

Abstract

Shape memory polymers are promising materials in many emerging applications due to their large extensibility and excellent shape recovery. However, practical application of these polymers is limited by their poor energy densities (up to ∼1 MJ/m3). Here, we report an approach to achieve a high energy density, one-way shape memory polymer based on the formation of strain-induced supramolecular nanostructures. As polymer chains align during strain, strong directional dynamic bonds form, creating stable supramolecular nanostructures and trapping stretched chains in a highly elongated state. Upon heating, the dynamic bonds break, and stretched chains contract to their initial disordered state. This mechanism stores large amounts of entropic energy (as high as 19.6 MJ/m3 or 17.9 J/g), almost six times higher than the best previously reported shape memory polymers while maintaining near 100% shape recovery and fixity. The reported phenomenon of strain-induced supramolecular structures offers a new approach toward achieving high energy density shape memory polymers.
Short abstract
We report an approach to achieve a high energy density shape memory polymer based on the formation of strain-induced supramolecular nanostructures, which immobilize stretched chains to store entropic energy.
Introduction
Many emerging applications, such as soft robotics, deployable hinges or space structures, sealants, and smart biomedical sutures and devices require high energy density, one-way shape memory materials capable of large-strain and hysteresis-free shape recovery.1−5 Shape memory polymers (SMPs) are a promising choice due to their excellent shape recovery and fixity, large extensibility, low density, and ease of processing.6 Furthermore, SMPs can be integrated with 3D or 4D printing, can be patterned or programmed into complex shapes, and exhibit locally controlled actuation, which greatly enhances their potential for broader application.7−9 However, SMPs ubiquitously suffer from poor energy density (<1 MJ/m3),10 limiting their use where performing mechanical work is required.
SMPs reversibly alternate between a temporary deformed state and an initial undeformed state through application of a stimulus, such as heat or light. Stabilization (or fixing) of the temporary state requires a controllable molecular level change (e.g., glass or melting transition,11−15 dynamic networks,16−21 strain-induced crystallization,22−25 or liquid crystal phase transition26−28) that can be selectively activated and deactivated. Upon deactivation, SMPs return to their original undeformed state, driven by the relaxation of deformed chains between network junctions (e.g., topological entanglements, chemical cross-links, or secondary interpenetrating networks) that preserve the material’s memory of its initial state via stored entropic energy.29
Achieving high energy density SMPs that simultaneously possess high recovery stress and large recoverable strain poses a significant challenge.6 In general, the recovery stress generated by an SMP as it returns to its initial state is determined by the stored entropic energy in the network, which is controlled by the density and strength of network junctions.10 Since junction density in an entangled network is predetermined by the entanglement molecular weight of the polymer, SMPs generally store entropic energy on the same order of magnitude as their entanglement plateau modulus (∼1 MPa), providing an upper limit on the achievable recovery stress.30 A high degree of chemical cross-linking can increase junction density (and thus increase recovery stress) but significantly reduces stretchability, decreasing overall energy density. Dynamic bonds offer a potential solution because their ability to break and reform allows for chain sliding, while still increasing junction density.31,32 However, if the dynamic bonds are too weak, recovery stress is minimally increased. If the dynamic bonds are too strong, their behavior resembles that of a cross-linked network with low stretchability.17 Strain-induced crystallization in SMPs has also improved energy density, but the formation of crystallites during extension is difficult to control.22,23,25 While composite materials (e.g., polymer-CNT) have been reported with higher energy densities, these composites have significantly worse shape fixity and recovery (<50%) and low recoverable strain (<50%), creating hysteresis issues.33 To date, none of these mechanisms have produced high energy density SMPs that simultaneously possess high recovery stress and large recoverable strain.
This work addresses this challenge by reporting a novel shape memory mechanism based on the formation of strain-induced supramolecular structures by polymer chains with dynamic bonds. Under strain, polymer chains align into stable and hierarchically organized supramolecular nanostructures, trapping the stretched polymer chains in a highly elongated state. Accordingly, large amounts of energy are stored (19.6 MJ/m3 or 17.9 J/g), six times higher than the best previously reported SMPs, while maintaining near 100% shape fixity and recovery.
Results and Discussion
The polymer employed here (denoted hereafter as PPG-MPU, Mn = 10 kDa, ĐM = 1.1, Figure 1a) was synthesized through a simple, one-pot synthesis of diamine-terminated poly(propylene glycol) (PPG) macromonomers (Jeffamine D400, Mn = 0.4 kDa) and 4,4′-methylenebis(phenyl isocyanate),32 which incorporates 4,4′-methylenebis(phenyl urea) (MPU) units into the polymer backbone, similar to other systems with bisurea-based dynamic bonds.21,34,35 PPG-MPU has a high loading of these MPU hydrogen-bonding units (∼38 wt %) and thus a large density of strong network junctions and much higher Tg of 50 °C (Figure S1) compared to PPG (−75 °C).36 Furthermore, both reagents are common components of the polyurethane industry and produced in millions of tons per year, giving PPG-MPU an estimated raw materials cost of less than $5/kg (see Note S1), making it attractive for large-scale applications.
Figure 1.

Achieving high energy density in shape memory polymers using strain-induced supramolecular structures. (a) Combining a flexible backbone polymer (polypropylene glycol, PPG) with a strong and directional hydrogen-bonding unit (methylene bisphenylurea, MPU) creates a tough and stretchable polymer with high network junction density. (b) When the polymer is stretched from its initial amorphous state, the alignment of the polymer chains induces the formation of supramolecular structures from the directional hydrogen bond as shown by the insets. The creation of these structures stabilizes the chains in a temporary state. Upon heating, the supramolecular structures disassemble and the film rapidly contracts to its initial state.
Our previous work has shown that polymer chains with periodically placed and directional dynamic bonds assemble spontaneously into supramolecular nanofibers if their overall number-average molecular weight (Mn) is below the polymer’s critical molecular weight of entanglement (Mc).37 Here, we exploit this understanding to design a high energy density SMP whose Mn > Mc (Mc, PPG ≈ 5–6 kDa).38,39 Initially (i.e., no strain), PPG-MPU adopts an amorphous structure without the presence of large supramolecular aggregates due to topological entanglements. When strained, polymer chains align, and the dynamic bonds reassemble into large, ordered supramolecular nanostructures (Figure 1b). These nanostructures trap the polymer backbones in a highly stretched state, increasing the amount of stored entropic energy compared to other reported SMPs. Furthermore, a high junction density of entanglements and dynamic bonds preserves the initial state of the polymer, allowing for full recovery to its original state upon heating. Thus, these strain-induced supramolecular cooperative assemblies enable high recovery stress and energy density without sacrificing extensibility, shape recovery, or shape fixity. Throughout this paper, we use both “supramolecular” and “dynamic” to describe the noncovalent bonds formed between urea groups, as the former highlights the intermolecular (or interchain) nature of the bond formation, while the latter underlines the ability of these interactions to reversibly associate and dissociate (distinct from the more specific class of dynamic covalent bonds).
