

Abstract
Inflammation is a host defense mechanism orchestrated through imperative factors - acute inflammatory responses mediated by cellular and molecular events leading to activation of defensive immune subsets - to marginalize detrimental injury, pathogenic agents and infected cells. These potent inflammatory events, if uncontrolled, may cause tissue damage by perturbing homeostasis equilibrium towards immune dysregulation. A parallel host mechanism operates to contain inflammatory pathways and facilitate tissue regeneration. Thus, resolution of inflammation is an effective moratorium on the pro-inflammatory pathway to avoid the tissue damage inside the host and leads to reestablishment of tissue homeostasis. Dysregulation of the resolution pathway can have a detrimental impact on tissue functionality and contribute to the diseased state. Multiple reports have suggested peculiar dynamics of miRNA expression during various pro- and anti-inflammatory events. The roles of miRNAs in the regulation of immune responses are well-established. However, understanding of miRNA regulation of the resolution phase of events in infection or wound healing models, which is sometimes misconstrued as anti-inflammatory signaling, remains limited. Due to the deterministic role of miRNAs in pro-inflammatory and anti-inflammatory pathways, in this review we have provided a broad perspective on the putative role of miRNAs in the resolution of inflammation and explored their imminent role in therapeutics.
Keywords: MicroRNA, Post-transcriptional regulation, Inflammation, Resolution, Pathogenesis, Wound healing
1. Introduction
Inflammation is a tightly regulated host response against a diverse gamut of biological and non-biological factors (viz., pathogens, damaged cells and toxic compounds, pathogenic insult) to reinstate tissue homeostasis [1, 2]. Typically, inflammation involves the following sequence of events: 1) induction phase, 2) inflammatory phase, and 3) resolution. The induction phase is comprised of activation of tissue resident innate immune subsets (macrophages and dendritic cells) after recognition of the biological/non-biological threat. Various lipid and protein mediators are released to recruit innate immune cells (polymorphonuclear cells {PMNs}) at the site of damage [1–4]. This is followed by the inflammatory phase during which the pathogen is phagocytosed, the derived antigens are presented to adaptive immune cells for further amplification of inflammatory processes, and antibody-producing B cells are activated. Upon removal of the danger signal, the resolution phase ensues and tissue homeostasis is restored. Each phase of inflammation should be well-coordinated, as failure to resolve the inflammation leads to multiple chronic inflammatory diseases: arthritis [5], colitis [6], or asthma [7] associated with irreversible tissue damage and high odds of developing cardiovascular disease, cancer, osteoporosis, etc. [8–10].
Multiple genes work in a concerted manner to regulate molecular pathways that guide each phase of inflammation, including resolution. In addition to protein coding genes, the human genome encodes for non-protein coding RNAs which participate as transcriptional and post-transcriptional regulators of host functions. MicroRNAs (miRNA) are a class of endogenous gene regulatory non-coding RNAs that perform central functions in various cellular processes including cell differentiation, apoptosis, immune response, cell proliferation, etc. In this pursuit, miRNA have been identified as molecular switches to fine-tune both inflammation and the resolution process [11]. Thus, miRNAs, either as mimics or antagonists, hold therapeutic promise as drugs, in mitigating inflammation or augmenting resolution.
In this review, we summarize the sequence of events involved in the process of inflammation and resolution, and their control by specific miRNAs. Furthermore, we will provide insights on miRNA expression dynamics and regulomics (involving miRNA mimics/antagonist) can be leveraged in inducing the resolution of uncontrolled inflammation, which might otherwise result in chronic inflammation and tissue damage.
2. Inflammation Process For The Protection From Biological/Non-Biological Insult
The initiation of inflammation occurs after local non-immune or immune cells sense biological/non-biological antigens through highly conserved receptors and generate a conducive microenvironment for the activation and infiltration of other immune cells [1–4]. Amongst various classes of Pattern Recognition Receptors (PRRs), including C-type lectin receptors (CLRs), retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), and NOD-like receptors (NLRs), Toll-Like Receptors (TLRs) are both the most studied and highly conserved receptors [12, 13]. Typically, TLR signaling is mediated through myeloid differentiation factor-88 (MyD88) [14, 15] and involves nuclear transport of transcription factors viz. activator protein-1 (AP-1) and NF-κB or interferon regulatory factor 3 (IRF3) in the nucleus for the production of pro-inflammatory molecules including cytokines/chemokines (interleukin-1 beta, interleukin-6, tumor necrosis factor alpha (TNF-α), etc.) and prostaglandins E2 (PGE2) [4, 14, 15]. Activated tissue resident cells secrete inflammatory mediators, which increase blood flow in the affected region to induce the redness (rubor) and increased heat (calor) characteristic of vasodilation in the inflammatory process. PRRs or Damage Associated Molecular Patterns (DAMPs) harboring macrophages and mastocytes release vasoactive amines (histamine and serotonin) as well as pro-inflammatory lipid mediators eicosanoids (viz. prostaglandin E2 and leukotriene B4) for the remodeling of local vasculature to increase the blood flow to the inflamed site [1–4, 16, 17]. Nitric oxide produced by the activated tissue resident macrophages further induces the permeability of blood vessels [18, 19], which results in the leakage of plasma proteins into the damaged tissue area (edema) and subsequent swelling (tumor) of the tissues [18–20]. Plasma proteins include complement, lysozyme, and antibodies to directly kill and facilitate opsonization of microbes, a perfect preparation for the imminent cellular phase [21]. When the inflammatory region is the wounded tissue, the exuded platelets, coagulants, plasmin and kinins induce clot formation around this area, thereby providing a structural scaffold in the form of a fibrin lattice to initiate wound repair events [22–25]. Some of the leaked tissue fluid containing bacteria is also reported to be funneled to the regional lymph nodes, wherein it prepares the host defense system to activate the adaptive immune response [26, 27]. Bradykinin released during this course also increases sensitivity to pain (hyperalgesia, dolor) in the affected region. Sometimes, loss of function (functio laesa) can be seen as a negative neurological reflex in response to pain [28].
Leukocytes, the cellular component of inflammation, play a pivotal role in clearing the origin of inflammation. Elicitation of adept innate and adaptive immune responses cannot be assured unless leukocyte extravasation occurs through blood vessels at the site of inflammation - the process known as diapedesis [29]. Activation of tissue resident macrophages induces secretion of cytokines (viz., IL-1 and TNF-α) and chemokines, which binds to proteoglycans located in the cell walls of inflamed tissue, passes through the injured/inflamed tissue and induces surface expression of P-selectin, E-selectin, Intercellular Adhesion Molecule 1 (ICAM1) and Vascular cell adhesion protein 1 (VCAM1) on endothelial cells [30]. Weak binding of carbohydrate ligands on the leukocyte cell membrane to these integrin molecules allows them to “roll” along the endothelial surface of the blood vessels - due to weak bond formation and breakage in a simultaneous manner. As a result, these leukocytes extravasate into the inflamed tissue to neutralize the antigenic stimuli and perform the following imperative functions: 1) phagocytic uptake of bacteria, viruses, and cellular debris, 2) release of enzymes to damage pathogenic invaders, and 3) secrete pro-inflammatory molecules to maintain the immune response until clearance of the pathogenic insult is achieved. Typically, granulocytes (PMN subtype; neutrophils) are known to induce acute inflammation, whilst monocytes and lymphocytes (T cells and B cells) are responsible for sustained inflammation. Activation of complement C3b, in conjunction with antibodies (produced by plasma cells) exuded into the inflamed tissue, augment phagocytosis and opsonization of microbial antigens by granulocytes and monocytes. These phagocytes also express opsonin receptors, Fc receptor (FcR) and complement receptor 1 (CR1), which orchestrate activation of the complement system in order to clear the microbes from the host [18–20, 31].
Interestingly, DAMPs can also be triggered in the immune system in the absence of an acute infectious insult or activation of PAMPs. This situation is dubbed as ‘non-infective sterile systemic chronic inflammation (SCI), which occurs more frequently with age. Typically, SCI promotes low-grade but persistent inflammation responsible for hypertension, hyperglycemia and dyslipidemia, type 2 diabetes, non-alcoholic fatty liver disease (NAFLD), cardiovascular disease (CVD), chronic kidney disease, depression, neurodegenerative and autoimmune diseases, osteoporosis and sarcopenia. Furthermore, the triggers of sterile inflammation have been classified as extracellular (originating from the extracellular matrix) and intracellular (including high-mobility group box 1 [HMGB1]), mitochondrial DNA and peptides, uric acid, and cellular chaperones (e.g., heat shock proteins)). A study involving murine renal IRI models provides the evidence that HMGB1 signaling via TLR4 is the key factor exacerbating the disease in these models. Also, more work is needed to understand the in-depth mechanism of sterile inflammation [32].
3. Chronic Inflammation Has The Potential To Damage Impacted Organs
Though inflammation has evolved as a protective response to neutralize foreign organisms/material, the safety and survival of the host also demands an efficiently operated, well-balanced regulatory system to induce the refurbishment of damaged tissue structures and function. Principally, PRR activation results in the activation of the following three signaling cascades: mitogen-activated protein kinase (MAPK), nuclear factor kappa-B (NF-κB), and Janus kinase (JAK)-signal transducer and activator of transcription (STAT). Therefore, the following four mechanisms are employed by the host to mitigate uncontrolled inflammation: 1) members of protein tyrosine phosphatase (PTP) family (SHP1/2 and CD45) negatively regulate tyrosine kinase (TK) signaling [33]. Further, MAPK kinase phosphatases (MKPs) of this family regulate the duration of TNF-α-mediated JNK activation [34]; 2) suppressors of cytokine signaling (SOCS), which function as ubiquitin ligases, are responsible for the negative feedback control of JAK–STAT signaling [35]; 3) ubiquitin ligase (A20) interferes with TLR and TNFR signaling and inhibits the NF-κB pathway [36]; and 4) anti-inflammatory mediators (IL-10) are induced by pro-inflammatory signaling pathways [37]. Häcker et al. reported that the gene expression equilibrium of TNF receptor (TNFR) associated factor (TRAF) −3 and −6, an upshot of TLR signaling, determines the expression of IL-10 [39]. Perturbation in any of the aforementioned mechanisms would result in chronic inflammation-mediated injuries to the host, wherein inflammation may persist for longer than the normal duration and may eventually cause tissue damage. Indeed, the progression of cardiovascular disease [40], lung diseases (COPD) [41], diabetes (T2D) [42], pancreatitis [43], intestinal inflammatory diseases (CD and UC) [44], liver diseases (alcoholic or nonalcoholic steatohepatitis, drug-induced liver injury, and ischemia/reperfusion) [45–47], kidney diseases, (glomerulonephritis, end-stage renal disease, or acute or chronic kidney disease) [49–50] have convincingly demonstrated organ-specific uncontrolled inflammatory events as central to these diseases. Table 1 lists the dysregulation of key inflammatory processes during the onset and progression of the aforementioned diseases.
Table 1:
Diseases associated with uncontrolled inflammation.
| Name of the organ | Name of the disease | Dysregulated immune signaling | Reference |
|---|---|---|---|
| Cardiovascular disease | atherosclerosis | Intracellular components TLR-mediated NF-κB signaling via necrotic cells | [40] |
| Pancreas | Type 2 diabetes | High levels of IL-1β, IL-6, TNF-α, CRP, fibrinogen, serum amyloid A, plasminogen activator inhibitor, and haptoglobin, sialic acid, IL-1 receptor antagonist (IL-1RA), activation of NF-κB, MAPK, and JAK-STAT pathways | [42] |
| Pancreas | Pancreatitis | Activation of macrophages, neutrophils, and granulocytes, inflammatory cytokines mediated pancreatic stellate cells (PSCs) activation | [43] |
| Kidney | glomerulonephritis, end-stage renal disease, or acute or chronic kidney disease (CKD | Activation of transcription factors (NF-κB or MAPK), DAMPs, and PAMPs, and Nod-like receptors (NLRs). | [49–50] |
| Liver | alcoholic or nonalcoholic steatohepatitis, drug-induced liver injury, and ischemia/reperfusion | DAMPs (during SI) and PAMPs activation and formation of inflammasome, high levels of of IL-1β and other pro-inflammatory cytokines, activation of Kupffer cell leading to hepatocyte damage, and/or cholestasis. | [45–47] |
| Lung | COPD, asthama | Asthama :Activation and uncontrolled infiltration of macrophage, neutrophil, and T lymphocyte into airways COPD: production of cytokines (TNF-α, IL-6 and IL-8) chemokines, oxygen radicals, proteases. | [41] |
| Intestine | Ulcerative colitis (UC) and Crohn disease (CD) | CD: IFN-γ/IL-17 and IL-12/IL-23 UC: IL-13 | [44] |
4. Resolution Phase of Inflammation
The persistence of immunological, physiological and tissue architectural changes during the orchestration of acute inflammation can be harmful in the absence of satisfactory regulation and therefore demands timely recession after the neutralization of the pathogen/tissue injury threat [51, 52]. Thus, the successful resolution of inflammation requires: 1) gradual cessation of immune cell extravasation (stop), 2) counter-regulation of pro-inflammatory chemokines and cytokines (sink), 3) downplay of immune cell activation signaling, 4) induction of apoptosis of immune effector cells (both innate and adaptive) and their efferocytosis (of neutrophils) by reprogrammed M2 macrophages (skew and kill), 5) induction of immune suppressive cells to control pro-inflammatory cell activation and function, and 6) the induction of tissue healing processes (heal). Therefore, similar to the initiation phase of inflammation, resolution also necessitates a well-coordinated resonance of these pathways [51, 52]. Failure of one or more of these cardinal steps will likely result in human chronic inflammatory diseases [41–50].
Resolution of inflammation is very different from anti-inflammatory effects. For instance, glucocorticoids are given to inhibit the action of pro-inflammatory cytokines (IL-1α, IL-1β, TNF-α), keratinocyte growth factor, TGF-(β1, β2, and β3), platelet-derived growth factors (PDGF) and their receptors, tenascin-C, stromelysin-2, and macrophage metalloelastase at the wounded skin which impacts the expression of important genes involved in wound healing [53]. Intriguingly, a novel cyclooxygenase-2 (COX-2) inhibitor prescribed to avoid pain and inflammation induces thrombosis [54]. Also, clinical reports evince that TNF-α-neutralizing therapy should not be given to patients with known cardiac history [55]. This therapy dampens host potential to combat various pathogens and the host become susceptible to various opportunistic infections (listeriosis, pneumonia and aspergillosis) and activation of latent tuberculosis [56]. Therefore, anti-inflammatory agents can ameliorate the actions of specific pro-inflammatory mediators that are involved in tissue damage, whilst resolution activates the cellular processes that eventually coordinate not only to control the damage but also to trigger the damage repair and tissue healing processes.
