

Short abstract
Content available: Author Audio Recording
Abbreviations
- BCAA
branched‐chain amino acid
- CI
confidence interval
- EASL
European Association for the Study of the Liver
- GABA
gamma aminobutyric acid
- HE
hepatic encephalopathy
- I.V.C.
inferior vena cava
- PSE
portosystemic encephalopathy
- v.
vein
Listen to an audio presentation of this article.

Nathan M. Bass
The association of maniacal behavior with jaundice was clearly described in antiquity by Hippocrates (ca. 460–370 BCE) in his Prognostics and Prorrhetics. 1 , 2 Celsus and Galen in Rome (first and third centuries CE, respectively) both similarly noted this syndrome. 2 In the Hippocratic Corpus, the author(s) observed that “those who are mad from phlegm are quiet and do not cry or make a noise; but those from bile [presumably yellow bile, the “choleric humor,” as shown in Fig. 1] are vociferous, malignant and will not be quiet.” 3 Meat intolerance of patients with cirrhosis had been observed previously in Anglo‐Saxon medicine 4 by Émile Jules Louis Parmentier (1860‐1940), a Parisian neurologist (Fig. 2), in his Doctoral Thesis in Medicine on cardiac cirrhosis. But as brought to attention in 1955 5 and again posthumously in 1978 by the quintessentially English physician William Hedley John Summerskill. (1926‐1977) 6 (Fig. 3) of later legendary Mayo fame, it was the Bard himself (aka William Shakespeare) who Summerskill first credited 5 , 7 with the poetic observation, in 1602, of the dietary precipitation of encephalopathy in the personality of the lush of Twelfth Night character Sir Andrew Aguecheek (Fig. 4), the dimwitted, vain, and clownish knight, who himself notes:
Methinks sometimes I have no more wit than
a Christian or an ordinary man has; but I
am a great eater of beef, and I believe that
does harm to my wit. (Act 1, Sc. III.)
FIG 1.

Image of woodcut from Physiognomische Fragmente zur Beförderung der Menschenkenntnis und Menschenliebe (1775‐1778) by Johann Kaspar Lavater; depictions of the four humors: phlegmatic and choleric, sanguine and melancholic.
FIG 2.

Photograph of Émile Jules Louis Parmentier in 1890.
FIG 3.

The incomparable William Hedley John Summerskill, Mayo’s celebrated hepatologist and Director of the Gastroenterology Unit until his untimely demise in 1977, 6 demonstrating the characteristic “flapping” hand tremor of HE. Photo reproduced with permission from Mayo Foundation for Medical Education and Research. All rights reserved. Watch here a video demonstrating asterixis. Reproduced with permission from New England Journal of Medicine. 59 Copyright 2010, Massachusetts Medical Society.
FIG 4.

Sir Andrew Aguecheek (left) with red stockings and doublet of despised yellow, standing next to his so‐called friend Sir Toby Belch (right) in the third act of William Shakespeare’s comedy Twelfth Night, or What You Will. Sir Toby, an aptronymically named flatulent drunkard and glutton, misused Sir Andrew’s generosity and gullibility and milked him for approximately two‐thirds of his stated income. Painting by Francis Wheatley (1771).
Well, perhaps.
A more rigorous exposition was provided in 1761 by Giovanni Battista Morgagni (Fig. 5), Professor of Theoretical Medicine in Padua, Italy, who wrote about the fate of an alcoholic Venetian nobleman, subsequently shown at postmortem to have cirrhosis of the liver, whose death was preceded by ascites, agitation, somnolence, violent delirium, and coma, thus providing the first recording of the progression of the disorder through a series of worsening stages. 1 , 2 In 1860, Friedrich Theodor von Frerichs, in his A Clinical Treatise on Diseases of the Liver 8 , 9 (Fig. 6A), described his experience with several patients diagnosed with cirrhosis—and some with acute liver failure—who manifested altered consciousness, “noisy” delirium, deep coma, and death. In his renowned first English textbook of liver disease (Fig. 6B), George Budd 10 also reported his familiarity with the phenomenon of liver disease patients who had cerebral symptoms, mostly associated with jaundice that in one way or another he attributed to reduced bile secretion and that he concluded was secondary to sudden and severe emotional distress in some cases, although by no means in all cases. In the second edition of his exhaustive textbook on liver disease (Fig. 6C), in particular in his Croonian Lecture XV at the Royal Society and Royal College of Physicians, Charles Murchison, 11 * the very same who had translated Frerich’s treatise on liver disease (Fig. 6A), discussed extensively, but inconclusively, the pathogenesis of the cerebral symptoms of advanced liver disease, which he referred to as “the typhoid state.” 11 Murchison’s uncertainty about the mechanism of cerebral dysfunction in acute or chronic liver disease even when jaundice was mild was in accord with Frerichs’s puzzlement before him, who noted: “It still remains for us to allude to the causes of the nervous symptoms.” 8 Indeed, that protracted journey had already commenced, although Frerichs narrowly missed discovering the role of ammonia in its pathogenesis despite having precipitated convulsions in dogs by injecting them with ammonium carbonate. At the beginning of the 19th century, the stage was actually set for understanding the pathophysiological basis of what was originally termed “hepatic coma,” when Scottish physician William Saunders (1743‐1817), Fellow of the Royal College of Physicians of the Royal Societies of London and Edinburgh and Senior Physician to Guy’s Hospital, inferred a relationship between the brain and the liver, as embodied in the observation in his textbook of liver diseases 12 (which was the substance of his 1792 Gulstonian Lecture to the Royal College of Physicians): “There seems much sympathy between the brain and the liver, and in maniacal persons, in whom there is generally a defection in the secretion of bile.” Aside from some isolated reports in 1833 to 1834, 13 , 14 a liver‐brain connection was not pursued until the second half of the 19th century.
FIG 5.

