

Studies aimed at identifying genetic factors predisposing to cytomegalovirus (CMV) infection following hematopoietic cell transplantation (HCT) have varied widely and have not been validated. Casto and colleagues report a study of 117 previously reported variants in donor and recipient pairs of more than 2000 CMV-positive HCT recipients. Of the 117 candidate genes, they implicate only ABCB1, which encodes P-glycoprotein, in CMV reactivation. The gene variant results in reduced efflux of calcineurin inhibitors from donor lymphocytes, likely exerting its effect by increasing immune suppression.
Key Points
A synonymous variant in P-glycoprotein is associated with the risk of CMV reactivation after allogeneic HCT.
Visual Abstract

Abstract
Human cytomegalovirus (CMV) reactivation is a frequent complication of allogeneic hematopoietic cell transplantation (HCT). Despite routine screening for CMV reactivation and early antiviral treatment, the rates of CMV-related complications after HCT remain high. Genetic variants in both the donor and recipient have been associated with the risk of CMV reactivation and disease after HCT, but these associations have not been validated, and their clinical importance remains unclear. In this study, we assessed 117 candidate variants previously associated with CMV-related phenotypes for association with CMV reactivation and disease in a cohort of 2169 CMV-seropositive HCT recipients. We also carried out a genome-wide association study (GWAS) for CMV reactivation and disease in the same cohort. Both analyses used a prespecified discovery and replication approach to control the risk of false-positive results. Among the 117 candidate variants, our analysis implicates only the donor ABCB1 rs1045642 genotype as a risk factor for CMV reactivation. This synonymous variant in P-glycoprotein may influence the risk of CMV reactivation by altering the efflux of cyclosporine and tacrolimus from donor lymphocytes. In the GWAS analysis, the donor CDC42EP3 rs11686168 genotype approached the significance threshold for association with CMV reactivation, although we could not identify a mechanism to explain this association. The results of this study suggest that most genomic variants previously associated with CMV phenotypes do not significantly alter the risk for CMV reactivation or disease after HCT.
Introduction
Allogeneic hematopoietic stem cell transplantation (HCT) is a life-saving intervention used in the treatment of hematologic disorders and malignancies. Infections remain a common cause of morbidity and mortality after HCT, and human cytomegalovirus (CMV) is one of the most common etiologic agents of such infections.1,2 Screening for CMV viremia and pharmacologic CMV prophylaxis are now a standard part of clinical management after HCT.3 Despite these preemptive interventions, adverse outcomes due to CMV are still commonly observed among post-HCT patients. Recent studies have shown that the incidence of any CMV reactivation, clinically significant CMV reactivation, and CMV disease are 75%, ∼50%, and 8%, respectively, in CMV-seropositive allogeneic HCT recipients.4,5 While not always symptomatic, CMV reactivation in HCT recipients is associated with poor clinical outcomes, including higher rates of mortality4.
While CMV disease following HCT can result from either primary CMV infection or reactivation of latent infection,6 reactivation is much more common than primary infection.7 The CMV serostatus of the donor and recipient remain the most important predictors of CMV disease after HCT, with seropositive recipients at the highest risk, seronegative recipients with seropositive donors at intermediate risk, and seronegative recipients with seronegative donors at the lowest risk.4 The chemotherapy regimen used for conditioning before HCT also affects the risk of CMV reactivation and disease, with the use of rabbit anti-thymocyte globulin,8 alemtuzumab,8-11 or reduced intensity conditioning4 conferring higher risk. The use of cord blood grafts12 and the development of graft-versus-host disease (GVHD) after HCT4 are also associated with higher risks for CMV reactivation and disease.
Genetic variants in the donor and recipient have been investigated for association with CMV reactivation and disease after HCT. Variants have been selected for analysis in these studies based on their association with other phenotypes or known biologic processes. While some variants, such as rs1800023 in CCR513,14 and rs12979860 in IFNL4,14-16 have been associated with the risk of CMV-related phenotypes after HCT in multiple studies, most such genotype-phenotype associations have been reported only once.
In this study, we sought to replicate the results of these previous analyses in a large HCT cohort for which genomic data from both donors and recipients is available. We conducted a candidate variant analysis, which includes single-nucleotide polymorphisms (SNPs) and insertion/deletion polymorphisms (indels) associated with CMV reactivation or disease after HCT, solid organ transplantation, or human immunodeficiency virus infection as well as polymorphisms associated with congenital CMV. We also conducted a genome-wide association study (GWAS) of CMV reactivation and CMV disease after HCT to identify additional SNPs associated with these phenotypes.
Methods
Study population
Blood samples were collected from recipients and donors before HCT following protocols approved by the Fred Hutchinson Cancer Research Center (FHCRC) Institutional Review Board. Recipients and donors signed consent forms allowing the use of clinical data and biospecimens for research. Research to evaluate genomic associations with clinical outcomes was approved by the FHCRC Institutional Review Board.
The samples and data used for this study came from a cohort of 2169 CMV-seropositive HCT recipients and their donors. Sixteen otherwise eligible patients who received an antiviral medication to prevent a first episode of CMV reactivation were not included in the study. With the use of CMV-seronegative or leukocyte-reduced transfusion products to prevent CMV infection, the incidence of CMV reactivation and CMV disease in CMV-seronegative recipients was too low to contribute information to the analysis, so they were also excluded.
To be considered evaluable for CMV reactivation, patients must have had at least 50% of expected weekly surveillance tests for CMV reactivation during the first 100 days after HCT (ie, at least 7 of 14 tests, except in cases of early death). They also were required to have at least 1 test during the first 4 weeks after HCT. Recipients had hematological malignancies or myelodysplasia and had a first allogeneic HCT at our center from 1990 through 2011. Some recipients had a single prior autologous HCT.