5. Key Pathways Involved In Accomplishing Successful Infection Resolution
Recent scientific reports unraveled the upregulation, release and coordination of immune mediators that have the potential to shift acute inflammation towards resolution. These molecules include pro-resolving lipid mediators [lipoxins (e.g., LXA4), resolvins (e.g., RvD1), protectins, and maresins] [57], proteins and peptides [e.g., annexin A1 (AnxA1), adrenocorticotropic hormone, chemerin peptides, and galectin-1] [58], gaseous mediators (e.g., H2S and CO) [59], a purine (adenosine) [60], as well as neuromodulators (acetylcholine and other neuropeptides) released under the control of the vagus nerve [61]. In this section, we will discuss the generation of the aforementioned mediators during various infection pathways.
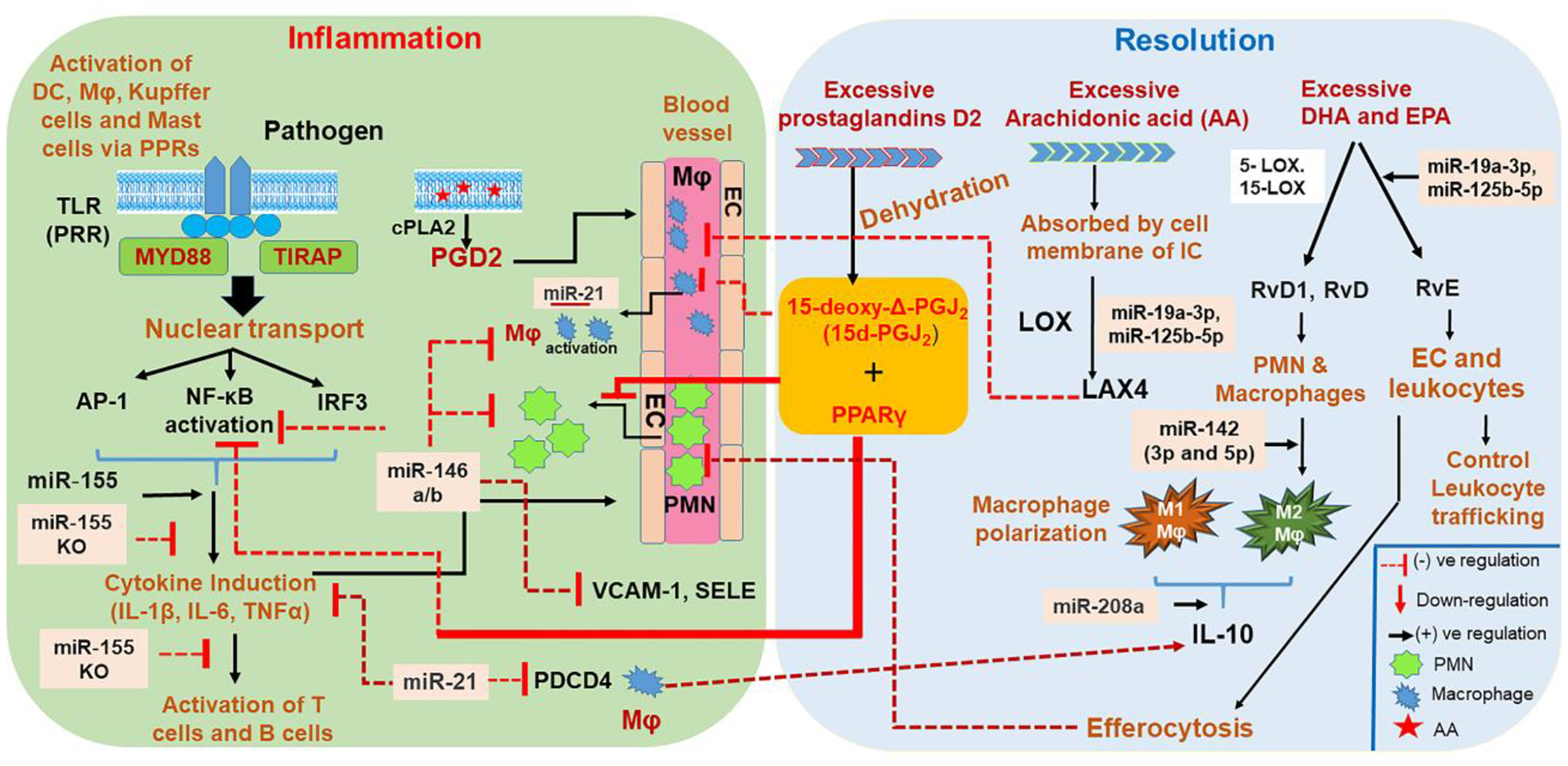
5A. Role of Prostaglandin Derived 15d-PGJ2 in the Resolution of Inflammation
During inflammation, COX-2 activity produces PGD2 as a major molecule from a variety of immune subsets viz., macrophages, DC, T cells, mast cells and platelets [62]. Dehydration of PGD2 yields sPGJ2, Δ-PGJ2 and 15-deoxy-Δ-PGJ2 (15d-PGJ2) [63]. Out of these metabolites, 15d-PGJ2 has demonstrated high affinity towards peroxisome proliferator-activated receptor gamma (PPARγ) and therefore suppresses the induction of the NF-κB, AP-1, and STATs inflammatory cascades [64]. 15d-PGJ2 also suppresses the inducible nitric oxide synthase, interleukin (IL)-1β, TNF-α and IL-12 in macrophages [65], microglial cells [66] and dendritic cells [67]. Importantly, 15d-PGJ2 preferentially circumvents monocyte trafficking rather than PMN recruitment at the site of inflammation. Therefore, these molecules can control chronic inflammatory processes without affecting the onset phase of acute inflammation. Also, the apoptosis-inducing attribute of this molecule in granulocytes, macrophages, and myofibroblasts makes it imperative in preventing the exacerbation of tissue damage during the late phase of inflammation [68]. Importantly, 15d-PGJ2 treatment is reported confer immune protection in the experimental models of ischemia reperfusion injury [69], inflammatory bowel disease [70], adjuvant-induced arthritis [71] and experimental autoimmune encephalomyelitis [72]. Furthermore, 15d-PGJ2 has shown dose-dependent immune protection in different cell types (peripheral blood monocytes-macrophages, J774 macrophages or A549 cells) [73]. It is also reported that Heme Oxygenase-1 (HO-1) expression, a stress-inducible enzyme that enhances degradation of heme to liberate free iron, carbon monoxide, biliverdin and bilirubin in mammalian cells, regulates resolution of 15d-PGJ2-mediated inflammation [74].
5B. Immune Cell-Derived Lipoxins and Resolvins Inhibit Leukocyte Recruitment, Efferocytosis and Apoptosis Induction by Macrophages
Recent progress in the field of lipidomics elucidated the presence of various eicosanoids in resolution exudates derived from mice. It has been shown that arachidonic acid [(AA; refers to all-cis-5,8,11,14-eicosatetraenoic acid, an omega-6 polyunsaturated fatty acid [PUFA])-derived eicosanoids can be released from the plasma membrane of activated immune cells expressing TNFR and TLR4 upon influx of Ca2+. This process is catalyzed by the cytosolic Ca2+-dependent enzyme Cytosolic Phospholipase A2 (cPLA2) [75, 76]. Conversely, cytosolic Ca2+-independent PLA2 (iPLA2) and specialized pro-resolving mediators (SPMs) catalyze the recycling of AA back into the plasma membrane to restore cellular hemostasis [77, 78]. Importantly, transcellular conversion of AA via the lipoxygenase (LOX) superfamily of enzymes in multiple immune cells converts it into the lipoxin (LX) family of eicosanoid metabolites, which inhibit the following vital aspects of inflammation: 1) chemotaxis, 2) PMN rolling and transmigration through endothelial cells of blood vessels, and 3) PMN-mediated increase in vascular permeability to restore tissue homeostasis [79]. Successful resolution of allergic pleural edema via LX and their analogues and enhancing non-phlogistic phagocytosis of monocyte-derived macrophages to clear the apoptotic PMNs substantiated their potential therapeutic role in the resolution of inflammation [80]. Inhibitory signaling induced by LXA4 and aspirin-triggered 15-epi-LXA4, and their other stable analogues, is initiated after their binding to high affinity G-protein-coupled receptors (ALXR or FPRL1) [80].
Down-regulation of CD11b/CD18 in human neutrophils can control inflammation as it impairs endothelial–leukocyte interactions [81]. Interestingly, a TNF-α-induced murine dorsal air-pouch model of acute inflammation initially showed induction of leukotriene B4, followed by PMN infiltration and PGE2 synthesis [82]. Later, a remarkable decrease in PMN infiltration was observed with the concomitant synthesis of 5-LOX-generated leukotriene B4 to 15-LOX-elicited pro-resolving LXA4 in the presence of PGE2. It was concluded that the presence of LXA4 in this experimental inflammation setting suggests that it may serve as a signature of controlled rather than uncontrolled inflammation [82].
In addition to AA-derived fatty-acid substrates, the potent anti-inflammatory products derived from docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), omega-3 fatty-acid constituents of fish oils, have also been detected in the resolving phase of aspirin-treated TNF-α-induced inflammation. These products are known as resolvin (Resolvin D and E). The synthesis of RvD1 and RvD2 necessitates the catalysis of 15-lipoxygenase (15-LOX) and 5-lipoxygenase (5-LOX), which can take place in mononuclear cells (viz., neutrophils and macrophages) and between cells, such as leukocytes-endothelial cells and neutrophils-macrophages. EPA-derived RvE1 and RvE2 are catalyzed by aspirin-COX2 and 5-LOX via interaction of endothelial cells and leukocytes [83, 84]. Unlike RevDs, RevEs can be generated by an aspirin-independent pathway via cytochrome P450-driven oxygenation of EPA. RvE3 is generated from 18-hydroxyeicosapentaenoic intermediate through the 12/15-LOX pathway in eosinophils [85]. RvD1 acts through ALX/FPR2 and an orphan receptor GPR32 RvE1 on leukocytes, RvE1 mediate its function by binding to ChemR23 and LTB4 receptor (BLT1), while chemerin peptide C15 exert their action at ChemR23. Collectively, all these resolvins limit neutrophil activation and infiltration and specifically stimulate non-phlogistic macrophage phagocytosis at the site of inflammation [85]. Thus, multiple resolvins act via their respective receptors to control the diverse set of immune reactions leading to attenuation of inflammation.
Multiple reports have demonstrated a direct correlation of various inflammatory diseases and production of AnxA1, lipoxins, and resolvins in IBD, in the uncontrolled pathogenesis of atherosclerosis [86], periodontitis [87], chronic liver disease [88] and asthma [89]. In addition to gastrointestinal inflammation resolution, downregulation of LXA4, its receptor FPR2, and 15-lipoxygenase was found to be associated with persistent airway inflammation (asthma) and wheezy infants as compared to control group [90]. Interestingly, LXs and their analogues turned out to be highly effective in the resolution of diseases in the following experimental models: immune-mediated glomerulonephritis [91], renal ischaemia-reperfusion injury [92], a variety of skin inflammatory-like diseases [93] and gastritis [94]. Importantly, the pro-resolving mediator RvE1 stimulates the production of LXA4, which further bolstered the resolution events [95]). Moreover, LXA4 or Resolvin E1 (RvE1)-treated trinitrobenzenesulphonate (TNBS)-induced colitis models show attenuated leukocyte infiltration, reduced pro-inflammatory cytokines and IgG serum levels in different studies [96, 97]. Interestingly, it has also been reported that pro-resolving mediators including AnxA1 and LXA4, induce the production of anti-inflammatory cytokines such as IL-10 [98].
5C. Perturbation of the NF-κB Pathway Induces Resolution
The NF-κB pathway plays a central role in inflammation by regulating the expression of pro-inflammatory cytokines/chemokines and anti-apoptotic genes that could protect the inflammatory leukocytes from programmed cell death. Therefore, the activation of endogenous pathways that could perturb the pro-inflammatory or anti-apoptotic gene networks of the NF-κB pathway would certainly have the potential to drive the host immune system towards the resolution of inflammation. Interestingly, 15d-PGJ2 acting along with PPARγ activation is demonstrated to modify the DNA binding of NF-κB by alkylating the Cys179 of IKKβ (located in the kinase activation loop, and Cys62 and Cys38 residues located in the DNA binding subunits of NF-κB and RELA, respectively) [99]. As a result, NF-κB is unable to bind at the promoters of various pro-inflammatory cytokine genes. 15d-PGJ2 also inhibits NF-κB-induced expression of BCL-XL (anti-apoptotic gene) in CD28 co-stimulated primary human CD4+ T cells in vitro, thereby arresting the effector function of T cells [100]. Similarly, 15d-PGJ2 directed inhibition of NF-κB plays a pivotal role by regulating the apoptosis of leukocytes and PMNs [101]. Häcker et al. has shown that TRAF3, a downstream signaling molecule of the TLR pathway, plays an important role in controlling the NF-kB-mediated inflammatory cascade by inducing IL-10. Overproduction of pro-inflammatory cytokines IL-12 and IL-6 in myeloid cells from Traf3−/− mice further substantiated this observation [39]. Overall, in conjunction with anti-inflammatory cytokines (IL-10, IL-1RA and TGF-β), lipid mediators facilitate timely activation of the resolution phase.
6. microRNA Biogenesis Pathways and Regulation by Inflammation
Since their discovery by Victor Ambros and colleagues in Caenorhabditis elegans as domineering gene regulators [102, 103], ~2500 miRNAs in humans have been cataloged with various degrees of inter-species conservation [104]. Interestingly, unlike transcription factors (TF), miRNAs are readily available effector molecules as they do not require translation, post-translation modifications, or translocation back into the nucleus to begin their effector functions. Burgeoning reports suggested that miRNAs play imperative roles in controlling key regulators of acute and chronic inflammation. Lately, the regulatory role of miRNAs has been demonstrated in controlling the development and functions of various lineages of immune cells [104–106]. Interestingly, expression dynamics of miRNAs have shown their potential to regulate host immune response outcomes - both in positive and negative manners [107, 108]. miRNAs can control immune reactions either by inhibiting pro-inflammatory molecules (negative feedback) or by reinforcing the inflammatory environment by repressing the expression of inhibitors (feed forward) [109, 110]. The effects of miR-155, miR-146a and miR-223 have been widely studied and shown to be implicated in a multitude of innate immune activities [109, 111, 112]. However, these miRNAs only represent a fraction of human-encoded miRNAs. To gain a holistic understanding of miRNA regulated gene networks, further studies on cell-specific, inflammation- or resolution phase-enriched miRNAs will provide finer details of their intricate yet integral functions. The following are the two known processes for miRNA biogenesis in the cell.