Shape Memory Properties
PPG-MPU exhibited clear shape programmability as shown in Figure 2a, in which the polymer is stretched over 300% into a fixed state and then returns to its original length after heating to 70 °C. The pristine stress–strain curve (Figure 2b, Figure S2) highlights the polymer’s high tensile strength (70 MPa), modulus (1.2 GPa), extensibility (500%), and toughness (270 MPa). The high extensibility of flexible polymers with dynamic bonds has been attributed to the continual breaking and reforming of dynamic bonds, which enables chain sliding.31,32 Two additional interesting phenomena were observed during tensile testing (Figure 2c). First, the polymer film changes from transparent to opaque white above ∼300% strain, indicating the emergence of large domains similar to the formation of large nematic domains in liquid crystals or stress whitening in semicrystalline polymers.40,41 This coincides with strain hardening as seen by the slope increase in the stress–strain curve at 300%. Additionally, instead of necking to a clean break, PPG-MPU often fractures by way of many tendrils ripping apart, with lengths up to 1 cm. This observation is consistent with the formation of large and highly connected supramolecular domains and helps to explain the high stretchability of PPG-MPU compared to other polymers that possess a high weight percent of dynamic bonds but exhibit poor extensibility.42
Figure 2.

Mechanical and shape memory characterization of PPG-MPU. (a) Images of PPG-MPU in its initial state, stretched over 300% to a temporary state and then heated to its recovered state (from top to bottom). The scale bar is 1 cm for all images. (b) Pristine stress–strain curve of PPG-MPU at a strain rate of 200% per minute. The shaded area under the curve shows the material’s high toughness of 270 MPa. (c) When strained above 300%, PPG-MPU transitions from transparent to opaque white, characteristic of the formation of large domains (left). When strained to failure, PPG-MPU fractures into multiple tendrils (right). The scale bar is 1 cm for both images. (d) Representative recovery stress versus temperature for PPG-MPU at 300% strain. (e) Representative free strain recovery experiment on PPG-MPU from 300% strain. (f) Literature comparison of the estimated energy density (y-axis) and the product of shape fixity and recovery (x-axis) plotted for different SMPs (see Table S1).
Next, we characterized the one-way shape memory behavior of the polymer through dynamic mechanical analysis. Remarkably, PPG-MPU exhibited a maximum recovery stress (σr,max) of 13.1 ± 0.4 MPa (Figure 2d, Figure S2) and a maximum recoverable strain (εr,max) of 300% (Figure 2e), corresponding to an estimated volumetric energy density (E = 1/2σr,maxεr,max) of 19.6 ± 0.6 MJ/m3. Furthermore, PPG-MPU has a shape fixity and recovery of Rf = 0.93 ± 0.05 and Rr = 0.99 ± 0.01, respectively (Figure 2e, Figure S2), allowing it to undergo multiple shape memory cycles with minimal hysteresis (Figure S3). Additionally, the low density (∼1.09 g/cm3) of PPG-MPU leads to a high specific energy density of 17.9 J/g, approximately 460 times the energy density of skeletal muscle (0.039 J/g).43
The combination of large extensibility with high recovery stress gives this polymer high energy density (almost six times greater than the previous highest energy density reported for an SMP25) as plotted in Figure 2f. Importantly, this high energy density is achieved simultaneously with high shape fixity and shape recovery (x-axis of Figure 2f, where unity is ideal). Each polymer in Figure 2f is classified based on the molecular level change that stabilizes the temporary state (e.g., strain-induced crystallization, glass or melting transition, dynamic networks, or liquid crystal, Table S1).
Thus, the polymer presented here represents the first example of an SMP that simultaneously possesses high energy density (19.6 MJ/m3 or 17.9 J/g), recovery stress (>10 MPa), extensibility (>500%), and shape fixity and recovery (>0.9). We next sought to compare these experimental recovery stress values to theoretical limits. The entropy change per volume (ΔS/V) associated with extending a network of Gaussian chains with junction density (vj) and extension ratio (α) is given by30,44
| 1 |
The attainable recovery stress (σr) based on the release of this stored entropic energy at a given temperature (T) is then given by
| 2 |
Equation 2 allows us to estimate the maximum achievable recovery stress
in our system, assuming the chains extend from a Gaussian state (<r> ≈ N1/2l) to a near fully extended state (<r> ≈ Nl) and a junction density equal to density over the backbone
molecular weight (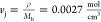 ). This results in an
estimated maximum
recovery stress (σr,max) of 20 MPa,
similar to the experimentally observed recovery stress of 13.1 MPa.
). This results in an
estimated maximum
recovery stress (σr,max) of 20 MPa,
similar to the experimentally observed recovery stress of 13.1 MPa.
In addition, we can use the estimated entropic energy required to achieve this high level of chain extension to determine the minimum enthalpic gain needed to stabilize the extended chains (ΔG = ΔH – TΔS < 0). From eq 1, we estimated the entropic change to be 0.12 J/cm3 K, which implies a minimum enthalpic gain of ∼15 kJ/mol. Previous studies have estimated the enthalpy associated with each urea–urea hydrogen bond to be a similar value (∼15 kJ/mol).45,46 Since MPU forms between two and four urea–urea hydrogen bonds, the estimated enthalpic gain from bond formation is 2–4 times higher (∼30–60 kJ/mol) than the required minimum amount, suggesting that the observed level of stored entropic energy is reasonable (∼25–50% of the maximum based on the bond strength of MPU). This analysis supports the attribution of PPG-MPU’s high energy density to the additional entropic energy stored due to the strain-induced formation of supramolecular nanostructures.
Structural Characterization
To better understand the molecular and microstructural origins of PPG-MPU’s advantageous shape memory properties, we collected 2D transmission small-angle X-ray scattering (SAXS) images of the polymer bulk film at 0% and 500% strain (Figure 3a,b). The strained film exhibits a much higher intensity and markedly anisotropic scattering pattern, similar to scattering patterns of oriented, uniaxially deformed polyethylene.47,48 The high-intensity regions lie in line with the stretching direction, suggesting that periodic structures orient perpendicular to strain as indicated in the inset. Averaging the intensity parallel to the stretching direction shows a consistent increase in intensity with increasing strain across the entire q-range below 0.2 Å–1 (Figure 3c). Furthermore, we can estimate the change in anisotropy as a function of strain by calculating Herman’s orientation parameter (f), with the stretching direction defined as the 0° axis:49
| 3 |
where <cos2 θ> is the mean-square cosine given by
 |
4 |
Figure 3.