The study was limited to recipients of European ancestry because the number of available non-European recipients was too small for a meaningful analysis. We used the minimum covariant determinant method described by Conomos et al17 to define European ancestry. Typing of HLA-A, B, C, DRB1, and DQB1 alleles out to 4 digits was used to evaluate HLA matching between donors and recipients.
Recipients were prepared for HCT with either myeloablative or nonmyeloablative conditioning regimens. Aspirated bone marrow cells or growth factor–mobilized blood cells were used for HCT. Patients underwent surveillance for CMV and received CMV prophylaxis/preemptive therapy for CMV reactivation according to the institutional protocol in place at the time of their transplants as described previously.18,19 Demographic, clinical, and transplant characteristics of recipient/donor pairs are shown in Table 1. Most patients in this study received cyclosporine or tacrolimus to prevent GVHD after HCT. Doses of both medications were adjusted to maintain blood concentrations within their respective therapeutic ranges.
Table 1.
Characteristics of the study cohort (N = 2169)
| Characteristic | n (%) |
|---|---|
| Number genotyped | |
| Recipients genotyped | 1930 (89) |
| Donors genotyped | 2025 (93) |
| Patient age at transplantation in years | |
| Median | 46 |
| Range | 0-75 |
| Diagnosis | |
| Acute leukemia | 907 (42) |
| Chronic myeloid leukemia | 487 (22) |
| Myelodysplastic syndromes or myeloproliferative neoplasms | 383 (18) |
| Chronic lymphocytic leukemia | 72 (3) |
| Malignant lymphoma or multiple myeloma | 320 (15) |
| Disease risk* | |
| Low | 445 (21) |
| Intermediate | 596 (27) |
| High | 1005 (46) |
| Not classified | 123 (6) |
| Donor-recipient gender combination | |
| Male to male | 659 (30) |
| Male to female | 538 (25) |
| Female to male | 476 (22) |
| Female to female | 494 (23) |
| Donor CMV serostatus | |
| Negative | 1168 (54) |
| Positive | 1000 (46) |
| Donor type | |
| Related, HLA-A, B, C, DRB1, DQB1-matched | 956 (44) |
| Related, HLA-A, B, C, DRB1, DQB1-mismatched | 63 (3) |
| Unrelated, HLA-A, B, C, DRB1, DQB1-matched | 746 (34) |
| Unrelated, HLA-A, B, C, DRB1, DQB1-mismatched | 404 (19) |
| Graft source | |
| Bone marrow | 1135 (52) |
| Mobilized blood cells | 1034 (48) |
| Conditioning regimen | |
| Myeloablative with <900 cGy total body irradiation | 788 (36) |
| Myeloablative with ≥900 cGy total body irradiation | 966 (45) |
| Nonmyeloablative | 415 (19) |
| Posttransplant immunosuppression | |
| Cyclosporine and methotrexate | 1141 (53) |
| Cyclosporine and mycophenolate mofetil | 378 (17) |
| Tacrolimus and methotrexate | 318 (15) |
| Other | 332 (15) |
Low risk includes chronic myeloid leukemia in chronic phase or myelodysplastic syndrome-refractory anemia; intermediate risk includes acute leukemia, chronic lymphocytic leukemia, or non-Hodgkin lymphoma in remission; high risk includes all others.
Literature search
On 16 March 2020, we conducted a PubMed search to identify published studies reporting significant associations between one or more human genetic variants and a CMV-related phenotype. Our search query was ([“cytomegalovirus” OR “CMV”) AND (“genotype*” OR “polymorphism*” OR “SNP*” OR “variant*”]), and we limited our search to journal reports or letters with a focus on human subjects, which were published in English and in Medline. The search yielded 1308 articles, which were then manually reviewed. From this review, we identified a list of 123 candidate genetic variants that had been associated with a CMV-related trait at the P ≤ .05 level in ≥1 previous study.
Sample preparation, genotyping, quality assurance/control, imputation
Details of sample preparation, genotyping, quality assurance and control, and imputation have been described previously.20 The GWAS screen encompassed 29 826 485 variants that passed quality control. No sample was available for 144 (7%) of the recipients and 239 (11%) of the donors in the cohort (Table 1).
End points
Three outcomes were evaluated in this study: (1) the first CMV reactivation indicated by antigenemia or viremia at any level; (2) high-level reactivation demonstrated by antigenemia at ≥10 positive cells per 200 000 blood leukocytes or viremia at ≥1000 copies/mL (equivalent to 312 IU/mL), used historically as an indication for antiviral treatment21; and (3) the first proven CMV-related disease in the recipient at any time after HCT. CMV antigenemia was defined by the detection of the CMV pp65 antigen in blood leukocytes, and viremia was defined by the detection of CMV DNA in the plasma by polymerase chain reaction (PCR). Screening for CMV reactivation gradually transitioned from antigen testing to PCR in 2007, and the PCR threshold for treatment of CMV viremia was set such that the percentage of patients treated for CMV reactivation after this transition was the same as it had been before the transition.19 Weekly CMV antigen or CMV PCR tests were done in the University of Washington Clinical Virology Laboratory according to standard clinical practices at the FHCRC/Seattle Cancer Care Alliance (SCCA) at the time. Proven CMV-related disease was defined according to the criteria in Ljungman et al.22 The 3 different outcomes and the 2 different genomes (ie, recipient and donor) yielded 6 genome-outcome combinations that we analyzed separately.
Statistical analysis
Candidate variants were assessed for association with CMV reactivation and disease under allelic, recessive genotypic, and dominant genotypic models. For a variant with major and minor alleles “a” and “b”, the allelic model tests whether the number of minor b alleles (0, 1, or 2) in the donor or recipient is associated with the risk of CMV reactivation or disease. The recessive genotypic model compares outcomes associated with “bb” genotype vs the combined “aa” and “ab” genotypes, while the dominant genotypic model compares outcomes with the combined "bb" and "ab" genotypes vs the "aa" genotype.
