

Abstract
Radiation is associated with tissue damage and increased risk of atherosclerosis but there are currently no treatments and a very limited mechanistic understanding of how radiation impacts tissue repair mechanisms. We uncovered that radiation significantly delayed temporal resolution programs that were associated with decreased efferocytosis in vivo. Resolvin D1 (RvD1), a known pro-resolving ligand, promoted swift resolution and restored efferocytosis in sublethally irradiated mice. Irradiated macrophages exhibited several features of senescence, including increased expression of p16INK4A and p21, heightened levels of SA- β-gal, COX-2, several pro-inflammatory cytokines/chemokines, and oxidative stress (OS) in vitro, and when transferred to mice exacerbated inflammation in vivo. Mechanistically, heightened OS in senescent macrophages led to impairment in their ability to carry out efficient efferocytosis and treatment with RvD1 reduced OS and improved efferocytosis. Sublethally irradiated Ldlr−/− mice exhibited increased plaque necrosis, p16INK4A cells and decreased lesional collagen compared with non-irradiated controls and treatment with RvD1 significantly reduced necrosis and increased lesional collagen. Removal of p16INK4A hematopoietic cells during advanced atherosclerosis with p16-3MR mice reduced plaque necrosis and increased production of key intraplaque resolving mediators. Our results demonstrate that sublethal radiation drives macrophage senescence and efferocytosis defects and suggest that RvD1 may be a new therapeutic strategy to limit radiation-induced tissue damage.
Keywords: radiation, senescence, inflammation-resolution, resolvin, macrophage, efferocytosis, atherosclerosis
Introduction
Inflammation-resolution, an active process that tempers pro-inflammatory factors and promotes tissue repair, is controlled by several endogenous mediators that include specialized pro-resolving mediators (SPM) such as lipoxins, resolvins, protectins, and maresins (1). SPMs are protective in vivo, act to control leukocyte trafficking, enhance the clearance of dead cells (i.e. efferocytosis) and promote tissue repair in a manner that does not compromise host defense (1). Dysregulated inflammation-resolution is associated with several prevalent human diseases including atherosclerosis and so understanding processes that may derail these programs are of clinical interest.
Ionizing radiation which is broadly used as a treatment for some types of cancers is associated with increased atherosclerosis (2-4). Documented radiation-induced cardiovascular disease (CVD) extends beyond cancer therapy and is also associated with environmental and occupational exposure (5). Radiation is thought to promote atherosclerosis through direct injury to endothelial and smooth muscle cells within the vasculature (2), yet other cellular players are likely involved. Along these lines, macrophages reside in nearly every tissue and, understanding how macrophages respond to radiation is of interest (6). Macrophages are also critical effectors of inflammation-resolution and how radiation impacts temporal resolution programs and efferocytosis are not known.
Radiation can arrest the proliferation of cancer cells but an off target effect is a maladaptive halt in the proliferation of otherwise healthy cells, a process called senescence (7). Radiation-induced senescence provokes the senescence-associated secretory phenotype (SASP) which is associated with increased oxidative stress (OS), aberrant metabolic programs, and the release of pro-inflammatory factors (8). There are major gaps in our understanding as to why radiation-induced SASP is left unchecked by our bodies and assessing the mechanisms associated with inflammation and resolution/repair in this context are highly underexplored.
Hematopoietic cells and macrophages are important players in the progression of atherosclerosis (9, 10). Sublethal radiation leads to hematopoietic stem cell (HSC) senescence and mimics several features of aging and atherosclerosis such as alterations in HSC functions like reduced clonogenicity and skewed differentiation toward myeloid lineages (11). Moreover, human macrophages acquire a pro-inflammatory phenotype when exposed to sublethal radiation (6, 12). Therefore, we posit that radiation promotes macrophage senescence/SASP and those senescent macrophages contribute to prolonged inflammation and atherosclerosis progression.
Here, we used a model of sublethal radiation and observed a significant delay in temporal inflammation-resolution in vivo that was associated with impaired efferocytosis. Treatment with the SPM called Resolvin D1 (RvD1) promoted inflammation-resolution and efferocytosis in sublethally irradiated mice. We found that macrophages do indeed undergo senescence when exposed to radiation and that radiation-induced senescence led to increased pro-inflammatory cytokines/chemokines and OS and defective efferocytosis, that was reversed with the treatment of RvD1 in vitro. necrosis, p16INK4A cells and decreased lesional collagen compared with non-irradiated controls and treatment with RvD1 significantly reduced necrosis and increased lesional collagen. Conditional removal of hematopoietic p16INK4A cells during advanced atherosclerosis reduced plaque necrosis and increased key SPMs in plaques. Together, we found that sublethal radiation promotes macrophage senescence and that RvD1 can be a new therapeutic strategy to combat radiation-induced damage.
Materials and Methods
Experimental Animals
Male 8-10 weeks old C57BL/6 mice were purchased from Taconic and 8 weeks old male Ldlr−/− mice were purchased from The Jackson Laboratory. Mice were housed in the Albany Medical College Animal Research Facility. All animal experiments were conducted in accordance with the Albany Medical College IACUC guidelines for animal care and were approved by the Animal Research Facility at Albany Medical College.
γ-radiation and Zymosan A-induced peritonitis
Male C57BL/6 mice (Taconic) were mock or sublethally γ-irradiated (7 grays) and were given a 3 month recovery period to induce bone marrow myeloid cell senescence (13). After 3 months, mice were intraperitoneally (i.p.) injected with 200 μg of ZymA (or ZymA, Sigma, Cat #Z4250) per mouse. For RvD1 treatment studies, mice were i.p. injected with 300 ng of RvD1 (Cayman Chemical, Cat# 10012554) and 200 μg of ZymA simultaneously and peritoneal exudates were collected by lavage 24 hrs post injection. For some experiments control or senescent (SC) macrophages (0.8 x106 cells/mouse) were i.p. injected simultaneously with ZymA and peritoneal cells were collected as described below. Peritoneal cells were harvested at the indicated time points and were then enumerated with a hemocytometer and Trypan blue exclusion. Remaining cells were washed in FACS buffer (PBS containing 5% (vol/vol) of FBS and labeled with FITC anti-Ly6G (BioLegend cat #127607) and APC anti-F4/80 (eBioscience, cat #17-4801-82) for 30 minutes at 4°C. Cells were then washed and resuspended in FACS buffer before performing flow-cytometric assessment. Flow-cytometry was carried out on a FACSCalibur (BD Biosciences), and data were analyzed by FlowJo software. Also, whole bone marrow was flushed from femurs and mRNA was extracted with a Qiagen RNeasy Mini kit (Cat #74106) and cDNA was synthesized using QuantiTect Reverse Transcription kit (Cat #205313) according to manufacturer’s instructions. Bone marrow mRNA was assessed for p16INK4A, p19ARF and p21 gene expression by qRT-PCR (details below). In parallel, experiments to determine PMN frequency, whole bone marrow was flushed from femurs and tibias and after RBC lysis, cell suspensions were plated and stained using the following antibodies (BioLegend): PE-Cy7-conjugated CD11b (M1/70, cat #101216), APC-Cy7-conjugated Ly6G (A18, cat # 127623) and Pacific Blue-conjugated Ly6C (HK1.4, cat # 128013). Surface stained cells were analyzed on an LSR II (BD Biosciences) with FACSDiva software and analyzed using FlowJo software (TreeStar, Ashland, OR).
Senescent Macrophages (SC-Macrophages)
Elicited peritoneal macrophages from C57BL/6 mice were collected by i.p. injection of ZymA (300 μg/mouse). Mice were sacrificed 48 hrs post ZymA injection and peritoneal cells were collected by lavage. Cells were enumerated as above and macrophages were plated (700 x103 cells/well in a 12-well tissue culture plate) in DMEM containing high glucose (4.5g/L) (Corning 10-013-CV), 10% FBS, 20% L-cell conditioned media (vol/vol) and 1% Penn Strept overnight. Media was refreshed the next day and the adherent peritoneal macrophages were subjected to 5 grays of γ-radiation (Gammacell 40 Exactor, MDS Nordion) and cultured for 3 additional days with the above media. For some experiments, SC-macrophages that had been exposed to γ-radiation for 2 days were then treated with either Vehicle (PBS) or 10 nM RvD1 for an additional 24 hrs.