Structural characterization of strain-induced supramolecular nanostructures. 2D SAXS images of PPG-MPU at 0% (a) and 500% (b) strain. Insets show the morphological change from initially amorphous to aligned supramolecular structures based on the change in scattering profiles. (c) 1D SAXS intensity curves parallel to the direction of strain (integrated over ±9°) at 0% (black), 100% (purple), 200% (blue), 300% (green), 400% (yellow), and 500% (orange) strain. Inset: plots Herman’s orientation parameter as a function of strain. Height AFM image of PPG-MPU at 0% (d) and 300% (e) strain. (f) 2D-FFT of the height AFM image in panel e. (g) 2D WAXS images of PPG-MPU unstrained (top) and strained (bottom). (h) 1D WAXS scattering profiles of strained PPG-MPU parallel (black) and perpendicular (blue) to strain. (i) Ratio of absorbance of urea N–H stretch (∼3350 cm–1) to the PPG backbone C–O stretch (∼1100 cm–1) as a function of polarizer angle for unstrained (black) and strained (blue) samples of PPG-MPU. For all parts, blue arrows indicate the stretching direction.
The inset of Figure 3c plots the orientation parameter as a function of strain, with f = 0 representing an isotropic state and f = 1 representing perfect alignment perpendicular to strain. Anisotropy increases with increasing strain, reaching almost 0.4 at 500% strain, a value similar to that of cold-drawn polyethylene.48 These data support the hypothesis that, during strain, polymer chains align along the stretching axis and simultaneously organize the dynamic bonds into large supramolecular nanostructures. These structures significantly increase both overall scattering intensity (due to the emergence of highly periodic structures) and anisotropy (due to their preferred orientation perpendicular to strain).
To visualize the supramolecular nanostructures, we performed atomic force microscopy (AFM). While PPG-MPU initially exhibits an isotropic and amorphous structure (Figure 3d), during straining, ordered nanostructures clearly emerge (Figure 3e, Figure S5). Performing a 2D fast Fourier transform (2D-FFT) on the AFM in Figure 3e (i.e., converting the real-space height profile into reciprocal space) allows for a qualitative comparison to the transmission SAXS data. Remarkably, the shape of the 2D-FFT image (Figure 3f) resembles that of the 2D-SAXS data (Figure 3b), suggesting that despite AFM being a surface technique, the observed nanostructures exist throughout the bulk film.50
Two-dimensional transmission wide-angle X-ray scattering (WAXS) data of unstrained PPG-MPU shows two broad peaks at 0.75 and 1.37 Å–1, corresponding to 8.4 and 4.6 Å, respectively (Figure 3g, Figure S4). The latter matches the expected urea–urea stacking distance for two hydrogen-bonded urea groups,51 while we attribute the former to the amorphous halo from the backbone PPG chains.52,53 Straining the sample results in an anisotropic scattering pattern that lacks any sharp peaks that would be expected for strain-induced crystallization of the polymer chains. The strained PPG-MPU sample exhibits a marked increase in the intensity of the urea–urea hydrogen-bond peak perpendicular to strain compared to parallel to strain, consistent with the alignment of periodic urea–urea hydrogen bonds oriented perpendicular to strain (Figure 3h). Thus, the WAXS data suggest that the periodic structures seen via SAXS are composed of aligned urea–urea hydrogen bonds. We further confirmed the molecular orientation of the polymer chains using polarized transmission Fourier transform infrared spectroscopy (FTIR), which shows a clear orientation of the N–H stretching (∼3350 cm–1) and C=O stretching (∼1720 cm–1) of the urea groups perpendicular to the straining axis compared to the intensity of the C–O stretch (∼1100 cm–1) of the PPG backbone (Figure 3i, Figure S4).
The above structural characterization reveals two key features of the strain-induced supramolecular structures: (1) uniform, periodic nanofibers oriented perpendicular to strain and (2) long fibrils oriented parallel to strain that arise between neighboring regions of banded nanofibers. These features are schematically illustrated in Figure 4a, which shows the proposed hierarchical organization of PPG-MPU at high strain. At the macroscopic scale, bulk films of strained PPG-MPU show a clearly oriented structure from both SAXS (Figure 3b) and polarized optical microscopy images (Figure 4b). At the micron-scale, elongated fibril regions consisting of banded nanofibers emerge in AFM images at high strain (Figure 4c, Figure S8). Even though the average chain has a Mn of only 10 kDa, which is less than 50 nm when fully extended, these well-defined fibrils extend over 10 μm in length or 200 chain contour lengths (Figure S8). The emergence of these fibrils is likely linked to the mechanical failure of the bulk film into macroscopic tendrils over ∼1 cm in length as shown in Figure 2c.
Figure 4.

Hierarchical nanostructure formation in highly strained PPG-MPU. (a) Schematic of hierarchical assembly. From left to right: (i) A bulk film of PPG-MPU is strained. (ii) Long, fibril shaped regions >10 μm in length (green) and ∼100 nm in width (orange) emerge parallel to strain. (iii) These long fibrils are comprised of stacked layers of nanofibers oriented perpendicular to strain that are ∼8 nm in diameter (purple) and separated by amorphous connecting regions. Each nanofiber is composed of multiple aligned nanorods. (iv) Nanorods are formed from stacked urea–urea hydrogen bonds (4.5 Å, yellow) between MPU units on the chain. The formation of these strain-induced supramolecular nanostructures traps stretched PPG backbone chains (∼2 nm, red) in an elongated state, resulting in a desirable shape memory behavior. (b) Polarized optical microscope images of a stretched PPG-MPU film. The orientation of the cross polarizer is shown in the top left. (c) Height AFM image of PPG-MPU at 500% strain. Dashed white box indicates the zoomed region. (d) Zoomed height AFM image shown in 3D (top). Height profile along the center of the fibril showing periodic fiber cross sections (bottom). For all parts, white arrows indicate the stretching direction.
