

Abstract
Cell based therapies including chimeric antigen receptor (CAR) T cells are promising for treating leukemias and solid cancers. At the same time, there is interest in enhancing the functionality of these cells via surface decoration with nanoparticles (backpacking). Magnetic nanoparticle cell labeling is of particular interest due to opportunities for magnetic separation, in vivo manipulation, drug delivery and magnetic resonance imaging (MRI). While modification of T cells with magnetic nanoparticles (MNPs) was explored before, we questioned whether MNPs are compatible with CAR-T cells when introduced during the manufacturing process. We chose highly aminated 120 nm crosslinked iron oxide nanoworms (CLIO NWs, ~36,000 amines per NW) that could efficiently label different adherent cell lines and we used CD123 CAR-T cells as the labeling model. The CD123 CAR-T cells were produced in the presence of CLIO NWs, CLIO NWs plus protamine sulfate (PS), or PS only. The transduction efficiency of lentiviral CD123 CAR with only NWs was ~23% lower than NW+PS and PS groups (~33% and 35%, respectively). The cell viability from these three transduction conditions was not reduced within CAR-T cell groups, though lower compared to non-transduced T cells (mock T). Use of CLIO NWs instead of, or together with cationic protamine sulfate for enhancement of lentiviral transduction resulted in comparable levels of CAR expression and viability but decreased the proportion of CD8+ cells and increased the proportion of CD4+ cells. CD123 CAR-T transduced in the presence of CLIO NWs, CLIO NWs plus PS, or PS only, showed similar level of cytotoxicity against leukemic cell lines. Furthermore, fluorescence microscopy imaging demonstrated that CD123 CAR-T cells labeled with CLIO NW formed rosettes with CD123+ leukemic cells as the non-labeled CAR-T cells, indicating that the CAR-T targeting to tumor cells has maintained after CLIO NW labeling. The in vivo trafficking of the NW labeled CAR-T cells showed the accumulation of CAR-T labeled with NWs primarily in the bone marrow and spleen. CAR-T cells can be magnetically labeled during their production while maintaining functionality using the positively charged iron oxide NWs, which enable the in vivo biodistribution and tracking of CAR-T cells.
Keywords: CAR-T, iron oxide, nanoworm, magnetic labeling, leukemia, cell trafficking
1. Introduction
Chimeric antigen receptor (CAR) T cells are genetically engineered immune cells that can rapidly attack and eliminate tumor cells, Currently, there are over 500 clinical trials using CAR-T cells registered with the National Institutes of Health (NIH). [1] CD19 antigen was the first successful clinical target for CAR-T cell therapy, [2] resulting in two CD19 CAR-T cell therapies: Kymriah® CAR-T for the treatment of B-cell precursor acute lymphoblastic leukemia (ALL) and large B-cell lymphomas; and Yescarta® CAR-T for the treatment of certain types of large B-cell lymphomas. [3] Later, two additional CD19 CAR-T therapies were approved: Tecartus® to treat adult patients with mantle cell lymphoma (MCL) [4]; and Breyanzi® to treat relapsed/refractory large B-cell lymphoma [5] followed by Abecma to treat B-cell maturation antigen (BCMA)-directed CAR-T cell therapy for the treatment of multiple myeloma. [6] Since CAR-T therapies have provided a revolutionary approach to treat blood cell related diseases, another hematologic malignancy called myelodysplastic syndrome (MDS) could also be treated with CAR-T cells. Currently available FDA approved treatment options for MDS are limited to supportive care and most patients eventually succumb to disease or progress to acute myeloid leukemia (AML). [7] Forman lab has developed T cells modified with CD123 CAR to have antitumor effects on AML [8] and it was found that CD123 is upregulated on the bone marrow stem cells of the high risk MDS patients during the disease progression [9]. Furthermore, it was demonstrated that the CD123 CAR-T cells could eliminate CD123+ MDS stem cells in a patient-derived xenograft model in vivo. [10]
Although several CAR-T products are clinically available, their in vivo trafficking and dynamic body distribution remains relatively unexplored. Detection and visualization of CAR-T cells in patients should help understand CAR-T behavior, monitor treatments, predict therapeutic outcomes and manage risks of CAR-T toxicity in clinical trials. [1, 11–13] There are several reporter system being used in preclinical and clinical trials of CAR-T cells that incorporate radioisotopes for positron emission tomography (PET) and single-photon emission computed tomography (SPECT), including enzymes [14, 15], transporters [16] and cell surface proteins [17–19]. Another radiolabeled compound (In111 labeled oxyquinoline) approved for autologous leukocyte radiolabeling, has also been used to label CAR-T cells in one of the first human trials of CAR-T therapy for colorectal cancer. [20]
Nanoparticles (NPs) have been extensively developed for many biomedical applications in the past decades. Besides serving as drug delivery platforms, NPs can also be used to enable cell tracking and detection in vitro and in vivo. [21] Magnetic nanoparticles (MNPs) have several advantages, due to their magnetic properties for cell manipulation and magnetic resonance imaging (MRI) [22]. Thus, stem cells could be efficiently labeled with MNPs and tracked in vivo with MRI to evaluate the stem cell therapy. [23–27] CAR-T cells were also labeled by poly-L-lysine coated ultrasmall superparamagnetic iron oxide (USPIO) NPs in vitro and detected using MRI in vivo according to one recent research report.[28] We previously reported surface modification and functionalization of SPIO NPs – nanoworms (NWs) to minimize immune clearance and achieve specific cell targeting. [29–31] In this study, we asked whether positively charged crosslinked iron oxide (CLIO) NWs could be introduced during CD123 CAR-T cell production resulting in CAR-T cell labeling with minimal interference with the CAR-T production process and its cytotoxicity towards tumor cells in vitro.
2. Methodology
2.1. Synthesis, Modifications and Characterizations of Iron Oxide Nanoworms
Superparamagnetic iron oxide (SPIO) nanoworms (NWs) were synthesized and modified according to our previous publications [29, 30]. Briefly, 100 g dextran 20K (Sigma-Aldrich, St. Louis, MO) was dissolved in 500 ml sterile double distilled water and mixed with 21 g Fe(III) chloride (Sigma-Aldrich) and 8.3 g Fe(II) chloride (Thermo-Fisher Scientific, Waltham, MA). Under stirring, 40 ml ammonium was added to form the SPIO NWs. The solution was continuously stirred at 80 °C in the oil bath for 6 h and then purified with the Minimate™ Tangential Flow Filtration System (Pall Corporation, New York, NY). SPIO NWs (30 ml of 10 mg Fe/ml) were mixed with 30 ml double distilled water, 30 ml epichlorohydrin (Sigma-Aldrich) and 10 g sodium hydroxide (Thermo-Fisher Scientific) for 1 h under slow heating from room temperature (RT) to 55 °C to form crosslinked iron oxide (CLIO) NWs. Aminated CLIO was prepared by the addition of 30 ml ammonium (25%) to the CLIO preparation and stirred overnight at RT. The aminated CLIO (CLIO-NH2) was purified by dialysis and ultrafiltration to remove excess epichlorohydrin, sodium hydroxide, ammonium and other side products. To label the CLIO NWs with fluorescence dyes, Cy3-NHS or Cy7-NHS (50 μg in 10 μl DMSO) was conjugated to CLIO-NH2 (0.1 mg of Fe in 200 μl 1X PBS) by mixing the dye molecule with the NWs at 1400 rpm in the cold room (4 °C) overnight. Excess dyes were removed by ultracentrifugation of NWs at 60,000 rpm for 10 min using a Beckman Optima Ultracentrifuge (Beckman Coulter Life Sciences, Indianapolis, IN). To determine the amine content, the excess Cy7 dye in the supernatant after ultracentrifugation was optically measured using a SpectraMax M5 microplate reader (Molecular Devices, San Jose, CA) to determine the unbound Cy7. The bound Cy7 was calculated by subtracting the unbound Cy7 from the total Cy7 added initially. The amine groups on aminated crosslinked NWs were equal to the bound dye molecules. All NW samples were filtered through 0.22 μm filters and stored in sterile vials. Size and zeta potential measurements of CLIO NWs were performed using a Zetasizer (Malvern Panalytical Ltd., Malvern, UK). Transmission electron microscopy (TEM) imaging of CLIO NWs were conducted using an FEI Tecnai G2 transmission electron microscope at a 100 kV working voltage.