Giovanni Battista Morgagni’s seminal work, De sedibus et causis morborum per anatomen indagatis (Seats and Causes of Disease Investigated by Means of Anatomy), published in Venice in 1761. Image Source: The Rare Book, Health Sciences Library System, University of Pittsburgh, Pittsburgh, PA.
FIG 6.

Title pages of historic textbooks on liver disease by A. Friedrich Theodor von Frerichs: 1860 German version translated by Charles Murchison, New York edition, 1879 (A); George Budd, London, 1845 (B); and Charles Murchison, second edition, London, 1877 (including the Croonian Lectures) (C).
The study of portosystemic shunting in the pathogenesis of hepatic encephalopathy (HE) was initiated in 1877 by a two‐page report from a 28‐year‐old Russian surgeon by the name of Nikolai Vladimirovich Eck (Fig. 7), who created total portocaval shunts in eight dogs,† seven of which died within a week. 9 , 16 , 17 The survivor lasted 2 and a half months and then bolted to freedom, never to be seen or heard of again. Undeterred by this largely dismal outcome, and, perhaps, in the first hint at the cynical aphorism, “the operation was a success but the patient died,” Eck declared the operation “perfectly safe” to be “carried out on human beings.” Although the eponymous “Eck fistula” assured the young surgeon his place in history, he made no observations pertinent to neurological impacts of the operation and performed no further experiments, as he had been called up to serve in the Russian army. Nonetheless, his work inspired a series of detailed, scientifically rigorous experiments carried out in the laboratory of Ivan Petrovich Pavlov (Fig. 8), better known for his work on digestive physiology and the “conditioned reflex” for which he received the Nobel Prize in Physiology or Medicine in 1904. Pavlov and his team 18 noted that following the fashioning of an Eck fistula in dogs, they became irritable, stuporose, and ataxic when fed protein, leading to the coining of the term “meat intoxication.” It was also noted that continued protein feeding led to death, whereas if it was stopped, they made a full recovery. Most notably, these investigators explored the role of nitrogen, and specifically ammonia, in the pathogenesis of the observed disorder by precipitating it with the administration of ammonium chloride or sodium carbamate. 9
FIG 7.

Nikolai Vladimirovich Eck.
FIG 8.

Ivan Petrovich Pavlov (1849‐1936). Courtesy of the National Library of Medicine (https://collections.nlm.nih.gov/catalog/nlm:nlmuid‐101426180‐img).
The first portocaval shunt, following Eck’s procedure, in a human was performed by Vidal in 1903 in a patient with unrelenting hematemesis. 17 , 19 Vidal’s commentary regarding the postoperative course is truly remarkable: “Elaboration of albuminoides in the digestive tract has produced as we know an abundant production of ammoniacal substances which are toxic for the organism when the liver is no longer there to guard against this danger and transform them into urea and other waste products. Even in limited amounts, these substances produce sweats, muscular trembling, intense anxiety, cardiac arrhythmias—briefly a presentation characteristic of ammonia intoxication. The subject must return to the almost exclusive absorption of fats and hydrocarbons (sugars, various cereals, potatoes, etc.). This is the principal danger of total exclusion.” In this brief paragraph, Vidal anticipated the systematic elucidation, over the next five decades, of the role of ammonia and the dietary management of HE. Key supportive elements for the hypothesis that ammonia from the gut escapes removal by the diseased liver were the observation by Matthews that feeding ammonium salts to dogs with Eck fistula induced neurological symptoms 20 and the appreciation of the liver’s role in converting ammonia to urea. 21
The description of the urea cycle in 1932 by Krebs and Henseleit 22 would further pave the way to a better understanding of the role of ammonia in the neuropsychiatric syndrome complicating liver disease and its mitigation by therapeutic intervention. The 1930s and 1940s saw a proliferation of studies describing the clinical manifestations of “hepatic coma” (the commonly used term at the time, coupled to the concept of “hepatic precoma”) in patients with cirrhosis, both with and without surgical portal systemic shunts, and explored the role of ammonia and its nitrogenous precursors in its pathogenesis. 2 , 9 In 1953, Adams and Foley 23 gave a detailed description of the behavioral, neuromuscular, and electroencephalographic features of hepatic coma. They noted hyperreflexia, increased neuromuscular tone, and the characteristic “flapping tremor” or “liver flap,” sometimes likened to the beating of a bird’s wing or flügelschlagen, although it was not unique to the patient with advanced liver disease (Fig. 9). After consulting with a Jesuit classics scholar friend from Boston over a glass or several of Metaxa, Foley formulated the term asterixis from the Greek, roughly translating as “an inability to maintain a fixed position.” 24 Here, it is worth mentioning that asterixis is technically not a tremor but a form of negative myoclonus in which there are irregular abrupt lapses of muscular tone that reflect involuntary pauses in electromyographic activity. Foley also described a fine tremor of the outstretched fingers, or mini‐asterixis, that is often overlooked by an inattentive examiner. In those exciting early days of astute clinical observations, much was made of the “fetor hepaticus,” a peculiar odor sensed on the breath of patients, that became associated with hepatic coma. 9 Several creative and rather fanciful descriptions of the fetor were proposed (Table 1), some actually leading to the implication, by the most gifted sniffers, of specific chemical substances in the neuropathogenesis of HE. The ability to accurately detect this olfactory clinical sign is highly variable among clinicians, and there is little reliance on it these days as a diagnostic aid. However, even the most nasally challenged would have no difficulty distinguishing a fetor hepaticus from the odor of rotting fish that characterizes the syndrome of trimethylaminuria recognized by none other than the Bard in 1610, in the person of the slave Caliban in The Tempest. The fish odor syndrome 25 is due to an autosomal recessive inherited deficiency of hepatic flavin monooxygenase 3, the vital enzyme for the metabolism of the malodorous trimethylamine, which is formed by colonic bacteria from odorless trimethylamine‐N‐oxide and other dietary precursors and enters the enterohepatic circulation to be converted to the odorless oxide.
FIG 9.