IMR-90 Fibroblast Senescence
Human fetal lung IMR-90 fibroblast cells were purchased from Coriell Institute for Medical Research, NJ (I90-83). Proliferating IMR-90 cells (passage 6-15) were cultured in Eagle’s Minimum Essential media (Corning 10-009-CV) with 5% FBS and 1% Penn Strept. until ~80% confluency. IMR-90 cells were detached with trypsin-EDTA (Sigma cat# 03690), enumerated, and plated in a 10cm cell culture dish (750x103 cells/dish). Cells were cultured overnight (~16 hrs) to allow for attachment and were then subjected to 10 grays of γ-radiation, and cultured (37°C, 5% CO2) for 10 days. Fresh media was replaced on day 2, 5 and 8. On day 10, senescent IMR-90 cells were treated with either Vehicle (PBS, Corning #21-040-CV) or 10nM Resolvin D1 (RvD1) in serum-free media for additional 24 hrs and end point analyses (all of which are described in detail below) were performed on day 11.
Quantitative Real-time PCR
The total RNA was extracted from control or SC-macrophages or IMR-90 cells using a Qiagen RNeasy Mini kit (Cat #74106) and cDNA was synthesized using QuantiTect Reverse Transcription kit (Cat #205313) according to manufacturer’s instruction. Expression of mRNA was assessed with PerfeCTa SYBR Green FastMix (QuantaBio, VWR, cat# 101414-288) and run on BioRad CFX Connect real time qRT-PCR machine. Relative expression (ΔCt) was normalized to housekeeping genes, and the (ΔΔCt) method was used. The sequence for human and murine primers are as follows: Murine housekeeping gene 18S forward: 5’- ATG CGG CGG CGT TAT TCC-3’ reverse: 5’-GCT ATC AAT CTG TCA ATC CTG TCC-3’, murine p16INK4A forward: 5’-AAT CTC CGC GAG GAA AGC-3’ reverse: 5’-GTC TGC AGC GGA CTC CAT-3’, murine p21 forward: 5’-TTG CCA GCA GAA TAA AAG GTG-3’, reverse: 5’-TTT GCT CCT GTG CGG AAC-3’, murine p19ARF forward: 5’-GCC GCA CCG GAA TCCT-3’ reverse: 5’-TTG AGC AGA AGA GCT GCT ACGT-3’, Human housekeeping gene 18S forward: 5’ATG GGC GGC GGA AAA TAGC-3’ reverse: 5’-TCT TGG TGA GGT CAA TGT CTGC-3’, human p16INK4A forward: 5’-CTT CGG CTG ACT GGC TGG-3’ reverse: 5’-TCA TCA TGA CCT GGA TCG GC-3’, human p21 forward: 5’-AGT CAG TTC CTT GTG GAG CC-3’, reverse: 5’-GAC ATG GCG CCT CCT CTG-3’, human p19ARF forward: 5’-CCC TCG TGC TGA TGC TAC TG-3’ reverse: 5’-ACC TGG TCT TCT AGG AAG CGG-3’.
Ki67 staining
Control or SC-macrophages were prepared as above and plated in an 8-well chambered coverslip (Lab-Tek). Three days post radiation, macrophages were fixed with 100% Methanol for 5 mins, washed with 1X PBS and incubated with 0.4% Titron-X-100 for an additional 10 mins at room temperature. Fixed cells were blocked with 5% BSA in PBS for 1 hr at room temperature and incubated with rabbit anti-mouse Ki67 primary antibody (Abcam ab15580) at 1:200 dilution overnight at 4°C. The following day macrophages were then washed with 1X PBS to remove unbound Ki67 antibody and incubated with Alexa Fluor-594 goat anti-rabbit secondary antibody (Invitrogen A-11037) at 1:250 dilution for 2 hrs at room temperature. Nuclei were stained with Hoechst for 10 mins and images were acquired immediately on a Leica SPE confocal microscope and 6-7 different fields were acquired per well per group. Macrophages whose Ki67 stain colocalized with the nucleus were considered a Ki67 positive cell. The total number of macrophages and Ki67 positive macrophages were counted and expressed as percentage of Ki67 positive cells.
CellROX staining
Control or SC-macrophages were stimulated with Vehicle, 10nM RvD1 or 10μM N-acetyl-L-cysteine (NAC, ROS inhibitor, Sigma cat#A7250) for 24 hrs. Then, cells were stained with 5μM CellROX Green staining solution for 30 mins as per manufacturer’s instructions (Invitrogen, cat# C10492), followed by a counterstain with DAPI, and images were immediately acquired on a Leica confocal microscope. 6-7 different visual fields per treatment were analyzed and the percent of CellROX positive cells per total number of cells (nuclear stain) per visual field was enumerated.
Metabolic flux analysis
Senescent IMR-90 cells or macrophages were treated with Vehicle or RvD1 as described above. Cells were seeded (30,000 cells/well) on a Seahorse XF96 cell culture plate (Agilent, #102601-100) on the day of the assay. For measuring glycolytic ATP production, IMR-90 cells were incubated in Seahorse XF DMEM media pH=7.4 (Agilent, #103575-100) supplemented with 1mM pyruvate (Sigma Cat# S8636), 10 mM glucose (Sigma, Cat # G8769), 2mM L-glutamine (Corning, Cat#25-005-Cl) at 37°C for 1 hr. Basal extracellular acidification rate (ECAR) was measured using the Seahorse XFe Real-Time ATP Rate assay kit (Agilent, #103592-100) and run in the Seahorse XF96 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA). After three basal ECAR measurements, the oligomycin A (1.5μM) and Rotenone/Antimycin A (0.5μM) were serially injected after every three measurements. Glyco ATP production rate was calculated according to equations described in the Agilent Seahorse XF Real-Time ATP Rate Assay user guide.
Macrophages were incubated in glucose-free, DMEM (Agilent Cat# 102353, pH=7.4 at 37°C), supplemented with 143mM NaCl and 2mM L-Glutamine for 1 hr for measuring glycolytic function. After three basal ECAR measurements, 20 mM Glucose (Sigma, cat # G8769) followed by 1μM oligomycin, (Sigma, cat#75351) and then 80 mM 2-deoxy glucose (Sigma, cat# D6134) were injected in a sequential manner. The ECAR after addition of glucose is reported as fold change.
Senescence-associated beta-galactosidase (SA-β-gal) assay
Bright field imaging
SA-β-gal activity was detected in IMR-90 cells using a SA-β-gal staining kit (Cell Signaling, #9860). IMR-90 cells (40,000 cells/well in a 12-well plate) were seeded in complete media, senescence was induced and treated with Vehicle or RvD1 as described above. The cells were fixed and incubated for 12-14 hrs at 37°C in the presence of the β-galactosidase staining. Next, cells were imaged on a Zeiss brightfield microscope, and 10-15 different fields were acquired per well/group. SA-β-gal+ cells were enumerated based on blue staining.
C12-FDG flow cytometric method
SA-β-gal activity in control or SC-macrophages was determined using flow cytometry as described in ref (14). Briefly, 700x103 macrophages/well were seeded into 12-well plates in DMEM media as described above, senescence was induced and treated with Vehicle or RvD1 as described above. The media was removed, and cells were incubated with 100nM bafilomycin A1 for 1 h in fresh cell culture medium. C12-FDG (33 μm) is a fluorescent β-gal substrate and was added for additional 1 hr at 37°C. Macrophages were detached using cell stripper (Corning, cat#23-25-056-CI-PK), centrifuged at 250 x g for 5 min at 4°C and resuspended in 0.4 mL of FACS buffer. Samples were acquired with an LSRII Flow cytometer and analyzed with FlowJo software.
COX-2 staining
Control and SC-macrophages were fixed with 4% PFA for 10 mins and permeabilized with 1x perm wash buffer (BD cat#51-2091KZ) for 30 mins at RT. Fixed cells were stained with rabbit anti-mouse COX-2-Alexa488 at 1:100 dilution in perm wash buffer (Cell signaling, cat #13596S) for 1 hr at RT. Stained cells were washed with FACS buffer (5% BSA in PBS) and 10,000 events were collected in BD FACS Calibur. Data was analyzed using FlowJo software. Results were expressed as fold change of mean fluorescence intensity of COX-2.
PGE2 ELISA
Supernatants from control or SC-macrophages or IMR90 cells were collected and subjected to PGE2 analysis by ELISA (Cayman Chemical).
In vitro Efferocytosis assay
Control or SC-macrophages were stimulated with Vehicle, 10nM RvD1 or 10μM NAC 24 hrs prior to performing efferocytosis. The next day, Jurkats were enumerated and then stained with PKH26 (Sigma) according to the manufacturer’s instructions. Excess dye was removed by washing and the Jurkats were resuspend in RPMI containing 10% FBS. To induce apoptosis, Jurkats were then exposed to UV radiation (0.16 Amps, 115 Volts, 254 nm wavelength) for 15 mins at room temperature and then placed in an incubator (37°C, 5% CO2) for 3 hrs (15). Apoptotic Jurkats were either co-cultured with macrophages in a 3:1 ratio or IMR-90 cells in 10:1 ratio, for an additional 1 hr or 2 hrs respectively in 37°C, 5% CO2. Excess apoptotic cells were removed by washing ~3x with PBS. The cells were then immediately fixed with 4% formalin and subjected to fluorescence imaging with a Biorad ZOE Fluorescent Cell Imager. Six-to-seven different fields were acquired per well/group and an efferocytic event was considered a macrophage containing red apoptotic cells. Results were expressed as the percent of efferocytosis per total macrophages.
