

Abstract
Chandipura virus (CHPV) is an emerging human pathogen of great clinical significance. In this study, we have investigated the susceptibility pattern of both normal and cancer cell lines of human origin to wild-type (wt) CHPV in order to explore the possibility of developing CHPV as an oncolytic vector (OV). Marked cytopathic effect along with enhanced virus output was observed in cancer cell lines (HeLa, A549, U-138, PC-3, and HepG2) in comparison to normal human adult dermal fibroblast (HADF) cells. At an MOI of 0.1, cancer cell lines were differentially susceptible to CHPV, with cells like HeLa and U-138 having pronounced cell death, while the PC-3 were comparatively resistant. All cell lines used in the study except U-138 restricted CHPV infection to varying degrees with IFN-β pre-treatment and supplementation of interferon (IFN) could neither activate the IFN signaling pathway in U-138 cells. Finally, U-138 tumor xenografts established in non-obese diabetic severe combined immunodeficiency (NOD/SCID) mice showed significant delay in tumor growth in the CHPV-challenged animals. Thus, targeted cytopathic effect in cancer cells at a very low dose with restricted replication in normal cells offers a rationale to exploit CHPV as an oncolytic vector in the future.
Keywords: Chandipura virus, cytopathic property, oncolytic vector, interferon-β, tumor growth and regression, U-138 glioma cells, xenograft model, anti-viral response, virus induced cytotoxicity
Graphical abstract
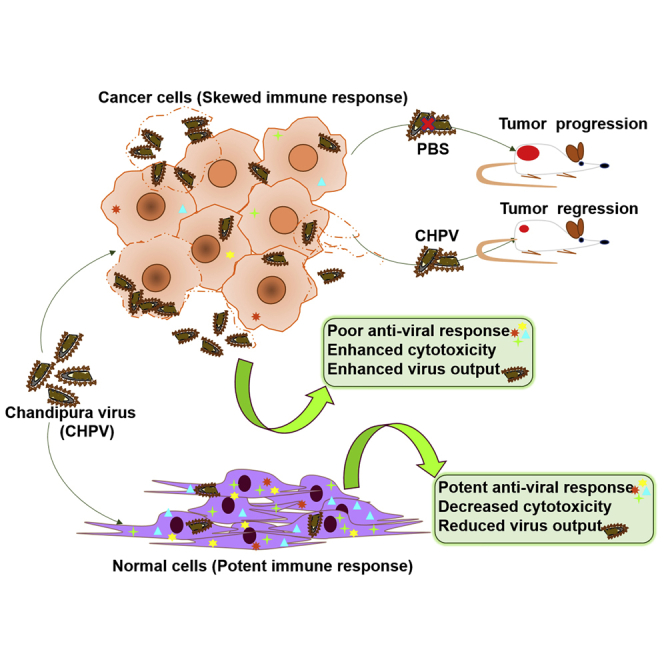
Dissecting the oncolytic property of uncharacterized viruses is pivotal in developing novel oncolytic vectors (OVs). Investigation into effects of Chandipura virus infection in cell lines of human origin and xenograft tumor model revealed marked susceptibility of U-138 glioma cells. Normal cells being relatively resistant offers the rationale for developing CHPV-OV.
Introduction
Chandipura virus (CHPV), a vector-borne vesiculovirus, is a potent neurotropic virus, with ∼55%–77% mortality reported in children.1, 2, 3, 4 Distantly related to vesicular stomatitis virus (VSV), CHPV harbors a negative-sense single-stranded RNA that encodes the glycoprotein (G), matrix protein (M), nucleoprotein (N), phosphoprotein (P), and large polymerase protein (L).5,6 CHPV has been shown to infect a range of cells of both insect and vertebrate origin. Insect cell lines from Aedes aegypti and Phlebotomus papatasi and vertebrate cell lines, Vero E6, rhabdomyosarcoma, and porcine stable kidney cells were found to be readily susceptible to CHPV. Although CHPV grew to significant titers in all the cell lines, a marked cytopathic effect was noticed in only vertebrate cells.7 The susceptibility of mouse and human neuroblastoma cells, Neuro-2a and SH-SY5Y, to CHPV and the histopathological analysis of brain sections of mouse challenged with CHPV suggest that CHPV can readily infect cells of the central nervous system, most notably the neurons.8, 9, 10 Being an emerging virus, many aspects of the molecular mechanism of CHPV pathogenesis, including cell susceptibility, mechanism of cell death, host antiviral response, and modulation of host responses by the virus are poorly understood. Our study focuses on understanding the cytopathic properties of CHPV in normal and various cancer cell lines of human origin and deciphering the possibility of exploiting this property of CHPV for oncolytic purposes.
The lytic property of cytopathic viruses is a pathogenic determinant essential for the replication and dissemination of these viruses. A rapidly evolving area is the utilization of cytopathic viruses to target cancers termed as “oncolytic virotherapy.” The employment of wild-type (wt) virus is the first important step in testing the feasibility of exploiting a particular virus for oncolytic therapy. Many wild-type viruses from different families have been tested for their oncolytic potential, which formed the basis for developing them into safe oncolytic vectors (OVs).11, 12, 13 The importance of oncolytic virotherapy is highlighted by many vectors that are in various stages of clinical trial or in the market.14 For example, a genetically engineered herpes simplex virus named Talimogene laherparepvec (T-VEC) is the first clinically approved oncolytic vector for use in the United States and European Union for the treatment against advanced melanoma.15 Currently, many viruses belonging to the Rhabdoviridae family are being exploited for their oncolytic potential. Because of their relatively small genome, which facilitates genetic manipulation with ease, simple life cycle restricted only to the cytoplasm, lack of pre-existing immunity in the majority of the human population, broad tropism, and the ability to produce a very high viral yield, rhabdoviruses are considered as potential candidates for oncolytic virotherapy. Investigations into the oncolytic potential of many rhabdoviruses, including VSV and Maraba virus (MV), were first focused on the wild-type virus. Several genetically modified VSV-based oncolytic vectors have been developed so far and have shown promising results in preclinical and clinical trials.16, 17, 18, 19 These observations form the basis of our rationale to investigate the oncolytic potential of CHPV. So far, a detailed investigation into the efficacy of CHPV as an oncolytic vector in cancer cell lines of human origin has not been carried out. All these necessitated an in-depth analysis of the oncolytic potential of CHPV in cancer cell lines of human origin.
Our results show that while cancer cell lines and normal cells of human origin are permissive to CHPV, not all cells are equally susceptible. CHPV was found to be highly cytopathic in human glioblastoma cells, U-138, while normal human adult dermal fibroblast (HADF) cells showed significant resistance. We correlated the increased cytopathicity observed in the susceptible cells to the differential antiviral responses induced by type I interferon (IFN) as reported earlier with VSV.20,21 A defect in IFN signaling in U-138 cells led to marked susceptibility of these cells to CHPV, as IFN-β pre-treatment had no protective effect while HADF cells showed remarkable resistance upon IFN-β sensitization. In an in vivo U-138 xenograft model in non-obese diabetic severe combined immunodeficiency (NOD/SCID) mice, tumor progression was significantly restricted and delayed in CHPV-challenged animals compared to the control animals. The selective sensitivity of the cells of glial origin opposed to the natural resistance of normal cells to CHPV infection at a low MOI offers the exciting proposition of exploiting CHPV as an oncolytic vector.
