

Abstract
Signaling through fibroblast growth factor receptor one (FGFR1) is a known inducer of proliferation in both embryonic and human adult mesenchymal stem cells (hMSCs) and positively regulates maintenance of stem cell viability. Leveraging the mitogenic potential of FGF2/FGFR1 signaling in stem cells for therapeutic applications necessitates a mechanistic understanding of how this receptor stimulates cell cycle progression. Using small interfering RNA (siRNA) depletion, antibody-inhibition, and small molecule inhibition, we establish that FGFR1 activity is rate limiting for self-renewal of hMSCs. We show that FGFR1 promotes stem cell proliferation through multiple mechanisms that unite to antagonize cyclin-dependent kinase (CDK) inhibitors. FGFR1 not only stimulates c-Myc to suppress transcription of the CDK inhibitors p21Waf1 and p27Kip1, thus promoting cell cycle progression but also increases the activity of protein kinase B (AKT) and the level of S-phase kinase-associated protein 2 (Skp2), resulting in the nuclear exclusion and reduction of p21Waf1. The in vivo importance of FGFR1 signaling for the control of proliferation in mesenchymal progenitor populations is underscored by defects in ventral mesoderm formation during development upon inhibition of its signaling. Collectively, these studies demonstrate that FGFR1 signaling mediates the continuation of MSC growth and establishes a receptor target for enhancing the expansion of mesenchymal progenitors while maintaining their multilineage potential.
Keywords: Adult stem cells, Fibroblast growth factor, Heparin-binding, Cell cycle, Cell expansion
Introduction
Mesenchymal stem cells (MSCs) remain quiescent in the bone marrow until they are stimulated to divide in response to particular growth factors, cytokines, or hormones. Such ligands initiate signaling cascades by binding and activating specific cell surface receptors; these trigger transit beyond a key cell cycle stage, the restriction (R) point, in the G1 phase. This trigger ultimately starts DNA replication during S phase and the completion of cell division at mitosis [1–4]. Fibroblast growth factors (FGFs) are known to be particularly potent at controlling cell growth and differentiation (see review [5]). Autocrine FGF-2, which is stored and protected in the extracellular matrix, is known to regulate MSC proliferation [6]. FGF-2 activity is dependent on its binding to one of four tyrosine kinase receptors (fibroblast growth factor receptor [FGFR1–4]) [7], with FGFR1 being particularly important for the control of MSC phenotype [8–16].
We have previously shown that FGFR1 signaling promotes MSC proliferation and negatively regulates osteogenic differentiation and matrix mineralization [8]. Further evidence also suggests that FGFR1 is a putative marker for naive MSCs in vivo [17]. Furthermore, when added during the in vitro expansion of MSCs, FGF2 increases growth rates while maintaining multilineage capability [15, 16, 18, 19], although osteogenic differentiation is inhibited in the continued presence of FGF2 [8, 20–22], and FGFR1 deficiency has been shown to enhance mineralization in mature osteoblasts [11]. Taken together, these results suggest that control over FGF2:FGFR1 signaling will be crucial for the development of MSC therapeutics.
FGFR1-stimulated cell cycle progression requires that cells pass through the restriction point (R-point) [3, 23, 24]. Passage beyond this checkpoint requires mitogenic signals from at least two different growth factors and renders cell cycle progression free of exogenously added mitogenic stimuli; loss of the R-point results in uncontrolled proliferation [3, 25]. The sequential phosphorylation/inactivation of retinoblastoma (Rb) by cyclin-dependent kinase (CDK) complexes leads to E2F release and the induction of E2F-dependent gene transcription. E2F-1 regulates the transcription of genes implicated in nucleotide synthesis, DNA replication, and the cell cycle control necessary for G1/S progression [26, 27]. In G1, the first complex to form is composed of D cyclins and CDK4/6 which phosphorylate Rb on serine 780, so impeding its binding to E2F-1 [28] leading to complete Rb inactivation and S-phase progression. How FGFR1 signaling controls the pRb/E2F dependent commitment for cell cycle progression at the R-point in MSCs has not been explored in significant detail.
FGFR1 signaling may induce cell proliferation by stimulating CDK activity and/or by inhibiting cyclin kinase inhibitors (CDKNs) to facilitate the CDK-mediated release of pRb from its activation of pRb/E2F complexes. Two classes of CDKNs have been identified: the INK4/CDKN2 family (p15, p16, p18, and p19) and the Cip/Kip/CDKN1 family (p21Waf1, p27Kip1, and p57Kip2). Proteins in the INK4 family block cyclin D-associated kinase activity [29], thus preventing activation of the CyclinE-CDK2 complexes and the initial phosphorylation of Rb. In contrast, the Cip/Kip family of proteins inhibit most of the CDK complexes, including cyclin D-CDK4/6, cyclin E-CDK2, and cyclin A-CDK2 [29–32]. In this study, we demonstrate that FGFR1 promotes human MSC (hMSC) proliferation by suppressing the expression of p21Waf1/CDKN1A and p27Kip1/CDKN1B expression through c-Myc/MYC, as well as by promoting the nuclear exclusion and degradation of these two CDKNs through the phosphoinositide 3-kinase (PI3K)/AKT pathway, so driving cell cycle progression through the restriction point. These findings provide a mechanistic rationale for targeting FGFR1 activity in therapeutic applications that require the modulation of either self-renewal capacity or the multilineage potential of hMSCs.
Materials and Methods
Reagents
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich (St. Louis, MO, http://www.sigmaaldrich.com).
Plasmids
Constitutively active FGFR1 (Fc-R1TK) was prepared by cloning the constant portion of human IgG1 (a generous gift from Mark Spanevello, Queensland Institute of Medical Research, Australia) to the N-terminus of the transmembrane and tyrosine kinase domains of FGFR1 IIIc (generously provided by Ong Siew Hwa and Graeme Guy, Institute of Cell and Molecular Biology, A*STAR, Singapore) into pcDNA3.1 (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) as previously described [33]. Full-length cDNAs encoding human p21 and p27 (a generous gift from Philip Kaldis, Institute of Cell and Molecular Biology, A*STAR, Singapore) were cloned into pEGFP-C1 (Clontech, Mountain View, CA, http://www.clontech.com). The fibroblast receptor substrate two (FRS2) expression construct was a generous gift from Graeme Guy. The cyclin-dependent kinase subunit one (Cks1) expression vector was purchased from Origene (Rockville, MD, http://www.origene.com). The pGL3–6xOSE-Luc and pGL3-p21prom-Luc constructs have been previously described [34, 35].
Cell Culture
hMSCs from adult bone marrow (Lonza, Walkersville, MD, http://www.lonza.com) were maintained in Dulbecco’s modified Eagle’s medium (1,000 mg/L glucose) supplemented with 10% fetal bovine serum (FBS) (Hyclone, Logan, UT, http://www.hyclone.com), penicillin/streptomycin, and 2 mM l-glutamine (maintenance media). Media was changed every third day unless stated otherwise. The pathway chemical inhibitors were purchased from Calbiochem (San Diego, http://www.emdbiosciences.com) and used at the following concentrations: 10 μM U0126, 25 μM SU5402, 10 μM LY294002, 10 μM Calphostin C, 10 μM SB202190, 100 μM U-73122, 30 μM Raf1 inhibitor 1, and the vehicle dimethyl sulfoxide (DMSO) (1:1,000) as the control. FGF2 was used at 2.5 ng/ml for the entire study. For serum starvation, cells were cultured in maintenance media supplemented with 0.2% FBS for 48 hours.
Cell Assays
Cell counts, cell cycle, and apoptosis assays were analyzed on a GUAVA PCA-96 benchtop flow cytometer following the manufacturer’s instructions (Millipore, Billerica, MA, http://www.millipore.com). For proliferation assays, cells were seeded at 3,000 cells per square centimeter in maintenance media and the following day the media was changed. The cells were grown for a further 6 days under the various treatment conditions and cell numbers were determined using the Viacount FLEX reagent and software (Millipore). For cell cycle assays, cells were either serum starved and released in the presence of a control antibody or an FGFR1-neutralizing antibody [20]. In parallel experiments, antibodies were added directly to cells in logarithmic growth phase. Cell cycle distribution was assessed by propidium iodide (PI) staining with the Guava cell cycle software. Cell viability was determined using green-fluorescent fluorescein diacetate (CellTracker Green carboxyfluorescein diacetate (CFDA), Invitrogen) when cells were at 50% confluence. The levels of cellular fluorescence were measured using a FACSarray bioanalyser (BD Bioscience, San Diego, CA, http://www.bdbiosciences.com).
Cell Cycle Progression
Cells in logarithmic growth phase were seeded at 15,000 cells per square centimeter and serum deprived (0.2% FBS) for 48 hours. Quiescent cells were then released into maintenance media containing vehicle or FGFR1 inhibitor (SU5402). For baseline determination, triplicates from each condition were harvested immediately prior to release into maintenance media (0 hour time point). Total RNA and/ or protein were harvested at various intervals after release.
Promoter Activity Assay
Cells were electroporated with 60 μg of the reporters and 600 ng of CMV-Renilla luciferase and 6 μg of either empty plasmid or the c-Myc expression plasmid as previously described [36] and seeded into 12-well plates containing either 25 μM SU5402 or DMSO. After 48 hours, the levels of firefly and renilla luciferase were measured using the Dual Luciferase Reporter Assay System (Promega, Madison, WI, http://www.promega.com). Results were normalized by dividing the firefly light units by the renilla light units. Transfection experiments were repeated in triplicate.
p21 and p27 Fusion Protein Localization
Cells were seeded at 15,000 cells per square centimeter in 12-well plates overnight then transfected using Lipofectamine LTX Plus reagent and 800 ng per well of pEGFP-p21 or pEGFP-p27 in OPTI-MEM media (Invitrogen). After 4 hours, the media was replaced with maintenance media alone or supplemented with DMSO, SU5402, LY294002, or 0.2% FBS media. Photomicrographs were taken after 24 hours with an Olympus BX51 microscope.