γ-radiation Induced Murine Atherosclerosis
Male Ldlr−/− mice (8 weeks old) were subjected to mock or 7 grays of γ-radiation and immediately fed a Western Diet (TD.88137, Envigo) for 12 weeks. Mice were socially housed in standard cages at 22°C under a 12 hrs light and 12 hrs dark cycle. During weeks 12–15, WD-fed Ldlr−/− mice were randomly assigned to receive Vehicle (i.e. 500 μL of sterile PBS) or RvD1 (100 ng/mouse) for an additional 3 weeks, while still on the WD. Mice were sacrificed at the end of 15 weeks. Lesion and necrotic area analysis were carried out on H&E stained lesional cross-sections, and lesional collagen was determined by picrosirius red staining as per the manufacturer’s instructions (Polysciences, cat#24901). Images were acquired using an Olympus camera and Olympus DP2-BSW software as previously described (16). Briefly, frozen specimens were immersed in OCT, cryosectioned and 10μm sections were placed on glass slides. Atherosclerotic lesion area, defined as the region from the internal elastic lamina to the lumen, was quantified by taking the average of 6 sections spaced ~24μm apart beginning at the base of the aortic root.
Murine lesion p16INK4A staining
γ-radiation induced murine atherosclerosis experiments were performed as described above. Frozen sections were fixed with ice-cold 100% methanol for 15 mins and washed with 1X PBS. Fixed sections were incubated with 0.3% triton 100 for 10 mins and then blocked with blocking buffer (1% BSA in PBS+0.3% triton X 100) for 1 hr at room temperature. Sections were incubated with rabbit anti-mouse CDKN2A/p16INK4A primary antibody (Abcam ab211542) at 1:100 dilution overnight at 4°C. The following day sections were then washed with 1X PBS and incubated with Alexa Fluor-647 goat anti-rabbit secondary antibody (Invitrogen 21246) at 1:500 dilution for 2 hrs at room temperature. Nuclei were stained with DAPI for 10 mins and images were acquired immediately on a Leica SPE confocal microscope. Five-six different fields were acquired per mouse section and p16INK4A positive cells were counted and expressed as percentage of total lesional cells.
Human plaques
Human coronary artery specimens with atherosclerotic lesions were selected from individuals enrolled in CVPath Institute Registry with atherosclerosis, a history of being diagnosed with cancer or a history of being diagnosed with cancer and treated with radiation therapy. Briefly, the artery segments were fixed in formalin, and 2 to 3 millimeter segments were embedded in paraffin. Cross-sections of 5μm thick were cut from each of the segments and mounted on slides. Slides were stained with CDKN2A/p16INK4A (Abcam ab54210) primary antibody at 1:8000 dilution for immunohistochemical analyses. Plaque classifications were determined according to our previously published criteria (17). Slides were scanned on Axio.Z1 Slide Scanner (Carl Zeiss, Germany), and image panels were prepared on HALO image analysis platform ver 3.0 (Indica Labs, Corrales, NM).
p16-3MR bone marrow transfers into Ldlr−/− mice
p16-3MR mice were from UNITY Biotechnology, Inc. Ldlr−/− mice were lethally γ-irradiated for complete ablation of bone marrow. Radiation was given in two doses, each dose being 4.75 grays with 4 hrs between doses. After the second dose of radiation, we i.v. injected bone marrow cells from the femurs and pelvis of either p16-3MR mice or C57BL/6 (WT) control mice into irradiated Ldlr−/− mice. The mice were given antibiotic water (SMZ and TMP, Aurobindo, cat #NDC 65862-496-47), and a 6 week recovery period. After 6 weeks, mice were fed WD for an additional 10 weeks to allow for the development of atherosclerosis. At the end of 10 weeks, mice were randomly assigned to receive i.p. injections of either Vehicle (PBS) or 5mg/kg/mouse Ganciclovir (GCV, Sigma cat#G2536) for the next 3 weeks while still on WD. Mice were sacrificed at the end of 13 weeks and necrotic core analysis was carried out on H&E-stained lesional cross sections as above.
Identification of Lipid Mediators by Targeted LC-MS/MS
In order to identify and quantify the lipid mediators in atherosclerotic aortas from Vehicle or GCV treated p16-3MR transplanted Ldlr−/− mice, a targeted liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based analysis was performed as described previously (18). Aortas were isolated and immediately placed in ice-cold methanol. Deuterium-labeled synthetic standards d8-5S-HETE, d5-LXA4, d5-RvD2, d4-LTB4 and d4-PGE2 (Cayman Chemical) were then added to each sample and the tissue was minced on ice. Supernatants were collected after centrifugation (13,000 rpm, 10 min, 4 °C) and acidified to pH 3.5. The samples were then subjected to solid phase extraction using Isolute C18 columns (Biotage). Lipid mediators were eluted from the column following addition of methyl formate and were then concentrated under N2 gas and resuspended in methanol:water (50:50). The samples were analyzed by LC-MS/MS using a Poroshell reverse-phase C18 column (100 mm × 4.6 mm × 2.7 μm; Agilent Technologies)-equipped high-performance liquid chromatography (HPLC) system (Shimadzu) coupled to a QTrap 5500 mass spectrometer (AB Sciex) operating in negative ionization mode and using scheduled multiple reaction monitoring (MRM). Lipid mediators were identified in the experimental samples by matching retention times to synthetic standards run in parallel. The abundance of each mediator was determined using standard curves generated with synthetic standards for each individual mediator and by accounting for the recovery of the internal deuterium-labeled standards, followed by normalization to total protein content.
Cytokine array
Elicited peritoneal macrophages were plated in a 6-well plate (2x106 cells/well) and then subjected to the senescence protocol as described above. Supernatants were collected, concentrated 2-fold with 3 KDa ultracentrifuge filters (Amicon) and subjected to a proteome profiler mouse cytokine array according to manufacturer’s instructions (R&D Systems, #ARY006). Immunoblots were analyzed with ImageJ and data were expressed as protein levels relative to control.
Cholesterol assay
WD-diet fed Ldlr−/− mice were sacrificed, and blood was collected in 10% EDTA by retro-orbital bleeding procedure. Immediately after collection, blood was centrifuged at full speed for 30 mins at 4°C and cholesterol assay (Wako, cat# 999-02601) was performed according to manufacturer’s instruction.
Statistical analysis
For all in vivo studies, mice were randomly assigned to their respective groups. Results are represented as mean ± S.E.M. Prism (GraphPad Inc., La Jolla, CA) software was used for statistical analysis and statistical differences were determined using the two-tailed Student’s t-test, One-way ANOVA or Two-way ANOVA with Tukey’s or Sidak’s multiple comparison post-hoc analysis. Details regarding statistical tests can be found in the figure legends.
