

Abstract
Myositis comprises a heterogeneous group of skeletal muscle disorders which converge on chronic muscle inflammation and weakness. Our understanding of myositis pathogenesis is limited, and many myositis patients lack effective therapies. Using muscle biopsy transcriptome profiles from 119 myositis patients (spanning major clinical and serological disease subtypes) and 20 normal controls, we generated a co-expression network of 8101 dynamically regulated transcripts. This network organized the myositis transcriptome into a map of gene expression modules representing interrelated biological processes and disease signatures. Universally myositis-upregulated network modules included muscle regeneration, specific cytokine signatures, the acute phase response, and neutrophil degranulation. Universally myositis-suppressed pathways included a specific subset of myofilaments, the mitochondrial envelope, and nuclear isoforms of the anti-apoptotic humanin protein. Myositis subtype-specific modules included type 1 interferon signaling and titin (dermatomyositis), RNA processing (antisynthetase syndrome), and vasculogenesis (inclusion body myositis). Importantly, therapies exist to target influential proteins in many myositis-dysregulated modules, and nearly all modules contained understudied proteins and non-coding RNAs – many of which were extraordinarily dysregulated in myositis and may represent novel therapeutic targets. Finally, we apply our network to patient classification, finding that a deep learning algorithm trained on patient-level network “images” successfully assigned patients to clinical groups and further into molecular subclusters. Altogether, we provide a global resource to probe and contextualize differential gene expression in myositis.
Keywords: Myositis, myopathy, transcriptome, co-expression, network, deep learning
INTRODUCTION
Myositis encompasses a heterogeneous family of autoimmune disorders characterized by chronic muscle weakness and inflammation. These disorders include subsets which share certain clinical or histopathological features, such as dermatomyositis (DM), sporadic inclusion body myositis (IBM), the antisynthetase syndrome (ASS), and immune-mediated necrotizing myopathy (IMNM). Moreover, most myositis patients have a single myositis-specific autoantibody (MSA) which further predicts aspects of disease course. For example, within DM, anti-Mi2+ patients tend to have severe muscle disease [27], while anti-MDA5+ patients tend to be clinically amyopathic [13].
The phenotypic diversity in myositis raises a fundamental question: to what extent do these disorders converge on muscle weakness through alterations of a shared set of molecular pathways? Previous studies in major clinical subsets of myositis have identified upregulation of inflammatory response genes and suppression of muscle-specific transcripts. However, studies comparing pathways across all major myositis subsets are rare, and it is challenging to assess modestly affected pathways in studies with relatively few patients per group. More broadly, classical differential expression and pathway analyses, which rely upon curated gene sets, may not detect highly coordinated and disease-relevant expression changes which reflect yet-unknown processes or regulatory programs.
To systematically probe the global signaling landscape in myositis muscle, we used RNA-sequencing profiles of 119 myositis and 20 control muscle biopsies to create a co-expression network comprising biological processes and disease signatures. We define dysregulated modules in clinical, MSA, and deep learning-derived myositis subsets and use network topology to evaluate putative hubs and novel components of myositis-altered pathways.
METHODS
Clinical samples and data
Written informed consent was provided by participants enrolled in institutional review board (IRB)-approved cohorts (National Institutes of Health IRB#91-AR-0196, Johns Hopkins Myositis Center IRB#NA_00007454, Clinic Hospital Barcelona IRB#HCB/2015/0479, Vall d’Hebron Hospital Barcelona IRB#PR(AG)68/2008). Detailed demographic, treatment, histopathology, and clinical data for the included patients are detailed in Online Resource Supplementary Table 1. Patients met criteria for IBM [19] or had one of the following MSAs: NXP2, Mi2, TIF1γ, MDA5, HMGCR, SRP or Jo1. DM and ASS patients had classic clinical features and no patients were included who met criteria for other related syndromes (e.g., lupus or systemic sclerosis). Autoantibody testing was performed as described for HMGCR [22] or by line blot (EUROLINE Myositis Profile 4). Normal tissue (NT) biopsies were from the Johns Hopkins Neuromuscular Pathology Laboratory (n=10) and University of Kentucky Skeletal Muscle Biobank (n=10). Strength, measured at hip flexors and deltoids, was converted to a 10-point scale [16] and serum creatine kinase was log2-transformed.