We conducted the analyses of candidate and genome-wide variants in separate discovery and replication phases. For this purpose, 60% of the overall study cohort was randomly assigned to the discovery cohort and the remainder to the replication cohort. In the analysis of candidate variants, donor or recipient variants associated with an outcome at a value of P < .01 in the discovery cohort under any of the 3 genetic models were evaluated in the replication cohort. Post-hoc power estimates for discovery in the analysis of candidate variants were calculated for a hazard ratio (HR) of 1.5 or 0.67, based on the estimated standard error of the log HR and a 2-sided .01 significance level. The analysis of genome-wide variants was limited to the allelic model, and associations with a value of P < 1.0 × 10−6 and minor allele frequency (MAF) ≥0.01 in the discovery cohort were evaluated in the replication cohort.
In replication testing of candidate and genome-wide discoveries, Bonferroni adjustments were applied separately to the 6 genome-outcome combinations. Different genetic models applied to the same variant-genome-outcome association and variants in linkage disequilibrium with r2 > 0.7 for the same genome-outcome association were counted only once. Post-hoc power estimates for replication were calculated for the discovery hazard ratio point estimate and the estimated standard error of the replication log hazard ratio, with a Bonferroni-corrected significance level reflecting the number of discovery findings. Genotype-phenotype associations replicated in a univariable analysis were tested in a multivariable analysis that adjusted for clinical covariates known to be associated with the end point, although such covariates cannot be confounders of a genotype-phenotype association unless they are also related to the genotype in question, which is generally implausible.
Results
Candidate variant rs1045642 in ABCB1 in donors is associated with CMV reactivation
The literature search identified 123 variants associated with a CMV-related phenotype in ≥1 population (supplemental Table 1). Supplemental Table 2 summarizes the quality control assessment of the candidate variants genotyped or imputed on the 3 platforms used for our study. One variant (rs2032582) has 3 alleles and is counted twice, and rs1619379 was used as a proxy for rs371194629, which was not available in our data set. Six variants were not analyzed; 2 did not pass quality control (rs5743708 and rs1071803) and had no proxies in high linkage disequilibrium, 2 were not included in our data set (rs368234815 and rs179009) but had proxies already included in the list from the literature (rs12979860 and rs179008, respectively), and 2 were not included in our data set (rs121917864 and rs2910164) and had no proxies in high linkage disequilibrium.
Results of testing the remaining candidate donor and recipient variants for association with any CMV reactivation, high-level CMV reactivation, or CMV disease in the discovery phase are summarized in supplemental Tables 3-5. In donors, only 2 candidate variants, rs1045642 in ABCB1 and rs16944 in IL1B, met the P < .01 threshold of significance in the discovery phase (Table 2). In recipients, 2 variants, rs3116496 in CD28 and rs3135932 in IL10RA, had values of P < .01 for association with CMV reactivation, and 4 were associated with CMV disease (rs231775 and rs3087243 in CTLA4, rs1061680 in LILRB1, and rs4253728 in PPARA) (Table 2). Estimated post-hoc power for discovery of candidate variants with HRs ≥1.5 or ≤0.67 for associations with CMV reactivation was 80% or better for 80% of variants evaluated in the allelic and dominant models but was much lower in the recessive model (Figure 1A). Power for discovery of variants with HRs ≥1.5 or ≤0.67 for associations with CMV disease was low in all 3 genetic models due to the low event rate (Figure 1B).
Table 2.
Association of candidate variants with CMV reactivation and CMV disease after allogeneic HCT
| Discovery results | Replication results | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr | Gene | SNP | Alleles* | Genome | End point | Model | MAF† | P value | HR | LB | UB | Bf-C | P value | HR | LB | UB | Power‡ |
| 7 | ABCB1 | rs1045642 | A/G | Donor | Any R | D | 0.47 | .002 | 0.74 | 0.62 | 0.89 | 0.05 | .03 | 0.79 | 0.64 | 0.97 | 80 |
| 2 | IL1B | rs16944 | G/A | Donor | HLR | A | 0.34 | .0006 | 0.68 | 0.55 | 0.85 | 0.05 | .27 | 0.86 | 0.66 | 1.12 | |
| 2 | IL1B | rs16944 | G/A | Donor | HLR | D | 0.34 | .004 | 0.67 | 0.51 | 0.88 | 0.05 | .29 | 0.83 | 0.59 | 1.17 | |
| 2 | CD28 | rs3116496 | T/C | Recipient | Any R | A | 0.19 | .003 | 1.26 | 1.09 | 1.46 | 0.025 | .28 | 1.11 | 0.92 | 1.33 | 60 |
| 2 | CD28 | rs3116496 | T/C | Recipient | Any R | D | 0.19 | .003 | 1.30 | 1.09 | 1.54 | 0.025 | .23 | 1.14 | 0.92 | 1.40 | 57 |
| 11 | IL10RA | rs3135932 | A/G | Recipient | Any R | R | 0.18 | .005 | 0.42 | 0.21 | 0.85 | 0.025 | .41 | 1.28 | 0.73 | 2.23 | 79 |
| 3 | PTX3 § | rs2305619 | G/A | Recipient | HLR | A | 0.49 | .002 | 1.37 | 1.12 | 1.67 | 0.05 | .98 | 1.00 | 0.79 | 1.26 | |
| 2 | CTLA4 | rs231775 | A/G | Recipient | Disease | A | 0.37 | .0001 | 1.48 | 1.21 | 1.81 | 0.01 | .02 | 0.71 | 0.53 | 0.95 | 52 |
| 2 | CTLA4 | rs231775 | A/G | Recipient | Disease | D | 0.37 | .0009 | 1.66 | 1.22 | 2.26 | 0.01 | .02 | 0.65 | 0.44 | 0.94 | 53 |
| 2 | CTLA4 | rs231775 | A/G | Recipient | Disease | R | 0.37 | .005 | 1.70 | 1.20 | 2.40 | 0.01 | .19 | 0.66 | 0.34 | 1.26 | 16 |
| 2 | CTLA4 | rs3087243 | G/A | Recipient | Disease | A | 0.46 | .004 | 0.75 | 0.61 | 0.92 | 0.01 | .25 | 1.17 | 0.89 | 1.53 | 32 |
| 2 | CTLA4 | rs3087243 | G/A | Recipient | Disease | D | 0.46 | .004 | 0.65 | 0.49 | 0.87 | 0.01 | .38 | 1.21 | 0.79 | 1.86 | 26 |
| 19 | LILRB1 | rs1061680 | T/C | Recipient | Disease | R | 0.27 | .004 | 0.38 | 0.18 | 0.82 | 0.01 | .42 | 1.34 | 0.68 | 2.66 | 56 |
| 22 | PPARA | rs4253728 | G/A | Recipient | Disease | A | 0.28 | .004 | 0.67 | 0.50 | 0.89 | 0.01 | .82 | 1.05 | 0.71 | 1.53 | 31 |
| 22 | PPARA | rs4253728 | G/A | Recipient | Disease | D | 0.28 | .001 | 0.57 | 0.40 | 0.80 | 0.01 | .72 | 1.09 | 0.68 | 1.74 | 42 |
Rows demarcated by lines indicate genome-phenotype combinations.
