

Abstract
Thrombotic complication has been an important symptom in critically ill patients with COVID-19. It has not been clear whether the virus spike (S) protein can directly induce blood coagulation in addition to inflammation. Heparan sulfate (HS)/heparin, a key factor in coagulation process, was found to bind SARS-CoV-2 S protein with high affinity. Herein, we found that the S protein can competitively inhibit the bindings of antithrombin and heparin cofactor II to heparin/HS, causing abnormal increase in thrombin activity. SARS-CoV-2 S protein at a similar concentration (~10 μg/mL) as the viral load in critically ill patients can cause directly blood coagulation and thrombosis in zebrafish model. Furthermore, exogenous heparin/HS can significantly reduce coagulation caused by S protein, pointing to a potential new direction to elucidate the etiology of the virus and provide fundamental support for anticoagulant therapy especially for the COVID-19 critically ill patients.
Keywords: SARS-CoV-2, Spike protein, Heparan sulfate, Coagulation, Anticoagulant therapy
1. Introduction
The new coronavirus pneumonia (COVID-19) caused by SARS-CoV-2 virus infection has become a major threat to human health. Apart from inflammation, increasing clinical data have emerged to indicate that thrombotic complications are an important feature in critically ill patients with COVID-19 [1], [2], [3], [4], [5]. However, the pathogenesis of coagulopathy in COVID-19 has not been fully elucidated. Severe infection and inflammation invariably lead to hemostatic abnormalities [6], [7], [8]. SARS-CoV-2 infection could induce a cytokine storm with the activation of leukocytes, endothelium and platelets, which would promote the upregulation of tissue factor, coagulation activation, thrombin generation, and fibrin formation [9]. Furthermore, severe hypoxemia may result in reducing blood flow and vasoconstriction in COVID-19 [10], [11]. However, it remains unclear whether SARS-CoV-2 is directly involved in the coagulation process.
Vascular endothelial damage is part of the pathogenesis of organ injury in severe COVID-19 [1], [12]. The endothelium covered with heparan sulfate (HS), a polydisperse glycosaminoglycan. Consistent with heparin, one of the important function of heparin sulfate on the endothelium of blood vessels is to regulate blood coagulation [13], [14], [15]. Under normal physiological conditions, hemostasis is exquisitely initiated, controlled, and terminated by a series of processes involving the action of different serine proteases (e.g. thrombin, and coagulation factor Xa) and anticoagulation factors, such as antithrombin (AT) and heparin cofactor II (HCII) [16], [17], [18], [19], [20], [21]. Thrombin, also called factor IIa, is the main effector protease of the coagulation cascade, which converts circulating fibrinogen to fibrin monomer before its polymerization to form fibrin, the fibrous matrix with platelets of blood clots [16]. Along with the clotting process, a series of serine protease inhibitors (e.g., AT and HCII) can inactivate thrombin to prevent excessive clotting [17], [18], [19]. In this overall complex process, HS/heparin play an important role as allosteric activators to increase the activities of AT and HCII, which exist in blood with usually low activity in a repressed reactivity state. Heparin can allosterically activate AT/HCII and increase their inhibitory ability by 1000–10,000 folds [19], [20], [21].
Recent reports have shown that SARS-CoV-2 spike (S) protein can bind heparin and heparan sulfate (HS) with high affinity [22], [23], [24], [25], [26]. A question is therefore raised on whether SARS-CoV-2 can interfere with the anticoagulation process by competing with HS in the vascular endothelium. To investigate whether SARS-CoV-2 can interfere directly with the anticoagulation process by binding to HS/heparin, we established competitive binding experiments on heparin/HS polysaccharide microarrays for the first time. The effects of S protein on thrombin activity and thrombus formation were investigated by in vitro anticoagulation experiments and in vivo experiments using the zebrafish embryo model.