Asterixis. With the patient instructed to hold their hands in a dorsiflexed position, a burst of rapid, involuntary flexion‐extension movements at the wrist is observed, corresponding to the sudden loss and recovery of the extensor muscle tone. The arrows show the direction of the “flapping” motion when the hand is dorsiflexed. Watch here, a video demonstrating asterixis. Reproduced with permission from New England Journal of Medicine. 59 Copyright 2010, Massachusetts Medical Society.
TABLE 1.
Fetor Hepaticus 9
| Fanciful descriptions: |
| “Pungent, like a small bit of decaying liver” |
| “Peculiar musty smell” |
| “Smelling like mice” |
| “The smell of a freshly opened corpse” |
| “A mixture of rotten eggs and garlic” |
| “A scorched, fruity odor” |
| Chemical substances suggested by the odor: |
| Amines |
| Indoles |
| Methyl mercaptan |
| Dimethyldisulfide |
| Short‐chain fatty acids |
A seminal series of contributions then came from the group in London led by arguably one of the most formidable hepatologists, physicians, researchers, and women of all time, Sheila Patricia Violet Sherlock, D.B.E., F.R.C.P., F.R.C.P.E., F.R.S., H.F.R.S.E., F.M.G.A., F.C.R.G.A. (Fig. 10). 26 The group around (the future Prof. Dame) Sheila Sherlock (1918‐2001), in which the young Bill Summerskill played a significant part, elucidated the role of the intestinal absorption of ammonia and its access to the systemic circulation through the impaired liver, via portosystemic collaterals, or both (Fig. 11). 2 , 27 , 28 Sherlock hence advanced the term “portosystemic encephalopathy” (PSE) to displace the term “hepatic coma,” noting that not all cases progressed to this extreme stage of impaired consciousness, and that it could also manifest as a chronic dementia. These authors also stressed the role of factors in the precipitation of episodic PSE, including dietary protein, gastrointestinal bleeding, and narcotics. In a fortunate melding of terms both ancient and modern, the succinct term hepatic encephalopathy (HE) prevails today.
FIG 10.

Prof. Dame Sheila Sherlock. Reproduced by kind permission from the Sheila Sherlock Centre, the Royal Free Hospital, London, UK.
FIG 11.