A, allelic; Any R, any reactivation; Bf-C, Bonferroni correction; Chr, chromosome; D, dominant; HLR, high-level reactivation; LB, lower boundary of the 95% CI; R, recessive; UB, upper boundary of the 95% CI.
Plus-strand major/minor alleles; allele designations may differ from those used in previous studies.
MAF in the combined donor and recipient samples used for the evaluation.
Power is based on the discovery HR point estimate and estimated standard error of the replication log HR, with a Bonferroni-corrected significance level reflecting the number of discovery findings.
Also VEPH1.
Figure 1.
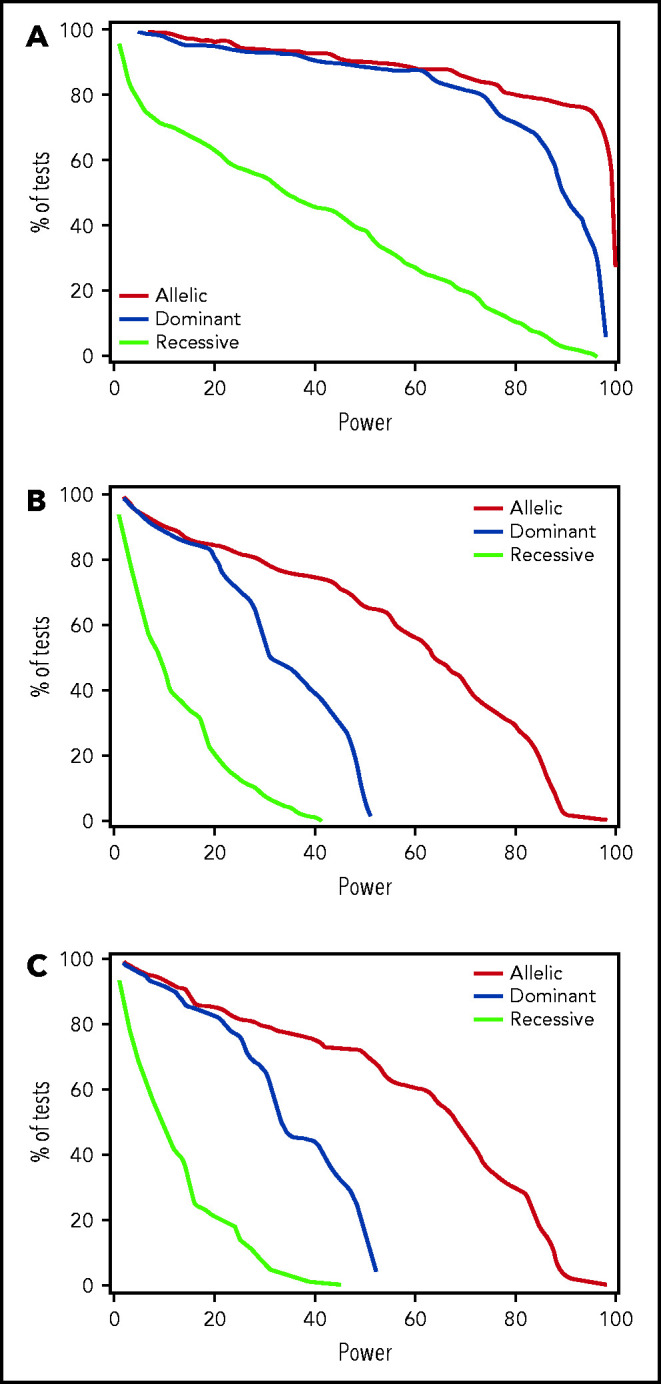
For candidate variants, discovery power is adequate for any CMV reactivation in the allelic and dominant genetic models but not in the recessive model, while discovery power is limited for high-level CMV activation and disease. Plots show the percentage of tested recipient variants with post-hoc power at the level on the x-axis with each of the 3 genetic models and an assumed clinically significant HR of 1.5 or 0.67, based on the observed discovery standard error and a .01 threshold of statistical significance for (A) CMV reactivation, (B) high-level CMV reactivation, and (C) CMV disease. Results for donor variants are essentially the same.