Results
Sublethal γ-radiation delays temporal inflammation-resolution
To test the impact of radiation on resolution, we first mock or sublethally irradiated (IR) male C57/BL/6 mice with 7 grays of ionizing radiation. The mice were given a 3 month recovery period to induce hematopoietic cell senescence (11, 13). Senescence is mediated by changes in the expression of cell cycle regulators like p16INK4A, p19ARF or p21 as examples. Consistent with the literature, we observed that bone marrow from IR mice had significantly higher expression of the senescence markers p16INK4A (Supplemental Fig. 1A), p19ARF (Supplemental Fig. 1B), and p21 (Supplemental Fig. 1C) compared with controls. Accordingly, we also observed other features of senescence and aging including a significant increase in long-term hematopoietic stem cells (LT-HSCs), a decrease in short-term HSC, a strong trend toward increased MMP2s (i.e. erythroid progenitors) (Supplemental Fig. 1D) and a significant decrease in lymphoid-biased MMP4s (Supplemental Fig. 1E). To directly test the role of sublethal radiation-induced senescence on the resolution response, we used the widely known in vivo model of Zymosan A (ZymA) induced sterile, self-limited inflammation (19). Control or IR mice were injected with 200 μg of ZymA per mouse and peritoneal exudates were collected by lavage 4, 24 and 48 hrs post injection. Leukocytes were enumerated and polymorphonuclear cell (PMN) were assessed by flow cytometry. The peak PMN response (4 hrs) is a measure of inflammation and the time from the peak to when the PMN reach half maximal (e.g. ~24 hrs) is a quantitative measure of resolution called the resolution interval or Ri (19). We found that the Ri for the mock (control) mice was 23 hrs whereas the Ri for the IR mice was 35 hrs (Fig. 1A), which suggests an overall delay in resolution by 12 hrs in the IR mice. We observed that the PMN numbers were significantly higher in the IR group at 24 hrs (Fig. 1B). Since the timely loss of PMN in tissues is associated with efficient resolution, we next quantified the percent retention of PMN from 4 to 24 hrs in the IR versus control mice. The IR mice had significantly higher PMN from 4 to 24 hrs (Fig. 1C), again suggesting an overall delay in tissue resolution in mice that received a sublethal dose of radiation. The modestly reduced influx of neutrophils to the peritoneum at early time points was not due to a loss of bone marrow neutrophils (Supplemental Fig. 1F). Indeed, bone marrow neutrophils in control mice had a significantly higher frequency 24 hrs post ZymA, followed by a significant decline by 48 hrs, whereas bone marrow neutrophil frequency in the IR mice did not change throughout the time course (Supplemental Fig. 1F). Moreover, peritoneal macrophage numbers were significantly higher in the control mice at 24 hrs post ZymA injection compared to IR mice (Supplemental Fig. 1G). To rule out non-specific effects of IR, we performed a bone marrow transfer (BMT) in which mice were given a lethal dose of γ-radiation followed by replenishment of otherwise healthy bone marrow cells from C57BL/6 mice. Lethal doses of γ-radiation also deplete peritoneal macrophages which are replenished by bone marrow precursors cells upon BMT (20). We found no significant differences in the resolution interval between BMT and non-irradiated control mice (Supplemental Fig. 1H). These results suggest that BMT and thus a replenishment of healthy bone marrow did not delay resolution.
Fig. 1. Inflammation-resolution is defective in sublethally irradiated mice.

(A) C57BL/6 mice were subjected to mock or sublethal radiation as described in the methods section. Peritoneal exudates were collected 4, 24 and 48 hrs post ZymA injection (200 μg/mouse). Cells were enumerated and PMN were analyzed by flow cytometry. Results are n = 4 separate cohorts, *p<0.05, **p<0.01, ns - non-significant, Two-way ANOVA with Sidak’s multiple comparison test. (B) PMN number at 24 hrs post ZymA injection were analyzed, **p<0.01, Student’s t-test. (C) The percentage of retained PMN in the peritoneum were calculated, ****p<0.0001, Student’s t-test. (D) Vehicle or 300 ng of RvD1 was i.p. injected simultaneously with ZymA and PMN number was enumerated 24 hrs post injection, *p<0.05, Student’s t-test. (E) Efferocytosis was assessed by calculating the ratio of free to associated PMN at 24 hrs post ZymA injection, ***p<0.001, One-way ANOVA with Tukey’s multiple comparison test. All results are expressed as mean ± S.E.M, and each symbol represents an individual mouse.
We next questioned whether administration of a key pro-resolving ligand, RvD1 to IR mice would improve resolution endpoints. Indeed, intraperitoneal injection of RvD1 (300 ng/mouse) given with ZymA, significantly decreased PMN numbers at 24 hrs post ZymA (Fig. 1D). Mechanistically, the loss of PMN from the peritoneum during the resolution phase is in part through efferocytosis (21). To determine efferocytosis in vivo, we assessed free PMN versus those associated with macrophages using flow cytometry. We observed a significant increase in the free:associated PMN ratio in the IR mice, compared with controls (Fig. 1E), which suggests a defect in efferocytosis. Together, these results provide evidence that sublethal radiation impairs temporal inflammation-resolution and deranges efferocytosis in vivo.
Sublethal γ-radiation promotes macrophage senescence/SASP and Resolvin D1 limits SASP
Certain peritoneal and elicited macrophages have a proliferative capacity and so we next questioned whether sublethal radiation maladaptively halted their proliferation to promote senescence/SASP. For these experiments, we harvested Zymosan-elicited peritoneal macrophages 48 hrs after ZymA injection. These macrophages were plated, then irradiated (5 grays) and cultured in the presence of L-cell conditioned media for an additional 3 days. Both control and irradiated macrophages excluded Trypan blue equally (not shown). Senescent cells are known to acquire a flattened shape in vitro and irradiated macrophages exhibited an enlarged and flattened shape compared with control macrophages (Fig. 2A). Importantly, irradiated macrophages also exhibited several features of cellular senescence including a significant reduction in proliferation as determined by Ki67 staining (Fig. 2B & Supplemental Fig. 2A), and a significant increase in p16INK4A (Fig. 2C) and p21 expression (Fig. 2D). The expression of p19ARF was not increased in irradiated macrophages compared with non-irradiated controls (Fig. 2E). SA-β-gal activity is another known marker of senescence, and we used a quantitative flow cytometric method in which control or irradiated macrophages were incubated for 1 hr with 5-dodecanoylaminofluorescein di-β-D-galactopyranoside (C12-FDG), a fluorescent substrate for β-galactosidase enzyme (14). Using this approach, we observed that the irradiated macrophages also had a significant increase in SA-β-gal activity compared with control macrophages (Fig. 2F). We also observed that irradiated macrophages exhibited other features of senescence and SASP including increased COX-2 levels as determined by intracellular staining by flow cytometry (Fig. 2G). Using metabolic flux analysis, we observed that irradiated macrophages had significantly higher extracellular acidification rates (ECAR) after the addition of glucose, consistent with an increase in aerobic glycolysis capacity (Fig. 2H). A representative ECAR tracing is shown on the left and the quantification after addition of glucose is shown on the right (Fig. 2H). Irradiated macrophages also had enhanced oxidative stress (OS) as determined by CellROX staining (Fig. 2I & Supplemental Fig. 2B) and increased PGE2 levels as determined by ELISA (Fig. 2J). Further, we assessed numerous cytokine/chemokines using a protein array and found sICAM-1, CXCL1, CXCL2 and TNFα were significantly increased in irradiated macrophages compared with control macrophages (Fig. 2K). A representative immunoblot array as well as quantification of other cytokines/chemokines that did not significantly change between irradiated and control macrophages are shown in Supplemental Fig. 2C. Together, these results strongly suggest that irradiated macrophages undergo senescence accompanied with a pro-inflammatory SASP and we will refer to these cells as senescent macrophages (SC-macrophages).
Fig. 2. RvD1 reduces macrophage SASP in vitro.

(A) Representative images of control or SC-macrophages. Cells are outlined by black dashed lines, Scale bar is 50μm. (B) SC-macrophages were treated with Vehicle or 10 nM RvD1 for 24hrs, stained with Ki67 and images were acquired on a Lecia confocal microscope and quantified as percentage of Ki67+ cells. (C-E) mRNA expression of p16INK4A, p21 and p19ARF was measured by qPCR. (F-G) Flow cytometic analysis of (F) C12-FDG staining or (G) COX-2 was performed. (H) Representative ECAR tracing (left) and ECAR quantification after addition of glucose (right) were assessed by a glycolysis stress test using an Agilent Seahorse XFe96 Analyzer. For G and H, macrophages pooled from 4 mice and performed 2 separate times. (I) SC-macrophages were treated with either 10nM RvD1 or 10mM NAC for 24 hrs. CellROX Green fluorescence was detected using Leica confocal microscope. Results are expressed as percentage of total cells. (J-K) Supernatants from control and SC-macrophages were subjected to (J) PGE2 ELISA analysis or (K) a cytokine array. Results are expressed as fold change to control. All results are mean ± S.E.M., analyzed by One-way ANOVA with Tukey’s post-hoc test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns - non-significant. n = 3 independent experiments unless otherwise specified.
To determine whether RvD1 could reduce senescence/SASP in these cells, we stimulated SC-macrophages with RvD1 (10 nM) 2 days post-radiation and cultured them for an additional 24 hrs. Treatment with RvD1 moderately increased the percentage of Ki67+ macrophages (Fig. 2B & Supplemental Fig. 2A) and slightly decreased p16INK4A expression that did not reach significance (Fig. 2C). Also, RvD1 did not modulate the expression of p21 (Fig. 2D) or p19ARF (Fig. 2E), which suggests that RvD1 did not significantly modulate the cell cycle and therefore did not reverse senescence once the macrophages were already committed to this program. We next questioned whether RvD1 modulated the phenotype of the senescent cells. Indeed, RvD1 significantly decreased SA-β-gal activity (Fig. 2F), COX-2 levels (Fig. 2G), aerobic glycolysis (Fig. 2H), OS (Fig. 2I & Supplemental Fig. 2B) and PGE2 (Fig. 2J). Indeed, RvD1 significantly reduced OS as potently as the ROS inhibitor, N-acetyl-L-cysteine (NAC) and the co-treatment of NAC and RvD1 did not further limit OS (Fig. 2I & Supplemental Fig. 2B). Both NAC and RvD1 decreased PGE2 (Fig. 2J), which suggest that OS may play a role in elevated PGE2 levels in SC-macrophages. Moreover, RvD1 significantly decreased levels of sICAM-1, CXCL1, M-CSF, MCP-1, CXCL2 and TNFα in SC-macrophages (Fig. 2K & Supplemental Fig. 2C). Together, these results suggest RvD1 limits the SASP in macrophages.