Immunoprecipitation
hMSCs were seeded at 15,000 cells per square centimeter in 6 cm dishes overnight. The following day, cells were transfected with 1.5 μg Fc-R1TK, 0.75 μg FRS2 and 0.75 μg Cks1 expression plasmids using Lipofectamine LTX (Invitrogen). After 24 hours, cells were washed with phosphate buffered saline (PBS), disrupted in lysis buffer (20 mM HEPES pH 7.5, 137 mM NaCl, 1% Triton X-100, 10% glycerol, 1.5 mM MgCl2, 1 mM ethylene glycol tetraacetic acid (EGTA), 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1× protease inhibitor cocktail), mixed with protein A magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany, http://www.miltenyibiotec.com) for 1 hour, and purified as per the manufacturer’s instructions.
Antibodies
Mouse anti-actin (MAb1501R) was purchased from Chemicon (Temecula, CA, http://www.chemicon.com). Rabbit anti-extracellular signal-regulated kinase (ERK)1/2 (M5670) and rabbit anti-phospho ERK1/2 (M8159) were from Sigma-Aldrich. Mouse anti-c-Myc (#9402), rabbit anti-AKT (#9272), rabbit anti-phospho AKT (#9271), mouse anti-CDK4 (#2906), rabbit anti-p15ink4 (#4822), rabbit anti-p16ink4 (#4824), rabbit anti-phospho FRS2 (#3864), and mouse anti-phospho FGFR1 (#3476) were purchased from Cell Signaling (Beverly, MA, http://www.cellsignal.com). Rabbit anti-FRS2 (Ab10425) was purchased from Abcam (Cambridge, U.K., http://www.abcam.com). Rabbit anti-Cks1 (sc-6238) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, http://www.scbt.com). Mouse anti-Skp2 (S-phase kinase-associated protein two) was from Invitrogen. Mouse anti-p27Kip1 (DCS-72.F6) was purchased from ThermoFisher Scientific (Rockford, IL, http://www.thermoscientific.com). Mouse anti-p21waf1 (#556430) was purchased from BD Biosciences (San Diego, CA, http://www.bdbiosciences.com). Donkey anti-human IgG (709–035-149), goat anti-mouse IgG-horseradish peroxidase (HRP) (115–035-062), and goat anti-rabbit IgG (111–035-045) were purchased from Jackson ImmunoResearch (West Grove, PA, http://www.jacksonimmuno.com).
Immunoblotting
Lysates were loaded into 4%–12% precast gels and separated at 200 V for 1 hour using the XCell system (Invitrogen). Proteins were transferred onto nitrocellulose membranes and blocked with TBST with 5% nonfat dry milk for 1 hour. The primary antibodies were incubated overnight on an orbital shaker, and secondary antibodies were incubated for 1 hour before detection with chemiluminescence (Pierce, Rockford, IL, http://www.piercenet.com). We typically probed for multiple antigens in parallel with the same samples, unless indicated otherwise in the figure legends (that is, the same blots were reprobed for different antigens).
siRNA Silencing
The siRNA against human FGFR1 (Gen-BankTM accession number NM_015850) was purchased from Qiagen (Hs_FGFR1_6 cat # 8102224677; Hilden, Germany, http://www1.qiagen.com). Cells were seeded at 15,000 cells per square centimeter (50% confluence) overnight and then transfected with siRNA using HiPerfect (Qiagen) following the manufacturer’s instructions. The cells were then incubated at 37°C for another 4 days to achieve maximum knockdown (70%−80%, data not shown). A luciferase siRNA (Sigma-Proligo) was used as a control in all experiments.
RNA Purification and Quantitative Polymerase Chain Reaction
RNA from the gene silencing experiment was purified using a Nucleospin II kit or Nucleospin RNA/Protein kit (Machery-Nagel, Germany, Düren, http://www.mn-net.com). Both RNA quality and concentration were assessed by spectrometry, and 0.5 μg was used for reverse transcription using Superscript III polymerase (Invitrogen). Quantitative polymerase chain reaction (PCR) was performed on 20 ng of cDNA/sample using specific Taqman primer probes [8] from Life Technologies, ABI, Carlsbad, CA, http://www.lifetechnologies.com (Histone H4: Hs00269118_s1, Cyclin B2: Hs00270424_m1, CDKN1A (p21waf): Hs00355782_m1, CDKN1B (p27Kip1): Hs00153277_m1, c-Myc: Hs00153408_m1, FGFR1: Hs00241111_m1.
Preparation of Xenopus laevis Embryos
Xenopus laevis eggs were fertilized, cleaned (dejellied), and prepared for injection in 1X MMR (Marc’s Modified Ringer) media supplemented with 5% Ficoll (Sigma). The anti-FGFR1 blocking solution was prepared at a 1:250 dilution in double-distilled water. Embryos were injected at the 4-cell stage with 6 nL per cell of anti-FGFR1 blocking solution. Embryos were either injected into both dorsal cells, both ventral cells or remained uninjected as controls. The embryos were allowed to recover for 2 hours in 1×MMR media supplemented with 5% Ficoll and were subsequently transferred to 0.1 × MMR media. The embryos were allowed to develop until stage 35/36 before being fixed in 3.7% formaldehyde in 1×MEM-salts containing 3-(N-morpholino)propanesulfonic acid (MOPS), EGTA, and MgSO4 at pH 7.4 and photographed under light microscopy.
Statistical Analysis
Error bars in the figures represent the mean and SD of at least three biological samples. Student’s t test was performed to evaluate whether the difference between two conditions was significant (p<0.05). Significant differences were marked with an asterisk (*).
Results
FGFR1 Signaling is Required for hMSC Proliferation
FGFR1 expression levels correlate with cell proliferation [37]. We therefore investigated the functional role of FGFR1 in cell proliferation by loss-of-function analysis using three different methods: siRNA depletion, antibody inhibition, or small molecule inhibition. We first depleted FGFR1 in hMSCs using siRNA and observed a 60% decrease in FGFR1 expression and a 25% decrease in cell growth (Fig. 1). In contrast, blocking the receptor with either an FGFR1-neutralizing antibody [20] or the chemical inhibitor SU5402 [38] decreased cell proliferation by approximately 80% without affecting the levels of FGFR1 expression (Fig. 1). The similarities in biological effects obtained with siFGFR1 as opposed to FGFR1 inhibition (using either an antibody or small molecule) indicate that the mitogenic functions of the receptor can be effectively perturbed by suppressing either FGFR1 activity or overall receptor levels on the cell surface. Because data obtained by the three different methods provide both mutually corroborating and biologically consistent outcomes, our results strongly confirm that FGFR1-specific signaling is necessary for the cell cycle progression of hMSCs [33].
Figure 1.
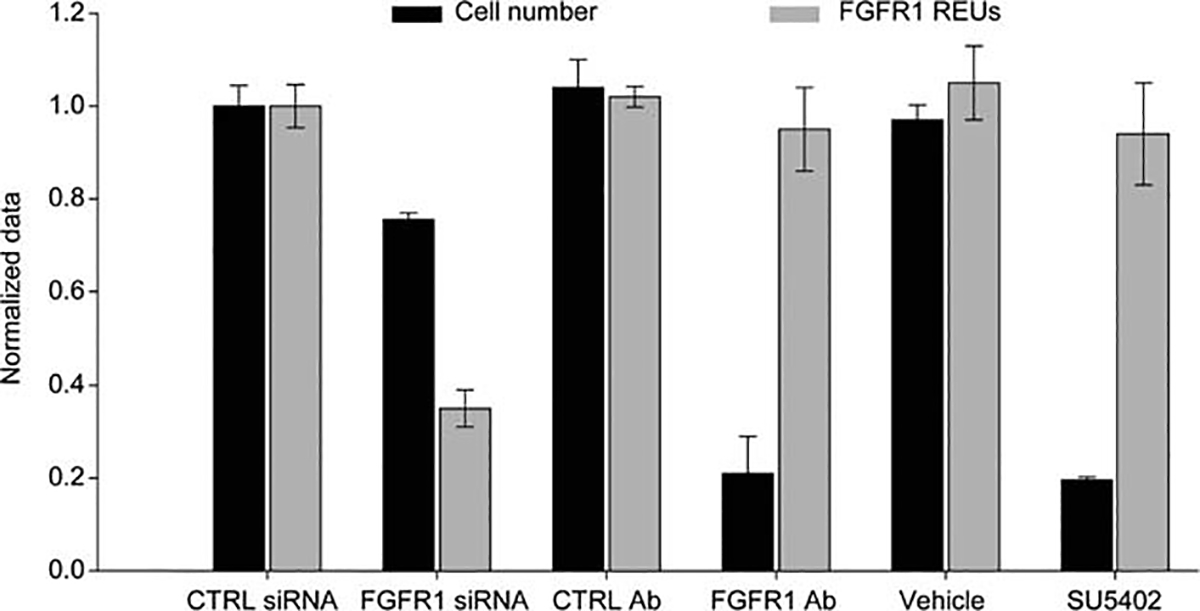
FGFR1 signaling is required for the normal cell cycle progression of human mesenchymal stem cells (hMSCs). Changes in FGFR1 expression and cell number following receptor inhibition by siRNA knockdown, neutralizing antibody challenge, or drug inhibition over 6 days. Abbreviations: FGFR1, fibroblast growth factor receptor 1; REU, relative expression unit; siRNA, small interfering RNA.
Next we determined the mechanistic contribution of FGFR1 signaling to hMSC growth. In accordance with our previous findings [8], hMSC proliferation could be inhibited by blocking key mediators of FGF signaling (Fig. 2A). Our data show that FGFR1 is a key upstream bottleneck in the FGF signaling pathway and, together with ERK and PI3K, is a major transducer of mitogenic signaling in hMSCs. Inhibition of PI3K signaling is highly effective in decelerating growth of hMSCs, suggesting that the PI3K/AKT pathway is a major cytosolic effector of those mitogenic signals that support autocrine FGF2/FGFR1-dependent proliferation of hMSCs.