(i). The Canonical miRNA Biogenesis Pathway:
This pathway involves RNA polymerase II-driven transcription of a primary miRNA transcript (pri-miRNA), which is then processed by the endoribonucleases Drosha/DGCR8 in the nucleus - termed as precursor miRNA (pre-miRNA) - and exported to the cytoplasm via the Exportin 5/RanGTP-dependent pathway. However, intron-derived miRNAs (mirtrons) use Exportin 1 instead of Exportin 5 for their nucleus-to-cytoplasm transport. Pre-miRNAs are then cleaved by Dicer (another endoribonuclease) at their looped end in the cytoplasm. The resultant short (20–25 bp), double-stranded RNA is the product of this cleavage - containing miRNA and its complementary sequence. Thereafter, the double stranded RNA duplex is unwound by Argonaute (AGO) and the mature miRNA (either 5p or 3p strand of the miRNA duplex) is loaded onto the miRNA-induced silencing complex (miRISC) [112].
(ii). Non-Dicer Dependent miRNA Biogenesis Pathway:
This alternative pathway of miRNA biogenesis solely depends on Ago2 to perform the final processing of the miRNA. Importantly, the secondary structure of the pre-miRNA (with distinct loop and hairpin structures) determines whether Dicer or Ago2 will act on the miRNA precursor. Multiple miRNAs have so far been reported to be processed by this pathway. miR-451 is the most well-characterized miRNA in this context. The precursor of miR-451 possesses a very small and distinctive loop structure [113].
miRNAs generated via either biogenesis process are loaded onto the RISC and guide the ribonucleoprotein complex to cognate mRNA sequences via Watson-Crick base-pairing rules and induce either translational repression and/or transcript degradation via mRNA deadenylation and decapping [114]. Increasing scientific reports affirm that the production of various inflammatory proteins during the process of acute inflammation either directly or indirectly impact the biogenesis of miRNAs and their target recognition. For instance, miR-21 maintains the contractile phenotype of human vascular smooth muscle cells by targeting PDCD4 (programmed cell death 4). It turned out that Transforming Growth Factor beta (TGF-β) and Bone Morphogenetic Proteins (BMP) signaling increases the processing of pre-miR-21 by assisting the crosstalk between SMAD signal transducers and RNA helicase p68 (also known as DDX5), a component of the DROSHA microprocessor complex [115].
Furthermore, Adenosine deaminase acting on double-stranded RNA 1 (ADAR1) is reported to impair pri-miRNA processing events. This enzyme catalyzes A-to-I edits in noncoding regions, particularly within the SINE family of retrotransposons that form long double-stranded RNAs, thus disabling their capacity to trigger the activators of innate immunity and resultant autoimmunity. The deficiency of ADAR1 results in development of a condition of sterile immunity – activation of innate immune cells via double-stranded RNA intermediates of various vital reactions for the cell. Yang et al., has demonstrated that ADAR1 has intrinsic capability to edit multiple pri-miRNAs including pri-miR-142, pri-miR-181a, pri-miR-200, etc. Amongst these, miR-142 and miR-181a have clearly been shown to be involved in T cell function [116].
7. microRNAs Regulate Innate And Adaptive Immune Signaling
In light of the above section, we can clearly state that miRNAs can potentially influence immune responses. Multiple miRNAs have been found to be important for the development, differentiation, survival, and function of both innate and adaptive immune cells. Various studies have revealed the expression modulation of miR-146a, miR-125b, miR-155, miR-9, etc. upon TLR activation [117–120]. Similarly, miR-155 expression dynamics were deciphered in Th1 and Th2 differentiation events. Rodriguez et al., reported Th1 to Th2 shift in CD4+ T cells complemented with the production of cytokines like IL-4, IL-5 and IL-10 [121]. In the absence of miR-155, a reduced proportion of germinal center B cells and extra-follicular B cells which failed to produce high affinity IgG1 antibodies were observed. Other studies, in this pursuit, have reported the role of miR-155 in the generation of plasma cells by directly repressing its target gene PU.1 [122]. The focus of this review is resolution phase regulation by miRNAs; therefore, we will not delve into miRNA-mediated control of inflammatory pathways. Nonetheless, we have catalogued multiple miRNAs involved in innate and adaptive immune signaling (Table 2). For a detailed mechanistic regulation of inflammation pathways by miRNAs, please refer to excellent review articles [106, 107].
Table 2:
miRNAs regulates various imperative events in inflammation by targeting critical genes involved in the pathway.
| miRNA | Targets | Process | References |
|---|---|---|---|
| TLR pathway | |||
| miR-9 | NF-κB1/p50 | Transcription by NF-κB is decreased | [123] |
| miR-16 | TNF-α | Decrease levels of inflammatory | [124] |
| miR-17–5p, 20a, 106a | Transcription factor CBF | Inhibition of monocyte maturation | [125] |
| miR-21 | PDCD4, IL12p35 | Derepression of IL-10 | [126] |
| miR-27b | PPARγ | [127] | |
| miR-105 | TLR2 | [128] | |
| miR-106 | IL-10 | Cooperates with RNA binding protein | [129] |
| miR-125b | TNF-α | to decrease IL-10 | [130] |
| miR-145 | MAL | Impedes TLR signaling | [131] |
| miR-146a | TRAF6, IRAK1, IRAK2 | Negative feedback regulator in TLR signaling | [132] |
| miR-155 | AID, MyD88, TAB2, IKKԑ, SHIP1, SOCS1, C/.EBPBβ | Proinflammatory in TLR signaling | [133] |
| miR-199 | IKKβ | TLR4/NF-κB p65/NGAL pathways | [134] |
| miR-221 | TNF-α | [135] | |
| miR-223 | TLR3, TLR4, IKKα | Granulopoiesis and monocyte activation | [136] |
| Let-7i and let7e | TLR4 | Downregulate inflammatory signaling | [137] |
| T cell development and function | |||
| miR-181a | SHP2 | T cells: strength of the transduced T-cell receptor signal. | [138] |
| miR-155 | SOCS1 | miR-155-deficient mice suffer impaired survival of Treg, T regs/Th17 ratio | [139] |
| miR-146a | STAT1 | miR-146a-deficient Treg (FoxP3) cannot inhibit hyperactive immune system. Upregulated in RA | [140] |
| miR-17~92 cluster | pten | immunoproliferative disorder and autoimmune disease | [141] |
| miR-326 | Ets-1 transcription factor | autoimmune inflammation | [142] |
| B cells development and function | |||
| miR-181a | Lin28/Mcl-1 | guided B-cell development | [143] |
| miR-150 | c-Myb and Foxp1 | B-cell development | [144] |
| miR-34a | c-Myb and Foxp1 | B cell development | [145] |
| miR-155 | Pu.1, SHIP1 and AID | regulate antibody secretion and class-switch recombination | [146] |
| miR-181b | reduce the class-switch recombination rates | [147] | |
8. microRNAs Regulate Resolution And Wound Healing
Burgeoning evidences have now demonstrated a critical role of miRNAs in resolution of the infection. Sashwati et al., have shown the direct involvement of miRNAs in the resolution of ischemic wound repair via regulation of the cellular redox environment in human microvascular endothelial cells (HMECs) [148]. Changes in miRNA profiles during the onset of T2D and insulin resistance were shown to interfere with wound healing [149]. Recchiuti et al., showed that the pro-resolving mediator RvD1 induces time-dependent expression of various miRNAs in exudates both in vivo and in vitro [150]. These miRNAs have been reported to target genes that have key roles in pro-inflammatory responses. We will discuss how miRNA expressional dynamics will impact the regulation of inflammation, resolution and tissue healing processes. Figure 1 illustrates how various miRNAs can shape initiation and resolution of inflammation by directly targeting genes involved in cytokine signaling, innate immune functions and lipid pathways.
Figure 1. MiRNA-mediated regulation of inflammation and resolution.
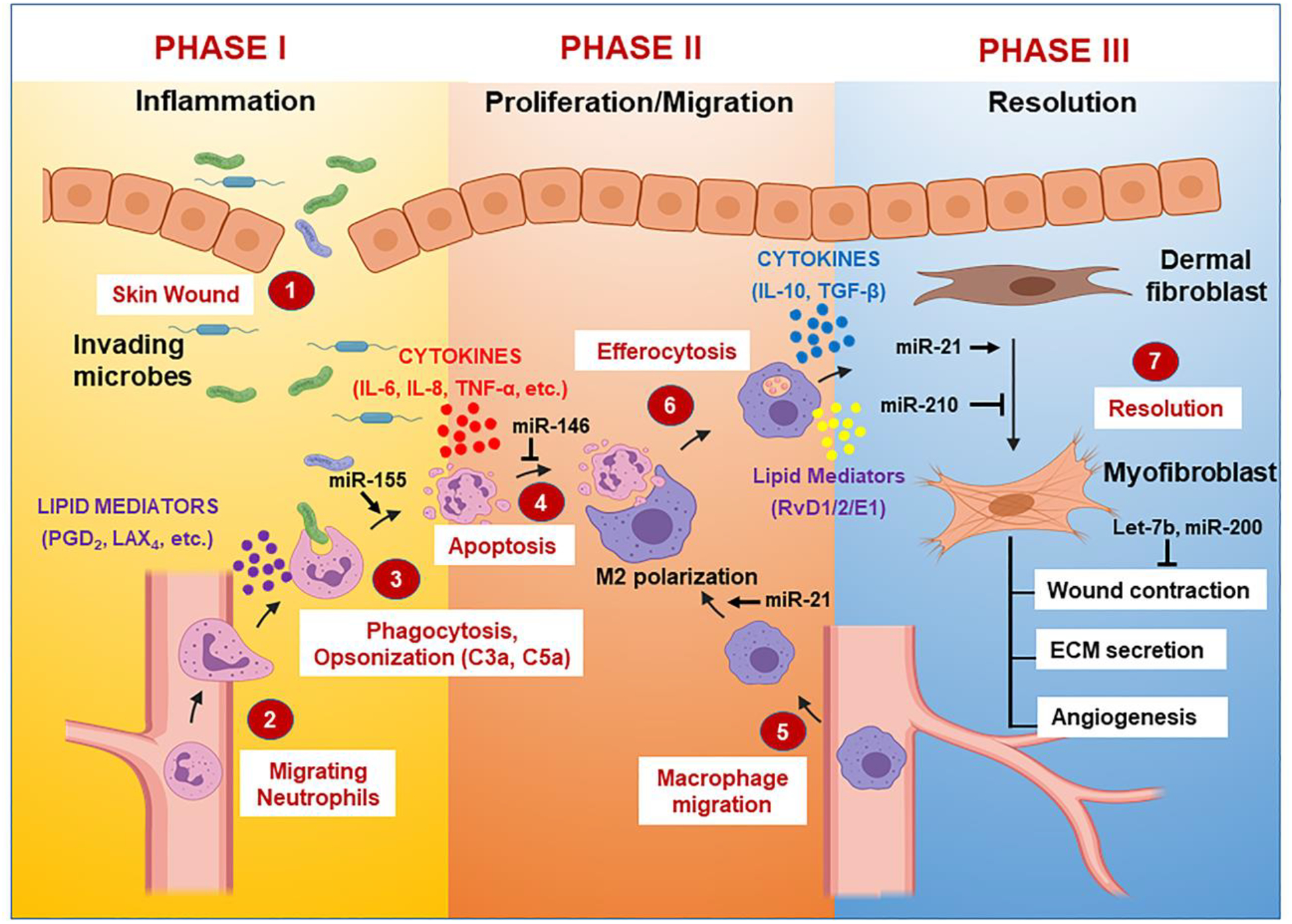
The key events involved in the inflammation and resolution are directly regulated by various miRNA expression.
8A. miRNA Regulation of the Resolution Phase
(i). miR-155 Knockout Induces The Resolution of Inflammatory Diseases
The miR-155 gene is located on chromosome 21 in the B-cell integration cluster (BIC) [151]. LPS or type I interferons (IFN) induce the expression of miR-155 both in monocytes and macrophages derived from humans and mice, thereby suggesting its pivotal role in both bacterial and viral infection. Interestingly, miR-155 is highly expressed in activated B and T cells and impacts expression of various cytokine genes [139,146]. Activation of MyD88, the TRIF pathways via LPS, or poly I:C stimulation can all induce the expression of miR-155. Interestingly, in LPS-stimulated macrophages, miR-155 binding with 3’UTR of the TNF-α transcript releases it from self-repression, thereby facilitating its translation [117]. In contrast, miR-155 expression is inhibited by anti-inflammatory cytokine IL-10 [152]. Treatment with IL-10 enhances the expression of the miR-155 target gene, Src homology2 (SH2) domain-containing inositol 5′-phosphatase 1 (SHIP1), which in turn negatively regulates TLR-induced responses [153].
miR-155 KO mice have been demonstrated to show a defective immune response against Salmonella typhimurium due to defective B- and T- cell action. Moreover, DCs were unable to present the antigen in this study due to altered Th1 responses [121]. In a separate study, miR-155 KO mice exhibit high levels of activation-induced cytidine diamine (AID), which which has high mutagenic potential during class switching and somatic hypermutation in B cells [154]. Typically, B-cells obtained from WT mice undergoing class switching express high but regulated levels of AID; however, mutation in the 3’UTR binding site of miR-155 has been shown to increase proportions of Myx-Igh translocations, thereby leading to disrupted affinity maturation of B cells [154].
Anti-inflammatory dietary compounds including resveratrol [155], curcumin [156], apigenin [157], and quercetin [158] are reported to facilitate the downregulation of miR-155. Similarly, polyunsaturated fatty acids, docosahexaenoic acid, and arachidonic acid significantly reduce the expression of miR-155 in LPS-stimulated murine macrophages [159]. Overall, these findings clearly demonstrate that restoration of miR-155 expression can both augment the resolution phase and concomitantly suppress inflammatory pathways.
(ii). miR-146 Functions as a miR-155 Antagonist in The Resolution Phase
The miR-146 family consists of two isoforms viz., miR-146a and miR-146b located on chromosome 5 and chromosome 10, respectively. Both isoforms share an exact seed sequence, 2–8 nts from the 5′ end of mature miRNA, suggesting that they target similar transcripts. miR-146a and miR-146b have been elucidated in negative regulation of inflammatory diseases and tumors (e.g. lymphoma) by targeting key molecules involved in the NF-κB pathway [160]. Furthermore, various studies have divulged the development of B cell lymphoma and other myeloid malignancies in aging miR-146a/b knockout (KO) mouse models [161, 162]. The B-cell lymphomas observed in miR-146a- and −146b–KO mice were different in terms of histological changes and malignancy. For example, the malignant rate of B cell lymphoma in miR-146b KO mice was lower than in miR-146a KO mice. Both of these miRNAs target TRAF6 and interleukin-1 receptor–associated kinase 1 (IRAK1), which are endogenous inhibitors of NF-κB activation [162]. Furthermore, cancer cells require constitutively active nuclear factor-κB (NF-κB) to maintain survival, proliferation, and metastatic potential. Therefore, the NF-κB signaling inhibition potential of miR-146 has potential therapeutic value in cancer resolution. In this pursuit, miR-146b overexpression has been shown to prevent the development breast cancer, glioma, gallbladder cancer, and large B-cell lymphoma [162–166].