Senescent macrophages are poor efferocytes and adoptive transfer of senescent macrophages prolongs inflammation in vivo
A key function of macrophages is their ability to clear dead cells, a process called efferocytosis. Previous studies have shown that increased OS and TNFα limits efferocytosis (22), and that intact ICAM-1 on macrophages promotes efferocytosis (23). Because SC-macrophages have elevated levels of intracellular ROS, increased TNFα and elevated sICAM-1, we next questioned whether SC-macrophages have impaired efferocytosis. Indeed, we observed that SC-macrophages have significantly less efferocytosis compared with healthy proliferating controls (Fig. 3A, B). Representative images of efferocytosis are shown in Fig. 3A. Importantly, RvD1 or NAC rescued defective efferocytosis (Fig. 3A, B), which suggests that elevated OS in SC-macrophages limits efficient efferocytosis. These results also suggest that RvD1 rescues efferocytosis through limiting OS in SC-macrophages.
Fig. 3. Senescent macrophages are poor efferocytes.

(A, B) Efferocytosis was performed as explained in the methods and (A) representative images are shown where macrophages are in brightfield and apoptotic cells are red. (B) Efferocytosis was quantified and the total number of macrophages with internalized apoptotic cells were counted as an efferocytic event and results are expressed as percentage of macrophages per visual field. Scale bar is 50 μm, n = 3 independent experiments performed in quadruplet. ***p<0.001, ****p<0.0001, ns-non-significant, One-way ANOVA with Tukey’s post-hoc test. (C) Simultaneous i.p. injection of ZymA with control or senescent macrophages was performed. PMN were collected 4 hrs post injection and enumerated by flow cytometry. **p<0.01, Student’s t-test. All results are expressed as mean ± S.E.M. and each symbol represents an individual mouse.
We next questioned whether SC-macrophages exhibited a pro-inflammatory and anti-resolution phenotype in vivo. For these experiments we adoptively transferred either control or SC-macrophages simultaneously with ZymA. Exudates were collected after 4 hrs and PMN were enumerated. We observed that the transfer of SC-macrophages had significantly higher PMN numbers at 4 hrs post ZymA injection compared with controls (Fig. 3C). These results suggest that SC-macrophages drive prolonged inflammation in vivo.
RvD1 limits SASP in senescent IMR-90 fibroblasts
IMR-90 fibroblasts are a well-accepted model to study senescence in vitro (7). Therefore, we also wanted to determine whether this commonly accepted senescence paradigm exhibited similar results as the SC-macrophages. We subjected IMR-90 cells to 10 grays of radiation and then monitored SASP and senescence markers 10 days after radiation (7). First, we found that similar to irradiated SC-macrophages, senescent IMR-90 fibroblasts also had a significant decrease in efferocytosis compared with controls (Fig. 4A). Consistent with our macrophage results, we observed that senescent IMR-90 cells had significantly higher SA-β-gal activity, which is almost completely abrogated by RvD1 (Fig. 4B). Representative images of SA- β-gal staining (blue color) in IMR-90 cells are shown in Fig. 4B and reveal that senescent cells have increased blue staining compared with controls and that RvD1 significantly limited the blue staining (Fig. 4B). Additionally, RvD1 dramatically reduced PGE2 levels almost to that of control cells (Fig. 4C), and significantly decreased aerobic glycolysis based on ECAR values (Fig. 4D) and ATP resulting from glycolysis (Fig. 4E). Lastly, we observed that that RvD1 moderately reduced the expression of the senescence genes, p16INK4A (Fig. 4F), p19ARF (Fig. 4G) and p21 (Fig. 4H). Together, these results suggest that RvD1 limits key components of the SASP in IMR-90 cells.
Fig. 4. RvD1 reduces senescence markers in IMR-90 senescent fibroblast.
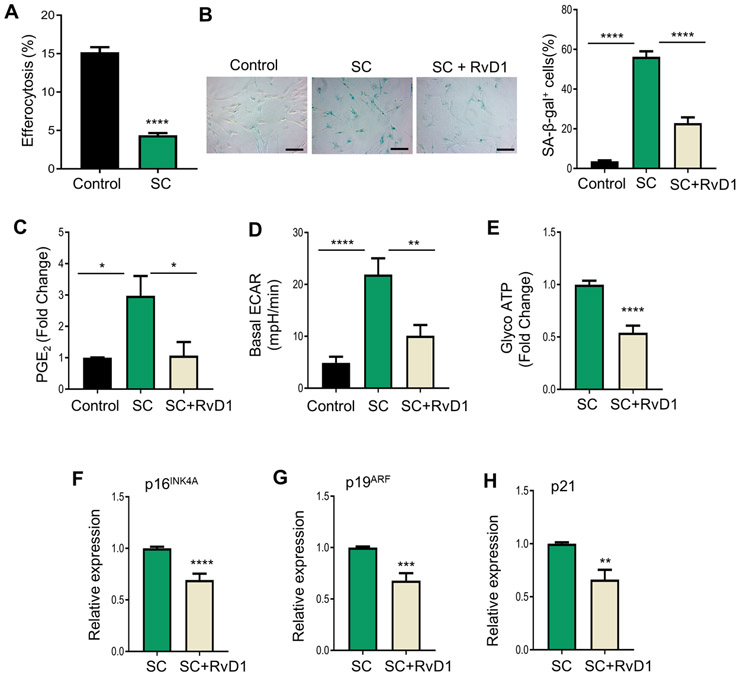
(A) Control or senescent cells (SC) were co-cultured with PKH26-labelled apoptotic Jurkats for 2 hrs in a 1:10 ratio. Apoptotic cells were washed off and images were acquired with a BioRad Zoe Fluorescence Cell Imager. n = 2 independent experiments performed in quadruplet, ****p<0.0001, Student’s t-test. (B) Control, SC or SC+RvD1 (10nM) were assessed for SA- β-gal and images were acquired on a Zeiss microscope and analyzed as percent of positively stained cells as shown on the right. The magnification is 20X, and the scalebar is 20 μm, n = 3 independent experiments, ****p<0.0001, One-way ANOVA with Tukey’s multiple comparison test. (C) PGE2 levels were measured by ELISA, n = 3 independent experiments, *p<0.05, One-way ANOVA with Tukey’s multiple comparison test. (D) ECAR was assessed by an ATP rate test using an Agilent Seahorse XFe96 Analyzer. n = 3 independent experiments, ****p<0.0001, One-Way ANOVA with Tukey’s multiple comparison test. (E) Glyco ATP was calculated from (D). (F-H) p16INK4A (F), p19ARF (G) and p21 (H) mRNA levels were measured by real-time qPCR. **p<0.01, ***p<0.001, ****p<0.0001, Student’s t-test. All results are mean ± S.E.M.
RvD1 limits necrosis and senescent cell accumulation in sublethally irradiated Ldlr−/− atherosclerotic mice
Impaired resolution programs such as defective efferocytosis and limited repair/remodeling are associated with atherosclerosis progression. A consequence of radiation therapy in humans is an increased risk for atherosclerosis (24) and murine models also reveal that radiation exacerbates atherosclerosis (25, 26). In agreement with the literature (25, 26), we also found that sublethal radiation significantly increased the percent of plaque necrosis per lesion area in Ldlr−/− mice, compared with mock controls (Fig. 5A, B). Lesion area, which was quantified using H&E sections as well as Oil Red O staining were not significantly different between the groups (Supplemental Fig. 3A, B). We also found that sublethal radiation significantly decreased the percent of plaque collagen per lesion area in Ldlr−/− mice, compared with mock controls (Fig. 5C, D), which overall suggests that sublethal radiation promotes less “stable” plaques. We next questioned whether treatment with RvD1 could limit plaque necrosis and promote repair in the context of sublethal radiation. For these studies, Ldlr−/− mice were mock or sublethally irradiated (IR) and then immediately placed on a Western Diet (WD) for 12 weeks. After 12 weeks on WD, mice were randomly assigned to receive Vehicle or RvD1 (100 ng/mouse) 3x/week for an additional 3 weeks while still on the WD. Mice were then sacrificed, and aortic root lesions were interrogated for plaque necrosis and collagen content. Treatment with RvD1 significantly decreased the percent of lesional necrosis per total lesion area compared with IR mice (Fig. 5A, B) and significantly increased plaque collagen content (Fig. 5C, D). Representative H&E images of lesional necrosis and fibrous cap thickness are shown in Fig. 5A and 5C respectively. There were no differences in lesion area between Vehicle vs RvD1 treated mice that received sublethal radiation (Supplemental Fig. 3A, B). Body weight (33.75 ± 0.90 grams for IR, 31.45 ± 0.87 grams for IR+RvD1), and plasma cholesterol levels (1009 ± 121.3 mg/dL for IR vs 897.1 ± 119.5 mg/dL for IR+RvD1) were not different between treatment groups.