Figure 2.
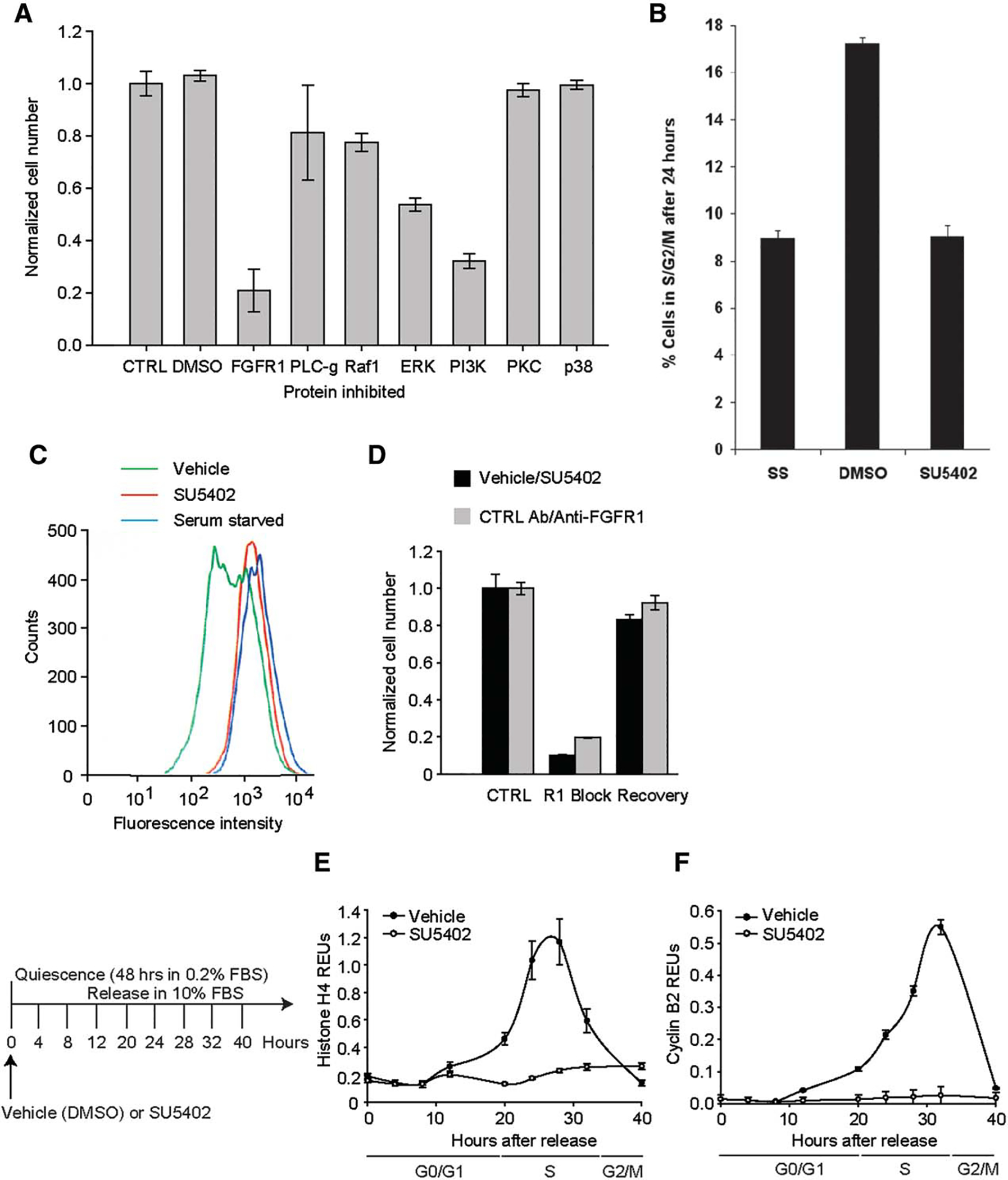
Mediators of human mesenchymal stem cell (hMSC) proliferation. (A): hMSC growth over 6 days after inhibition of known FGFR signal transducers. (B): Cell cycle progression in hMSCs under FGFR1 inhibition. (C): Analysis of cell division after 48 hours using CFDA fluorescence. (D): Recovery of hMSC growth following FGFR1 inhibition after 6 days. (E, F): The effect of FGFR1 inhibition on Histone H4 (E) and Cyclin B2 (F) mRNA expression. Abbreviations: DMSO, dimethyl sulfoxide; ERK, extracellular signal-regulated kinase; FBS, fetal bovine serum; FGFR1, fibroblast growth factor receptor 1; PI3K, phosphatidylinositide 3-kinases; PKC, protein kinase C.
To elucidate the mechanistic role of FGFR1 in cell cycle progression, we first synchronized hMSCs into a quiescent state (G0) by serum deprivation for 48 hours and then measured their re-entry into the cell cycle and subsequent progression into S phase of the cell cycle with or without SU5402 supplementation (Fig. 2B). Within the first 24 hours, none of the cells cultured with SU5402 reached S phase; the percentage of cells in G1/G0 remained similar to that observed for cells maintained in serum-deprivation media (Fig. 2B). We confirmed the quiescent nature of these cells by staining them with CFDA and monitoring the resultant decrease in fluorescence over a 48-hour culture period (Fig. 2C). Importantly, untreated control cells (vehicle; green) readily divided over the assay timeframe, leading to a notable decrease in cellular fluorescence (Fig. 2C). Meanwhile, cells cultured in the presence of SU5402 (red) failed to divide, with their levels of fluorescence matching the profile obtained for hMSCs maintained in low serum conditions (blue). When cultured for 4 days under FGFR1 inhibition, no further loss of cellular fluorescence was observed (Supporting Information Fig. 1). Thus, FGFR1 signaling is mandatory for cell cycle progression in hMSCs.
Having established the requirement for FGFR1 signaling during cell cycle progression, we next examined whether normal growth could be restored in cells previously treated with an FGFR1 blocking agent. Using either SU5402 or the neutralizing FGFR1 antibody, we first inhibited FGFR1 signaling for 6 days, then removed the inhibitor, and allowed the cells to restore their competence for cell growth for another 6 days. As shown in Figure 2D, hMSCs were able to resume cell cycle progression and proliferate normally following removal of the blocking agent, with a similar number of cells being obtained in the treated/recovered and the control groups. Because the antibody showed more interassay variability than the chemical inhibitor, subsequent experiments were conducted with SU5402. The data in Figure 2 indicate that FGFR1 inhibition is reversible which may thus allow its use for manipulation of MSC renewal and lineage differentiation.
FGFR1 Signaling is Required to Traverse Beyond the G1/S-Phase Transition
To map the progression of hMSCs through the cell cycle, we measured mRNA expression profiles in hMSCs 40 hours after their release from serum-starved conditions (Fig. 2E, 2F). To verify that the cells were arrested prior to S phase, histone H4 and cyclin B2 expression levels were measured. Histone mRNA levels begin to accumulate at the beginning of S phase reaching a peak in mid-S-phase [39, 40], while cyclin B levels begin to accumulate in G2 with maximal levels at mitosis [41]. Inhibition of FGFR1 signaling during serum stimulation of cell cycle progression was achieved by supplementation with SU5402 (compared to vehicle treated cells). In the presence of SU5402 neither histone H4 (Fig. 2E) nor cyclin B2 (Fig. 2F) expression changed, indicating that FGFR1 signaling positively regulates the ability of hMSCs to progress into S phase (20–24 hours).
FGFR1 Negatively Regulates the Levels of p21Waf1 and p27Kip1
As high levels of cyclin inhibitors such as p21waf1/CDKN1A and p27Kip1/CDKN1B (herein collectively referred to as CDKNs) are known to result in cell cycle arrest at the restriction point, we next assessed whether FGFR1 signaling influenced the expression of CDKNs. Even in serum-starved conditions (cell cycle arrest, t=0 hour), inhibition of FGFR1 signaling significantly increased the mRNA levels of both inhibitors compared to control (Fig. 3A, 3B). In contrast, when FGFR1 signaling was inhibited for 48 hours in asynchronous hMSCs there was an increase in the levels of both transcripts (Fig. 3C, 3D). Interestingly, addition of FGF-2 decreased p21 mRNA levels but had no effect on p27.
Figure 3.