2. Materials and methods
2.1. Materials
The SARS-CoV-2 S trimer with His-tag, and S1 subunit with mouse IgG2a Fc-tag (mFc-tag) were obtained from ACRO Biosystems (Beijing, China). These proteins are expressed from human 293 cells (HEK293) with the tag at C-terminus. AT and HCII with His-tag were obtained from Sino Biologicals (Beijing, China). The AlexaFluor (AF) 488-labeled goat anti-mouse IgG and AF 647-labeled rabbit anti-His-tag were purchased from Cell Signaling Technology (Danvers, USA). AT, Xa, thrombin and chromogenic substrates for anticoagulation experiment were purchased from Adhoc International Technologies (Beijing, China). HCII, Texas Red and low melting agarose were obtained from Thermo Fisher Scientific (Rockford, USA). Unfractionated heparin was generously provided by Qilu Pharmaceutical (Jinan, China). Heparan sulfate (HS), MS-222, 1-phenyl-2-thiourea, o-dianisidine and Fluorescein isothiocyanate-dextran and BSA was from Millipore-Sigma (St. Louis, USA). Bivalirudin was obtained from Shenzhen Salubris pharmaceuticals (Shenzhen, China). NHS glass slides (Schott Nexterion H) were purchased from Schott Glaswerke AG (Jena, Germany). For in vivo experiment in zebrafish embryos, the buffer of SARS-CoV-2 S trimer was replaced with sterile PBS by centrifugal filter (Amicon Ultra-0.5, 30 kDa), and the protein concentration was determined by Mico BCA™ protein assay kit (Thermo Fisher Scientific, Rockford, USA). The embryos of 48 hpf were obtained through natural mating (AB line) and maintained at 28.5 °C in Nantong University Zebrafish Facility. The study conducted is fully compliant with the local institutional and the Chinese national regulation for the animal protection. Embryonic stages were defined as described in our previous work [27]. The Tg(fli1a:EGFP-CAAX) was generated in Nantong University and Tg(gata1:DesRed) lines were used as previously described [28].
2.2. Heparin microarray binding assays of SARS-CoV-2 S1, AT and HCII
Microarray printing was carried out onto NHS-funtionalized glass slides (Schott Nexterion H; Jena, Germany) as a 16-subarray layout. Heparin was dissolved in sodium phosphate buffer (100 mmol/L, pH 7.5) at a concentration of 0.5 mg/mL and printed in eight replicates using sciFLEXARRAYER (SCIENION, Berlin, Germany). The printed heparin was left on slide at 25 °C overnight with 80% humidity. Before binding the slides were rinsed with TBST (20 mM Tris, 150 mM NaCl, 0.05% Tween 20, pH 7.4) for 2 min to remove the excess heparin and blocked with 1% BSA in TBST for 30 min. Binding assay were carried out essentially using protocols published previously [29]. In brief, for binding and competition of binding analyses, 100 μL protein solution in 1% BSA were applied to each subarray and the slide was left for 1.5 h at room temperature. After washing for three times with TBST, the slide was incubated with 100 μL diluted corresponding fluorescent antibody for 1 h at room temperature. The slide was washed three times with TBST, three times with distilled water, and dried with centrifugation. To assess the effect of the mFc tag of the mouse IgG2a Fc-labeled S1 on heparin binding, three Fc-containing fluorescent antibodies, goat anti-mouse IgG, rabbit anti-His-tag and a mouse IgG2a Fc-containing anti-GST, were as the controls. No heparin binding signals were observed in any of the three control experiments (data now shown).
2.3. Competitive binding of SARS-CoV-2 S1 and AT or HCII
Competitive binding assays were carried out by three ways (Table 1 ). i) The pre-mixed solution of S1 and AT (or HCII) was incubated on microarray for simultaneously competitive binding; ii) AT or HCII was incubated with heparin on microarray for 1.5 h. After washing with TBST for three times, S1 was incubated for a further 1.5 h; iii) S1 was incubated for 1.5 h before AT or HCII was incubated for a further 1.5 h. As controls, S1, AT and HCII were also incubated respectively in parallel. AF488 labeled anti-mouse IgG was added and incubated for 1 h before addition of AF647 labeled anti-His-tag and incubation for another hour. The final binding signals of 8 (spots) × 4 (rows) repeating heparin spots in both competition experiments and control experiments were readout and averaged. The ratio of the mean value of competition experiment to that of the control experiment was calculated shown as percentage as a measure to indicate the degree of S protein competition.
Table 1.
Experimental design of on-slide competitive binding assay.
| Incubation time | Competition assay |
Control |
|||||
|---|---|---|---|---|---|---|---|
| i | ii | iii | 1 | 2 | 3 | 4 | |
| 1.5 h | S1 + AT/HCII | AT/HCII | S1 | S1 | 1% BSA | AT/HCII | 1% BSA |
| 1.5 h | 1% BSA | S1 | AT/HCII | 1% BSA | S1 | 1% BSA | AT/HCII |
| 1 h | AF488 labeled anti-mouse IgG | AF488 labeled anti-mouse IgG | |||||
| 1 h | AF 647 labeled anti-His-tag | AF 647 labeled anti-His-tag | |||||
Concentration of His-AT in competitive binding is 1 μg/mL, and His-HCII of 2 μg/mL. The corresponding concentration of mFc-S1 were 2 μg/mL and 4 μg/mL. The concentration of control proteins was the same as that of competitive binding.
