

Abstract
Background: Gene therapy cannot be yet considered a far perspective, but a tangible therapeutic option in the field of retinal diseases. Although still confined in experimental settings, the preliminary results are promising and provide an overall scenario suggesting that we are not so far from the application of gene therapy in clinical settings. The main aim of this review is to provide a complete and updated overview of the current state of the art and of the future perspectives of gene therapy applied on retinal diseases.
Methods: We carefully revised the entire literature to report all the relevant findings related to the experimental procedures and the future scenarios of gene therapy applied in retinal diseases. A clinical background and a detailed description of the genetic features of each retinal disease included are also reported.
Results: The current literature strongly support the hope of gene therapy options developed for retinal diseases. Although being considered in advanced stages of investigation for some retinal diseases, such as choroideremia (CHM), retinitis pigmentosa (RP), and Leber's congenital amaurosis (LCA), gene therapy is still quite far from a tangible application in clinical practice for other retinal diseases.
Conclusions: Gene therapy is an extremely promising therapeutic tool for retinal diseases. The experimental data reported in this review offer a strong hope that gene therapy will be effectively available in clinical practice in the next years.
Keywords: inherited retinal dystrophies, gene therapy, Stargardt disease, retinitis pigmentosa, choroideremia, X-linked retinoschisis
Introduction
Inherited retinal diseases (IRDs), also referred to as inherited retinal dystrophies, are a clinically and genetically heterogeneous group of neurodegenerative disorders, primarily involving photoreceptors, retinal pigment epithelium (RPE), and/or the choroid. Taken as a whole, IRDs have an estimated global prevalence of about 1 in 2,000 individuals, affecting more than two million people worldwide, and standing out as the leading cause of blindness in the Western working-age population (1).
Inherited Retinal Diseases are classified according to different criteria, including the primarily diseased retinal cell type (rod-dominated disease, cone-dominated disease, generalized retinal degenerations, and vitreoretinal disorders), the age of onset, the progression of visual impairment over time (stationary or progressive), and the presence or absence of associated systemic features (isolated or syndromic IRDs).
Since the identification of the first gene responsible for an IRD back in 1988 (2), enormous progress has been made in the field of molecular testing, leading to the identification of over 270 disease-causing genes (https://sph.uth.edu/retnet/sum-dis.htm).
Nevertheless, until very recent times, these major diagnostic advances did not go hand in hand with the development of vision-sparing or vision-restoring therapeutic strategies and IRDs have been long accounted as largely incurable diseases.
Over the last decades, this view has changed, as novel therapeutic options started to be explored in preclinical studies, with some of them transitioning to the clinical setting, including gene therapy, cell therapy (3), retinal prosthetics (4), and even direct brain stimulation (5).
In this context, gene-based therapies stand out as one of the most promising frontiers of IRD treatment and the introduction of voretigene neparvovec-rzyl (Luxturna), the first FDA- and EMA-approved gene therapy treatment, paved the way for further research.
The first section of this review is aimed at making the reader familiar with the basic concepts and nomenclature used in the field in retinal gene therapy, while the second section explores in detail those IRDs for which gene-based therapy approaches have made it to the human trial stage. Both sections adopt a combined descriptive and analytic approach, in order to provide a broad overview of the state of the art of gene therapy in IRDs, including discussion of current obstacles and research gaps, as well as a description of the most promising strategies that are being developed to overcome these obstacles and to fill these gaps.
Methods
We searched all English language and human subject articles using keywords search of MEDLINE library. The keywords included the following: Inherited retinal dystrophies; gene therapy; Stargardt disease (STGD); Retinitis pigmentosa (RP); chorioderemia; X-linked retinoschisis (XLRS); Leber's congenital amaurosis (LCA). All the references were carefully examined by two expert researchers (Alessandro Arrigo, Alessia Amato) which collected and ordered all the relevant information, considering the main topic of this review as expressed in the manuscript title.
Section 1: Basics Concepts in Retinal Gene Therapy
Gene therapy is the treatment of a disease through genetic material (DNA or RNA), that is transferred into the cells of the patient in order to modify gene expression. Since 1990, when the first gene therapy trial was performed in two children with adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID) (6), this approach has been studied for and applied to a variety of inherited and acquired disorders, with more than 20 gene therapies officially approved for clinical uses by the drug regulatory agencies from different countries.
The Eye as an Ideal Target for Gene Therapy
Since the dawn of gene therapy, the human eye has always presented itself as an appealing target for a number of reasons.
First, owing to the presence of the so-called blood-retinal barrier (BRB), made up of the tight junctions between the endothelial cells of retinal microvasculature (i.e., inner BRB) and between RPE cells (i.e., outer BRB), the retina is an immune-privileged site, meaning that the introduction of foreign substances is less likely to cause an inflammatory reaction.
Second, the eye is a relatively small and enclosed compartment, which in turn has two important implications: lower doses of therapeutics are needed and the risk of systemic dissemination of the locally administered vector is generally negligible (which, again, minimizes the risk of immune responses).
Moreover, since they are paired organs, it is possible to treat one eye and use the fellow eye as an ideal control to assess the efficacy and safety of the treatment.
Finally, the eye is an easily accessible district, from both a surgical [via subretinal or intravitreal injections (IVIs)] and a diagnostic standpoint, so that non-invasive studies can be performed to monitor function and structure of the treated retinas.
Gene Delivery Systems
With regards to gene therapy, it is crucial to differentiate between ex vivo approaches, where patients' cells are collected, cultured, modified, and transplanted back to the same individual (7), and in vivo approaches, where a gene-therapy vector is directly administered to a living organism. Though some attempts are being made in the preclinical setting with transplant of gene corrected cells (8), ocular gene therapy relies on an in vivo approach, since the genetic material is administered directly into the patient's eye by means of a subretinal or IVI.
Another important distinction is in the way nucleic acids are delivered to their target cells. DNA and RNA are, in fact, large in size and negatively charged molecules, two features that hinder their ability to cross cell membranes. This obstacle is overcome by employing a variety of gene delivery systems, which can be divided into two main categories: non-viral and viral systems.
Viral Delivery Systems
Viruses are the most used vectors and the process by which they infect and release their genetic content into target cells is termed transduction. Several different recombinant viruses, that are replication deficient, can be used to deliver therapeutic nucleic acids, with differences in terms of cargo limits, integration capabilities, transduction efficiency, cellular tropism, and risk of immune responses.
Adenoviruses (Ads) are a family of DNA viruses that can infect quiescent and dividing cells and replicate in the host nucleus without integrating their genome. Adenoviruses have been largely tested as gene therapy vectors, mainly due to their cargo capacity (approximately 8–36 kb) and ability to transduce many cell types. As far as IRDs are concerned, however, conventional Adenoviral vectors (AVs), constructed by substituting the E1 region with the transgene cassette of interest (9), had limited success, owing to the expression of some viral genes in the infected target cells, which enhanced immunogenicity and undermined treatment longevity, even in the immune-privileged environment of the human eye. These issues have been partially addressed with second- and third-generation vectors, characterized by progressive stripping of all viral coding sequences and implementation of helper-dependent AVs. However, problems with contaminating helper viruses, vector instability, and replication-competent AVs have been reported (10, 11).
Adeno-Associated Viruses (AAVs) are defective single-strand (ss) DNA parvoviruses with more than 20 integration sites in the human genome. Recombinant AAVs (rAAVs) vectors are by far the most frequently used ones in gene therapy approaches for IRDs, because of their lack of pathogenicity, favorable immunologic profile (since, unlike AVs, they do not carry any virus open reading frame), non-integrating nature in the absence of rep protein (which minimizes the risk of insertional mutagenesis, unlike LVs), ability to provide a stable transgene expression and extended retinal tropism. To date, 13 naturally occurring serotypes of AAV have been isolated from primates (AAV1–AAV13): different serotypes have a different capsid conformation and different properties, especially as far as tropism is concerned. Moreover, AAVs can be modified in several ways, for example by packaging the viral genome bordering the transgene into the capsid of a different AAV serotype, process known as pseudotyping or cross-packaging (e.g., an AAV2/8 vector is a pseudotype in which the genome of AAV2 serotype is packaged into an AAV8 capsid) (12). Both serotype and pseudotype choice are important to optimize vector design for the target disease. To date, the serotypes and pseudotypes that have been used in clinical trials for IRDs include AAV2/5, AAV2/8, and AAV8. The major disadvantage of rAAV vectors is their limited cargo capacity, which cannot exceed 4.7 kb. Although with an apparently reduced photoreceptor transduction efficiency, dual AAV vectors—each of which contains half of a large transgene expression cassette—have been shown to improve retinal phenotype in murine models of IRDs (13–15).
Lentiviruses (LVs) are retroviruses with a larger packing capacity (8 kb), which makes them a compelling alternative to AAV vectors for those IRDs whose causative gene coding sequence exceeds the 4.7 kb limit, such as ABCA4-related Stargardt's disease and MYO7A-related Usher's syndrome type 1B. So far, the retroviral variant of human immunodeficiency virus type 1 (HIV-1) and the equine infectious anemia virus (EIAV) have been studied for IRDs. Lentivirus vectors have two main drawbacks. First, LVs are integrating in nature and genomic integration, if on the one hand leads to a sustained expression of the foreign DNA, on the other hand carries a risk of insertional mutagenesis (16, 17). Such risk may not be justified in the case of IRDs, since in post-mitotic tissues, like the retina, a stable transduction can be achieved even by lentiviral episomes. This limitation can be overcome by employing integration-deficient lentiviral vectors (IDLVs), which have been successfully used in a rodent model of retinal degeneration (18). Second, LVs are capable of effectively transducing RPE cells and only to a lesser extent, which is generally insufficient for therapeutic purposes, differentiated photoreceptors (19, 20).
Non-viral Delivery Systems
Non-viral delivery systems have some advantages over viral delivery systems, including potentially unlimited cargo capacity, simultaneous conveyance of multiple therapeutics, low immunogenicity, and inexpensive manufacturing procedures.
Non-viral delivery systems use physicochemical agents to compact the DNA and/or transport it across the membranes.
Physical methods include, for instance, sonoporation (21), and electroporation (22) (i.e., the use of ultrasound or electricity to temporarily increase cell permeability) and direct injection of DNA into target cells (23), respectively, offering no protection from enzymatic degradation of therapeutic nucleic acids.
Chemical agents, which protect the payload from the action of nucleases, include, among others, cationic liposomes, lipopolyplex, and nanoparticles (NPs) (24–27).
Though some preclinical gene therapy studies have successfully used non-viral DNA systems (28–30), these agents are hard to export to an in vivo clinical setting, mainly because of transiency of gene expression (31, 32), ultimately resulting in a relatively inefficient delivery (9).
Administration Routes
At least in part, the success of gene therapy approach depends on its administration route. So far, the ongoing clinical trials for IRDs have relied on two injection modalities (i.e., subretinal and intravitreal), both of which have their strengths and weaknesses.
Subretinal injection (SRI) is adopted in most clinical trials, since it allows for the administration of the vector in close proximity to the most common cell target site (i.e., RPE and photoreceptors). Moreover, SRI places the therapeutic material in a closed immune-privileged compartment, thus diminishing the risk of immune reactions. Of course, SRI is a delicate procedure, requiring a vitrectomy, retinotomy, and the development of a transitory iatrogenic neuroretinal displacement and it is potentially associated with a number of complications, including retinal tears, cataract progression, or retinal/choroidal hemorrhages. With respect to SRI, vitreoretinal subspecialists have reported the utility of in vivo real-time monitor of the surgical act through integrated optical coherence tomography (OCT) operating microscope (33).
Intravitreal injection is certainly less invasive and technically challenging and can be performed in a clinic setting, thereby extending the accessibility of gene-based therapies to larger populations. However, while adequately transducing inner retinal cells, such as retinal ganglion cells (RGCs), the intravitreally-administered vectors are far less effective on outer retinal layers, due to dilution in the vitreous cavity and to the inner limiting membrane (ILM) barrier, which is particularly thick in primates. Therefore, in order to compensate for these obstacles and observe a significant therapeutic effect on target cells, much higher doses would have to be injected in a non-immune-privileged site. This, in turn, brings about a significant risk of immunogenicity, not only in the form of potential adverse inflammatory reactions, but also in the form of neutralizing antibodies, accounting for the need of repeated injections in intrinsically frail retinas.
Apart from these two main administration routes, a third one is currently being studied, that is suprachoroidal delivery, whereby therapeutics are conveyed in the space located between the sclera and the choroid. Though preclinical studies and clinical trials showed a good safety, comparable to that of IVIs, spreading of vectors into the systemic circulation is a potential risk (34–37). Finally, preclinical studies have attempted a sub- ILM approach (38).
Gene-Based Therapies in IRDS: the Strategies
Gene-based therapies can rely on different strategies, depending on the features, and molecular pathogenesis of the diseases being addressed, which can be schematically divided into two main categories:
Autosomal recessive (AR) or X-linked recessive (XLR) diseases; when a disease-causing mutation abrogates the normal gene function, it is defined as loss-of-function mutation. In this case, the mechanism underlying the associated disorder is caused by a loss of function, whereby a single copy of the gene (in case of AR inheritance) or the absence of functional alleles (in case of XLR inheritance in hemizygous males) is not sufficient to guarantee sufficient levels of the protein. The best way to address recessively inherited retinal dystrophies is by gene augmentation (or gene replacement).
Autosomal dominant (AD) diseases; these diseases are usually caused by gain-of-function mutations, whereby an aberrant protein is formed, resulting in disruption of cellular or tissue activity, or by a dominant negative effect, in which a defective subunit poisons a macromolecular complex. Gene augmentation alone is not enough to address AD IRDs, which require more sophisticated approaches, broadly classified as forms of gene silencing (or knockdown). Gene silencing can be associated with gene replacement, often by means of dual AAV vectors.
Gene Augmentation
Since its initial conceptualization back in the 1960s (39), the idea of gene therapy was based on the straightforward assumption that monogenic recessive disorders could be cured by replacing a faulty gene with a normal copy of it delivered through therapeutic vectors. This approach is called gene augmentation (or gene replacement) because the synthesis of the protein is augmented and its function, at least partially, restored. The gene of interest can be delivered as DNA or mRNA. Though having the advantage of not requiring delivery into the nucleus, thereby reducing the risk of integration into the host genome, the mRNA platform is far more immunogenic and less stable than the DNA platform, which is therefore the preferred one for ocular gene therapy (40). Most clinical gene therapy trials, as well as the first, and currently only, FDA- and EMA-approved treatment for an IRD, voretigene neparvovec-rzyl (Luxturna), rely on gene replacement, that does not require modification of native DNA and therefore is particularly compelling, owing to its simple design and relative ease of investigation.
Gene Silencing
Gene augmentation is an established approach for recessive monogenic disorders, but it is not suited to AD diseases resulting from pathological gain-of function mutations. In these cases, the therapeutic goal is to prevent the altered gene from expressing and encoding an aberrant protein that would interfere with normal cell function. To do so, it is possible to adopt several different approaches, which, schematically, can act at three levels: DNA, mRNA, and the intermediate process in between them (i.e., transcription).
In all of these cases, the host nucleic acids can be targeted in two ways, that is in a mutation-dependent fashion, whereby specific allele inhibition is sought in order to allow the expression of the wild-type copy of the gene, or in a mutation-independent fashion, in which a combined approach with gene augmentation is mandatory, since both copies of the gene (the mutated and the functional one) are silenced and replaced by a non-silenced wild-type form of it. Although allele-specific strategies do not disrupt the endogenous wild-type genome, allele-independent approaches are more far more practical since they don't have to be customized for specific disease-causing mutations. Allele-independent strategies, however, require the expression of both nucleic acid molecules in the same vector and are therefore limited by packaging issues.
DNA-Based Therapies (Genome Editing)
To date, the CRISPR/Cas9 system is considered the most advanced genome editing tool. This system consists of the Cas9 endonuclease, delivered into target cells in conjunction with a guide RNA (gRNA), which is able to cut the genome at any desired genomic location. The double-strand breaks (DSBs) created by the enzyme subsequently activate one of the DNA repair pathways: non-homologous end joining (NHEJ), homology-directed repair (HDR), or microhomology-mediated and joining (40). The development of the homology-independent targeted integration (HITI) strategy, that relies on NHEJ rather than HDR, enabled gene editing in the retina, since post-mitotic cells lack HDR, and was first used in a rat model of MERTK-related RP, with structural and functional improvements (41).
Other than for strict genome editing, the CRISPR/Cas9 can also be used as part of a gene silencing strategy to inactivate mutant alleles causing a toxic gain-of-function, or as part of a splicing modulation approach to prevent the inclusion of pseudoexons (i.e., deep-intronic sequences erroneously recognized as exons due to DNA mutations) that would result in the synthesis of an aberrant protein. The CRISPR/Cas9-based transcript degradation approach has been successfully used in studies on autosomal dominant retinitis pigmentosa (adRP) associated with rhodopsin mutations (RHO-adRP) (42–45), while the latter strategy of restoring splice defects has been applied to a deep intronic mutation of CEP290 in preclinical models (46).
mRNA Silencing
mRNA silencing strategies rely on a variety of antisense inhibitors (i.e., nucleic acid molecules that are complementary to and hybridize with protein-coding mRNAs) and function either by clearing mRNA molecules or by repressing their translation. mRNA silencing strategies include:
Ribozymes.
Ribozymes are RNA molecules able to catalyze a chemical reaction in the absence of proteins, which can be used to promote site-specific cleavage of a target phosphodiester bond in order to inhibit gene expression (47). Ribozymes have been the first RNA-based therapeutic strategy investigated in IRDs (48). This approach, however, has been largely abandoned and replaced by newer RNA-based technologies, mainly because the recognition sequence of these molecules are highly represented in the human genome, with a consequent risk of off-target effects, and because of the computational complexity of identifying mRNA cleavage sites (49).
Small Interfering RNAs.
RNA interference (RNAi) is another post-transcriptional gene silencing technology based on an evolutionary conserved pathway (50, 51). The effectors of RNAi, called small interfering RNAs (siRNAs), are 21–23 nucleotide-long double-stranded (ds) RNA molecules able to inhibit gene expression by binding to specific mRNAs. These siRNAs can be either naturally obtained in the presence of long pieces of dsRNA, which—for gene therapy purposes—are delivered by DNA vectors and cleaved by an RNase III enzyme called Dicer (52), or can be synthetically produced and directly introduced into the cells (51), the latter approach being less immunogenic, since long ds-RNAs can trigger an innate immune response (53). Unlike ribozymes, whose nucleolytic activity is independent of proteins, siRNAs do not directly take part to complementary mRNA degradation. Instead, siRNAs, once in the cytoplasm of the target cell, are incorporated in the so-called RNAi-induced silencing complex (RISC), which contains both the helicase that unwinds the ds-siRNA into its sense and antisense strands and the endonuclease Argonaute-2. The latter enzyme is in charge of clearing the sense strand of the siRNA molecule and the target mRNA sequence, the access to which is guided by the complementarity to the antisense strand (54). Just like with ribozymes, off-target effects are a major obstacle in the translation to the human clinics, since they can induce a toxic phenotype in target cells, especially in the presence of specific motifs (55); new computational methods able to screen candidate siRNAs can help overcome such obstacles (56). Another possible side effect is the elicitation of immune responses, which is more likely to occur when certain sequences are present (57). Both the above-described post-transcriptional silencing strategies (i.e., ribozymes and RNAi) have been successfully used in animal models of IRDs, with particular reference to adRP associated with rhodopsin mutations (58–61), providing proof-of-concept for RNA-based retinal gene therapies.
Antisense Oligonucleotides.
Antisense oligonucleotides (AONs) are small DNA or RNA molecules that can be designed complementary to their target mRNAs (62). Over the last years, AONs have been the object of increasing interest among retinal gene therapists, since they represent the only approach, other than gene augmentation, that has made it to the clinical setting (see section 2: Gene therapy in IRDS).
Depending on their chemical properties, AONs can display two distinct mechanisms.