Muscle regeneration models
Primary human skeletal muscle myoblasts (Lonza) were cultured per manufacturer’s protocol. At 80% confluence, differentiation was induced by changing media to DMEM with 2% horse serum and L-glutamine. Timepoints had biological duplicate. Mouse muscle injury via cardiotoxin was achieved as described [21]. Injured TA muscles were harvested at days 3 (n=2), 5 (n=2), 7 (n=2), 10 (n=4), 14 (n=4), and 28 (n=3) post-injury. Contralateral (uninjured) TA muscles were also collected (n=9).
Co-expression network creation, clustering, and topology analysis
RNA sequencing was performed as previously described [2]. Networks were visualized in Cytoscape [32]. Transcripts were considered for the network if they were expressed at least >1 FPKM in at least 5% of samples. This threshold was chosen to reduce noise from genes expressed at very low absolute levels or genes expressed in few samples, which would be expected to have outlier effects on fold-change-based visualizations and Pearson Correlation Coefficients (PCC), respectively [38]. We then created an unweighted network using a soft threshold (absolute magnitude of PCC > 0.6) which included approximately the top percentile of all correlations. We chose the unweighted approach because many genes correlated with their known co-functional genes at PCC values near 0.6 or 0.7, but also correlated with inflammatory markers (e.g. leukocyte-specific transcripts) at similar or higher values. This reflects the greater variance in inflammatory gene expression, where expression varies on several orders of magnitude from high-inflammation samples to low-inflammation samples, as opposed to universally expressed genes which often have much more muted changes, even if dynamically regulated. Because these correlations are still likely meaningful, we did not choose a strictly rank-based network but rather did not use correlation magnitude as an edge weight when determining network position. Our network was clustered using the Markov algorithm using the default settings. GSEA [36] of modules was performed using the Molecular Signature Database [18] as described [1]. Finally, the network was globally organized using the Compound Spring Embedder layout with manual repositioning to improve visibility where needed. Degree centrality for a given gene was defined as the number of direct connections between that gene and other genes in the same Markov cluster. Relative degree centrality was then the degree centrality of an individual gene divided by the average degree centrality of other members of that cluster. We note that no substantial differences existed when using an alternative measure of centrality, betweenness centrality.
Deep learning and hierarchical clustering
Each patient’s gene expression profile was calculated as a log2 fold-change relative to the median normal control biopsy, imposed onto the network structure in Cytoscape [32], and saved as an image. The convolutional neural network was implemented using the modules Tensorflow and Keras in Python using common settings. Briefly, we created a sequential model with three convolutional layers using the rectified linear unit (ReLU) activation function. A dropout of 50% of neurons was used to prevent overfitting. Image data augmentation via the ImageDataGenerator function was used during training to alter original networks (width, height, zoom, and brightness) and increase the diversity of images on which features could be trained. Because the desired endpoint of the model was its features rather than maximum external generalizability, 90% of images stratified by clinical subtype were used to train the model (i.e. create and refine features) while 10% of samples served as the validation set used to tune hyperparameters. Minibatch size was 8. Categorical cross-entropy (five categories: NT, DM, ASS, IBM, and IMNM) was the loss function and rmsprop was the optimizer. Training was stopped when accuracy on the validation set stabilized for 50 epochs. Finally, the output (classification) layer was removed so that the outcome of the model was a 64-feature vector. Each original image was then passed through the network to get a 139 sample × 64 feature matrix before hierarchical clustering and visualization using the cluster.hierarchy.dendrogram function in scipy. We emphasize that this method is not intended to define ground truth patient subgroups in myositis, but simply to group patients which have the greatest overall transcriptional network similarity. This process was repeated several times with reshuffled training and validation sets to ensure that the features were robust to which samples were selected for training. Source code for our model and the weights determined through our training and validation process are available at github.com/mendillolab.