Results
Normal fibroblast cells show delayed susceptibility to CHPV compared to cancer cells
CHPV being cytopathic in nature, the susceptibility of a range of cancer cells to CHPV was compared against normal HADF cells. Preliminary investigations revealed that an MOI of 0.1 was sufficient to induce significant morphological changes in most of the cancer cell lines under investigation. The cancer cell lines chosen based on their origin included U-138 (brain), A549 (lung), HeLa (cervical), HepG2 (liver), PC-3 (prostate), and HADF (normal cells from human skin). Upon attaining 80% confluency, cells were infected with CHPV at an MOI of 0.1. Post-infection, morphological changes induced by CHPV were monitored for 48 h, and phase-contrast images were acquired at hourly intervals. At 48 hours post infection (hpi), most of the cancer cells showed remarkable cell rounding with loss of adherence, which was less pronounced in the case of HADF cells (Figure 1A). The onset and progression of morphological changes post-infection varied even among the cancer cell lines. The cytopathic effect of CHPV was more pronounced in U-138 and HeLa cells, with rounding of cells observed as early as 6 and 9 hpi, respectively. Among the other cancer cell lines, visible cytopathic effects appeared around 9 hpi for A549 and HepG2 cells, while the same was observed around 15 hpi in PC-3 cells. CHPV-induced cytopathic effects were largely delayed in HADF cells, with visible cell rounding occurring by 24 h. Surprisingly, only 20%–30% of cells had altered morphology even at 48 hpi (Figure 1A).
Figure 1.

Chandipura virus (CHPV) infection shows differential susceptibility and reduced cell viability among various cell lines
(A) Human cancer cell lines U-138, A549, HeLa, HepG2, PC-3, and a primary normal cell line HADF (indicated on the left side of each panel) were infected with 0.1 MOI of CHPV for 48 h, and the morphological changes were monitored using bright-field microscopy. Left panel indicates mock-infected cells and the right panel that of cells infected with 0.1 MOI of CHPV. Note the marked cell rounding and the complete disruption of the monolayer in the CHPV-infected cancer cells in comparison to HADF cells. Images were acquired at a magnification of ×100 and scale bars represent 100 μm. (B and C) Cell viability assay to quantify the cytolytic potential of CHPV. In vitro cell viability assay was carried out on all the selected cells after infecting the cells with 0.1, 1, 5, and 10 MOI of CHPV. Percentage of cell viability was calculated at 24 h (B) and 48 h (C) post-infection with respect to the mock-infected control. The degree of cytotoxicity was varied with the type of cell, MOI of the virus, and the time after infection (see also Figure S1). The bar represents the standard error of mean (SEM) of two independent experiments performed in triplicate; ∗p ≤ 0.05, ∗∗p ≤ 0.01, and ∗∗∗p ≤ 0.001.
To test whether the morphological alterations corresponded to CHPV-induced cytotoxicity, cell viability (MTT) assay was carried out at MOIs of 0.1, 1, 5, and 10 for 24 and 48 h. At the 24 h time point, cytotoxicity was found to be directly dependent on the MOI in the case of A549, HeLa, PC-3, and HADF cells, while U-138 and HepG2 cells did not follow the pattern. At an MOI of 0.1, >80% cell survival was observed in all the cell lines except U-138 at 24 hpi. Interestingly, at 24 hpi, while HeLa cells recorded <10% cell viability at all the MOIs tested other than 0.1, ∼60% of cells were viable in U-138 cells at all MOIs (Figure 1B). By 48 h, the highest degree of resistance was noticed in HADF cells followed by HepG2 and PC-3 when compared to U-138, A549, and HeLa cells (Figure 1C). To further confirm CHPV-induced cytotoxicity, live-dead cell imaging assay was carried out on all the cell lines at an MOI of 0.1, 1, 5, and 10. Fluorescent microscopic analysis of the cells at 48 h post-infection showed that the cytotoxic effects of CHPV matched our observations with phase-contrast microscopy and MTT assay (Figure S1). The observation that U-138 cells are susceptible to CHPV at a low MOI of 0.1 suggests that tumor cells of glial origin are highly susceptible to CHPV.
CHPV exhibits delayed growth kinetics in normal cells compared to cancer cells
Having observed that CHPV-induced morphological changes were significant in cancer cell lines when compared to HADF, both single- and multi-step growth kinetics of CHPV were assessed in these cells. For the multi-step growth curve, supernatants collected after infecting the respective cells at 0.1 MOI for 3, 6, 9, 12, and 24 h were used, while the supernatants from the 5 MOI infections were used for single-step growth curve analysis. Plaque assay results showed that the growth kinetics of CHPV in PC-3 and HADF cells followed a typical single- and multi-step growth pattern, unlike the other cell lines. Culture supernatants from U-138, A549, HeLa, and HepG2 cells had saturating limits of CHPV at both the MOIs by ∼12–24 h post-infection, although at 0.1 MOI the levels of virus generated were relatively lower in A549, HeLa, and HepG2 cells than at 5 MOI. Interestingly, significant difference in virus titer was observed between HADF and the cancer cell lines at 0.1 MOI compared to 5 MOI. At the 24 h time point, among the cancer cell lines the viral titers were found to be consistently high at both the MOIs; however, marked differences were noticed in U-138 and HeLa cells. The growth kinetics besides correlating with microscopic observations also clearly suggest that cancer cells support CHPV growth to a greater extent than that of normal cells (Figure 2).
Figure 2.

Growth kinetics of CHPV in different cell lines
A comparative analysis of the replication pattern of CHPV in different cell lines was performed by plaque assay. Viral titer was estimated at different time points (3, 6, 9, 12, and 24 hpi) after infection with a low (0.1) and high MOI (5) of CHPV followed by the generation of single- and multi-step growth curves. A marked difference in the replication pattern of CHPV was observed among the various cell lines, noticed most prominently in the growth curves of PC-3 and HADF cells. Each data point represents the mean ± SEM of three independent experiments.
Induction of apoptosis by CHPV is different across cell lines
The mechanism of CHPV-mediated cytotoxicity was investigated by probing for apoptotic markers caspase-3 and PARP1 in mock- and CHPV-infected cells by immunoblotting. Generation of cleaved caspase-3 from procaspase-3 in the CHPV-infected cells was not found to be uniform across cell lines. While U-138 and HeLa cells showed substantial conversion of procaspase-3 at both 12 and 24 hpi, only a modest conversion was observed in A549 and HepG2 cells, with no notable change observed in the case of both PC-3 and HADF (Figure 3).
Figure 3.

Apoptosis contributes to CHPV-induced cytotoxicity
Mock- and CHPV-infected lysates at 12 and 24 h (for cancer cells) and 12, 24, 36, and 48 h (for HADF cells) were collected and probed with antibodies against select apoptotic markers by western blotting. Conversion of PARP1 into cleaved PARP1 is clearly evident in all the cell lines infected with CHPV. Compared to cancer cells, a significant delay in PARP cleavage can be visualized in HADF cells. Conversion of caspase-3 into the cleaved caspase-3 in the CHPV-infected cells varied across cell lines. M indicates the matrix protein of CHPV, and β-actin was used as the loading control. The blots are representative of three independent experiments. The role of apoptosis in CHPV-induced cytotoxicity could be established in the HeLa cells using HeLa Bcl-2 cell line (Figure S2),
In contrast, conversion of PARP1 into cleaved PARP1 was observed in all the cell lines infected with CHPV. Almost complete conversion of PARP1 into its cleaved form was observed in A549 and HeLa cells at 24 hpi. While CHPV-infected HADF cells also demonstrated PARP1 cleavage, there was a substantial delay, with detectable cleavage occurring as late as 36 hpi.
The appearance of the cleaved products of the apoptotic markers corresponded with the levels of CHPV matrix protein (M). Thus, at an MOI of 0.1, CHPV induces apoptotic-pathway-dependent cytotoxicity in all the cancer cells tested; however, this effect was significantly delayed in HADF cells.