2.4. Microarray data readout and analysis
Microarray slides were scanned with GenePix 4300A fluorescence scanner (Molecular Devices, Union City, CA) with red laser channel (scan wavelength at 635 nm and 488 nm); 5 μm resolution; 100% laser power; 500–600 PMT gain. Quantitation of the fluorescence was performed using GenePix Pro Software 7.3 (Molecular Devices). The fluorescence intensity for each spot was quantified by subtraction of the local background. The parameters for recording the fluorescence images were selected considering the signal to noise ratio, and saturation of the signal in the different experiments. The results were processed using the GrapPad PRISM (Graph-Pad). The error bars show the average percentage error for all data points reported in the figures.
2.5. Effect of heparin/HS and bivalirudin on the inactivation of factor IIa or Xa
The anti-Xa and anti-thrombin activities of heparin were estimated by published methods [30]. The final concentrations of reactants included 0.1 IU/mL AT or 68 nM HCII, 1 IU/mL thrombin or 1.42 nkat/mL factor Xa and different amounts (between 0 and 1000 μg/mL) heparin in 200 μL TS/PEG buffer (0.02 M Tris-HCl, 0.15 M NaCl, and 1.0 mg/mL polyethylene glycol 6000, pH 7.4). Incubations were performed in 96-well plates and a 120 μL mixture of heparin/HS and AT or HCII was incubated at 37 °C for 2 min. Then, 40 μL Xa or thrombin was added. After incubation for 5 min, the residual amidolytic activity of factor Xa or IIa was measured by the addition of 40 μL of 0.625 mM Xa chromogenic substrate S-2765 or 1 mM thrombin chromogenic substrate S-2238.
For bivalirudin, the final concentrations of reactants included 1 IU/mL thrombin and different amounts (between 0 and 1000 μg/mL) of bivalirudin. A 160 μL mixture of thrombin and bivalirudin with series concentrations was incubated at 37 °C for 5 min. The residual amidolytic activity of thrombin was measured by 1 mM thrombin chromogenic substrate S-2238. After 20 min, the absorbance at 405 nm was recorded. The IC50 values of heparin in each pathway were obtained from the inhibitory effect curves.
2.6. Inhibition of S trimer with AT, HCII or bivalirudin
Inhibitory experiment for S trimeric protein was similarly carried out except the following: 40 μL S trimer with serial concentrations were incubated with 40 μL heparin at the concentrations of 0.04 μg/mL for AT-thrombin pathway, 0.1 μg/mL for AT-Xa pathway, 2.0 μg/mL for HCII-thrombin pathway, 30 μg/mL HS, or 0.16 μg/mL enoxaparin for AT-thrombin pathway at room temperature for 10 min. Aliquot (80 μL) of each mixture was incubated with 40 μL AT or HCII in 96-well plates. And the follow up steps is the same as the above. 40 μL S trimer with serial concentrations were incubated with 40 μL bivalirudin (20 μg/mL). Then 80 μL thrombin were added. After incubated at 37 °C for 5 min, the chromogenic substrate S-2238 was added.
For estimation of the amounts of trimeric S protein used in the inhibition and also in vivo zebrafish (see below) experiments, SARS-CoV-2 viral loads ranged from 6.41 × 102 to 1.34 × 1011 copies/mL in clinical sputum samples [31] and 100 spike proteins in each viral particle [32], based on the previous reports, were used for calculation. The highest viral load is therefore equivalent to 1.34 × 1013 S trimer/mL or in molar concentration 2.23 × 10−11 mol/mL which can be converted to protein mass concentration of ~15 μg/mL (S trimer: 650 kDa),
2.7. Microscopic imaging and o-dianisidine staining
Photos were taken with an Olympus DP70 camera on an Olympus stereomicroscope MVX10. For confocal imaging of zebrafish embryos, they were anesthetized with E3 solution/0.16 mg/mL MS-222/1% 1-phenyl-2-thiourea and embedded in 0.8% low melting agarose. Florescence image and movie were recorded using a confocal microscope Nikon TI2-E-A1RHD25. The confocal images were analyzed using Nikon NIS-Elements AR-SP software. O-Dianisidine staining for globin was carried out as described previously [28]. In brief, the dechorionated zebrafish embryos were stained for around 15 min with a solution consisting of o-dianisidine (0.6 mg/mL), sodium acetate (0.01 mol/L), 0.65% hydrogen peroxide, and 40% (v/v) ethanol in a petri dish in the dark. The embryos were then used directly for imaging analysis.