Interestingly, miR-146 plays an imperative role in counteracting miR-155-induced targets during inflammation via targeting NF-κB activity. To counterbalance the pro-inflammatory effect of miR155, expression of miR-146 accumulates concomitantly and targets IRAK1 and TRAF6 to suppress NF-κB activation. Luly et al., has shown that the inhibition of miR-146a in lipopolysaccharide (LPS)-stimulated macrophages obtained from cystic fibrosis patients results in increased IL-6 production, thereby substantiating the role of miR-146 in inflammation resolution [167]. In congruence, Cheng et al, highlighted the role of miR-146a and miR-146b in restraining the extent and duration of endothelial cell activation in response to pro-inflammatory cytokine stimulation and recruitment of leukocytes in response to IL-1β treatment. The group unequivocally showed that miR-146a over-expression controls the expression of VCAM-1 and SELE (endothelial activation markers for leukocyte recruitment), and facilitates the reduction of THP-1 monocytes that would otherwise remain adhered to IL-1β-treated endothelial cells [168]. Interestingly, Chatzikyriakidou et al., demonstrated a clear association between a polymorphism in the 3′UTR of miR-146a target IRAK1 with susceptibility to Rheumatoid Arthritis (RA) [169]. Similarly, Tang et al., elucidated that under-expression of miR-146a and over-activation of the type I IFN pathway could contribute to the pathogenesis of SLE [170]. These studies clearly highlight the significance of miRNA regulatory networks that counterbalance reciprocal functions. In this regard, miR-155 and miR-146 mimic pro-inflammatory (IL-1, IL-6 and IL-8) and anti-inflammatory (IL-10 and IL-1RA) cytokines that antagonize each other’s function in a pleiotropic manner. In addition, both can have autocrine and paracrine effects thus reflecting their widespread impact. Thus, miRNAs and effector immune mediators work in concert to reinforce their purpose and perturbation in their functional axis can have a detrimental impact on timely inflammation resolution.
(iii). miR-21 Controls Inflammation Through IL-10 Induction
Recent reports have identified miR-21 as an anti-inflammatory miRNA which curbs excessive inflammatory responses and initiates the resolution phase. miR-21 is shown to regulate the expression of IL-10 (a potent anti-inflammatory cytokine) in LPS-induced macrophages by targeting PDCD4, a negative regulator of IL-10 [171, 172]. A study by Lu et al., has elucidated the role of this miRNA in controlling the production of pro-inflammatory IL-12 in dendritic cells [173].
Similar to miR-146, upregulation of miR-21 expression occurs upon challenge with LPS or the Gram-negative bacterium V. anguillarum, which subsequently suppresses the expression of LPS-induced pro-inflammatory cytokines by targeting IL-1 receptor-associated kinase 4 (IRAK4). Therefore, the expression of both miR-146 and miR-21 could be a plausible counter-regulatory mechanism to control excessive inflammatory damage by NF-κB signaling [175].
Multiple studies have shown induction of miR-21 expression after treatment of monocytes with: 1) all-trans retinoic acid (to generate neutrophils) [172], 2) GM-CSF/IL-4 (to generate immature DCs) [176], and 3) LPS-mediated B-cell activation [177]. Upregulation of miR-21 in epithelial cells and in mesenchymal cells plays a crucial role in the initiation of cell migration and during wound healing, respectively [178]. Adequate expression of this miRNA has been considered as one of the decisive factors during tissue remodeling after pulmonary and cardiac injury [180].
(iv). miR-208, miR-219, miR-19a-3p and miR-125b-5p Regulate Leukotriene Biogenesis in Resolution
As described earlier, leukotrienes are central in the execution of inflammation resolution. PUFA-generated RvD1 is shown to up-regulate miR-208a and miR-219 in exudates isolated from ALX/FPR2 transgenic mice (a self-limited peritonitis model) [150]. In this study, RvD1 (100 mg/mouse) treatment was associated with the upregulation of miR-208a and miR-219 in exudates isolated from ALX/FPR2 transgenic mice compared with control littermates. RvD1-treated mice also showed 50% reduction in PMN infiltration in the murine dorsal air-pouch model. In this study, the overexpression of miR-208a in human macrophages was associated with the up-regulation of IL-10 [150]. miR-219 overexpression in macrophages led to significant reduction in CD14, TNF receptor II, phospholipase C2, arachidonate 5-LOX and leukotrienes. Interestingly, leukotriene-generating enzyme 5-LO was shown to be regulated by miR-19a-3p and miR-125b-5p, thereby suggesting tight regulation of the resolution process by a subset of co-expressed miRNAs [150].
(iv). miR-142 Exerts Pleiotropic Inhibition of Inflammatory Pathways
The mir142 gene is a highly conserved mammalian miRNA located on chromosome 17. The pre-miR-142 encodes for two mature isoforms: miR-142-3p and miR-142-5p, each with distinct target subsets. These miRNAs are broadly expressed in hematopoietic stem cell-derived lineages and play a central role in the differentiation of megakaryocytes, eosinophils, neutrophils, mast cells, dendritic cells, monocytes/macrophages, erythrocytes, T/B cells, natural killer (NK) cells and innate lymphoid cells (ILCs) [181–183]. In addition, miR-142-3p and -5p regulate numerous cellular and effector functions of immune cells including cytoskeletal rearrangement, innate and adaptive immune responses, cell polarization, apoptosis, inflammatory signaling cascades, etc. [181–184]. Studies from our lab have unequivocally demonstrated multiple functional aspects of miR-142-3p in myeloid cell functions. Both miR-142-3p and -5p are downregulated during the differentiation of monocyte-derived macrophages and dendritic cells. Interestingly, this precedes the functional acquisition of differentiated cells as reflected by remarkable induction of surface markers [182, 183]. Enforced expression of miR-142-3p attenuated phagocytosis, antigen uptake and processing, and innate immune responses, primarily by targeting protein kinase C alpha (PRKCA) transcripts in myeloid cells [182, 184, 185]. Challenge with multiple inflammatory stimuli (e.g., microbes or IgG coated beads) causes miR-142-3p suppression with concomitant upregulation of inflammatory pathways in the initial phase of inflammation. We have recently observed that downregulation of transcription factor PU.1, a TF that regulates miR-142 expression, upon bacterial challenge and simultaneous induction of pro-inflammatory miR-155, which directly targets PU.1, together inhibit miR-142-3p expression [Valverde et al., Unpublished results]. Thus, it is possible that miR-155-mediated suppression of miR-142 is required to elicit a potent immune response and the gradual induction of miR-142-3p reinforces anti-inflammatory miR-146 to counterbalance the pro-inflammatory activity of miR-155.
An immuno-protective function of miR-142 has been demonstrated in maintaining regulatory immune cells. For instance, H3 lysine-4 tri-methylation (H3K4me3) at the Mir142 locus is mediated by super enhancer bound by the forkhead box P3 (FOXP3), a Treg lineage–determining TF. Higher expression of miR-142-5p in Tregs downregulates its cognate mRNA, phosphodiesterase-3b (PDE3B), which controls cellular cAMP/cGMP levels required for maintaining Treg function [186]. miR-142−/− CD4+ T cells had higher AMP/GMP levels and produced higher levels of cytokines (IL-2, lL-4, IL-17, IFN-γ) that facilitated effector T cells functions. Interestingly, Tregs from miR-142 KO mice failed to suppress proliferation of effector T cells - strongly supporting a central role of miR-142 in shaping immune-suppressive pathways. Myeloid-derived suppressive cells (MDSC), similar to Tregs, perform immunosuppressive functions by attenuating effector T cell function and proliferation. Enforced expression of miR-142 in bone marrow cells predominantly impaired CD11b+Gr-1lo-neg cell subset generations composed of mature macrophages [187]. IL-6-mediated induction of MDSC is mediated by higher LAP* levels which promotes macrophage differentiation. By directly binding to the UTR of gp130 and C/EBPβ LAP*, miR-142-3p dampens signaling cascades required for macrophage differentiation and promotes granulocytic cells. This function of miR-142-3p, in conjunction with adoptive transfer of tumor-specific cytotoxic T lymphocyte (CTLs), blocked tumor-associated macrophage (TAM) differentiation and impaired tumor growth in vivo. These data demonstrate a broad function of miR-142 in attenuating tumor inflammation and highlight its potential as a therapeutic target.
Global transcriptome analysis of miR-142-3p-overexpressing dendritic cells showed a wide range of pathways regulated by miRNA including cell adhesion, cell movement, cytoskeletal homeostasis, immune pathways, etc. [188]. Consistent with this, miR-142-3p-transfected cells exhibit loss of cell polarity and disrupted actin polymerization status. Surface electron microscopy and confocal microscopy further shows smaller cell size of miR-142-3p overexpressing cells. Chapnik et al., identified various actin pathway-related gene targets in Mir142−/− mice, further supporting its role in cytoskeletal dynamics; miR-142-deficient mice also failed to execute actin-dependent proplatelet formation [181]. Chemotaxis, phagocytosis, cytokine production and antigen presentation are key aspects of leukocytes, all of which rely on robust cytoskeletal function. Dysregulation of miR-142 can severely perturb a wide spectrum of cell-intrinsic leukocyte functions. Not surprisingly, altered expression of miR-142-3p/5p is commonly reported in various immune-mediated diseases including multiple sclerosis, systemic lupus erythematosus, periodontitis, Sjögren’s syndrome, etc. [189–192]. Exploiting the diagnostic and therapeutic potential of immune-suppressive miR-142-3p/5p may provide new treatment modalities for a wide spectrum of immune disorders.
8B. Role of miRNAs in Wound Healing: Lessons from Skin Model
Multiple groups strived to work on the skin wound healing process in order to divulge the sequence of events involved. Herein, we are presenting various steps in wound healing and their miRNA-mediated regulation. Broadly, skin wound healing involves three overlapping phases: inflammation, proliferation/migration, and maturation (Figure 2). During the inflammatory phase, neutrophils transmigrate from blood vessels at the wound margin along with a fibrin clot and engulf microbes via NADPH oxidase pathway driven respiratory burst [193]. Neutrophils produce fibrillar networks at the wound margin to avoid the undesirable spread of antimicrobial peptides/enzymes to prevent the spread of infection [194]. Subsequently, the proliferation phase ensues which attracts macrophages to the wound site. Macrophages at the wound site perform: 1) efferocytosis of apoptotic neutrophil population, and 2) secrete biologically active substances which stimulate epithelial cells and wound-infiltrated fibroblasts [195] to cover the wound surface (Figure 2). The phenomenon of efferocytosis – an important component of the resolution phase- is regulated by miR-21 by inducing M1 to M2 macrophage phenotype switch [174]. Both professional and non-professional phagocytes are involved in elimination of apoptotic neutrophils, which are a predominant population of circulating white blood cells (WBCs) in humans and function as critical players in host defense against bacterial and fungal infections. Spleen, liver, and bone marrow are the prime sites for neutrophil efferocytosis events. Nucleotides, lysophosphatidylcholine, and sphingosine-1-phosphate are a few important chemoattractants that signal the location of apoptosis-undergoing cells to phagocytes (M2 macropahges). Furthermore, apoptotic cells also express resolvin E1 (RvE1), protectin D1 (Ref. 101), lactoferrin and annexin A1, to restrict further recruitment of granulocytes and thus, favor the preponderance of PMNs- a necessary factor for resolution and optimal wound healing. Interestingly, these apoptotic cells lose expression of ‘don’t-eat-me’ cell-surface molecules viz CD31 and CD47 and concomitantly upregulate expression of various pro-phagocytic signals e.g., phospholipids, nucleotides and phosphatidylserine. The crosstalk between tissue-resident M2 macrophages and dying PMNs harboring pro-phagocytic signals ensures the successful removal of apoptotic cells before they can perpetuate another cycle of inflammation by releasing histotoxic mediators upon membrane rupture. Engulfment of PMNs undergoing apoptosis triggers the immune-regulatory or pro-resolution phenotype viz., PDL1 and ICOS ligand expression, secretion of anti-inflammatory cytokines IL-10 and TGFβ, production of PCNA-associated factor, production of PGE2 and cAMP, or inhibition of the secretion of pro-inflammatory cytokines such as TNF, GM-CSF, IL-1β, IL-12 and IL-18 to limit the potential collateral tissue damage [3, 51–53]. Dermal fibroblasts transdifferentiate into myofibroblasts which are involved in wound contraction, secretion of extracellular matrix (ECM) including collagens, proteoglycans, and adhesive glycoproteins for the formation of granulation tissue, and induce endothelial cells to form blood vessels within the granulation tissue [196].
Figure 2. Sequence of events in resolution of inflammation.
Schematic depicting the three phases involved in skin wound healing following injury/trauma. The coordinated and synergistic temporal events during these phases are modulated by various cytokines, lipid mediators and spatiotemporal miRNA expression.
The decisive role of miRNA expression dynamics has been independently reported by various groups in improving the efficiency and quality of skin wound healing. Yang et al, has demonstrated that downregulation of miR-155 via antisense oligodeoxynucleotides (AS-ODN) at wound sites reduces fibrosis [197]. In addition to this, another group has shown that over-expression of miR-21 at the wound sites using a lentivirus vector facilitated the murine skin wound healing process [198]. Furthermore, expression of miR-146a and miR-106b during skin wound healing has been shown to prevent transepidermal water loss at biofilm-infected chronic skin wound sites. Inhibition of miR-210 via administration of lipid nanoparticles containing encapsulated miR-210 results in enhanced re-epithelialization. Interestingly, inhibition of miRNA function via the antisense ODNs miR-15b AS ODN, miR26a AS ODN, light-inducible anti-miR-92a, miR-200b AS ODN, and miR-132 mimic have shown therapeutic implications during skin repair in diabetic wounds. Also, miR-142 plays a critical role in clearing S. aureus infected-skin wound sites via neutrophils [199]. Table 3 elucidates the roles of various miRNA in regulating important events in the skin-wound healing process. Together, these studies demonstrate a requirement of miRNAs in the skin wound healing model and therapeutic targeting of miRNAs may restore or augment the normal repair/regeneration process.