To further test if CHPV-induced cytotoxicity is via apoptosis, cell viability assays were performed on CHPV-infected HeLa cells and compared against Bcl-2 overexpressing HeLa cells. Bcl-2 is an apoptosis regulator protein, and the overexpression of the same can inhibit the mechanism of apoptosis. The cells were infected with CHPV at an MOI of 1, and the viability was calculated at 24 hpi. Compared to the regular HeLa cells, HeLa-Bcl-2 cells were found to be more viable, with greater than 50% reduction in the cell death (Figure S2). These observations thus highlight that CHPV-induced cytotoxicity is in part dependent on apoptosis.
IFN-β pre-treatment promotes resistance to CHPV infection differentially in cancer and normal cells
Sensitivity to IFN (α and β)-mediated responses has been well established in the prototypic rhabdovirus VSV. Having observed that the panel of cell lines tested is sensitive to CHPV to varying degrees, it was imperative to test if supplementation of IFN-β prior to CHPV infection can confer protection to these cells from the virus. Thus, cells were treated with 1,000 U of IFN-β for 24 h followed by infection with 0.1 MOI of CHPV, and the morphological changes were observed at 48 hpi using phase-contrast microscopy (Figure 4A).
Figure 4.

IFN-β pre-treatment differentially protects cells from CHPV infection
The protective effect of IFN-β on cells during CHPV infection was studied using microscopy and MTT assay. (A) Phase-contrast microscopy. Images acquired 48 hpi show the protective effect of IFN-β pre-treatment in all the cell lines infected with CHPV except U-138 cells. Note the marked reduction in the alterations in morphology of IFN-β-treated cells (right panels) in comparison to the untreated (left panels), while no difference is observed in the case of U-138 cells (left top two panels). HADF cells can be seen virtually unaffected by CHPV post-IFN-β treatment (right, bottom panel). Images were acquired at a magnification of ×100, and scale bars represent 100 μm. (B) MTT assay. All the cells as indicated on the x axis were infected with 0.1 MOI of CHPV either with or without IFN-β treatment followed by cell viability assay 48 hpi. The percentage of metabolically active cells was significantly higher in the IFN-β pre-treated cells compared to the control CHPV-infected cells. Data represent the average of six data points ± SD from two independent experiments (see also Figure S3). (C) Plaque assay. Cell culture supernatant collected 48 hpi was subjected to plaque assay to estimate the viral titer. Supernatants from the IFN-β pre-treated groups (gray bars) had significantly less virus output compared to the untreated (black bars) except in the case of U-138 cells. Data represent the mean ± SD of three independent experiments. ∗∗∗p ≤ 0.001,∗∗p ≤ 0.01, ∗p ≤ 0.05; ns indicates non-significance.
IFN-β pre-treatment had a profound effect on most of the cancer cells as well as HADF cells, while the control A549, HeLa, HepG2, and PC-3 cells showed significant changes in morphology. Post-CHPV infection, the cells primed with IFN resisted the cytopathic effects of CHPV to a great extent. This was marked by the presence of a sizeable population of adherent cells with normal morphology, while very few cells had altered morphology. While the cytopathic effect of 0.1 MOI of CHPV on HADF was minimal, IFN-β pre-treatment further increased the resistance of these cells to CHPV. Among all the cell lines tested, U-138 was distinct, as IFN-β pre-treatment had little to no effect in protecting the cells from CHPV, where most of the cells showed altered morphology with extensive cell rounding and shrinkage similar to CHPV-infected cells. (Figure 4A).
IFN-β pre-treated cells were infected with CHPV (0.1 MOI) for 48 h, and cell viability assay was carried out. The percentage of metabolically active cells was normalized against the corresponding mock-infected control (Figure 4B). Cell viability assay results mirrored the microscopic observations in Figure 4A. Most of the IFN-β pre-treated cell types showed >80% cell viability upon CHPV infection. HADF cells were found to be the most viable, with >95% cell viability in both the IFN-β untreated and pre-treated cells. In contrast, IFN-β pre-treatment did not have any effect on U-138 cells. Live-dead staining assay results correlated with the MTT assay results (Figure S3).
To further investigate if viability of the IFN-β pre-treated cells was due to restricted virus replication and output, the culture supernatant was collected from 24 hpi and subjected to plaque assay to estimate the viral titer. A comparative analysis of the viral titer between the untreated and the IFN-β pre-treated groups of all the cell lines except that of U-138 cells showed significant reduction in the viral titer in the case of treated cells (Figure 4C). The log difference (log 10) between the groups in the case of A549, HeLa, HepG2, PC-3, and HADF was found to be 6, 4, 4, 7, and 6. The difference between these individual groups was found to be statistically significant except in U-138, where it was insignificant. Upon comparing the viral titer in the IFN-β pre-treated, CHPV-challenged HADF cells to other cancer cell lines with similar treatment, the viral titer was significantly higher in cancer cells. Thus, sensitization of cancer cells with IFN-β prior to CHPV challenge can restrict virus replication in a cell-line-specific manner.
Sensitivity of U-138 cells to CHPV infection is due to impaired IFN response and STAT1 activation
Results obtained from our previous experiments clearly demonstrate a strong dependency on IFN to limit CHPV infection with marked variability among the cancer cells studied. While IFN-β pre-treatment did not effectively restrict CHPV replication and cell death in U-138 cells, the opposite effect was seen in the normal HADF cells. The lack of protection of U-138 cells from CHPV, even after the administration of exogenous IFN-β, prompted us to investigate the status of the IFN signaling in those cells. Phosphorylation of STAT1 is a critical step in the IFN signaling pathway.
We tested the natural response of the cells under investigation to IFN treatment in the absence of a bona fide CHPV infection. The whole-cell lysates collected with and without IFN-β treatment were probed for STAT1 and phospho-STAT1 (pSTAT1) level. Differential levels of phosphorylated STAT1 were observed upon IFN-β treatment in all the cell lines except U-138 (Figure 5A).
Figure 5.

IFN-β-mediated STAT1 activation is arrested in U-138 cells
(A) Monolayers of the indicated cells were treated with 1,000 U/mL IFN-β for 24 h, and the levels of STAT1 and pSTAT1 were detected by immunoblotting. IFN-β treatment resulted in the conversion of STAT1 into pSTAT1 in all the cells except U-138 cells. (B) The change in expression levels of STAT1 and conversion into pSTAT1 was also checked in CHPV-infected cells by immunoblotting. The + above the panel indicates lanes containing CHPV-infected lysates, while − indicates those from uninfected lysates. Since HADF cells are slow to respond to CHPV infection, an additional 48 hpi sample, indicated by the encircled +, was used for the assay. β-actin served as the loading control for both the experiments, and the blots are representative images of three independent experiments.
The enhanced phosphorylation of STAT1 in PC-3 and HADF cells in response to IFN-β treatment correlated well with the decreased virus output and increased survival in these cells upon sensitization with IFN-β. We also tested the phosphorylation status of the STAT1 protein in the infected lysates to check whether the cells can activate the IFN pathway in response to CHPV infection. As expected, CHPV infection did not induce phosphorylation of STAT1 in U-138 cells (Figure 5B). Other cells that exhibited a similar pattern included HeLa and HepG2 cells, while all the other cell lines showed STAT1 phosphorylation. Normal HADF cells showed a delay in the activation of STAT1 that correlated with the time of detection of CHPV proteins. The low levels of endogenous STAT1 and the absence of IFN-β induced pSTAT1 during CHPV infection sets apart U-138 cells as an excellent candidate for testing the oncolytic potential of CHPV.