2.8. Analysis of hemostasis
The procedure of hemostasis analysis was based on the preciously described work [33] with minor modifications. The embryos with visibly circulating blood cells were selected for this procedure. The staged embryos were anesthetized with 0.02% MS-222 in E3 solution, transferred to the microinjection plate (2% agarose) using a transfer pipet. The PBS, S protein (0.25, 0.50, 0.75, 1.0 ng; equivalent to viral loads of 1011 copies/mL in the total embryo's blood volume of 60 nL [34]), BSA and the mixed reagents (0.40 ng S protein and 0.20 ng heparin/HS) were microinjected into the common cardinal vein (CCV) together with fluorescein isothiocyanate-dextran or neutral dextran (MW 70,000) labeled with Texas Red for confirmation of the successful injection. The tip of a fine needle was used to pierce the posterior caudal tail vein adjacent to the urogenital opening before gently pulling away. Bleeding time was recorded immediately after blood loss can be visualized from the wound and stopped when blood loss from the wound ceased.
3. Results and discussion
3.1. SARS-CoV-2 spike protein binding to heparin polysaccharide
We initially used carbohydrate microarrays [35] to investigate the effect of SARS-CoV-2 S protein on AT and HCII binding to heparin. We prepared heparin polysaccharide microarrays by printing 0.5 mg/mL heparin onto NHS-functionalized glass slides [36]. To simultaneously detect the respective binding signals, S1 protein was tagged with mFc and detected by fluorescence 488 nm while AT and HCII were labeled with His-tags and detected by 635 nm. As shown in Fig. 1A, the intense binding signals indicated strong binding avidity to heparin from all three proteins. Before further competition experiment, the binding curves of the three individual proteins were obtained. The binding signals were essentially saturated at protein concentrations of >1 μg/mL for AT (Fig. 1B) and >2 μg/mL for HCII (Fig. 1C). Under saturated concentrations, all the binding sites of heparin immobilized on the microarray surface are considered to be occupied by AT or HCII, and under this condition, the competitive effect of the S protein becomes apparent and could be readily assessed. However, the mFc-S1 binding signals were saturated only at much higher concentration (~40 μg/mL, Fig. 1D).
Fig. 1.

Different binding signals of His-AT, His-HCII and mFc-S1 on heparin microarray. (A) Fluorescence binding signals of His-tagged AT and HCII (635 nm) and mFc-tagged SARS-CoV-2 S1 (488 nm). Binding curves of His-AT (B), His-HCII (C) and mFc-S1 (D) with serial concentrations were obtained from the fluorescence intensities of microspot images.
3.2. Competitive binding of AT or HCII to heparin by SARS-CoV-2 S1
In competition experiment, the percentage of binding signals compared to the controls were used to estimate the competition ability (Table 2 ). The data showed stronger heparin binding avidity of AT than S1 (96.4% and 62.0%, respectively). However, when S1 binding performed first, the binding signals of AT reduced to 76.3%, indicating that S1 can inhibit AT binding to heparin. S1 could bind heparin under all experimental conditions, even after AT 100% binding. Compared with AT, the competition effect of S1 on HCII binding to heparin is more apparent. With pre-mixed S1 and HCII, 73.3% of binding signals from HCII and 25.8% binding signals from S1 remained, indicating that the S1 binds to heparin less strongly. When S1 binding to heparin occurred first, a very high S1 binding signal was observed (92.7%). These results indicated that S protein of SARS-CoV-2 can interfere with anticoagulation activity of AT and HCII by competing with their binding to heparin, especially when S protein occupies heparin binding sites first.
Table 2.
Competitive binding of SARS-CoV-2 spike protein S1, anticoagulation factors AT and HCII to heparin on microarrays.
| Competitive binding experiments | Residual bindinga |
Residual bindingb |
||
|---|---|---|---|---|
| AT | S1 | HCII | S1 | |
| i) with premixed S1 & AT/HCII | 96.4% ± 23.1% | 62.0% ± 12.5% | 73.3% ± 15.6% | 25.8% ± 0.5% |
| ii) with AT/HCII followed by S1 | 104.8% ± 18.7% | 24.3% ± 1.7% | 76.9% ± 13.0% | 11.4% ± 2.0% |
| iii) with S1 followed by AT/HCII | 76.3% ± 21.0% | 67.0% ± 27.8% | 78.5% ± 11.4% | 92.7% ± 18.0% |
Means and standard deviations were obtained from 8 replicate points and three parallel experiments. The ratio of the mean value of the competition experiment to that of the control experiment was calculated shown as percentage to indicate the degree of S protein competition.
Concentrations used: S1 (2 μg/mL) and AT (1 μg/mL).
Concentration used: S1 (4 μg/mL) and HCII (2 μg/mL).