2.2. Cell Labeling using CLIO NWs and Imaging
Different cancer cell lines including glioma cells AM38, breast cancer cells SKBR3, MCF7 and MDA-MB 231 were used to test the cell labeling of CLIO NWs. The cells were seeded in 96-well plates and after 24 h, NWs labeled with Cy7 were added to cells for another 24 h incubation. The concentration of NWs incubated with cells was 100 μg Fe/ml. Cells were washed with 1X PBS three times, fixed with 4% paraformaldehyde and imaged with a Zeiss Fluorescence Microscope. Both fluorescence and bright field images were recorded and merged.
2.3. Production of CD123 CAR-T cells using NWs
CD123 CAR constructs and lentiviral vectors were generated as described previously [8, 10]. Fresh peripheral blood mononuclear cells (PBMCs) were obtained from healthy donors under the IRB protocol 06–0720 approved by the University of Colorado Institutional Review Board. T cells were isolated from PBMCs using Ficoll-Paque Premium (GE Healthcare, Chicago, IL) and then selected using human CD3 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Purified CD3+ T cells were cultured in X-VIVO™ 15 medium (Lonza Group, Basel, Switzerland) supplemented with 10% heat-inactivated fetal bovine serum (Corning Inc., Corning, NY), 50 units/ml interleukin (IL)-2 (PeproTech, Rocky Hill, NJ) and 0.5 ng/ml IL-15 (PeproTech) and were stimulated with CD3/CD28 Dynabeads (Gibco Biosciences, Ireland). Transduction of T cells with lentiviral CD123 CAR were conducted using three conditions: protamine sulfate (PS, 10 μg/ml), PS with NW (PS: 10 μg/ml; NW: 100 μg/ml of Fe), and NW (100 μg/ml of Fe). The T cells without CAR-Transduction were left in culture as the mock group. After 2 days of transduction, the medium was changed to remove the leftover lentiviral vector. T cells remained in culture for another 6 days (fresh medium was changed 2 times) and then the Dynabeads were removed and CD123 CAR expression in T cells was analyzed using flow cytometry with FITC anti-CD3 antibody (BioLegend, San Diego, CA), anti-human EGFRt (BioLegend), biotin-Protein L (GeneScript, China), streptavidin-PE (BD Biosciences, San Diego, CA) and Live/Dead Fixable Near-IR Dead Cell Stain Kit (Invitrogen, Thermo Fisher Scientific). The total cell number after Trypan blue staining were also counted for each group of T cells.
2.4. CAR-T cell Cytotoxicity Assays
To evaluate the cytotoxicity of CD123 CAR-T cells produced from PS, PS+NW and NW conditions, CAR-T cells (5 × 104) were co-cultured with equal number of tumor cells (MDS-L cells or human MOLM13 cells) in X-VIVO™ 15 medium supplemented with 10% heat-inactivated FBS for 48 h at 37 °C and 5% CO2 in the incubator. The expression of CD123 and CD3 was analyzed by flow cytometry. The number of tumor cells and CAR-T cells was determined using APC anti-human CD123 antibody (BD Biosciences) and FITC anti-human CD3 antibody (BioLegend). Tumor cells co-cultured with mock T cells and with no T cells were used as the control groups.
2.5. CAR-T Cell Labeling in vitro with Iron Oxide Nanoworms
After CAR-T cell production using lentiviral CD123 CAR in the condition of PS, Cy3 labeled CLIO NWs were incubated with the CD123 CAR-T or mock T cells (1 × 106 cells/ml) at the concentrations of 0, 4, 10 and 20 μg/ml of Fe for 1, 24 and 48 h in the X-VIVO™ 15 medium with 10% heat-inactivated FBS at 37 °C and 5% CO2 in an incubator. After incubation, the cells were washed 3 times with 1X PBS by centrifugation at 500 g for 2 min. The cell viability was determined using a DAPI / Annexin V-FITC apoptosis detection kit (Thermo Fisher Scientific). The percentage of NW+ cells was analyzed by flow cytometry by detecting Cy3 signals on the NW+ cells.
2.6. Microscopy Imaging of Nanoworm Labeled Cells
CD123 CAR-T cells (1 × 106 cells/ml) were incubated with Cy3 or Cy7 labeled CLIO NWs at 25 or 50 μg/ml of Fe for 2 h at room temperature (RT) and quickly washed 5X with 1% bovine serum albumin (BSA) in 1X PBS. The cells were cytospined onto glass slides and fixed with 4% paraformaldehyde for fluorescence microscopy imaging. Cy3 conjugated CLIO NW labeled CD123 CAR-T cells were co-cultured with GFP overexpressed MOLM13 cells for 24 h and then added onto a glass slide for cell imaging.
2.7. Biodistribution of Nanoworm Labeled CAR-T cell in a Leukemia Mouse Model
To establish a leukemia animal model, MOLM13 cells were injected intravenously (i.v.) to NSG mice, 3 × 105 cells for each mouse. After 10 days, CD123 CAR-T cells labeled with Cy7-NWs (3 × 106 cells per mouse) were injected i.v. to the MOLM13 bearing NSG mice. After 3 days, the mice were sacrificed and organs including liver, spleen, kidneys, heart, lungs and bones were collected, scanned using the Li-COR Odyssey near-infrared fluorescence scanner (LI-COR Biosciences, Lincoln, NE). Cy7 intensities from organs were quantified using ImageJ to show the body distribution of CAR-T cells. Bone marrow cells were isolated, stained with PE anti-human CD45 and APC anti-human CD4 antibodies (BioLegend), and analyzed using flow cytometry. To image the CAR-T cells and Cy7-NWs, organs were fixed using 10% neutral buffered formalin (Thermo-Fisher Scientific) for 24 h, immersed into 30% sucrose (Thermo-Fisher Scientific overnight, embedded in the optimal cutting temperature (OCT) compound (Thermo-Fisher Scientific) and sectioned using a Leica CM1850 cryostat (Leica Biosystems Inc., Buffalo Grove, IL). The slides were divided into two groups of staining, one was immunostaining for fluorescence imaging while the other was Prussian blue staining for light microscopy imaging. CAR-T cells were immuno-stained with APC anti-human CD4 antibody (BioLegend). Cell nucleus was stained with Hoechst 33342 (Thermo-Fisher Scientific). Prussian blue staining and Nucleus Fast Red staining was conducted to detect NWs in the tissues with blue color and nucleus showed red color.
2.8. Cell Imaging
The fluorescence microscopy imaging was conducted using a Zeiss fluorescence microscope with 10x and 20x objective lenses. The light microscopy imaging was performed using an Olympus light microscope with a 20x objective lens.
2.9. Flow Cytometry
All flow cytometry analyses of in vitro experiments were performed with the BD FACSCelesta flow cytometer (BD Biosciences) and mouse bone marrow cells from in vivo experiments were analyzed using Guava easyCyte HT flow cytometer (Merck KGaA, Darmstadt, Germany). The flow data were further analyzed and plotted using FlowJo software (FlowJo LLC., Ashland, OR).