CHPV suppresses tumor growth in U-138 xenografts
In order to test the efficacy of CHPV in restricting the tumor growth, we established a U-138 xenograft tumor model in male NOD/SCID mice. Initial standardization experiments were carried out to identify the number of cells and the time required to generate palpable tumors and optimize the concentration of CHPV required. Most animals developed palpable tumors within 2 weeks of injecting 5 × 105 U-138 cells subcutaneously. On day 15, when the tumors had a mean volume of ∼250 mm3, a single dose of 1 × 106 pfu of wild-type CHPV was administered intra-tumorally in the test mice, while the control animals received PBS only. Tumor size was measured daily, and the overall health of the animals was recorded. A steady increase in tumor volume was noted in the PBS control group, while the tumor volume appeared to be maintained around 300 mm3 in the CHPV-challenged group. By day 9 post-challenge, the mean tumor volume in the control group was found to be as high as 1,500 mm3, while that in the CHPV-challenged group remained unchanged. Thus, CHPV challenge limited tumor growth significantly (Figures 6A and 6B).
Figure 6.

CHPV restricts tumor progression in a U-138 xenograft tumor model
(A) Schematic depicting the experimental steps involved and the timeline. U-138 xenograft tumors were established by injecting 5 × 105 cells in the right flank of NOD/SCID mice. Day 15 post-implantation, and upon the tumors reaching a palpable size, they were challenged either with 106 pfu of CHPV or the vehicle PBS alone. (B) Tumor volume curve. Tumor volume was measured daily until the 9th day, and the curve was constructed by plotting the tumor volume against the day measured. The endpoint was set as the day when the tumors in the control animals reached the ethical cutoff volume of 1,500 mm3. ∗∗p ≤ 0.01. (C) Images of the resected tumors from 3 different animals from both groups. A marked difference in the tumor size between the two groups is evident, with the tumors in control animals significantly larger than those of the CHPV-challenged animals. (D) Plaque assay. Plaque assays were carried out on the homogenates of the excised tumors. The data represent the average ± SD of the virus titer from the tumors of six individual animals. ## indicates the absence of virus in the PBS control group.
On day 24, when the average tumor volume in the PBS-treated control animals corresponded to the Institutional Animal Ethics Committee (IAEC)-approved ethical endpoint (1,500 mm3), all the mice from both the groups were euthanized, and the tumors were resected out. Even though variation in tumor size was noticed in the virus-challenged group despite receiving an equal amount of virus, overall, CHPV administration restricted tumor growth in all the mice in the group (Figure 6C).
In order to check if the CHPV-challenged tumors retained the virus on the day of sacrifice, tumor tissues were homogenized, and plaque assays were carried out with the tissue homogenates. Interestingly, on an average, a titer of 1 × 107 pfu/mL/mg of tissue could be recovered (Figure 6D). The results so far suggest that CHPV continued to replicate in the tumor, significantly minimizing the tumor growth, while the tumors in the PBS-treated group grew unchecked.
CHPV challenge ameliorates/limits histopathological changes in tumor in mice
The tumor tissues from the virus-treated and control animals were dissected after euthanasia and subjected to histopathological analysis. Hematoxylin and eosin staining was performed to assess the pathological status of the tissues. The sections of PBS-treated tumors revealed large, tightly packed histologically discernible tumor cells having multiple nucleoli and vacuolations in the cytoplasm. The border of the tumor was found to be encapsulated with fibroblasts. Neovascularization was extremely prominent on both the outer and inner core zones of the tumor (Figures 7A–7C).
Figure 7.

Histopathological analysis of the tumor tissue
(A–C) Hematoxylin and eosin staining of the tumor samples from the PBS-treated control mice. The tumor sections reveal large, tightly packed histologically discernible tumor cells with multiple nucleoli and vacuolations in the cytoplasm. The black arrows indicate severe neovascularization. (D–F) Histology sections of tumors challenged with CHPV. Moderate to severe necrosis can be observed in all these sections highlighted with asterisks. The images also show that CHPV infection mediated loss of cell architecture with prominent remnants of pyknotic nuclei. The images from both groups are from three individual animals. Images were acquired at a magnification of ×100, and scale bars represent 100 μm.
Compared to the control tumors, stark differences were observed in the pathological features of the CHPV-treated tumors. The inner core zones of the CHPV-challenged tumors revealed moderate to severe necrosis (Figure 7D). The cell architecture was completely lost and had remnants of pyknotic nuclei. The necrotic zone was filled with fibroblasts showing an amorphous and hyalinized texture. This was synonymous with the central zone of the tumor mass, the actual site of CHPV treatment. The necrotic inner zone was surrounded by a thin layer of intact tumor cells and mild neovascularization followed by a fibroblastic encapsulation (Figures 7D–7F). Thus, a distinguishable difference in tumor pathology was found between the groups, with the CHPV-treated group having extensive areas of necrosis and less tumor neo-blood vessels compared to the untreated tumor group.
Discussion
Among the rhabdoviruses, CHPV is considered to be one of the emerging viruses identified to cause sporadic outbreaks of pediatric encephalitis.1, 2, 3,22 Unlike other vesiculoviruses such as VSV, Maraba, and Carajas, not many studies have been performed to exploit the cytopathic potential of CHPV.23, 24, 25 Understanding the nature of susceptibility of cancer cells of various origin and the mechanism of cell death is of paramount importance to exploit CHPV’s cytopathic potential. Our results indicate that, in vitro, irrespective of the site of origin, cancer cells were susceptible to the cytopathic effects of CHPV, albeit to varying degrees. Cell rounding, a characteristic feature of virus-induced morphological change in the infected cell, can be considered as an early indicator of cell death. CHPV-induced cell rounding was rapid and more prominent in cancer cells compared to the normal fibroblast cells. The enhanced susceptibility of cancer cells to CHPV can be attributed to the natural alterations in the antiviral responses of these cells, making them easily prone to viral infections.26,27
Even though viruses exhibit remarkable tropism, cancer cells, irrespective of the species or the tissue of origin, are easily susceptible to many viruses, as documented in the case of poxviruses.28,29 At an MOI of 0.1, the primary HADF cells were found to restrict CHPV infection significantly. Such restriction is usually generated due to the potent antiviral responses induced during virus infection in normal cells, which limits further spread of the virus. Although the cancer cell lines compared to HADF were readily permissive to CHPV, marked differences existed among the cancer cell lines tested. While some cancer cell lines, including U-138, HeLa, and A549, showed remarkable susceptibility, others like PC-3 were less permissive to CHPV, as was observed in the case of VSV.30 However, elaborate studies are required to interpret the differences in susceptibility to CHPV.
Unimpeded virus replication in the cell of interest is vital for the generation of progeny virion and virus dissemination.31 Differences in the levels of virus infectivity and output have been reported in the case of wild-type and attenuated strains of rabies virus (RABV) and VSV. While the wild-type RABV productively infected the cells, marked restriction was noticed in the attenuated RABV strain (SAD-L16).32, 33, 34, 35 Thus, besides cell-specific restriction, natural or engineered changes in viruses can impede their replication. This is critical in the generation of oncolytic vectors, as it is important that engineered mutations do not affect the overall growth and cytopathic property of the virus. Even though microscopic analysis revealed that all the cancer cell lines did not support CHPV infection equally, a comparative analysis of the virus output showed that CHPV yielded measurable titers in all the cell lines, including HADF. The highlight of these findings is that, among the cancer cell lines, select cell lines of specific tissue origin are more susceptible to CHPV, which is ideal in terms of exploiting CHPV for its oncolytic potential. The other significant highlight is that even at a low MOI of 0.1, CHPV is capable of inducing potent cytopathic effect in most cancer cell lines tested, which highlights that CHPV can be used to target cancer cells even at a low dose. The limited susceptibility of normal cells to CHPV supports the rationale that an attenuated CHPV can specifically target tumor cells while effectively being restricted by normal cells.