Figure 4d shows a zoomed section of the fibril from the AFM image in Figure 4c along with the corresponding height profile, highlighting the periodic nanofibers oriented perpendicular to the long axis of the fibrils. We can estimate the median diameter of the nanofibers to be ∼8–9 nm (Figure S6), which is significantly greater than the expected distance between neighboring MPU hydrogen-bonding units along the polymer chain when the PPG backbone chain is near full extension (∼2 nm), suggesting that the nanofibers comprise at least 3–5 nanorods (i.e., rods of aligned MPU units), consistent with our previously reported results.37 A similar value for the fiber diameter (5.9 nm) can be obtained by fitting the SAXS data of strained PPG-MPU (Figure S7), which provides an excellent power-law fit with I ∝ q–3.65 up to an onset point of 0.107 Å–1, corresponding to the fiber diameter. We attribute the power-law scattering to surface fractal behavior created by interfaces of staggered nanofibers, similar to surface fractal behavior observed in polyethylene crystals and block copolymers.54,55 The obtained fractal dimension (D = 2.35) suggests that the interface is moderately rough compared to a perfectly flat surface (D = 2) or a fully crumpled surface (D = 3) and is close to the independently obtained surface fractal dimension from AFM (D = 2.3) (Figure S7).
We hypothesize that the alternating behavior between structured nanofibers and amorphous connecting regions arises from the localization of entanglement points between neighboring nanofibers, similar to recent MD simulations that have shown nonaffine clustering of entanglements between homopolymer chains during elongation.56 We posit that during strain, sections of the polymer chain between entanglement points are kinetically free to lengthen along the stretching axis, and chain sliding between neighboring chains aligns these lengthened sections into supramolecular nanofibers while simultaneously clustering the kinetically restricted entanglement junctions into the amorphous connecting regions between nanofibers. This hypothesis corresponds nicely with the observed nanofiber diameter, since the entanglement molecular weight (Me) of PPG is ∼2–3 kDa (Mc ≈ 2–3Me),57 which corresponds to about 3–5 repeat units of PPG-MPU (0.65 kDa each) or a length of ∼6–10 nm.
In summary, the structural characterization data presented above suggest a clear hierarchical structure as illustrated in Figure 4a. Along the axis of strain, long fibrils (>10 μm in length, > 100 nm in diameter) emerge comprising banded nanofibers (∼8 nm in diameter) that are separated by amorphous connecting domains. Each of these nanofibers consists of aligned rods of hydrogen-bonded urea groups (4.5 Å) connected by stretched PPG backbone chains (∼2 nm). The resulting hierarchical structure spans over five orders of magnitude, from stacked urea groups 4.5 Å apart to ordered fibrils over 10 μm in length, and results in uniform films at the macroscale (>1 cm).
Artificial Muscle Actuation
Finally, we showed the potential of PPG-MPU to be used as a strong and fast, soft, one-way actuator in a variety of demonstrations. Movie S1 shows a prestrained and twisted PPG-MPU thin film (1 mg) lifting and spinning a quarter (5.6 g) by 3.6 cm in less than a second upon heating with a heat gun (Figure 5a,b). The polymer lifts over 5000 times its own weight with a work output of 2 J/g or a power output of 2 W/g. Movie S2 presents another prestrained PPG-MPU film (25 mg) raising a 70 g weight by 5.1 cm, corresponding to a work output of 1.4 J/g (Figure 5c,d), and Movie S3 shows a prestrained PPG-MPU film undergoing rapid, load-free shape recovery after being placed on a hot plate. We compare this performance to other polymer actuators in Figure 5g (Table S2), which plots the ratio of load mass to actuator mass on the y-axis and the distance lifted on the x-axis. The specific work output (W, J/g) can then be calculated directly:
| 5 |
where ml is the load mass, ma is the actuator mass, d is the displacement, and g is the acceleration of gravity. Figure 5g highlights the differences between materials that possess similar work output but achieve it through different combinations of heavy loads versus large displacements. Compared to other actuators, PPG-MPU simultaneously achieves both a large actuation distance and high work output. It is important to note that as a one-way shape memory polymer PPG-MPU must be reprogrammed after each cycle, limiting its potential applications compared to reversible actuators, which do not need to be reprogrammed during cycling. Previous work has shown, through careful material processing and programming, that two-way shape memory can be achieved,58,59 which could enable PPG-MPU to be reversible without reprogramming; however, this is outside the scope of the current work.
Figure 5.

Demonstration of PPG-MPU as a high energy density actuator. (a, b) A prestrained film of PPG-MPU (1 mg) lifts a quarter (5.6 g, 5000 times the polymer’s weight) by 3.6 cm upon heating, corresponding to a work output of 2 J/g. (c, d) A prestrained film of PPG-MPU (25 mg) lifts a 70 g weight (2800 times the polymer’s weight) by 5.1 cm upon heating, corresponding to a work output of 1.4 J/g. (e, f) An artificial muscle comprised of prestrained films of PPG-MPU (3.8 g) actuates a full-size mannequin arm (0.6 kg) upon heating. For scale, the total length of the arm is 0.75 m, and the forearm (elbow to the end of the hand) is 0.45 m. (g) Comparison of PPG-MPU (yellow stars) to other polymer-based actuators (gray triangles) based on the mass of the load lifted divided by the mass of the actuator (y-axis) versus the achieved displacement of the load (x-axis). The dashed lines represent constant specific work density, increasing from 10–4 J/g (dark blue, bottom left) to 10 J/g (green, top right).
Lastly, to demonstrate both the high energy density of PPG-MPU as well as its inexpensive and scalable synthesis, we actuated a full-size human mannequin arm (0.75 m in length, Figure 5e,f, Figure S9) using an artificial muscle comprising prestrained PPG-MPU films (Movie S5). When heated, the fibers in the relaxed PPG-MPU muscle (3.8 g) contract to lift and hold the full-size arm (0.6 kg). These demonstrations highlight the combination of high energy density and ideal shape recovery achieved in the PPG-MPU polymer through the formation of strain-induced supramolecular nanostructures. The combination of these properties presents exciting future opportunities including integration with 3D or 4D printing, more complex patterning or programming, and locally controlled actuation.7−9
Conclusion
This work presents the first report of a shape memory polymer based on supramolecular nanostructures that achieves record-high energy density of 19.6 MJ/m3 with shape fixity and recovery above 90%. This performance is achieved through the novel mechanism of strain-induced supramolecular nanostructures, which fix flexible polymer chains in a highly elongated state, increasing the amount of stored entropic energy. Furthermore, the polymer reported here, which is synthesized using a simple one-pot synthesis, is low cost (<$5/kg for raw materials), single-component, solution soluble, and has a low density. These properties, combined with the excellent shape memory properties and high energy density of PPG-MPU, demonstrate the appeal of using strain-induced supramolecular structures to achieve high energy density one-way shape memory polymers.
Methods
Materials
Difunctional (primary amine) poly(propylene glycol) (H2N-PPG-NH2) with a molecular weight of 400 g/mol was obtained from Huntsman (Jeffamine D-400). All other chemicals and solvents were purchased from Sigma-Aldrich. All reagents were used as received without further purification. No unexpected or unusually high safety hazards were encountered.