In replication testing, the risk of any CMV reactivation was lower with the combined donor AG or GG genotypes of rs1045642 than with the AA genotype (HR, 0.79; 95% confidence interval [CI], 0.6-1.0; P = .03) (Figure 2A). This association did not change after multivariable adjustment for clinical risk factors, including patient age, the use of anti-thymocyte globulin or a nonmyeloablative regimen for conditioning before HCT, use of an unrelated donor or a CMV-seropositive donor, and time-dependent onset of grade II to IV GVHD (HR, 0.75; 95% CI, 0.6-0.9; P = .01). Further evaluation of rs1045642 did not show an association with high-level CMV reactivation in the replication cohort (HR, 1.03; 95% CI, 0.7-1.5; P = .89). In replication testing, the donor rs16944 genotype was not significantly associated with the risk of high-level CMV reactivation (Table 2).
Figure 2.
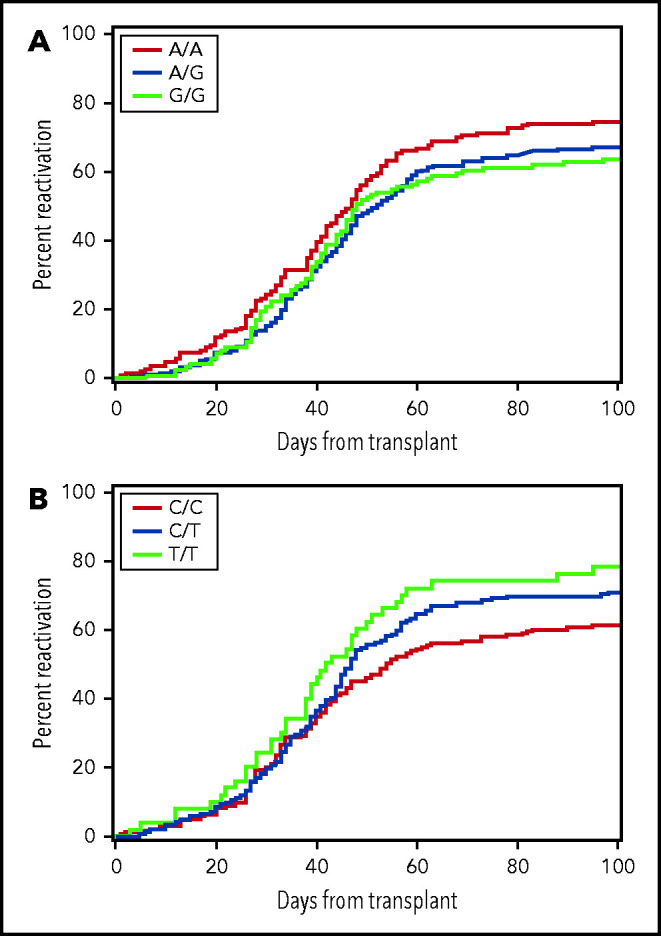
The risk of CMV reactivation is associated with donor genotypes. Plot (A) shows data for donor rs1045462 genotype and (B) shows data for donor rs11686168 genotype. The data represent the replication cohort and do not provide a clear indication of the genetic model for either of these associations.
In replication testing, the P value of .02 with the allelic and dominant models for the association of recipient rs231775 CTLA4 genotypes with CMV disease was close to the Bonferroni-corrected threshold P value of .01, but the HRs for these associations were >1.0 in the discovery cohort and <1.0 in the replication cohort. Associations of recipient rs3087243, rs1061680, and rs4253728 genotypes with CMV disease were not statistically significant in the replication cohort, although power for replication of these results was low.
ABCB1 encodes P-glycoprotein-1 (P-gp), which transports drugs from the intracellular compartment to the extracellular compartment. Cyclosporine, tacrolimus, methotrexate, mycophenolate mofetil, and prednisone are all substrates of P-gp. In addition, cyclosporine and tacrolimus act as inhibitors of P-gp. Therefore, we tested whether the association of rs1045642 genotypes with CMV reactivation differed between patients who received cyclosporine vs tacrolimus to prevent GVHD after HCT. In the combined discovery and replication cohorts, the HR for the association of rs1045642 with CMV reactivation in the dominant model (AG or GG combined vs AA) was 0.77 (95% CI, 0.65-0.90) (n = 1048) for patients who received cyclosporine and 0.73 (95% CI, 0.55-0.96) (n = 342) for those who received tacrolimus.
Association between rs11686168 in CDC42EP3 in donors and CMV reactivation approached significance in GWAS
In the discovery phase, a total of 65 variants were associated with CMV reactivation, high-level reactivation or disease in donors or recipients with a value of P ≤ 1 × 10−6 (supplemental Tables 6 and 7). Although statistical power was adequate, none of the associations tested for replication met the Bonferroni-adjusted threshold of significance (results not shown). The association of the donor CDC42EP3 rs11686168 genotype with any CMV reactivation, however, approached the 0.008 threshold of significance (HR, 1.26; 95% CI, 1.1-1.5; P = .009) (Figure 2B). This association did not change after multivariable adjustment for clinical risk factors as described above (HR, 1.30; 95% CI, 1.1-1.6; P = .003). Further evaluation of rs11686168 did not show a statistically significant association with high-level CMV reactivation in the replication cohort (HR, 1.22; 95% CI, 0.9-1.7; P = .21), although the HR point estimate is similar to the HR point estimate for any CMV reactivation in the replication cohort. The association of the rs11686168 genotype with CMV reactivation did not differ between CMV-seropositive and seronegative donors. In the combined discovery and replication cohorts, the HR was 1.35 (95% CI, 1.2-1.6) with CMV-seropositive donors and 1.35 (95% CI, 1.2-1.6) with CMV-seronegative donors.