Apoptosis is a well-orchestrated set of molecular events culminating in cell death via the extrinsic or intrinsic pathways and plays a crucial role in host-pathogen interactions.36,37 In VSV-infected Jurkat T cells, PARP cleavage and caspase-3 and caspase-7 activation resulted in apoptosis, whereas in H4 cells stably transfected with Bcl-2 and Bcl-xL the effect was reversed, suggesting an apoptosis-dependent cell death.38, 39, 40 Whether CHPV infection induces apoptosis differentially in different cancer cell lines required further investigation. In normal neuronal cells, CHPV has been shown to induce apoptosis via the Fas-dependent extrinsic pathway.8 Experiments by probing molecular markers of apoptosis, including cleaved PARP and cleaved caspase-3 from infected lysates, suggest that CHPV induces apoptosis differently in the selected cell lines. Although in HeLa cells a role for apoptosis in CHPV-induced cytotoxicity maybe attributed (Figure S2), whether other apoptotic or necrotic pathways also contribute to cell death in CHPV-infected cells requires further investigation. The resistance of HADF cells to wild-type CHPV infection and delayed induction of apoptosis in these cells demonstrates the possibility of utilizing an attenuated CHPV as an oncolytic vector.
'This is further substantiated by the remarkable resistance of HADF cells to CHPV after being sensitized with IFN-β. Antiviral signaling pathways, especially those that involve type I IFNs, have a significant role in limiting viral spread in many viruses, including influenza, polio, and herpes simplex virus.41 Among the rhabdoviruses, the importance of type I IFN in limiting virus infection has been elegantly demonstrated in VSV and RABV by using antibodies against IFN-α/β, IFN-β cloned into virus, or mice defective in IFN signaling.42, 43, 44 In approaches involving the use of oncolytic vectors, IFNs have an important role in restricting the infection in normal host cells. However, impaired or skewed antiviral responses, including IFN signaling, are advantageous, as they can allow selective replication of the vectors in cancer cells.44 In our study, IFN-β pre-treatment had a protective effect on both cancer and normal cells against CHPV infection. Increased cell viability, a significant reduction in viral titer, and the cytopathic effect upon IFN-β pre-treatment highlight the importance of IFN in limiting CHPV infection. Among the cancer cell lines investigated, the glial-origin U-138 cells stood apart with respect to IFN response. IFN response proceeds via the JAK-STAT pathway, wherein the phosphorylation of STAT1 is important for the downstream signaling.45 The failure in the phosphorylation of STAT-1 in U-138 cells corroborated with the reduced sensitivity of these cells to IFN-β, while the high degree of phosphorylation in PC-3 and HADF cells correlated well with the restricted replication of CHPV in these cells. IFN-induced upregulation of STATs in specific cell types has already been reported.46 Therefore, the alteration in STAT1 expression and phosphorylation can be key determinants in the choice of a candidate cancer type for oncolytic therapy. All our results suggest that each cell type responds to IFN-β differently; in U-138 cells this pathway is impaired, and exogenous addition of IFN-β could not protect these cells from CHPV infection, which indicates that further investigation is needed in this area. Type I IFN, including IFNα, β, and ω genes, has been mapped to the short arm of chromosome 9, and structural abnormalities in this region are present in most of the gliomas, which is indicative of the high susceptibility of U-138 cells to CHPV, as observed in our study.47
The origin of the U-138 cell line can be traced to glioblastoma multiforme, an aggressive brain cancer that is hard to treat and cure.48 Current standard treatment regimens include chemotherapy with a DNA-alkylating agent temozolomide (TMZ) concomitant with surgery and/or radiation therapy.49 Still, the prognosis is poor, and the median survival of patients is only 12–15 months. Temozolomide resistance is a primary concern, and at least 50% of TMZ-treated patients do not respond to the treatment.50 Previous studies have shown that U-138 cells are highly resistant to TMZ chemotherapy.51 The glial origin and skewed IFN response of U-138 cells make them excellent candidates for testing the oncolytic potential of the neurotropic CHPV.
After having identified a target cell line, the feasibility of exploiting the oncolytic property of CHPV to restrict tumor growth in vivo was tested using the U-138 xenograft tumor model in the male NOD/SCID mouse. It is quite evident from our results that the growth of established tumors was severely restricted upon CHPV challenge compared to PBS treatment (Figure 6). These observations are further supported by a previous study in which baby hamster kidney cells (BHK) were used to establish tumors in nude mice, and the data show tumor regression and enhanced lifespan in the wild-type CHPV-challenged group compared to the control.52 Our study focused on cancer cell lines of human origin, extensive dissection of the growth and cytopathic properties of CHPV in these cell lines, and the in vivo validation using the U-138 tumor explant model in NOD/SCID mice. Rational application of our findings is possible only after due attenuation of CHPV, as it is a potent human pathogen. Prior studies have shown that understanding the oncolytic potential of wild-type viruses is of great importance to generate attenuated oncolytic vectors. As early as 1949, the oncolytic potential of viruses was demonstrated in mouse tumor models using wild-type Russian Far East encephalitis virus. Even though the infection led to fatal encephalitis in the mice, effective tumor regression was also observed.53,54 These observations paved the way for exploitation of viruses for oncolytic virotherapy. Preliminary studies using wild-type vesiculoviruses, including VSV and Maraba virus, demonstrated the potential of using these viruses as oncolytic vectors.21,55 Engineered attenuations of the wild type and subsequent arming have resulted in the development of potent and safe VSV based oncolytic vectors, which are in different stages of clinical trials. In conclusion, the data represented here demonstrate that CHPV, by virtue of its inherent oncolytic potential, can further be modified as a safe and effective oncolytic vector to target hard-to-treat cancers.
Materials and methods
Ethics statement
All animal ethical guidelines were followed with regard to the use of animals for this study. Animal experiments were performed after obtaining approval from the IAEC of Rajiv Gandhi Centre for Biotechnology (RGCB) as per the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Department of Animal Husbandry and Dairying, Ministry of Agriculture and Farmers Welfare, Government of India. Due approval was obtained from the Institutional Biosafety Committee, RGCB, for the handling of CHPV.
Cells and cell culture conditions
Cell lines Vero, A549, U-138, PC-3, HepG2, and HeLa were kindly provided by Prof. Griffith D. Parks (University of Central Florida, Orlando, FL, USA), while HADF cells (HiFi Adult Dermal Fibroblasts, cat. no. CL005) were procured from HiMedia, India. A549, U-138, HepG2, and HeLa cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Sigma, St. Louis, MO, USA), 2 mM L-glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin (Thermo Scientific) at 37°C in a humidified CO2 incubator (5%). HeLa-Bcl-2 cells were a kind gift from Prof. Douglas Lyles (Wake Forest School of Medicine, Winston-Salem, NC, USA) and were maintained under conditions similar to that of HeLa cells. Vero cells were maintained in minimum essential medium (MEM, Invitrogen) supplemented with 10% heat-inactivated FBS, and PC-3 cells were maintained in DMEM/nutrient mixture F-12 (DMEM/F-12, Invitrogen) supplemented with 10% heat-inactivated FBS. HADF cells were maintained in a proprietary media supplemented with growth factors (cat. no. AL525; HiMedia, India) as per the manufacturer’s instruction.
Virus
CHPV (NIV no. 653514, M/K no. M-1461, deposit: 28/X/1987) was a kind gift from the Director of the National Institute of Virology, Pune, India. Viral stocks were prepared in Vero cells, and the stock titer was estimated using a standard plaque assay.