Synthesis and Film Preparation
This synthesis is a modification of a previously described synthesis for PDMS-based, amine-terminated macromonomers.32 A solution of H2N-PPG-NH2 (Mn ≈ 400 g/mol, Jeffamine D400) and anhydrous dichloromethane (8.0 g in 150 mL, 0.13 mM) was prepared under a N2 atmosphere. Methylenebis(phenyl isocyanate) (MPU) was added in a 1:1 molar ratio of amine/isocyanate functional groups (5.0 g, 0.13 mM). The resulting mixture was stirred for 72 h at room temperature, until the solution gelled and partially precipitated. The synthesized polymer was quenched in methanol and then fully precipitated by adding an excess of hexane. The recovered polymer was subjected to vacuum evaporation for 2 h at 90 °C. Molecular weight according to GPC: Mw = 12.0 kDa, Mn = 10.6 kDa, ĐM = 1.1 (Figure S10). 1H NMR (400 MHz, d2-C2D2Cl4, δ/ppm): 7.75 (br, 4H), 7.30 (br, 4H), 7.03 (br, 4H), 3.93 (br, 2H), 3.81 (br, 1.5H), 3.45 (br, 17H), 1.09 (br, 17–18H) (assignments given in Figure S11). Elemental analysis data: Analytical calculations for (C39H62N4O8)n: C, 65.5; H, 8.7; N, 7.8; O, 17.9. Found: C, 64.3; H, 8.5; N, 8.3; O, 18.9 (remaining).
Film samples were prepared by drop casting 100 mg/mL solutions in CHCl3 onto SiO2 wafers treated with a monolayer of octadecyltrichlorosilane (OTS) to allow for easy removal of the film and dried for over 12 h at room temperature and then again at 70 °C for at least 24 h. Higher quality films were obtained using lower concentrations (e.g., 50 mg/mL compared to 100 mg/mL), though the resulting drop-casted films were much thinner for lower concentrations, due to the decreased solution viscosity.
Mechanical Characterization (Instron)
Room temperature tensile tests were conducted on an Instron 5565 Instrument at a constant strain rate of 200% per minute. Rectangular samples with an approximate dimension of 10 mm × 4 mm × 0.05 mm were cut from the substrate and loaded onto the extensometer with pressurized grips.
Dynamic Mechanical Analysis
Dynamic mechanical analysis (DMA) was performed on a DMA Q800 instrument. Shape memory experiments were preprogrammed into the DMA, with the polymer initially deformed to 300% strain at 50% strain per minute, held at fixed strain for 30 min, released to 0.001 N, and then heated at 5 °C per minute to 70 °C. Recovery stress experiments were performed by first straining the polymer film to 300% strain at 50% strain per minute and then holding for 30 min. The samples were released to 0.001 N and then held under constant strain, while the temperature was ramped at 5 °C per minute.
Small-Angle X-ray Scattering Methods
Small-angle X-ray scattering (SAXS) was conducted in transmission mode on bulk polymer films at beamline 4–2 at Stanford Synchrotron Radiation Lightsource (SSRL) of SLAC National Accelerator Laboratory (SLAC, Menlo Park, CA). Bulk polymer films were tested as free-standing films with a thickness of 0.03 mm on a custom-built uniaxial stretcher. The X-ray wavelength was 0.827 Å (beam energy 15 keV) with a sample-to-detector distance of 3.512 m. The Pilatus 1M fast detector was used for 2D scattering data acquisition, and reduction into scattering intensity profiles as a function of the scattering vector q was done using a customized code at the beamline. Otherwise, IgorPro (Wavemetrics Inc.) was used for plotting and processing the 1D and 2D data. For each sample, 30 frames of 1 s exposure were averaged to improve the signal-to-noise ratio. Measurements were performed in ambient air.
Wide-Angle X-ray Scattering Methods
Wide-angle X-ray scattering (WAXS) was conducted in transmission mode on bulk polymer films at beamline 11–3 at Stanford Synchrotron Radiation Lightsource (SSRL) of SLAC National Accelerator Laboratory (SLAC, Menlo Park, CA). Bulk polymer films were tested as free-standing films. The X-ray wavelength was 0.974 Å (beam energy 12.735 keV) with a sample-to-detector distance of 250 mm. 2D scattering data were exported with the NIKA package60 and calibrated in Wxdiff software using images from a LaB6 standard. Integrations 90–120° (out-of-plane) and 0–30° (in-plane) were taken for each sample from five separate exposures in the same position and averaged together to reduce background noise. Background subtraction was performed by fitting a linear baseline. Measurements were performed in ambient air.
Fourier Transform Infrared Spectroscopy
Infrared spectra were collected on a Nicolet iS50 FT-IR spectrometer in transmission mode at room temperature. Polarization-dependent spectra were collected by varying the angle of the built-in polarizer.
Polarized Optical Microscopy
Films were imaged using a Leica DM4000 M LED optical microscope equipped with a cross polarizer. Samples were rotated 45° under the cross-polarized light to view birefringence.
Size Exclusion Chromatography
Size exclusion chromatography (SEC) analysis was performed using a Tosoh EcoSEC Ambient (Room Temp)-GPC equipped with two TSK gel GPC columns (G3000Hhr and G4000Hhr; 7.8 mm I.D. × 30 cm, 5 μm) calibrated with a conventional calibration curve using monodisperse polystyrene standards. THF (40 °C) was used as a carrier solvent at the flow rate of 1.0 mL/min. Samples were prepared at 1 mg/mL in 50% CHCl3 and 50% THF by volume, by first dissolving in CHCl3 and then THF.
Atomic Force Microscopy
Height and phase images were collected via atomic force microscopy (AFM) on a Nanoscope IIII Multimode AFM in tapping mode with Tap300AI-G probes (radius < 10 nm). All images, statistical distribution extraction, surface fractal analysis, and 2D-FFT calculations related to AFM data were processed using Gwyddion software.
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) thermal analysis was performed on a TA Instruments Q2000 DSC. Approximately 10 mg of polymer was placed in sealed aluminum pans and then heated to 120 °C for 5 min and cooled to −50 °C for 5 min. Samples were then ramped from −50 to 120 °C at a rate of 20 °C/min. Glass transition temperatures were extracted using TA Universal Analysis software.
Acknowledgments
C.B.C. acknowledges support from the Department of Defense (DoD) through the National Defense Science & Engineering Graduate (NDSEG) Fellowship Program. This work is in part supported by the Army Research Office Materials Design Program (Grant No. W911NF-21-1-0092). Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under Award ECCS-1542152. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory for SAXS and WAXS experiments is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The authors thank Ivan Rajkovic and Thomas Weiss for their support during SAXS beamtime and Chris Tassone for support at WAXS beamtime.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.1c00829.