First, they can act as authentic gene silencers, by mediating, similarly to siRNAs, degradation of target transcripts in a RNase H1-dependent fashion (63). This mechanism has been studied in vitro for a NR2E3 variant underlying adRP (64) and in vivo for RHO-adRP, both in the preclinical and clinical settings (65).
Other than for knockdown purposes, AONs can also be used as pre-mRNA splicing modulators, an interesting application in the field of IRDs, since up to 15% of all retinal degeneration-causing mutations affect the splicing machinery (66). In this context, AONs generally promote exon skipping, meaning that they bind to target pre-mRNA sequences, and block the recruitment of splicing factors. This approach proves particularly useful when exclusion of pseudoexons is sought, as in the case of CEP290-related LCA (see section 2: Gene therapy in IRDS), which at present stands out as the most advanced application of this technology, having reached phase III of clinical evaluation (NCT03913143). Though promising proof-of-concept studies have been developed for many other genes, such as OPA1, CHM and ABCA4 (67–72), the only other IRD-causing gene whereby the AON-based approach has been translated to the clinics is USH2A (73), for which—following the success of the phase 1/2 trial (NCT03913143)—two final stage registration trials are planned to start by the end of 2021.
Despite the unquestionable advantages of RNA therapeutics over other gene-based strategies, such as the titratability and affordability of the employed molecules, the reversibility of their effects, and the non-genome altering approach, some major challenges still lie ahead of this field. One of such challenges is related to the instability of naked nucleic acids, which are promptly degraded by endonucleases (74), resulting in a time-limited effect. To avoid enzymatic clearance of antisense molecules, two strategies can be adopted. The first strategy is chemical modification of the nucleic acids, in order to make them endonuclease-resistant, as it has been done with both siRNAs and with first-, second-, and third- AONs (75–80). The second approach is to package these therapeutic molecules inside vectors, either viral (81), thus requiring a SRI, or non-viral (82), allowing for repeated, though sufficiently long-lasting, IVIs. As a matter of fact, in a recent study on a mouse model of CEP290-LCA, IVI injection of second-generation AONs compared to SRI of AAV-packaged AONs, while exhibiting comparable duration of effect (approximately 1 month), turned out to be more effective (81). This is probably due to the fact that chemically modified AONs are more efficiently taken up by cells compared to AAVs, further underscoring the importance of developing IVI-based approaches.
Transcriptional Silencing
Therapeutic strategies for gain-of-function mutations mainly rely on DNA-based technologies, such as the CRISPR/Cas system, and on RNA silencing, thereby acting upstream and downstream of the transcription process.
Over the last decade, efforts have been made to target the intermediate step between DNA and RNA, that is the process by which genetic sequences are used as templates to assembly pre-mRNA transcripts.
In the field of IRDs, three different mutation-independent transcriptional repression strategies have been developed and successfully applied to preclinical models of RHO-adRP.
The first of these strategies employed zinc finger-based artificial transcription factors (ZF-ATFs) targeting the human rhodopsin promoter to achieve, in a mouse model of adRP, transcriptional silencing of both wild-type and mutated RHO alleles in a mutation-independent fashion, which was followed by gene replacement of the endogenous RHO copies (83).
The same group showed that in vivo AAV-mediated delivery of a modified version of a synthetic transcription factor (TF), uncoupled from its repressor domain and designed to bind a 20-bp DNA sequence motif (ZF6-cis sequence), could block RHO expression in living porcine retina without significant genome-wide transcript perturbations (84).
Based on these results, the authors went on to further unveil the potential of TF-based transcription silencing, this time by delivering to rods an ectopic TF (i.e., a TF which is normally not expressed by rods) with a DNA-binding preference for the ZF6-cis sequence photoreceptors of pigs, resulting in suppression of RHO with limited off-target effects in a mouse and porcine retinas (85).
Taken together, these data support the role of transcriptional silencing as a promising novel mode to treat gain-of-function mutations in autosomal dominantly inherited IRDs.
Non-targeted Gene Therapies
With over 270 genes associated with IRDs, developing mutation-specific or even gene-specific approaches becomes challenging. In this context, the role of non-targeted gene therapy is to provide alternative strategies, aimed at improving vision independently of the causative gene (86). Attempts have been made by delivering via AAV vectors molecules capable of prolonging photoreceptor cell survival, including neurotrophic factors, such as ciliary neurotrophic factor (CNTF), glial cell line-derived neurotrophic factor, basic fibroblast growth factor and rod-derived con viability factor, and anti-apoptotic agents, such as X-linked inhibitor of apoptosis (87–92). Other than CNF, whose efficacy was rather modest (93), none of these approaches have been tested in humans so far.
As an alternative mutation-independent strategy, optogenetics delivered as non-targeted gene therapy for advanced RP is also being tested. In this case, the assumption is that inducing expression of light sensitive opsins in bipolar or RGCs could activate the visual pathway even in the absence of viable photoreceptors (94–97). There are three ongoing clinical trials using optogenetics in RP patients, RST-001 (NCT02556736), GS030 (NCT03326336), and BS01 (NCT04278131), while one phase 1/2 clinical trial, vMCO-I, has been recently completed (NCT04919473).
Section 2: Gene Therapy in IRDS
Gene Therapy in Leber Congenital Amaurosis
Leber congenital amaurosis represents a group of IRDs, with an estimated prevalence of 2–3/100,000, characterized by four clinical milestones: severe and early visual impairment (usually occurring by the 6th week of life), sensory nystagmus (an indirect manifestation of the low fixation ability), amaurotic pupils (an expression of the poor retinal sensitivity from the retina to the brainstem), and non-recordable electroretinography (ERG) responses (98). Other frequent phenotypic features include high refractive errors, photophobia, nyctalopia, and the so-called oculodigital sign of Franceschetti, consisting of a repetitive, deep rubbing of the eyes. Association between LCA and keratoconus and cataracts, as well as with a wide range of systemic manifestations, including intellectual disability, olfactory dysfunction, stereotypical movements, and behaviors has also been reported (98–102). From a clinical standpoint, LCA exhibits an extremely heterogeneous phenotype, ranging from an essentially normal retina to variable degree of vessel attenuation, bone spicule pigmentation, pseudopapilledema, macular coloboma, salt and pepper pigmentation, yellow confluent peripheral spots, white retinal spots, preserved para-arteriolar RPE (PPRPE) and Coats reaction, with some gene-specific features (98–105).
A milder form of the same disease spectrum has been described using several different expressions, including early-onset severe retinal dystrophy (EOSRD), severe early childhood-onset retinal dystrophy (SECORD) and early-onset RP (106). Unlike LCA, which is present at birth or within the first weeks of life and is associated with nystagmus, poor pupillary responses and abolished ERGs, EOSRD/SECORD has a slightly later onset (after infancy but before 5 years of age) and is characterized by a better residual visual function and ERG responses (106). Nevertheless, large genotypic overlap exists between these two disease entities, though certain genes are more frequently associated with LCA and others with EOSRD/SECORD (106).
Genetic Features
Leber congenital amaurosis is mostly inherited in an AR fashion, though for some genes, like CRX, AD patterns have been reported (107). So far, more than 25 genes, overall accounting for at least 80% of all LCA cases, have been described, the most common of which are listed in Table 1 (106–126). Accordingly with the current literature, the most common LCA-causing genes are, in descending order, GUCY2D, CEP290, CRB1, RDH12, and RPE65 (1, 3, 4). LCA-associated genes encode proteins, whose functions can be divided into four main categories: phototransduction (e.g., GUCY2D), photoreceptor morphogenesis (e.g., CRB1 and CRX), retinoid cycle (e.g., RDH12 and RPE65), and ciliary transport processes (e.g., CEP290) (98).
Table 1.
Main genes associated with the onset of LCA.
| Gene | Protein | Function | Molecular weight (kDa) | Reference |
|---|---|---|---|---|
| IMPDH1 | Inosine 5′-monophosphate dehydrogenase 1 | Guanine synthesis | ~55 | (5) |
| CRX | Cone–rod homeobox | Photoreceptor morphogenesis | ~32 | (6) |
| CRB1 | Crumbs homolog 1 | Photoreceptor morphogenesis | ~154 | (7) |
| GDF6 | Growth differentiation factor 6 | Photoreceptor morphogenesis | ~14 | (8) |
| SPATA7 | Spermatogenesis-associated protein 7 | Photoreceptor ciliary transport | ~68 | (9) |
| LCA5 | Libercilin | Photoreceptor ciliary transport | ~80 | (10) |
| RPGRIP1 | Retinitis pigmentosa GTPase regulator-interacting protein 1 | Photoreceptor ciliary transport | ~147 | (11) |
| CEP290 | Tubby-like protein | Photoreceptor ciliary transport | ~290 | (12) |
| TULP1 | Tubby-like protein | Photoreceptor ciliary transport | ~70 | (13) |
| CLUAP1 | Clusterin associated protein 1 | Photoreceptor ciliary transport | ~48 | (14) |
| IQCB1 | Intraflagellar transport 140 chlamydomonas homolog protein | Photoreceptor ciliary transport | ~69 | (15) |
| IFT140 | Intraflagellar transport 140 chlamydomonas homolog protein | Photoreceptor ciliary transport | ~165 | (16) |
| ALMS1 | ALMS Protein | Photoreceptor ciliary transport | ~460 | (17) |
| GUCY2D | Guanylate cyclase-1 | Phototransduction | ~120 | (18) |
| AIPL1 | Aryl-hydrocarbon-interacting-protein-like 1 | Phototransduction/protein biosynthesis | ~43 | (19) |
| RD3 | Protein RD3 | Protein trafficking | ~70 | (20) |
| RPE65 | Retinoid isomerase | Retinoid cycle | ~65 | (21) |
| RDH12 | Retinol dehydrogenase 12 | Retinoid cycle | ~38 | (22) |
| LRAT | Lecithin:retinol acyl transferase | Retinoid cycle | ~25 | (23) |
Gene Therapy in RPE65-LCA
The RPE65 gene product plays a critical role in the retinoid cycle, so that RPE65 mutations affect visual function before photoreceptor structure. Therefore, in contrast with many other IRDs in which visual dysfunction results from rods and cones death, RPE65-LCA patients retain viable cells for years before significant degeneration becomes evident. This structure-function dissociation makes RPE65-related retinal dystrophies a particularly compelling target for gene replacement strategies.
Proof of principle for retinal gene therapy came from the pioneering studies conducted in the early 2000's on a peculiar canine model (127, 128). These preclinical studies employed a subset of Briard dogs with a homozygous 4-bp deletion in the RPE65 gene resulting in a premature stop codon, thereby appearing to be an excellent spontaneous model for human RPE65-related LCA (129). These studies reported, after a single SRI of AAV-mediated RE65, an improvement in blue light stimulated dark-adapted ERGs and cone flicker, pupillometry, and VEP in the injected eyes and in qualitative behavioral assessments in the treated dogs, which were stable 3 years after the procedure (127, 128). Further evidence of the efficacy of this approach came from the naturally Rpe65-mutated rd12 murine model and from the genetically built Rpe65−/− knockout mouse (130, 131). Following the success of animal studies, clinical trials were initiated in 2007 by groups from the UK and the US (132–134), culminating in the first FDA- and EMA-approved AAV-based retinal gene therapy drug, voretigene neparvovec-rzyl (Luxturna) (135). Follow-up studies revealed stable improvements in most patients, peaking at 6–12 months after injection (136–138), but observational trials aimed at evaluating the long-term effects of Luxturna are still ongoing (NCT03602820, NCT01208389).
Gene Therapy in CEP290-LCA
The protein encoded by the CEP290 gene localizes to the photoreceptor connecting cilium and, besides microtubule-associated transport across the cilium, is required for outer segment (OS) regeneration and phototrandusction. The most common CEP290 mutation is the so called IVS26 c.2991+1655A>G mutation (p.Cys998X), an adenine to guanine point mutation located within intron 26 creating a novel splice donor site, which results in the inclusion of a pseudoexon in the mRNA and in the consequent creation of a premature codon stop. This mutation has been addressed by means of two innovative approaches.
The first strategy relies on a CRISPR/Cas9 system, called EDIT-101, consisting of an AAV5 vector used to deliver the Sthaphylococcus aureus Cas9 and CEP290-specific gRNAs with no identified off-targets. EDIT-101, or a non-human primate (NHP) surrogate vector, were shown to restore normal splicing in vitro (in photoreceptor-containing retinal explants) and in vivo (in mice and NHPs) with no serious adverse events (139).
The second strategy exploits the AON technology to remove the 128-bp pseudoexon included in the IVS26-mutated CEP290 mRNA transcript. Preclinical evidence of the efficacy of the AON designed to restore IVS26 splicing defects, called QR-110, came from in vitro studies on LCA10 fibroblasts (140).
In the wake of these results, IVI of this oligonucleotide was successfully attempted in NHP s (141), finally reaching the clinical setting with a phase I/II trial showing vision improvement at 3 months with no complications in LCA type10 patients treated with multiple doses of intravitreal QR-110 (142). A phase II/III multiple-dose clinical trial is still ongoing and is aimed at evaluating efficacy, safety, tolerability, and systemic exposure of QR-110 administered via IVI in patients with LCA type 10 due to CEP290 c.2991+1655A>G mutation after 24 months of treatment (NCT03913143).
Gene Therapy in Other Forms of LCA
Although so far only two LCA-associated genes have made it to the human trial stage, for many other disease-causing genes, including GUCY2D, CRB1, and RDH12, preclinical studies are underway, showing promising results (143–148).
Gene Therapy in Retinitis Pigmentosa
The term RP refers to a heterogeneous group of IRDs, with variable phenotypes—ranging from mild nyctalopia to total blindness—and genotypes—with over 100 identified RP-causing genes. All inheritance patterns are possible, including AD, AR, or X-linked disorders, whereas maternal (mitochondrial) inheritance is very rare in RP (149). The estimated prevalence of this multiform condition is 1 in 3,000–7,000 individuals (149, 150). In the initial stages, rod photoreceptors degenerate, resulting in night blindness, with difficulty seeing in dim light and adapting to changes in light sensitivity, and in visual field (VF) constriction, starting from the mid-periphery and extending toward the center, eventually leading to complete loss of peripheral vision, the so-called “tunnel vision” (151). With disease progression, also cones become affected and visual acuity (VA) declines. From a clinical standpoint, the fundus appearance of RP features a typical triad, consisting of attenuated retinal vessels, intraretinal pigment deposits with a bone spicule configuration, and optic disc pallor (Figure 1).
Figure 1.
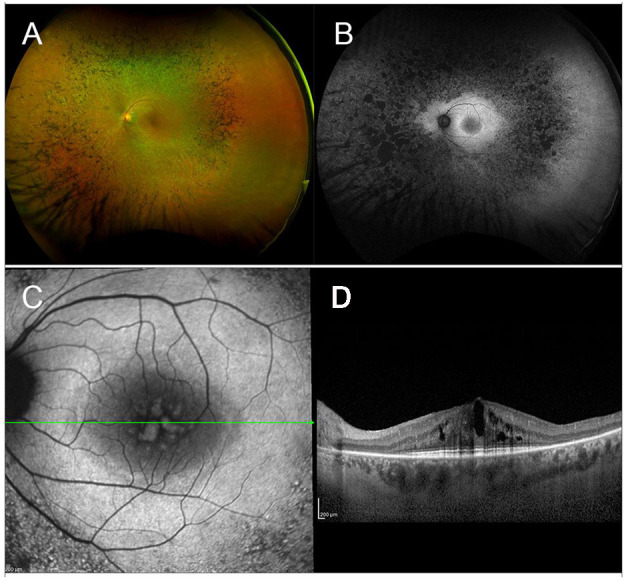
Multimodal imaging in RP. (A) Ultra-wide field retinography displays the typical triad: optic disc pallor, vessel attenuation, and bone spicule pigmentation. (B) Fundus autofluorescence shows the typical perifoveal ring of hyperautofluorescence and multiple hypoautofluorescent regions, corresponding to the pigment deposits, and to the areas of RPE atrophy. (C) Blue fundus autofluorescence shows a petaloid hyperautofluorescent pattern, compatible with a cystoid macular edema. (D) Structural OCT confirms the presence of intraretinal cysts.
Though ERG has been long used to help diagnose and monitor RP, nowadays multimodal imaging is of crucial importance for both initial assessment and follow-up of RP patients. Fundus autofluorescence (FAF), shows a coexistence of hypoautofluorescent regions (correlated to the masking effect of pigment deposits or to the presence of areas of RPE atrophy) and hyperautofluorescent regions (usually in the form of an hyperautofluorescent perifoveal ring) (152, 153). Optical Coherence Tomography shows decreased thickness of the outer nuclear layer (ONL) and loss of external limiting membrane (ELM) and ellipsoid zone (EZ), all of which were shown to correlate well with VF defects (154, 155). More recently, a novel imaging technique, optical coherence tomography angiography (OCTA), has been implemented to explore the existence and potential clinical relevance of different retinal and choroidal vascular patterns in RP patients (156, 157).
Considering the heterogeneity of RP, specific genetic features and currently ongoing clinical trials will be discussed separately for each relevant RP-associated gene.
RHO-RP
Genetic Features
The rhodopsin (RHO) gene was the first identified RP-causing gene (158, 159). Human RHO is a 6.7 kb-long DNA sequence, containing five exons and mapping on the long arm of chromosome 3 (3q22.1). It encodes rhodopsin, a 348-aa light-sensitive G-protein coupled receptor (GPCR) expressed from rod OSs disks. Rhodopsin is the protein that initiates the phototransduction cascade upon absorption of photons by its chromophore, 11-cis retinal. The vast majority of RHO mutations show an AD inheritance pattern (RHO-adRP), accounting for 25% of adRP cases and leading to RP with a toxic gain-of-function or a dominant-negative effect of the mutated protein (160). However, few recessively inherited mutations are described and have been reported to cause a milder phenotype (161).
Gene Therapy
As previously discussed (see Section 1: Basics concepts in retinal gene therapy), IRDs related to a toxic gain-of-function cannot be treated with a gene replacement approach. In these cases, in fact, there is a double therapeutic goal of silencing the mutant allele and increasing the wild type to mutant gene expression ratio. These goals can be achieved in an allele-specific or in a mutation-independent fashion, both of which have their pros and cons (see Section 1: Basics concepts in retinal gene therapy). In Section 1: Basics concepts in retinal gene therapy, we provided an overview of the current gene therapy strategies, and we described the three main targets of gene silencing approaches, all of which have been investigated in RHO-adRP, as reported in the excellent paper by Meng et al. (162).
As far as DNA-based therapies are concerned, the CRISPR/Cas9 system technology has been successfully applied to animal models and human retinal explants of RHO-adRP, both in an allele-specific (43–45) and in a mutation-independent way (42, 163).
Post-transcriptional RNA-based silencing strategies have perhaps been the most promising for RHO-adRP.
A dual vector short hairpin RNA (shRNA) suppression and replacement therapeutic strategy for RHO-adRP, named RHONova, proved to restore function and preserve morphology in a murine model of the disease independently of the mutation and received orphan drug designation in Europe and in the US, although there has been no publicly available updates on its clinical development (162–166). More recently, another RNAi-based mutation-independent strategy has been attempted, this time by means of a single AAV2/5 vector expressing both a shRNA targeting human RHO and a healthy copy of the gene, modified so as to be shRNA-resistant, with encouraging morpho-functional results (167). Further preclinical studies are currently being conducted on this gene therapy product candidate, now called IC-100, with a phase 1/2 clinical trial expected to begin by the end of 2021.
Antisense oligonucleotides have been used to promote allele-specific knock-down of P23H-mutant mRNA in a murine model of RHO-adRP, without affecting wild-type RHO expression. This approach yielded excellent preclinical results and transitioned to the clinical stage, with a phase 1/2 clinical trial currently ongoing and is scheduled to conclude in October 2021.
Finally, transcriptional repression strategies have also been attempted in the preclinical setting (83–85), as reported in Section 1: Basics concepts in retinal gene therapy of this review.