To evaluate whether the rs11686168 association could be explained by any other variant, we combined data from the discovery and replication cohorts to evaluate P values and r2 values for all variants within a 400-kb window on chromosome 2 centered on rs11686168 as shown in Figure 3. This analysis did not identify any variants having r2 > 0.8 with rs11686168, and for those with r2 values between 0.6 and 0.8, the P values for association with CMV reactivation were appreciably larger than the P value for rs11686168. This result implicates rs11686168 as the driver of the association signal, although it remains possible that the signal is due to another linked variant that was not genotyped in our cohort.
Figure 3.

Locus-zoom plot shows −log10(P values) for association with CMV reactivation in the combined discovery and replication cohorts as a function of position in a 0.2-Mb region on either side of rs11686168 on chromosome 2. Correlation coefficient r2 values for linkage disequilibrium with rs11686168 are coded according to the inset. The blue plot and the right-side scale show recombination rates as a function of chromosomal position. Genes within this region are displayed in the lower panel.
Discussion
In this study, we found evidence supporting an association of the donor ABCB1 rs1045642 genotype with the risk of CMV reactivation after allogeneic HCT, but the results did not show evidence supporting an association of any other candidate variants with the risk of CMV reactivation or disease in our cohort. In our genome-wide analysis, the donor CDC42EP3 rs11686168 genotype came very close to meeting the P value threshold for significance established for replication in our study. In the overall cohort, this association also met the standard 5 × 10−8 threshold of statistical significance for a GWAS discovery. However, inclusion of the discovery cohort in this analysis introduces bias, so this result remains to be tested for validation in an independent cohort.
At least 3 studies have shown that the rs1045642 A allele is associated with higher intracellular concentrations of tacrolimus.23-25 At least one study has shown that it is also associated with higher intracellular concentrations of cyclosporine,26 although other studies have not confirmed this effect.24,27 Our results are consistent with results reported by Cattaneo et al28 showing that the recipient rs1045642 A allele is associated with a higher risk for CMV reactivation in renal transplant recipients.
The synonymous rs1045742 variant is theorized to affect phenotype by substituting a commonly used codon for a rarely used one, which alters the folding of P-gp that occurs simultaneously with translation.29 Accordingly, it was suggested that the protein encoded by the A allele (designated as the T allele in some previous studies) has lower efflux activity, leading to higher intracellular cyclosporine concentrations, greater inhibition of lymphocyte function, and greater risk of CMV reactivation.28 The association of the donor rs1045642 A allele with a higher risk of CMV reactivation in our study is consistent with this hypothesis. Our results also show a similar effect in patients treated with tacrolimus, suggesting that the rs1045642 variant does not alter the substrate specificity of P-gp.
In the current study, dosing of calcineurin inhibitors was adjusted to target whole-blood concentrations within a range that was not informed by the donor rs1045642 genotype. The donor rs1045642 genotype did not have any detectable effect on whole-blood concentrations of either cyclosporine or tacrolimus during the first 21 days after HCT (data not shown). At any given whole-blood concentration, however, the intracellular concentration of calcineurin inhibitors in leukocytes will be higher in patients with donor rs1045642 genotypes that have low P-gp activity indicated by the homozygous A allele than in those with higher P-gp activity indicated by the presence of ≥1 G allele. Taken together, our results suggest that CMV reactivation is an exquisitely sensitive indicator of impaired antiviral immunity.
Much less is known about rs11686168, a regulatory region variant in CDC42EP3. This gene encodes CDC42 effector protein 3, which is part of a protein family regulated by CDC42 and is involved in the organization of actin/septin cytoskeletons.30 CDC42EP3 lacks identifiable enzymatic active sites and is thought to act as a structural protein.31 Pertinent to our results, the US28 protein encoded by CMV hijacks the physiological regulation of CDC42 by the adenosine triphosphate binding cassette transporter ABCA1 and directly activates CDC42, which induces actin/septin remodeling and reorganization of lipid rafts at the cell surface.32 The advantage gained by CMV from remodeling of rafts has not been established, but the possibilities include enhanced immune evasion,33 increased exocytosis of viral proteins, or enhanced cell-to-cell transmission of the virus,34 a known mechanism of CMV spread in humans.35 Although rs11686168 variants in CDC42EP3 could affect the downstream effects of CDC42 activation by CMV US28, the lack of any difference between CMV-seropositive and seronegative donors rules out this mechanism for an association with CMV reactivation.
Our analyses have some limitations. First, we were not able to evaluate copy-number variants that have been implicated as important determinants of CMV reactivation and disease in some studies. Second, the sample sets of up to 1167 recipients and up 1209 donors used for discovery are small relative to many of the data sets used for GWAS analyses. Accordingly, the 1 × 10−6 threshold of statistical significance that we used for discovery is much less stringent than the 5 × 10−8 threshold typically used for genome-wide significance. Third, our analysis is also subject to false-positive results that are frequently observed by chance. We attempted to control false-positive results by using a discovery and replication approach in both our candidate variant and GWAS analyses. Our results indicate a plausible association of the donor rs1045642 genotype with the risk of CMV reactivation after allogeneic HCT through a mechanism involving the efflux activity of P-gp, but we could not identify a mechanism to explain the association of the donor rs11686168 genotype with CMV reactivation. Finally, it is possible that our analysis missed CMV-associated variants due to power limitations. To overcome this limitation, we provide the overall discovery plus replication cohort-wide statistical analysis of donor and recipient variants with MAF >1% and P ≤ 1 × 10−6 for association with CMV reactivation, high-level reactivation, or disease with the allelic model in supplemental Tables 8 (results) and 9 (quality control) for metanalysis with results from other cohorts.