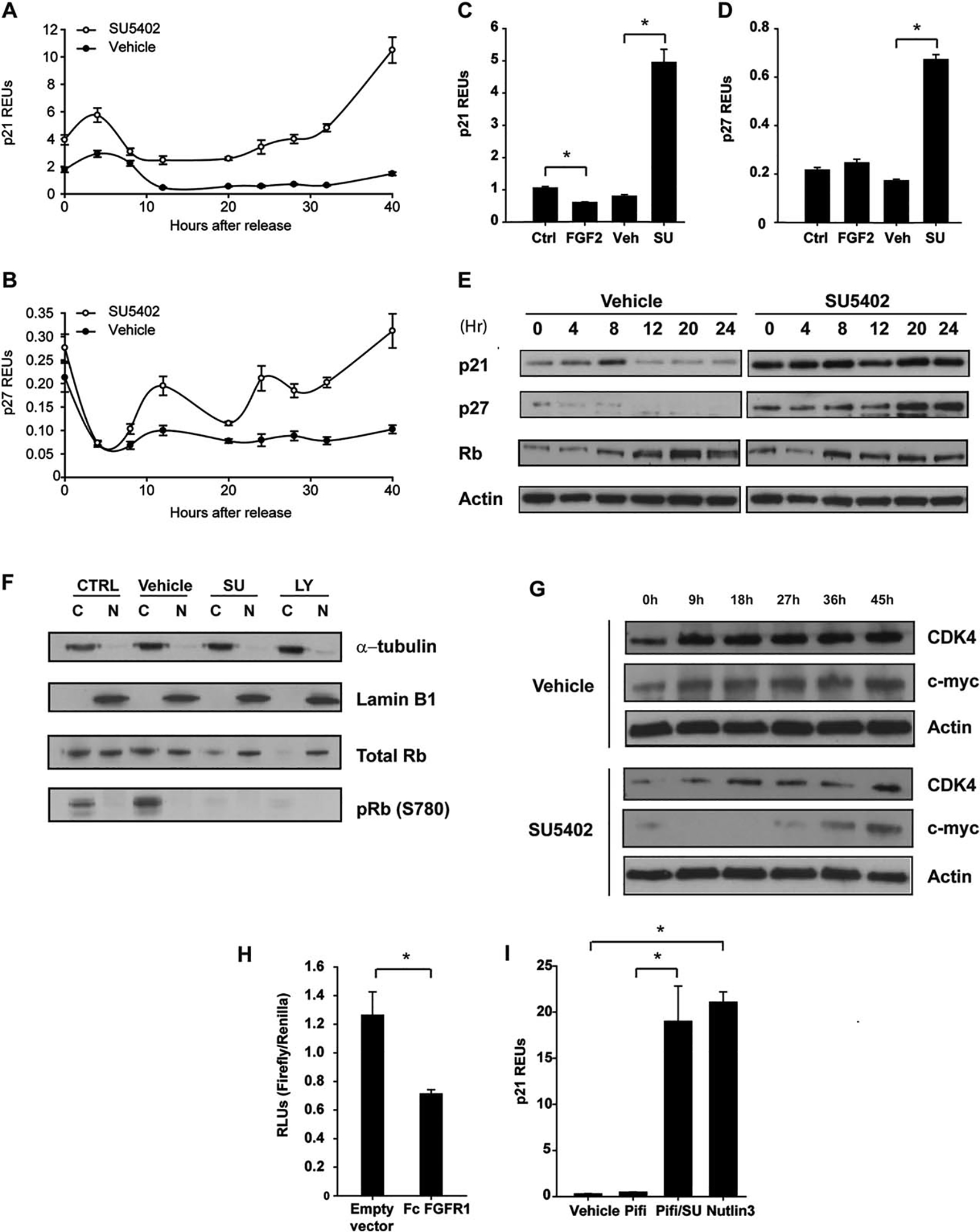
FGFR1 signaling in human mesenchymal stem cells (hMSCs) controls the expression of p21Waf1 and p27Kip1. The effect of FGFR1 inhibition on the mRNA expression of p21Waf1 (A) and p27Kip1 (B). The effect of FGF2 stimulation or FGFR1 inhibition on p21Waf1 (C) and p27Kip1 (D) in nonsynchronized cells for 48 hours. (E): Western blot analysis of p21Waf1, p27Kip1, and Rb following FGFR1 inhibition. (F): Western blot analysis of cytoplasmic and nuclear proteins following FGFR1 or PI3K inhibition for 16 hours. (G): Western blot analysis of CDK4 and c-Myc expression in SU5402-treated hMSCs. (H): p21 reporter assay in cells cotransfected with constitutively active FGFR1. (I): Polymerase chain reaction analysis of p21 expression in hMSCs subjected to the p53 inhibitor Pifithrin alone or in combination with SU5402, or the p53 activator Nutlin 3 alone. (C), (D) and (H), the asterisk (*) was to indicate that there was significant difference between the two groups. Abbreviations: CDk4, cyclin dependent kinase; FGF2, fibroblast growth factor; FGFR1, fibroblast growth factor receptor 1; Rb, retinoblastoma; REU, relative expression unit.
To determine the relationship between FGFR1 signaling and CDKN protein levels during S-phase progression, we next synchronized hMSCs by serum deprivation and released them in the media with serum or serum plus SU5402 over a 24-hour period. Levels of p21 remained consistently high with SU5402 and increased for p27 (Fig. 3E) suggesting that FGFR1 signaling normally inhibits the mRNA and protein levels of these CDKNs to facilitate cell cycle progression. To examine the relationship between FGF signaling and Rb phosphorylation, SU5402-treated hMSCs were synchronized and released into serum with or without inhibition for 16 hours (mid G1), and nuclear and cytoplasmic proteins were harvested (Fig. 3F). The absence of phosphorylation on serine 780 of Rb in the presence of SU5402 suggests a lack of cyclin D-CDK4/6 activity, thus preventing a key initiating event in cell cycle progression. There were no changes in the levels of cyclin D1 between the treated and control cells (data not shown), although when the cells were released with serum in the presence of SU5402, CDK4 levels were low compared to the control (Fig. 3G). Low CDK4 levels may limit formation of active cyclin D-CDK4/6 complexes because the balance between CDK complexes and CDKNs shifts in favor of the latter, thus preventing Rb phosphorylation.
Both p21 and p27 are believed to have dual roles as inhibitors and activators [42] of cyclin D complexes [42] and that the phosphorylation of p27 on tyrosines 88/89 results in the inactivation of its inhibitory activity [43]. Cyclin D-CDK4/6 complexes thus act as a reservoir for CDKNs, abrogating the inhibitory activity of CDKNs on cyclin E-CDK2 complexes [24, 29]. In the absence of FGFR1 signaling, it is possible that CDKNs remain inhibitory, leading to the absence of pRb. Interestingly, pRb was not detected with either SU5402 or LY294004 treatment (Fig. 3F); we only observed pRb in the cytoplasm of control cells (Fig. 3F) in accordance with a recent study [44].
As SU5402 had greater effects on p21 expression, we next assessed whether FGFR1 signaling affects the activity of the p21 promoter. We transiently cotransfected hMSCs with a p21 promoter/reporter construct [34] and a vector expressing a constitutively active FGFR1 [33]. We observed a 50% decline in basal promoter activity in response to FGFR1 signaling (Fig. 3H), indicating that active FGFR1 signaling inhibits p21 expression. As p21 is one of the major targets of p53 signaling [45], we next examined whether p53 was required for the increase in p21 mRNA observed with FGFR1 inhibition (Fig. 3C). Cells were cultured for 48 hours in the presence of either the p53 inhibitor Pifithrin A (10 μM), Pifithrin A with SU5402, or the p53 activator Nutlin3 (10 μM), and changes in p21 mRNA expression levels were monitored. Blocking FGFR1 in the presence of Pifithrin A failed to inhibit p21 mRNA expression, suggesting a p53-independent mechanism (Fig. 3I). Notably, inhibition of FGFR1 resulted in an increase in p21 levels similar to the p53 activator Nutlin 3.
FGFR1 Sustains c-Myc Expression
The transcription factor c-Myc has been shown to negatively regulate the expression of both p21 and p27 [46, 47]. ERK1/2 is a major cytosolic transducer of FGFR1-induced mitogenicity (Fig. 2A) that results in c-Myc stabilization [48]; previous work has shown that FGF-2 increases the stability of c-Myc [49]. To investigate this link further, we blocked FGFR1 activity to examine the effect on c-Myc expression (Fig. 4A). We observed a gradual, albeit modest, decrease in c-Myc mRNA transcripts in the presence of SU5402, suggesting that FGFR1 signaling is required to sustain expression. Levels of c-Myc protein were also reduced in cells treated with SU5402 for the initial 27 hours, however, the levels rebounded thereafter, presumably due to the reduced effectiveness of the drug (Fig. 3G). Interestingly, c-Myc levels were extremely low in serum-starved cells when combined with the PI3K inhibitor (LY294002) that quickly recovered to control levels upon release. This low c-Myc expression (in PI3K inhibited cells) was strongly negatively correlated with the expression of p21 and p27 (Supporting Information Figs. 2A–2E; r2 = .912 for p21 and r2 = .8797 for p27, Supporting Information Figs. 2D, 2E). These results suggest that in hMSCs, c-Myc may also inhibit the expression of CDKN.
Figure 4.
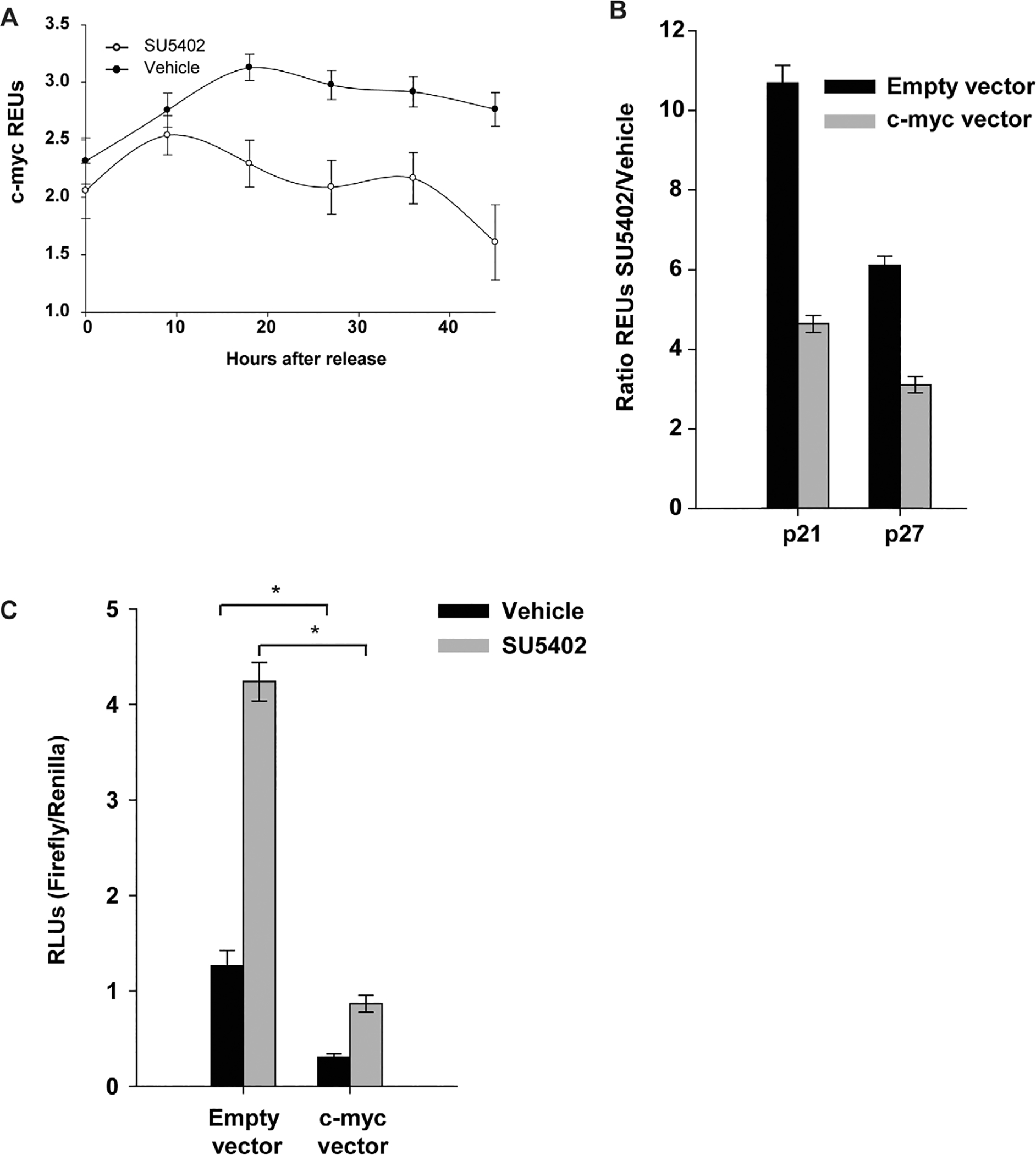
Fibroblast growth factor receptor 1 (FGFR1) signaling regulates the expression of p21 and p27 through c-Myc (A): Polymerase chain reaction (PCR) analysis of c-Myc expression upon reentry into the cell cycle in the presence of the FGFR1 inhibitor. (B): Expression of p21 and p27 calculated as a ratio of treatment with SU5402 as compared to vehicle in human mesenchymal stem cells (hMSCs) overexpressing c-Myc. (C): The effect of SU5402 on the p21 promoter in c-Myc-overexpressing hMSCs over a 48-hour period. The asterisk (*) was to indicate that there was significant difference between the two groups. Abbreviations: REU, relative expression unit; RLU, relative light unit.