RPGR-XLRP
Genetic Features
X-linked retinitis pigmentosa (XLRP) is responsible for 5–20% of all RP cases. So far, three disease-causing genes have been identified: RP GTPase Regulator (RPGR) at the RP3 locus, retinitis pigmentosa 2 (RP2) at the RP2 locus and the oral-facial-digital syndrome type 1 (OFD1) (168–170).
The RPGR gene, whose mutations account for 70–90% of XLRP cases, encodes RPGR, a key protein in photoreceptor ciliary function. RPGR transcripts undergo a complex splicing process and generate constitutive variants, expressed by most tissues, and ORF15 variants, which are highly specific for the retina, by using alternative polyadenylation sites and splicing sites (171, 172). While mutations in the exons unique to the constitutive variant are almost exclusive of XLRP, mutations in the ORF15 exon, considered a mutational hot spot, are also found in cone dystrophy (COD) and cone-rod dystrophy (CORD) pedigrees (173).
X-linked retinitis pigmentosa is regarded as the most aggressive genetic subtype of RP, with hemizygous males exhibiting a particularly severe phenotype, characterized by early onset and rapid progression, eventually resulting in legal blindness by the end of the third decade of life. Heterozygous female carriers usually show some degree of fundus and FAF alterations, with an associated visual function that can range from 20/20 BCVA to no light perception (174–178). The variable extent of retinal involvement in female carriers could be explained by the dominant nature of some RPGR mutations or could be the result of a random skewed X inactivation phenomenon.
Gene Therapy
X-linked retinitis pigmentosa is the IRD with the highest number of ongoing gene therapy clinical trials, all of which rely on a AAV-mediated gene replacement strategy.
Before it was possible to transition to the clinics, RPGR canine models of XLRP (XLPRA1 and XLPRA2) treated with subretinal AAV2/5 full-length human RPGRex1-ORF15 provided preclinical evidence of the beneficial effects of this approach (179).
Moving on to the clinical setting, Nightstar Therapeutics/Biogen recently published the initial results at 6 months of its phase 1/2 dose escalation trial (NCT03116113) (180). Eighteen patients divided in six cohorts of three patients received increasing concentrations of AAV8.coRPGR vector (from 5 × 1,010 to 5 × 1,012 gp/ml) by means of a SRI. The primary outcome of the study was safety and initial results showed no significant concerns aside from subretinal inflammation in patients at the higher doses, that resolved after steroid treatment. Moreover, some secondary endpoints suggest sustained reversal of VF loss.
Another ongoing phase 1/2 trial, sponsored by MeiraGTx, is employing a SRI of an AAV2/5 vector as part of an open label, non-randomized, dose-escalation intervention followed by randomized dose confirmation against a control arm (NCT03252847). The same company recently initiated a phase 3 trial (NCT04671433).
Applied Genetic Technologies Corp. is sponsoring a phase 1/2 clinical trial (NCT03316560) to evaluate the safety and efficacy of SRI of rAAV2tYF-GRK1-RPGR and a phase 2/3 trial, which is scheduled to begin in the second half of 2021 (NCT04850118).
Finally, 4D Molecular Therapeutics launched the first clinical trial attempting to treat XLRP through a single intravitreal delivery of 4D-125, a drug product developed for gene therapy, which comprises an AAV capsid variant (4D-R100) carrying a codon-optimized human RPGPR transgene (NCT04517149).
PDE6B-RP
Genetic Features
PDE6B encodes the beta-subunit of the rod cGMP-phosphodiesterase, an enzyme that plays a key role during phototransduction. Mutations in PDE6B cause 2–5% of autosomal recessive retinitis pigmentosa (arRP) and rarely AD congenital stationary night blindness (CSNB) (181, 182).
Gene Therapy
After preclinical evidence that intraocular administration of the normal PDE6B gene preserved retinal morphology and functions in a mouse model of RP, a phase 1/2 clinical trial for subretinal administration of AAV2/5-hPDE6B was recently initiated (NCT03328130).
PDE6A-RP
Genetic Features
PDE6A encodes the alpha-subunit of the rod cGMP-phosphodiesterase. The loss of this enzyme function leads to chronically elevated cGMP levels, which cause an increased calcium inflow into the cell and thereby the hyperactivation of cell death pathways. Mutations in PDE6A cause 2–5% of arRP (183).
Gene Therapy
Patients with biallelic mutations of the PDE6A genes usually exhibit a mild to moderate phenotype, with an elevated degree of symmetry between the two eyes and with a relatively slow diseases course, though most patients have constricted VF by their fourth decade of life (184). Considering these features, PDE6A-related RP stands out as a compelling candidate for those gene therapy approaches requiring viable rods.
At the end of 2020, a phase 1/2 clinical trial sponsored by STZ eyetrial was commence and is currently open to enrolment (NCT04611503).
RLBP1-RP
Genetic Features
RLBP1 (also known as CRALBP) encodes cellular retinaldehyde-binding protein, which acts primarily as an acceptor of 11-cis retinal during the isomerization step of the visual cycle. Mutations in RLBP1 can cause three early-onset forms of arRP: retinitis punctata albescens, characterized by round punctate white deposits scattered throughout the entire retina in young patients with progression to more severe phenotypes in older individuals, Newfoundland rod-COD and Bothnia dystrophy, the latter two associated with a more severe prognosis. RLBP1 mutations can also cause fundus albipunctatus, which is considered as a subtype of CSNB (185–188).
Gene Therapy
Proof of concept of the efficacy of gene replacement in RLBP1-related RP came from a study conducted on a mouse model of the disease, in which self-complementary AAV8 vector carrying the gene for human RLBP1 under control of a short RLBP1 promoter (scAAV8-pRLBP1-hRLBP1, or CPK850) was delivered via SRI, resulting in an improved electroretinographic response (189). The success of the preclinical study, followed by the publication of non-clinical safety data by the same group of authors (190), paved the way to clinical trials. To date, there is one ongoing phase 1/2 trial opened for recruiting, aimed at exploring the maximum tolerated dose, safety, and potential efficacy of CPK850 delivered through a single SRI. The trial is scheduled to end in 2026.
MERTK-RP
Genetic Features
MERTK encodes the widely expressed tyrosine-protein kinase Mer, a receptor tyrosine kinase involved in a signal transduction pathway that regulates numerous cellular processes. In the retina, it is expressed in the RPE and it is involved in the phagocytosis of rod OSs. MERTK mutations are responsible for arRP, with onset within the second decade of life and progressive decline of VA, which is often reduced to light perception before age 50 (191, 192).
Gene Therapy
An open-label, dose escalation phase 1 trial of AAV2-mediated gene augmentation therapy for RP caused by MERTK mutation was conducted in Saudi Arabia (193). The SRI of the vector was not associated with major side effects, and 50% of patients (three out of six) demonstrated improved VA, though only one of them maintained this improvement after 2 years of follow-up.
Gene Therapy in Usher Syndrome (RP)
Usher syndrome is defined by the association of AR deafness (most commonly congenital) and retinopathy indistinguishable from typical RP (Figure 2). Usher syndrome is the most common RP-associated syndrome, accounting for almost 20% of all RP patients. Depending on the severity of the hearing loss, Usher syndrome is divided into three clinical subtypes (194).
Figure 2.
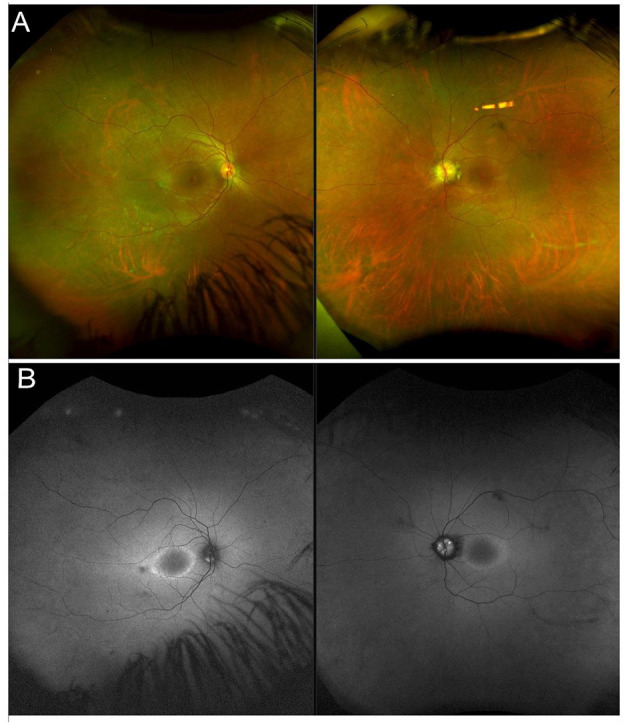
Multimodal imaging in a patient with a non-syndromic USH2A-related form of RP. (A) Ultra-wide field retinography shows near complete absence of pigment bone spicules. (B) Fundus autofluorescence displays the typical perifoveal ring of hyperautofluorescence.
Usher syndrome type 1 (the most common form) is characterized by profound, congenital sensorineural deafness (with consequent prelingual deafness or severe speech impairment), vestibular symptoms and childhood onset retinopathy.
Usher syndrome type 2 presents with congenital partial, non-progressive deafness, absence of vestibular symptoms, and milder and later-onset retinopathy.
Finally, Usher syndrome type 3 is characterized by progressive deafness starting in the second to fourth decade, adult-onset retinopathy and hypermetropic astigmatism.
Genetic Features
To date, 16 genes associated to Usher syndrome have been identified, two of which are good candidates for gene therapy and deserve a more detailed description.
The first of such genes is MYO7A. MYO7A encodes myosin VIIA, involved in in transport of melanosomes and phagosomes along actin filaments in the RPE and of opsin and other phototransduction proteins in photoreceptors (195). Mutations in MYO7A are associated with Usher Syndrome type 1B (USH1B).
The second relevant gene is USH2A, which encodes usherin. Mutations in USH2A, besides being the commonest association with type 2 Usher syndrome (80%), are the most frequent cause of AR non-syndromic RP (10–15%) (196–199). Clear genotype-phenotype correlations for USH2A mutations are not easy to establish. Generally, however, nonsense mutations, frameshifts mutations, or canonical splice site mutations in USH2A, either biallelic or combined with one missense allele, are associated with Usher syndrome type II, whereas the association of two missense mutations tends to result in non-syndromic RP (200). Of note, a peculiar feature of both non-syndromic and syndromic USH2A-related retinopathy is the fact that fundoscopy generally shows mild or no pigment deposits.
Gene Therapy
The MYO7A gene has 49 exons and spans approximately 87 kb of genomic sequence on chromosome 11q13.5, therefore significantly exceeding the cargo capacity of AAV vectors. For this reason, attempts have been made to deliver MYO7A in the shaker1 mouse model of Usher syndrome type 1B by means of UshStat, an EIAV lentiviral vector carrying the wild-type gene (EIAV-CMV-MYO7A) (201). Later on, Sanofi sponsored two phase 1/2 clinical trials, one of which is currently ongoing (NCT02065011), while the other has been stopped not for safety reasons (NCT01505062).
As far as USH2A is concerned, though being associated with recessive IRDs, a number of factors, including poor understanding of the physiological function of usherin and the USH2A gene size (15 kb), stand in the way of gene replacement strategies. Therefore, in order to develop an alternative approach, great interest was focused on a subset of mutations on exon 13 resulting in aberrant pre-mRNA splicing, which leads to the inclusion of a pseudoexon in the mature USH2A transcript. Since exon 13 consists of a multiplier of three nucleotides, skipping this exon does not disturb the open reading frame and likely results in the synthesis of a shorter protein with predicted residual function. This is the rationale behind the employment of AONs in an AR IRD. Encouraging results came from preclinical studies (73, 202) and ProQR Therapeutics sponsored the STELLAR phase 1/2 clinical trial (NCT03780257), whose purpose is to evaluate the safety and tolerability of a single intravitreal administration of AONs (QR-421a) in subjects with RP due to mutations in exon 13 of the USH2A gene. Enrolled patients receive one single IVT injection of QR-421a or sham-procedure in one eye (subject's worse eye) and are then followed up for 24 months. In March 2021, ProQR announced positive results from clinical trial of QR-421a and planned to start two phase 2/3 trials (SIRIUS and CELESTE).
Gene Therapy in Choroideremia
Choroideremia (CHM) is a rare XLR IRD, characterized by a progressive, centripetal, retinal degeneration, with a prevalence of 1:50,000 cases (203, 204). Vision loss progresses from nyctalopia in childhood to VF constriction in early adulthood and, ultimately, to legal blindness by the fifth decade of life (205, 206). From a pathogenic standpoint, Müller cells are the site of first damage, which is followed by outer retinal degeneration and, finally, by inner retinal thinning (207). Multimodal imaging findings include extended retinal hypoautofluorescence, with exclusive sparing of a central islet that allows patients to retain a good central vision. Structural OCT outside of this central region usually shows complete outer retinal atrophy (i.e., involving both photoreceptors and RPE cells), outer retinal tubulations and thinning of inner retinal layers and choroid (208, 209). Optical coherence tomography angiography displays an almost preserved superficial capillary plexus and choriocapillaris, with a significantly compromised deep capillary plexus (DCP), in the central islet, surrounded by completely absent choriocapillaris in the rest of the retina (210) (Figure 3).
Figure 3.
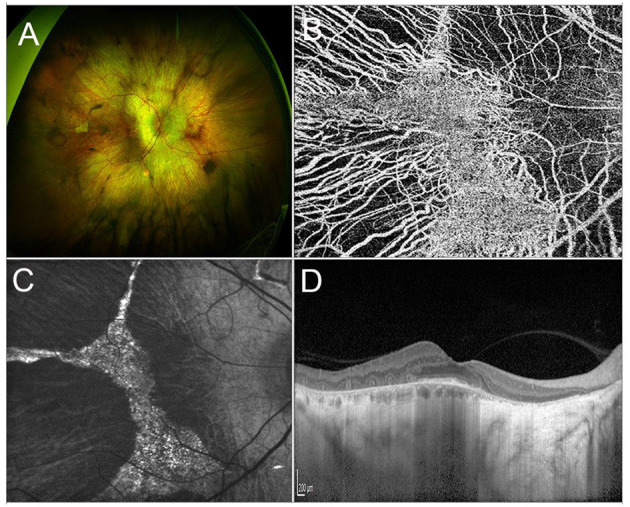
Multimodal imaging in CHM. (A) Ultra-wide field retinography shows a widespread chorioretinal atrophy, with a central islet of retinal sparing. (B) OCTA detects the sparing of the choriocapillaris vascular layer limited to the central islet. (C) Fundus autofluorescence displaying a central hyperautofluorescent region surrounded by hyporeflective atrophic tissue. (D) Structural OCT scan passing shows chorioretinal atrophy and outer retinal tubulations in the parafovea and a relative sparing of the photoreceptor layer in the fovea.
Genetic Features
Choroideremia is caused by mutations in the CHM gene, which encodes component A of Rab geranylgeranyl-transferase, referred to as Rab escort protein 1 (REP1), a key mediator of post-translational lipidation (prenylation) and subcellular localization of a family of intracellular protein trafficking regulators, known as the Rab GTPases (211). The CHM gene is ubiquitously expressed, but in most tissues, including adrenal gland, brain, and thyroid, the homolog REP2 protein partially counterbalances REP1 deficiency. The reasons why REP2 does not prevent disease manifestation in the eye are yet to be elucidated. In rare instances, CHM may be part of a contiguous gene syndrome involving Xq21. Indeed, males with large interstitial deletions of an additional X-chromosome portion, other than Xq21, may develop CHM together with birth defects (cleft lip and palate and agenesis of the corpus callosum) and severe cognitive deficits (212). Moreover, previous reports described cases of males with a small deletion of Xq21 presenting with CHM, mixed sensorineural and conductive hearing deficits (in case of deletion of POU3F4), and varying degrees of cognitive deficits (in case of deletion of RSK4) (213). In addition, a previous report described the case of a female patient affected by CHM, sensorineural deafness, and primary ovarian failure secondary to a balanced X-4 translocation (214). A large study, involving more than 70 patients affected by this disease, reported a 94% rate of identification of a CHM mutation (204). Mutations of uncertain significance included non-contiguous duplications, insertion, deletion, point mutations, and aberrant splicing (204). It is worth of notice the absence of disease-causing missense mutations, in contrast to the majority of human genetic diseases, which are mainly determined by such mutations (204). To date, no defined genotype–phenotype correlation has been identified for CHM.
Gene Therapy
Choroideremia is a promising candidate for gene therapy since the 1.9 kb CHM cDNA is small enough to fit the size capacity of AAV vectors. Preclinical proof-of-concept studies on the feasibility of CHM gene replacement have been conducted both in vitro, by inducing the CHM gene in pluripotent stem cells (iPSCs) from patients with CHM, and in vivo, by delivering the AAV2-CHM virus in normal sighted mice and zebrafish, with no evidence of toxicity (215, 216), paving the way to clinical trials on CHM patients. The results of the first-in-human clinical trial date back to 2014, when MacLaren et al. reported phase I safety and efficacy data on six patients treated with low-dose subretinal AAV2-REP administered subfoveally, demonstrating an improvement in BCVA and retinal sensitivity for up to 3.5 years after treatment (217). These findings were confirmed by the 24-month data coming from the phase II trial (218). Less encouraging results came from another phase I clinical trial using the same vector at higher doses in six patients, since one of the six untreated eyes exhibited an improvement of >15 ETDRS letters, prompting the authors to conclude that VA should not be used as a primary outcome measure for future CHM gene therapy trials (219). So far, several clinical trials employing SRI of AAV2.REP1 have been conducted (NCT02671539, NCT02077361, NCT02553135, NCT02341807, NCT03496012). Combining together their results, overall 40 patients have been treated with a median gain of 1.5 ±7.2 SD in ETDRS letters, highly variable between the different trials (220). Although some issues still need to be addresses, such as the identification of proper endpoints and the development of safety enhancements to facilitate subretinal gene delivery, gene therapy for CHM has reached phase III clinical trials, providing real promise for patients. It is worth of notice the launch of the first phase I CHM clinical trial aimed at evaluating safety, tolerability, and preliminary efficacy of a single IVI of a rAAV gene therapy, 4D-110, in male patients with genetically confirmed CHM (NCT04483440).
Gene Therapy in X-Linked Retinoschisis
X-linked retinoschisis is an IRD caused by mutations in the RS1 gene on Xp22.1. With an estimated prevalence ranging between 1 in 15,000 and 1 in 30,000, it is the most common form of juvenile-onset retinal degeneration in males, whereas heterozygous female carriers usually do not display any symptoms (221, 222). From a clinical standpoint, the typical feature of the disease, present in 98–100% of cases, is the foveal schisis, often seen as a spokewheel pattern of folds radiating out from the fovea, with peripheral retinoschisis being encountered in about 50% of patients (223–225) (Figure 4). Visual acuity generally starts declining in the first two decades of life, followed by a very slow progression of macular atrophy until the fifth or sixth decade, with possible evolution to legal blindness (226, 227). However, patients may also have a better prognosis, as long as the most common complications (i.e., retinal detachment and vitreous hemorrhage) do not occur.
Figure 4.