3.3. Negative effect of spike protein on anticoagulation
We next investigated the effect of the trimeric S protein on coagulation pathways by in vitro heparin anticoagulation assays (Fig. 2 ). Heparin IC50 of anti-Xa and anti-thrombin activities were estimated as described [30]. With increased heparin concentrations, the activities of thrombin and factor Xa decreased and were almost completely inhibited via the AT and HCII pathways. The IC50 values of heparin on AT are low (~0.01 μg/mL for thrombin and ~0.025 μg/mL for Xa, Fig. 2A). However, the IC50 for HCII is relatively high (~0.5 μg/mL, Fig. 2D). When S protein was added and incubated together with heparin at concentration of IC50 value, heparin lost its anticoagulation-regulating ability in an S protein concentration-dependent manner. S protein at 8 μg/mL, estimated to be equivalent to viral concentrations of ~1010 copies/mL, the viral load in the sputum of critically ill patients [31], [32], could completely restore the coagulation effect of thrombin by the heparin-AT-thrombin pathway, and could restore 90% of Xa activity by the heparin-AT-Xa pathway (Fig. 2B, C). For the heparin-HCII-thrombin pathway, S trimer at 32 μg/mL could completely inhibit HCII binding (Fig. 2E). Compared with the AT pathway, HCII was less sensitive to the S protein as higher heparin concentration was required for activating itself. Further assays indicated that a higher concentration of HS (IC50 ~ 7.5 μg/mL, Fig. 2F) than that of heparin was needed to inhibit the activity of thrombin. However, a similar concentration of S protein (8 μg/mL) can restore the activity of thrombin by competitive binding to HS (Fig. 2G).
Fig. 2.

Anticoagulation activities of heparin, HS and bivalirudin and the inhibitory effect S protein of SARS-CoV-2. (A) Heparin inactivates thrombin and Xa activities through AT with IC50 at ~0.01 μg/mL and ~0.025 μg/mL respectively. (B) S trimer inhibits the effect of AT on thrombin by competitive binding to heparin. (C) S trimer inhibits the effect of AT on Xa by binding to heparin. (D) Heparin inactivates thrombin activity through HCII with IC50 at ~0.5 μg/mL. (E) S trimer inhibits the effect of HCII by binding to heparin. (F) HS inactivates thrombin activity through AT with IC50 at ~7.5 μg/mL. (G) S trimer inhibits the effect of AT by binding to HS. (H) Bivalirudin inactivates thrombin activities with IC50 at ~5 μg/mL. (I) S trimer cannot reverse inhibitory effect of bivalirudin on thrombin.
To further demonstrate the promoting effect of S protein on thrombin activity by competitive binding with heparin/HS rather than direct action on thrombin, a thrombin inhibitor, bivalirudin, was used in the in vitro experiment. The results showed S trimer at 32 μg/mL cannot eliminate the inhibitory effect of bivalirudin (Fig. 2H, I).
3.4. Spike protein promotes blood coagulation and thrombosis in vivo
We further looked at the effect of SARS-CoV-2 S protein on blood coagulation in vivo using zebrafish embryos, which have been widely used for investigation of blood vessel formation, hematopoietic development, hemostasis and thrombosis [37], [38], [39]. We microinjected S protein into the CCV before mechanical injury at the posterior cardinal vein (PCV). The positions of microinjection and the injury, and the timeline are illustrated in Fig. 3A, B. We found that 0.75 ng and 1 ng S protein in the whole blood of an embryo (equivalent to viral concentration ~ 1011 copies/mL) significantly reduced the bleeding time in response to the mechanical injury (Fig. 3C) compared with the inactivity of 1 ng BSA.
Fig. 3.
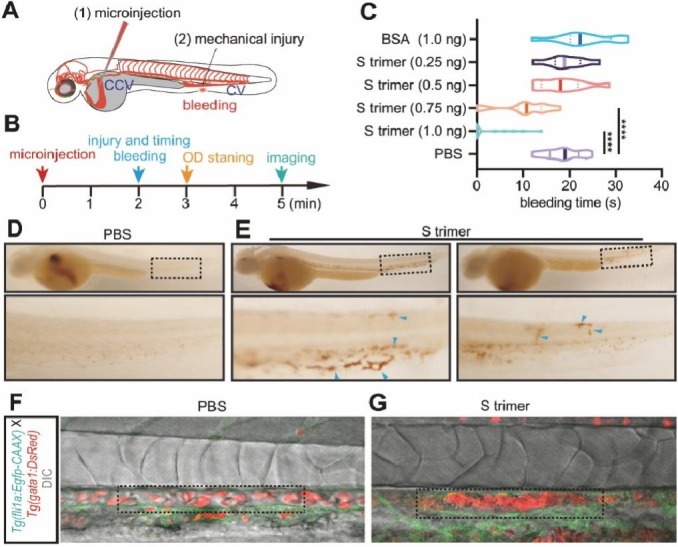
SARS-CoV-2 S protein causes coagulation and thrombosis in zebrafish. (A, B) The positions and the timeline of microinjection and the mechanical injury on zebrafish embryos. (C) The effect of S trimer on bleeding time in response to the mechanical injury; ****, P < 0.0001. (D, E) Imaging analysis of o-dianisidine stained embryos microinjected with PBS and S trimer. (F, G) Confocal imaging analysis of Tg(gatal:DsRed:fli1a:Egfp-CAAX) embryos microinjected with PBS and S trimer.