Table 3:
miRNA expression patterns during skin wound healing and their regulation of various aspects of the process.
| miRNA | Process | References |
|---|---|---|
| miR-155 | Down regulation via AS ODNs can improve efficiency and quality of skin wound healing by reducing inflammation | [190] [191] |
| miR-21 | (a) Upregulated in epithelial cells at the initiation of beginning of cell migration at wound site. (b) Down-regulation delayed wound healing from 1–14 days after injury. (c) Over-expression via lentivirus facilitated the improved healing in rat model targeting Pten gene |
[178],[192] |
| miR-146 a miR-106b | (a) Prevent the skin infection during wound healing | [193] |
| miR-200c | (a) Overexpression delayed re-epithelialization in human skin model | [194] |
| miR-210 | (a) Downregulation embargo skin re-epithelialization | [195] |
| miR-15b, miR26a, miR-92a, miR-200b, | (a) Down regulation Improved skin healing in diabetes patients | [196] |
| miR-132, miR27b | (a) Downregulation improved skin wound healing in diabetes patients | [197][198] |
| miR29b and miR-1908 | (a) Overexpression inhibit scarring | [199] [200] |
| miR-132 | (a) enhance the transition from inflammation to proliferation during wound healing | [201] |
| miR-142 | (a) neutrophils to clear S. aureus infected-skin wound sites | [202] |
| Let-7b | (a) overexpression exhibit a delay in re-epithelialization at wound site | [203] |
| miR-198 | (a) inhibits keratinocytes migration and proliferation | [211], [212] |
| miR-23b | (a) cutaneous wound healing via inhibition of inflammatory reactions | [213] |
| miR-483-3p | (a) controls proliferation in wounded epithelial cells | [214] |
9. Diagnostic and Therapeutic Use of miRNAs in Disease Resolution
Induction of resolution events is now considered a holy grail of treatment of acute and chronic inflammation. Instead of inducing individual miRNAs or treating the host with multiple miRNAs using complicated delivery vehicles, Recchiuti et al. treated the self-limited murine peritonitis model with RvD1 (300 ng/mouse or 15 μg kg−1). Interestingly, they observed significant modulation of expression of multiple miRNAs including miR-21, miR-146b, miR-208a, miR-203, miR-142, miR-302d, and miR-219, in resolving exudates of this murine model. Their findings corroborate previous studies showing temporal expression changes in these miRNAs. Interestingly, miR-21 and miR-203 were upregulated only after 12 h post-RvD treatment [150]. It was proposed that an initial subset of miRNAs might be involved in the induction of resolution, whilst a later group of miRNAs may participate in the maintenance or final events associated with inflammation resolution. A remarkable reduction (25–50%) in zymosan-elicited neutrophil infiltration into the peritoneum was observed, suggesting a pro-resolving function of RvD1. In addition, this group further treated human macrophages overexpressing recombinant RvD1 receptors ALX/FPR2 or GPR32 with RvD1 (at concentrations as low as 10 nM) in vitro and observed similar changes in miRNA profiles, strongly supporting their in vivo findings. RvD1 treatment attenuates nuclear translocation of NF-κB and SMAD and reduces phosphorylation of IκB, an endogenous inhibitor of NF-κB. Recchiuti et al., found that RvD1-dependent miRNA changes were solely based on the expression of its receptor and dose; therefore, the group also expressed the above miRNAs (appeared in the resolution exudates) via expression vectors and found significant downregulation of their gene targets. For example, human macrophages overexpressing miR-146b showed downregulation of S100A12, LPS binding protein (LBP), C-reactive protein, complement component 8 α polypeptide (C8a), Toll-like receptor (TLR) 9 and 10, and peptidoglycan recognition protein (PGLYRP) 1 and 2. miR-208a-overexpressing human macrophages showed reduced expression of CD14, CD40 ligand (CD40L), prostacyclin I2 receptor (PTGIR), thromboxane A2 receptor (TBXA2R), and programmed cell death 4 (PDCD4). Also, miR-146b overexpression led to significant down-regulation of IL-8, IL-10, IL-12 receptor β2, chemokine CC motif receptor 3 (CCR3), interferon (IFN) α1 and IFNβ1, and members of the IL-1 family. Expression of miR-219 resulted in significant downregulation of CD14, TNF receptor II, phospholipase Cγ2, and arachidonate 5-lipoxygenase [150]. Therefore, miRNA treatment has demonstrated a wide gamut of regulatory functions in vivo compared to RvD alone.
Resolvin D1 has also proven effective in the resolution of the long-term lung inflammation-associated with P. aeruginosa. RvD1 treatment considerably reduced the P. aeruginosa load, leukocyte infiltration, lung tissue damage and inflammatory signaling by affecting the gene expression of TLR (and their downstream genes) and miR-21 and −155 [200]. Research in the area of therapeutic targeting of miRNA to potentiate disease resolution is still in its infancy and demands extensive studies to understand the role of RvD-induced miRNAs in treating various other chronic inflammatory diseases associated with different organs based on their endogenous RvD receptors. In vivo delivery of both resolvins coupled with miRNA over-expression vectors could be a promising stratagem to induce the sequence of events involved in resolution. Furthermore, expression of the aforementioned miRNAs in the tissue exudates can be explored for diagnosis of the resolution phase, a reasonable strategy to be embarked upon in the impending future.
10. Conclusions
In summary, resolution of inflammation is a highly regulated sequence of events that eventually restores tissue homeostasis and stymies the development of chronic inflammatory disease. Numerous studies have extensively delineated the underlying role of miRNAs in regulating inflammation and resolution events to maintain tissue homeostasis. Despite compelling evidence, exploiting the knowledge of expressional dynamics of selected miRNA to achieve desired inflammation resolution during chronic inflammatory diseases remains a challenge. Over-expression of miR-21, miR-142, miR-146 and miR-208, and concomitant down-regulation of miR-155, may provide convincing evidences to exacerbate the pro-resolution molecular circuitry in acute/chronic inflammatory disease. Additionally, miRNAs can also be used in conjunction with lipid mediators of resolution. Synergistic targeting of two distinct yet mutually inclusive pathways may drive resolution in a robust manner. The crux of inflammation and resolution is wound healing; therefore, further identification and elucidation of miRNA-mediated precise regulation of the skin wound healing process will advance our understanding of the pathway. It is possible that information from inflammation resolution can be translated to treat cancers, despite an immunosuppressive microenvironment, and may involve dysregulation of similar repertoire of miRNAs. In the last decade, miRnome analysis in various immune-mediated diseases and cancers have identified a subset of miRNAs with potential diagnostic and prognostic functions. However, translating this knowledge remains a daunting task. In vivo targeted delivery of miRNAs, their long-term expression, and safety issues remain as major limitations. Selecting appropriate and effective delivery strategies for multiple miRNA/antagomirs will allow simultaneous modulation of multiple host genes (under direct influence of miRNAs) to mitigate inflammatory diseases and prevent tissue damage. Researchers anticipate that development and standardization of miRNA-based strategies will curtail the dreaded effects of multiple chronic inflammatory conditions, which so far remain incurable.
ACKNOWLEDGMENT
We are thankful to Samantha Schaller for critical reading of the manuscript.
FUNDING SOURCE
This work was funded by the NIH/NIDCR R03 DE027147 and R01DE027980 to ARN.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OR INTEREST
The authors have no conflict of interest to declare.
References
- [1].Medzhitov R, Origin and physiological roles of inflammation, Nature 454 (2008) 428–435. [DOI] [PubMed] [Google Scholar]
- [2].Aggarwal BB, Van Kuiken ME, Iyer LH, Harikumar KB, Sung B, Review Molecular targets of nutraceuticals derived from dietary spices: potential role in suppression of inflammation and tumorigenesis, Exp. Biol. Med 234 (2009) 825–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nathan C, Neutrophils and immunity: challenges and opportunities, Nat. Rev. Immunol 6 (2006) 173–182. [DOI] [PubMed] [Google Scholar]
- [4].Yao C, Narumiya S, Prostaglandin-cytokine crosstalk in chronic inflammation, Br. J. Pharm 176 (2019) 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Probert L, Eugster HP, Akassoglou K, Bauer J, Frei K, Lassmann H, Fontana A, TNFR1 signalling is critical for the development of demyelination and the limitation of T-cell responses during immune-mediated CNS disease, Brain 123 (2000) 2005–2019. [DOI] [PubMed] [Google Scholar]
- [6].Houslay MD, Schafer P, Zhang KY, Keynote review: phosphodiesterase-4 as a therapeutic target, Drug Dis. Today 10 (2005) 503–1519. [DOI] [PubMed] [Google Scholar]
- [7].Schett G, Elewaut D, McInnes IB, Dayer JM, Neurath MF, How cytokine networks fuel inflammation: toward a cytokine-based disease taxonomy, Nat. Med 19 (2013) 822–824. [DOI] [PubMed] [Google Scholar]
- [8].McInnes IB, Schett G, Pathogenetic insights from the treatment of rheumatoid arthritis, Lancet 389 (2017) 2328–2337. [DOI] [PubMed] [Google Scholar]
- [9].Nielsen OH, Ainsworth MA, Tumor necrosis factor inhibitors for inflammatory bowel disease, N. Engl. J. Med 369 (2013) 754–762. [DOI] [PubMed] [Google Scholar]
- [10].Pua KH, Chew CL, Lane DP, Tergaonkar V, Inflammation-associated genomic instability in cancer, Genome Instab. Dis 1 (2020) 1–9. [Google Scholar]
- [11].Alam MM, O’Neill LA, MicroRNAs and the resolution phase of inflammation in macrophages, Eur. J. Immunol 41 (2011) 2482–2485. [DOI] [PubMed] [Google Scholar]
- [12].Kotas ME, Medzhitov R, Homeostasis, inflammation, and disease susceptibility, Cell 160 (2015) 816–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Takeuchi O, Akira S, Pattern recognition receptors and inflammation, Cell 140 (2010) 805–820. [DOI] [PubMed] [Google Scholar]
- [14].Iwasaki A, Medzhitov R, Control of adaptive immunity by the innate immune system, Nat. Immunol 16 (2015) 343–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kawai T, Akira S, Toll-like receptors and their crosstalk with other innate receptors in infection and immunity, Immunity 34 (2011) 637–650. [DOI] [PubMed] [Google Scholar]
- [16].Pober JS, Sessa WC, Inflammation and the blood microvascular system, Cold Spring Harb. Perspect. Biol 7 (2014), a016345, 10.1101/cshperspect.a016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Stuart LM, Lacy-Hulbert A, De-mystifying microbicidal killing, Nat. Immunol 16 (2015) 1107–1118. [DOI] [PubMed] [Google Scholar]
- [18].Förstermann U, Sessa WC, Nitric oxide synthases: regulation and function, Eur. Heart J 33 (2012) 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Claesson-Welsh L, Vascular permeability–the essentials, Ups. J. Med Sci 120 (2015) 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Baer C, Squadrito ML, Iruela-Arispe ML, De Palma M, Reciprocal interactions between endothelial cells and macrophages in angiogenic vascular niches, Exp. Cell Res 319 (2013) 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dunkelberger JR, Song WC, Complement and its role in innate and adaptive immune responses, Cell Res. 20 (2010) 34–50. [DOI] [PubMed] [Google Scholar]
- [22].Koh TJ, DiPietro LA, Inflammation and wound healing: the role of the macrophage, Expert Rev. Mol. Med 13 (2011), e23, 10.1017/S1462399411001943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Diegelmann RF, Evans MC, Wound healing: an overview of acute, fibrotic and delayed healing, Front. Biosci 9 (2004) 283–289. [DOI] [PubMed] [Google Scholar]
- [24].Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M, Growth factors and cytokines in wound healing, Wound Repair Regen. 6 (2008) 585–601. [DOI] [PubMed] [Google Scholar]
- [25].Hofman Z, de Maat S, Hack CE, Maas C, Bradykinin: inflammatory product of the coagulation system, Clin. Rev. Allergy Immunol 51 (2016) 152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell, 4th Ed., Garland Science, New York, 2002. 〈https://www.ncbi.nlm.nih.gov/books/NBK26921/〉. [Google Scholar]
- [27].Hampton HR, Chtanova T, Lymphatic migration of immune cells, Front. Immunol 10 (2019) 1168, 10.3389/fimmu.2019.01168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Xu M, Bennett DLH, Querol LA, Wu LJ, Irani SR, Watson JC, Pittock SJ, Klein CJ, Pain and the immune system: emerging concepts of IgG-mediated autoimmune pain and immunotherapies, J. Neurol. Neurosurg. Psychiatry 91 (2020) 177–188, 10.1136/jnnp-2018-318556. Epub 2018 Sep 17. [DOI] [PubMed] [Google Scholar]
- [29].Filippi MD, Mechanism of diapedesis: importance of the transcellular route, Adv. Immunol 129 (2016) 25–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Haraldsen G, Kvale D, Lien B, Farstad IN, Brandtzaeg P, Cytokine-regulated expression of E-selectin, intercellular adhesion molecule-1 (ICAM-1), and vascular cell adhesion molecule-1 (VCAM-1) in human microvascular endothelial cells, J. Immunol 156 (1996) 2558–2565. [PubMed] [Google Scholar]
- [31].Mosser DM, Zhang X, Measuring opsonic phagocytosis via Fcγ receptors and complement receptors on macrophages, Curr. Protoc. Immunol 95 (2011), 10.1002/0471142735.im1427s95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rabadi MM, Ghaly T, Goligorksy MS, Ratliff BB, HMGB1 in renal ischemic injury, Am. J. Physiol. Ren. Physiol 303 (2012) F873–F885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lorenz U, SHP-1 and SHP-2 in T cells: two phosphatases functioning at many levels, Immunol. Rev 228 (2009) 342–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M, Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases, Cell 120 (2005) 649–661. [DOI] [PubMed] [Google Scholar]