Mechanism of portal systemic encephalopathy. This diagram shows how a source of nitrogen (i.e., ammonia), derived from origins in the intestine, bypasses removal by the diseased liver, enters the systemic circulation, and reaches the brain. It is now recognized that urea is converted to ammonia by intestinal bacteria. Reproduced with permission from Liver Transplantation. 26 Copyright 2002, American Association for the Study of Liver Diseases.
In a productive collaboration with Sheila Sherlock, the neurologist Basil G. Parsons‐Smith 28 gave more detailed attention to electroencephalographic changes and first proposed a grading system for the clinical severity of PSE. Harold Conn and his colleagues, 9 , 29 to use Conn’s own term, “purified” the grading system proposed by Parsons‐Smith, while investigating the therapeutic efficacy of lactulose. The location of Conn’s storied academic home for half a century (Yale University) is in New Haven, Connecticut, but the system for assessing HE that incorporated alterations in the state of consciousness, intellectual function, behavior, and neuromuscular function and came to be known as the “West‐Haven” grading scale or criteria, derived its moniker from the location of the West Haven Connecticut Veterans Affairs Hospital, where Conn practiced for many decades and conducted his extensive clinical research in hepatology. Conn and Lieberthal, in their famed monograph, 9 made it quite clear that the grading of HE depended on criteria that were fluid across the proposed grades, thus creating a significant challenge in comparing the status among individual patients. Of substantial importance was the appreciation of the difference in natural history between HE accompanying cirrhosis and that observed in severe, acute (fulminant) liver failure. 30 At the other extreme emerged the detection of a more subtle level of HE, not captured by standard clinical assessment, but revealed only on neurophysiological testing. 31 This entity of “subclinical HE” eventually morphed into “minimal HE.” Further evolution in grading concepts now groups minimal and grade 1 HE into a “covert” category, and grades 2 through 4 as “overt,” which can be episodic or persistent. 32 Most important, the disturbing impact of minimal HE on complex intellectual performance and ability to drive a vehicle are now well recognized. 33 The need for a less cumbersome, more reproducible scale across observers and institutions conducting clinical trials has seen recent refinements to the West‐Haven scale, using well‐defined criteria and precise algorithms. 34 , 35
Technical difficulties in blood ammonia measurement, which have been portrayed as “a flight after a Will‐o'‐th'‐wisp,” 36 as well as substantial individual variation, account, in part, for a long‐recognized unreliable correlation with the clinical severity of HE among patients. 37 Forsooth, it has been suggested ironically that “blood ammonia levels probably cause as much confusion in those requesting their measurement as in the patients in whom they are being measured.” 7 Further, given that even typical encephalopathy is not necessarily pathognomonic for liver disease, it should go without saying that other reasons for hyperammonemic encephalopathy should be considered, such as large spontaneous portosystemic shunts 38 and genetic metabolic deficiencies (e.g., ornithine transcarbamylase deficiency 39 ) among a catalogue of inherited and acquired aberrations of ammonia metabolism, 40 while not forgetting other general (Fig. 12) and mechanistically more specific causes of cerebral dysfunction 41 that must always be excluded before concluding that encephalopathy is necessarily HE, even in patients with obvious liver disease. It is fitting to mention here the pediatric metabolic hyperammonemic entity of Reyes syndrome, 40 which was attributed to salicylate (aspirin) toxicity in children and adolescents and is now almost historic seemingly because of aspirin avoidance in this age group. It is thought that Reyes is not a single entity but a hodgepodge that includes both inherited and likely acquired imperfections in fatty acid oxidation or the urea cycle. There have even been rare cases of decompensated cirrhosis in which overzealous diuresis and circulatory volume contraction induce hypercarbia for which, uncommonly, compensatory hypoventilation supervenes that results in coma because of severe carbon dioxide retention, but not hyperammonemia. Measuring blood ammonia levels may be justified, therefore, in the diagnosis of patients in whom there is no other evidence of liver disease, or when a urea‐cycle defect is suspected, and in patients with acute liver failure because the relationship between arterial ammonia levels with cerebral edema and brain herniation 42 dictates a heightened vigilance for the detection and treatment of intracranial hypertension, especially for ammonia values greater than 150 to 200 μmol/L. 43 , 44
FIG 12.
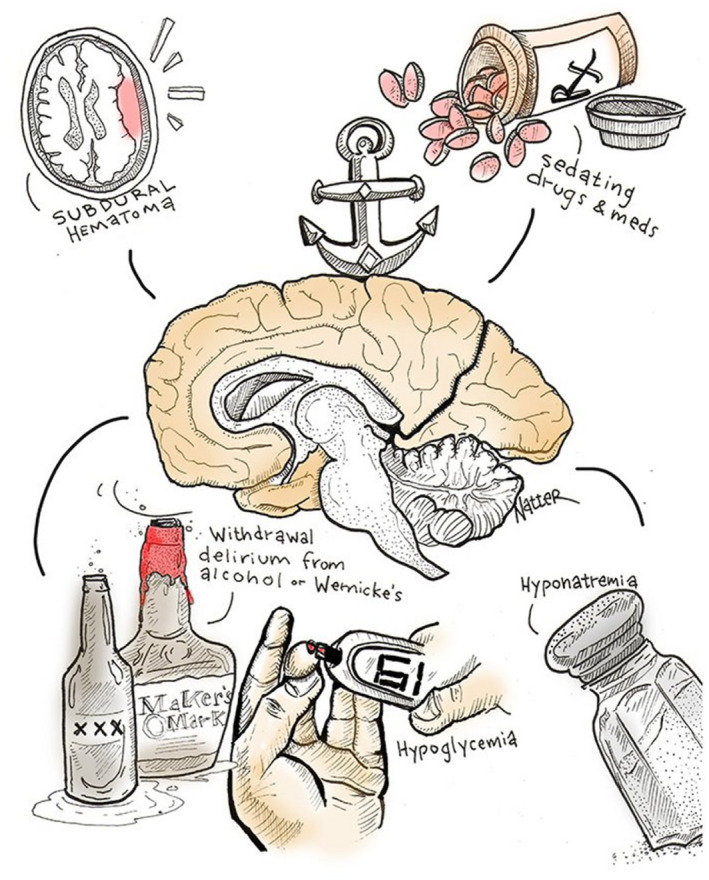