2.10. Data Plotting and Statistical Analysis
The data plotting was performed using a GraphPad Prism software (GraphPad Software, San Diego, CA). Statistical significance of data was determined with unpaired, two-tailed t tests using the GraphPad Prism software (GraphPad Software). P values < 0.05 were considered as statistically significant.
3. Results and Discussion
3.1. Surface Modifications, Characterizations of Iron Oxide Nanoworms for Cell Labeling in vitro
The SPIO NWs were coated with dextran layers that were chemically crosslinked by epichlorohydrin in a strong basic condition. The crosslinked iron oxide (CLIO) NWs could be aminated using ammonium in order to further conjugate fluorescence dyes onto the NWs via amine reactions with NHS esters. The modification procedures of NWs are shown in Figure 1A. The morphology of CLIO NWs was visualized under TEM (Figure 1B). The hydrodynamic sizes and zeta-potentials of CLIO NWs were characterized, and the NWs were all positively charged with or without Cy3 or Cy7 conjugation as well as similarly sized with ~120 nm average diameter (Figure 1C and 1D). The total amine groups were quantified by subtracting the unreacted Cy7 dyes from the total dyes added initially. Approximately, there are ~36,000 amine groups available on the CLIO-NH2 surface. These amine groups give us the capacity to further functionalize our NWs with different molecules such as antibodies targeting to cancer cells [30] or complement inhibitors to reduce immune recognition of NWs [31] or click-chemistry tools to eliminate excess antibodies from plasma [32]. In this study, we aimed to explore the possibility of using the positively charged aminated CLIO for CAR-T cell labeling.
Figure 1. Modifications and characterizations of dextran-coated superparamagetic iron oxide (SPIO) nanoworms (NWs).
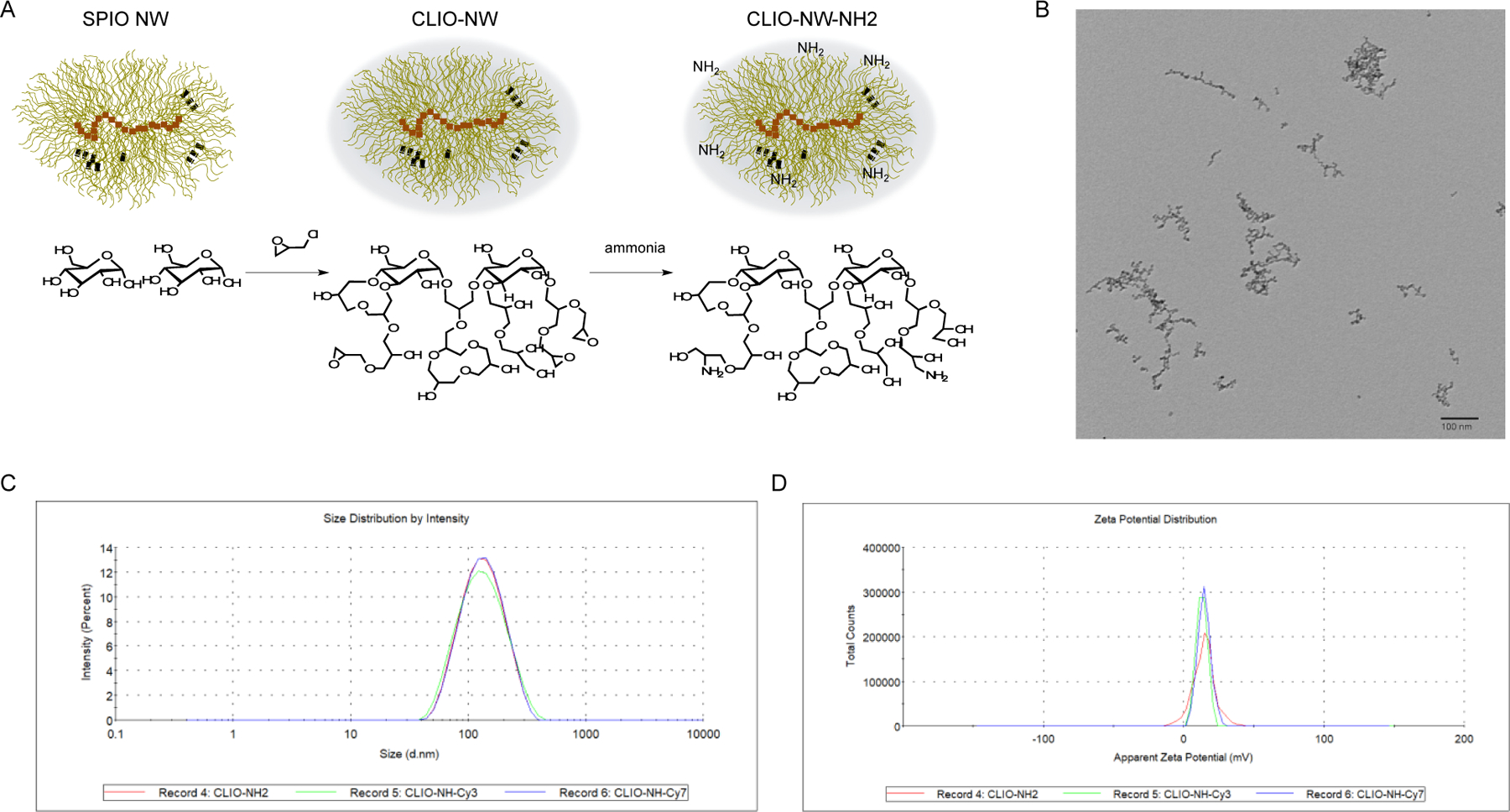
(A) Schematic of modifications of NWs. SPIO NWs consist of iron oxide cores surrounded by a dextran shell and the dextran layer was crosslinked by epihalohydrin to form crosslinked iron oxide (CLIO) NWs. CLIO NWs were further modified with amine groups, which could be further conjugated with fluorescence molecules as Cy3 and Cy7.
(B) Transmission Electron Microscopy image of CLIO NWs. The scale bar is 100 nm.
(C) Size measurements of CLIO-NH2 NWs and Cy3 or Cy7 conjugated CLIO NWs. The hydrodynamic sizes of CLIO NH2, CLIO-NH-Cy3 and CLIO-NH-Cy7 are 141.3 ± 57.89 nm, 142.0 ± 64.21 nm and 142.5 ± 57.93 nm, respectively.
(D) Zeta potential measurements of CLIO-NH2 NWs and Cy3 or Cy7 conjugated CLIO NWs. The zeta potentials of CLIO NH2, CLIO-NH-Cy3 and CLIO-NH-Cy7 are 14.4 ± 7.82 mV,13.0 ± 3.96 mV and 14.7 ± 4.60 mV, respectively.
3.2. NW Labeling on Mammalian Cell Lines
In this study, we firstly demonstrated the cell labeling of CLIO NWs in different cell lines, including glioma cell line AM38, breast cancer cell lines SKBR-3, MCF-7 and MDA-MB231. The imaging of cells incubated with Cy7 labeled CLIO NWs was taken by both fluorescence and bright field settings (Figure 2). All four cell lines were labeled with NWs that were able to be detected by fluorescence imaging. The positively charged NWs mostly adhered to the cell surface due to their interactions with negative charged cell membranes. By using this universal cell labeling mechanism, our CLIO-NH2 NWs could serve as a cell labeling reagent for a broad range of mammalian cells.
Figure 2. CLIO NW labeling on different human cell lines.