Virus purification
Sucrose gradient-purified CHPV was used for in vivo experiments. Briefly, Vero cells were infected with CHPV at an MOI of 1 for 1 h, and the infection media was replaced with virus growth media. After 24 h, the culture supernatant was collected and clarified by centrifugation at 3,200 × g, 4°C, for 10 min. The supernatant was then subjected to ultracentrifugation at 82,600 × g for 3 h at 4°C in a Beckman centrifuge using the SW28 rotor. The pellet thus obtained was reconstituted in 500 μL NTE buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA [pH 8.0]) and was layered on a continuous 15%–60% (wt/vol) sucrose gradient prepared in NTE buffer and centrifuged at 150,000 × g at 4°C for 12 h using an SW41 rotor. The viral band was collected and made up to 13 mL with ice-cold NTE and subjected to further centrifugation at 150,000 × g at 4°C for 1 h in an SW41 rotor. The pellet thus obtained was resuspended in plain DMEM, aliquoted, snap-frozen, and stored at −80°C.
Phase-contrast microscopy
The susceptibility of the above-mentioned cell lines to CHPV infection was monitored by phase-contrast microscopy. Equal numbers of cells were seeded in 35 mm dishes and incubated for 24–48 h until the cells were 80%–90% confluent. The cells were infected with CHPV at an MOI of 0.1 and 5 for 1 h. The inoculum was removed, and the cells were supplemented with virus growth media. Phase-contrast images were captured at every 3 h interval up to 48 h using an inverted fluorescence IXT3 microscope (Olympus, Japan).
Live-dead cell imaging assay
The live and dead cell population post CHPV infection (Figures S1 and S3) was detected using Live/Dead Cell Imaging kit (Thermo Scientific, Waltham, MA, USA) according to the manufacturer’s instruction. Briefly monolayers of cells in chamber well glass slides (Thermo Scientific, Waltham, MA, USA) were infected with CHPV at an MOI of 0.1, 1, 5, and 10. 48 h post-infection, equal volumes of a 2× stock of the live cell component A and the dead cell component B were added to the chamber wells and incubated for 15 min at room temperature in the dark. Fluorescent images were acquired at 488/570 nm using an Olympus Inverted Fluorescence IXT3 microscope, and the images were merged using the cellSens software (Olympus, Japan).
Cell viability assay
The viability of the cells upon CHPV infection was assessed using the MTT reagent (Promega, Madison, WI, USA) according to the manufacturer’s instruction. Briefly, 24 h post-seeding in a 96-well plate, the cells were infected at an MOI of 0.1, 1, 5, and 10. At the indicated time points (24 and 48 hpi), 15 μL of MTT reagent was added to each well and incubated for 4 h until the purple formazan precipitate was visible. After the incubation, 100 μL of the solubilization/stop solution provided in the kit was added, mixed, and incubated for a further period of 1 h, and the absorbance was measured at 570 nm with a multimode plate reader (Tecan Spark 10M, Switzerland). The percent viability was calculated by normalizing the infected cells against the corresponding mock-infected control.
Regular HeLa and HeLa-Bcl-2 cells were infected with CHPV at an MOI of 1, and cell viability was performed at 24 hpi using the same protocol mentioned above.
Growth kinetics assay
Cells were grown to ∼80%–90% confluency in 12 well plates and infected with CHPV at an MOI of 0.1 (low MOI) and 5 (high MOI). After incubation at 37°C for 1 h, the inoculum was removed, and cells were washed thrice with PBS and replaced with media containing 2% FBS. At specific time points post-infection (3, 6, 9, 12, and 24 h), the supernatants were collected and subjected to titration by plaque assay. Briefly, monolayers of Vero cells were infected with serially diluted culture supernatant and incubated at 37°C for 1 h. After the infection, the medium was replaced with an overlay medium (DMEM, 2% FBS, 1× Penicillin-Streptomycine-Glutamine (PSG), 2% methylcellulose, 5 mM HEPES). The plates were incubated for 48 h at 37°C, fixed with 3.7% formaldehyde, and stained with 0.2% crystal violet. The plaques were counted, and the viral titer was plotted against their corresponding time using GraphPad Prism software version 5.01
Immunoblotting
Cells were infected with 0.1 MOI CHPV, and the cell lysates were harvested at 12 and 24 hpi. The lysates were subjected to immunoblotting and were probed for apoptotic markers PARP1 and caspase-3 and their cleaved form (Cell Signaling Technology). CHPV infection was detected using a mouse polyclonal antibody generated in-house, while β-actin served as the loading control (Sigma, St. Louis, MO, USA). The phosphorylation status of STAT1 in cancer cells was assessed by treating the cells with 1,000 U of IFN-β for 24 h. The cell lysate collected from IFN-β-challenged and control-media-treated cells were subjected to immunoblotting to assess the levels of STAT1 (cat. no. 14994) and p-STAT1 (Tyr701, cat. no. 9167S) (Cell Signaling Technology). The phosphorylation status of STAT1 in CHPV-infected lysates were also assessed by using the same antibodies, STAT1 and pSTAT1. All the blots were detected with WestPico Chemiluminescent substrate (Thermo Pierce, Rockford, IL, USA).
Interferon-β pre-treatment and CHPV infection
Equal numbers of cells seeded in 35 mm dishes were treated with 1,000 U/mL of recombinant human IFN-β (PeproTech, East Windsor, NJ, USA) for 24 h, while the control cells were maintained in culture medium. The cells were then infected with 0.1 MOI of CHPV, and the morphological changes were observed at different time points using a phase-contrast microscope (Olympus IXT3, Japan). Cell viability assay was carried out at 48 hpi, and supernatants collected 24 hpi were used for viral titer estimation by plaque assay.
Subcutaneous xenograft tumor model
Subcutaneous tumors were established by injecting U-138 cells (5 × 105 cells/100 μL PBS) in the right flank of 6- to 8-week-old male NOD/SCID mice. When the tumor volume attained a predetermined size of 150–250 mm3, 1 × 106 pfu of wild-type CHPV (suspended in 50 μL PBS) was injected intra-tumorally in the test mice, while the control tumors were challenged with PBS. A total of 6 mice were allocated per group.
The tumor size was measured daily using a Vernier caliper, and the volume was calculated using the formula (L × W2)/2, where L = tumor length and W = tumor width. The animals were monitored for weight loss, piloerection, morbidity, respiratory distress, and hindlimb paralysis. When the tumor volume exceeded a size of 1,500 mm3, the IAEC-approved ethical endpoint, the mice were sacrificed and the tumor mass was excised. A portion of the tumor stored in DMEM containing 0.75% bovine serum albumin fraction V was homogenized using a manual Dounce homogenizer (Wheaton, NJ, USA), and the homogenates were used for determining the viral titer. The other half was fixed in 10% neutral buffered formalin for histopathological analysis. Tumor tissues collected were homogenized, and plaque assays were carried out by serially diluting the tissue homogenates.
Histopathological analysis
Animals were sacrificed, and the tumor tissues were collected and fixed with 10% neutral buffered formalin. Tissues were then embedded in paraffin, and 5 μm sections were generated using Leica HistoCore Semi-motorized Microtome Histocore Multicut and mounted onto charged glass slides (Corning, Corning, NY, USA). The mounted sections were stained using hematoxylin and eosin. Pathological changes were analyzed by a certified pathologist who was blinded to the treatment groups of the experiments.
Statistical analysis
All the data were plotted and analyzed using GraphPad Prism 5.01 software. Statistical significance was calculated using the Student’s t test.