Notes: Estimated raw materials cost. Data: DSC thermal analysis of PPG-MPU. Additional mechanical tests for PPG-MPU. Cycling data for PPG-MPU. Orientation of urea–urea H-bond stacking. AFM images of PPG-MPUunder various strains. AFM estimate of fiber diameter. Surface fractal analysis and SAXS fiber diameter estimate. Additional AFM images. Scale reference for size of mannequin arm used in demo. GPC data for PPG-MPU. 1H NMR labeled spectra. Tables: Energy density comparison for various shape memory polymers. Demonstrated work output for various actuators (PDF)
Movie: fast actuation of a quarter (MP4)
Movie: Actuation of a 70 g weight (MP4)
Movie: Rapid load-free shape recovery (MP4)
Movie: Actuation of a full-size human mannequin arm (MOV)
Author Contributions
# C.B.C. and S.N. made an equal contribution.
The authors declare no competing financial interest.
Supplementary Material
References
- Lendlein A.; Kelch S. Shape-Memory Polymers. Angew. Chem., Int. Ed. 2002, 41 (12), 2034–2057. 10.1002/1521-3773(20020617)41:12<2034::AID-ANIE2034>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Hu J.; Zhu Y.; Huang H.; Lu J. Recent Advances in Shape-Memory Polymers: Structure, Mechanism, Functionality, Modeling and Applications. Prog. Polym. Sci. 2012, 37 (12), 1720–1763. 10.1016/j.progpolymsci.2012.06.001. [DOI] [Google Scholar]
- Roth P. J.; Lowe A. B. Stimulus-Responsive Polymers. Polym. Chem. 2017, 8 (1), 10–11. 10.1039/C6PY90169G. [DOI] [Google Scholar]
- Lendlein A.; Gould O. E. C. Reprogrammable Recovery and Actuation Behaviour of Shape-Memory Polymers. Nature Reviews Materials 2019, 4 (2), 116–133. 10.1038/s41578-018-0078-8. [DOI] [Google Scholar]
- Xia Y.; He Y.; Zhang F.; Liu Y.; Leng J. A Review of Shape Memory Polymers and Composites: Mechanisms, Materials, and Applications. Adv. Mater. 2021, 33 (6), 2000713. 10.1002/adma.202000713. [DOI] [PubMed] [Google Scholar]
- McCracken J. M.; Donovan B. R.; White T. J. Materials as Machines. Adv. Mater. 2020, 32 (20), 1906564. 10.1002/adma.201906564. [DOI] [PubMed] [Google Scholar]
- Biswas M. C.; Chakraborty S.; Bhattacharjee A.; Mohammed Z. 4D Printing of Shape Memory Materials for Textiles: Mechanism, Mathematical Modeling, and Challenges. Adv. Funct. Mater. 2021, 31 (19), 2100257. 10.1002/adfm.202100257. [DOI] [Google Scholar]
- Jin B.; Song H.; Jiang R.; Song J.; Zhao Q.; Xie T. Programming a Crystalline Shape Memory Polymer Network with Thermo- and Photo-Reversible Bonds toward a Single-Component Soft Robot. Science Advances 2018, 4 (1), eaao3865 10.1126/sciadv.aao3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Guo X.; Ye J.; Zheng N.; Kohli P.; Choi D.; Zhang Y.; Xie Z.; Zhang Q.; Luan H.; et al. Freestanding 3D Mesostructures, Functional Devices, and Shape-Programmable Systems Based on Mechanically Induced Assembly with Shape Memory Polymers. Adv. Mater. 2019, 31 (2), 1805615. 10.1002/adma.201805615. [DOI] [PubMed] [Google Scholar]
- Anthamatten M.; Roddecha S.; Li J. Energy Storage Capacity of Shape-Memory Polymers. Macromolecules 2013, 46 (10), 4230–4234. 10.1021/ma400742g. [DOI] [Google Scholar]
- Tian M.; Gao W.; Hu J.; Xu X.; Ning N.; Yu B.; Zhang L. Multidirectional Triple-Shape-Memory Polymer by Tunable Cross-Linking and Crystallization. ACS Appl. Mater. Interfaces 2020, 12 (5), 6426–6435. 10.1021/acsami.9b19448. [DOI] [PubMed] [Google Scholar]
- Lewis C. L.; Meng Y.; Anthamatten M. Well-Defined Shape-Memory Networks with High Elastic Energy Capacity. Macromolecules 2015, 48 (14), 4918–4926. 10.1021/acs.macromol.5b00763. [DOI] [Google Scholar]
- Nguyen N. A.; Meek K. M.; Bowland C. C.; Barnes S. H.; Naskar A. K. An Acrylonitrile-Butadiene-Lignin Renewable Skin with Programmable and Switchable Electrical Conductivity for Stress/Strain-Sensing Applications. Macromolecules 2018, 51 (1), 115–127. 10.1021/acs.macromol.7b02336. [DOI] [Google Scholar]
- Wang W.; Ping P.; Chen X.; Jing X. Polylactide-Based Polyurethane and Its Shape-Memory Behavior. Eur. Polym. J. 2006, 42 (6), 1240–1249. 10.1016/j.eurpolymj.2005.11.029. [DOI] [Google Scholar]
- Lin T.; Tang Z.; Guo B. New Design Strategy for Reversible Plasticity Shape Memory Polymers with Deformable Glassy Aggregates. ACS Appl. Mater. Interfaces 2014, 6 (23), 21060–21068. 10.1021/am505937p. [DOI] [PubMed] [Google Scholar]
- Jiang L.; Liu Z.; Lei Y.; Yuan Y.; Wu B.; Lei J. Sustainable Thermosetting Polyurea Vitrimers Based on a Catalyst-Free Process with Reprocessability, Permanent Shape Reconfiguration and Self-Healing Performance. ACS Appl. Polym. Mater. 2019, 1 (12), 3261–3268. 10.1021/acsapm.9b00672. [DOI] [Google Scholar]
- Zhang G.; Zhao Q.; Zou W.; Luo Y.; Xie T. Unusual Aspects of Supramolecular Networks: Plasticity to Elasticity, Ultrasoft Shape Memory, and Dynamic Mechanical Properties. Adv. Funct. Mater. 2016, 26 (6), 931–937. 10.1002/adfm.201504028. [DOI] [Google Scholar]