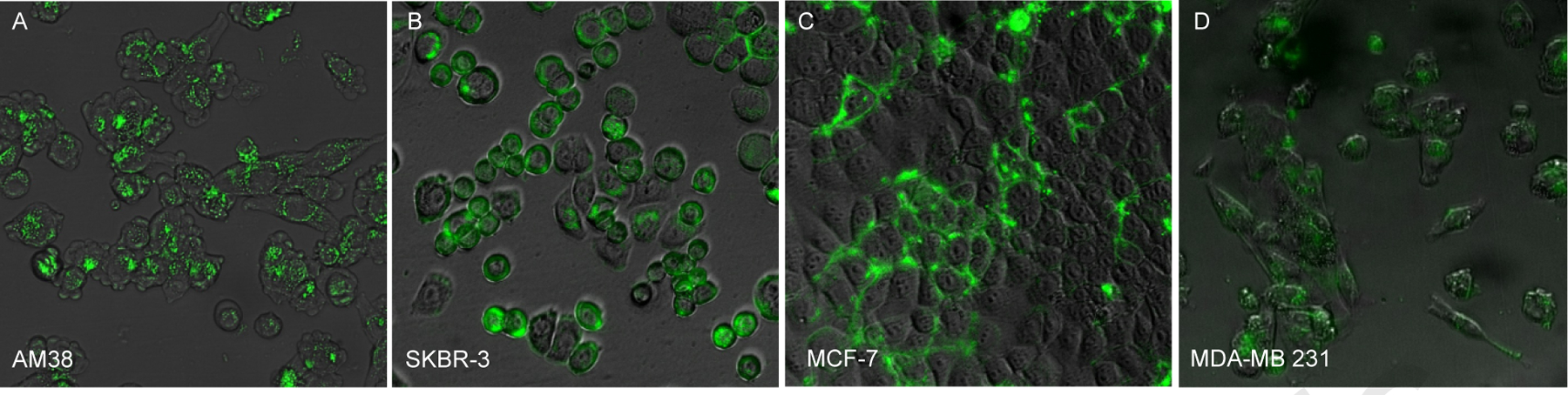
CLIO NWs conjugated with Cy7 were incubated with cells cultured in 96-well plates at 100 μg/ml of Fe for 24 h and washed, fixed and imaged using a fluorescence microscope. Both fluorescence and bright-field images were taken, and CLIO NWs were detected by the near-infrared channel (green color) in the cells (A) human glioma AM38; (B) human breast cancer SKBR-3; (C) human breast cancer MCF-7; (D) human breast cancer MDA-MB 231.
3.3. Nanoworm-mediated CAR-T Production and CAR-T Cytotoxicity on Tumor Cells in vitro
Since CLIO-NH2 NWs are positively charged, we compared the effect NWs had on CD123 CAR-T production with positively charged protamine sulfate (PS). We further evaluated the effects of NW addition on CAR expressions and T cell viability. The production procedure of CD123 CAR-T cells followed previous reports [8, 10]. The transduction of the lentiviral anti-CD123 CAR was conducted under three conditions: PS, PS+NW and NW (Figure 3A) and the transduction efficiency was evaluated with flow cytometry by detecting the EGFRt in CD3+ T cells (Figure 3B). The CAR-T transduction efficiency of NW plus PS (33%) was similar to that in the condition of PS alone (~35%). CLIO NW alone decreased transduction efficiency to ~23% (Figure 3C). Cell viability was similar (25 to 30%) among the three groups (Figure 3C) upon completion of CAR-T production on day 8. Viral-mediated gene-transfer was tested at different concentrations of PS by Cornetta et al, showing that 10 μg/ml PS, the current concentration used in CAR-T transductions, yielded the highest gene-transfer (>80%) [33]. CLIO-NH2 NWs and PS are positively charged suggesting that this property of NWs contributes to its role in lentiviral CAR-transduction, though the transduction via NWs was lower than PS.
Figure 3. Evaluation of CAR-T production in the conditions of CLIO NWs.
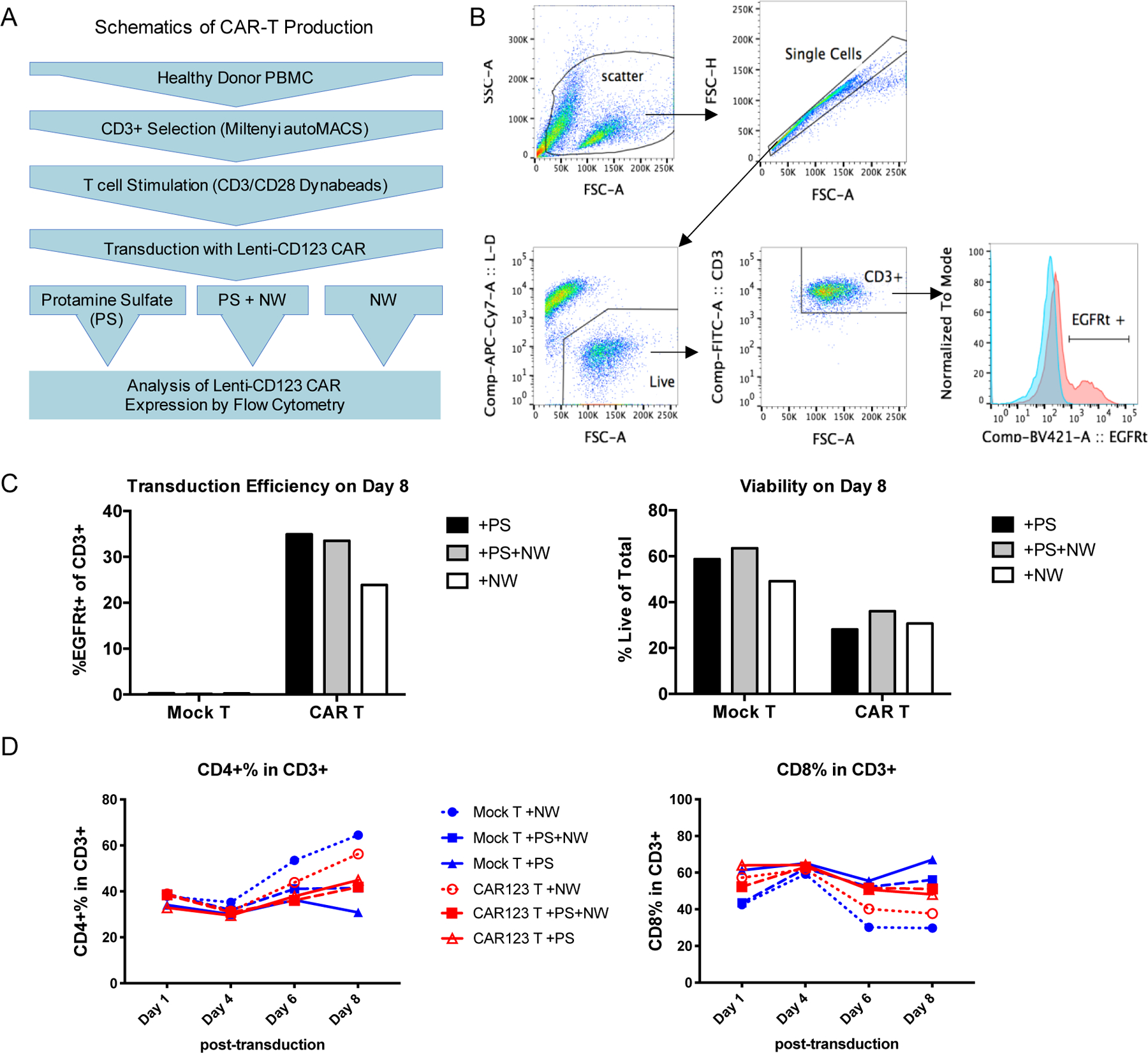
(A) Schematic of CAR-T cell production mediated by protamine sulfate (PS), NW or both.