Acknowledgments
We thank Prof. Chandrabhas Narayana, Director, RGCB for his constant support. Dr. D.T. Mourya, Director, NIV and Dr. A. Basu are duly acknowledged for providing Chandipura virus that was used in this study. We thank Dr. Jackson James, Dr. Ani V. Das, Dr. Ruby John Anto, and Dr. R.C. Arunkumar for their critical comments and valuable suggestions during the discussions. We acknowledge the personnel of RGCB Animal Research Facility for the excellent services rendered. We thank Ms. S. Viji, Dr. Ramaswami Karthik, Mr. K.G. Anurup, Ms. Surabhi, and Mr. Tilak of RGCB and Dr. Gunveen Sachdeva, Department of Laboratory Medicine, AIIMS, New Delhi for their assistance with histopathology and confocal microscopy. This work was supported in part by aid from the Department of Biotechnology-Ramalingaswami Fellowship (BT/RLF/Re-entry/29/2012), Ministry of Science and Technology, India and the Department of Biotechnology-Rajiv Gandhi Centre for Biotechnology intramural funds (to J.B.J.), and Council for Scientific and Industrial Research (CSIR), India for the fellowship 09/716(0166)/2015-EMR-I (to R.K.M.).
Author contributions
R.K.M.: Formal analysis, investigation, methodology, validation, visualization, writing – original draft, and writing – review and editing; A.A.K: investigation, methodology, and visualization; J.N.: investigation, methodology, visualization, and writing – review and editing; V.S.J.: methodology, resources, validation, and visualization; U.K., N.A.K., and S.M.S.: methodology, analysis, validation, and writing – review and editing; A.R.: resources and supervision; J.B.J.: conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, writing – original draft, and writing – review and editing.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.09.009.
Supplemental information
References
- 1.Dwibedi B., Sabat J., Hazra R.K., Kumar A., Dinesh D.S., Kar S.K. Chandipura virus infection causing encephalitis in a tribal population of Odisha in eastern India. Natl. Med. J. India. 2015;28:185–187. [PubMed] [Google Scholar]
- 2.Gurav Y.K., Tandale B.V., Jadi R.S., Gunjikar R.S., Tikute S.S., Jamgaonkar A.V., Khadse R.K., Jalgaonkar S.V., Arankalle V.A., Mishra A.C. Chandipura virus encephalitis outbreak among children in Nagpur division, Maharashtra, 2007. Indian J. Med. Res. 2010;132:395–399. [PubMed] [Google Scholar]
- 3.Chadha M.S., Arankalle V.A., Jadi R.S., Joshi M.V., Thakare J.P., Mahadev P.V., Mishra A.C. An outbreak of Chandipura virus encephalitis in the eastern districts of Gujarat state, India. Am. J. Trop. Med. Hyg. 2005;73:566–570. [PubMed] [Google Scholar]
- 4.Rao B.L., Basu A., Wairagkar N.S., Gore M.M., Arankalle V.A., Thakare J.P., Jadi R.S., Rao K.A., Mishra A.C. A large outbreak of acute encephalitis with high fatality rate in children in Andhra Pradesh, India, in 2003, associated with Chandipura virus. Lancet. 2004;364:869–874. doi: 10.1016/S0140-6736(04)16982-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cherian S.S., Gunjikar R.S., Banerjee A., Kumar S., Arankalle V.A. Whole genomes of Chandipura virus isolates and comparative analysis with other rhabdoviruses. PLoS ONE. 2012;7:e30315. doi: 10.1371/journal.pone.0030315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basak S., Mondal A., Polley S., Mukhopadhyay S., Chattopadhyay D. Reviewing Chandipura: a vesiculovirus in human epidemics. Biosci. Rep. 2007;27:275–298. doi: 10.1007/s10540-007-9054-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jadi R.S., Sudeep A.B., Kumar S., Arankalle V.A., Mishra A.C. Chandipura virus growth kinetics in vertebrate cell lines, insect cell lines & embryonated eggs. Indian J. Med. Res. 2010;132:155–159. [PubMed] [Google Scholar]
- 8.Ghosh S., Dutta K., Basu A. Chandipura virus induces neuronal death through Fas-mediated extrinsic apoptotic pathway. J. Virol. 2013;87:12398–12406. doi: 10.1128/JVI.01864-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balakrishnan A., Mishra A.C. Immune response during acute Chandipura viral infection in experimentally infected susceptible mice. Virol. J. 2008;5:121. doi: 10.1186/1743-422X-5-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anukumar B., Shahir P. Immune regulation in Chandipura virus infection: characterization of CD4+ T regulatory cells from infected mice. Virol. J. 2011;8:259. doi: 10.1186/1743-422X-8-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudin C.M., Poirier J.T., Senzer N.N., Stephenson J., Jr., Loesch D., Burroughs K.D., Reddy P.S., Hann C.L., Hallenbeck P.L. Phase I clinical study of Seneca Valley Virus (SVV-001), a replication-competent picornavirus, in advanced solid tumors with neuroendocrine features. Clin. Cancer Res. 2011;17:888–895. doi: 10.1158/1078-0432.CCR-10-1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Au G.G., Lindberg A.M., Barry R.D., Shafren D.R. Oncolysis of vascular malignant human melanoma tumors by Coxsackievirus A21. Int. J. Oncol. 2005;26:1471–1476. doi: 10.3892/ijo.26.6.1471. [DOI] [PubMed] [Google Scholar]
- 13.Tai C.K., Kasahara N. Replication-competent retrovirus vectors for cancer gene therapy. Front. Biosci. 2008;13:3083–3095. doi: 10.2741/2910. [DOI] [PubMed] [Google Scholar]
- 14.Eissa I.R., Bustos-Villalobos I., Ichinose T., Matsumura S., Naoe Y., Miyajima N., Morimoto D., Mukoyama N., Zhiwen W., Tanaka M. The Current Status and Future Prospects of Oncolytic Viruses in Clinical Trials against Melanoma, Glioma, Pancreatic, and Breast Cancers. Cancers (Basel) 2018;10:E356. doi: 10.3390/cancers10100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andtbacka R.H., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 16.Jenks N., Myers R., Greiner S.M., Thompson J., Mader E.K., Greenslade A., Griesmann G.E., Federspiel M.J., Rakela J., Borad M.J. Safety studies on intrahepatic or intratumoral injection of oncolytic vesicular stomatitis virus expressing interferon-beta in rodents and nonhuman primates. Hum. Gene Ther. 2010;21:451–462. doi: 10.1089/hum.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandez M., Porosnicu M., Markovic D., Barber G.N. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J. Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stephenson K.B., Barra N.G., Davies E., Ashkar A.A., Lichty B.D. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012;19:238–246. doi: 10.1038/cgt.2011.81. [DOI] [PubMed] [Google Scholar]
- 19.Felt S.A., Grdzelishvili V.Z. Recent advances in vesicular stomatitis virus-based oncolytic virotherapy: a 5-year update. J. Gen. Virol. 2017;98:2895–2911. doi: 10.1099/jgv.0.000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lichty B.D., Power A.T., Stojdl D.F., Bell J.C. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol. Med. 2004;10:210–216. doi: 10.1016/j.molmed.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 21.Stojdl D.F., Lichty B., Knowles S., Marius R., Atkins H., Sonenberg N., Bell J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 22.Tandale B.V., Tikute S.S., Arankalle V.A., Sathe P.S., Joshi M.V., Ranadive S.N., Kanojia P.C., Eshwarachary D., Kumarswamy M., Mishra A.C. Chandipura virus: a major cause of acute encephalitis in children in North Telangana, Andhra Pradesh, India. J. Med. Virol. 2008;80:118–124. doi: 10.1002/jmv.21041. [DOI] [PubMed] [Google Scholar]