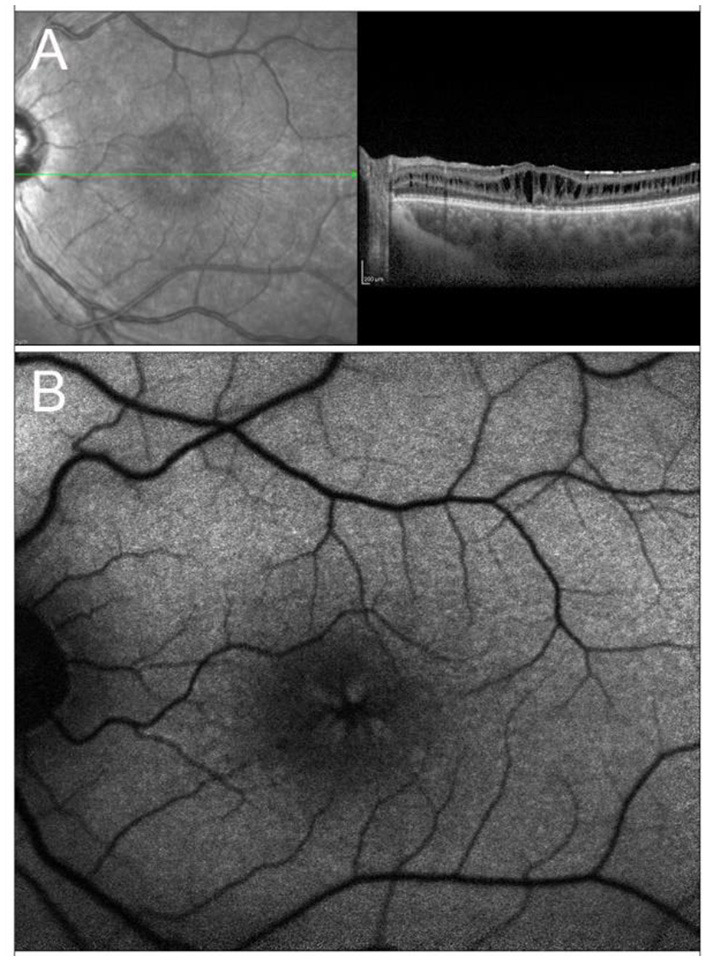
Multimodal imaging in X-linked retinoschisis. (A) Combined infrared reflectance imaging and structural OCT. Note the spokewheel pattern of folds radiating out from the fovea, which corresponds to the foveal schitic cavities seen on structural OCT. (B) Fundus autofluorescence also shows the spokewheel pattern due to the overlying foveal schisis.
Electroretinography is helpful in the diagnosis of XLRS, since there is a typically reduced b-wave amplitude, with a relatively preserved a-wave amplitude (the so-called “negative” waveform) (228).
Multimodal imaging findings include visualization of macular schisis on structural OCT and detection of foveal vascular impairment at the DCP level upon OCTA (229).
Genetic Features
RS1 encodes retinoschisin, a secretory protein exclusively expressed in retinal photoreceptors and bipolar cells, that can however be detected in all neuroretinal layers (230–232). Retinoschisin is found in a homo-oligomeric forms and, more specifically, it is an octamer made up of eight identical discoidin domains joined by intramolecular disulphide bonds (233). Mutations in the RS1 sequence disrupt subunit assembly, thus interfering with retinoschin's role in retinal cell adhesion and organization of retinal architecture (233, 234).
Gene Therapy
Preclinical studies have shown that IVI administration of AAV8-scRS/IBPhRS vector, as well as SRI of the AAV5-mOPs-RS1 resulted in significant morpho-functional improvement in the retinoschisin knockout (Rs1-KO) mouse, with evidence of good tolerability in rabbits (235–238). Building upon these encouraging results, two phase 1/2 trials were initiated with different constructs, administered via IVI, since XLRS is an inner retinal pathology. For the first of such trials (NCT02317887), sponsored by the National Eye Institute (NEI), employing AAV8-scRS/IRBPhRS in adults (≥18 years old), promising initial findings have already been reported (239). The second trial (NCT02416622) uses rAAV2tYF-CHhRS1, intravitreally injected in adults (≥18 years old) in the first dose-escalation phase, with subsequent enrolment of individuals ≥6 years of age, after the maximum tolerated dose is identified.
Gene Therapy in Stargardt Disease
Stargardt disease, the most common hereditary macular dystrophy, is characterized by a progressive, centrifugal, macular degeneration, associated with different patterns of peripheral retinal alterations (240). The prevalence is of 1:8,000–10,000 individuals (240). From a clinical standpoint, one of the most typical findings in STGD are flecks, namely debris accumulations resulting from the progressive degeneration of RPE cells. Flecks have been described as hyperautofluorescent, corresponding to lipofuscin accumulations, and hypoautofluorescent, resulting from debris absorption and outer retinal atrophy onset (241). Other fundus findings include complete hypoautofluorescent central atrophy, surrounded by a halo of patchy, mottled hypoautofluorescence (242) (Figure 5). More recently, different STGD patterns have been described, characterized by progressively wider involvement of mid and extreme retinal periphery, as assessed by ultrawide field imaging (243). Furthermore, quantitative multimodal imaging and OCTA allowed to categorize STGD eyes accordingly to the amount of involvement of the retinal vascular and choroidal networks, highlighting different morpho-functional features and progression rates (156, 244, 245).
Figure 5.
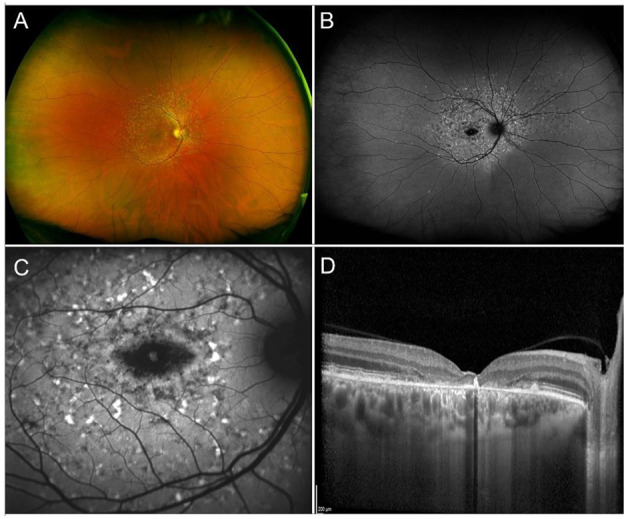
Multimodal imaging in Stargardt disease. (A) Ultra-wide field retinography shows numerous flecks inside and outside of the vascular arcades. (B) Ultra-wide field fundus autofluorescence displays the flecks as mixed hypo- and hyper-autofluorescent dots and shows an area of central atrophy, better highlighted by 30° fundus autofluorescence (C). (D) Structural OCT shows chorioretinal atrophy and clumping of hyperreflective lipofuscin-rich material corresponding to flecks.
Genetic Features
Stargardt disease can be distinguished in three different forms: (I) STGD1, the most common form, displays an AR homozygous or compound heterozygous transmission and is caused by mutations in the ABCA4 gene; (II) STGD3, which is determined by AD mutations in the ELOVL4 gene; (III) STGD4, a rare AD form associated with mutations in the PROM1 gene.
ABCA4, whose mutations are responsible for STGD1, is a 150 kb gene encoding an ATP-binding cassette transporter localized along the rims of photoreceptor OSs (246). This ATP-dependent flippase importer transports phosphatidylethanolamine (PE) and the all-trans-retinal (atRAL)/PE Schiff base (N-Ret-PE) into the cytosol, where atRAL is converted to all-trans-retinol (atROL) by retinol dehydrogenase RDH8 and RDH12. The absence of ABCA4 results in the accumulation of photo-toxic bisretinoids (A2E) with lipofuscin buildup in the RPE.
STGD3-causing ELOVL4 gene contains six exons and plays a fundamental role in the synthesis of very long-chain polyunsaturated fatty acids (247).
Finally, the PROM1 gene contains 23 exons distributed within a genomic sequence of more than 50 kb and encodes a protein involved in the organization of the plasma membrane and in the biogenesis of photoreceptor disks (248).
Genotype-phenotype correlations are challenging due to the heterogeneous disease manifestations. The most common genotypic classification includes three groups: genotype A (carriers of two or more deleterious variants); genotype B (one deleterious variant and >1 missense or in-frame insertion/deletion variants); genotype C (two or more missense or in-frame insertion or null variants) (249). Previous investigations highlighted how deleterious variants tend to be associated with more aggressive forms of the disease, while missense mutations yield to milder phenotypes (249–252). On the other hand, null alleles result in more severe STGD forms with an earlier onset (253). Other causes of severe phenotypes include truncating and severely misfolding mutations, deletions, stop codons, and insertions (254, 255). Furthermore, hypomorphic and deep intronic variants influencing the splicing process, have been also described (72, 256–258).
Gene Therapy
The main obstacles to the development of gene therapy approaches for STGD regard the dimension of the ABCA4 gene, whose coding sequence exceeds the cargo capacity of AAV, and the extreme complexity of deleterious variants, as previously described.
In the wake of the success of animal studies using lentiviral gene therapy to deliver the corrected ABCA4 gene (259), starting from 2011, Sanofi sponsored a phase I/II clinical trial (NCT01367444) to test the efficacy of a SRI of SAR422459, a recombinant lentiviral vector (EIAV) transporting a modified form of the ABCA4 gene. Notwithstanding the encouraging preliminary findings, in 2020, the sponsor decided to stop development of the product for non-safety reasons.
In 2019, the Applied Genetic Technologies Corporation announced the development of a hybrid AAV dual vector and published preclinical data supporting the potential of this technology in STGD (260).
More recently, promising results have been reported in animal models with a non-viral technique relying on subretinal delivery of self-assembled NPs (261, 262). Compared to viral vectors, non-viral delivery systems have unlimited payload, low immunogenicity, and minimal side effects, features that may allow to circumvent the obstacles which are currently standing in the way of STGD gene therapy (263).
Gene Therapy in Achromatopsia
Achromatopsia (ACHM) is an AR cone dysfunction affecting approximately 1 in 30,000 individuals (264, 265). Achromatopsia is a phenotypically and genotypically heterogeneous disease that can present in a complete or incomplete form. Complete ACHM is characterized by a totally abolished cone function, with a BCVA that is usually no >20/200 and with a total absence of color perception. In incomplete ACHM, residual cone function is present and patients have a higher VA and some degree of color discrimination (266). Prominent features of both complete and incomplete ACHM include photophobia, pendular nystagmus, central scotomata and high refractive (usually hypermetropic) errors (266). Electroretinography shows non-recordable cone-mediated responses with normal o near-normal rod responses (267). Fundus examination is normal or features non-specific alterations, such as central pigment mottling or attenuation of foveal reflex (268). On OCT, ACHM patients may display a variable phenotype, ranging from a normal to disrupted or absent EZ, which can sometimes be replaced by a hyporeflective cavitation, to complete outer retinal atrophy including the RPE (269). Fundus autofluorescence can be normal or show hyperautofluorescence, usually in zones of preserved EZ and likely preceding photoreceptor loss, or hypoautofluorescence, localized to areas of photoreceptor loss or RPE atrophy (270). Interestingly, adaptive optics scanning laser ophthalmoscopy (AOSLO) has shown the presence of cones in all ACHM patients, albeit reduced in number and with a highly variable density, regardless of OCT appearance, including areas of absent EZ reflectivity (271). This has important applications when it comes to patient selection for gene therapy trials.
Genetic Features
Mutations in six genes are responsible for over 90% of all ACHM cases, five of which are involved in the phototransduction process.
CNGA3 and CNGB3, encoding for the a- and b-subunit of the cyclic nucleotide-gated (CNG) cation channel 3 found in cones OSs, together account for 70–80% of cases of ACHM worldwide (272).
GNAT2 encodes the catalytic a-subunit of the G-protein transducin and is responsible for an infrequent form (<2%) of ACHM (273).
Equally rare subtypes are those caused by mutations in the PDE6C and PDE6H gene, encoding for the catalytic a- and inhibitory g-subunits of the photoreceptor-specific phosphodiesterase (274, 275).
Most recently, ATF6 has been identified as a sixth ACHM-associated gene. ATF6 encodes a transmembrane TFs ubiquitously expressed and involved in endoplasmic reticulum homeostasis (276). The frequent finding of foveal hypoplasia in ATF6-ACHM lead to the suggestion that this gene may be crucial for foveal development (276).
Gene Therapy
Promising preclinical results have been reported for ACHM caused by mutations in the CNGA3, CNB3, and GNAT2 genes. Taken together, these studies, which have been conducted on knock-out mouse models and naturally occurring mouse, sheep, and canine models, suggest that gene replacement approaches are effective and durable in ACHM, especially if administered early in life (277–284).
Other than for these encouraging data, ACHM seems particularly suited for gene augmentation for a number of reasons, including the presence of viable cones in all patients, as demonstrated by means of AOSLO, and its stationary or slowly progressive nature, which provides a wide window of opportunity.
The first phase I/II clinical trial commenced in November 2015 (NCT02610582) in Germany to assess the safety and efficacy of SRI of rAAV.hCNGA3 in patients with CNGA3-ACHM, using a dose-escalation protocol. Short after, another phase I/II clinical trial for CNGA-related ACHM (NCT02935517) was initiated in the US and in Israel and is still ongoing.
As far as CNGB3-ACHM is concerned, a phase I/II dose-escalation trial has been conducted in the UK (NCT03001310) to test the efficacy and safety of subretinal delivery of AAV2/8-hCARp.hCNGB3 and another similar multicentric phase I/II trial is still ongoing in the US and in Israel (NCT02599922).
Final Remarks
In the present review, we tried to resume all the relevant findings and the present status of gene therapy in IRDs. On the basis of the above-described data (all the quoted clinical trials are listed in Table 2), a clinically applicable gene therapy represents a tangible perspective more than a still far target. Some IRDs seem to be closer to an upcoming definitive gene therapy treatment, whereas further studies are warranted for other ones. Overall considering all the techniques and approaches under investigation, the main current limitations include the safety profile of gene therapy, especially regarding the surgically-related risks for the retina, and sometimes the need of repeated treatments. The intravitreal route of administration might provide higher safety and feasibility profiles, although limiting the penetration of the treatment and drug concentrations effectively reaching retinal targets, if compared with subretinal approaches, turning out to be powerful but potentially riskier. Furthermore, it is known that each IRD may be characterized by extremely heterogeneous genotypic-phenotypic relationship. This is quite challenging to be evaluated both in clinical practice and in research contexts. We may assume that the different phenotypic expression of the mutated gene might have an influence not only on the morpho-functional status of the patients, but also on the clinical effect of gene therapy. From this point of view, future prospective studies should be focused on deeper assessments of genotypic-phenotypic features of each IRD, on new classification strategies and on the meanings that these advances in knowledge might have on gene therapy planning.
Table 2.
Clinical trials on IRDs.
| N | Trial ID | Study title | Disease | Drug | Start date | Stop date | Phase | Status |
|---|---|---|---|---|---|---|---|---|
| 1 | NCT03913143 | Double-masked, randomized, controlled, multiple-dose study to evaluate efficacy, safety, tolerability, and syst. exposure of QR-110 in Leber's congenital amaurosis (LCA) due to C.2991+1655A>G mutation (p.Cys998X) in the CEP290 gene | Leber congenital amaurosis | Sepofarsen (QR-110) | 2019 | Ongoing | Phase I/II | Active, not recruiting |
| 2 | NCT02556736 | Phase I/IIA, open-label, dose-escalation study of safety, and tolerability of intravitreal RST-001 in patients with advanced retinitis pigmentosa (RP) | Retinitis pigmentosa | RST-001 | 2015 | Ongoing | Phase I/II | Active, not recruiting |
| 3 | NCT03326336 | A phase 1/2a, open-label, non-randomized, dose-escalation study to evaluate the safety, and tolerability of GS030 in subjects with retinitis pigmentosa | Retinitis pigmentosa | GS030-DP | 2017 | Ongoing | Phase I/II | Recruiting |
| 4 | NCT04278131 | Phase 1/2, safety, and efficacy trial of BS01, a recombinant adeno-associated virus vector expressing chronosFP in patients with retinitis pigmentosa | Retinitis pigmentosa | BS01 | 2020 | Ongoing | Phase I/II | Recruiting |
| 5 | NCT04919473 | A phase I/IIa open label, dose-escalation study to evaluate the safety, and tolerability of intravitreal vMCO-I in patients with advanced retinitis pigmentosa | Retinitis pigmentosa | vMCO-I | 2019 | 2020 | Phase I/II | Completed |
| 6 | NCT03602820 | A long-term follow-up study in subjects who received an adenovirus-associated viral vector serotype 2 containing the human RPE65 gene (AAV2-hRPE65v2, voretigene neparvovec-rzyl) administered via subretinal injection | Leber congenital amaurosis | AAV2-hRPE65v2 | 2015 | Ongoing | N/A | Active, not recruiting |
| 7 | NCT01208389 | A follow-on study to evaluate the safety of re-administration of adeno-associated viral vector containing the gene for human RPE65 [AAV2-hRPE65v2] to the contralateral eye in subjects with leber congenital amaurosis (LCA) previously enrolled in a phase 1 study | Leber congenital amaurosis | AAV2-hRPE65v2 | 2010 | Ongoing | Phase I/II | Active, not recruiting |
| 8 | NCT03913143 | Double-masked, randomized, controlled, multiple-dose study to evaluate efficacy, safety, tolerability, and syst. exposure of QR-110 in Leber's congenital amaurosis (LCA) due to c.2991+1655A>G mutation (p.Cys998X) in the CEP290 gene | Leber Congenital Amaurosis | Sepofarsen (QR-110) | 2019 | Ongoing | Phase II/III | Active, not recruiting |
| 9 | NCT03116113 | A dose escalation (phase 1), and dose expansion (phase 2/3) clinical trial of retinal gene therapy for X-linked retinitis pigmentosa using an adeno-associated viral vector (AAV8) encoding retinitis pigmentosa GTPase regulator (RPGR) | Retinitis pigmentosa | BIIB112 | 2017 | 2020 | Phase I/II | Completed |
| 10 | NCT03252847 | An open label, multi-center, phase I/II dose escalation trial of a recombinant adeno-associated virus vector (AAV2-.RPGR) for gene therapy of adults and children with X-linked retinitis pigmentosa owing to defects in retinitis pigmentosa GTPase regulator (RPGR) | Retinitis pigmentosa | AAV2/5-RPGR | 2017 | Ongoing | Phase I/II | Active, not recruiting |
| 11 | NCT04671433 | Phase 3 randomized, controlled study of AAV5-RPGR for the treatment of X-linked retinitis pigmentosa associated with variants in the RPGR gene | Retinitis pigmentosa | AAV5-RPGR | 2021 | Ongoing | Phase III | Recruiting |
| 12 | NCT03316560 | A phase 1/2 open-label dose escalation study to evaluate the safety and efficacy of AGTC-501 (rAAV2tYF-GRK1-RPGR) and a phase 2 randomized, controlled, masked, multi-center study comparing two doses of AGTC-501 in male subjects with x-linked retinitis pigmentosa confirmed by a pathogenic variant in the RPGR gene | Retinitis pigmentosa | rAAV2tYF-GRK1-RPGR | 2018 | Ongoing | Phase I/II | Recruiting |
| 13 | NCT04850118 | A phase 2/3, randomized, controlled, masked, multi-center study to evaluate the efficacy, safety, and tolerability of two doses of AGTC-501, a Recombinant adeno-associated virus vector expressing RPGR (rAAV2tYF-GRK1-RPGR), compared to an untreated control group in male subjects with X-linked Retinitis pigmentosa confirmed by a pathogenic variant in the RPGR gene | Retinitis pigmentosa | rAAV2tYF-GRK1-hRPGRco | 2021 | Ongoing | Phase II/III | Not yet recruiting |
| 14 | NCT04517149 | An open-label, phase 1/2 trial of gene therapy 4D-125 in males with X-linked retinitis pigmentosa (XLRP) caused by mutations in the RPGR gene | Retinitis pigmentosa | 4D-125 | 2020 | Ongoing | Phase I/II | Recruiting |
| 15 | NCT03328130 | Safety and efficacy of a unilateral subretinal administration of HORA-PDE6B in patients with retinitis pigmentosa harboring mutations in the PDE6B gene leading to a defect in PDE6ß expression | Retinitis pigmentosa | AAV2/5-hPDE6B | 2017 | Ongoing | Phase I/II | Recruiting |
| 16 | NCT04611503 | PIGMENT—PDE6A gene therapy for retinitis pigmentosa | Retinitis pigmentosa | rAAV.hPDE6A | 2019 | Ongoing | Phase I/II | Recruiting |
| 17 | NCT02065011 | An open-label study to determine the long-term safety, tolerability, and biological activity of SAR421869 in patients with usher syndrome type 1B | Retinitis pigmentosa | SAR421869 | 2013 | Ongoing | Phase I/II | Active, not recruiting |
| 18 | NCT01505062 | A phase I/IIA dose escalation safety study of subretinally injected SAR421869, administered to patients with retinitis pigmentosa associated with usher syndrome type 1B | Retinitis pigmentosa | SAR421869 | 2012 | 2019 | Phase I/II | Terminated for non-safety reasons |
| 19 | NCT03780257 | A first-in-human study to evaluate the safety and tolerability of QR-421a in subjects with retinitis pigmentosa (RP) due to mutations in exon 13 of the USH2A gene | Retinitis Pigmentosa | QR-421a | 2019 | Ongoing | Phase I/II | Active, not recruiting |
| 20 | NCT02671539 | THOR—tübingen choroideremia gene therapy trial open label phase 2 clinical trial using an adeno-associated viral vector (AAV2) encoding rab-escort protein 1 (REP1) | Choroideremia | rAAV2.REP1 | 2016 | 2018 | Phase II | Completed |
| 21 | NCT02077361 | An open label clinical trial of retinal gene therapy for choroideremia using an adeno-associated viral vector (AAV2) encoding Rab-escort protein-1 (REP1) | Choroideremia | rAAV2.REP1 | 2015 | Ongoing | Phase I/II | Active, not recruiting |
| 22 | NCT02553135 | An open label phase 2 clinical trial of retinal gene therapy for choroideremia using an adeno-associated viral vector (AAV2) encoding Rab-escort protein 1 (REP1) | Choroideremia | AAV2-REP1 | 2015 | 2018 | Phase II | Completed |
| 23 | NCT02341807 | A phase 1/2 safety study in subjects with CHM (choroideremia) gene mutations using an adeno-associated virus serotype 2 vector to deliver the normal human CHM gene [AAV2-hCHM] to the retina | Choroideremia | AAV2-hCHM | 2015 | Ongoing | Phase I/II | Active, not recruiting |
| 24 | NCT03496012 | A randomized, open label, outcomes-assessor masked, prospective, parallel controlled group, phase 3 clinical trial of retinal gene therapy for choroideremia using an adeno-associated viral vector (AAV2) encoding Rab escort protein 1 (REP1) | Choroideremia | AAV2-REP1 | 2017 | 2020 | Phase III | Completed |
| 25 | NCT04483440 | Phase 1 Open-label, dose-escalation study of the safety, tolerability, and preliminary efficacy of intravitreal 4D-110 in patients with choroideremia | Choroideremia | 4D-110 | 2020 | Ongoing | Phase I | Recruiting |
| 26 | NCT02317887 | A phase I/IIa study of RS1 ocular gene transfer for X-linked retinoschisis | X-linked retinoschisis | RS1 AAV | 2015 | Ongoing | Phase I/II | Recruiting |
| 27 | NCT02416622 | A multiple-site, phase 1/2, safety, and efficacy trial of a recombinant adeno-associated virus vector expressing retinoschisin (rAAV2tYF-CB-hRS1) in patients with X-linked retinoschisis | X-linked retinoschisis | rAAV2tYF-CB-hRS1 | 2015 | Ongoing | Phase I/II | Active, not recruiting |
| 28 | NCT01367444 | A phase I/IIA dose escalation safety study of subretinally injected SAR422459, administered to patients with Stargardt's macular degeneration | Stargardt's disease | SAR422459 | 2011 | 2019 | Phase I/II | Terminated for non-safety reasons |
| 29 | NCT02610582 | Safety and efficacy of a bilateral single subretinal injection of rAAV.hCNGA3 in adult and minor patients with CNGA3-linked achromatopsia investigated in a randomized, wait list controlled, observer-masked trial | Achromatopsia | rAAV.hCNGA3 | 2015 | Ongoing | Phase I/II | Recruiting |
| 30 | NCT02935517 | A multiple-site, phase 1/2, safety, and efficacy trial of AGTC 402, a recombinant adeno-associated virus vector expressing CNGA3, in patients with congenital achromatopsia caused by mutations in the CNGA3 gene | Achromatopsia | AGTC-402 | 2017 | Ongoing | Phase I/II | Recruiting |
| 31 | NCT03001310 | An open label, multi-center, phase I/II dose escalation trial of a recombinant adeno-associated virus vector (AAV2/8-hCARp.hCNGB3) for gene therapy of adults and children with achromatopsia owing to defects in CNGB3 | Achromatopsia | AAV-CNGB3 | 2017 | 2019 | Phase I/II | Completed |
| 32 | NCT02599922 | A multiple-site, phase 1/2, safety, and efficacy trial of a recombinant adeno-associated virus vector expressing CNGB3 in patients with congenital achromatopsia caused by mutations in the CNGB3 Gene | Achromatopsia | rAAV2tYF-PR1.7-hCNGB3 | 2016 | Ongoing | Phase I/II | Recruiting |
The list follows the order of appearance in the manuscript (from https://clinicaltrials.gov/).