c-Myc Inhibits the Expression of p21 and p27
To determine whether c-Myc suppresses expression of KIP/CDKN1 factors, hMSCs were transiently transfected with c-Myc and cultured in the presence of SU5402 for 48 hours, and the transcript levels of p21 and p27 were measured. When FGFR1 signaling was inhibited, p21 levels were >10 times higher than with vehicle but only fivefold higher when c-Myc was over-expressed (Fig. 4B). Similar results were observed with p27 expression where the SU5402/vehicle ratio decreased from sixfold to threefold. Thus, c-Myc can negatively regulate the expression of p21 and p27 but is not sufficient to completely abrogate the effects of blocking FGFR1. This is in agreement with the results presented in Figure 3G where c-Myc levels increase after 36 hours in SU5402 but does not inhibit CDKNs expression (Supporting Information Figs. 2A, 2B). Using a p21 promoter reporter (Fig. 4C), we then verified whether c-Myc overexpression could reverse the transcriptional activation of p21 that was observed when FGFR1 is inhibited (Fig. 4B). When c-Myc was overexpressed, there was a 75% reduction in p21 levels, an effect that was greatly exacerbated by reduced FGFR1 signaling (Fig. 4C). These results confirm that c-Myc is a potent inhibitor of p21 expression and that increased p21 expression in the absence of FGFR1 signaling may result from the lack of c-Myc expression. Notably, blocking FGFR1 signaling increased the activity of the p21 promoter despite c-Myc overexpression, suggesting that other factors controlled by FGFR1 may antagonize p21 expression.
FGFR1 Controls the Localization of CDKNs
FGFR signaling has been shown to exert its effects through the PI3K/AKT pathway in a cell-dependent manner [50]. Given that blocking PI3K had an antiproliferative effect similar to SU5402 (Fig. 2), we next investigated whether FGF-2 stimulation could induce AKT phosphorylation in hMSCs, and whether inhibition of FGFR1 could prevent such phosphorylation. FGF-2 noticeably increased AKT phosphorylation, and blocking either FGFR1 (SU5402) or PI3K (LY294002) completely negated this effect (Fig. 5A). Both p21 and p27 can be phosphorylated by AKT within their nuclear localization signal, which results in their retention in the cytoplasm [51, 52], as well as increased availability for ubiquitination and degradation. To examine the effect of FGFR1 on localization of CDKNs, we transfected eGFP-p21 and eGFP-p27 expression vectors into hMSCs. Transfected cells were then cultured with or without inhibitors (SU5402 or LY294002) for 24 hours. In untreated cells (control or vehicle), the fusion proteins were evenly distributed throughout the cells, with very few cells having either more protein in the nucleus or exclusive nuclear expression (Fig. 5B). When the cells were cultured with inhibitors, the fusion proteins were found mostly in the nucleus (arrows), suggesting that FGFR1 signaling is responsible for CDKNs cytoplasmic relocalization following AKT activation. As blocking PI3K does not increase p21 or p27 expression (Supporting Information Figs. 2A, 2B), the antiproliferative effect of LY294002 may be due to increased levels of nuclear CDKNs.
Figure 5.
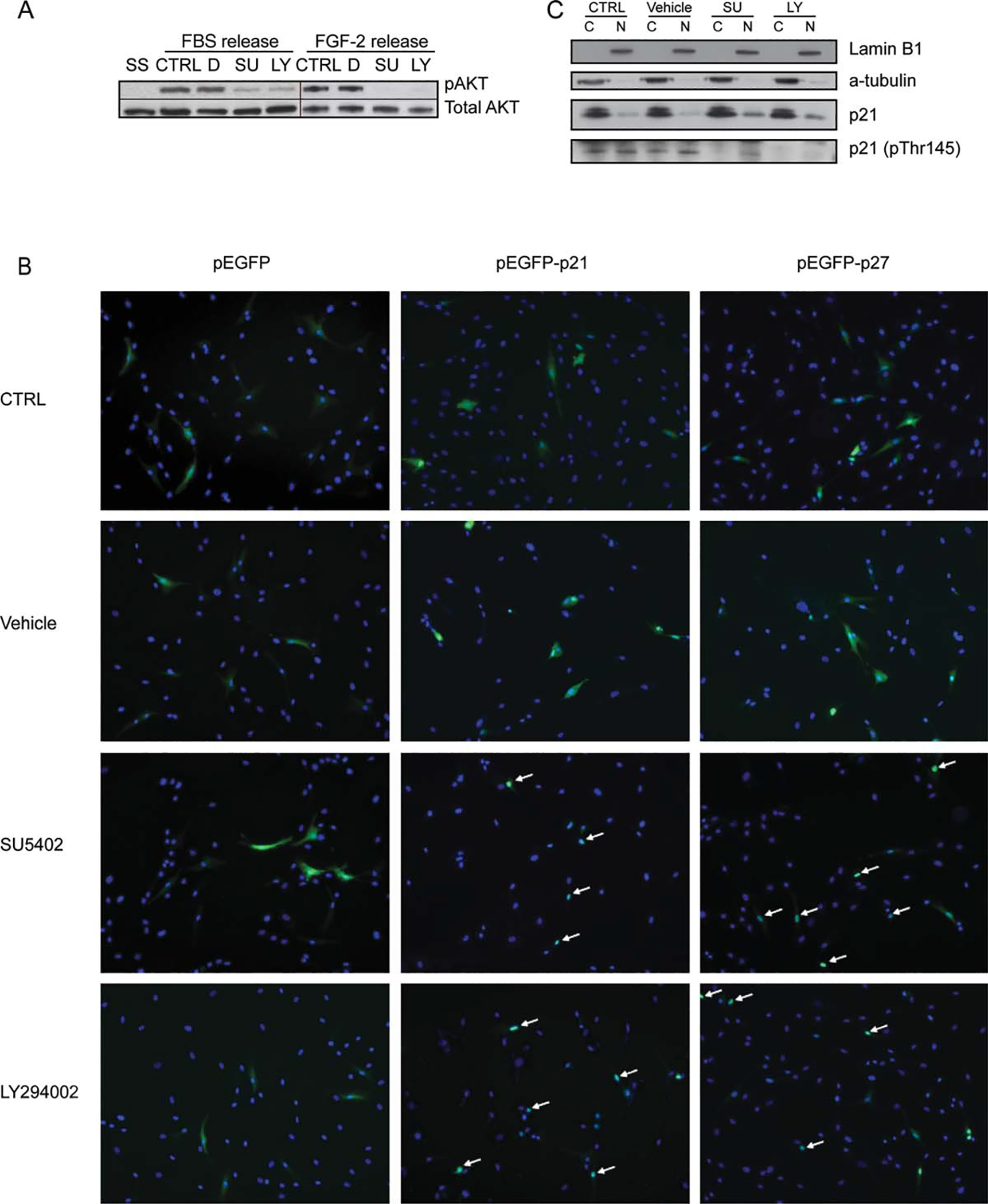
FGFR1 regulates the subcellular localization of p21 and p27. (A): Western blot analysis of AKT phosphorylation in hMSCs released from SS into FBS or FGF2 supplemented with DMSO (D), SU, or LY. For FGF-2 release condition, the blot was reprobed for total AKT after probing for phospho-AKT. (B): Cells were transfected with eGFP alone, or eGFP fused to p21 or p27, and the subcellular localization of p21 was determined by fluorescence microscopy after 24 hours. (C): Western blot analysis of p21 phosphorylation (pTHr145) in the cytoplasmic (C) and nuclear (N) fractions of cells cultured in the conditions described in (A) for 24 hours. Lamin B1 is the loading control for the nuclear fraction and a-tubulin for the cytoplasmic fraction. Abbreviations: AKT, protein kinase B; D, DMSO; EGFP, enhanced green fluorescent protein; FBS, fetal bovine serum; FGF, fibroblast growth factor; LY, LY294002; SU, SU5402; SS, serum starvation.