We then investigated whether S protein could cause thrombosis. After microinjection of S protein, o-dianisidine staining was performed (Fig. 3B). The results showed that thrombosis took place in the PCV, dorsal aorta (DA) and capillaries, including intersegmental vessels (ISVs), dorsal lateral anastomotic vessel (DLAV), and caudal vein plexus (CVP) (Fig. 3D, E). Confocal imaging of the Tg(gatal:DsRed:fli1a:Egfp-CAAX) line, in which the red blood cells were labeled with red fluorescence and the endothelial cells with membrane-bound green fluorescent protein, revealed that injection of S protein resulted in plaque formation in the DA wall (Fig. 3F, G, and Video A, Video B). It is important to note that thrombosis occurred in a very short time (3–5 min, Fig. 3B, and Video A, Video B), suggesting a direct effect of the S protein on thrombosis rather than an inflammation-induced clotting.
3.5. HS/heparin treatment of coagulation caused by S protein
Finally, we examined whether HS/heparin could alleviate the effect of S protein on blood coagulation and thrombosis in zebrafish. We co-injected S protein and HS or heparin into the CCV and subsequently injured at the PCV mechanically. We found that co-injection of S protein with heparin/HS significantly extended the bleeding time compared with injection of S protein only (Fig. 4A, B). Co-injection of S protein with HS also reduced the total frequency (F) of thrombosis, caused by injection of S protein only as the control, from 100% to 42% in artery (DA), vein (PCV) and capillaries (ISV, DLAV, and CVP) (Fig. 4C). More specifically, the thrombosis frequency was found to be reduced from 53% (19 + 34%) to 13% (5 + 8%) in large vessels (DA and PCV: DP), from 81% (47 + 34%) to 37% (29 + 8%) in capillaries (DLAV, ISV and CVP: DIC), and from 34% to 8% in both DIC and DP. These combined results suggest that HS/heparin could neutralize blood coagulation and thrombosis caused by S protein.
Fig. 4.

HS/heparin treatment can reduce the coagulation caused by S protein. (A, B) The effect of HS and heparin (Hep) co-injection with S trimer on bleeding time; ****, P < 0.0001. (C) The frequency (F) of thrombosis happened in capillaries (DLAV, ISV and CVP), large vessels including artery and vein (DA, PCV), and both capillaries and large vessels (DIC + DP).
4. Conclusion
In this study, we demonstrated that the SARS-CoV-2 spike protein can compete with anticoagulation factors AT and HCII bindings to heparin by blocking the inactivation process of thrombin or factor Xa. Once HS is bound by S protein, its dissociation is difficult, and HS can no longer interact with AT/HC II. Therefore, with the increased virus concentration in certain parts of the organs/tissues, such as microvascular, local HS can be exhausted, leading to exacerbated coagulation and other adverse consequences, especially in critically ill patients. This rapid coagulation response may be an additional independent factor for the inflammatory storm of severe COVID-19 patients.
The following are the supplementary data related to this article.
Blood cells flow with microinjection of BSA.
Blood cells flow with microinjection of S protein.
CRediT authorship contribution statement
Yi Zheng: Methodology, Investigation, Formal analysis, Writing – original draft. Jinxiang Zhao: Methodology, Investigation, Writing – original draft. Jiaqi Li: Methodology, Investigation, Writing – original draft. Zhimou Guo: Formal analysis. Jiajing Sheng: Methodology, Investigation. Xianlong Ye: Methodology, Investigation. Gaowa Jin: Formal analysis. Chaoran Wang: Methodology, Investigation. Wengang Chai: Formal analysis, Writing – review & editing, Funding acquisition. Jingyu Yan: Writing – review & editing, Funding acquisition, Conceptualization, Supervision. Dong Liu: Writing – review & editing, Funding acquisition, Conceptualization, Supervision. Xinmiao Liang: Writing – review & editing, Funding acquisition, Conceptualization, Supervision.
Declaration of competing interest
The authors declare no competing financial interests.
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (21934005, 22074143, 81870359), Dalian Science and Technology Innovation Fund of COVID-19 Emergency Project (2020), Wellcome Trust Biomedical Resource grants (218304/Z/19/Z), the March of Dimes Prematurity Research Center grant (22-FY18-82), and Natural Science Foundation of Jiangsu Province (BK20180048).