(B) Gating strategy of flow cytometry analysis on Lentiviral CD123 CAR transduction efficiency.
(C) Bar graphs of transduction efficiency and viability in CAR-T cell products mediated by PS, NW or both.
(D) Analysis of CD4+ and CD8+ sub-populations in the total CD3+ T cell population
Interestingly, CLIO-NH2 NW increased CD4+ compartment and decreased CD8+ compartment in both mock T cells and CAR-T cells (Figure 3D). The anti-tumor efficacy of CAR-T in vivo could be related to the CD4+ to CD8+ ratio in CAR-T populations after production. Turtler et al. reported that B-ALL patients treated with CAR-T consisted with a 1:1 ratio of CD4+ to CD8+ had the highest rate of remission [34]. Lee et al. demonstrated that anti-CD19 CAR-T with a 70:30 ratio of CD4+ to CD8+ had better tumor inhibition in Raji tumor NSG mice than the group of 1:1 ratio of CD4+ to CD8+ cells [35]. The CD4:CD8 ratios for the CD123 CAR-T cells produced in the PS, PS+NW and NW groups were 45:48, 42:51 and 56:38, respectively. Thus, we further investigated if the changes of CD4:CD8 ratios of CAR-T would influence the cytotoxicity on tumor cells in vitro. The CAR-T cells were co-cultured with CD123+ tumor cells MDS-L or MOLM13 at 1:1 cell number ratio of T cells to tumor cells. After 48 h, the CD123 CAR-T cells would bind and kill the tumor cells, which was analyzed with flow cytometry (Figure 4A). The percentage of CD123+ tumor cells co-cultured with CD123 CAR-T cells was significantly reduced compared to those with mock T cells or without T cells (Figure 4B, P<0.05 for MDS-L and P<0.01 for MOLM13). CAR-T cells produced in the presence of CLIO-amine NWs showed a similar level of cytotoxicity to tumor cells as the CAR-T cells produced in the presence of PS (Figure 4B), suggesting that CLIO-NH2 NWs did not alter CAR-T antitumor activity, though the efficiency for NW-mediated transduction appeared lower than PS groups (Figure 3C). SPIO NPs have been clinically used for treating iron-deficiency anemia in patients with chronic kidney disease or MRI of livers and inflamed tissues. [36, 37] Here we explored the new application of SPIO as lentiviral CAR-Transduction reagent for the first time. Our results showed low toxicity of NWs on the CAR-T cells and no side effects of NWs on the CAR-T cell cytotoxicity on tumor cells. The interactions of NWs with T cells need more systemic investigations to understand the dynamic effects on CAR-T cells during the production process.
Figure 4. Kill assay of tumor cells by NW-transduced CAR-T cells.
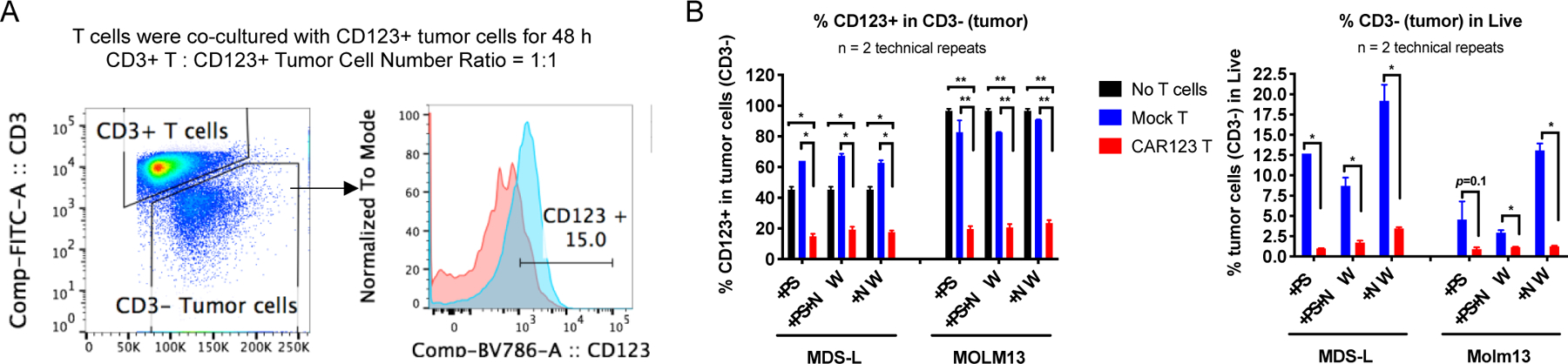
(A) Gating strategy of flow cytometry analyses on CD123+ tumor cells (MDS-L) after co-cultured with mock or CAR-T cells.
(B) Statistical analyses of percentage of CD123+ cells in CD3-tumor cells after co-cultured with mock or CAR T cells produced with protamine sulfate (PS), NW or PS+NW. *P<0.05, **P<0.01.
3.4. Labeling of CAR-T cells using Nanoworms and CAR-T Targeting to Tumor Cells in vitro
The CD123 CAR or mock T cells were incubated with Cy3 conjugated CLIO-NH2 NWs at 4, 10 and 20 μg/ml of Fe for 1, 24, and 48 h in cell culture medium and then the cells were washed with 1X PBS by centrifugation. Analysis of Cy3-NW labeled cells showed increased labeling with increased NW concentration. (Figure 5A). At 20 μg/ml concentration of the NWs, nearly 25% CAR-T cells were labeled after 1 h incubation. The percentage of NW positive cells decreased at 24 and 48 h after NW addition to T cells due to T cell proliferation (Figure 5A).
Figure 5. Direct labeling of CAR-T cells with NWs and CAR-T targeting to MOLM13 cells in vitro.