To verify if similar effects are observed for endogenous p21, synchronized cells were stimulated (as in Fig. 3F) prior to harvesting cytoplasmic and nuclear proteins. There was a marked increase in p21 levels in the nuclei of cells treated with either inhibitor, yet the level observed in the cytoplasmic fraction was similar (Fig. 5C). No differences were observed in the localization of p27 proteins (data not shown). The cytoplasmic relocation of p21 is attributable to phosphorylation of p21 on threonine residue 145 (Thr145) by AKT, which is known to alter the subcellular localization of p21 [53]. No phosphorylation was observed in the cytoplasmic fractions from samples treated with either inhibitor and reduced phosphorylation in the nucleus fraction by SU5402 or diminished phosphorylation in the nucleus fraction by LY294002 (Fig. 5C), further reinforcing the hypothesis that FGFR1 signaling controls the localization of p21 through AKT activation.
Degradation of CDKNs is Impaired in the Absence of FGFR1 Signaling***
Skp2 and Cks1 are part of the SCF/Skp2 complex. This complex regulates cell entry into S-phase by initiating the degradation of p21 and p27. It has recently been shown that Ras/mitogen-activated protein kinase (MAPK)-dependent control of p27 expression occurs by regulating the stability of Skp2 [54]. As the Ras/MAPK pathway is one of the major downstream effectors of the FGF-2/FGFR1 axis [55], it is possible that FGFR1 signaling controls the degradation of CDKNs through the Ras/MAPK pathway. Moreover, Cks1 has been shown to bind the unphosphorylated form of FRS2 and is released upon its activation by FGF receptor signaling [56]; this links tyrosine kinase receptor activation and cell cycle progression. Therefore, we investigated whether Cks1 was a component of the FGFR1/FRS-2 complex. hMSCs were transfected with a constitutively active FGFR1 [33], wild type (wt) FRS2, and wt Cks1 by electroporation. Transfected cells were cultured in media containing DMSO or SU5402 to prevent any activation of the receptor upon translation. FGFR1 precipitation confirmed that 25 μM SU5402 prevented activation of an otherwise active receptor (Fig. 6A). Furthermore, while FRS2 coprecipitated with FGFR1 regardless of the treatment, we could not precipitate Cks1, possibly because FRS2-Cks1 may not be complexed with FGFR1. To verify this, we performed a similar transfection experiment; however, FRS2 was pulled down instead. Similarly, Cks1 also did not precipitate with FRS2 under these conditions (data not shown); this could be due to differences with the insect cell system that was used previously to prove this interaction [56]. Interestingly, Skp2 expression was greatly impaired in the absence of FGFR1 signaling (Fig. 6B). It has been shown that Skp2 is a target of E2F1 [57], so its absence in cells treated with the FGFR1 inhibitor further supports the possibility that Rb remains active and prevents E2F1-induced transcription. It may also reflect the fact that Skp2 expression is regulated by AKT and MAPK [58], two pathways activated by FGFR1 signaling. Thus it is plausible that FGFR1 signaling partially controls the degradation of CDKNs by participating in their cytoplasmic localization and enabling the formation of the SCF/Skp2 complex in late G1 for their degradation. This proposed mechanism would be parallel to the effects of FGFR1 signaling on CDKN expression levels (Fig. 6C).
Figure 6.
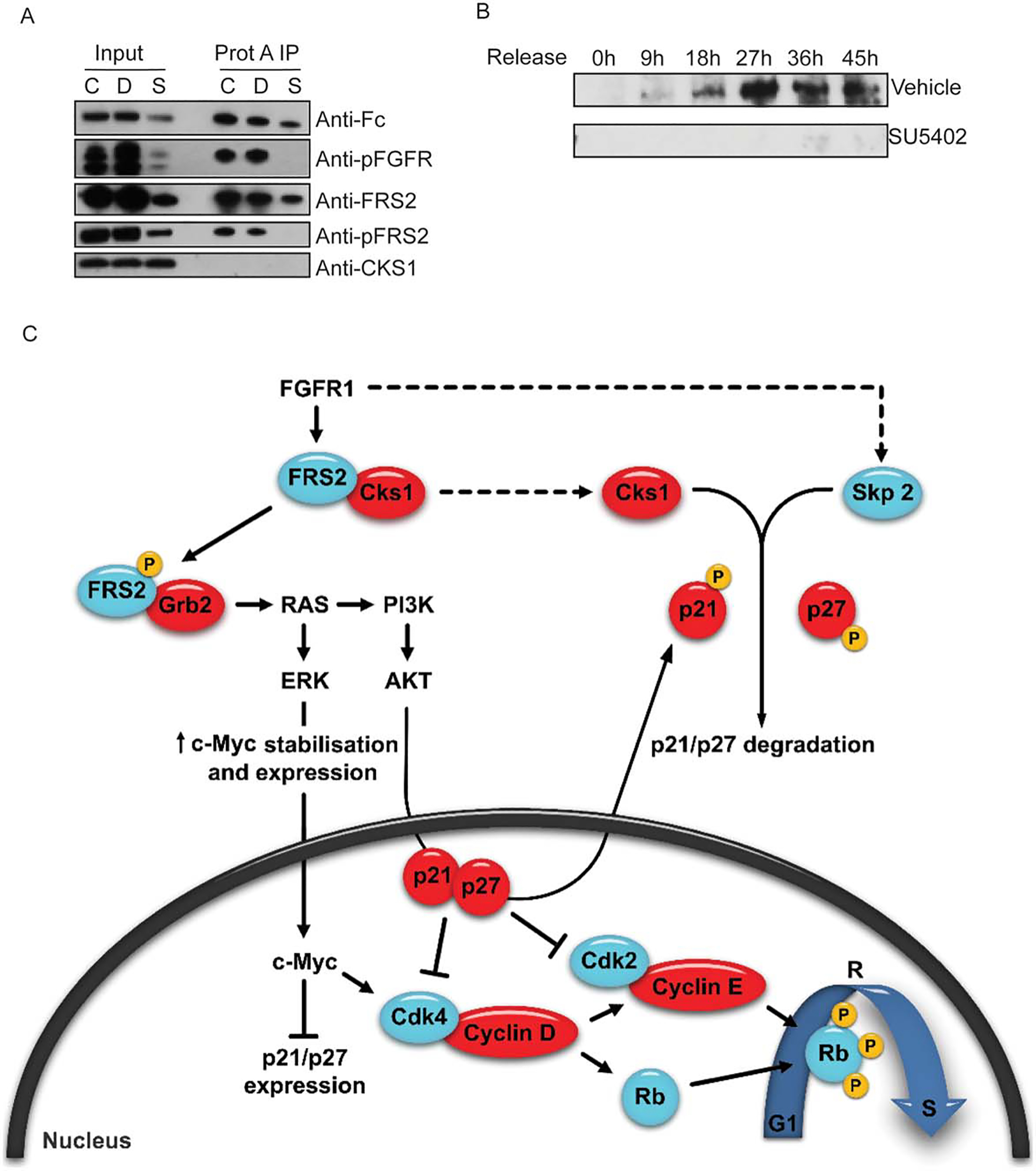
FGFR1 signaling influences the expression of S-phase kinase-associated protein two (Skp2) but not the release of Cks1. (A): Cells were transfected with vectors coexpressing a constitutively active form of FGFR1 (Fc-FGFR1), wild type (wt) FRS2, and wt Cks1. Blot depicts immunoprecipitation of Fc-R1TK (constitutively active FGFR1) 24 hours after cell synchronization and release into maintenance media alone (CTRL), or supplemented with DMSO vehicle (D) or SU5402 (SU). (B): Western blot analysis of Skp2 expression after cell synchronization and release into DMSO vehicle or SU5402 supplemented media. (C): Hypothesized pathways for FGFR1 control of cell cycle progression in hMSCs. Abbreviations: AKT, protein kinase B; Cks1, cyclin-dependent kinase subunit one; CDK, cyclin dependent kinase; FRS, fibroblast receptor substrate; FGFR1, fibroblast growth factor; ERK, extracellular signal-regulated kinase; PI3K, phosphatidylinositide 3-kinases; Rb, retinoblastoma.
FGFR1 Signaling is Necessary for Mesoderm Development in Xenopus
The role of FGF signaling in mesoderm induction has been well-established in Xenopus [59] where it has been shown that a dominant-negative form of xFGFR1 can block such induction [53]. Moreover, it has been shown that xFRS2 phosphorylation is essential for early mesodermal induction and is part of a complex including FGFR1 [60]. As FGFR1 signaling is essential for the proliferation of cultured hMSCs, we next examined whether inactivating the receptor in vivo affects mesoderm development. Dorsal or ventral cells at the four-cell stage in Xenopus embryos were injected with an FGFR1-neutralizing antibody and allowed to develop until stage 35/36 (Fig. 7). Injecting ventral cells with the antibody negatively affected mesoderm development, whereas injection of dorsal cells had only mild effects, mostly on eye development. This finding confirms that FGFR1 signaling is important for mesoderm induction, in a manner consistent with an absence of progenitor cell proliferation.
Figure 7.