Exploring the myositis network interactively
The myositis network is made available as a network session which can be altered in the free and intuitive program Cytoscape [32]. Notably, all individual patient-level data is removed from this session for anonymity. The following are brief instructions on common use cases. The position, connections (edges), relative expression, and cluster memberships of each gene (node) can be queried using the interactive search function (upper right) and the filtering tab (far left). Group-level expression aggregates can be toggled using the style tab. To overlay a custom expression profile, add a standard (e.g., comma-separated value) file containing the gene identifiers as one column and the gene expression (preferably normalized to a control or standardized, and centered at 0) via the “import table from file” function. Then, in the style tab, change the node fill color column from “Myositis” to your column. A simple screenshot taken of this network is suitable for analysis by our neural network model, which can provide an estimated diagnosis and show where the biopsy clusters amongst our cohort of patients. Code and further instructions on how to perform custom analyses are available at github.com/mendillolab.
RESULTS
An atlas of coordinated gene expression programs in healthy and diseased skeletal muscle
To generate a map of the myositis transcriptome, we built a co-expression network [38]. RNA-sequencing was performed on muscle biopsy samples from 20 healthy controls (normal tissue; NT) and 119 patients with myositis, 39 with DM (11 anti-Mi2+, 12 anti-NXP2+, 11 anti-TIF1γ+, and 5 anti-MDA5+), 49 with IMNM (9 anti-SRP+ and 40 anti-HMGCR+), 18 with anti-Jo1+ ASS, and 13 with IBM (Fig. 1a, Online Resource Supplementary Table 1). The expression level of each expressed gene was then correlated with every other gene across the 139 profiles. Of 10,203 eligible genes, 8101 had at least one strong correlation (Pearson Correlation Coefficient >0.6 or <−0.6). These genes were then included in our network as nodes and were connected by all correlations (edges) meeting this threshold (1.58 million edges, 1.5% of possible correlations).
Figure 1: A map of gene expression programs in healthy and diseased skeletal muscle biopsies.