Fig. 5. RvD1 limits necrosis and p16INK4A positive cells and increases collagen in progressing plaques from sublethally irradiated Ldlr−/− mice.

(A, B) Representative H&E image and quantification of percent necrosis of lesion area in mock, Vehicle (IR) or RvD1 (IR+RvD1) treated sublethally irradiated-Ldlr−/− mice. Magnification is 20X, and the scale bar is 50μm. (C, D) Representative picrosirius red images and quantification of percent picrosirius red area in mock, IR or IR+RvD1 mice. The magnification is 10X and the scale bar is 100μm. (E, F) Representative p16INK4A immunofluorescence images and quantification of p16INK4A+ lesion cells are shown. White stars represent p16INK4A+ cells and p16INK4A is shown in magenta, whereas nuclei are stained in blue with DAPI. Magnification is 40X and the scale bar is 50μm. (G, H) Immunohistochemistry of p16INK4A in human coronary atherosclerotic lesions from non-cancer patients (NC), patients diagnosed with cancer but not treated with radiation (NR) and those with cancer and radiation treatment (R). (G) Whole artery cross-sections are shown on the top, and a higher magnification (within the boxed region) is shown on the bottom. The scale bars are 1mm and 200μm, respectively and display p16INK4A+ cells in brown. (H) Quantification of the percent of p16INK4A cells per total lesional cells is shown from five different arterial beds from 3 separate patients per group. (B, D, F) Each symbol represents an individual mouse. All results are expressed as mean ± S.E.M., analyzed by One-way ANOVA with Tukey’s post-hoc test. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, ns – non-significant.
We next questioned if sublethal radiation increased intraplaque p16INK4A cells. We performed immunofluorescence staining on murine plaques and found that there were significantly more p16INK4A cells in the IR plaques compared with controls (Fig. 5E, F). Moreover, RvD1 treatment reduced the percentage of lesional p16INK4A cells (Fig. 5E, F). Representative immunofluorescence images of lesional p16INK4A cells are shown in Fig. 5E, and clearly depict reduced p16INK4A staining (shown as pink) in mice treated with RvD1. These results together with our in vitro findings in Fig. 2 and 4 suggest that RvD1 may limit initiation of new p16INK4A cells or promote their removal in tissues. Together, these results suggest that RvD1 mitigates sublethal radiation-induced atherosclerosis.
Next, we assessed whether radiation leads to increased p16INK4A cells in human plaques. We obtained human coronary artery specimens with atherosclerotic lesions from individuals enrolled in the CVPath Institute Registry and assessed plaques from patients with atherosclerosis and no history of cancer (NC), plaques from patients who had cancer, but no radiation and (NR), plaques from patients who had cancer and radiation treatment (R). These human plaque sections were stained with p16INK4A and representative lesional p16INK4A images are shown in Fig. 5G. First, we found immunohistochemical analysis revealed that human atherosclerotic plaques exhibited p16INK4A cells (Fig. 5G, H), which is consistent with the literature (27) and suggests that atherosclerosis and aging promotes the accumulation of senescent cells in plaques. Importantly, patients who had received radiation therapy had significantly more p16INK4A cells in the plaques (Fig. 5G, H). Therefore, these results suggest that irradiation further promotes the accumulation of senescent cells in human plaques. Collectively, these results suggest that senescent cells accumulate in advanced plaques and are elevated in the context of sublethal irradiation.
Conditional removal of p16INK4A positive hematopoietic cells limits necrosis and promotes key SPM in advanced atherosclerotic plaques.
To determine the impact of hematopoietic cell senescence and SPM synthesis in plaques, we took advantage of the p16INK4A driven 3MR (trimodality reporter) fusion protein, which contains functional domains of a synthetic Renilla luciferase (LUC), monomeric red fluorescent protein (mRFP), and truncated herpes simplex virus 1 (HSV-1) thymidine kinase (HSV-TK) (Fig. 6A) (28). The HSV-TK allows killing of p16INK4A positive cells by ganciclovir (GCV), a nucleoside analog that has a high affinity for HSV-TK but low affinity for the cellular TK. We performed a bone marrow transfer (BMT) from p16-3MR mice into Ldlr−/− mice and after 6 weeks of recovery, mice were placed on WD for 10 weeks. After 10 weeks, mice were randomly assigned to receive Vehicle or GCV (5mg/kg, 3x/week) i.p. injections for an additional 3 weeks while still on WD (Fig. 6A). Representative H&E images of aortic root cross sections display less necrosis in the GCV-treated plaques as outlined by dashed lines compared with Vehicle controls (Fig. 6B, left panel). We also found that there was a significant decrease in lesion area (Vehicle – 48693 ± 1700 μm2 and GCV - 25838 ± 3700 μm2) as well as necrosis:lesion area (Vehicle – 0.4102 ± 0.02 and GCV - 0.2931 ± 0.04) in GCV-treated mice compared with Vehicle. There was no significant change in body weight (Vehicle: 35.8 ± 0.75 grams and GCV: 34.39 ± 1.01grams), plasma cholesterol (Vehicle: 1187 ± 110.8 mg/dL and GCV: 960.8 ± 77.62 mg/dL) or blood glucose levels (Vehicle: 183.1 ± 9.80 mg/dL and GCV: 169.9 ± 5.81 mg/dL) between the groups. Importantly, we transplanted C57BL/6 marrow in Ldlr−/− mice and carried out experiments as in Fig. 6A. We found that GCV treatment did not alter lesion area (Vehicle - 54152 ± 2229 μm2 and GCV- 55128.16 ± 3467μm2) or necrotic area (Vehicle - 19125 ± 714.2 μm2 GCV- 19134 ± 284.8 μm2). A representative H&E image is shown in Supplemental Fig. 3C. Body weight and plasma cholesterol levels were non-significantly different between the groups (data not shown). These results suggest that the GCV treatment did not exert any actions on plaque from mice that were transplanted with C57BL/6 bone marrow. Together, these results suggest that hematopoietic senescent cells drive atheroprogression.
Fig. 6. Removal of p16INK4A positive cells during advanced atherosclerosis limits necrosis and promotes key SPMs.
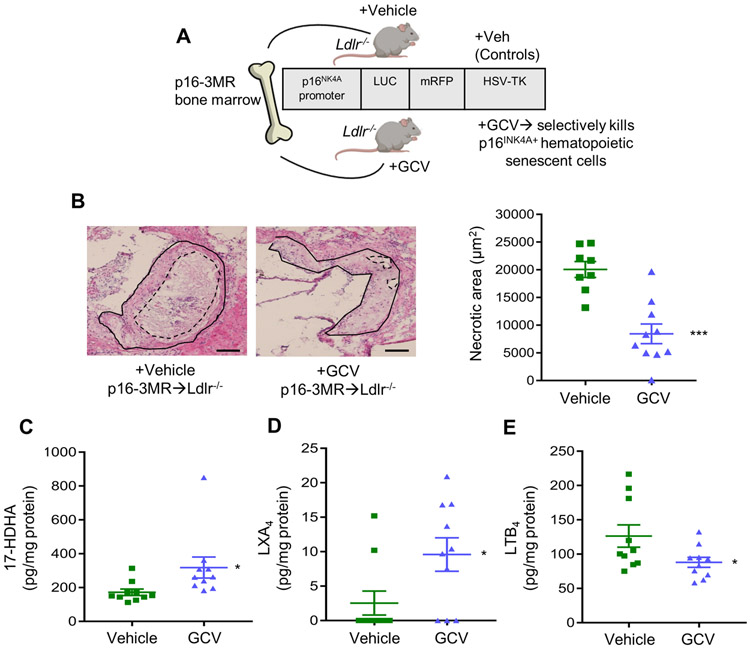
(A) Scheme depicting the p16-3MR bone marrow transfer. (B) Representative H&E stained aortic root images of Vehicle or Ganciclovir (GCV, 5mg/kg, 3x/week, i.p.) treated p16-3MR→Ldlr−/− are shown on the left. Lesions are outlined with black solid lines and necrotic regions are outlined with blacked dashed lines. The magnification is 20X, and the scale bar is 50μm. Quantification of lesional necrosis is shown on the right, ***p<0.001, Student’s t-test. Aortic (C) 17-HDHA, (D) LXA4 or (E) LTB4 were quantified by LC-MS/MS analysis, and lipid mediators are expressed in pg/mg of protein, *p<0.05, Student’s t-test. For all experiments, each symbol represents an individual mouse and data are shown as mean ± S.E.M.