References
- 1.Ackermann M., Verleden S.E., Kuehnel M., Haverich A., Welte T., Laenger F., et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N. Engl. J. Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Connors J.M., Levy J.H. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033–2040. doi: 10.1182/blood.2020006000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Middeldorp S., Coppens M., van Haaps T.F., Foppen M., Vlaar A.P., Müller M.C.A., et al. Incidence of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020;18(8):1995–2002. doi: 10.1111/jth.14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang N., Li D., Wang X., Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020;18:844–847. doi: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helms J., Tacquard C., Severac F., Leonard-Lorant I., Ohana M., Delabranche X., et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med. 2020;46:1089–1098. doi: 10.1007/s00134-020-06062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levi M., van der Poll T., Buller H.R. Bidirectional relation between inflammation and coagulation. Circulation. 2004;109(22):2698–2704. doi: 10.1161/01.CIR.0000131660.51520.9A. [DOI] [PubMed] [Google Scholar]
- 7.Manne B.K., Denorme F., Middleton E.A., Portier I., Rowley J.W., Stubben C., et al. Platelet gene expression and function in COVID-19 patients. Blood. 2020;136(11):1317–1329. doi: 10.1182/blood.2020007214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varga Z., Flammer A.J., Steiger P., Haberecker M., Andermatt R., Zinkernagel A.S., et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelmann B., Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat. Rev. Immunol. 2013;13(1):34–45. doi: 10.1038/nri3345. [DOI] [PubMed] [Google Scholar]
- 10.Grimmer B., Kuebler W. The endothelium in hypoxic pulmonary vasoconstriction. J. Appl. Physiol. 2017;123:1635–1646. doi: 10.1152/japplphysiol.00120.2017. [DOI] [PubMed] [Google Scholar]
- 11.Shi C., Wu T., Li J., Sullivan M.A., Wang C., Wang H., et al. Comprehensive landscape of heparin therapy for COVID-19. Carbohydr. Polym. 2021;254 doi: 10.1016/j.carbpol.2020.117232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dupont A., Rauch A., Staessens S., Moussa M., Rosa M., Corseaux D., et al. Vascular endothelial damage in the pathogenesis of organ injury in severe COVID-19. Arterioscler. Thromb. Vasc. Biol. 2021;41:1760–1773. doi: 10.1161/ATVBAHA.120.315595. [DOI] [PubMed] [Google Scholar]
- 13.Bombeli T., Mueller M., Haeberli A. Anticoagulant properties of the vascular endothelium. Thromb. Haemost. 1997;77(3):408–423. [PubMed] [Google Scholar]
- 14.Bourin M.C., Lindahl U. Glycosaminoglycans and the regulation of blood coagulation. Biochem. J. 1993;289(Pt 2):313–330. doi: 10.1042/bj2890313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Munoz E.M., Linhardt R.J. Heparin-binding domains in vascular biology. Arterioscler. Thromb. Vasc. Biol. 2004;24(9):1549–1557. doi: 10.1161/01.ATV.0000137189.22999.3f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lane D.A., Philippou H., Huntington J.A. Directing thrombin. Blood. 2005;106(8):2605–2612. doi: 10.1182/blood-2005-04-1710. [DOI] [PubMed] [Google Scholar]
- 17.Pike R.N., Buckle A.M., le Bonniec B.F., Church F.C. Control of the coagulation system by serpins. Getting by with a little help from glycosaminoglycans. FEBS J. 2005;272(19):4842–4851. doi: 10.1111/j.1742-4658.2005.04880.x. [DOI] [PubMed] [Google Scholar]
- 18.Rau J.C., Mitchell J.W., Fortenberry Y.M., Church F.C. Heparin cofactor II: discovery, properties, and role in controlling vascular homeostasis. Semin. Thromb. Hemost. 2011;37:339–348. doi: 10.1055/s-0031-1276582. [DOI] [PubMed] [Google Scholar]
- 19.Verhamme I.M., Bock P.E., Jackson C.M. The preferred pathway of glycosaminoglycan-accelerated inactivation of thrombin by heparin cofactor II. J. Biol. Chem. 2004;279(11):9785–9795. doi: 10.1074/jbc.M313962200. [DOI] [PubMed] [Google Scholar]
- 20.Bick R.L., Walenga J., Fareed J., Hoppensteadt D.A. Unfractionated heparin, low molecular weight heparins, and pentasaccharide: basic mechanism of actions, pharmacology, and clinical use. Hematol. Oncol. Clin. North Am. 2005;19(1):1–51. doi: 10.1016/j.hoc.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Casu B., Lindahl U. Structure and biological interactions of heparin and heparan sulfate. Adv. Carbohydr. Chem. Biochem. 2001;57:159–206. doi: 10.1016/s0065-2318(01)57017-1. [DOI] [PubMed] [Google Scholar]