- [35].Alexander WS, Hilton DJ, The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response, Annu. Rev. Immunol 22 (2004) 503–529. [DOI] [PubMed] [Google Scholar]
- [36].Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, McNally E, Pickart C, Ma A, The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses, Nat. Immunol 5 (2004) 1052–1060. [DOI] [PubMed] [Google Scholar]
- [37].Iyer SS, Cheng G, Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease, Crit. Rev. Immunol 32 (2012) 23–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cevey ÁC, Penas FN, Alba Soto CD, Mirkin GA, Goren NB, IL-10/STAT3/SOCS3 axis is involved in the anti-inflammatory effect of benznidazole. Front. Immunol 10 (2019) 1267, 10.3389/fimmu.2019.01267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Häcker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Häcker G, Mann M, Karin M, Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6, Nature 439 (2006) 204–207. [DOI] [PubMed] [Google Scholar]
- [40].Cesari M, Penninx BW, Newman AB, Kritchevsky SB, Nicklas BJ, Sutton-Tyrrell K, Rubin SM, Ding J, Simonsick EM, Harris TB, Inflammatory markers and onset of cardiovascular events, Circulation 108 (2003) 2317–2322. [DOI] [PubMed] [Google Scholar]
- [41].Mroz RM, Noparlik J, Chyczewska E, Braszko JJ, Holownia A, Molecular basis of chronic inflammation in lung diseases: new therapeutic approach, J. Physiol. Pharm 58 (2007) 453–460. [PubMed] [Google Scholar]
- [42].Low Wang CC, Hess CN, Hiatt WR, Goldfine AB, Clinical update: cardiovascular disease in diabetes mellitus: atherosclerotic cardiovascular disease and heart failure in type 2 diabetes Mellitus - mechanisms, management, and clinical considerations, Circulation 133 (2016) 2459–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yadav D, Lowenfels AB, The epidemiology of pancreatitis and pancreatic cancer, Gastroenterology 144 (2013) 1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Strober W, Fuss I, Mannon P, The fundamental basis of inflammatory bowel disease, J. Clin. Invest 117 (2007) 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Brenner C, Galluzzi L, Kepp O, Kroemer G, Decoding cell death signals in liver inflammation, J. Hepatol 59 (2013) 583–594. [DOI] [PubMed] [Google Scholar]
- [46].Leitão HS, Doblas S, Garteiser P, D’Assignies G, Paradis V, Mouri F, Geraldes CF, Ronot M, Van Beers BE, Hepatic fibrosis, inflammation, and steatosis: influence on the MR viscoelastic and diffusion parameters in patients with chronic liver disease, Radiology 283 (2016) 98–1107. [DOI] [PubMed] [Google Scholar]
- [47].Ramadori G, Moriconi F, Malik I, Dudas J, Physiology and pathophysiology of liver inflammation, damage and repair, J. Physiol. Pharm 59 (2008) 107–117. [PubMed] [Google Scholar]
- [48].Ernandez T, Mayadas TN, The changing landscape of renal inflammation, Trends Mol. Med 22 (2016) 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Poveda J, Sanz AB, Rayegomateos S, Ruizortega M, Carrasco S, Ortiz A, Sanchezniño MD, NFκBiz protein downregulation in acute kidney injury: modulation of inflammation and survival in tubular cells, BBA Mol. Basis Dis 1862 (2016) 635–646. [DOI] [PubMed] [Google Scholar]
- [50].Sanz A, Sanchez-Niño M, Ramos A, Moreno J, Santamaria B, Ruiz-Ortega M, Egido J, Ortiz A, NF-kappaB in renal inflammation, J. Am. Soc. Nephrol 21 (2010) 1254–1262. [DOI] [PubMed] [Google Scholar]
- [51].Serhan CN, Chiang N, Van Dyke TE, Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators, Nat. Rev. Immunol 8 (2008) 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Headland SE, Norling LV, The resolution of inflammation: principles and challenges, Semin Immunol. 27 (2015) 149–160. [DOI] [PubMed] [Google Scholar]
- [53].Grose R, Werner S, Kessler D, Tuckermann J, Huggel K, Durka S, Reichardt HM, Werner S, A role for endogenous glucocorticoids in wound repair, EMBO Rep. 3 (2002) 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bhala N, Emberson J, Merhi A, Abramson S, Arber N, et al. , Coxib and traditional NSAID Trialists’ (CNT) Collaboration, Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials, Lancet 382 (2013) 769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sarzi-Puttini P, Atzeni F, Shoenfeld Y, Ferraccioli G, TNF-alpha, rheumatoid arthritis, and heart failure: a rheumatological dilemma, Autoimmun. Rev 4 (2005) 153–161. [DOI] [PubMed] [Google Scholar]
- [56].Ali T, Kaitha S, Mahmood S, Ftesi A, Stone J, Bronze MS, Clinical use of anti-TNF therapy and increased risk of infections, Drug Health Patient Saf. 5 (2013) 79–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Serhan CN, Chiang N, Dalli J, Levy BD, Lipid mediators in the resolution of inflammation, Cold Spring Harb. Perspect. Biol 7 (2015), a016311, 10.1101/cshperspect.a016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Perretti M, D’Acquisto F, Annexin A1 and glucocorticoids as effectors of the resolution of inflammation, Nat. Rev. Immunol 9 (2009) 62–70. [DOI] [PubMed] [Google Scholar]
- [59].Wallace JL, Ianaro A, Flannigan KL, Cirino G, Gaseous mediators in resolution of inflammation, Semin. Immunol 27 (3) (2015) 227–233, 10.1016/j.smim.2015.05.004. [DOI] [PubMed] [Google Scholar]
- [60].Hasko G, Cronstein B, Regulation of inflammation by adenosine, Front. Immunol 4 (2013) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mirakaj V, Dalli J, Granja T, Rosenberger P, Serhan CN, Vagus nerve controls resolution and pro-resolving mediators of inflammation, J. Exp. Med 211 (2014) 1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Urade Y, Hayaishi O, Prostaglandin D synthase: structure and function, Vitam. Horm 58 (2000) 89–120. [DOI] [PubMed] [Google Scholar]
- [63].Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K, 15-deoxy-delta 12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes, J. Biol. Chem 277 (2002) 10459–10466. [DOI] [PubMed] [Google Scholar]
- [64].Bell-Parikh LC, Ide T, Lawson JA, McNamara P, Reilly M, FitzGerald GA, Biosynthesis of 15-deoxy-delta12,14-PGJ2 and the ligation of PPARgamma, J. Clin. Invest 112 (2003) 945–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jiang C, Ting AT, Seed B, PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines, Nature 391 (1998) 82–86. [DOI] [PubMed] [Google Scholar]
- [66].Petrova TV, Akama KT, Van Eldik LJ, Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14-prostaglandin J2, Proc. Natl. Acad. Sci. USA 96 (1999) 4668–4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Gosset P, Bureau F, Angeli V, Pichavant M, Faveeuw C, Tonnel AB, Trottein F, Prostaglandin D2 affects the maturation of human monocyte-derived dendritic cells: consequence on the polarization of naive Th cells, J. Immunol 170 (2003) 4943–4952. [DOI] [PubMed] [Google Scholar]
- [68].Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, Fitzgerald D, Yaqoob MM, Gilroy DW, Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyΔ12–14 PGJ2, Proc. Natl. Acad. Sci. USA 104 (2007) 20979–20984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chatterjee P, The cyclopentenone prostaglandin 15-deoxy-Delta(12,14)-prosta-glandin J(2) ameliorates ischemic acute renal failure, Cardiovasc. Res 61 (2004) 630–643. [DOI] [PubMed] [Google Scholar]
- [70].Cuzzocrea S, Ianaro A, Wayman NS, Mazzon E, Pisano B, Dugo L, Serraino I, Di Paola R, Chatterjee PK, Di Rosa M, Caputi AP, Thiemermann C, The cyclopentenone prostaglandin 15-deoxy-delta(12,14)-PGJ2 attenuates the development of colon injury caused by dinitrobenzene sulphonic acid in the rat, Br. J. Pharm 138 (2003) 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K, Kohno M, Yamada R, Hla T, Sano H, 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats, J. Clin. Invest 106 (2000) 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Matsuoka T, Prostaglandin D2 as a mediator of allergic asthma, Science 287 (2000) 2013–2017. [DOI] [PubMed] [Google Scholar]
- [73].Lee TS, Tsai HL, Chau LY, Induction of heme oxygenase-1 expression in murine macrophages is essential for the anti-inflammatory effect of low dose 15-deoxy-delta 12,14-prostaglandin J2, J. Biol. Chem 278 (2003) 19325–19330. [DOI] [PubMed] [Google Scholar]
- [74].Ryter SW, Otterbein LE, Morse D, Choi AM, Heme oxygenase/carbon monoxide signaling pathways: regulation and functional significance, Mol. Cell Biochem 234 (2002) 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G, Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention, Chem. Rev 111 (2011) 6130–6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Brash AR, Arachidonic acid as a bioactive molecule, J. Clin. Investig 107 (2001) 1339–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Philippe R, Urbach V, Specialized pro-resolving lipid mediators in cystic fibrosis, Int. J. Mol. Sci 19 (2018) 2865, 10.3390/ijms19102865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Nelson AJ, Stephenson DJ, Cardona CL, Lei X, Almutairi A, White TD, Tusing YG, Park MA, Barbour SE, Chalfant CE, Ramanadham S, Macrophage polarization is linked to Ca2+-independent phospholipase A2β-derived lipids and cross-cell signaling in mice, J. Lipid Res 61 (2020) 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Sala A, Folco G, Murphy RC, Transcellular biosynthesis of eicosanoids, Pharm. Rep 62 (2010) 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Mitchell S, Thomas G, Harvey K, Cottell D, Reville K, Berlasconi G, Petasis NA, Erwig L, Rees AJ, Savill J, Brady HR, Godson C, Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo, J. Am. Soc. Nephrol 13 (2002) 2497–2507. [DOI] [PubMed] [Google Scholar]
- [81].Papayianni A, Serhan CN, Brady HR, Lipoxin A4 and B4 inhibit leukotriene-stimulated interactions of human neutrophils and endothelial cells, J. Immunol 156 (1996) 2264–2272. [PubMed] [Google Scholar]
- [82].Payá M, Terencio MC, Ferrándiz ML, Alcaraz MJ, Involvement of secretory phospholipase A2 activity in the zymosan rat air pouch model of inflammation, Br. J. Pharm 117 (1996) 1773–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kohli P, Levy BD, Resolvins and protectins: mediating solutions to inflammation, Br. J. Pharm 158 (2009) 960–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K, Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing, J. Exp. Med 192 (2000) 1197–11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Li C, Wu X, Liu S, Shen D, Zhu J, Liu K, Role of resolvins in the inflammatory resolution of neurological diseases, Front Pharm. 11 (2020) 612, 10.3389/fphar.2020.00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Brezinski DA, Nesto RW, Serhan CN, Angioplasty triggers intracoronary leukotrienes and lipoxin A4. Impact of aspirin therapy, Circulation 86 (1992) 56–63. [DOI] [PubMed] [Google Scholar]
- [87].Van Dyke TE, Serhan CN, Resolution of inflammation: a new paradigm for the pathogenesis of periodontal diseases, J. Dent. Res 82 (2002) 82–90. [DOI] [PubMed] [Google Scholar]
- [88].Clària J, Titos E, Jiménez W, Ros J, Ginès P, Arroyo V, Rivera F, Rodés J, Altered biosynthesis of leukotrienes and lipoxins and host defense disorders in patients with cirrhosis and ascites, Gastroenterology 115 (1998) 147–156. [DOI] [PubMed] [Google Scholar]
- [89].Levy BD, De Sanctis GT, Devchand PR, Kim E, Ackerman K, Schmidt BA, Szczeklik W, Drazen JM, Serhan CN, Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4), Nat. Med 8 (2002) 1018–1023. [DOI] [PubMed] [Google Scholar]
- [90].Gagliardo R, Gras D, La Grutta S, Chanez P, Di Sano C, Albano GD, Vachier I, Montalbano AM, Anzalone G, Bonanno A, Riccobono L, Gjomarkaj M, Profita M, Airway lipoxin A4/formyl peptide receptor 2-lipoxin receptor levels in pediatric patients with severe asthma, J. Allergy Clin. Immunol 137 (2016) 1796–1806. [DOI] [PubMed] [Google Scholar]
- [91].Munger KA, Montero A, Fukunaga M, Uda S, Yura T, Imai E, Kaneda Y, Valdivielso JM, Badr KF, Transfection of rat kidney with human 15-lipoxygenase suppresses inflammation and preserves function in experimental glomerulonephritis, Proc. Natl. Acad. Sci. USA 96 (1999) 13375–13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Leonard MO, 15-Epi-16-(para-fluorophenoxy)-lipoxin A(4)-methyl ester, a synthetic analogue of 15-epi-lipoxin A(4), is protective in experimental ischemic acute renal failure, J. Am. Soc. Nephrol 13 (2002) 1657–1662. [DOI] [PubMed] [Google Scholar]
- [93].Schottelius AJ, Giesen C, Asadullah K, Fierro IM, Colgan SP, Bauman J, Guilford W, Perez HD, Parkinson JF, An aspirin-triggered lipoxin A4 stable analog displays a unique topical anti-inflammatory profile, J. Immunol 169 (2002) 7063–7070. [DOI] [PubMed] [Google Scholar]
- [94].Souza MH, de Lima OM, Zamuner SR, Fiorucci S, Wallace JL, Gastritis increases resistance to aspirin-induced mucosal injury via COX-2-mediated lipoxin synthesis, Am. J. Physiol. Gastrointest. Liver Physiol 285 (2003) G54–G61. [DOI] [PubMed] [Google Scholar]
- [95].Haworth O, Cernadas M, Yang R, Serhan CN, Levy BD, Resolvin E1 regulates interleukin 23, interferon-gamma and lipoxin A4 to promote the resolution of allergic airway inflammation, Nat. Immunol 9 (2008) 873–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Fiorucci S, Wallace JL, Mencarelli A, Distrutti E, Rizzo G, Farneti S, Morelli A, Tseng JL, Suramanyam B, Guilford WJ, Parkinson JF, A beta-oxidation-resistant lipoxin A4 analog treats hapten-induced colitis by attenuating inflammation and immune dysfunction, Proc. Natl. Acad. Sci. USA 101 (2004) 15736–15741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, Blumberg RS, Serhan CN, Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis, Proc. Natl. Acad. Sci. USA 102 (2005) 7671–7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Souza DG, Fagundes CT, Amaral FA, Cisalpino D, Sousa LP, Vieira AT, Pinho V, Nicoli JR, Vieira LQ, Fierro IM, Teixeira MM, The required role of endogenously produced lipoxin A4 and annexin-1 for the production of IL-10 and inflammatory hyporesponsiveness in mice, J. Immunol 179 (2007) 8533–8543. [DOI] [PubMed] [Google Scholar]