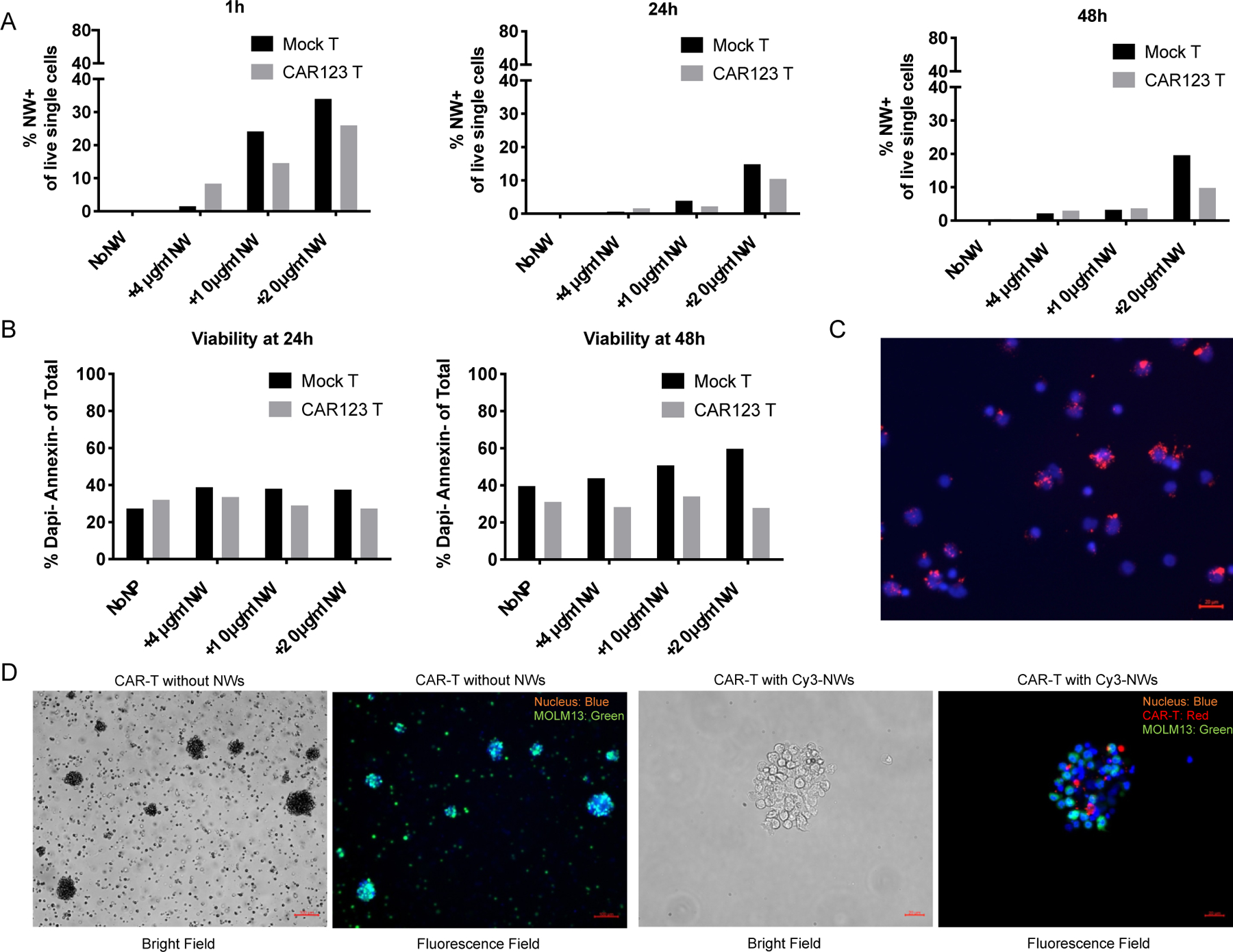
(A) Mock or CAR T cells were cultured at 1×106/ml and Cy3 conjugated NWs were added to cells at 4, 10 and 20 μg/ml. Labeling of NW on T cells were examined by flow cytometry 1, 24 and 48 h post-incubation. Percentage of NW+ cells in live single cells were calculated and plotted in bar graphs.
(B) Cell viability of mock and CAR T cells were analyzed with DAPI/Annexin using flow cytometry.
(C) Fluorescence images of Cy3 conjugated NW labeled T cells. NWs were incubated with 1×106/ml T cells at 25 μg/ml for 2 h at RT. Cells were washed 5X with 1X PBS containing 1% bovine serum albumin (BSA) and cytospined or just applied onto glass slides for fluorescence microscopy imaging. Cell nuclei were stained using Hoechst (blue color). Cy3-NWs showed red color.
(D) Representative images of CAR-T cells loaded with Cy3 conjugated NWs (red color) and Hoechst (blue color) staining after co-cultured with MOLM13 cells expressing green fluorescent protein (GFP) for 24 h.
T cell viability was analyzed after 24 and 48 h incubation with Cy3-NWs. The CD123 CAR-T cells showed ~25–30% viability after NW labeling, which was lower than mock T cells which were ~30–50% viability (Figure 5B). These results indicated the CD123 CAR-T cells are more sensitive to NW labeling than mock T cells. The Cy3-NW labeled CAR-T cells were detected under a fluorescence microscope (Figure 5C). Furthermore, we tested the Cy3-NW labeled CD123 CAR-T targeting to the human leukemia MOLM13 cells in vitro. The CAR-T without Cy3-NW labeling or labeled with Cy3-NWs showed the binding to MOLM13 cells (Figure 5D), indicating the direct in vitro labeling of CAR-T cells with the NWs did not influence the T cell targeting to tumor cells.
3.5. Body Distribution of Nanoworm Labeled CAR-T cells in a Leukemia Mouse Model
The body distribution of Cy7-CLIO labeled CAR-T cells in the MOLM13 leukemia NSG mouse model was studied. CD123 CAR-T cells were incubated with Cy7-NWs for 2 h at 50 μg/ml Fe concentrations to increase the Cy7 intensities in T cells for in vivo detection. The Cy7-NW labeled CAR-T cells (3 million cells per mouse) were then injected intravenously into MOLM13 bearing NSG mice (n=2). The mice were sacrificed 3-day post-injection of CAR-T cells and organs were scanned by the Li-COR Odyssey near-infrared fluorescence imager (Figure 6A). The fluorescence intensities of Cy7 were quantified in bone marrow (BM), heart, lungs, liver, spleen and kidneys. The Cy7 signals in the liver had the highest level followed by lungs and spleen, whereas the BM, heart and kidney had the lowest levels of Cy7 (Figure 6B). The BM cells were isolated and analyzed with flow cytometry to detect Cy7-NW labeled CAR-T cells. The percentage of Cy7+, CD4+, CD45+ and GFP-cells was ~10% in the total BM cell populations (Figure 6C), indicating the BM accumulation of CD123 CAR-T cells after 3 days of injection. Histology analysis of mouse organs was conducted by preparing frozen sections for immunostaining and Prussian blue staining. CAR-T cells were detected by staining with APC anti-human CD4 antibody and the CAR-T cells were colocalized with Cy7-NWs in the spleen, whereas no colocalization of CD4 stained CAR-T cells with Cy7-NWs were observed in the liver (Figure 6D). This observation suggested that the NWs were dissociated from CAR-T during the transportation across the liver sinusoidal endothelial cells but remain attached with CAR-T when passing through spleen non-sinusoidal endothelia cells, which also indicated that most NWs were on the surface of CAR-T cells. However, it is very likely that NWs could detach from CAR-T cells in human spleens because of the sinusoidal endothelium in human spleens, similar to the liver sinusoidal endothelium in both human and mice[38].
Figure 6. In Vivo biodistribution of NW labeled CD123 CAR-T cells in the MOLM13 NSG mouse model.