- 22.Clausen T.M., Sandoval D.R., Spliid C.B., Pihl J., Perrett H.R., Painter C.D., et al. SARS-CoV-2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183(4):1043–1057. doi: 10.1016/j.cell.2020.09.033. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim S.Y., Jin W., Sood A., Montgomery D.W., Grant O.C., Fuster M.M., et al. Characterization of heparin and severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) spike glycoprotein binding interactions. Antivir. Res. 2020;181 doi: 10.1016/j.antiviral.2020.104873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon P.S., Oh H., Kwon S.J., Jin W., Zhang F., Fraser K., et al. Sulfated polysaccharides effectively inhibit SARS-CoV-2 in vitro. Cell Discov. 2020;6:50. doi: 10.1038/s41421-020-00192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan L., Song Y., Xia K., He P., Zhang F., Chen S., et al. Heparan sulfates from bat and human lung and their binding to the spike protein of SARS-CoV-2 virus. Carbohydr. Polym. 2021;260 doi: 10.1016/j.carbpol.2021.117797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta Y., Maciorowski D., Zak S.E., Kulkarni C.V., Herbert A.S., Durvasula R., et al. Heparin: a simplistic repurposing to prevent SARS-CoV-2 transmission in light of its in-vitro nanomolar efficacy. Int. J. Biol. Macromol. 2021;183:203–212. doi: 10.1016/j.ijbiomac.2021.04.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X., Ling C., Li L., Qin Y., Qi J., Liu X., et al. MicroRNA-10a/10b represses a novel target gene mib1 to regulate angiogenesis. Cardiovasc. Res. 2016;110(1):140–150. doi: 10.1093/cvr/cvw023. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J., Qi J., Wu S., Peng L., Shi Y., Yang J., et al. Fatty acid binding protein 11a is required for brain vessel integrity in zebrafish. Front. Physiol. 2017;8:214. doi: 10.3389/fphys.2017.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao T., Yan J., Liu C., Palma A.S., Guo Z., Xiao M., et al. Chemoenzymatic synthesis of O-mannose glycans containing sulfated or nonsulfated HNK-1 epitope. J. Am. Chem. Soc. 2019;141(49):19351–19359. doi: 10.1021/jacs.9b08964. [DOI] [PubMed] [Google Scholar]
- 30.Pomin V.H. How to analyze the anticoagulant and antithrombotic mechanisms of action in fucanome and galactanome? Glycoconj. J. 2014;31:89–99. doi: 10.1007/s10719-013-9509-3. [DOI] [PubMed] [Google Scholar]
- 31.Pan Y., Zhang D., Yang P., Poon L.L.M., Wang Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 2020;20(4):411–412. doi: 10.1016/S1473-3099(20)30113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bar-On Y.M., Flamholz A., Phillips R., Milo R. Science forum: SARS-CoV-2 (COVID-19) by the numbers. elife. 2020;9 doi: 10.7554/eLife.57309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clay H., Coughlin S.R. Mechanical vessel injury in zebrafish embryos. J. Vis. Exp. 2015;96 doi: 10.3791/52460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Craig M.P., Gilday S.D., Dabiri D., Hove J.R. An optimized method for delivering flow tracer particles to intravital fluid environments in the developing zebrafish. Zebrafish. 2012;9(3):108–119. doi: 10.1089/zeb.2012.0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukui S., Feizi T., Galustian C., Lawson A.M., Chai W. Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions. Nat. Biotechnol. 2002;20:1011–1017. doi: 10.1038/nbt735. [DOI] [PubMed] [Google Scholar]
- 36.Stowell S.R., Arthur C.M., McBride R., Berger O., Razi N., Heimburg-Molinaro J., et al. Microbial glycan microarrays define key features of host-microbial interactions. Nat. Chem. Biol. 2014;10:470–476. doi: 10.1038/nchembio.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gore A.V., Monzo K., Cha Y.R., Pan W., Weinstein B.M. Vascular development in the zebrafish. Cold Spring Harb. Perspect. Med. 2012;2(5) doi: 10.1101/cshperspect.a006684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jagadeeswaran P., Cooley B.C., Gross P.L., Mackman N. Animal models of thrombosis from zebrafish to nonhuman primates: use in the elucidation of new pathologic pathways and the development of antithrombotic drugs. Circ. Res. 2016;118(9):1363–1379. doi: 10.1161/CIRCRESAHA.115.306823. [DOI] [PubMed] [Google Scholar]
- 39.Jing L., Zon L.I. Zebrafish as a model for normal and malignant hematopoiesis. Dis. Model. Mech. 2011;4(4):433–438. doi: 10.1242/dmm.006791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Blood cells flow with microinjection of BSA.
Blood cells flow with microinjection of S protein.