A further consideration is that the analysis was restricted to European ancestry samples, since they made up the predominant ancestral group (86%) in our study. Though unlikely, including a relatively small number of samples from multiple other non-European or admixed populations could lead to false-positive or false-negative results due to nongenetic associations of ancestry with CMV infection. Deidentified individual donor and recipient genomic and disease data from our study are available from the National Center for Biotechnology Information database of Genotypes and Phenotypes (accession number phs001918) to be combined with data from other centers for analyses of other ancestral groups.
In summary, our results suggest that the G allele for rs1045642 in ABCB1 in donors reduces the risk of CMV reactivation by ∼20%, comparable to the magnitude of the effect observed for donor CMV serostatus. The validity of this association as a true-positive result is supported by the existence of a plausible biologic mechanism for this result given the effects of rs1045642 on intracellular concentrations of cyclosporine and tacrolimus. In principle, therapeutic blood concentration ranges for calcineurin inhibitors could be set at slightly lower levels in patients whose donors have the homozygous rs1045642 AA genotype to compensate for the lower P-gp activity and higher intracellular calcineurin inhibitor concentrations that increase the risk of CMV reactivation in patients who are not treated with antiviral agents. We acknowledge though that the adoption of new prevention strategies that replace current preemption approaches could lessen the future clinical relevance of this finding. Our results also suggest that all other variants previously implicated as mediators of CMV-related phenotypes do not significantly affect the risk of CMV reactivation or disease after HCT. This result throws doubt on the usefulness of donor or recipient genotype as a means of predicting the likelihood of CMV-related complications in HCT patients.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
This work was supported by National Institutes of Health (NIH), National Institute of Allergy and Infectious Diseases grants AI33484, AI149213, and T32AI118690; NIH, National Cancer Institute grants CA015704 and CA18029; and NIH, National Heart, Lung, and Blood Institute grants HL087690, HL088201, HL094260, HL105914, and K24HL093294.
Footnotes
Deidentified data for the donors and recipients used in this study are available under accession number phs001918 in the National Center for Biotechnology Information online database.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: D.M.L. and B.E.S. were responsible for data acquisition, imputation, informatics analyses, quality control, and statistical analyses; all authors other than J.A.H. revised draft manuscripts and approved the final version of the manuscript; and all authors contributed to the study concept and design, interpretation of results, and writing of the manuscript..
Conflict-of-interest disclosure: The authors declare no competing financial interests.
John A. Hansen died on 31 July 2019.
Correspondence: Amanda M. Casto, Department of Medicine, Division of Allergy and Infectious Diseases, University of Washington, 850 Republican St, S130, Seattle, WA 98109; e-mail: amcasto@uw.edu.
REFERENCES
- 1.Osarogiagbon RU, Defor TE, Weisdorf MA, Erice A, Weisdorf DJ. CMV antigenemia following bone marrow transplantation: risk factors and outcomes. Biol Blood Marrow Transplant. 2000;6(3):280-288. [DOI] [PubMed] [Google Scholar]
- 2.Ljungman P. CMV infections after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2008;42(Suppl 1):S70-S72. [DOI] [PubMed] [Google Scholar]
- 3.Tomblyn M, Chiller T, Einsele H, et al. ; Centers for Disease Control and Prevention . Guidelines for preventing infectious complications among hematopoietic cell transplantation recipients: a global perspective. Biol Blood Marrow Transplant. 2009;15(10):1143-1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.George B, Pati N, Gilroy N, et al. Pre-transplant cytomegalovirus (CMV) serostatus remains the most important determinant of CMV reactivation after allogeneic hematopoietic stem cell transplantation in the era of surveillance and preemptive therapy. Transpl Infect Dis. 2010;12(4):322-329. [DOI] [PubMed] [Google Scholar]
- 5.Lindsay J, Othman J, Kerridge I, et al. Cytomegalovirus (CMV) management in allogeneic hematopoietic cell transplantation: Pre-transplant predictors of survival, reactivation, and spontaneous clearance. Transpl Infect Dis. 2021;23(3):e13548. [DOI] [PubMed] [Google Scholar]
- 6.Meesing A, Razonable RR. New developments in the management of cytomegalovirus infection after transplantation. Drugs. 2018;78(11):1085-1103. [DOI] [PubMed] [Google Scholar]
- 7.Bhat V, Joshi A, Sarode R, Chavan P. Cytomegalovirus infection in the bone marrow transplant patient. World J Transplant. 2015;5(4):287-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.George B, Kerridge IH, Gilroy N, et al. A risk score for early cytomegalovirus reactivation after allogeneic stem cell transplantation identifies low-, intermediate-, and high-risk groups: reactivation risk is increased by graft-versus-host disease only in the intermediate-risk group. Transpl Infect Dis. 2012;14(2):141-148. [DOI] [PubMed] [Google Scholar]
- 9.O’Brien S, Ravandi F, Riehl T, et al. Valganciclovir prevents cytomegalovirus reactivation in patients receiving alemtuzumab-based therapy. Blood. 2008;111(4):1816-1819. [DOI] [PubMed] [Google Scholar]
- 10.Nguyen DD, Cao TM, Dugan K, Starcher SA, Fechter RL, Coutre SE. Cytomegalovirus viremia during Campath-1H therapy for relapsed and refractory chronic lymphocytic leukemia and prolymphocytic leukemia. Clin Lymphoma. 2002;3(2):105-110. [DOI] [PubMed] [Google Scholar]