Conclusions
Inherited retinal diseases are significantly disabling conditions affecting young, working-age populations. Despite the provision of low-vision aids and assistance from specialist services, to date the management of these disorders remains largely suboptimal and the development of definitive therapies should be regarded as a priority. Based on preclinical data and on an ever-growing body of clinical evidence, gene-based strategies can now be looked at with cautious optimism. However, whilst gene therapy holds great hope for the treatment of a wide range of IRDs in the future, there are caveats to be considered, which are mainly related to the careful selection of appropriate target diseases, patients, and outcome measures and to the surgical challenges of vector delivery. Natural history studies, long term follow-up of treated patients and advances in the field of genetic testing and molecular diagnostics are among the lines of research that can be pursued to address these issues and to expand the spectrum of IRDs that can be treated with this potentially revolutionary approach.
Author Contributions
AAr and AAm: review design, data analysis, data interpretation, and manuscript draft. EA, AS, and MM: data acquisition and data analysis. MB and FB: data interpretation, manuscript revision, and study supervision.
Conflict of Interest
FB consultant for Alcon (Fort Worth, Texas, USA), Alimera Sciences (Alpharetta, Georgia, USA), Allergan Inc. (Irvine, California, USA), Farmila-Thea (Clermont-Ferrand, France), Bayer Shering-Pharma (Berlin, Germany), Bausch and Lomb (Rochester, New York, USA), Genentech (San Francisco, California, USA), Hoffmann-La-Roche (Basel, Switzerland), NovagaliPharma (Évry, France), Novartis (Basel, Switzerland), Sanofi-Aventis (Paris, France), Thrombogenics (Heverlee, Belgium), and Zeiss (Dublin, USA). The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
- 1.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. (2010) 29:335–75. 10.1016/j.preteyeres.2010.03.004 [DOI] [PubMed] [Google Scholar]
- 2.Mitchell GA, Brody LC, Looney J, Steel G, Suchanek M, Dowling C, et al. An initiator codon mutation in ornithine-delta-aminotransferase causing gyrate atrophy of the choroid and retina. J Clin Invest. (1988) 81:630–3. 10.1172/JCI113365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terrell D, Comander J. Current stem-cell approaches for the treatment of inherited retinal degenerations. Semin Ophthalmol. (2019) 34:287–92. 10.1080/08820538.2019.1620808 [DOI] [PubMed] [Google Scholar]
- 4.Yue L, Weiland JD, Roska B, Humayun MS. Retinal stimulation strategies to restore vision: fundamentals and systems. Prog Retin Eye Res. (2016) 53:21–47. 10.1016/j.preteyeres.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 5.Bosking WH, Beauchamp MS, Yoshor D. Electrical stimulation of visual cortex: relevance for the development of visual cortical prosthetics. Annu Rev Vis Sci. (2017) 3:141–66. 10.1146/annurev-vision-111815-114525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blaese RM, Culver KW, Miller AD, Carter CS, Fleisher T, Clerici M, et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science. (1995) 270:475–80. 10.1126/science.270.5235.475 [DOI] [PubMed] [Google Scholar]
- 7.Tang R, Xu Z. Gene therapy: a double-edged sword with great powers. Mol Cell Biochem. (2020) 474:73–81. 10.1007/s11010-020-03834-3 [DOI] [PubMed] [Google Scholar]
- 8.Bashar AE, Metcalfe AL, Viringipurampeer IA, Yanai A, Gregory-Evans CY, Gregory-Evans K. An ex vivo gene therapy approach in X-linked retinoschisis. Mol Vis. (2016) 22:718–33. [PMC free article] [PubMed] [Google Scholar]
- 9.Athanasopoulos T, Munye MM, Yanez-Munoz RJ. Nonintegrating gene therapy vectors. Hematol Oncol Clin North Am. (2017) 31:753–70. 10.1016/j.hoc.2017.06.007 [DOI] [PubMed] [Google Scholar]
- 10.O'Neal WK, Zhou H, Morral N, Langston C, Parks RJ, Graham FL, et al. Toxicity associated with repeated administration of first-generation adenovirus vectors does not occur with a helper-dependent vector. Mol Med. (2000) 6:179–95. 10.1007/BF03402113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinwaerder DS, Carlson CA, Lieber A. Generation of adenovirus vectors devoid of all viral genes by recombination between inverted repeats. J Virol. (1999) 73:9303–13. 10.1128/JVI.73.11.9303-9313.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu ML, Edwards TL, O'Hare F, Hickey DG, Wang JH, Liu Z„, et al. Gene therapy for inherited retinal diseases: progress and possibilities. Clin Exp Optom. (2021) 104:444–54. 10.1080/08164622.2021.1880863 [DOI] [PubMed] [Google Scholar]
- 13.Ong T, Pennesi ME, Birch DG, Lam BL, Tsang SH. Adeno-associated viral gene therapy for inherited retinal disease. Pharm Res. (2019) 36:34. 10.1007/s11095-018-2564-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trapani I, Colella P, Sommella A, Iodice C, Cesi G, de Simone S, et al. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol Med. (2014) 6:194–211. 10.1002/emmm.201302948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McClements ME, Barnard AR, Singh MS, et al. An AAV Dual vector strategy ameliorates the stargardt phenotype in adult Abca4(−/−) Mice. Hum Gene Ther. (2019) 30:590–600. 10.1089/hum.2018.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trapani I, Puppo A, Auricchio A. Vector platforms for gene therapy of inherited retinopathies. Prog Retin Eye Res. (2014) 43:108–28. 10.1016/j.preteyeres.2014.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E, et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. (2003) 348:255–6. 10.1056/NEJM200301163480314 [DOI] [PubMed] [Google Scholar]
- 18.Yáñez-Muñoz RJ, Balaggan KS, MacNeil A, Howe SJ, Schmidt M, Smith AJ, et al. Effective gene therapy with nonintegrating lentiviral vectors. Nat Med. (2006) 12:348–53. 10.1038/nm1365 [DOI] [PubMed] [Google Scholar]
- 19.Cashman SM, Sadowski SL, Morris DJ, Frederick J, Kumar-Singh R. Intercellular trafficking of adenovirus-delivered HSV VP22 from the retinal pigment epithelium to the photoreceptors–implications for gene therapy. Mol Ther. (2002) 6:813–23. 10.1006/mthe.2002.0806 [DOI] [PubMed] [Google Scholar]
- 20.Ziccardi L, Cordeddu V, Gaddini L, Matteucci A, Parravano M, Malchiodi-Albedi F, et al. Gene therapy in retinal dystrophies. Int J Mol Sci. (2019) 20:5722. 10.3390/ijms20225722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fechheimer M, Boylan JF, Parker S, Sisken JE, Patel GL, Zimmer SG. Transfection of mammalian cells with plasmid DNA by scrape loading and sonication loading. Proc Natl Acad Sci USA. (1987) 84:8463–7. 10.1073/pnas.84.23.8463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann E, Schaefer-Ridder M, Wang Y, Hofschneider PH. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. (1982) 1:841–5. 10.1002/j.1460-2075.1982.tb01257.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capecchi MR. High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell. (1980) 22:479–88. 10.1016/0092-8674(80)90358-X [DOI] [PubMed] [Google Scholar]
- 24.Fraley R, Subramani S, Berg P, Papahadjopoulos D. Introduction of liposome-encapsulated SV40 DNA into cells. J Biol Chem. (1980) 255:10431–5. 10.1016/S0021-9258(19)70482-7 [DOI] [PubMed] [Google Scholar]
- 25.Gao X, Huang L. Potentiation of cationic liposome-mediated gene delivery by polycations. Biochemistry. (1996) 35:1027–36. 10.1021/bi952436a [DOI] [PubMed] [Google Scholar]
- 26.Conley SM, Cai X, Naash MI. Nonviral ocular gene therapy: assessment and future directions. Curr Opin Mol Ther. (2008) 10:456–63. [PMC free article] [PubMed] [Google Scholar]
- 27.Trigueros S, Domenech EB, Toulis V, Marfany G. In vitro gene delivery in retinal pigment epithelium cells by plasmid dna-wrapped gold nanoparticles. Genes (Basel). (2019) 10:289. 10.3390/genes10040289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farjo R, Skaggs J, Quiambao AB, Cooper MJ, Naash MI. Efficient non-viral ocular gene transfer with compacted DNA nanoparticles. PLoS ONE. (2006) 1:e38. 10.1371/journal.pone.0000038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai X, Nash Z, Conley SM, Fliesler SJ, Cooper MJ, Naash MI, et al. A partial structural and functional rescue of a retinitis pigmentosa model with compacted DNA nanoparticles. PLoS ONE. (2009) 4:e5290. 10.1371/journal.pone.0005290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Read SP, Cashman SM, Kumar-Singh R. POD nanoparticles expressing GDNF provide structural and functional rescue of light-induced retinal degeneration in an adult mouse. Mol Ther. (2010) 18:1917–26. 10.1038/mt.2010.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dezawa M, Takano M, Negishi H, Mo X, Oshitari T, Sawada H. Gene transfer into retinal ganglion cells by in vivo electroporation: a new approach. Micron. (2002) 33:1–6. 10.1016/S0968-4328(01)00002-6 [DOI] [PubMed] [Google Scholar]
- 32.Matsuda T, Cepko CL. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci USA. (2004) 101:16–22. 10.1073/pnas.2235688100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vasconcelos HM, Jr, Lujan BJ, Pennesi ME, Yang P, Lauer AK. Intraoperative optical coherence tomographic findings in patients undergoing subretinal gene therapy surgery. Int J Retina Vitreous. (2020) 6:13. 10.1186/s40942-020-00216-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olsen TW, Feng X, Wabner K, Conston SR, Sierra DH, Folden DV, et al. Cannulation of the suprachoroidal space: a novel drug delivery methodology to the posterior segment. Am J Ophthalmol. (2006) 142:777–87. 10.1016/j.ajo.2006.05.045 [DOI] [PubMed] [Google Scholar]
- 35.Einmahl S, Savoldelli M, D'Hermies F, Tabatabay C, Gurny R, Behar-Cohen F. Evaluation of a novel biomaterial in the suprachoroidal space of the rabbit eye. Invest Ophthalmol Vis Sci. (2002) 43:1533–9. [PubMed] [Google Scholar]
- 36.Mandelcorn ED, Kitchens JW, Fijalkowski N, Moshfeghi DM. Active aspiration of suprachoroidal hemorrhage using a guarded needle. Ophthalmic Surg Lasers Imaging Retina. (2014) 45:150–2. 10.3928/23258160-20140306-09 [DOI] [PubMed] [Google Scholar]
- 37.Goldstein DA, Do D, Noronha G, Kissner JM, Srivastava SK, Nguyen QD. Suprachoroidal corticosteroid administration: a novel route for local treatment of noninfectious uveitis. Transl Vis Sci Technol. (2016) 5:14. 10.1167/tvst.5.6.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gamlin PD, Alexander JJ, Boye SL, Witherspoon CD, Boye SE. SubILM injection of AAV for gene delivery to the retina. Methods Mol Biol. (2019) 1950:249–62. 10.1007/978-1-4939-9139-6_14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogers S, Pfuderer P. Use of viruses as carriers of added genetic information. Nature. (1968) 219:749–51. 10.1038/219749a0 [DOI] [PubMed] [Google Scholar]
- 40.Vazquez-Dominguez I, Garanto A, Collin RWJ. Molecular therapies for inherited retinal diseases-current standing, opportunities and challenges. Genes (Basel). (2019) 10:654. 10.3390/genes10090654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. (2016) 540:144–9. 10.1038/nature20565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai YT, Wu WH, Lee TT, Wu WP, Xu CL, Park KS, et al. Clustered regularly interspaced short palindromic repeats-based genome surgery for the treatment of autosomal dominant retinitis pigmentosa. Ophthalmology. (2018) 125:1421–30. 10.1016/j.ophtha.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li P, Kleinstiver BP, Leon MY, Prew MS, Navarro-Gomez D, Greenwald SH, et al. Allele-specific CRISPR-Cas9 genome editing of the single-base P23H mutation for rhodopsin-associated dominant retinitis pigmentosa. CRISPR. (2018) 1:55–64. 10.1089/crispr.2017.0009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clement K, Rees H, Canver MC, Gehrke JM, Farouni R, Hsu JY, et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat Biotechnol. (2019) 37:224–6. 10.1038/s41587-019-0032-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bakondi B, Lv W, Lu B, Jones MK, Tsai Y, Kim KJ, et al. In Vivo CRISPR/Cas9 gene editing corrects retinal dystrophy in the S334ter-3 rat model of autosomal dominant retinitis pigmentosa. Mol Ther. (2016) 24:556–63. 10.1038/mt.2015.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garanto A, van Beersum SE, Peters TA, Roepman R, Cremers FP, Collin RW. Unexpected CEP290 mRNA splicing in a humanized knock-in mouse model for Leber congenital amaurosis. PLoS ONE. (2013) 8:e79369. 10.1371/journal.pone.0079369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewin AS, Hauswirth WW. Ribozyme gene therapy: applications for molecular medicine. Trends Mol Med. (2001) 7:221–8. 10.1016/S1471-4914(01)01965-7 [DOI] [PubMed] [Google Scholar]
- 48.Hauswirth WW, Lewin AS. Ribozyme uses in retinal gene therapy. Prog Retin Eye Res. (2000) 19:689–710. 10.1016/S1350-9462(00)00007-0 [DOI] [PubMed] [Google Scholar]
- 49.Moore SM, Skowronska-Krawczyk D, Chao DL. Emerging concepts for RNA therapeutics for inherited retinal disease. Adv Exp Med Biol. (2019) 1185:85–9. 10.1007/978-3-030-27378-1_14 [DOI] [PubMed] [Google Scholar]
- 50.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. (1998) 391:806–11. 10.1038/35888 [DOI] [PubMed] [Google Scholar]
- 51.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. (2001) 411:494–8. 10.1038/35078107 [DOI] [PubMed] [Google Scholar]
- 52.Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, Plasterk RH. Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. (2001) 15:2654–9. 10.1101/gad.927801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edition. Br J Pharmacol. (2011) 164(Suppl 1):S1–324. 10.1111/j.1476-5381.2011.01649_1.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guzman-Aranguez A, Loma P, Pintor J. Small-interfering RNAs (siRNAs) as a promising tool for ocular therapy. Br J Pharmacol. (2013) 170:730–47. 10.1111/bph.12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fedorov Y, Anderson EM, Birmingham A, Reynolds A, Karpilow J, Robinson K, et al. Off-target effects by siRNA can induce toxic phenotype. RNA. (2006) 12:1188–96. 10.1261/rna.28106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong R, Kim J, Kim HS, Kim M, Lum L, Levine B, et al. Computational detection and suppression of sequence-specific off-target phenotypes from whole genome RNAi screens. Nucleic Acids Res. (2014) 42:8214–22. 10.1093/nar/gku306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. (2005) 11:263–70. 10.1038/nm1191 [DOI] [PubMed] [Google Scholar]
- 58.Lewin AS, Drenser KA, Hauswirth WW, Nishikawa S, Yasumura D, Flannery JG, et al. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med. (1998) 4:967–71. 10.1038/nm0898-967 [DOI] [PubMed] [Google Scholar]
- 59.Chadderton N, Millington-Ward S, Palfi A, O'Reilly M, Tuohy G, Humphries MM, et al. Improved retinal function in a mouse model of dominant retinitis pigmentosa following AAV-delivered gene therapy. Mol Ther. (2009) 17:593–9. 10.1038/mt.2008.301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hernan I, Gamundi MJ, Planas E, Borras E, Maseras M, Carballo M. Cellular expression and siRNA-mediated interference of rhodopsin cis-acting splicing mutants associated with autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. (2011) 52:3723–9. 10.1167/iovs.10-6933 [DOI] [PubMed] [Google Scholar]
- 61.O'Reilly M, Millington-Ward S, Palfi A, Chadderton N, Cronin T, McNally N, et al. A transgenic mouse model for gene therapy of rhodopsin-linked retinitis pigmentosa. Vision Res. (2008) 48:386–91. 10.1016/j.visres.2007.08.014 [DOI] [PubMed] [Google Scholar]
- 62.Collin RW, Garanto A. Applications of antisense oligonucleotides for the treatment of inherited retinal diseases. Curr Opin Ophthalmol. (2017) 28:260–6. 10.1097/ICU.0000000000000363 [DOI] [PubMed] [Google Scholar]
- 63.Bennett CF, Swayze EE. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. (2010) 50:259–93. 10.1146/annurev.pharmtox.010909.105654 [DOI] [PubMed] [Google Scholar]
- 64.Naessens S, Ruysschaert L, Lefever S, Coppieters F, De Baere E. Antisense oligonucleotide-based downregulation of the G56R pathogenic variant causing NR2E3-associated autosomal dominant retinitis pigmentosa. Genes (Basel). (2019) 10:363. 10.3390/genes10050363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murray SF, Jazayeri A, Matthes MT, Yasumura D, Yang H, Peralta R, et al. Allele-specific inhibition of rhodopsin with an antisense oligonucleotide slows photoreceptor cell degeneration. Invest Ophthalmol Vis Sci. (2015) 56:6362–75. 10.1167/iovs.15-16400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu MM, Zack DJ. Alternative splicing and retinal degeneration. Clin Genet. (2013) 84:142–9. 10.1111/cge.12181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bonifert T, Gonzalez Menendez I, Battke F, Theurer Y, Synofzik M, Schöls L, et al. Antisense oligonucleotide mediated splice correction of a deep intronic mutation in OPA1. Mol Ther Nucleic Acids. (2016) 5:e390. 10.1038/mtna.2016.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garanto A, van der Velde-Visser SD, Cremers FPM, Collin RWJ. Antisense oligonucleotide-based splice correction of a deep-intronic mutation in CHM underlying choroideremia. Adv Exp Med Biol. (2018) 1074:83–9. 10.1007/978-3-319-75402-4_11 [DOI] [PubMed] [Google Scholar]