Common causes of cerebral dysfunction and reduced consciousness that are not specific to liver disease, depicted in cartoon form. Reproduced with permission from Michael Natter, MD (Instagram: @mike.natter; Twitter: @mike_natter), published by Medscape (https://www.medscape.com/), “15 Cartoons That Explain a Medical Career Better Than Words Can,” 2019. Available at: https://www.medscape.com/slideshow/medical‐career‐cartoons‐6011872#11.
The seemingly fickle consistency of evidence for the role of ammonia based on its peripheral assay in clinical HE helped to spur a diligent and creative search for other players involved in the pathogenesis of HE. 3 , 9 , 45 , 46 , 47 , 48 A concise list of these is shown in Table 2. There was often much excitement attending each of these innovations in our understanding of HE, and several led to trials of new therapies based on the attendant hypotheses. Alas, to paraphrase Shakespeare’s Hamlet, most strutted and fretted their hour upon the stage and then were heard no more. Some, including more recent ideas on the role of inflammation, 49 hold definite agency, at least in exacerbating the problem, but ammonia remains essentially undisplaced as the prime neurotoxin, possibly aided and abetted, to some as yet undetermined extent, by other players in the dramatis personae. Cerebral edema, a critical, often fatal process in HE accompanying acute liver failure 30 —and also in severe episodes of chronic HE and acute‐on‐chronic liver failure—clearly appears to correlate with blood ammonia levels. 42
TABLE 2.
| Mediator | Intervention | Example |
|---|---|---|
| Ammonia | Substrate reduction | Dietary protein restriction |
| Nonabsorbed disaccharides | Lactulose, lactitol | |
| Alpha‐glucosidase inhibition | Acarbose | |
| Poorly absorbed antibiotics | Neomycin, rifaximin | |
| Biochemical ammonia scavenging | Sodium benzoate, sodium phenylacetate, glycerol phenylbutyrate, l‐ornithine phenylacetate, l‐ornithine‐l‐aspartate | |
| Adsorbents | AST‐120 (carbon adsorbent) | |
| Competitive alteration of gut microbiota | Probiotics | |
| Synergistic neurotoxins | ||
| Mercaptans | ||
| Short‐chain fatty acids | Enhanced mitochondrial transport | l‐Carnitine |
| Phenol | ||
| Metals | ||
| Manganese excess | ||
| Zinc deficiency | Zinc supplements | |
| False neurotransmitters | ||
| Impaired dopaminergic neurotransmission | Dopamine receptor agonist | Bromocriptine |
| Increased ratio of plasma aromatic to BCAAs | Reversal of abnormal neurotransmitter/precursor ratio | Oral or intravenous BCAAs |
| Biogenic amines (e.g., octopamine) | Enhance dopamine synthesis in the brain | l‐Dopa |
| GABA receptor agonists | ||
| GABA | ||
| Endogenous and natural benzodiazepines | Benzodiazepine antagonist | Flumazenil |
| Neurosteroids | Neurosteroid antagonists | GR3027 |
| Systemic and neuroinflammation | Prevention and treatment of infectious/inflammatory complications of end‐stage liver disease | Antibiotics, experimental anti‐inflammatory therapies |
The field of therapy for HE has developed rationally in accord with the best efforts at understanding its pathogenesis (Table 2). The most consistently successful therapies have generally upheld the abiding nature of the ammonia hypothesis, with a focus on the role of gut bacteria in its production from urea. Dietary protein restriction, popular in the early days of management, 50 has given way to a more careful maintenance of protein intake to counter muscle loss (sarcopenia) and the loss of a key site of ammonia conversion to glutamine. 51 The minimally absorbed aminoglycoside antibiotic, neomycin, was initially used as a means of reducing the colonic flora responsible for the generation of ammonia from urea. Its use was attended by a risk for severe ototoxicity and renal and intestinal adverse effects. 29 , 52 Other antibiotics (e.g., metronidazole and vancomycin) were also explored, but their long‐term use was also attended by occasional unhappy toxicity, adding to the burden of symptoms plaguing an already unfortunate patient population. 9 , 48 Lactulose emerged initially from the thoughtful hypothesis and demonstration by Johannes Bircher 53 ‡ and subsequent clinical trials in the 1960s and 1970s 9 , 29 as an effective, albeit unpleasant, therapy with the rationale that this nonabsorbed disaccharide, fermented by colonic bacteria, achieved several impacts on the intestinal generation and elimination of ammonia. The medical treatment of HE has, alas, been monumentally stagnant compared with the explosion of progress in other areas of hepatology. At least, however, the audacious, some would say reckless, surgical approach to exclude colonic bacteria (i.e., colectomy), which had been used occasionally to treat chronic HE after portosystemic shunt surgery, 54 has long since been abandoned. Lactulose was approved by the US Food and Drug Administration in 1977. It remained the only such licensed agent for the treatment of HE until rifaximin, a far safer and better tolerated minimally absorbed antibiotic, achieved that status in 2010—more than 30 years later 55 (Fig. 13). There have been none since as of this writing.
FIG 13.

