017-02399-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer RJ, Shah N, Valm A, Paster B, Dewhirst F, Inui T, Cisar JO. 2017. Interbacterial adhesion networks within early oral biofilms of single human hosts. Appl Environ Microbiol 83:e00407-17. 10.1128/AEM.00407-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu H, Sobue T, Thompson A, Xie Z, Poon K, Ricker A, Cervantes J, Diaz PI, Dongari-Bagtzoglou A. 2014. Streptococcal co-infection augments Candida pathogenicity by amplifying the mucosal inflammatory response. Cell Microbiol 16:214–231. 10.1111/cmi.12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Souza JGS, Bertolini M, Thompson A, Mansfield JM, Grassmann AA, Maas K, Caimano MJ, Barao VAR, Vickerman MM, Dongari-Bagtzoglou A. 2020. Role of glucosyltransferase R in biofilm interactions between Streptococcus oralis and Candida albicans. ISME J 14:1207–1222. 10.1038/s41396-020-0608-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jakubovics NS, Kerrigan SW, Nobbs AH, Strömberg N, Dolleweerd CJV, Cox DM, Kelly CG, Jenkinson HF. 2005. Functions of cell surface-anchored antigen I/II family and Hsa polypeptides in interactions of Streptococcus gordonii with host receptors. Infect Immun 73:6629–6638. 10.1128/IAI.73.10.6629-6638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roky M, Trent JO, Demuth DR. 2020. Identification of functional domains of the minor fimbrial antigen involved in the interaction of Porphyromonas gingivalis with oral streptococci. Mol Oral Microbiol 35:66–77. 10.1111/omi.12280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolenbrander PE, Palmer RJ, Periasamy S, Jakubovics NS. 2010. Oral multispecies biofilm development and the key role of cell-cell distance. Nat Rev Microbiol 8:471–480. 10.1038/nrmicro2381. [DOI] [PubMed] [Google Scholar]
- 13.Ramsey MM, Whiteley M. 2009. Polymicrobial interactions stimulate resistance to host innate immunity through metabolite perception. Proc Natl Acad Sci USA 106:1578–1583. 10.1073/pnas.0809533106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dutton LC, Paszkiewicz KH, Silverman RJ, Splatt PR, Shaw S, Nobbs AH, Lamont RJ, Jenkinson HF, Ramsdale M. 2016. Transcriptional landscape of trans-kingdom communication between Candida albicans and Streptococcus gordonii. Mol Oral Microbiol 31:136–161. 10.1111/omi.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hendrickson EL, Beck DAC, Miller DP, Wang Q, Whiteley M, Lamont RJ, Hackett M. 2017. Insights into dynamic polymicrobial synergy revealed by time-coursed RNA-Seq. Front Microbiol 8:261. 10.3389/fmicb.2017.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jakubovics NS, Gill SR, Iobst SE, Vickerman MM, Kolenbrander PE. 2008. Regulation of gene expression in a mixed-genus community: stabilized arginine biosynthesis in Streptococcus gordonii by coaggregation with Actinomyces naeslundii. J Bacteriol 190:3646–3657. 10.1128/JB.00088-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mutha NVR, Mohammed WK, Krasnogor N, Tan GYA, Choo SW, Jakubovics NS. 2018. Transcriptional responses of Streptococcus gordonii and Fusobacterium nucleatum to coaggregation. Mol Oral Microbiol 33:450–464. 10.1111/omi.12248. [DOI] [PubMed] [Google Scholar]
- 18.Mutha NVR, Mohammed WK, Krasnogor N, Tan GYA, Wee WY, Lee Y, Choo SW, Jakubovics NS. 2019. Transcriptional profiling of coaggregation interactions between Streptococcus gordonii and Veillonella parvula by Dual RNA-Seq. Sci Rep 9:7664. 10.1038/s41598-019-43979-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levin-Sparenberg E, Shin JM, Hastings EM, Freeland M, Segaloff H, Rickard AH, Foxman B. 2016. High-throughput quantitative method for assessing coaggregation among oral bacterial species. Lett Appl Microbiol 63:274–281. 10.1111/lam.12622. [DOI] [PubMed] [Google Scholar]
- 20.Palmer RJ, Jr, Gordon SM, Cisar JO, Kolenbrander PE. 2003. Coaggregation-mediated interactions of streptococci and actinomyces detected in initial human dental plaque. J Bacteriol 185:3400–3409. 10.1128/JB.185.11.3400-3409.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cisar JO, Kolenbrander PE, McIntire FC. 1979. Specificity of coaggregation reactions between human oral streptococci and strains of Actinomyces viscosus or Actinomyces naeslundii. Infect Immun 24:742–752. 10.1128/iai.24.3.742-752.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhagwat SP, Nary J, Burne RA. 2001. Effects of mutating putative two-component systems on biofilm formation by Streptococcus mutans UA159. FEMS Microbiol Lett 205:225–230. 10.1111/j.1574-6968.2001.tb10952.x. [DOI] [PubMed] [Google Scholar]
- 24.Mattos-Graner RO, Duncan MJ. 2017. Two-component signal transduction systems in oral bacteria. J Oral Microbiol 9:1400858. 10.1080/20002297.2017.1400858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jimenez JC, Federle MJ. 2014. Quorum sensing in group A streptococcus. Front Cell Infect Microbiol 4:127. 10.3389/fcimb.2014.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohammed WK, Krasnogor N, Jakubovics NS. 2018. Streptococcus gordonii Challisin protease is required for sensing cell-cell contact with Actinomyces oris. FEMS Microbiol Ecol 94:fiy043. 10.1093/femsec/fiy043. [DOI] [PubMed] [Google Scholar]