- 69.Albert S, Garanto A, Sangermano R, Khan M, Bax NM, Hoyng CB, et al. Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying Stargardt disease. Am J Hum Genet. (2018) 102:517–27. 10.1016/j.ajhg.2018.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bauwens M, Garanto A, Sangermano R, Naessens S, Weisschuh N, De Zaeytijd J, et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet Med. (2019) 21:1761–71. 10.1038/s41436-018-0420-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garanto A, Duijkers L, Tomkiewicz TZ, Collin RWJ. Antisense oligonucleotide screening to optimize the rescue of the splicing defect caused by the recurrent deep-intronic ABCA4 variant c.4539+2001G>A in Stargardt disease. Genes (Basel). (2019) 10:452. 10.3390/genes10060452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sangermano R, Garanto A, Khan M, Runhart EH, Bauwens M, Bax NM, et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet Med. (2019) 21:1751–60. 10.1038/s41436-018-0414-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Slijkerman RW, Vaché C, Dona M, García-García G, Claustres M, Hetterschijt L, et al. Antisense oligonucleotide-based splice correction for USH2A-associated retinal degeneration caused by a frequent deep-intronic mutation. Mol Ther Nucleic Acids. (2016) 5:e381. 10.1038/mtna.2016.89 [DOI] [PubMed] [Google Scholar]
- 74.Saleh AF, Arzumanov AA, Gait MJ. Overview of alternative oligonucleotide chemistries for exon skipping. Methods Mol Biol. (2012) 867:365–78. 10.1007/978-1-61779-767-5_23 [DOI] [PubMed] [Google Scholar]
- 75.Hall AH, Wan J, Shaughnessy EE, Ramsay Shaw B, Alexander KA. RNA interference using boranophosphate siRNAs: structure-activity relationships. Nucleic Acids Res. (2004) 32:5991–6000. 10.1093/nar/gkh936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gaglione M, Messere A. Recent progress in chemically modified siRNAs. Mini Rev Med Chem. (2010) 10:578–95. 10.2174/138955710791384036 [DOI] [PubMed] [Google Scholar]
- 77.Chiu YL, Rana TM. siRNA function in RNAi: a chemical modification analysis. RNA. (2003) 9:1034–48. 10.1261/rna.5103703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. (2016) 44:6549–63. 10.1093/nar/gkw533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dias N, Stein CA. Antisense oligonucleotides: basic concepts and mechanisms. Mol Cancer Ther. (2002) 1:347–55. [PubMed] [Google Scholar]
- 80.Devi GR, Beer TM, Corless CL, Arora V, Weller DL, Iversen PL. In vivo bioavailability and pharmacokinetics of a c-MYC antisense phosphorodiamidate morpholino oligomer, AVI-4126, in solid tumors. Clin Cancer Res. (2005) 11:3930–8. 10.1158/1078-0432.CCR-04-2091 [DOI] [PubMed] [Google Scholar]
- 81.Garanto A, Chung DC, Duijkers L, Corral-Serrano JC, Messchaert M, Xiao R, et al. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum Mol Genet. (2016) 25:2552–63. 10.1093/hmg/ddw118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Turchinovich A, Zoidl G, Dermietzel R. Non-viral siRNA delivery into the mouse retina in vivo. BMC Ophthalmol. (2010) 10:25. 10.1186/1471-2415-10-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mussolino C, Sanges D, Marrocco E, Bonetti C, Di Vicino U, Marigo V, et al. Zinc-finger-based transcriptional repression of rhodopsin in a model of dominant retinitis pigmentosa. EMBO Mol Med. (2011) 3:118–28. 10.1002/emmm.201000119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Botta S, Marrocco E, de Prisco N, Curion F, Renda M, Sofia M, et al. Rhodopsin targeted transcriptional silencing by DNA-binding. Elife. (2016) 5:e12242. 10.7554/eLife.12242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Botta S, de Prisco N, Marrocco E, Renda M, Sofia M, Curion F, et al. Targeting and silencing of rhodopsin by ectopic expression of the transcription factor KLF15. JCI Insight. (2017) 2:e96560. 10.1172/jci.insight.96560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Trapani I, Auricchio A. Seeing the light after 25 years of retinal gene therapy. Trends Mol Med. (2018) 24:669–81. 10.1016/j.molmed.2018.06.006 [DOI] [PubMed] [Google Scholar]
- 87.Liang FQ, Dejneka NS, Cohen DR, Krasnoperova NV, Lem J, Maguire AM, et al. AAV-mediated delivery of ciliary neurotrophic factor prolongs photoreceptor survival in the rhodopsin knockout mouse. Mol Ther. (2001) 3:241–8. 10.1006/mthe.2000.0252 [DOI] [PubMed] [Google Scholar]
- 88.McGee Sanftner LH, Abel H, Hauswirth WW, Flannery JG. Glial cell line derived neurotrophic factor delays photoreceptor degeneration in a transgenic rat model of retinitis pigmentosa. Mol Ther. (2001) 4:622–9. 10.1006/mthe.2001.0498 [DOI] [PubMed] [Google Scholar]
- 89.Buch PK, MacLaren RE, Durán Y, Balaggan KS, MacNeil A, Schlichtenbrede FC, et al. In contrast to AAV-mediated Cntf expression, AAV-mediated Gdnf expression enhances gene replacement therapy in rodent models of retinal degeneration. Mol Ther. (2006) 14:700–9. 10.1016/j.ymthe.2006.05.019 [DOI] [PubMed] [Google Scholar]
- 90.Schuettauf F, Vorwerk C, Naskar R, Orlin A, Quinto K, Zurakowski D, et al. Adeno-associated viruses containing bFGF or BDNF are neuroprotective against excitotoxicity. Curr Eye Res. (2004) 29:379–86. 10.1080/02713680490517872 [DOI] [PubMed] [Google Scholar]
- 91.Byrne LC, Dalkara D, Luna G, Fisher SK, Clérin E, Sahel JA, et al. Viral-mediated RdCVF and RdCVFL expression protects cone and rod photoreceptors in retinal degeneration. J Clin Invest. (2015) 125:105–16. 10.1172/JCI65654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yao J, Feathers KL, Khanna H, Thompson D, Tsilfidis C, Hauswirth WW, et al. XIAP therapy increases survival of transplanted rod precursors in a degenerating host retina. Invest Ophthalmol Vis Sci. (2011) 52:1567–72. 10.1167/iovs.10-5998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Birch DG, Bennett LD, Duncan JL, Weleber RG, Pennesi ME. Long-term follow-up of patients with retinitis pigmentosa receiving intraocular ciliary neurotrophic factor implants. Am J Ophthalmol. (2016) 170:10–4. 10.1016/j.ajo.2016.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bi A, Cui J, Ma YP, Olshevskaya E, Pu M, Dizhoor AM, et al. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron. (2006) 50:23–33. 10.1016/j.neuron.2006.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cehajic-Kapetanovic J, Eleftheriou C, Allen AE, Milosavljevic N, Pienaar A, Bedford R, et al. Restoration of vision with ectopic expression of human rod opsin. Curr Biol. (2015) 25:2111–22. 10.1016/j.cub.2015.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lin B, Koizumi A, Tanaka N, Panda S, Masland RH. Restoration of visual function in retinal degeneration mice by ectopic expression of melanopsin. Proc Natl Acad Sci USA. (2008) 105:16009–14. 10.1073/pnas.0806114105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tomita H, Sugano E, Yawo H, Ishizuka T, Isago H, Narikawa S, et al. Restoration of visual response in aged dystrophic RCS rats using AAV-mediated channelopsin-2 gene transfer. Invest Ophthalmol Vis Sci. (2007) 48:3821–6. 10.1167/iovs.06-1501 [DOI] [PubMed] [Google Scholar]
- 98.den Hollander AI, Roepman R, Koenekoop RK, Cremers FP. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. (2008) 27:391–419. 10.1016/j.preteyeres.2008.05.003 [DOI] [PubMed] [Google Scholar]
- 99.Heher KL, Traboulsi EI, Maumenee IH. The natural history of Leber's congenital amaurosis. Age-related findings in 35 patients. Ophthalmology. (1992) 99:241–5. 10.1016/S0161-6420(92)31985-2 [DOI] [PubMed] [Google Scholar]
- 100.Dagi LR, Leys MJ, Hansen RM, Fulton AB. Hyperopia in complicated Leber's congenital amaurosis. Arch Ophthalmol. (1990) 108:709–12. 10.1001/archopht.1990.01070070095043 [DOI] [PubMed] [Google Scholar]
- 101.Lorenz B, Gyürüs P, Preising M, Bremser D, Gu S, Andrassi M, et al. Early-onset severe rod-cone dystrophy in young children with RPE65 mutations. Invest Ophthalmol Vis Sci. (2000) 41:2735–42. [PubMed] [Google Scholar]
- 102.Koenekoop RK. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol. (2004) 49:379–98. 10.1016/j.survophthal.2004.04.003 [DOI] [PubMed] [Google Scholar]
- 103.Dharmaraj S, Leroy BP, Sohocki MM, Koenekoop RK, Perrault I, Anwar K, et al. The phenotype of Leber congenital amaurosis in patients with AIPL1 mutations. Arch Ophthalmol. (2004) 122:1029–37. 10.1001/archopht.122.7.1029 [DOI] [PubMed] [Google Scholar]
- 104.Galvin JA, Fishman GA, Stone EM, Koenekoop RK. Clinical phenotypes in carriers of Leber congenital amaurosis mutations. Ophthalmology. (2005) 112:349–56. 10.1016/j.ophtha.2004.08.023 [DOI] [PubMed] [Google Scholar]
- 105.Koenekoop RK, Cremers FP, den Hollander AI. Leber congenital amaurosis: ciliary proteins on the move. Ophthalmic Genet. (2007) 28:111–2. 10.1080/13816810701537457 [DOI] [PubMed] [Google Scholar]
- 106.Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. (2017) 101:1147–54. 10.1136/bjophthalmol-2016-309975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sohocki MM, Sullivan LS, Mintz-Hittner HA, Birch D, Heckenlively JR, Freund CL, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. (1998) 63:1307–15. 10.1086/302101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xu Y, Guan L, Xiao X, Zhang J, Li S, Jiang H, et al. ALMS1 null mutations: a common cause of Leber congenital amaurosis and early-onset severe cone-rod dystrophy. Clin Genet. (2016) 89:442–7. 10.1111/cge.12617 [DOI] [PubMed] [Google Scholar]
- 109.Xu M, Yang L, Wang F, Li H, Wang X, Wang W, et al. Mutations in human IFT140 cause non-syndromic retinal degeneration. Hum Genet. (2015) 134:1069–78. 10.1007/s00439-015-1586-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sweeney MO, McGee TL, Berson EL, Dryja TP. Low prevalence of lecithin retinol acyltransferase mutations in patients with Leber congenital amaurosis and autosomal recessive retinitis pigmentosa. Mol Vis. (2007) 13:588–93. [PMC free article] [PubMed] [Google Scholar]
- 111.Sun W, Gerth C, Maeda A, Lodowski DT, Van Der Kraak L, Saperstein DA, et al. Novel RDH12 mutations associated with Leber congenital amaurosis and cone-rod dystrophy: biochemical and clinical evaluations. Vision Res. (2007) 47:2055–66. 10.1016/j.visres.2007.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Soens ZT, Li Y, Zhao L, Eblimit A, Dharmat R, Li Y, et al. Hypomorphic mutations identified in the candidate Leber congenital amaurosis gene CLUAP1. Genet Med. (2016) 18:1044–51. 10.1038/gim.2015.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ramamurthy V, Roberts M, van den Akker F, Niemi G, Reh TA, Hurley JB. AIPL1, a protein implicated in Leber's congenital amaurosis, interacts with and aids in processing of farnesylated proteins. Proc Natl Acad Sci USA. (2003) 100:12630–5. 10.1073/pnas.2134194100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Perrault I, Hanein S, Gerard X, Delphin N, Fares-Taie L, Gerber S, et al. Spectrum of SPATA7 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. (2010) 31:E1241–50. 10.1002/humu.21203 [DOI] [PubMed] [Google Scholar]
- 115.Nichols LL, 2nd, Alur RP, Boobalan E, Sergeev YV, Caruso RC, Stone EM, et al. Two novel CRX mutant proteins causing autosomal dominant Leber congenital amaurosis interact differently with NRL. Hum Mutat. (2010) 31:E1472–1483. 10.1002/humu.21268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mataftsi A, Schorderet DF, Chachoua L, Boussalah M, Nouri MT, Barthelmes D, et al. Novel TULP1 mutation causing leber congenital amaurosis or early onset retinal degeneration. Invest Ophthalmol Vis Sci. (2007) 48:5160–7. 10.1167/iovs.06-1013 [DOI] [PubMed] [Google Scholar]
- 117.Lotery AJ, Jacobson SG, Fishman GA, Weleber RG, Fulton AB, Namperumalsamy P, et al. Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol. (2001) 119:415–20. 10.1001/archopht.119.3.415 [DOI] [PubMed] [Google Scholar]
- 118.Li T. Leber congenital amaurosis caused by mutations in RPGRIP1. Cold Spring Harb Perspect Med. (2014) 5:a017384. 10.1101/cshperspect.a017384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gerber S, Hanein S, Perrault I, Delphin N, Aboussair N, Leowski C, et al. Mutations in LCA5 are an uncommon cause of Leber congenital amaurosis (LCA) type II. Hum Mutat. (2007) 28:1245. 10.1002/humu.9513 [DOI] [PubMed] [Google Scholar]
- 120.Estrada-Cuzcano A, Koenekoop RK, Coppieters F, Kohl S, Lopez I, Collin RW, et al. IQCB1 mutations in patients with leber congenital amaurosis. Invest Ophthalmol Vis Sci. (2011) 52:834–9. 10.1167/iovs.10-5221 [DOI] [PubMed] [Google Scholar]
- 121.Chao DL, Burr A, Pennesi M. RPE65-related leber congenital amaurosis/early-onset severe retinal dystrophy. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews((R)). Seattle, WA: University of Washington; (1993). [PubMed] [Google Scholar]
- 122.Burnight ER, Wiley LA, Drack AV, Braun TA, Anfinson KR, Kaalberg EE, et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. (2014) 21:662–72. 10.1038/gt.2014.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Boye SE. Leber congenital amaurosis caused by mutations in GUCY2D. Cold Spring Harb Perspect Med. (2014) 5:a017350. 10.1101/cshperspect.a017350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bowne SJ, Sullivan LS, Mortimer SE, Hedstrom L, Zhu J, Spellicy CJ, et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest Ophthalmol Vis Sci. (2006) 47:34–42. 10.1167/iovs.05-0868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Azadi S, Molday LL, Molday RS. RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc Natl Acad Sci USA. (2010) 107:21158–63. 10.1073/pnas.1010460107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Asai-Coakwell M, March L, Dai XH, Duval M, Lopez I, French CR, et al. Contribution of growth differentiation factor 6-dependent cell survival to early-onset retinal dystrophies. Hum Mol Genet. (2013) 22:1432–42. 10.1093/hmg/dds560 [DOI] [PubMed] [Google Scholar]
- 127.Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. (2001) 28:92–5. 10.1038/ng0501-92 [DOI] [PubMed] [Google Scholar]
- 128.Acland GM, Aguirre GD, Bennett J, Aleman TS, Cideciyan AV, Bennicelli J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. (2005) 12:1072–82. 10.1016/j.ymthe.2005.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Aguirre GD, Baldwin V, Pearce-Kelling S, Narfstrom K, Ray K. Acland GM. Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Mol Vis. (1998) 4:23. [PubMed] [Google Scholar]
- 130.Pang JJ, Chang B, Hawes NL, Hurd RE, Davisson MT, Li J, et al. Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA). Mol Vis. (2005) 11:152–62. [PubMed] [Google Scholar]
- 131.Rohrer B, Goletz P, Znoiko S, Ablonczy Z, Ma JX, Redmond TM, et al. Correlation of regenerable opsin with rod ERG signal in Rpe65−/− mice during development and aging. Invest Ophthalmol Vis Sci. (2003) 44:310–5. 10.1167/iovs.02-0567 [DOI] [PubMed] [Google Scholar]
- 132.Bainbridge JW, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. (2008) 358:2231–9. 10.1056/NEJMoa0802268 [DOI] [PubMed] [Google Scholar]
- 133.Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. (2008) 19:979–90. 10.1089/hum.2008.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. (2008) 358:2240–8. 10.1056/NEJMoa0802315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Patel U, Boucher M, de Leseleuc L, Visintini S. Voretigene neparvovec: an emerging gene therapy for the treatment of inherited blindness. In: CADTH Issues in Emerging Health Technologies. Ottawa, ON: Canadian Agency for Drugs and Technologies in Health; (2016). p. 1–11. [PubMed] [Google Scholar]
- 136.Drack AV, Bennett J, Russell S, High KA, Yu Z-F, Tillman A, et al. How long does gene therapy last? 4-year follow-up phase 3 voretigene neparvovec trial in RPE65-associated LCA/inherited retinal disease. J AAPOS. (2019) 23:e7. 10.1016/j.jaapos.2019.08.018 [DOI] [Google Scholar]
- 137.Testa F, Maguire AM, Rossi S, Pierce EA, Melillo P, Marshall K, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital Amaurosis type 2. Ophthalmology. (2013) 120:1283–91. 10.1016/j.ophtha.2012.11.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bainbridge JW, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, et al. Long-term effect of gene therapy on Leber's congenital amaurosis. N Engl J Med. (2015) 372:1887–97. 10.1056/NEJMoa1414221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med. (2019) 25:229–33. 10.1038/s41591-018-0327-9 [DOI] [PubMed] [Google Scholar]
- 140.Biasutto P, Dulla K, Adamson P, Schulkens I, Schmidt I, Lane A, et al. QR-110 treatment for leber's congenital amaurosis type 10: restoration of CEP290 mRNA levels and ciliation in LCA10 iPSC-derived optic cups. Invest Ophthalmol. (2017) 58:249. [Google Scholar]
- 141.Dulla K, Aguila M, Lane A, Jovanovic K, Parfitt DA, Schulkens I, et al. Splice-modulating oligonucleotide QR-110 restores CEP290 mRNA and function in human c.2991+1655A>G LCA10 models. Mol Ther Nucleic Acids. (2018) 12:730–40. 10.1016/j.omtn.2018.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cideciyan AV, Jacobson SG, Drack AV, Ho AC, Charng J, Garafalo AV, et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat Med. (2019) 25:225–8. 10.1038/s41591-018-0295-0 [DOI] [PubMed] [Google Scholar]