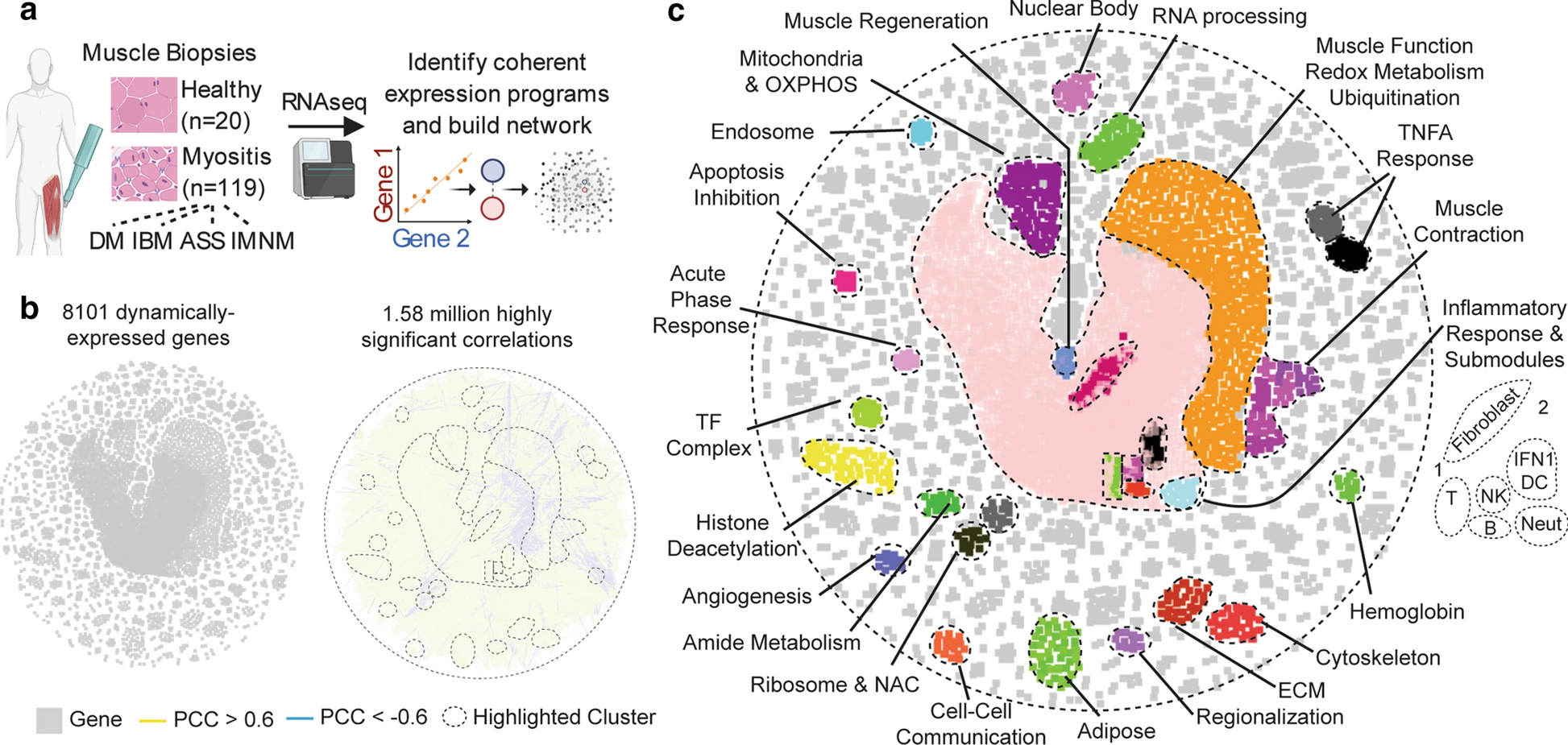
(a) Schematic of approach. (b) A global similarity network of dynamically expressed transcripts in normal or myositis patient biopsies. All Pearson correlation coefficients (PCC) with an absolute magnitude greater than 0.6 are included as unweighted edges. (c) Network modules revealed by Markov clustering, Gene Set Enrichment Analysis, and the imposition of cell-type-specific transcripts.
The resulting network revealed a map of modular gene expression programs dynamically regulated in healthy and diseased muscle (Fig. 1b). Supporting the validity of this approach, many network modules represented specific biological processes (Fig. 1c). The largest module, inflammation, also contained sub-clusters enriched for genes expressed specifically in non-skeletal muscle cell types. For example, genes highly expressed in T cells tended to colocalize, as did B cell, NK cell, neutrophil, and fibroblast genes (Fig. 1c). Notably, while most network connections were positive (95.7%), many anticorrelations localized to the interface between inflammatory markers and muscle function genes (Fig. 1b–c). Thus, our network successfully distilled the myositis transcriptome into a map of coherent gene expression programs.
Core gene expression program alterations in myositis
To identify core gene expression program alterations which span myositis subtypes, we imposed the relative gene expression profile of myositis patients – as compared with healthy controls – onto our network (Fig. 2a). Consistent with prior data, the central inflammation module was robustly overexpressed in myositis, as was a module representing TNFα and NFKB signaling (for highlighted cluster examples, see Fig. 2b). Another pan-myositis induced module comprised genes involved in muscle regeneration. This module contained MYOG and MYOD1, critical myogenic factors upregulated in damaged and regenerating muscle, including in myositis [24]. Beyond canonical factors, the myogenesis module included the long non-coding RNA (lncRNA) LINCMD1, which controls differentiation timing [9]; leiomyodin (LMOD1), a smooth muscle regulator [12] and the only cluster anticorrelate; and the understudied C11orf1 and DNAJB5-AS1.
Figure 2: Core gene expression alterations in myositis patients.