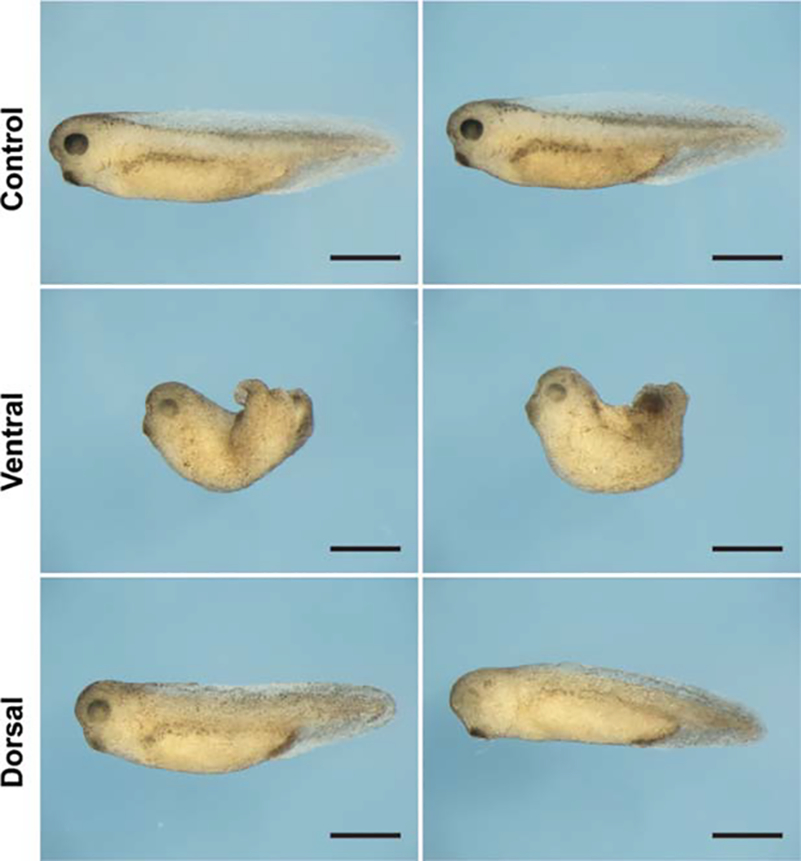
FGFR1 inhibition of Xenopus laevis embryos adversely impacts mesoderm development. Fibroblast growth factor receptor 1-neutralizing antibody was injected into either both ventral cells or both dorsal cells at the four cell stage and embryos were allowed to develop to stage 35/36. Uninjected embryos served as controls. Photomicrographs show duplicates. Scale bar ≈1 mm.
Discussion
Understanding how growth factors trigger the proliferative expansion of hMSCs is an important step for the harnessing of their therapeutic potential, especially for their promise in skeletal tissue regeneration. Even though it is known that endogenous FGF-2 production plays an important role in the proliferation of hMSCs [6], the mechanism underlying this effect is poorly understood. Here we further dissected the molecular pathways that are important for the mitogenic effect of FGF-2.
Because FGFR1 is a prominent receptor on MSCs during active cell proliferation and the most abundant member of the four FGFRs, we first demonstrated that blocking FGFR1 signaling has a deleterious effect on hMSC proliferation. FGFR1 signals through a multitude of pathways; the results obtained here using specific small molecule inhibitors identified the PI3K pathway as a key transducer of the mitogenic signal. Blocking FGFR1 signaling leads to a complete growth arrest, while reestablishing FGFR1 signaling is sufficient for the cells to resume normal growth. The importance of FGFR1 signaling for cell cycle progression in hMSCs suggests that it may play a role in the balance between self-renewal and lineage-commitment in multipotent stem cells. Notably, previous studies in our lab with mouse embryonic stem cells and rat MSCs revealed that blocking FGFR1 significantly increased differentiation [8, 61]. Conditional FGFR1 knockout in osteoblasts leads to increased FGFR3 expression and accelerates differentiation [11]. Our study reveals that FGFR1 is a negative regulator of CDKN function in hMSCs via c-Myc expression. These findings together suggest that the expression of FGFR1 promotes the self-renewal of uncommitted mesenchymal cells and maintains the multipotent stem cell state.
Because CDK function is essential for cell cycle progression, we investigated whether the two CDK inhibitors p21 and p27 are involved in FGFR1-mediated proliferation. Our results clearly show that basal FGFR1 signaling is sufficient to lower the expression and protein levels of both inhibitors, and that exogenous FGF-2 supplementation further decreases p21 expression. Importantly, inhibiting FGFR1 signaling leads to a drastic decrease in the CDK4 protein level. Consequently, we found that in the absence of FGFR1 signaling, Rb protein remains hypophosphorylated and no CDK-dependent phosphorylation of pRb on S780 could be observed. In essence, loss of FGFR1 signaling selectively eliminates the S780 phosphorylated isoform of pRb in the cytoplasm, while retaining hypophosphorylated Rb in the nucleus to suppress cell cycle progression. When combined, our data clearly indicate that FGF2/FGFR1 signaling controls the levels of cyclin, CDKs, and CDKNs to modulate the phosphorylation of Rb, which together is permissive for cell cycle progression through E2F-related transcriptional mechanisms.
Because FGF-2 supplementation inhibits p21 expression, we overexpressed a constitutively active FGFR1 and showed that it substantially decreases p21 promoter activity, an effect that was shown to be independent of p53 activity. FGF2 stimulation of FGFR1 leads to the phosphorylation of AKT and activation of c-Myc as a target of AKT signaling that suppresses the activity of CDK inhibitors. Our results indicate that FGFR1 signaling is important for the steady state of c-Myc RNA levels, and that there is a strong inverse correlation between the expression of c-Myc and those of p21 and p27. Importantly, overexpressing c-Myc in the presence of the FGFR1 inhibitor leads to a level of p21 promoter activity similar to that observed in the control cells, suggesting c-Myc can rescue an inhibition of FGFR1 signaling. Interestingly the c-Myc protein levels increased from 27 to 45 hours in the presence of the FGFR1 inhibitor despite the fact that mRNA levels where on a downward trend suggesting that there may be translational control mechanisms that are independent of FGFR1 signaling.
This study demonstrates that FGFR1 signaling does not only act on the levels of p21 but that it also modifies the localization of p21 and p27, most likely through their AKT-mediated phosphorylation leading to their nuclear export and potential degradation. Although some phosphorylated p21 could be detected in the nucleus of SU5402-treated cells, this form was not found in the cytoplasm suggesting exportation may be slower or impaired in the absence of FGFR1 signaling. FGFR1 signaling may also have a direct influence on the rate of CDK inhibitor degradation, because blocking FGFR1 signaling drastically reduces Skp2 protein levels upon cell cycle re-entry and progression. The importance of FGFR1 signaling for mesodermal precursor cell proliferation was corroborated by our in vivo results, whereby mesoderm induction was compromised in the Xenopus embryo model. Mesoderm specification during ventral tissue development is necessary for the formation of a large number of ventral and posterior tissues. Inhibition of FGFR1 ventrally interferes with ventral mesoderm specification through several mechanisms including the transcription-inducing activity of VegT and Xbra which is necessary for mesoderm specification [62]. Complete inhibition of FGF signaling will entirely abrogate Xbra expression [63]. The severity of any disruption to this signaling pathway is significantly amplified due to the fact that this also directly disrupts an FGF-based positive feedback loop that regulates the activity of Xbra, eFGF, and FGF4 [62, 64]. Subsequent developmental indicators of interference with this pathway are clearly evident in the restricted posterior tissue specification in embryos (Fig. 7) as evidenced by the absence of true tail formation. This role for FGF signaling can be found as early as blastula/gastrula stages via signaling to marginal zone cells which initiates mesoderm specification [65].
Conclusion
We have demonstrated that FGFR1 signaling in MSCs modulates CDK function and cell cycle progression by controlling the levels and localization of CDK inhibitors. This mechanistic understanding provides a rational basis for FGFR1-reversible inhibition and a route for the stimulation of self-renewal of human MSCs, a strategy with great promise for tissue regenerative therapies.
Supplementary Material
Acknowledgments
We acknowledge grant support from Singapore’s Agency for Science, Technology and Research (A*STAR), the Biomedical Research Council of Singapore, the Institute of Medical Biology, Singapore, and the National Medical Research Council of Singapore, as well as the National Institutes of Health Grant R01 AR049069 (A.v.W.).