Time to first breakthrough episode of HE (primary endpoint in the intention‐to‐treat population), according to study group, in a double‐blind, placebo‐controlled randomized clinical trial of rifaximin in patients who were in remission from recurrent HE. 55 Symbols represent patients for whom data were censored. The P values were calculated by means of the log rank test, with stratification according to geographic region. Reproduced with permission from New England Journal of Medicine. 55 Copyright 2010, Massachusetts Medical Society.
Ultimately, the dearth of treatment options and the intractability of the problem has provided the key motivation to develop technologies to provide extracorporeal “liver dialysis” devices to manage severe HE in acute and acute‐on‐chronic liver failure. 34 , 47 Suffice it to say, this remains a challenging work in progress. But all is not yet lost in the field of liver support devices 56 and with respect to alternative forms of liver renewal, 57 as Alexander Pope would have us believe in his 1732 Essay on Man, that “hope springs eternal in the human breast.” In contrast, definitive replacement of the acutely or chronically failed liver, pioneered by Thomas Starzl in the United States and Sir Roy Calne in the United Kingdom, has proved the most resoundingly successful treatment to date. 58 The obstacle here, of course, is the restrictive nature of donor organ availability. Given current trends, it is likely that, as with the poor (Matthew 26:11), HE will be with us always, but with the hope that there will be substantial progress in the medical management of this difficult complication of advanced liver disease.
Series Editor’s Postscript
In common with several other renowned authors in this series, Dr. Nathan Bass, the author of this essay on HE, is a peripatetic hepatologist. He journeyed from his birthplace in Kaapstad, the Mother City of South Africa (otherwise known as Cape Town), via London, UK, to settle in The City by the Bay, as San Francisco, California, is known colloquially. In San Francisco, his career blossomed under the mentorship of the formidable Rudi Schmid and the scholarly gentleman Bob Ockner, and he rose to full professorship. From 1997 until 1998, he served as the Acting Chief of the Division of Gastroenterology in the Department of Medicine at the University of California, San Francisco, and subsequently, from 1998 to 2008, as the Medical Director of the Liver Transplant Program and Director of the Hepatology Fellowship Training Program. If that were not enough, he lists 40 honors and awards, upward of 150 invited presentations and lectures worldwide and regionally, 250 peer‐reviewed and other publications, and more commentaries and letters than you can shake a stick at.§ He retired from clinical practice in 2011.
Dr. Bass is known as Tony, whose sobriquet derives from the nickname of a maternal uncle, Nathan Cohen (see photograph below from his family archives), a star rugby player who played for Southwest Africa (modern‐day Namibia) and whose fans cheered him on as “Coney,” abbreviated from his last name, which morphed into “Tony.” The first Tony was a victim of a German sniper in Northern Tuscany in 1945, and he is buried in the picturesque military cemetery of San Jacopo al Girone outside Florence, on the banks of the Arno.