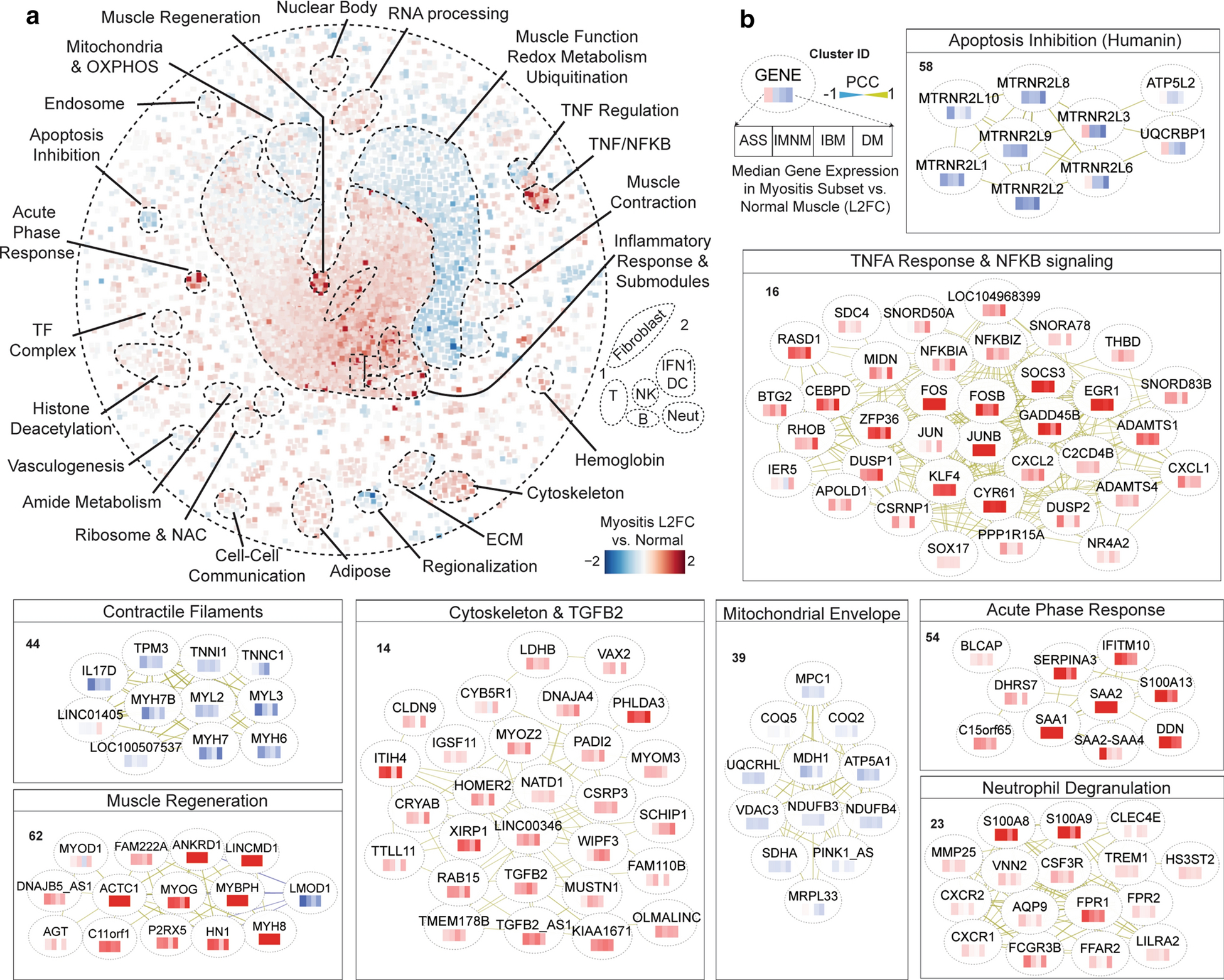
(a) Relative expression of network genes in the median myositis patient biopsy as compared with the median normal control patient. Fold changes are as indicated, i.e. red is overexpressed. (b) Example clusters altered similarly across clinical subsets of myositis. Nodes contain a heatmap showing the expression of genes in each major myositis subset; see key. Networks follow a force-directed layout with minimal rearrangements for legibility. For more information on individual clusters (numbered), see Online Resource Supplementary Table 2.