- 27.Jensen A, Scholz CFP, Kilian M. 2016. Re-evaluation of the taxonomy of the Mitis group of the genus Streptococcus based on whole genome phylogenetic analyses, and proposed reclassification of Streptococcus dentisani as Streptococcus oralis subsp. dentisani comb. nov., Streptococcus tigurinus as Streptococcus oralis subsp. tigurinus comb. nov., and Streptococcus oligofermentans as a later synonym of Streptococcus cristatus. Int J Syst Evol Microbiol 66:4803–4820. 10.1099/ijsem.0.001433. [DOI] [PubMed] [Google Scholar]
- 28.Lee JH, Lee J. 2010. Indole as an intercellular signal in microbial communities. FEMS Microbiol Rev 34:426–444. 10.1111/j.1574-6976.2009.00204.x. [DOI] [PubMed] [Google Scholar]
- 29.Chu W, Zere TR, Weber MM, Wood TK, Whiteley M, Hidalgo-Romano B, Valenzuela E, Jr, McLean RJ. 2012. Indole production promotes Escherichia coli mixed-culture growth with Pseudomonas aeruginosa by inhibiting quorum signaling. Appl Environ Microbiol 78:411–419. 10.1128/AEM.06396-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimazaki J, Furukawa S, Ogihara H, Morinaga Y. 2012. l-Tryptophan prevents Escherichia coli biofilm formation and triggers biofilm degradation. Biochem Biophys Res Commun 419:715–718. 10.1016/j.bbrc.2012.02.085. [DOI] [PubMed] [Google Scholar]
- 31.Brandenburg KS, Rodriguez KJ, McAnulty JF, Murphy CJ, Abbott NL, Schurr MJ, Czuprynski CJ. 2013. Tryptophan inhibits biofilm formation by Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:1921–1925. 10.1128/AAC.00007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inaba T, Oura H, Morinaga K, Toyofuku M, Nomura N. 2015. The Pseudomonas quinolone signal inhibits biofilm development of Streptococcus mutans. Microbes Environ 30:189–191. 10.1264/jsme2.ME14140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nomura M, Gourse R, Baughman G. 1984. Regulation of the synthesis of ribosomes and ribosomal components. Annu Rev Biochem 53:75–117. 10.1146/annurev.bi.53.070184.000451. [DOI] [PubMed] [Google Scholar]
- 34.Bao K, Bostanci N, Thurnheer T, Belibasakis GN. 2017. Proteomic shifts in multi-species oral biofilms caused by Anaeroglobus geminatus. Sci Rep 7:4409. 10.1038/s41598-017-04594-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng WL, Kazmierczak KM, Robertson GT, Gilmour R, Winkler ME. 2003. Transcriptional regulation and signature patterns revealed by microarray analyses of Streptococcus pneumoniae R6 challenged with sublethal concentrations of translation inhibitors. J Bacteriol 185:359–370. 10.1128/JB.185.1.359-370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merritt J, Niu G, Okinaga T, Qi F. 2009. Autoaggregation response of Fusobacterium nucleatum. Appl Environ Microbiol 75:7725–7733. 10.1128/AEM.00916-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz PI, Chalmers NI, Rickard AH, Kong C, Milburn CL, Palmer RJ, Jr, Kolenbrander PE. 2006. Molecular characterization of subject-specific oral microflora during initial colonization of enamel. Appl Environ Microbiol 72:2837–2848. 10.1128/AEM.72.4.2837-2848.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katharios-Lanwermeyer S, Xi C, Jakubovics NS, Rickard AH. 2014. Mini-review: microbial coaggregation: ubiquity and implications for biofilm development. Biofouling 30:1235–1251. 10.1080/08927014.2014.976206. [DOI] [PubMed] [Google Scholar]
- 39.Abeygunawardana C, Bush CA, Tjoa SS, Fennessey PV, McNeil MR. 1989. The complete structure of the capsular polysaccharide from Streptococcus sanguis 34. Carbohydr Res 191:279–293. 10.1016/0008-6215(89)85071-2. [DOI] [PubMed] [Google Scholar]
- 40.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jakubovics NS. 2015. Intermicrobial interactions as a driver for community composition and stratification of oral biofilms. J Mol Biol 427:3662–3675. 10.1016/j.jmb.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anders S, Pyl PT, Huber W. 2015. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. 2019. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47:D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1 and S2, Fig. S1 and S2. Download AEM.01558-21-s0001.pdf, PDF file, 0.1 MB (240.6KB, pdf)
Data Availability Statement
Raw sequence reads were deposited in the NCBI Sequence Read Archive (SRA) database under accession numbers SRR12650300, SRR12650301, SRR12650302, SRR12650303, SRR12650304, SRR12650305, SRR12650306, SRR12650307, SRR12650308, SRR12650309, SRR12650310, SRR12650311, SRR12650312, SRR12650313, SRR12650314, and SRR12650315. The genome sequence of S. oralis 34 can be accessed in the GenBank database under accession number JAHKGX000000000.