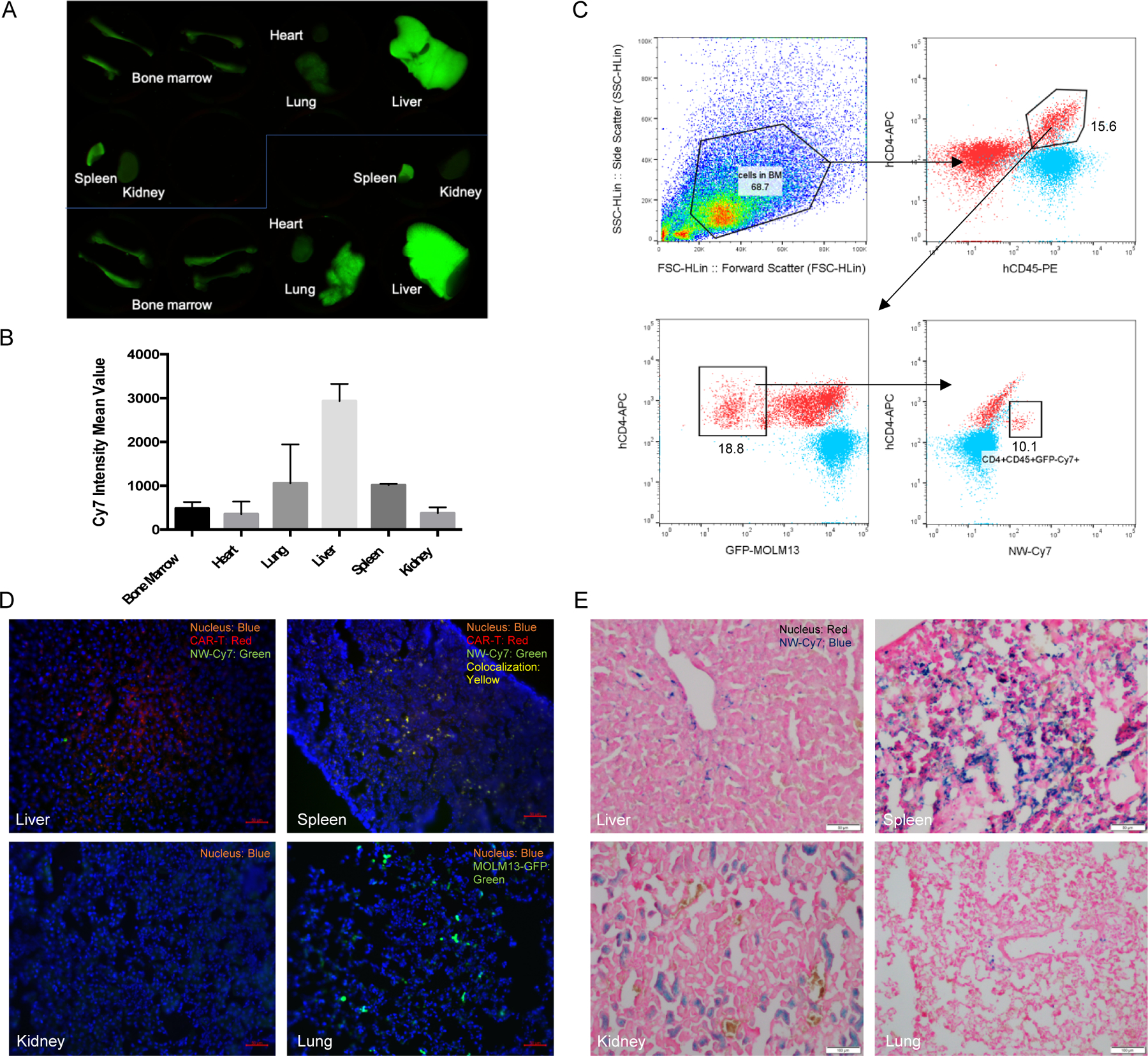
CD123 CAR-T cells were firstly labeled with Cy7 conjugated NWs. Cells were washed 3X with 1X PBS to remove free NWs. CAR-T cells (3 million) were then intravenously injected into MOLM13 bearing NSG mice (10-day postinjection of 3 × 105 MOLM13 cells).
(A) Organ scan image showed CAR-T distribution in mice (n=2). Organs were collected after 3 day-postinjection of CAR-T cells and scanned using the Li-COR Odyssey near-infrared fluorescence imager.
(B) CAR-T biodistribution levels in different organs. Cy7 intensity mean values in the organ scan image were measured using ImageJ and data was processed using Prism 6 software.
(C) Analysis of bone marrow (BM) cell population using flow cytometry. BM cells were stained using anti-human PE-CD45 and APC-CD4 antibodies. Samples were analyzed using the Guava easyCyte HT flow cytometer and the data was analyzed using FlowJo software.
(D) Fluorescence histology images of CAR-T cells and Cy7-NWs in different organs. Organs were fixed using 4% paraformaldehyde, embedded and sectioned using the Leica CM1850 cryostat. CAR-T cells were immunostained with APC anti-human CD4 antibody. CAR-T cells showed red color while Cy7-NWs showed green color in liver and spleen, whereas in the lung, the green color may come from MOLM13 cells expressing GFP.
(E) Prussian blue stained histology images of NWs in different organs. SPIO NWs showed blue color after Prussian blue staining and cell nuclei showed red color after nucleus fast red staining.
GFP overexpressed MOLM13 cells were detected in lungs but no CAR-T cells or Cy7-NWs were detected in lungs and the kidney (Figure 6D). Prussian blue staining was used to detect NWs in the frozen sections. Both liver and spleen tissues were found with NW accumulations (Figure 6E). The blue-color areas in the kidney were not from NW signals but could be from the free iron of NWs during the degradation process. These in vivo data have demonstrated the CAR-T body distribution and detection using NWs as the cell labeling reagent.
Another aspect that could influence CAR-T cells in vivo is the complement system which can help T cell proliferation in vivo [39, 40] as well as contribute to the complement-dependent cytotoxicity when using the specific antibody to eliminate CAR-T cells to reduce the toxicity of the CAR-T therapy [41, 42]. Our previous studies have shown that the NWs used in the present study to label CAR-T cells have minimum complement activation themselves in mouse due to the chemical crosslinking modifications [29], while in human the NWs have complement protein binding though the cellular uptake by blood immune cells is much less compared to the non-crosslinked NWs [43]. Further studies are needed to evaluate the complement protein interactions with the NW labeled CAR-T cells.
There are few reports on labeling CAR-T cells using NPs. Most recently, ultra-small SPIO (USPIO) NPs were used to label CAR-T cells in vitro and the CAR-T cells could be tracked using MRI and target the glioblastoma cells in vivo. [28] The USPIO needs to mix with poly-L-lysine to label CAR-T cells. This study has showed the potential to monitor CAR-T accumulation into the brain tumors and validate the efficacy of CAR-T therapy using USPIO NPs. Our NWs could also label mouse T cells which were detected in the glioma site in a mouse model (data not shown). Our future studies will continue exploring the possibilities of using the NWs to monitor CAR-T trafficking and engage CAR-T cell antitumor efficacy in vivo.
4. Conclusions
Our interest on the iron oxide nanomaterials inspired us to conduct this pilot study to investigate CAR-T cell labeling with our engineered iron oxide nanoparticles named nanoworms (NWs). We for the first time demonstrated that the iron oxide nanomaterial could be used for lentiviral transduction of T cells though the transduction efficiency was lower compared to protamine sulfate (PS). The positively charges on the NWs and the iron concentration could be adjusted to improve the transduction of CAR-T cells. The CAR-T cells produced with NWs had similar cytotoxicity on tumor cells as those produced using PS. The direct in vitro CAR-T labeling with NWs is straightforward and simple. However, the cell incubation conditions with NWs need improvement to achieve more efficient cell labeling. The NW labeled CAR-T cells showed similar level of binding to targeted tumor cells compared to NW free ones. The in vivo biodistribution data indicated that some CAR-T cells retained Cy7-NWs up to 72 h post-injection in a leukemia mouse model. Further studies are needed to understand the NW labeled CAR-T targeting to tumor cells in vivo and more investigations will focus on the CAR-T labeling for therapy outcome prediction and validation.
Highlights.
Iron oxide nanoworms with positive charges could contribute to both CAR-T manufacturing and labeling in vitro
Iron oxide nanoworms do not interfere with the capacity of CAR-T cells on targeting and killing tumor cells after labeling CAR-T cells.
CAR-T cells could be detected in vivo using iron oxide nanoworms as the cell labeling reagent.