- 23.Connor J.H., Naczki C., Koumenis C., Lyles D.S. Replication and cytopathic effect of oncolytic vesicular stomatitis virus in hypoxic tumor cells in vitro and in vivo. J. Virol. 2004;78:8960–8970. doi: 10.1128/JVI.78.17.8960-8970.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Travassos da Rosa A.P., Tesh R.B., Travassos da Rosa J.F., Herve J.P., Main A.J., Jr. Carajas and Maraba viruses, two new vesiculoviruses isolated from phlebotomine sand flies in Brazil. Am. J. Trop. Med. Hyg. 1984;33:999–1006. doi: 10.4269/ajtmh.1984.33.999. [DOI] [PubMed] [Google Scholar]
- 25.Bhatt P.N., Rodrigues F.M. Chandipura: a new Arbovirus isolated in India from patients with febrile illness. Indian J. Med. Res. 1967;55:1295–1305. [PubMed] [Google Scholar]
- 26.Pitha P.M. Introduction: interferon’s connection to cancer. Semin. Cancer Biol. 2000;10:69–72. doi: 10.1006/scbi.2000.0309. [DOI] [PubMed] [Google Scholar]
- 27.Stojdl D.F., Lichty B.D., tenOever B.R., Paterson J.M., Power A.T., Knowles S., Marius R., Reynard J., Poliquin L., Atkins H. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4:263–275. doi: 10.1016/s1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 28.McFadden G. Poxvirus tropism. Nat. Rev. Microbiol. 2005;3:201–213. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeh H.J., Bartlett D.L. Development of a replication-selective, oncolytic poxvirus for the treatment of human cancers. Cancer Gene Ther. 2002;9:1001–1012. doi: 10.1038/sj.cgt.7700549. [DOI] [PubMed] [Google Scholar]
- 30.Carey B.L., Ahmed M., Puckett S., Lyles D.S. Early steps of the virus replication cycle are inhibited in prostate cancer cells resistant to oncolytic vesicular stomatitis virus. J. Virol. 2008;82:12104–12115. doi: 10.1128/JVI.01508-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thorley J.A., McKeating J.A., Rappoport J.Z. Mechanisms of viral entry: sneaking in the front door. Protoplasma. 2010;244:15–24. doi: 10.1007/s00709-010-0152-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hastie E., Cataldi M., Marriott I., Grdzelishvili V.Z. Understanding and altering cell tropism of vesicular stomatitis virus. Virus Res. 2013;176:16–32. doi: 10.1016/j.virusres.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian B., Zhou M., Yang Y., Yu L., Luo Z., Tian D., Wang K., Cui M., Chen H., Fu Z.F., Zhao L. Lab-Attenuated Rabies Virus Causes Abortive Infection and Induces Cytokine Expression in Astrocytes by Activating Mitochondrial Antiviral-Signaling Protein Signaling Pathway. Front. Immunol. 2018;8:2011. doi: 10.3389/fimmu.2017.02011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ray N.B., Power C., Lynch W.P., Ewalt L.C., Lodmell D.L. Rabies viruses infect primary cultures of murine, feline, and human microglia and astrocytes. Arch. Virol. 1997;142:1011–1019. doi: 10.1007/s007050050136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamilton D.H., Moyer R.W., Moyer S.A. Characterization of the non-permissive infection of rabbit cornea cells by vesicular stomatitis virus. J. Gen. Virol. 1980;49:273–287. doi: 10.1099/0022-1317-49-2-273. [DOI] [PubMed] [Google Scholar]
- 36.Hay S., Kannourakis G. A time to kill: viral manipulation of the cell death program. J. Gen. Virol. 2002;83:1547–1564. doi: 10.1099/0022-1317-83-7-1547. [DOI] [PubMed] [Google Scholar]
- 37.James E.R., Green D.R. Manipulation of apoptosis in the host-parasite interaction. Trends Parasitol. 2004;20:280–287. doi: 10.1016/j.pt.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Koyama A.H. Induction of apoptotic DNA fragmentation by the infection of vesicular stomatitis virus. Virus Res. 1995;37:285–290. doi: 10.1016/0168-1702(95)00026-m. [DOI] [PubMed] [Google Scholar]
- 39.Hobbs J.A., Schloemer R.H., Hommel-Berrey G., Brahmi Z. Caspase-3-like proteases are activated by infection but are not required for replication of vesicular stomatitis virus. Virus Res. 2001;80:53–65. doi: 10.1016/s0168-1702(01)00350-1. [DOI] [PubMed] [Google Scholar]
- 40.Desforges M., Despars G., Bérard S., Gosselin M., McKenzie M.O., Lyles D.S., Talbot P.J., Poliquin L. Matrix protein mutations contribute to inefficient induction of apoptosis leading to persistent infection of human neural cells by vesicular stomatitis virus. Virology. 2002;295:63–73. doi: 10.1006/viro.2001.1329. [DOI] [PubMed] [Google Scholar]
- 41.Marcovistz R., Galabru J., Tsiang H., Hovanessian A.G. Neutralization of interferon produced early during rabies virus infection in mice. J. Gen. Virol. 1986;67:387–390. doi: 10.1099/0022-1317-67-2-387. [DOI] [PubMed] [Google Scholar]
- 42.Faul E.J., Wanjalla C.N., McGettigan J.P., Schnell M.J. Interferon-beta expressed by a rabies virus-based HIV-1 vaccine vector serves as a molecular adjuvant and decreases pathogenicity. Virology. 2008;382:226–238. doi: 10.1016/j.virol.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meraz M.A., White J.M., Sheehan K.C., Bach E.A., Rodig S.J., Dighe A.S., Kaplan D.H., Riley J.K., Greenlund A.C., Campbell D. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- 44.Müller U., Steinhoff U., Reis L.F., Hemmi S., Pavlovic J., Zinkernagel R.M., Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 45.Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lehtonen A., Matikainen S., Julkunen I. Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J. Immunol. 1997;159:794–803. [PubMed] [Google Scholar]
- 47.Olopade O.I., Jenkins R.B., Ransom D.T., Malik K., Pomykala H., Nobori T., Cowan J.M., Rowley J.D., Diaz M.O. Molecular analysis of deletions of the short arm of chromosome 9 in human gliomas. Cancer Res. 1992;52:2523–2529. [PubMed] [Google Scholar]
- 48.Hanif F., Muzaffar K., Perveen K., Malhi S.M., Simjee ShU. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017;18:3–9. doi: 10.22034/APJCP.2017.18.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maher E.A., Furnari F.B., Bachoo R.M., Rowitch D.H., Louis D.N., Cavenee W.K., DePinho R.A. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–1333. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 50.Lee S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016;3:198–210. doi: 10.1016/j.gendis.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ryu C.H., Yoon W.S., Park K.Y., Kim S.M., Lim J.Y., Woo J.S., Jeong C.H., Hou Y., Jeun S.S. Valproic acid downregulates the expression of MGMT and sensitizes temozolomide-resistant glioma cells. J. Biomed. Biotechnol. 2012;2012:987495. doi: 10.1155/2012/987495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balakrishnan A., Malik N. Chandipura Virus’ Oncolytic Potential in Experimentally Induced Tumor in Mice. Intervirology. 2020;1-4:1–4. doi: 10.1159/000512299. [DOI] [PubMed] [Google Scholar]
- 53.Moore A.E. Inhibition of growth of five transplantable mouse tumors by the virus of Russian Far East encephalitis. Cancer. 1951;4:375–382. doi: 10.1002/1097-0142(195103)4:2<375::aid-cncr2820040227>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 54.Moore A.E. The destructive effect of the virus of Russian Far East encephalitis on the transplantable mouse sarcoma 180. Cancer. 1949;2:525–534. doi: 10.1002/1097-0142(194905)2:3<525::aid-cncr2820020317>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 55.Balachandran S., Barber G.N. Vesicular stomatitis virus (VSV) therapy of tumors. IUBMB Life. 2000;50:135–138. doi: 10.1080/713803696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.