- [99].Castrillo A, Diaz-Guerra MJ, Hortelano S, Martin-Sanz P, Bosca L, Inhibition of IkappaB kinase and IkappaB phosphorylation by 15-deoxy-delta(12,14)-pros-taglandin J(2) in activated murine macrophages, Mol. Cell Biol 20 (2000) 1692–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Khoshnan A, Tindell C, Laux I, Bae D, Bennett B, Nel AE, The NF-kappa B cascade is important in Bcl-xL expression and for the anti-apoptotic effects of the CD28 receptor in primary human CD4+ lymphocytes, J. Immunol 165 (2000) 1743–1754. [DOI] [PubMed] [Google Scholar]
- [101].Prasad R, Giri S, Singh AK, Singh I, 15-deoxy-delta12,14-prostaglandin J2 attenuates endothelial-monocyte interaction: implication for inflammatory diseases, J. Inflamm 5 (2008) 14, 10.1186/1476-9255-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Lee RC, Feinbaum RL, Ambros V, The C elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14, Cell 75 (1993) 843–854. [DOI] [PubMed] [Google Scholar]
- [103].Wightman B, Ha I, Ruvkun G, Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans, Cell 75 (1993) 855–862. [DOI] [PubMed] [Google Scholar]
- [104].Kozomara A, Griffiths-Jones S, miRBase: annotating high confidence microRNAs using deep sequencing data, Nucl. Acids Res 42 (2014) D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Alivernini S, Gremese E, McSharry C, Tolusso B, Ferraccioli G, McInnes IB, Kurowska-Stolarska M, MicroRNA-155-at the critical interface of innate and adaptive immunity in arthritis, Front. Immunol 8 (2018) 1932, 10.3389/fimmu.2017.01932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].O’Connell RM, Rao DS, Baltimore D, microRNA regulation of inflammatory responses, Annu Rev. Immunol 30 (2012) 295–312. [DOI] [PubMed] [Google Scholar]
- [107].Self-Fordham JB, Naqvi AR, Uttamani JR, Kulkarni V, Nares S, MicroRNA: dynamic regulators of macrophage polarization and plasticity, Front. Immunol 8 (2017) 1062, 10.3389/fimmu.2017.01062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Mann M, Mehta A, Zhao JL, Lee K, Marinov GK, Garcia-Flores Y, Lu LF, Rudensky AY, Baltimore D, Author correction: an NF-κB-microRNA regulatory network tunes macrophage inflammatory responses, Nat. Commun 9 (1) (2018) 3338, 10.1038/s41467-018-05720-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Lu X, Yu Y, Tan S, The role of the miR-21–5p-mediated inflammatory pathway in ulcerative colitis, Exp. Ther. Med 19 (2020) 981–989, 10.3892/etm.2019.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Luly FR, Lévêque M, Licursi V, Cimino G, Martin-Chouly C, Théret N, Negri R, Cavinato L, Ascenzioni F, Del Porto P, MiR-146a is over-expressed and controls IL-6 production in cystic fibrosis macrophages, Sci. Rep 9 (2019) 16259, 10.1038/s41598-019-52770-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Yuan X, Berg N, Lee JW, Le TT, Neudecker V, Jing N, Eltzschig H, MicroRNA miR-223 as regulator of innate immunity, J. Leukoc. Biol 104 (2018) 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Gebert LFR, MacRae IJ, Regulation of microRNA function in animals, Nat. Rev. Mol. Cell Biol 20 (2019) 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Yang Shiuan Jr., Lai Eric C., Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants, Mol. Cell 43 (2011) 892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Fabian MR, Sonenberg N, Filipowicz W, Regulation of mRNA translation and stability by microRNAs, Annu. Rev. Biochem 79 (2010) 351–379. [DOI] [PubMed] [Google Scholar]
- [115].Davis BN, Hilyard AC, Lagna G, Hata A, SMAD proteins control DROSHA-mediated microRNA maturation, Nature 454 (2008) 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Yang W, Chendrimada TP, Wang Q, Higuchi M, Seeburg PH, Shiekhattar R, Nishikura K, Modulation of microRNA processing and expression through RNA editing by ADAR deaminases, Nat. Struct. Mol. Biol 13 (2006) 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, Fabbri M, Alder H, Liu CG, Calin GA, Croce CM, Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock, J. Immunol 179 (8) (2007) 5082–5089. [DOI] [PubMed] [Google Scholar]
- [118].Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M, Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to pro-inflammatory signals, Proc. Natl. Acad. Sci. USA 106 (2009) 5282–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Jurkin J, Schichl YM, Koeffel R, Bauer T, Richter S, Konradi S, Gesslbauer B, Strobl H, miR-146a is differentially expressed by myeloid dendritic cell subsets and desensitizes cells to TLR2-dependent activation, J. Immunol 184 (2010) 4955–4965. [DOI] [PubMed] [Google Scholar]
- [120].O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D, MicroRNA-155 is induced during the macrophage inflammatory response, Proc. Natl. Acad. Sci. USA 104 (2007) 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A, Requirement of bic/microRNA-155 for normal immune function, Science 316 (2007) 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Smith KG, Rada C, Enright AJ, Toellner KM, Maclennan IC, Turner M, MicroRNA-155 regulates the generation of immunoglobulin class-switched plasma cells, Immunity 27 (2007) 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M, Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals, Proc. Natl. Acad. Sci. USA 106 (2009) 5282–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Carballo E, Lai WS, Blackshear PJ, Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin, Science 281 (1998) 1001–1005. [DOI] [PubMed] [Google Scholar]
- [125].Fontana L, Pelosi E, Greco P, Racanicchi S, Testa U, Liuzzi F, Croce CM, Brunetti E, Grignani F, Peschle C, MicroRNAs 17–5p–20a–106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation, Nat. Cell Biol 9 (2007) 775–787. [DOI] [PubMed] [Google Scholar]
- [126].Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, O’Neill LAJ, Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21, Nat. Immunol 11 (2010) 141–147. [DOI] [PubMed] [Google Scholar]
- [127].Portius D, Sobolewski C, Foti M, MicroRNAs-dependent regulation of PPARs in metabolic diseases and cancers, PPAR Res. 2017 (2017) 1–19, 10.1155/2017/7058424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].He X, Jing Z, Cheng G, MicroRNAs: new regulators of toll-like receptor signalling pathways, BioMed. Res. Int 2014 (2014) 1–14, 10.1155/2014/945169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Sharma A, Kumar M, Aich J, Hariharan M, Brahmachari SK, Agrawal A, Ghosh B, Posttranscriptional regulation of interleukin-10 expression by hsa-miR-106a, Proc. Natl. Acad. Sci. USA 106 (2009) 5761–5766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Ma H, Wang X, Ha T, Gao M, Liu L, Wang R, Yu K, Kalbfleisch JH, Kao RL, Williams DL, Li C, MicroRNA-125b prevents cardiac dysfunction in polymicrobial sepsis by targeting TRAF6-mediated nuclear factor κB activation and p53-mediated apoptotic signaling, J. Infect. Dis 214 (2016) 1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, Hirst M, Hogge D, Marra M, Wells RA, Buckstein R, Lam W, Humphries RK, Karsan A, Identification of miR-145 and miR-146a as mediators of the 5q-syndrome phenotype, Nat. Med 16 (2010) 49–58. [DOI] [PubMed] [Google Scholar]
- [132].Nahid MA, Benso LM, Shin JD, Mehmet H, Hicks A, Ramadas RA, TLR4, TLR7/8 agonist-induced miR-146a promotes macrophage tolerance to MyD88-dependent TLR agonists, J. Leukoc. Biol 100 (2016) 339–349. [DOI] [PubMed] [Google Scholar]
- [133].Taganov KD, Boldin MP, Chang KJ, Baltimore D, NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses, Proc. Natl. Acad. Sci. USA 103 (2006) 12481–12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I, Leiser A, Schwartz PE, Rutherford T, Mor G, Regulation of IKKβ by miR-199a affects NF-κB activity in ovarian cancer cells, Oncogene 27 (2008) 4712–4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].El Gazzar M, McCall CE, MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance, J. Biol. Chem 285 (2010) 20940–20951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].He X, Jing Z, Cheng G, MicroRNAs: new regulators of Toll-like receptor signalling pathways, BioMed. Res. Int 2014 (2014) 1–14, 10.1155/2014/945169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Muxel SM, Acuña SM, Aoki JI, Zampieri RA, Floeter-Winter LM, Toll-like receptor and miRNA-let-7e expression alter the inflammatory response in Leishmania amazonensis-infected macrophages, Front. Immunol 9 (2018) 2792, 10.3389/fimmu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Li G, Yu M, Lee WW, Tsang M, Krishnan E, Weyand CM, Goronzy JJ, Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity, Nat. Med 18 (10) (2012) 1518–1524, 10.1038/nm.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Wang D, Tang M, Zong P, Liu H, Zhang T, Liu Y, Zhao Y, MiRNA-155 regulates the Th17/Treg ratio by targeting SOCS1 in severe acute pancreatitis, Front. Physiol 9 (9) (2018) 686, 10.3389/fphys.2018.00686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY, Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses, Cell 142 (2010) 914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Xiao C, Srinivasan L, Calado DP, Patterson HC, Zhang B, Wang J, Henderson JM, Kutok JL, Rajewsky K, Lymphoproliferative disease and autoimmunity in mice with increased miR-17–92 expression in lymphocytes, Nat. Immunol 9 (2008) 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, Li Z, Wu Z, Pei G, MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis, Nat. Immunol 10 (2009) 1252–1259. [DOI] [PubMed] [Google Scholar]
- [143].Chen CZ, Li L, Lodish HF, Bartel DP, MicroRNAs modulate hematopoietic lineage differentiation, Science 303 (2004) 83–86. [DOI] [PubMed] [Google Scholar]
- [144].Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K, MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb, Cell 131 (2007) 146–159. [DOI] [PubMed] [Google Scholar]
- [145].Rao DS, O’Connell RM, Chaudhuri AA, Garcia-Flores Y, Geiger TL, Baltimore D, MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1, Immunity 33 (1) (2010) 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Vigorito E, Perks KL, Abreu-Goodger C, Bunting S, Xiang Z, Kohlhaas S, Das PP, Miska EA, Rodriguez A, Bradley A, Smith KGC, Rada C, Enright AJ, Toellner KM, MacLennan ICM, Turner M, microRNA-155 regulates the generation of immunoglobulin class-switched plasma cells, Immunity 27 (2007) 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Glaesener S, Jaenke C, Habener A, Geffers R, Hagendorff P, Witzlau K, Imelmann E, Krueger A, Meyer-Bahlburg A, Decreased production of class-switched antibodies in neonatal B cells is associated with increased expression of miR-181b, PLoS One 13 (2) (2018), e0192230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Roy S, Sen CK, miRNA in wound inflammation and angiogenesis, Microcirculation 19 (2012) 224–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Mirra P, Raciti GA, Nigro C, Fiory F, D’Esposito V, Formisano P, Beguinot F, Miele C, Circulating miRNAs as intercellular messengers, potential biomarkers and therapeutic targets for type 2 diabetes, Epigenomics 7 (2015) 653–667. [DOI] [PubMed] [Google Scholar]
- [150].Recchiuti A, Krishnamoorthy S, Fredman G, Chiang N, Serhan CN, MicroRNAs in resolution of acute inflammation: identification of novel resolvin D1-miRNA circuits, FASEB J 25 (2011) 544–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Sethupathy P, Borel C, Gagnebin M, Grant GR, Deutsch S, Elton TS, Hatzigeorgiou AG, Antonarakis SE, Human microRNA-155 on chromosome 21 differentially interacts with its polymorphic target in the AGTR1 3’ untranslated region: a mechanism for functional single-nucleotide polymorphisms related to phenotypes, Am. J. Hum. Genet 81 (2007) 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].McCoy CE, Sheedy FJ, Qualls JE, Doyle SL, Quinn SR, Murray PJ, O’Neill LA, IL-10 inhibits miR-155 induction by toll-like receptors, J. Biol. Chem 285 (2010) 20492–204928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D, Inositol phosphatase SHIP1 is a primary target of miR-155, Proc. Natl. Acad. Sci. USA 28 (2009) 7113–7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, Papavasiliou FN, MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase, Immunity 28 (2008) 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Tili E, Michaille JJ, Adair B, Alder H, Limagne E, Taccioli C, Ferracin M, Delmas D, Latruffe N, Croce CM, Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD, Carcinogenesis 31 (2010) 1561–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Ma F, Liu F, Ding L, You M, Yue H, Zhou Y, Hou Y, Anti-inflammatory effects of curcumin are associated with down regulating microRNA-155 in LPS-treated macrophages and mice, Pharm. Biol 55 (2017) 1263–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Arango D, Diosa-Toro M, Rojas-Hernandez LS, Cooperstone JL, Schwartz SJ, Mo X, Jiang J, Schmittgen TD, Doseff AI, Dietary apigenin reduces LPS-induced expression of miR-155 restoring immune balance during inflammation, Mol. Nutr. Food Res 59 (2015) 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Boesch-Saadatmandi C, Loboda A, Wagner AE, Stachurska A, Jozkowicz A, Dulak J, Döring F, Wolffram S, Rimbach G, Effect of quercetin and its metabolites isorhamnetin and quercetin-3-glucuronide on inflammatory gene expression: role of miR-155, J. Nutr. Biochem 22 (2011) 293–299. [DOI] [PubMed] [Google Scholar]