Used with permission from Tony Bass.
Tony’s family journeyed from Eastern Europe to South Africa via the East End of London, UK, toward the end of the 19th century. Tony obtained his medical degree from the University of Cape Town, where he also obtained a Ph.D. investigating structural aspects of the glutathione‐S‐transferase phase II enzyme family, formerly known as ligandin and thought to be the intracellular carrier of bilirubin. He cut his hepatological teeth under the watchful guidance of Ralph Kirsch and Stuart Saunders in Cape Town and the redoubtable legendary Prof. Sheila Sherlock (later Dame Sheila Sherlock since 1978; see photograph below and Fig. 10), at the Royal Free Hospital, London, UK, before migrating west to San Francisco in 1981.

Shelia Sherlock, EASL. Reproduced with permission from the European Association for the Study of the Liver.
In this eloquent essay on the evolution of knowledge of HE, in which he cites William Shakespeare no less than three times, Tony traces the recognition of confusion in patients with liver injury by the ancient Anglo‐Saxons, Greeks, and Romans, through 18th‐century Padua in Italy and 19th‐century London, Berlin, and Saint Petersburg.
The 20th century saw the identification of gut‐derived ammonia as the cause of what was mechanistically termed PSE, a culprit that endured despite repeated competition from other potential mediators, and even in the face of obstacles to measurement that cast doubt on the ammonia hypothesis. After dwelling on the clinical intricacies of the HE/PSE syndromes, the author discusses treatment that was dominated for decades by patient‐unfriendly lactulose (which is formed by heat‐induced isomerization of the natural milk sugar, lactose). The turning point came on May 25, 2010, when the US Food and Drug Administration approved the oral use of the nonsystemic antibiotic rifaximin to reduce the risk for recurrence of HE in patients with advanced liver disease, shortly before the publication of the results in the New England Journal of Medicine 59 of the successful pivotal double‐blind, placebo‐controlled, randomized clinical trial spearheaded by now Professor Emeritus Dr. Nathan “Tony” Michael Bass, of the University of California, San Francisco.
Supporting information
Video S1
Potential conflict of interest: Nothing to report.
Footnotes
Fellow of the Royal College of Physicians; President of the Pathological Society of London; Physician and Lecturer on the Principles and Practice of Medicine, St. Thomas’s Hospital; Vice‐President and Consulting Physician, London Fever Hospital; Examiner in Medicine, University of London; formerly Physician and Lecturer on Medicine, Middlesex Hospital; and on Medical Staff HM Bengal Army.
This episode in the surgical history of portal hypertension is discussed in the current series 15 by the esteemed liver surgeon Michael Henderson and his collaborator Christopher Anderson, both of the University of Mississippi Medical Center, Jackson, Mississippi.
Bircher's older relative, the Swiss physician Maximilian Bircher‐Benner, around the turn of the century introduced a diet of raw foods and fruit to the patients in his chalet‐style sanatorium in Zürichberg. In particular, he popularized a supper of raw oats, fruits and nuts, and yoghurt, which was known as Birchermüesli, or simply Müesli. Of course, Müesli (or d’Spys—“the dish,” as Bircher‐Benner called it), is now widely popular as a Swiss breakfast dish that contrasts with granola that is sweetened and baked.
It is no longer considered poor form to end a sentence with a preposition, contrary to Winston Churchill’s objection that “this is the type of errant pedantry up with which I will not put.”
References
- 1. Amodio P. Hepatic encephalopathy: historical remarks. J Clin Exp Hepatol 2014;5:S4‐S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Summerskill WHJ, Davidson EA, Sherlock S, et al. The neuropsychiatric syndrome associated with hepatic cirrhosis and an extensive portal collateral circulation. Q J Med 1956;25:245‐266. [PubMed] [Google Scholar]
- 3. Hippocrates. The sacred disease . Adams F (trans.). The genuine works of Hippocrates: translated from the Greek with a preliminary discourse and annotations. London: The Sydenham Society; 1849. [Google Scholar]
- 4. Parmentier E. Études cliniques et anatomo‐pathologiques sur le foie cardiaque. Paris: G.Steinheil; 1890. [Google Scholar]
- 5. Summerskill WH. Aguecheek's disease. Lancet 1955;2:288‐289. [DOI] [PubMed] [Google Scholar]
- 6. Wolstenholme G, Luniewska V. William Hedley John Summerskill. Royal College of Physicians. Available at: HYPERLINK "sps:urlprefix::https" https://history.rcplondon.ac.uk/inspiring‐physicians/william‐hedley‐john‐summerskill.
- 7. Reuben A. There is nothin’ like a dame. Hepatology 2002;35:983‐985. [DOI] [PubMed] [Google Scholar]
- 8. Frerichs FT. A Clinical Treatise on Diseases of the Liver (Murchison C, Trans.). Vol. 1. London: New Sydenham Society; 1860. [Google Scholar]
- 9. Conn HO, Lieberthal MM. The Hepatic Coma Syndromes and Lactulose. Baltimore: Williams & Wilkins; 1978. [Google Scholar]
- 10. Budd G. Chapter III. Fatal jaundice: Section I. On Diseases of the Liver. London: John Churchill; 1845. [Google Scholar]
- 11. Murchison C. Clinical lectures on diseases of the liver, jaundice and abdominal dropsy – Including the croonian lectures on functional derangements of the liver, delivered at The Royal College of Physicians in 1874. Lauder Brunton T, Fayrer J, eds. London: Longmans, Green, and Co.; 1885:655‐693. [Google Scholar]
- 12. Saunders W. A Treatise on the Structure, Economy, and Diseases of the Liver; Together With an Enquiry Into the Properties and Component Parts of the Bile and Biliary Concretions. London: sold by G. G. and J. Robinson; J. Murray; J. Johnson; and T. Cox, Borough; 1793. [Google Scholar]
- 13. Griffin W. On what marked state does the occurrence of coma and sudden death in jaundice depend? London Med Gaz 1833. ‐1834;13:801‐805. [Google Scholar]
- 14. Aldis CJB. Case of jaundice with cerebral affection terminating fatally. London Med Gaz 1833. ‐1834;13:833‐834. [Google Scholar]
- 15. Henderson JM, Anderson CD. The surgical treatment of portal hypertension. Clin Liver Dis (Hoboken) 2020;15(suppl 1):S52‐S63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Konstantinov IE. Eck‐Pavlov shunt: the 120th anniversary of the first vascular anastomosis. Surgery 1997;121:640‐645. [DOI] [PubMed] [Google Scholar]
- 17. Donovan AJ, Covey PC. Early history of the portacaval shunt in humans. Surg Gynecol Obstet 1978;147:423‐430. [PubMed] [Google Scholar]
- 18. Hahn M, Massen VN, Nenski M, et al. Die Eck’sche Fistel zwischen der unteren hohlvene und der pfortader und ihre folgen fur den organismus. Arch Exp Pathol Pharmakol 1893;32:161‐210. [Google Scholar]
- 19. Vidal ME. Traitement chirurgical des ascites dans les cirrhosis du foie. 16th Congress Français de Chirurgie 1903;16:294‐304. [Google Scholar]
- 20. Matthews SA. Ammonia, a causative factor in meat poisoning in Eck fistula dogs. Am J Physiol 1922;59:459‐460. [Google Scholar]