We next investigated SPMs in aortic plaques from Vehicle or GCV treated p16-3MR transplanted Ldlr−/− mice by targeted LC-MS/MS analysis. We found that 17-HDHA, i.e. a biosynthetic pathway biomarker to RvD1 (Fig. 6C) and LXA4 (Fig. 6D), which is an SPM that binds and signals through the same G-protein coupled receptor as RvD1, was significantly increased, whereas the pro-inflammatory lipid mediator, LTB4 (Fig. 6E) was significantly decreased in the GCV-treated plaques compared with Vehicle controls. A complete list of identified lipid mediators are shown in Supplemental Table I. These results suggest that removal of p16INK4A positive cells during advanced atherosclerosis promotes key SPMs. Collectively, these results suggest a maladaptive role of senescent hematopoietic cells on inflammation-resolution responses in atherosclerosis progression.
Discussion
The findings of this study provide a previously unappreciated link between senescence and inflammation-resolution programs which may provide a new framework to approach treatment strategies in contexts where senescent cells accumulate. Our human plaque data suggests atherosclerosis is associated with p16INK4A cells, which is consistent with the literature (27). Moreover, we also found that radiation further increases p16INK4A cells in human plaques. A long-term consequence of mediastinal radiation is coronary artery disease. Radiation-induced vascular injury is thought to be a major factor that drives long-term occlusive atherosclerotic disease, but there are likely other aspects as well (29). Mediastinal radiation can impact the bone marrow, which is not surprising given that sterna and vertebrae are major sites of hematopoiesis in adults (30). Therefore, it is also possible that even focal thoracic radiation may impact the bone marrow to progress atherosclerosis. Nevertheless, more detailed human studies on bone marrow changes, radiation and atherosclerosis need to be investigated.
Additionally, atherosclerotic plaques have long been associated with the presence of senescent cells, namely endothelial and smooth muscle cells (31, 32). Recent data suggests that plaque macrophages may also become senescent (33) and our data herein strongly suggests that senescent hematopoietic cells and macrophages contribute to the progression of atherosclerosis in part but not limited to a defect in SPM synthesis. Along these lines, macrophages are known to proliferate in progressing atherosclerotic plaques, but their proliferation appears to be restricted to a few cycles (34, 35). How senescence is involved in the eventual arrest of proliferation of plaque macrophages remains to be explored, but our results provide evidence that removal of senescent hematopoietic cells during advanced atherosclerosis dampens atheroprogression. These findings, along with suggest bone marrow cells and macrophages are critical effectors of plaque progression (36, 37).
Along these lines, sublethal radiation in mice mimics several features of hematopoietic aging (38). Indeed, a major risk factor for atherosclerosis is age and how aging impacts the development and progression of atherosclerosis is a critically underexplored arena (39). A previous study showed that bone marrow transfer from aged mice into young Ldlr−/− mice produced larger plaques than Ldlr−/− mice who received young marrow (40). These findings suggest that the bone marrow from aged mice promotes atherosclerosis and a deeper understanding as to which cell types and cues within the aged bone marrow drives atheroprogression are of interest.
Previous reports suggested that macrophages reveal features of senescence, but context and function remained underdeveloped (33, 41, 42). Macrophages are highly responsive to their local tissue microenvironment and can exhibit pro-inflammatory, pro-fibrotic or even pro-resolving and pro-regenerative functions. A recent study observed that resident peritoneal macrophages from p16INK4A-activated mice exhibited several features of senescence and had increased uptake of Zymosan particles (42). In the context of progressing atherosclerotic lesions, macrophages also exhibited features of senescence, albeit function was not determined (33). Our work herein, suggests that radiation-induced senescence of macrophages leads to an impairment in efferocytosis which was associated with increased levels of ROS, sICAM-1 and TNFα. Increased ROS can lead to activated of ADAM17 on macrophages (43), which is an enzyme that facilitates the release of activated TNFα and sICAM-1 (44). ADAM17 also promotes the cleavage of MerTK (43), which is a critical efferocytosis receptor on macrophages. We previously found that released factors from senescent cells cleave MerTK to limit efferocytosis on otherwise heathy macrophages (15). Therefore, it is possible that released factors from radiation-induced senescent macrophages may act in an autocrine manner to limit efferocytosis. Nevertheless, context is critical and the manner in which a cell is driven toward senescence may drive its ultimate phenotype/function.
Along these lines, in the context of aging and advanced atherosclerosis, the accumulation of senescent cells and their resulting SASP is maladaptive (45, 46). However, senescence can also be protective since this is a program that can limit cancer, promote cutaneous wound healing and facilitate patterning during embryogenesis (8, 47). Recent reports suggest that senescent cells in arthritic joints can facilitate healing (48). Several questions remain in our understanding as to what drives senescent cells toward a tissue reparative versus tissue destructive phenotype and a deeper understanding of senescent cell markers and animal models are needed.
Lastly, we offer a proof-of-concept that RvD1 limits sublethal radiation-induced accumulation of senescent cells in atherosclerosis. Currently, there are limited options to quell the SASP or remove senescent cells from tissues. Senotherapeutics are emerging as intriguing new strategies to limit senescent cells in humans (49). Senolytics, for example, inactivate pro-survival mechanisms of senescent cells to promote apoptosis and subsequent clearance. Because efferocytosis is impaired in aging and atherosclerosis (33, 50), an increase in apoptotic cells overtime may limit the benefit of senolytics as these apoptotic cells can undergo secondary necrosis. The work presented herein provides an entirely new strategy to limit the most deleterious aspect of senescent cells, i.e., the SASP and suggests that RvD1 may act as a novel senotherapeutic in the context of age-related pathologies. SPMs in general are not immunosuppressive and act to promote tissue repair and regeneration (1) and may be a promising strategy to limit senescent cells in advanced atherosclerosis.
Supplementary Material
Key Points:
Senescent macrophages are poor efferocytes and RvD1 rescues this function.
RvD1 limits necrosis and senescent cells in plaques from irradiated Ldlr−/− mice.
Hematopoietic-derived senescent cells promote plaque necrosis and decrease SPM.
Acknowledgements
General: The authors thank Justin Heinz and Nicholas Rymut for their technical expertise.
Source of Funding
This work was supported by NIH grants HL141127 (G.F.), HL153019 (G.F.), HL142807 (D.J.), and R35GM131842 (K.J.M). HL106173 (M.S.), and GM095467 (M.S.). L.G. and A.V.F. are supported by Leducq Foundation Transatlantic Networks of Excellence Grant (18CVD02) to PlaqOmics Research Network.
Footnotes
Conflict of interests: The authors have declared that no conflicts of interest exist.