- Li J.; Viveros J. A.; Wrue M. H.; Anthamatten M. Shape-Memory Effects in Polymer Networks Containing Reversibly Associating Side-Groups. Adv. Mater. 2007, 19 (19), 2851–2855. 10.1002/adma.200602260. [DOI] [Google Scholar]
- Fang Z.; Zheng N.; Zhao Q.; Xie T. Healable, Reconfigurable, Reprocessable Thermoset Shape Memory Polymer with Highly Tunable Topological Rearrangement Kinetics. ACS Appl. Mater. Interfaces 2017, 9 (27), 22077–22082. 10.1021/acsami.7b05713. [DOI] [PubMed] [Google Scholar]
- Burnworth M.; Tang L.; Kumpfer J. R.; Duncan A. J.; Beyer F. L.; Fiore G. L.; Rowan S. J.; Weder C. Optically Healable Supramolecular Polymers. Nature 2011, 472 (7343), 334–337. 10.1038/nature09963. [DOI] [PubMed] [Google Scholar]
- Krajovic D. M.; Anthamatten M. Melt-Processable Shape-Memory Elastomers Containing Bisurea Segments. ACS Appl. Polym. Mater. 2021, 3 (4), 2082–2087. 10.1021/acsapm.1c00129. [DOI] [Google Scholar]
- Meng Y.; Jiang J.; Anthamatten M. Body Temperature Triggered Shape-Memory Polymers with High Elastic Energy Storage Capacity. J. Polym. Sci., Part B: Polym. Phys. 2016, 54 (14), 1397–1404. 10.1002/polb.23990. [DOI] [Google Scholar]
- Fritzsche N.; Pretsch T. Programming of Temperature-Memory Onsets in a Semicrystalline Polyurethane Elastomer. Macromolecules 2014, 47 (17), 5952–5959. 10.1021/ma501171p. [DOI] [Google Scholar]
- Li G.; Wang A. Cold, Warm, and Hot Programming of Shape Memory Polymers. J. Polym. Sci., Part B: Polym. Phys. 2016, 54 (14), 1319–1339. 10.1002/polb.24041. [DOI] [Google Scholar]
- Zhang P.; Li G. Structural Relaxation Behavior of Strain Hardened Shape Memory Polymer Fibers for Self-Healing Applications. J. Polym. Sci., Part B: Polym. Phys. 2013, 51 (12), 966–977. 10.1002/polb.23295. [DOI] [Google Scholar]
- Kim H.; Boothby J. M.; Ramachandran S.; Lee C. D.; Ware T. H. Tough, Shape-Changing Materials: Crystallized Liquid Crystal Elastomers. Macromolecules 2017, 50 (11), 4267–4275. 10.1021/acs.macromol.7b00567. [DOI] [Google Scholar]
- Saed M. O.; Ambulo C. P.; Kim H.; De R.; Raval V.; Searles K.; Siddiqui D. A.; Cue J. M. O.; Stefan M. C.; Shankar M. R.; Ware T. H. Molecularly-Engineered, 4D-Printed Liquid Crystal Elastomer Actuators. Adv. Funct. Mater. 2019, 29 (3), 1806412. 10.1002/adfm.201806412. [DOI] [Google Scholar]
- Lee K. M.; Bunning T. J.; White T. J. Autonomous, Hands-Free Shape Memory in Glassy, Liquid Crystalline Polymer Networks. Adv. Mater. 2012, 24 (21), 2839–2843. 10.1002/adma.201200374. [DOI] [PubMed] [Google Scholar]
- Xie T. Recent Advances in Polymer Shape Memory. Polymer 2011, 52 (22), 4985–5000. 10.1016/j.polymer.2011.08.003. [DOI] [Google Scholar]
- Hornat C. C.; Yang Y.; Urban M. W. Quantitative Predictions of Shape-Memory Effects in Polymers. Adv. Mater. 2017, 29 (7), 1603334. 10.1002/adma.201603334. [DOI] [PubMed] [Google Scholar]
- Li C.-H.; Wang C.; Keplinger C.; Zuo J.-L.; Jin L.; Sun Y.; Zheng P.; Cao Y.; Lissel F.; Linder C.; You X.-Z.; Bao Z. A Highly Stretchable Autonomous Self-Healing Elastomer. Nat. Chem. 2016, 8 (6), 618–624. 10.1038/nchem.2492. [DOI] [PubMed] [Google Scholar]
- Kang J.; Son D.; Wang G.-J. N.; Liu Y.; Lopez J.; Kim Y.; Oh J. Y.; Katsumata T.; Mun J.; Lee Y.; Jin L.; Tok J. B.-H.; Bao Z. Tough and Water-Insensitive Self-Healing Elastomer for Robust Electronic Skin. Adv. Mater. 2018, 30 (13), 1706846. 10.1002/adma.201706846. [DOI] [PubMed] [Google Scholar]
- Miaudet P.; Derré A.; Maugey M.; Zakri C.; Piccione P. M.; Inoubli R.; Poulin P. Shape and Temperature Memory of Nanocomposites with Broadened Glass Transition. Science 2007, 318 (5854), 1294–1296. 10.1126/science.1145593. [DOI] [PubMed] [Google Scholar]
- Colombani O.; Barioz C.; Bouteiller L.; Chanéac C.; Fompérie L.; Lortie F.; Montès H. Attempt toward 1D Cross-Linked Thermoplastic Elastomers: Structure and Mechanical Properties of a New System. Macromolecules 2005, 38 (5), 1752–1759. 10.1021/ma048006m. [DOI] [Google Scholar]
- Appel W. P. J.; Portale G.; Wisse E.; Dankers P. Y. W.; Meijer E. W. Aggregation of Ureido-Pyrimidinone Supramolecular Thermoplastic Elastomers into Nanofibers: A Kinetic Analysis. Macromolecules 2011, 44 (17), 6776–6784. 10.1021/ma201303s. [DOI] [Google Scholar]
- Wypych G.PPG Polypropylene Glycol. In Handbook of Polymers, 2nd ed.; Wypych G., Ed.; ChemTec Publishing, 2016; pp 517–519. 10.1016/B978-1-895198-92-8.50160-9. [DOI] [Google Scholar]
- Cooper C. B.; Kang J.; Yin Y.; Yu Z.; Wu H.-C.; Nikzad S.; Ochiai Y.; Yan H.; Cai W.; Bao Z. Multivalent Assembly of Flexible Polymer Chains into Supramolecular Nanofibers. J. Am. Chem. Soc. 2020, 142 (39), 16814–16824. 10.1021/jacs.0c07651. [DOI] [PubMed] [Google Scholar]
- Heinrich G.; Alig I.; Donth E. A Model for the Onset of Entanglements of Transient Hydrogen-Bonded Intermolecular Structures in Oligomeric Poly(Propylene Glycol). Polymer 1988, 29 (7), 1198–1202. 10.1016/0032-3861(88)90044-4. [DOI] [Google Scholar]