Footnotes
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Cantley LC, Auger KR, Carpenter C et al. Oncogenes and signal transduction. Cell 1991;64:281–302. [DOI] [PubMed] [Google Scholar]
- 2.Norbury C, Nurse P. Animal cell cycles and their control. Annu Rev Biochem 1992;61:441–470. [DOI] [PubMed] [Google Scholar]
- 3.Pardee AB. G1 events and regulation of cell proliferation. Science 1989;246:603–608. [DOI] [PubMed] [Google Scholar]
- 4.Seedorf K. Intracellular signaling by growth factors. Metabolism 1995; 44:24–32. [DOI] [PubMed] [Google Scholar]
- 5.Marie PJ. Fibroblast growth factor signaling controlling osteoblast differentiation. Gene 2003;316:23–32. [DOI] [PubMed] [Google Scholar]
- 6.Rider DA, Dombrowski C, Sawyer AA et al. Autocrine FGF2 increases the multipotentiality of human adipose-derived mesenchymal stem cells. Stem Cells 2008;26:1598–1608. [DOI] [PubMed] [Google Scholar]
- 7.Ornitz DM, Xu J, Colvin JS et al. Receptor specificity of the fibroblast growth factor family. J Biol Chem 1996;271:15292–15297. [DOI] [PubMed] [Google Scholar]
- 8.Dombrowski C, Song SJ, Chuan P et al. Heparan sulfate mediates the proliferation and differentiation of rat mesenchymal stem cells. Stem Cells Dev 2009;18:661–670. [DOI] [PubMed] [Google Scholar]
- 9.Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res 1999;5:1063–1071. [PubMed] [Google Scholar]
- 10.Hase T, Kawashiri S, Tanaka A et al. Correlation of basic fibroblast growth factor expression with the invasion and the prognosis of oral squamous cell carcinoma. J Oral Pathol Med 2006;35:136–139. [DOI] [PubMed] [Google Scholar]
- 11.Jacob AL, Smith C, Partanen J et al. Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev Biol 2006;296:315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev 2002;16:1446–1465. [DOI] [PubMed] [Google Scholar]
- 13.Quarto N, Longaker MT. Differential expression of specific FGF ligands and receptor isoforms during osteogenic differentiation of mouse adipose-derived stem cells (mASCs) recapitulates the in vivo osteogenic pattern. Gene 2008;424:130–140. [DOI] [PubMed] [Google Scholar]
- 14.Shimada A, Yabusaki M, Niwa H et al. Maternal-zygotic medaka mutants for fgfr1 reveal its essential role in the migration of the axial mesoderm but not the lateral mesoderm. Development 2008;135:281–290. [DOI] [PubMed] [Google Scholar]
- 15.Tsutsumi S, Shimazu A, Miyazaki K et al. Retention of multilineage differentiation potential of mesenchymal cells during proliferation in response to FGF. Biochem Biophys Res Commun 2001;288:413–419. [DOI] [PubMed] [Google Scholar]
- 16.Zaragosi LE, Ailhaud G, Dani C. Autocrine fibroblast growth factor 2 signaling is critical for self-renewal of human multipotent adipose-derived stem cells. Stem Cells 2006;24:2412–2419. [DOI] [PubMed] [Google Scholar]
- 17.Coutu DL, Francois M, Galipeau J. Inhibition of cellular senescence by developmentally regulated FGF receptors in mesenchymal stem cells. Blood 2011;117:6801–6812. [DOI] [PubMed] [Google Scholar]
- 18.Bianchi G, Banfi A, Mastrogiacomo M et al. Ex vivo enrichment of mesenchymal cell progenitors by fibroblast growth factor 2. Exp Cell Res 2003;287:98–105. [DOI] [PubMed] [Google Scholar]
- 19.Solchaga LA, Penick K, Porter JD et al. FGF-2 enhances the mitotic and chondrogenic potentials of human adult bone marrow-derived mesenchymal stem cells. J Cell Physiol 2005;203:398–409. [DOI] [PubMed] [Google Scholar]
- 20.Ling L, Murali S, Dombrowski C et al. Sulfated glycosaminoglycans mediate the effects of FGF2 on the osteogenic potential of rat calvarial osteoprogenitor cells. J Cell Physiol 2006;209:811–825. [DOI] [PubMed] [Google Scholar]
- 21.Quarto N, Longaker MT. FGF-2 inhibits osteogenesis in mouse adipose tissue-derived stromal cells and sustains their proliferative and osteogenic potential state. Tissue Eng 2006;12:1405–1418. [DOI] [PubMed] [Google Scholar]
- 22.Quarto N, Wan DC, Longaker MT. Molecular mechanisms of FGF-2 inhibitory activity in the osteogenic context of mouse adipose-derived stem cells (mASCs). Bone 2008;42:1040–1052. [DOI] [PubMed] [Google Scholar]
- 23.Pledger WJ, Stiles CD, Antoniades HN et al. Induction of DNA synthesis in BALB/c 3T3 cells by serum components: Reevaluation of the commitment process. Proc Natl Acad Sci USA 1977;74:4481–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts JM, Koff A, Polyak K et al. Cyclins, Cdks, and cyclin kinase inhibitors. Cold Spring Harb Symp Quant Biol 1994;59:31–38. [DOI] [PubMed] [Google Scholar]
- 25.Blagosklonny MV, Pardee AB. The restriction point of the cell cycle. Cell Cycle 2002;1:103–110. [PubMed] [Google Scholar]
- 26.Bracken AP, Ciro M, Cocito A et al. E2F target genes: Unraveling the biology. Trends Biochem Sci 2004;29:409–417. [DOI] [PubMed] [Google Scholar]
- 27.Muller H, Bracken AP, Vernell R et al. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev 2001;15:267–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitagawa M, Higashi H, Jung HK et al. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J 1996;15:7060–7069. [PMC free article] [PubMed] [Google Scholar]
- 29.Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995;9:1149–1163. [DOI] [PubMed] [Google Scholar]
- 30.Harper JW, Adami GR, Wei N et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993; 75:805–816. [DOI] [PubMed] [Google Scholar]
- 31.Polyak K, Lee MH, Erdjument-Bromage H et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994;78:59–66. [DOI] [PubMed] [Google Scholar]
- 32.Xiong Y, Hannon GJ, Zhang H et al. p21 is a universal inhibitor of cyclin kinases. Nature 1993;366:701–704. [DOI] [PubMed] [Google Scholar]
- 33.Burgar HR, Burns HD, Elsden JL et al. Association of the signaling adaptor FRS2 with fibroblast growth factor receptor 1 (Fgfr1) is mediated by alternative splicing of the juxtamembrane domain. J Biol Chem 2002;277:4018–4023. [DOI] [PubMed] [Google Scholar]
- 34.Westendorf JJ, Zaidi SK, Cascino JE et al. Runx2 (Cbfa1, AML-3) interacts with histone deacetylase 6 and represses the p21(CIP1/WAF1) promoter. Mol Cell Biol 2002;22:7982–7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaidi SK, Sullivan AJ, Medina R et al. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J 2004;23:790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helledie T, Nurcombe V, Cool SM. A simple and reliable electroporation method for human bone marrow mesenchymal stem cells. Stem Cells Dev 2008;17:837–848. [DOI] [PubMed] [Google Scholar]
- 37.Ozen M, Giri D, Ropiquet F et al. Role of fibroblast growth factor receptor signaling in prostate cancer cell survival. J Natl Cancer Inst 2001;93:1783–1790. [DOI] [PubMed] [Google Scholar]
- 38.Mohammadi M, McMahon G, Sun L et al. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 1997;276:955–960. [DOI] [PubMed] [Google Scholar]
- 39.Baumbach LL, Stein GS, Stein JL. Regulation of human histone gene expression: Transcriptional and posttranscriptional control in the coupling of histone messenger RNA stability with DNA replication. Biochemistry 1987;26:6178–6187. [DOI] [PubMed] [Google Scholar]
- 40.Plumb M, Stein J, Stein G. Coordinate regulation of multiple histone mRNAs during the cell cycle in HeLa cells. Nucleic Acids Res 1983; 11:2391–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pines J, Hunter T. Isolation of a human cyclin cDNA: Evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell 1989;58:833–846. [DOI] [PubMed] [Google Scholar]
- 42.Cheng M, Olivier P, Diehl JA et al. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J 1999;18:1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.James MK, Ray A, Leznova D et al. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol 2008;28:498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacob C, Grabner H, Atanasoski S et al. Expression and localization of Ski determine cell type-specific TGFbeta signaling effects on the cell cycle. J Cell Biol 2008;182:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.el-Deiry WS, Harper JW, O’Connor PM et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res 1994;54:1169–1174. [PubMed] [Google Scholar]
- 46.Chandramohan V, Mineva ND, Burke B et al. c-Myc represses FOXO3a-mediated transcription of the gene encoding the p27(Kip1) cyclin dependent kinase inhibitor. J Cell Biochem 2008;104:2091–2106. [DOI] [PubMed] [Google Scholar]
- 47.Gartel AL, Ye X, Goufman E et al. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci USA 2001;98:4510–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sears R, Nuckolls F, Haura E et al. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000;14: 2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lepique AP, Moraes MS, Rocha KM et al. c-Myc protein is stabilized by fibroblast growth factor 2 and destabilized by ACTH to control cell cycle in mouse Y1 adrenocortical cells. J Mol Endocrinol 2004;33:623–638. [DOI] [PubMed] [Google Scholar]
- 50.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2000; 103:211–225. [DOI] [PubMed] [Google Scholar]
- 51.Liang J, Zubovitz J, Petrocelli T et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med 2002;8:1153–1160. [DOI] [PubMed] [Google Scholar]
- 52.Zhou BP, Liao Y, Xia W et al. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 2001;3:245–252. [DOI] [PubMed] [Google Scholar]
- 53.Amaya E, Musci TJ, Kirschner MW. Expression of a dominant negative mutant of the FGF receptor disrupts mesoderm formation in Xenopus embryos. Cell 1991;66:257–270. [DOI] [PubMed] [Google Scholar]
- 54.Motti ML, De Marco C, Califano D et al. Loss of p27 expression through RAŜBRAF^MAP kinase-dependent pathway in human thyroid carcinomas. Cell Cycle 2007;6:2817–2825. [DOI] [PubMed] [Google Scholar]
- 55.Kouhara H, Hadari YR, Spivak-Kroizman T et al. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 1997;89:693–702. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Lin Y, Bowles C et al. Direct cell cycle regulation by the fibroblast growth factor receptor (FGFR) kinase through phosphorylation-dependent release of Cks1 from FGFR substrate 2. J Biol Chem 2004;279:55348–55354. [DOI] [PubMed] [Google Scholar]
- 57.Zhang L, Wang C. F-box protein Skp2: A novel transcriptional target of E2F. Oncogene 2006;25:2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Auld CA, Caccia CD, Morrison RF. Hormonal induction of adipogenesis induces Skp2 expression through PI3K and MAPK pathways. J Cell Biochem 2007;100:204–216. [DOI] [PubMed] [Google Scholar]
- 59.Slack JM, Isaacs HV, Song J et al. The role of fibroblast growth factors in early Xenopus development. Biochem Soc Symp 1996;62: 1–12. [PubMed] [Google Scholar]
- 60.Kusakabe M, Masuyama N, Hanafusa H et al. Xenopus FRS2 is involved in early embryogenesis in cooperation with the Src family kinase Laloo. EMBO Rep 2001;2:727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woei Ng K, Speicher T, Dombrowski C et al. Osteogenic differentiation of murine embryonic stem cells is mediated by fibroblast growth factor receptors. Stem Cells Dev 2007;16:305–318. [DOI] [PubMed] [Google Scholar]
- 62.Fletcher RB, Harland RM. The role of FGF signaling in the establishment and maintenance of mesodermal gene expression in Xenopus. Dev Dyn 2008;237:1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amaya E, Stein PA, Musci TJ et al. FGF signalling in the early specification of mesoderm in Xenopus. Development 1993;118: 477–487. [DOI] [PubMed] [Google Scholar]
- 64.Isaacs HV, Pownall ME, Slack JM. eFGF regulates Xbra expression during Xenopus gastrulation. EMBO J 1994;13:4469–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bottcher RT, Niehrs C. Fibroblast growth factor signaling during early vertebrate development. Endocr Rev 2005;26:63–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.