- 143.Mihelec M, Pearson RA, Robbie SJ, Buch PK, Azam SA, Bainbridge JW, et al. Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of leber congenital amaurosis caused by guanylate cyclase-1 deficiency. Hum Gene Ther. (2011) 22:1179–90. 10.1089/hum.2011.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Haire SE, Pang J, Boye SL, Sokal I, Craft CM, Palczewski K, et al. Light-driven cone arrestin translocation in cones of postnatal guanylate cyclase-1 knockout mouse retina treated with AAV-GC1. Invest Ophthalmol Vis Sci. (2006) 47:3745–53. 10.1167/iovs.06-0086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Boye SE, Boye SL, Pang J, Ryals R, Everhart D, Umino Y, et al. Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS ONE. (2010) 5:e11306. 10.1371/journal.pone.0011306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Boon N, Wijnholds J, Pellissier LP. Research models and gene augmentation therapy for CRB1 retinal dystrophies. Front Neurosci. (2020) 14:860. 10.3389/fnins.2020.00860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alves CH, Wijnholds J. AAV Gene augmentation therapy for CRB1-associated retinitis pigmentosa. Methods Mol Biol. (2018) 1715:135–51. 10.1007/978-1-4939-7522-8_10 [DOI] [PubMed] [Google Scholar]
- 148.Feathers KL, Jia L, Perera ND, Chen A, Presswalla FK, Khan NW, et al. Development of a gene therapy vector for RDH12-associated retinal dystrophy. Hum Gene Ther. (2019) 30:1325–35. 10.1089/hum.2019.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tsang SH, Sharma T. Retinitis pigmentosa (non-syndromic). Adv Exp Med Biol. (2018) 1085:125–30. 10.1007/978-3-319-95046-4_25 [DOI] [PubMed] [Google Scholar]
- 150.Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. (2018) 66:157–86. 10.1016/j.preteyeres.2018.03.005 [DOI] [PubMed] [Google Scholar]
- 151.Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis. (2006) 1:40. 10.1186/1750-1172-1-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Popovic P, Jarc-Vidmar M, Hawlina M. Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. (2005) 243:1018–27. 10.1007/s00417-005-1186-x [DOI] [PubMed] [Google Scholar]
- 153.Robson AG, Saihan Z, Jenkins SA, Fitzke FW, Bird AC, Webster AR, et al. Functional characterisation and serial imaging of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Br J Ophthalmol. (2006) 90:472–9. 10.1136/bjo.2005.082487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fischer MD, Fleischhauer JC, Gillies MC, Sutter FK, Helbig H, Barthelmes D, et al. new method to monitor visual field defects caused by photoreceptor degeneration by quantitative optical coherence tomography. Invest Ophthalmol Vis Sci. (2008) 49:3617–21. 10.1167/iovs.08-2003 [DOI] [PubMed] [Google Scholar]
- 155.Lupo S, Grenga PL, Vingolo EM. Fourier-domain optical coherence tomography and microperimetry findings in retinitis pigmentosa. Am J Ophthalmol. (2011) 151:106–11. 10.1016/j.ajo.2010.07.026 [DOI] [PubMed] [Google Scholar]
- 156.Arrigo A, Bordato A, Romano F, Aragona E, Grazioli A, Bandello F, et al. Choroidal patterns in retinitis pigmentosa: correlation with visual acuity and disease progression. Transl Vis Sci Technol. (2020) 9:17. 10.3390/jcm8091388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Arrigo A, Romano F, Albertini G, Aragona E, Bandello F, Battaglia Parodi M. Vascular patterns in retinitis pigmentosa on swept-source optical coherence tomography angiography. J Clin Med. (2019) 8:1425. 10.3390/jcm8091425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. (1990) 343:364–6. 10.1038/343364a0 [DOI] [PubMed] [Google Scholar]
- 159.Dryja TP, McGee TL, Hahn LB, Cowley GS, Olsson JE, Reichel E, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med. (1990) 323:1302–7. 10.1056/NEJM199011083231903 [DOI] [PubMed] [Google Scholar]
- 160.Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. (2005) 11:177–85. 10.1016/j.molmed.2005.02.007 [DOI] [PubMed] [Google Scholar]
- 161.Kumaramanickavel G, Maw M, Denton MJ, John S, Srikumari CR, Orth U, et al. Missense rhodopsin mutation in a family with recessive RP. Nat Genet. (1994) 8:10–1. 10.1038/ng0994-10 [DOI] [PubMed] [Google Scholar]
- 162.Meng D, Ragi SD, Tsang SH. Therapy in rhodopsin-mediated autosomal dominant retinitis pigmentosa. Mol Ther. (2020) 28:2139–49. 10.1016/j.ymthe.2020.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Diner BA, Das A, Nayak R, Flinkstrom Z, Tallo T, DaSilva J, et al. Dual AAV-based “Knock-out-and-replace” of RHO as a therapeutic approach to treat RHO-associated autosomal dominant retinitis pigmentosa (RHO adRP). Mol Ther. (2020) 28:108–9. [Google Scholar]
- 164.O'Reilly M, Palfi A, Chadderton N, Millington-Ward S, Ader M, Cronin T, et al. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am J Hum Genet. (2007) 81:127–35. 10.1086/519025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Millington-Ward S, Chadderton N, O'Reilly M, Palfi A, Goldmann T, Kilty C, et al. Suppression and replacement gene therapy for autosomal dominant disease in a murine model of dominant retinitis pigmentosa. Mol Ther. (2011) 19:642–9. 10.1038/mt.2010.293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Kiang AS, Palfi A, Ader M, Kenna PF, Millington-Ward S, Clark G, et al. Toward a gene therapy for dominant disease: validation of an RNA interference-based mutation-independent approach. Mol Ther. (2005) 12:555–61. 10.1016/j.ymthe.2005.03.028 [DOI] [PubMed] [Google Scholar]
- 167.Cideciyan AV, Sudharsan R, Dufour VL, Massengill MT, Iwabe S, Swider M, et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc Natl Acad Sci USA. (2018) 115:E8547–56. 10.1073/pnas.1805055115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Meindl A, Dry K, Herrmann K, Manson F, Ciccodicola A, Edgar A, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet. (1996) 13:35–42. 10.1038/ng0596-35 [DOI] [PubMed] [Google Scholar]
- 169.Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. (1998) 19:327–32. 10.1038/1214 [DOI] [PubMed] [Google Scholar]
- 170.Webb TR, Parfitt DA, Gardner JC, Martinez A, Bevilacqua D, Davidson AE, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet. (2012) 21:3647–54. 10.1093/hmg/dds194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kirschner R, Rosenberg T, Schultz-Heienbrok R, Lenzner S, Feil S, Roepman R, et al. RPGR transcription studies in mouse and human tissues reveal a retina-specific isoform that is disrupted in a patient with X-linked retinitis pigmentosa. Hum Mol Genet. (1999) 8:1571–8. 10.1093/hmg/8.8.1571 [DOI] [PubMed] [Google Scholar]
- 172.Vervoort R, Lennon A, Bird AC, Tulloch B, Axton R, Miano MG, et al. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. (2000) 25:462–6. 10.1038/78182 [DOI] [PubMed] [Google Scholar]
- 173.Pelletier V, Jambou M, Delphin N, Zinovieva E, Stum M, Gigarel N, et al. Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype-phenotype correlations and impact on genetic counseling. Hum Mutat. (2007) 28:81–91. 10.1002/humu.20417 [DOI] [PubMed] [Google Scholar]
- 174.Grover S, Fishman GA, Anderson RJ, Lindeman M. A longitudinal study of visual function in carriers of X-linked recessive retinitis pigmentosa. Ophthalmology. (2000) 107:386–96. 10.1016/S0161-6420(99)00045-7 [DOI] [PubMed] [Google Scholar]
- 175.Talib M, van Schooneveld MJ, Van Cauwenbergh C, Wijnholds J, Ten Brink JB, Florijn RJ, et al. The spectrum of structural and functional abnormalities in female carriers of pathogenic variants in the RPGR gene. Invest Ophthalmol Vis Sci. (2018) 59:4123–33. 10.1167/iovs.17-23453 [DOI] [PubMed] [Google Scholar]
- 176.Ogino K, Oishi M, Oishi A, Morooka S, Sugahara M, Gotoh N, et al. Radial fundus autofluorescence in the periphery in patients with X-linked retinitis pigmentosa. Clin Ophthalmol. (2015) 9:1467–74. 10.2147/OPTH.S89371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Comander J, Weigel-DiFranco C, Sandberg MA, Berson EL. Visual function in carriers of X-linked retinitis pigmentosa. Ophthalmology. (2015) 122:1899–906. 10.1016/j.ophtha.2015.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Salvetti AP, Nanda A, MacLaren RE. RPGR-related X-linked retinitis pigmentosa carriers with a severe “male pattern.”. Ophthalmologica. (2021) 244:60–7. 10.1159/000503687 [DOI] [PubMed] [Google Scholar]
- 179.Beltran WA, Cideciyan AV, Lewin AS, Iwabe S, Khanna H, Sumaroka A, et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci USA. (2012) 109:2132–7. 10.1073/pnas.1118847109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Cehajic-Kapetanovic J, Xue K, Martinez-Fernandez de la Camara C, Nanda A, Davies A, Wood LJ, et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med. (2020) 26:354–9. 10.1038/s41591-020-0763-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. (1993) 4:130–4. 10.1038/ng0693-130 [DOI] [PubMed] [Google Scholar]
- 182.Gal A, Orth U, Baehr W, Schwinger E, Rosenberg T. Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat Genet. (1994) 7:551. 10.1038/ng0894-551c [DOI] [PubMed] [Google Scholar]
- 183.Huang SH, Pittler SJ, Huang X, Oliveira L, Berson EL, Dryja TP. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat Genet. (1995) 11:468–71. 10.1038/ng1295-468 [DOI] [PubMed] [Google Scholar]
- 184.Kuehlewein L, Zobor D, Andreasson SO, Ayuso C, Banfi S, Bocquet B, et al. Clinical phenotype and course of PDE6A-associated retinitis pigmentosa disease, characterized in preparation for a gene supplementation trial. JAMA Ophthalmol. (2020) 138:1241–50. 10.1001/jamaophthalmol.2020.4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Fishman GA, Roberts MF, Derlacki DJ, Grimsby JL, Yamamoto H, Sharon D, et al. Novel mutations in the cellular retinaldehyde-binding protein gene (RLBP1) associated with retinitis punctata albescens: evidence of interfamilial genetic heterogeneity and fundus changes in heterozygotes. Arch Ophthalmol. (2004) 122:70–5. 10.1001/archopht.122.1.70 [DOI] [PubMed] [Google Scholar]
- 186.Eichers ER, Green JS, Stockton DW, Jackman CS, Whelan J, McNamara JA, et al. Newfoundland rod-cone dystrophy, an early-onset retinal dystrophy, is caused by splice-junction mutations in RLBP1. Am J Hum Genet. (2002) 70:955–64. 10.1086/339688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Burstedt MS, Sandgren O, Holmgren G, Forsman-Semb K. Bothnia dystrophy caused by mutations in the cellular retinaldehyde-binding protein gene (RLBP1) on chromosome 15q26. Invest Ophthalmol Vis Sci. (1999) 40:995–1000. [PubMed] [Google Scholar]
- 188.Naz S, Ali S, Riazuddin SA, Farooq T, Butt NH, Zafar AU, et al. Mutations in RLBP1 associated with fundus albipunctatus in consanguineous Pakistani families. Br J Ophthalmol. (2011) 95:1019–24. 10.1136/bjo.2010.189076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Choi VW, Bigelow CE, McGee TL, Gujar AN, Li H, Hanks SM, et al. AAV-mediated RLBP1 gene therapy improves the rate of dark adaptation in Rlbp1 knockout mice. Mol Ther Methods Clin Dev. (2015) 2:15022. 10.1038/mtm.2015.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.MacLachlan TK, Milton MN, Turner O, Tukov F, Choi VW, Penraat J, et al. Nonclinical safety evaluation of scAAV8-RLBP1 for treatment of RLBP1 retinitis pigmentosa. Mol Ther Methods Clin Dev. (2018) 8:105–20. 10.1016/j.omtm.2017.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. (2000) 26:270–1. 10.1038/81555 [DOI] [PubMed] [Google Scholar]
- 192.Tschernutter M, Jenkins SA, Waseem NH, Saihan Z, Holder GE, Bird AC, et al. Clinical characterisation of a family with retinal dystrophy caused by mutation in the Mertk gene. Br J Ophthalmol. (2006) 90:718–23. 10.1136/bjo.2005.084897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ghazi NG, Abboud EB, Nowilaty SR, Alkuraya H, Alhommadi A, Cai H, et al. Treatment of retinitis pigmentosa due to MERTK mutations by ocular subretinal injection of adeno-associated virus gene vector: results of a phase I trial. Hum Genet. (2016) 135:327–43. 10.1007/s00439-016-1637-y [DOI] [PubMed] [Google Scholar]
- 194.Fishman GA, Kumar A, Joseph ME, Torok N, Anderson RJ. Usher's syndrome. Ophthalmic and neuro-otologic findings suggesting genetic heterogeneity. Arch Ophthalmol. (1983) 101:1367–74. 10.1001/archopht.1983.01040020369005 [DOI] [PubMed] [Google Scholar]
- 195.Lopes VS, Gibbs D, Libby RT, Aleman TS, Welch DL, Lillo C, et al. The Usher 1B protein, MYO7A, is required for normal localization and function of the visual retinoid cycle enzyme, RPE65. Hum Mol Genet. (2011) 20:2560–70. 10.1093/hmg/ddr155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. (1998) 280:1753–7. 10.1126/science.280.5370.1753 [DOI] [PubMed] [Google Scholar]
- 197.Weston MD, Eudy JD, Fujita S, Yao S, Usami S, Cremers C, et al. Genomic structure and identification of novel mutations in usherin, the gene responsible for Usher syndrome type IIa. Am J Hum Genet. (2000) 66:1199–210. 10.1086/302855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rivolta C, Berson EL, Dryja TP. Paternal uniparental heterodisomy with partial isodisomy of chromosome 1 in a patient with retinitis pigmentosa without hearing loss and a missense mutation in the Usher syndrome type II gene USH2A. Arch Ophthalmol. (2002) 120:1566–71. 10.1001/archopht.120.11.1566 [DOI] [PubMed] [Google Scholar]
- 199.Seyedahmadi BJ, Rivolta C, Keene JA, Berson EL, Dryja TP. Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp Eye Res. (2004) 79:167–73. 10.1016/j.exer.2004.03.005 [DOI] [PubMed] [Google Scholar]
- 200.Hartel BP, Löfgren M, Huygen PL, Guchelaar I, Lo-A-Njoe Kort N, Sadeghi AM, et al. A combination of two truncating mutations in USH2A causes more severe and progressive hearing impairment in Usher syndrome type IIa. Hear Res. (2016) 339:60–8. 10.1016/j.heares.2016.06.008 [DOI] [PubMed] [Google Scholar]
- 201.Zallocchi M, Binley K, Lad Y, Ellis S, Widdowson P, Iqball S, et al. EIAV-based retinal gene therapy in the shaker1 mouse model for usher syndrome type 1B: development of UshStat. PLoS ONE. (2014) 9:e94272. 10.1371/journal.pone.0094272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Dulla K, Slijkerman R, van Diepen HC, Albert S, Dona M, Beumer W, et al. Antisense oligonucleotide-based treatment of retinitis pigmentosa caused by USH2A exon 13 mutations. Mol Ther. (2021) 29:2441s.o. 10.1101/2020.10.06.320499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Moosajee M, Ramsden SC, Black GC, Seabra MC, Webster AR. Clinical utility gene card for: choroideremia. Eur J Hum Genet. (2014) 22:4. 10.1038/ejhg.2013.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Simunovic MP, Jolly JK, Xue K, Edwards TL, Groppe M, Downes SM, et al. The spectrum of CHM gene mutations in choroideremia and their relationship to clinical phenotype. Invest Ophthalmol Vis Sci. (2016) 57:6033–9. 10.1167/iovs.16-20230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.MacDonald IM, Hume S, Zhai Y, Xu M. Choroideremia. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews((R)). Seattle, WA: University of Washington; (1993). [Google Scholar]
- 206.Roberts MF, Fishman GA, Roberts DK, Heckenlively JR, Weleber RG, Anderson RJ, et al. Retrospective, longitudinal, and cross sectional study of visual acuity impairment in choroideraemia. Br J Ophthalmol. (2002) 86:658–62. 10.1136/bjo.86.6.658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Jacobson SG, Cideciyan AV, Sumaroka A, Aleman TS, Schwartz SB, Windsor EA, et al. Remodeling of the human retina in choroideremia: rab escort protein 1 (REP-1) mutations. Invest Ophthalmol Vis Sci. (2006) 47:4113–20. 10.1167/iovs.06-0424 [DOI] [PubMed] [Google Scholar]
- 208.Romano F, Arrigo A, MacLaren RE, Charbel Issa P, Birtel J, Bandello F, et al. Hyperreflective foci as a pathogenetic biomarker in choroideremia. Retina. (2020) 40:1634–40. 10.1097/IAE.0000000000002645 [DOI] [PubMed] [Google Scholar]
- 209.Arrigo A, Romano F, Parodi MB, Charbel Issa P, Birtel J, Bandello F, et al. Reduced vessel density in deep capillary plexus correlates with retinal layer thickness in choroideremia. Br J Ophthalmol. (2021) 105:687–93. 10.1136/bjophthalmol-2020-316528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Battaglia Parodi M, Arrigo A, MacLaren RE, Aragona E, Toto L, Mastropasqua R, et al. Vascular alterations revealed with optical coherence tomography angiography in patients with choroideremia. Retina. (2019) 39:1200–5. 10.1097/IAE.0000000000002118 [DOI] [PubMed] [Google Scholar]
- 211.Seabra MC, Brown MS, Goldstein JL. Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl transferase. Science. (1993) 259:377–81. 10.1126/science.8380507 [DOI] [PubMed] [Google Scholar]
- 212.Schwartz M, Rosenberg T. Prenatal diagnosis of choroideremia. Acta Ophthalmol Scand Suppl. (1996) (219):33–6. 10.1111/j.1600-0420.1996.tb00381.x [DOI] [PubMed] [Google Scholar]
- 213.Yntema HG, van den Helm B, Kissing J, van Duijnhoven G, Poppelaars F, Chelly J, et al. A novel ribosomal S6-kinase (RSK4; RPS6KA6) is commonly deleted in patients with complex X-linked mental retardation. Genomics. (1999) 62:332–43. 10.1006/geno.1999.6004 [DOI] [PubMed] [Google Scholar]
- 214.Lorda-Sanchez IJ, Ibañez AJ, Sanz RJ, Trujillo MJ, Anabitarte ME, Querejeta ME, et al. Choroideremia, sensorineural deafness, and primary ovarian failure in a woman with a balanced X-4 translocation. Ophthalmic Genet. (2000) 21:185–9. 10.1076/1381-6810(200009)2131-ZFT185 [DOI] [PubMed] [Google Scholar]
- 215.Vasireddy V, Mills JA, Gaddameedi R, Basner-Tschakarjan E, Kohnke M, Black AD, et al. AAV-mediated gene therapy for choroideremia: preclinical studies in personalized models. PLoS ONE. (2013) 8:e61396. 10.1371/journal.pone.0061396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Moosajee M, Tracey-White D, Smart M, Weetall M, Torriano S, Kalatzis V, et al. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC-414 in human choroideremia fibroblasts and the nonsense-mediated zebrafish model. Hum Mol Genet. (2016) 25:3416–31. 10.1093/hmg/ddw184 [DOI] [PubMed] [Google Scholar]