- 11.Laurenti L, Piccioni P, Cattani P, et al. Cytomegalovirus reactivation during alemtuzumab therapy for chronic lymphocytic leukemia: incidence and treatment with oral ganciclovir. Haematologica. 2004;89(10):1248-1252. [PubMed] [Google Scholar]
- 12.Teira P, Battiwalla M, Ramanathan M, et al. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: a CIBMTR analysis. Blood. 2016;127(20):2427-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loeffler J, Steffens M, Arlt E-M, et al. Polymorphisms in the genes encoding chemokine receptor 5, interleukin-10, and monocyte chemoattractant protein 1 contribute to cytomegalovirus reactivation and disease after allogeneic stem cell transplantation. J Clin Microbiol. 2006;44(5):1847-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corrales I, Giménez E, Solano C, et al. Incidence and dynamics of active cytomegalovirus infection in allogeneic stem cell transplant patients according to single nucleotide polymorphisms in donor and recipient CCR5, MCP-1, IL-10, and TLR9 genes. J Med Virol. 2015;87(2):248-255. [DOI] [PubMed] [Google Scholar]
- 15.Annibali O, Piccioni L, Tomarchio V, et al. Impact of IFN lambda 3/4 single nucleotide polymorphisms on the cytomegalovirus reactivation in autologous stem cell transplant patients. PLoS One. 2018;13(7):e0200221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bravo D, Solano C, Giménez E, et al. Effect of the IL28B Rs12979860 C/T polymorphism on the incidence and features of active cytomegalovirus infection in allogeneic stem cell transplant patients. J Med Virol. 2014;86(5):838-844. [DOI] [PubMed] [Google Scholar]
- 17.Conomos MP, Laurie CA, Stilp AM, et al. Genetic diversity and association studies in US Hispanic/Latino populations: applications in the Hispanic Community Health Study/Study of Latinos. Am J Hum Genet. 2016;98(1):165-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamae H, Kirby KA, Sandmaier BM, et al. Effect of conditioning regimen intensity on CMV infection in allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2009;15(6):694-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green ML, Leisenring W, Stachel D, et al. Efficacy of a viral load-based, risk-adapted, preemptive treatment strategy for prevention of cytomegalovirus disease after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2012;18(11):1687-1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin PJ, Levine DM, Storer BE, Nelson SC, Dong X, Hansen JA. Recipient and donor genetic variants associated with mortality after allogeneic hematopoietic cell transplantation. Blood Adv. 2020;4(14):3224-3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Green ML, Leisenring W, Xie H, et al. Cytomegalovirus viral load and mortality after haemopoietic stem cell transplantation in the era of pre-emptive therapy: a retrospective cohort study. Lancet Haematol. 2016;3(3):e119-e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ljungman P, Boeckh M, Hirsch HH, et al. ; Disease Definitions Working Group of the Cytomegalovirus Drug Development Forum . Definitions of cytomegalovirus infection and disease in transplant patients for use in clinical trials. Clin Infect Dis. 2017;64(1):87-91. [DOI] [PubMed] [Google Scholar]
- 23.Vafadari R, Bouamar R, Hesselink DA, et al. Genetic polymorphisms in ABCB1 influence the pharmacodynamics of tacrolimus. Ther Drug Monit. 2013;35(4):459-465. [DOI] [PubMed] [Google Scholar]
- 24.Wang R, Sun X, Deng Y-S, Qiu X-W. Effects of MDR1 1236C > T-2677G > T-3435C > T polymorphisms on the intracellular accumulation of tacrolimus, cyclosporine A, sirolimus and everolimus. Xenobiotica. 2019;49(11):1373-1378. [DOI] [PubMed] [Google Scholar]
- 25.Capron A, Mourad M, De Meyer M, et al. CYP3A5 and ABCB1 polymorphisms influence tacrolimus concentrations in peripheral blood mononuclear cells after renal transplantation. Pharmacogenomics. 2010;11(5):703-714. [DOI] [PubMed] [Google Scholar]
- 26.Crettol S, Venetz J-P, Fontana M, et al. Influence of ABCB1 genetic polymorphisms on cyclosporine intracellular concentration in transplant recipients. Pharmacogenet Genomics. 2008;18(4):307-315. [DOI] [PubMed] [Google Scholar]
- 27.Ansermot N, Rebsamen M, Chabert J, et al. Influence of ABCB1 gene polymorphisms and P-glycoprotein activity on cyclosporine pharmacokinetics in peripheral blood mononuclear cells in healthy volunteers. Drug Metab Lett. 2008;2(2):76-82. [DOI] [PubMed] [Google Scholar]
- 28.Cattaneo D, Ruggenenti P, Baldelli S, et al. ; Mycophenolate Steroids Sparing (MYSS) Genetics Study Group . ABCB1 genotypes predict cyclosporine-related adverse events and kidney allograft outcome. J Am Soc Nephrol. 2009;20(6):1404-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimchi-Sarfaty C, Oh JM, Kim I-W, et al. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315(5811):525-528. [DOI] [PubMed] [Google Scholar]
- 30.Funakoshi Y, Wang Y, Semba T, et al. Comparison of molecular profile in triple-negative inflammatory and non-inflammatory breast cancer not of mesenchymal stem-like subtype. PLoS One. 2019;14(9):e0222336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farrugia AJ, Calvo F. The Borg family of Cdc42 effector proteins Cdc42EP1-5. Biochem Soc Trans. 2016;44(6): 1709-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Low H, Mukhamedova N, Cui HL, et al. Cytomegalovirus restructures lipid rafts via a US28/CDC42-mediated pathway, enhancing cholesterol efflux from host cells. Cell Rep. 2016;16(1):186-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fessler MB, Parks JS. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J Immunol. 2011;187(4):1529-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sviridov D, Bukrinsky M. Interaction of pathogens with host cholesterol metabolism. Curr Opin Lipidol. 2014;25(5):333-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sviridov D, Mukhamedova N. Cdc42 - A tryst between host cholesterol metabolism and infection. Small GTPases. 2018;9(3):237-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.