- [159].Roessler C, Kuhlmann K, Hellwing C, Leimert A, Schumann J, Impact of polyunsaturated fatty acids on miRNA profiles of monocytes/macrophages and endothelial cells—a pilot study, Int. J. Mol. Sci 18 (2017) 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Xie Y, Shu R, Jiang S, Song Z, Guo Q, Dong J, Lin Z, miRNA-146 negatively regulates the production of pro-inflammatory cytokines via NF-κB signalling in human gingival fibroblasts, J. Inflamm 11 (2014) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, Garcia-Flores Y, Luong M, Devrekanli A, Xu J, Sun G, Tay J, Linsley PS, Baltimore D, miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice, J. Exp. Med 208 (2011) 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Mitsumura T, Ito Y, Chiba T, Matsushima T, Kurimoto R, Tanaka Y, Kato T, Uchida K, Ito T, Yamamoto K, Eishi Y, Kitagawa M, Miyazaki Y, Inase N, Asahara H, Ablation of miR-146b in mice causes hematopoietic malignancy, Blood Adv 2 (2018) 3483–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Bhaumik D, Scott GK, Schokrpur S, Patil CK, Campisi J, Benz CC, Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells, Oncogene 27 (42) (2008) 5643–5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Xia H, Qi Y, Ng SS, Chen X, Li D, Chen S, Ge R, Jiang S, Li G, Chen Y, He ML, Kung H, Lai L, Lin MC, microRNA-146b inhibits glioma cell migration and invasion by targeting MMPs, Brain Res 1269 (2009) 158–165. [DOI] [PubMed] [Google Scholar]
- [165].Cai J, Xu L, Cai Z, Wang J, Zhou B, Hu H, MicroRNA-146b-5p inhibits the growth of gallbladder carcinoma by targeting epidermal growth factor receptor, Mol. Med. Rep 12 (2015) 1549–1555. [DOI] [PubMed] [Google Scholar]
- [166].Wu PY, Zhang XD, Zhu J, Guo XY, Wang JF, Low expression of microRNA-146b-5p and microRNA-320d predicts poor outcome of large B-cell lymphoma treated with cyclophosphamide, doxorubicin, vincristine, and prednisone, Hum. Pathol 45 (2014) 1664–1673. [DOI] [PubMed] [Google Scholar]
- [167].Luly FR, Lévêque M, Licursi V, Cimino G, Martin-Chouly C, Théret N, Negri R, Cavinato L, Ascenzioni F, Del Porto P, MiR-146a is over-expressed and controls IL-6 production in cystic fibrosis macrophages, Sci. Rep 9 (2019) 16259, 10.1038/s41598-019-52770-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Cheng HS, Sivachandran N, Lau A, Boudreau E, Zhao JL, Baltimore D, Delgado-Olguin P, Cybulsky MI, Fish JE, MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways, EMBO Mol. Med 5 (2013) 1017–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Chatzikyriakidou A, Voulgari PV, Georgiou I, Drosos AA, A polymorphism in the 3′-UTR of interleukin-1 receptor-associated kinase (IRAK1), a target gene of miR-146a, is associated with rheumatoid arthritis susceptibility, Jt. Bone Spine 77 (2010) 411–413. [DOI] [PubMed] [Google Scholar]
- [170].Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, Huang X, Zhou H, de Vries N, Tak PP, Chen S, Shen N, MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins, Arthritis Rheum. 60 (2009) 1065–1075. [DOI] [PubMed] [Google Scholar]
- [171].Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’Leary JJ, Ruan Q, Johnson DS, Chen Y, O’Neill LAJ, Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21, Nat. Immunol 11 (2010) 141–147. [DOI] [PubMed] [Google Scholar]
- [172].Sheedy FJ, Turning 21: induction of miR-21 as a key switch in the inflammatory response, Front Immunol 6 (2015) 19, 10.3389/fimmu.2015.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Lu TX, Munitz A, Rothenberg ME, MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression, J. Immunol 182 (2009) 4994–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Das A, Ganesh K, Khanna S, Sen CK, Roy S, Engulfment of apoptotic cells by macrophages: a role of MicroRNA-21 in the resolution of wound inflammation, J. Immunol 192 (2014) 1120–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Chu Q, Yan X, Liu L, Xu T, The Inducible microRNA-21 negatively modulates the inflammatory response in teleost fish via targeting IRAK4, Front. Immunol 10 (2019) 1623, 10.3389/fimmu.2019.01623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Cekaite L, Clancy T, Sioud M, Increased miR-21 expression during human monocyte differentiation into DCs, Front. Biosci. (Elite Ed.) 2 (2010) 818–828, 10.2741/E143. [DOI] [PubMed] [Google Scholar]
- [177].Thapa DR, Bhatia K, Bream JH, D’Souza G, Rinaldo CR, Wolinsky S, Detels R, Martínez-Maza O, B-cell activation induced microRNA-21 is elevated in circulating B cells preceding the diagnosis of AIDS-related non-Hodgkin lymphomas, AIDS 26 (2012) 1177–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Wang T, Feng Y, Sun H, Zhang L, Hao L, Shi C, Wang J, Li R, Ran X, Su Y, Zou Z, miR-21 regulates skin wound healing by targeting multiple aspects of the healing process, Am. J. Pathol 181 (2012) 1911–1920. [DOI] [PubMed] [Google Scholar]
- [179].Jiang C, Guo Y, Yu H, Lu S, Meng L, Pleiotropic microRNA-21 in pulmonary remodeling: novel insights for molecular mechanism and present advancements, Allergy Asthma Clin. Immunol 15 (2019) 33, 10.1186/s13223-019-0345-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Sen CK, Roy S, MicroRNA 21 in tissue injury and inflammation, Cardiovasc. Res 96 (2) (2012) 230–233, 10.1093/cvr/cvs222. [DOI] [Google Scholar]
- [181].Chapnik E, Rivkin N, Mildner A, Beck G, Pasvolsky R, Metzl-Raz E, Birger Y, Amir G, Tirosh I, Porat Z, Israel LL, Lellouche E, Michaeli S, Lellouche JPM, Izraeli S, Jung S, Hornstein E, miR-142 orchestrates a network of actin cytoskeleton regulators during megakaryopoiesis, Elife 3 (2014), e01964, 10.7554/eLife.01964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Naqvi AR, Fordham JB, Nares S, miR-24, miR-30b, and miR-142–3p regulate phagocytosis in myeloid inflammatory cells, J. Immunol 194 (4) (2015) 1916–1927, 10.4049/jimmunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Berrien-Elliott MM, Sun Y, Neal C, Ireland A, Trissal MC, Sullivan RP, Wagner JA, Leong JW, Wong P, Mah-Som AY, Wong TN, Schappe T, Keppel CR, Cortez VS, Stamatiades EG, Li MO, Colonna M, Link DC, French AR, Cooper MA, Wang WL, Boldin MP, Reddy P, Fehniger TA, MicroRNA-142 is critical for the homeostasis and function of Type 1 innate lymphoid cells, Immunity 51 (3) (2019) 479–490.e6, 10.1016/j.immuni.2019.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Fordham JB, Naqvi AR, Nares S, Regulation of miR-24, miR-30b, and miR-142–3p during macrophage and dendritic cell differentiation potentiates innate immunity, J. Leukoc. Biol 98 (2) (2015) 195–207, 10.1189/jlb.1A1014-519RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Naqvi AR, Fordham JB, Ganesh B, Nares S, miR-24, miR-30b and miR-142–3p interfere with antigen processing and presentation by primary macrophages and dendritic cells, Sci. Rep 6 (2016) 32925, 10.1038/srep32925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Anandagoda N, Willis JCD, Hertweck A, Roberts LB, Jackson I, Gökmen MR, Jenner RG, Howard JK, Lord GM, microRNA-142-mediated repression of phosphodiesterase 3B critically regulates peripheral immune tolerance, J. Clin. Invest 129 (3) (2019) 1257–1271, 10.1172/JCI124725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Sonda N, Simonato F, Peranzoni E, Calì B, Bortoluzzi S, Bisognin A, Wang E, Marincola FM, Naldini L, Gentner B, Trautwein C, Sackett SD, Zanovello P, Molon B, Bronte V, miR-142–3p prevents macrophage differentiation during cancer-induced myelopoiesis, Immunity 38 (6) (2013) 1236–1249, 10.1016/j.immuni.2013.06.004. [DOI] [PubMed] [Google Scholar]
- [188].Valverde A, Nares S, Naqvi AR, Impaired cell migration and structural defects in myeloid cells overexpressing miR-30b and miR-142–3p, Biochim. Biophys. Acta Gene Regul. Mech 1863 (11) (2020), 194628, 10.1016/j.bbagrm.2020.194628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Mandolesi G, De Vito F, Musella A, Gentile A, Bullitta S, Fresegna D, Sepman H, Di Sanza C, Haji N, Mori F, Buttari F, Perlas E, Ciotti MT, Hornstein E, Bozzoni I, Presutti C, Centonze D, miR-142–3p is a key regulator of IL-1β-dependent synaptopathy in neuroinflammation, J. Neurosci 37 (3) (2017) 546–561, 10.1523/JNEUROSCI.0851-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Ding S, Liang Y, Zhao M, Liang G, Long H, Zhao S, Wang Y, Yin H, Zhang P, Zhang Q, Lu Q, Decreased microRNA-142–3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus, Arthritis Rheum 64 (9) (2012) 2953–2963, 10.1002/art.34505. [DOI] [PubMed] [Google Scholar]
- [191].Naqvi AR, Brambila MF, Martínez G, Chapa G, Nares S, Dysregulation of human miRNAs and increased prevalence of HHV miRNAs in obese periodontitis subjects, J. Clin. Periodontol 46 (1) (2019) 51–61, 10.1111/jcpe.13040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Cortes-Troncoso J, Jang SI, Perez P, Hidalgo J, Ikeuchi T, Greenwell-Wild T, Warner BM, Moutsopoulos NM, Alevizos I, T cell exosome–derived miR-142–3p impairs glandular cell function in Sjögren’s syndrome, JCI Insight 5 (9) (2020), e133497, 10.1172/jci.insight.133497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Nathan C, Neutrophils and immunity: challenges and opportunities, Nat. Rev. Immunol 6 (2006) 173–182. [DOI] [PubMed] [Google Scholar]
- [194].Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD, Diabetes primes neutrophils to undergo NETosis, which impairs wound healing, Nat. Med 21 (2015) 815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Shaw TJ, Martin P, Wound repair: a showcase for cell plasticity and migration, Curr. Opin. Cell Biol 42 (2016) 29–37. [DOI] [PubMed] [Google Scholar]
- [196].Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA, Myofibroblasts and mechano-regulation of connective tissue remodelling, Nat. Rev. Mol. Cell Biol 3 (2002) 349–363. [DOI] [PubMed] [Google Scholar]
- [197].Yang LL, Liu JQ, Bai XZ, Fan L, Han F, Jia WB, Su LL, Shi JH, Tang CW, Hu DH, Acute downregulation of miR-155 at wound sites leads to a reduced fibrosis through attenuating inflammatory response, Biochem. Biophys. Res. Commun 453 (2014) 153–159. [DOI] [PubMed] [Google Scholar]
- [198].Yang X, Wang J, Guo SL, Fan KJ, Li J, Wang YL, Teng Y, Yang X, miR-21 promotes keratinocyte migration and re-epithelialization during wound healing, Int. J. Biol. Sci 7 (2011) 685–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Codagnone M, Cianci E, Lamolinara A, Mari VC, Nespoli A, Isopi E, Mattoscio D, Arita M, Bragonzi A, Iezzi M, Romano M, Recchiuti A, Resolvin D1 enhances the resolution of lung inflammation caused by long-term Pseudomonas aeruginosa infection, Mucosal Immunol. 11 (2018) 35–49. [DOI] [PubMed] [Google Scholar]
- [200].Winkler MA, Dib C, Ljubimov AV, Saghizadeh M, Targeting miR-146a to treat delayed wound healing inhuman diabetic organ-cultured corneas, PLoS One 9 (2014), e114692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Aunin E, Broadley D, Ahmed MI, Mardaryev AN, Botchkareva NV, Exploring a role for regulatory miRNAs in wound healing during ageing: involvement of miR-200c in wound repair, Sci. Rep 7 (2017) 3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Biswas S, Roy S, Banerjee J, Hussain SRA, Khanna S, Meenakshisundaram G, Kuppusamy P, Friedman A, Sen CK, Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds, Proc. Natl. Acad. Sci. USA 107 (2010) 6976–6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Banerjee J, Sen CK, MicroRNAs in skin and wound healing, Methods Mol. Biol 936 (2013) 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Li X, Li D, Wang A, Chu T, Lohcharoenkal W, Zheng X, Grünler J, Narayanan S, Eliasson S, Herter EK, Wang Y, Ma Y, Ehrström M, Eidsmo L, Kasper M, Pivarcsi A, Sonkoly E, Catrina SB, Ståhle M, Xu Landén N, MicroRNA-132 with therapeutic potential in chronic wounds, J. Invest. Dermatol 137 (2017) 2630–2638. [DOI] [PubMed] [Google Scholar]
- [205].Ozdemir D, Feinberg MW, MicroRNAs in diabetic wound healing: pathophysiology and therapeutic opportunities, Trends Cardiovasc. Med 29 (2019) 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Guo J, Lin Q, Shao Y, Rong L, Zhang D, miR-29b promotes skin wound healing and reduces excessive scar formation by inhibition of the TGF-β1/Smad/CTGF signaling pathway, Can. J. Physiol. Pharm 95 (2017) 437–442. [DOI] [PubMed] [Google Scholar]
- [207].Xie C, Shi K, Zhang X, Zhao J, Yu J, MiR-1908 promotes scar formation post-burn wound healing by suppressing Ski-mediated inflammation and fibroblast proliferation, Cell Tissue Res 366 (2016) 371–380. [DOI] [PubMed] [Google Scholar]
- [208].Li X, Li D, Wang A, Chu T, Lohcharoenkal W, Zheng X, Grünler J, Narayanan S, Eliasson S, Herter EK, Wang Y, Ma Y, Ehrström M, Eidsmo L, Kasper M, Pivarcsi A, Sonkoly E, Catrina SB, Ståhle M, Xu Landén N, MicroRNA-132 with therapeutic potential in chronic wounds, J. Invest. Dermatol 137 (2017) 2630–2638. [DOI] [PubMed] [Google Scholar]
- [209].Tanaka K, Kim SE, Yano H, Matsumoto G, Ohuchida R, Ishikura Y, Araki M, Araki K, Park S, Komatsu T, Hayashi H, Ikematsu K, Tanaka K, Hirano A, Martin P, Shimokawa I, Mori R, MiR-142 is required for staphylococcus aureus clearance at skin wound sites via small GTPase-mediated regulation of the neutrophil actin cytoskeleton, J. Invest. Dermatol 137 (2017) 931–940. [DOI] [PubMed] [Google Scholar]
- [210].Liu J, Luo C, Yin Z, Li P, Wang S, Chen J, He Q, Zhou J, Downregulation of let-7b promotes COL1A1 and COL1A2 expression in dermis and skin fibroblasts during heat wound repair, Mol. Med. Rep 13 (2016) 2683–2688. [DOI] [PubMed] [Google Scholar]
- [211].Sundaram GM, Common JEA, Gopal FE, Srikanta S, Lakshman K, Lunny DP, Lim TC, Tanavde V, Lane EB, Sampath P, ‘See-saw’ expression of microRNA-198 and FSTL1 from a single transcript in wound healing, Nature 495 (2013) 103–106. [DOI] [PubMed] [Google Scholar]
- [212].Wang J, Dan G, Shangguan T, Hao H, Tang R, Peng K, Zhao J, Sun H, Zou Z, miR-198 represses the proliferation of HaCaT cells by targeting cyclin D2, Int. J. Mol. Sci 16 (2015) 17018–17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Li H, Han X, Zuo K, Li L, Liu J, Yuan X, Shen Y, Shao M, Pang D, Chu Y, Zhao B, miR-23b promotes cutaneous wound healing through inhibition of the inflammatory responses by targeting ASK1, Acta Biochim. Biophys. Sin. (Shanghai) 50 (11) (2018) 1104–1113. [DOI] [PubMed] [Google Scholar]
- [214].Bertero T, Gastaldi C, Bourget-Ponzio I, Imbert V, Loubat A, Selva E, Busca R, Mari B, Hofman P, Barbry P, Meneguzzi G, Ponzio G, Rezzonico R, miR-483–3p controls proliferation in wounded epithelial cells, FASEB J. 25 (2011) 3092–3105. [DOI] [PubMed] [Google Scholar]