- 21. Salaskin S. Über das ammoniak in physiologischer und pathologischer hinsicht und die rolle im stoffwechsel stickstoffhaltiger substanzen. Z Physiol Chem 1898;25:449‐491. [Google Scholar]
- 22. Krebs HA, Henseleit K. Untersuchungen über die harnstoffbildung im tierkörper. Z Physiol Chem 1932;210:33‐66. [Google Scholar]
- 23. Adams RD, Foley JM. The neurological disorder associated with liver disease. Res Publ Assoc Res Nerv Ment Dis 1953;32:198‐237. [PubMed] [Google Scholar]
- 24. Pal G, Lin M, Laureno R. Asterixis—history and terminology. Neurology 2015;84(14 suppl):S44.004. [Google Scholar]
- 25. Messenger J, Clark S, Massick S, et al. A review of trimethylaminuria (fish odor syndrome). J Clin Aesthet Dermatol 2013;6:45‐48. [PMC free article] [PubMed] [Google Scholar]
- 26. Williams R. A personal reflection on the life and work of Dame Sheila Sherlock. Liver Transpl 2002;8:191‐192. [Google Scholar]
- 27. Sherlock S, Summerskill WH, White LP, et al. Portal‐systemic encephalopathy; neurological complications of liver disease. Lancet 1954;264:453‐457. [PubMed] [Google Scholar]
- 28. Parsons‐Smith BG, Summerskill WH, Dawson AM, et al. The electroencephalograph in liver disease. Lancet 1957;273:867‐871. [DOI] [PubMed] [Google Scholar]
- 29. Conn HO, Leevy CM, Vlahcevic ZR, et al. Comparison of lactulose and neomycin in the treatment of chronic portal‐systemic encephalopathy. A double blind controlled trial. Gastroenterology 1977;72:573‐583. [PubMed] [Google Scholar]
- 30. Ware AJ, D'Agostino AN, Combes B. Cerebral edema: a major complication of massive hepatic necrosis. Gastroenterology 1971;61:877‐884. [PubMed] [Google Scholar]
- 31. Gitlin N, Lewis DC, Hinkley L. The diagnosis and prevalence of subclinical hepatic encephalopathy in apparently healthy, ambulant, non‐shunted patients with cirrhosis. J Hepatol 1986;3:75‐82. [DOI] [PubMed] [Google Scholar]
- 32. Bajaj JS. Hepatic encephalopathy: classification and treatment. J Hepatol 2018;68:838‐839. [DOI] [PubMed] [Google Scholar]
- 33. Nardone R, Taylor AC, Höller Y, et al. Minimal hepatic encephalopathy: a review. Neurosci Res 2016;111:1‐12. [DOI] [PubMed] [Google Scholar]
- 34. Hassanein T, Blei AT, Perry W, et al. Performance of the hepatic encephalopathy scoring algorithm in a clinical trial of patients with cirrhosis and severe hepatic encephalopathy. Am J Gastroenterol 2009;104:1392‐1400. [DOI] [PubMed] [Google Scholar]
- 35. Bajaj JS, Frederick RT, Bass NM, et al. Overt hepatic encephalopathy: development of a novel clinician reported outcome tool and electronic caregiver diary. Metab Brain Dis 2016;31:1081‐1093. [DOI] [PubMed] [Google Scholar]
- 36. White LP, Phear EA, Summerskill WHJ, et al. Ammonium tolerance in liver disease: observations based on catheterization of the hepatic veins. J Clin Invest 1955;34:158‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bajaj JS, Bloom PP, Chung RT, et al. Variability and lability of ammonia levels in healthy volunteers and patients with cirrhosis: implications for trial design and clinical practice. Am J Gastroenterol 2020;115:783‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Praktiknjo M, Simón‐Talero M, Römer J, et al. Total area of spontaneous portosystemic shunts independently predicts hepatic encephalopathy and mortality in liver cirrhosis. J Hepatol 2020;72:1140‐1150. [DOI] [PubMed] [Google Scholar]
- 39. Walker V. Severe hyperammonaemia in adults not explained by liver disease. Ann Clin Biochem 2012;49:214‐228. [DOI] [PubMed] [Google Scholar]
- 40. Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest 2007;132:1‐19. [DOI] [PubMed] [Google Scholar]
- 41. Hughes CG, Patel MB, Pandharipande PP. Pathophysiology of acute brain dysfunction: what’s the cause of all this confusion? Curr Opin Crit Care 2012;18:518‐526. [DOI] [PubMed] [Google Scholar]
- 42. Clemmesen JO, Larsen FS, Kondrup J, et al. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999;29:648‐653. [DOI] [PubMed] [Google Scholar]
- 43. Wijdicks EFM. Hepatic encephalopathy. N Engl J Med 2016;375:1660‐1670. [DOI] [PubMed] [Google Scholar]
- 44. Stravitz RT, Lee WM. Acute liver failure. Lancet 2019;394:869‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jones EA, Schafer DF. Hepatic encephalopathy: a neurochemical disorder. In: Popper HP, Schaffner F, eds. Progress in Liver Diseases. Vol. 8. New York: Grune & Stratton; 1986:525‐540. [PubMed] [Google Scholar]
- 46. Butterworth RF. Neurosteroids in hepatic encephalopathy: novel insights and new therapeutic opportunities. J Steroid Biochem Mol Biol 2016;160:94‐97. [DOI] [PubMed] [Google Scholar]
- 47. Rahimi RS, Rockey DC. Hepatic encephalopathy: pharmacological therapies targeting ammonia. Semin Liver Dis 2016;36:48‐55. [DOI] [PubMed] [Google Scholar]
- 48. Ferenci P. Hepatic encephalopathy: Pathogenesis. In: Runyon BA (ed.). UpToDate. Philadelphia: Wolters Kluwer; 2019. Available at: HYPERLINK "sps:urlprefix::https" https://www.uptodate.com/contents/hepatic‐encephalopathy‐pathogenesis. Accessed December 28, 2020. [Google Scholar]
- 49. Jalan R, Olde Damink SWM, Hayes PC, et al. Pathogenesis of intracranial hypertension in acute liver failure: inflammation, ammonia and cerebral blood flow. J Hepatol 2004;41:613‐620. [DOI] [PubMed] [Google Scholar]
- 50. Silen W, Harper HA, Mawdsley DL, et al. Effect of antibacterial agents on ammonia production within the intestine. Proc Soc Exp Biol Med 1955;88:138‐140. [DOI] [PubMed] [Google Scholar]
- 51. Wijarnpreecha K, Werlang M, Panjawatanan P, et al. Association between sarcopenia and hepatic encephalopathy: a systematic review and meta‐analysis. Ann Hepatol 2020;19:245‐250. [DOI] [PubMed] [Google Scholar]
- 52. Ferenci P. Hepatic encephalopathy in adults: Treatment. In: Runyon BA (ed.), UpToDate. Philadelphia: Wolters Kluwer; 2020. Available at: HYPERLINK "sps:urlprefix::https" https://www.uptodate.com/contents/hepatic‐encephalopathy‐in‐adults‐treatment?topicRef=1257&source=see_link. Accessed December 28, 2020. [Google Scholar]
- 53. Bircher J, Muller J, Guggenheim P, et al. Treatment of chronic portal‐systemic encephalopathy with lactulose. Lancet 1966;1:890‐892. [DOI] [PubMed] [Google Scholar]
- 54. McDermott WV Jr, Victor M, Point WW. Exclusion of the colon in the treatment of hepatic encephalopathy. N Engl J Med 1962;267:850‐854. [Google Scholar]
- 55. Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med 2010;362:1071‐1081. [DOI] [PubMed] [Google Scholar]
- 56. Garcia Martinez JJ, Bendjelid K. Artificial liver support systems: what is new over the last decade? Ann Intensive Care 2018;8:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nicolas CT, Hickey RD, Chen HS, et al. Liver regenerative medicine: from hepatocyte transplantation to bioartificial livers and bioengineered grafts. Stem Cells 2017;35:42‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ahluwalia V, Wade JB, White MB, et al. Liver transplantation significantly improves global functioning and cerebral processing. Liver Transpl 2016;22:1379‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mendizabal M, Silva MO. Asterixis. N Engl J Med 2010;363:e14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1