Acknowledgments
We acknowledge the support from NIH 5R01CA194058, Evans MDS Foundation funding, and Jordan Lab from Division of Hematology, Department of Medicine, University of Colorado Anschutz Medical Campus. We also thank Dr. Stephen J. Forman from City of Hope Comprehensive Cancer Center for providing the CD123 CAR constructs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
The authors declare that they have no known conflict of interest, financial or otherwise.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Krebs S, et al. , CAR Chase: Where Do Engineered Cells Go in Humans? Front Oncol, 2020. 10: p. 577773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sadelain M, CAR therapy: the CD19 paradigm. J Clin Invest, 2015. 125(9): p. 3392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmad A, Uddin S, and Steinhoff M, CAR-T Cell Therapies: An Overview of Clinical Studies Supporting Their Approved Use against Acute Lymphoblastic Leukemia and Large B-Cell Lymphomas. Int J Mol Sci, 2020. 21(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voelker R, CAR-T Therapy Is Approved for Mantle Cell Lymphoma. JAMA, 2020. 324(9): p. 832. [DOI] [PubMed] [Google Scholar]
- 5.Mullard A, FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discov, 2021. 20(3): p. 166. [DOI] [PubMed] [Google Scholar]
- 6.Voelker R, Cell-Based Gene Therapy Is New Option for Multiple Myeloma. JAMA, 2021. 325(17): p. 1713. [DOI] [PubMed] [Google Scholar]
- 7.Germing U, et al. , Refinement of the international prognostic scoring system (IPSS) by including LDH as an additional prognostic variable to improve risk assessment in patients with primary myelodysplastic syndromes (MDS). Leukemia, 2005. 19(12): p. 2223–31. [DOI] [PubMed] [Google Scholar]
- 8.Mardiros A, et al. , T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood, 2013. 122(18): p. 3138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stevens BM, et al. , Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes. Nat Commun, 2018. 9(1): p. 3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stevens BM, et al. , CD123 CAR T cells for the treatment of myelodysplastic syndrome. Exp Hematol, 2019. 74: p. 52–63 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krebs S, et al. , Imaging of CAR T-Cells in Cancer Patients: Paving the Way to Treatment Monitoring and Outcome Prediction. J Nucl Med, 2019. 60(7): p. 879–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perrin J, et al. , Cell Tracking in Cancer Immunotherapy. Front Med (Lausanne), 2020. 7: p. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krekorian M, et al. , Imaging of T-cells and their responses during anti-cancer immunotherapy. Theranostics, 2019. 9(25): p. 7924–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yaghoubi SS, et al. , Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat Clin Pract Oncol, 2009. 6(1): p. 53–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sellmyer MA, et al. , Imaging CAR T Cell Trafficking with eDHFR as a PET Reporter Gene. Mol Ther, 2020. 28(1): p. 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emami-Shahri N, et al. , Clinically compliant spatial and temporal imaging of chimeric antigen receptor T-cells. Nat Commun, 2018. 9(1): p. 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minn I, et al. , Imaging CAR T cell therapy with PSMA-targeted positron emission tomography. Sci Adv, 2019. 5(7): p. eaaw5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vedvyas Y, et al. , Longitudinal PET imaging demonstrates biphasic CAR T cell responses in survivors. JCI Insight, 2016. 1(19): p. e90064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krebs S, et al. , Antibody with Infinite Affinity for In Vivo Tracking of Genetically Engineered Lymphocytes. J Nucl Med, 2018. 59(12): p. 1894–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hege KM, et al. , Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer, 2017. 5: p. 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kircher MF, Gambhir SS, and Grimm J, Noninvasive cell-tracking methods. Nat Rev Clin Oncol, 2011. 8(11): p. 677–88. [DOI] [PubMed] [Google Scholar]
- 22.Kolosnjaj-Tabi J, et al. , Cell labeling with magnetic nanoparticles: opportunity for magnetic cell imaging and cell manipulation. J Nanobiotechnology, 2013. 11 Suppl 1: p. S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu CW, et al. , Bifunctional magnetic silica nanoparticles for highly efficient human stem cell labeling. Nano Lett, 2007. 7(1): p. 149–54. [DOI] [PubMed] [Google Scholar]
- 24.Suh JS, et al. , Efficient labeling of mesenchymal stem cells using cell permeable magnetic nanoparticles. Biochem Biophys Res Commun, 2009. 379(3): p. 669–75. [DOI] [PubMed] [Google Scholar]
- 25.Jasmin, et al. , Labeling stem cells with superparamagnetic iron oxide nanoparticles: analysis of the labeling efficacy by microscopy and magnetic resonance imaging. Methods Mol Biol, 2012. 906: p. 239–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andreas K, et al. , Highly efficient magnetic stem cell labeling with citrate-coated superparamagnetic iron oxide nanoparticles for MRI tracking. Biomaterials, 2012. 33(18): p. 4515–25. [DOI] [PubMed] [Google Scholar]
- 27.Zhou S, et al. , Labeling adipose derived stem cell sheet by ultrasmall super-paramagnetic Fe3O4 nanoparticles and magnetic resonance tracking in vivo. Sci Rep, 2017. 7: p. 42793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie T, et al. , Non-invasive monitoring of the kinetic infiltration and therapeutic efficacy of nanoparticle-labeled chimeric antigen receptor T cells in glioblastoma via 7.0-Tesla magnetic resonance imaging. Cytotherapy, 2021. 23(3): p. 211–222. [DOI] [PubMed] [Google Scholar]
- 29.Wang G, et al. , High-relaxivity superparamagnetic iron oxide nanoworms with decreased immune recognition and long-circulating properties. ACS Nano, 2014. 8(12): p. 12437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang G, et al. , In Vitro and In Vivo Differences in Murine Third Complement Component (C3) Opsonization and Macrophage/Leukocyte Responses to Antibody-Functionalized Iron Oxide Nanoworms. Front Immunol, 2017. 8: p. 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaikwad H, et al. , Complement Inhibitors Block Complement C3 Opsonization and Improve Targeting Selectivity of Nanoparticles in Blood. Bioconjug Chem, 2020. 31(7): p. 1844–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith WJ, et al. , Accelerated Blood Clearance of Antibodies by Nanosized Click Antidotes. ACS Nano, 2018. 12(12): p. 12523–12532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cornetta K and Anderson WF, Protamine sulfate as an effective alternative to polybrene in retroviral-mediated gene-transfer: implications for human gene therapy. J Virol Methods, 1989. 23(2): p. 187–94. [DOI] [PubMed] [Google Scholar]
- 34.Turtle CJ, et al. , CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest, 2016. 126(6): p. 2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee DH, et al. , Improved Expansion and Function of CAR T Cell Products from Cultures Initiated at Defined CD4:CD8 Ratios. Blood, 2018. 132. [Google Scholar]
- 36.Wang YX and Idee JM, A comprehensive literatures update of clinical researches of superparamagnetic resonance iron oxide nanoparticles for magnetic resonance imaging. Quant Imaging Med Surg, 2017. 7(1): p. 88–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suciu M, et al. , Applications of superparamagnetic iron oxide nanoparticles in drug and therapeutic delivery, and biotechnological advancements. Beilstein J Nanotechnol, 2020. 11: p. 1092–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shetty S, Lalor PF, and Adams DH, Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol, 2018. 15(9): p. 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Afshar-Kharghan V, The role of the complement system in cancer. J Clin Invest, 2017. 127(3): p. 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwan WH, van der Touw W, and Heeger PS, Complement regulation of T cell immunity. Immunol Res, 2012. 54(1–3): p. 247–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rafiq S, Hackett CS, and Brentjens RJ, Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol, 2020. 17(3): p. 147–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brandt LJB, et al. , Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front Immunol, 2020. 11: p. 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Inturi S, et al. , Modulatory Role of Surface Coating of Superparamagnetic Iron Oxide Nanoworms in Complement Opsonization and Leukocyte Uptake. ACS Nano, 2015. 9(11): p. 10758–68. [DOI] [PMC free article] [PubMed] [Google Scholar]