- 217.MacLaren RE, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, Seymour L, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. (2014) 383:1129–37. 10.1016/S0140-6736(13)62117-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lam BL, Davis JL, Gregori NZ, MacLaren RE, Girach A, Verriotto JD, et al. Choroideremia gene therapy phase 2 clinical trial: 24-month results. Am J Ophthalmol. (2019) 197:65–73. 10.1016/j.ajo.2018.09.012 [DOI] [PubMed] [Google Scholar]
- 219.Dimopoulos IS, Hoang SC, Radziwon A, Binczyk NM, Seabra MC, MacLaren RE. Two-year results after AAV2-mediated gene therapy for choroideremia: the alberta experience. Am J Ophthalmol. (2018) 193:130–42. 10.1016/j.ajo.2018.06.011 [DOI] [PubMed] [Google Scholar]
- 220.Abbouda A, Avogaro F, Moosajee M, Vingolo EM. Update on gene therapy clinical trials for choroideremia and potential experimental therapies. Medicina (Kaunas) (2021) 57:64. 10.3390/medicina57010064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sikkink SK, Biswas S, Parry NR, Stanga PE, Trump D. X-linked retinoschisis: an update. J Med Genet. (2007) 44:225–32. 10.1136/jmg.2006.047340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Vainio-Mattila B, Eriksson AW, Forsius H. X-chromosomal recessive retinoschisis in the Region of Pori. An ophthalmo-genetical analysis of 103 cases. Acta Ophthalmol (Copenh). (1969) 47:1135–48. 10.1111/j.1755-3768.1969.tb02513.x [DOI] [PubMed] [Google Scholar]
- 223.Kellner U, Brummer S, Foerster MH, Wessing A. X-linked congenital retinoschisis. Graefes Arch Clin Exp Ophthalmol. (1990) 228:432–7. 10.1007/BF00927256 [DOI] [PubMed] [Google Scholar]
- 224.Mitamura Y, Miyanishi K, Shizukawa N, Tashimo A, Nakamura Y, Tagawa H, et al. A case of X-linked retinoschisis diagnosed in an infant. Retina. (2003) 23:731–2. 10.1097/00006982-200310000-00030 [DOI] [PubMed] [Google Scholar]
- 225.Peachey NS, Fishman GA, Derlacki DJ, Brigell MG. Psychophysical and electroretinographic findings in X-linked juvenile retinoschisis. Arch Ophthalmol. (1987) 105:513–6. 10.1001/archopht.1987.01060040083038 [DOI] [PubMed] [Google Scholar]
- 226.Riveiro-Alvarez R, Trujillo-Tiebas MJ, Gimenez-Pardo A, Garcia-Hoyos M, Lopez-Martinez MA, Aguirre-Lamban J, et al. Correlation of genetic and clinical findings in Spanish patients with X-linked juvenile retinoschisis. Invest Ophthalmol Vis Sci. (2009) 50:4342–50. 10.1167/iovs.09-3418 [DOI] [PubMed] [Google Scholar]
- 227.Apushkin MA, Fishman GA, Rajagopalan AS. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina. (2005) 25:612–8. 10.1097/00006982-200507000-00012 [DOI] [PubMed] [Google Scholar]
- 228.Tanimoto N, Usui T, Takagi M, Hasegawa S, Abe H, Sekiya K, et al. Electroretinographic findings in three family members with X-linked juvenile retinoschisis associated with a novel Pro192Thr mutation of the XLRS1 gene. Jpn J Ophthalmol. (2002) 46:568–76. 10.1016/S0021-5155(02)00539-7 [DOI] [PubMed] [Google Scholar]
- 229.Romano F, Arrigo A, Ch'ng SW, Battaglia Parodi M, Manitto MP, Martina E, et al. Capillary network alterations in X-linked retinoschisis imaged on optical coherence tomography angiography. Retina. (2019) 39:1761–7. 10.1097/IAE.0000000000002222 [DOI] [PubMed] [Google Scholar]
- 230.Grayson C, Reid SN, Ellis JA, et al. Retinoschisin, the X-linked retinoschisis protein, is a secreted photoreceptor protein, and is expressed and released by Weri-Rb1 cells. Hum Mol Genet. (2000) 9:1873–9. 10.1093/hmg/9.12.1873 [DOI] [PubMed] [Google Scholar]
- 231.Molday LL, Hicks D, Sauer CG, Weber BH, Molday RS. Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Invest Ophthalmol Vis Sci. (2001) 42:816–25. [PubMed] [Google Scholar]
- 232.Reid SN, Yamashita C, Farber DB. Retinoschisin, a photoreceptor-secreted protein, and its interaction with bipolar and muller cells. J Neurosci. (2003) 23:6030–40. 10.1523/JNEUROSCI.23-14-06030.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Wu WW, Wong JP, Kast J, Molday RS. RS1, a discoidin domain-containing retinal cell adhesion protein associated with X-linked retinoschisis, exists as a novel disulfide-linked octamer. J Biol Chem. (2005) 280:10721–30. 10.1074/jbc.M413117200 [DOI] [PubMed] [Google Scholar]
- 234.Wang T, Zhou A, Waters CT, O'Connor E, Read RJ, Trump D. Molecular pathology of X linked retinoschisis: mutations interfere with retinoschisin secretion and oligomerisation. Br J Ophthalmol. (2006) 90:81–6. 10.1136/bjo.2005.078048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bush RA, Zeng Y, Colosi P, Kjellstrom S, Hiriyanna S, Vijayasarathy C, et al. Preclinical dose-escalation study of intravitreal AAV-RS1 gene therapy in a mouse model of X-linked retinoschisis: dose-dependent expression and improved retinal structure and function. Hum Gene Ther. (2016) 27:376–89. 10.1089/hum.2015.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Janssen A, Min SH, Molday LL, Tanimoto N, Seeliger MW, Hauswirth WW, et al. Effect of late-stage therapy on disease progression in AAV-mediated rescue of photoreceptor cells in the retinoschisin-deficient mouse. Mol Ther. (2008) 16:1010–7. 10.1038/mt.2008.57 [DOI] [PubMed] [Google Scholar]
- 237.Min SH, Molday LL, Seeliger MW, Dinculescu A, Timmers AM, Janssen A, et al. Prolonged recovery of retinal structure/function after gene therapy in an Rs1h-deficient mouse model of x-linked juvenile retinoschisis. Mol Ther. (2005) 12:644–51. 10.1016/j.ymthe.2005.06.002 [DOI] [PubMed] [Google Scholar]
- 238.Marangoni D, Bush RA, Zeng Y, Wei LL, Ziccardi L, Vijayasarathy C, et al. Ocular and systemic safety of a recombinant AAV8 vector for X-linked retinoschisis gene therapy: GLP studies in rabbits and Rs1-KO mice. Mol Ther Methods Clin Dev. (2016) 5:16011. 10.1038/mtm.2016.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Cukras C, Wiley HE, Jeffrey BG, Sen HN, Turriff A, Zeng Y, et al. Retinal AAV8-RS1 gene therapy for X-linked retinoschisis: initial findings from a phase I/IIa trial by intravitreal delivery. Mol Ther. (2018) 26:2282–94. 10.1016/j.ymthe.2018.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Tanna P, Strauss RW, Fujinami K, Michaelides M. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol. (2017) 101:25–30. 10.1136/bjophthalmol-2016-308823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Voigt M, Querques G, Atmani K, Leveziel N, Massamba N, Puche N, et al. Analysis of retinal flecks in fundus flavimaculatus using high-definition spectral-domain optical coherence tomography. Am J Ophthalmol. (2010) 150:330–7. 10.1016/j.ajo.2010.04.001 [DOI] [PubMed] [Google Scholar]
- 242.Gomes NL, Greenstein VC, Carlson JN, Tsang SH, Smith RT, Carr RE, et al. A comparison of fundus autofluorescence and retinal structure in patients with Stargardt disease. Invest Ophthalmol Vis Sci. (2009) 50:3953–9. 10.1167/iovs.08-2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Klufas MA, Tsui I, Sadda SR, Hosseini H, Schwartz SD. Ultrawidefield autofluoresence in Abca4 Stargardt disease. Retina. (2018) 38:403–15. 10.1097/IAE.0000000000001567 [DOI] [PubMed] [Google Scholar]
- 244.Arrigo A, Romano F, Aragona E, di Nunzio C, Sperti A, Bandello F, et al. OCTA-based identification of different vascular patterns in Stargardt disease. Transl Vis Sci Technol. (2019) 8:26. 10.1167/tvst.8.6.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Arrigo A, Grazioli A, Romano F, Aragona E, Marchese A, Bordato A, et al. Multimodal evaluation of central and peripheral alterations in Stargardt disease: a pilot study. Br J Ophthalmol. (2020) 104:1234–8. 10.1136/bjophthalmol-2019-315148 [DOI] [PubMed] [Google Scholar]
- 246.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. (1997) 15:236–46. 10.1038/ng0397-236 [DOI] [PubMed] [Google Scholar]
- 247.Yi J, Li S, Jia X, Xiao X, Wang P, Guo X, et al. Evaluation of the ELOVL4, PRPH2 and ABCA4 genes in patients with Stargardt macular degeneration. Mol Med Rep. (2012) 6:1045–9. 10.3892/mmr.2012.1063 [DOI] [PubMed] [Google Scholar]
- 248.Zhang X, Ge X, Shi W, Huang P, Min Q, Li M, et al. Molecular diagnosis of putative Stargardt disease by capture next generation sequencing. PLoS One. (2014) 9:e95528. 10.1371/journal.pone.0095528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Fujinami K, Lois N, Mukherjee R, McBain VA, Tsunoda K, Tsubota K, et al. A longitudinal study of Stargardt disease: quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Invest Ophthalmol Vis Sci. (2013) 54:8181–90. 10.1167/iovs.13-12104 [DOI] [PubMed] [Google Scholar]
- 250.Riveiro-Alvarez R, Lopez-Martinez MA, Zernant J, Aguirre-Lamban J, Cantalapiedra D, Avila-Fernandez A, et al. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: retrospective analysis in 420 Spanish families. Ophthalmology. (2013) 120:2332–7. 10.1016/j.ophtha.2013.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Fujinami K, Sergouniotis PI, Davidson AE, Mackay DS, Tsunoda K, Tsubota K, et al. The clinical effect of homozygous ABCA4 alleles in 18 patients. Ophthalmology. (2013) 120:2324–31. 10.1016/j.ophtha.2013.04.016 [DOI] [PubMed] [Google Scholar]
- 252.Cella W, Greenstein VC, Zernant-Rajang J, Smith TR, Barile G, Allikmets R, et al. G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull's eye maculopathy. Exp Eye Res. (2009) 89:16–24. 10.1016/j.exer.2009.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lambertus S, van Huet RA, Bax NM, Hoefsloot LH, Cremers FP, Boon CJ, et al. Early-onset stargardt disease: phenotypic and genotypic characteristics. Ophthalmology. (2015) 122:335–44. 10.1016/j.ophtha.2014.08.032 [DOI] [PubMed] [Google Scholar]
- 254.Simonelli F, Testa F, Zernant J, Nesti A, Rossi S, Allikmets R, et al. Genotype-phenotype correlation in Italian families with Stargardt disease. Ophthalmic Res. (2005) 37:159–67. 10.1159/000086073 [DOI] [PubMed] [Google Scholar]
- 255.Gemenetzi M, Lotery AJ. Phenotype/genotype correlation in a case series of Stargardt's patients identifies novel mutations in the ABCA4 gene. Eye (Lond). (2013) 27:1316–9. 10.1038/eye.2013.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zernant J, Lee W, Collison FT, Fishman GA, Sergeev YV, Schuerch K, et al. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J Med Genet. (2017) 54:404–12. 10.1136/jmedgenet-2017-104540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Braun TA, Mullins RF, Wagner AH, Andorf JL, Johnston RM, Bakall BB, et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet. (2013) 22:5136–45. 10.1093/hmg/ddt367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Bauwens M, De Zaeytijd J, Weisschuh N, Kohl S, Meire F, Dahan K, et al. An augmented ABCA4 screen targeting noncoding regions reveals a deep intronic founder variant in Belgian Stargardt patients. Hum Mutat. (2015) 36:39–42. 10.1002/humu.22716 [DOI] [PubMed] [Google Scholar]
- 259.Kong J, Kim SR, Binley K, Pata I, Doi K, Mannik J, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. (2008) 15:1311–20. 10.1038/gt.2008.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Dyka FM, Molday LL, Chiodo VA, Molday RS, Hauswirth WW. Dual ABCA4-AAV vector treatment reduces pathogenic retinal A2E accumulation in a mouse model of autosomal recessive Stargardt disease. Hum Gene Ther. (2019) 30:1361–70. 10.1089/hum.2019.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Sun D, Sun W, Gao SQ, Wei C, Naderi A, Schilb AL, et al. Formulation and efficacy of ECO/pRHO-ABCA4-SV40 nanoparticles for nonviral gene therapy of Stargardt disease in a mouse model. J Control Release. (2021) 330:329–40. 10.1016/j.jconrel.2020.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Sun D, Schur RM, Sears AE, Gao SQ, Vaidya A, Sun W, et al. Non-viral gene therapy for Stargardt disease with ECO/pRHO-ABCA4 self-assembled nanoparticles. Mol Ther. (2020) 28:293–303. 10.1016/j.ymthe.2019.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Glover DJ, Lipps HJ, Jans DA. Towards safe, non-viral therapeutic gene expression in humans. Nat Rev Genet. (2005) 6:299–310. 10.1038/nrg1577 [DOI] [PubMed] [Google Scholar]
- 264.Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol. (2004) 88:291–7. 10.1136/bjo.2003.027102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Aboshiha J, Dubis AM, Carroll J, Hardcastle AJ, Michaelides M. The cone dysfunction syndromes. Br J Ophthalmol. (2016) 100:115–21. 10.1136/bjophthalmol-2014-306505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Hirji N, Aboshiha J, Georgiou M, Bainbridge J, Michaelides M. Achromatopsia: clinical features, molecular genetics, animal models and therapeutic options. Ophthalmic Genet. (2018) 39:149–57. 10.1080/13816810.2017.1418389 [DOI] [PubMed] [Google Scholar]
- 267.Andreasson S, Tornqvist K. Electroretinograms in patients with achromatopsia. Acta Ophthalmol (Copenh). (1991) 69:711–6. 10.1111/j.1755-3768.1991.tb02048.x [DOI] [PubMed] [Google Scholar]
- 268.Thiadens AA, Slingerland NW, Roosing S, van Schooneveld MJ, van Lith-Verhoeven JJ, van Moll-Ramirez N, et al. Genetic etiology and clinical consequences of complete and incomplete achromatopsia. Ophthalmology. (2009) 116:1984.e1–9.e1. 10.1016/j.ophtha.2009.03.053 [DOI] [PubMed] [Google Scholar]
- 269.Sundaram V, Wilde C, Aboshiha J, Cowing J, Han C, Langlo CS, et al. Retinal structure and function in achromatopsia: implications for gene therapy. Ophthalmology. (2014) 121:234–45. 10.1016/j.ophtha.2013.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Aboshiha J, Dubis AM, Cowing J, Fahy RT, Sundaram V, Bainbridge JW, et al. A prospective longitudinal study of retinal structure and function in achromatopsia. Invest Ophthalmol Vis Sci. (2014) 55:5733–43. 10.1167/iovs.14-14937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Scoles D, Flatter JA, Cooper RF, Langlo CS, Robison S, Neitz M, et al. Assessing photoreceptor structure associated with ellipsoid zone disruptions visualized with optical coherence tomography. Retina. (2016) 36:91–103. 10.1097/IAE.0000000000000618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Remmer MH, Rastogi N, Ranka MP, Ceisler EJ. Achromatopsia: a review. Curr Opin Ophthalmol. (2015) 26:333–40. 10.1097/ICU.0000000000000189 [DOI] [PubMed] [Google Scholar]
- 273.Aligianis IA, Forshew T, Johnson S, Michaelides M, Johnson CA, Trembath RC, et al. Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the alpha subunit of cone transducin (GNAT2). J Med Genet. (2002) 39:656–60. 10.1136/jmg.39.9.656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Thiadens AA, den Hollander AI, Roosing S, Nabuurs SB, Zekveld-Vroon RC, Collin RW, et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am J Hum Genet. (2009) 85:240–7. 10.1016/j.ajhg.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Kohl S, Coppieters F, Meire F, Schaich S, Roosing S, Brennenstuhl C, et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am J Hum Genet. (2012) 91:527–32. 10.1016/j.ajhg.2012.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kohl S, Zobor D, Chiang WC, Weisschuh N, Staller J, Gonzalez Menendez I, et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat Genet. (2015) 47:757–65. 10.1038/ng.3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Mühlfriedel R, Tanimoto N, Schön C, Sothilingam V, Garcia Garrido M, Beck SC, et al. AAV-mediated gene supplementation therapy in achromatopsia type 2: preclinical data on therapeutic time window and long-term effects. Front Neurosci. (2017) 11:292. 10.3389/fnins.2017.00292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Michalakis S, Mühlfriedel R, Tanimoto N, Krishnamoorthy V, Koch S, Fischer MD, et al. Restoration of cone vision in the CNGA3−/− mouse model of congenital complete lack of cone photoreceptor function. Mol Ther. (2010) 18:2057–63. 10.1038/mt.2010.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Carvalho LS, Xu J, Pearson RA, Smith AJ, Bainbridge JW, Morris LM, et al. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum Mol Genet. (2011) 20:3161–75. 10.1093/hmg/ddr218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Pang JJ, Deng WT, Dai X, Lei B, Everhart D, Umino Y, et al. AAV-mediated cone rescue in a naturally occurring mouse model of CNGA3-achromatopsia. PLoS ONE. (2012) 7:e35250. 10.1371/journal.pone.0035250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Alexander JJ, Umino Y, Everhart D, Chang B, Min SH, Li Q, et al. Restoration of cone vision in a mouse model of achromatopsia. Nat Med. (2007) 13:685–7. 10.1038/nm1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Gootwine E, Abu-Siam M, Obolensky A, Rosov A, Honig H, Nitzan T, et al. Gene augmentation therapy for a missense substitution in the cGMP-binding domain of ovine CNGA3 gene restores vision in day-blind sheep. Invest Ophthalmol Vis Sci. (2017) 58:1577–84. 10.1167/iovs.16-20986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Banin E, Gootwine E, Obolensky A, Ezra-Elia R, Ejzenberg A, Zelinger L, et al. Gene augmentation therapy restores retinal function and visual behavior in a sheep model of CNGA3 achromatopsia. Mol Ther. (2015) 23:1423–33. 10.1038/mt.2015.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Komáromy AM, Rowlan JS, Corr AT, Reinstein SL, Boye SL, Cooper AE, et al. Transient photoreceptor deconstruction by CNTF enhances rAAV-mediated cone functional rescue in late stage CNGB3-achromatopsia. Mol Ther. (2013) 21:1131–41. 10.1038/mt.2013.50 [DOI] [PMC free article] [PubMed] [Google Scholar]