- Wool R. P. Polymer Entanglements. Macromolecules 1993, 26 (7), 1564–1569. 10.1021/ma00059a012. [DOI] [Google Scholar]
- Kojima M. Stress Whitening in Crystalline Propylene-Ethylene Block Copolymers. J. Macromol. Sci., Part B: Phys. 1981, 19 (3), 523–541. 10.1080/00222348108015316. [DOI] [Google Scholar]
- Wei P.; Huang J.; Lu Y.; Zhong Y.; Men Y.; Zhang L.; Cai J. Unique Stress Whitening and High-Toughness Double-Cross-Linked Cellulose Films. ACS Sustainable Chem. Eng. 2019, 7 (1), 1707–1717. 10.1021/acssuschemeng.8b05485. [DOI] [Google Scholar]
- Korley L. T. J.; Pate B. D.; Thomas E. L.; Hammond P. T. Effect of the Degree of Soft and Hard Segment Ordering on the Morphology and Mechanical Behavior of Semicrystalline Segmented Polyurethanes. Polymer 2006, 47 (9), 3073–3082. 10.1016/j.polymer.2006.02.093. [DOI] [Google Scholar]
- Madden J. D. W.; Vandesteeg N. A.; Anquetil P. A.; Madden P. G. A.; Takshi A.; Pytel R. Z.; Lafontaine S. R.; Wieringa P. A.; Hunter I. W. Artificial Muscle Technology: Physical Principles and Naval Prospects. IEEE J. Oceanic Eng. 2004, 29 (3), 706–728. 10.1109/JOE.2004.833135. [DOI] [Google Scholar]
- Rubinstein M.; Colby R. H.. Polymer Physics; Oxford University Press: Oxford, 2003. [Google Scholar]
- Masunov A.; Dannenberg J. J. Theoretical Study of Urea and Thiourea. 2. Chains and Ribbons. J. Phys. Chem. B 2000, 104 (4), 806–810. 10.1021/jp993078e. [DOI] [Google Scholar]
- Morrison C. A.; Siddick M. M. Determining the Strengths of Hydrogen Bonds in Solid-State Ammonia and Urea: Insight from Periodic DFT Calculations. Chem. - Eur. J. 2003, 9 (3), 628–634. 10.1002/chem.200390067. [DOI] [PubMed] [Google Scholar]
- Romo-Uribe A.; Manzur A.; Olayo R. Synchrotron Small-Angle x-Ray Scattering Study of Linear Low-Density Polyethylene under Uniaxial Deformation. J. Mater. Res. 2012, 27 (10), 1351–1359. 10.1557/jmr.2012.83. [DOI] [Google Scholar]
- López-Barrón C. R.; Zeng Y.; Schaefer J. J.; Eberle A. P. R.; Lodge T. P.; Bates F. S. Molecular Alignment in Polyethylene during Cold Drawing Using In-Situ SANS and Raman Spectroscopy. Macromolecules 2017, 50 (9), 3627–3636. 10.1021/acs.macromol.7b00504. [DOI] [Google Scholar]
- Roe R.-J.Methods of X-Ray and Neutron Scattering in Polymer Science; Oxford University Press: New York, 2000. [Google Scholar]
- Henry C. K.; Sandoz-Rosado E.; Roenbeck M. R.; Magagnosc D. J.; Palmese G. R.; Strawhecker K. E.; Alvarez N. J. Direct Measure of Crystalline Domain Size, Distribution, and Orientation in Polyethylene Fibers. Polymer 2020, 202, 122589. 10.1016/j.polymer.2020.122589. [DOI] [Google Scholar]
- Swaminathan S.; Craven B. M.; McMullan R. K. The Crystal Structure and Molecular Thermal Motion of Urea at 12, 60 and 123 K from Neutron Diffraction. Acta Crystallogr., Sect. B: Struct. Sci. 1984, 40 (3), 300–306. 10.1107/S0108768184002135. [DOI] [Google Scholar]
- Halasa A. F.; Wathen G. D.; Hsu W. L.; Matrana B. A.; Massie J. M. Relationship between Interchain Spacing of Amorphous Polymers and Blend Miscibility as Determined by Wide-Angle X-Ray Scattering. J. Appl. Polym. Sci. 1991, 43 (1), 183–190. 10.1002/app.1991.070430115. [DOI] [Google Scholar]
- Zhao J.; Chen P.; Lin Y.; Chang J.; Lu A.; Chen W.; Meng L.; Wang D.; Li L. Stretch-Induced Crystallization and Phase Transitions of Poly(Dimethylsiloxane) at Low Temperatures: An in Situ Synchrotron Radiation Wide-Angle X-Ray Scattering Study. Macromolecules 2018, 51 (21), 8424–8434. 10.1021/acs.macromol.8b01872. [DOI] [Google Scholar]
- Ogawa T.; Miyashita S.; Miyaji H.; Suehiro S.; Hayashi H. Fractal Properties of Polymer Crystals. J. Chem. Phys. 1989, 90 (3), 2063–2067. 10.1063/1.455997. [DOI] [Google Scholar]
- Xie R.; Yang B.; Jiang B. Surface Fractals in Block Copolymers. Macromolecules 1994, 27 (8), 1997–2001. 10.1021/ma00086a003. [DOI] [Google Scholar]
- Hsu H.-P.; Kremer K. Clustering of Entanglement Points in Highly Strained Polymer Melts. Macromolecules 2019, 52 (17), 6756–6772. 10.1021/acs.macromol.9b01120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetters L. J.; Lohse D. J.; Colby R. H.. Chain Dimensions and Entanglement Spacings. In Physical Properties of Polymers Handbook; Mark J. E., Ed.; Springer: New York, NY, 2007; pp 447–454. 10.1007/978-0-387-69002-5_25. [DOI] [Google Scholar]
- Chung T.; Romo-Uribe A.; Mather P. T. Two-Way Reversible Shape Memory in a Semicrystalline Network. Macromolecules 2008, 41 (1), 184–192. 10.1021/ma071517z. [DOI] [Google Scholar]
- Li J.; Rodgers W. R.; Xie T. Semi-Crystalline Two-Way Shape Memory Elastomer. Polymer 2011, 52 (23), 5320–5325. 10.1016/j.polymer.2011.09.030. [DOI] [Google Scholar]
- Ilavsky J. Nika: Software for Two-Dimensional Data Reduction. J. Appl. Crystallogr. 2012, 45 (2), 324–328. 10.1107/S0021889812004037. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.