References
- 1.Serhan CN, and Levy BD. 2018. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest 128: 2657–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stewart FA, Hoving S, and Russell NS. 2010. Vascular damage as an underlying mechanism of cardiac and cerebral toxicity in irradiated cancer patients. Radiat Res 174: 865–869. [DOI] [PubMed] [Google Scholar]
- 3.Mertens AC, Liu Q, Neglia JP, Wasilewski K, Leisenring W, Armstrong GT, Robison LL, and Yasui Y. 2008. Cause-specific late mortality among 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst 100: 1368–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darby SC, Ewertz M, McGale P, Bennet AM, Blom-Goldman U, Bronnum D, Correa C, Cutter D, Gagliardi G, Gigante B, Jensen MB, Nisbet A, Peto R, Rahimi K, Taylor C, and Hall P. 2013. Risk of ischemic heart disease in women after radiotherapy for breast cancer. N Engl J Med 368: 987–998. [DOI] [PubMed] [Google Scholar]
- 5.Baselet B, Rombouts C, Benotmane AM, Baatout S, and Aerts A. 2016. Cardiovascular diseases related to ionizing radiation: The risk of low-dose exposure (Review). Int J Mol Med 38: 1623–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meziani L, Deutsch E, and Mondini M. 2018. Macrophages in radiation injury: a new therapeutic target. Oncoimmunology 7: e1494488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodier F, Coppé J-P, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, and Campisi J. 2009. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nature cell biology 11: 973–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiley CD, and Campisi J. 2016. From ancient pathways to aging cells—connecting metabolism and cellular senescence. Cell metabolism 23: 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swirski FK, and Nahrendorf M. 2016. Bone Marrow Takes Center Stage in Cardiovascular Disease. Circ Res 119: 701–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moore KJ, and Tabas I. 2011. Macrophages in the pathogenesis of atherosclerosis. Cell 145: 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shao L, Feng W, Li H, Gardner D, Luo Y, Wang Y, Liu L, Meng A, Sharpless NE, and Zhou D. 2014. Total body irradiation causes long-term mouse BM injury via induction of HSC premature senescence in an Ink4a- and Arf-independent manner. Blood 123: 3105–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teresa Pinto A, Laranjeiro Pinto M, Patricia Cardoso A, Monteiro C, Teixeira Pinto M, Filipe Maia A, Castro P, Figueira R, Monteiro A, Marques M, Mareel M, Dos Santos SG, Seruca R, Adolfo Barbosa M, Rocha S, and Jose Oliveira M. 2016. Ionizing radiation modulates human macrophages towards a pro-inflammatory phenotype preserving their pro-invasive and pro-angiogenic capacities. Sci Rep 6: 18765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Schulte BA, LaRue AC, Ogawa M, and Zhou D. 2006. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood 107: 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Debacq-Chainiaux F, Erusalimsky JD, Campisi J, and Toussaint O. 2009. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nature protocols 4: 1798. [DOI] [PubMed] [Google Scholar]
- 15.Rymut N HJ, Sadhu S, Hosseini Z, Riley CO, Marinello M, Maloney J, MacNamara KC, Spite M, Fredman G. 2019. Resolvin D1 promotes efferocytosis in aging by limiting senescent cell-induced MerTK cleavage. FASEB J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, and Tabas I. 2016. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun 7: 12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Virmani R, Kolodgie FD, Burke AP, Farb A, and Schwartz SM. 2000. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol 20: 1262–1275. [DOI] [PubMed] [Google Scholar]
- 18.Dalli J, Colas RA, Walker ME, and Serhan CN. 2018. Lipid Mediator Metabolomics Via LC-MS/MS Profiling and Analysis. Methods Mol Biol 1730: 59–72. [DOI] [PubMed] [Google Scholar]
- 19.Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, Hong S, and Serhan CN. 2005. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol 174: 4345–4355. [DOI] [PubMed] [Google Scholar]
- 20.Cain DW, O'Koren EG, Kan MJ, Womble M, Sempowski GD, Hopper K, Gunn MD, and Kelsoe G. 2013. Identification of a tissue-specific, C/EBPbeta-dependent pathway of differentiation for murine peritoneal macrophages. J Immunol 191: 4665–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwab JM, Chiang N, Arita M, and Serhan CN. 2007. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 447: 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McPhillips K, Janssen WJ, Ghosh M, Byrne A, Gardai S, Remigio L, Bratton DL, Kang JL, and Henson P. 2007. TNF-alpha inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase A2 and oxidant-dependent mechanisms. J Immunol 178: 8117–8126. [DOI] [PubMed] [Google Scholar]
- 23.Wiesolek HL, Bui TM, Lee JJ, Dalal P, Finkielsztein A, Batra A, Thorp EB, and Sumagin R. 2020. Intercellular Adhesion Molecule 1 Functions as an Efferocytosis Receptor in Inflammatory Macrophages. Am J Pathol 190: 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Min SS, and Wierzbicki AS. 2017. Radiotherapy, chemotherapy and atherosclerosis. Curr Opin Cardiol 32: 441–447. [DOI] [PubMed] [Google Scholar]
- 25.Stewart FA, Heeneman S, Te Poele J, Kruse J, Russell NS, Gijbels M, and Daemen M. 2006. Ionizing radiation accelerates the development of atherosclerotic lesions in ApoE−/− mice and predisposes to an inflammatory plaque phenotype prone to hemorrhage. Am J Pathol 168: 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoving S, Heeneman S, Gijbels MJ, te Poele JA, Russell NS, Daemen MJ, and Stewart FA. 2008. Single-dose and fractionated irradiation promote initiation and progression of atherosclerosis and induce an inflammatory plaque phenotype in ApoE−/− mice. International Journal of Radiation Oncology* Biology* Physics 71: 848–857. [DOI] [PubMed] [Google Scholar]
- 27.Holdt LM, Sass K, Gabel G, Bergert H, Thiery J, and Teupser D. 2011. Expression of Chr9p21 genes CDKN2B (p15(INK4b)), CDKN2A (p16(INK4a), p14(ARF)) and MTAP in human atherosclerotic plaque. Atherosclerosis 214: 264–270. [DOI] [PubMed] [Google Scholar]
- 28.Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge R-M, Vijg J, Van Steeg H, and Dollé ME. 2014. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Developmental cell 31: 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weintraub NL, Jones WK, and Manka D. 2010. Understanding radiation-induced vascular disease. J Am Coll Cardiol 55: 1237–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geerman S, Hickson S, Brasser G, Pascutti MF, and Nolte MA. 2015. Quantitative and Qualitative Analysis of Bone Marrow CD8(+) T Cells from Different Bones Uncovers a Major Contribution of the Bone Marrow in the Vertebrae. Front Immunol 6: 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, and Bennett M. 2006. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res 99: 156–164. [DOI] [PubMed] [Google Scholar]
- 32.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, and Komuro I. 2002. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation 105: 1541–1544. [DOI] [PubMed] [Google Scholar]
- 33.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, and van Deursen JM. 2016. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354: 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, Zavitz CC, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R, and Swirski FK. 2013. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med 19: 1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams JW, Zaitsev K, Kim KW, Ivanov S, Saunders BT, Schrank PR, Kim K, Elvington A, Kim SH, Tucker CG, Wohltmann M, Fife BT, Epelman S, Artyomov MN, Lavine KJ, Zinselmeyer BH, Choi JH, and Randolph GJ. 2020. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat Immunol 21: 1194–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, and Tall AR. 2010. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science 328: 1689–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poller WC, Nahrendorf M, and Swirski FK. 2020. Hematopoiesis and Cardiovascular Disease. Circ Res 126: 1061–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young K, Borikar S, Bell R, Kuffler L, Philip V, and Trowbridge JJ. 2016. Progressive alterations in multipotent hematopoietic progenitors underlie lymphoid cell loss in aging. J Exp Med 213: 2259–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tyrrell DJ, and Goldstein DR. 2021. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol 18: 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du W, Wong C, Song Y, Shen H, Mori D, Rotllan N, Price N, Dobrian AD, Meng H, Kleinstein SH, Fernandez-Hernando C, and Goldstein DR. 2016. Age-associated vascular inflammation promotes monocytosis during atherogenesis. Aging Cell 15: 766–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, Leonova K, Polinsky A, Chernova OB, and Gudkov AV. 2016. Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 8: 1294–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu JY, Souroullas GP, Diekman BO, Krishnamurthy J, Hall BM, Sorrentino JA, Parker JS, Sessions GA, Gudkov AV, and Sharpless NE. 2019. Cells exhibiting strong p16 (INK4a) promoter activation in vivo display features of senescence. Proc Natl Acad Sci U S A 116: 2603–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, and Tabas I. 2011. Shedding of the MER tyrosine kinase receptor is mediated by ADAM17 through a pathway involving reactive oxygen species, protein kinase δ, and P38 map kinase. Journal of Biological Chemistry: jbc. M111. 263020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheller J, Chalaris A, Garbers C, and Rose-John S. 2011. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends in immunology 32: 380–387. [DOI] [PubMed] [Google Scholar]
- 45.Campisi J, and Robert L. 2014. Cell senescence: role in aging and age-related diseases. Interdiscip Top Gerontol 39: 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pignolo RJ, Passos JF, Khosla S, Tchkonia T, and Kirkland JL. 2020. Reducing Senescent Cell Burden in Aging and Disease. Trends Mol Med 26: 630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, Hoeijmakers JH, de Bruin A, Hara E, and Campisi J. 2014. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31: 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Montero-Melendez T, Nagano A, Chelala C, Filer A, Buckley CD, and Perretti M. 2020. Therapeutic senescence via GPCR activation in synovial fibroblasts facilitates resolution of arthritis. Nat Commun 11: 745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kirkland JL, Tchkonia T, Zhu Y, Niedernhofer LJ, and Robbins PD. 2017. The Clinical Potential of Senolytic Drugs. J Am Geriatr Soc 65: 2297–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabas I 2010. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 10: 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.