Two other highly upregulated modules represented inflammatory processes less explored in myositis. The first module contained the stress-responsive serum acute-phase response proteins SAA1, SAA2, and α1-Antichymotrypsin (SERPINA3). This module may have a multifaceted role in myositis pathogenesis, as muscle SAA1 induction augments atrophy signaling [39] whereas SERPINA3 induction protects myofibers from proteases to attenuate myodegeneration [37]. The second module represented neutrophil degranulation, a process upregulated in many inflammatory disorders [17] including in certain types of myositis, where a subset of neutrophils (low-density granulocytes; LDGs) are thought to access affected muscles and deposit pathogenic neutrophil extracellular traps (NETs) [31]. While the precise role for neutrophils/LDGs in myositis muscle still requires further elucidation, our data suggest that neutrophil dysregulation is conserved across myositis, including in novel subtypes (IBM and IMNM). Of note, this module contains several neutrophil degranulation-associated genes which are strikingly altered in myositis patients – such as the NET potentiator TREM1 and chemoattract receptor FRP1 – which can be targeted by existing clinical or preclinical therapies [7, 30].
Beyond inflammation, several other clusters were dysregulated across myositis. The large muscle function, redox metabolism, and ubiquitination module was universally suppressed. Intriguingly, however, not all muscle contraction modules were altered. Rather, one contractile filament module, containing specific myosin, tropomyosin, and troponin isoforms, was particularly suppressed. These filamentous changes, which strongly correlated with clinical muscle weakness (r=0.35, P=0.0003; Online Resource Supplementary Table 2), likely represent a shared upstream mechanism of weakness in myositis. The myofilament cluster also provides intriguing novel correlates of weakness. For example, IL17D, a muscle-secreted cytokine which decreases proliferation of myeloid precursors [34], was underexpressed in a manner analogous to these contractile filaments. Additionally, two lncRNAs clustered with the underexpressed filaments, including LOC100507537, the recently discovered DWORF peptide which augments sarcoplasmic reticulum calcium reuptake [23], a critical step in muscle contraction which is altered in myositis [2].
Several other commonly altered clusters mapped to processes with relevance to myositis pathogenesis. A cytoskeleton remodeling module, including the fibrosis master regulator Transforming Growth Factor Beta-2 (TGFB2), was robustly upregulated. Notably, a neutralizing monoclonal antibody specific for TGFB2 (versus other family members) attenuates fibrosis in a mouse model of diabetic nephropathy, where TGFB2 is also preferentially induced [14]. Elsewhere, a mitochondrial subcluster reflecting the mitochondrial envelope was suppressed across myositis groups, which may relate to the muscle endurance deficits common in myositis patients [3]. A cluster of nuclear isoforms of the humanin protein, which inhibit apoptosis and broadly promote stress resilience [6], was also consistently suppressed. Finally, several dysregulated clusters did not map to a known gene ontology (see Online Resource Supplementary Table 2) – such gene sets may be relevant to yet unknown processes or regulatory programs active in myositis. Altogether, we identify novel molecular pathways and pathway components convergently altered across major subtypes of myositis.
Myositis subtype-specific gene expression programs and molecular heterogeneity
While myositis patients converge on muscle dysfunction, myositis subtypes differ at the level of muscle biopsy histology, extra-muscular manifestations, treatment response, and overall phenotypic severity. To investigate divergent gene expression programs which may represent unique upstream pathogenic features in myositis subtypes, we imposed the relative gene expression profile of each clinical myositis subset – as compared with all other myositis patients – onto our global network (Fig. 3a).
Figure 3: Unique transcriptional programs in clinical myositis subtypes.
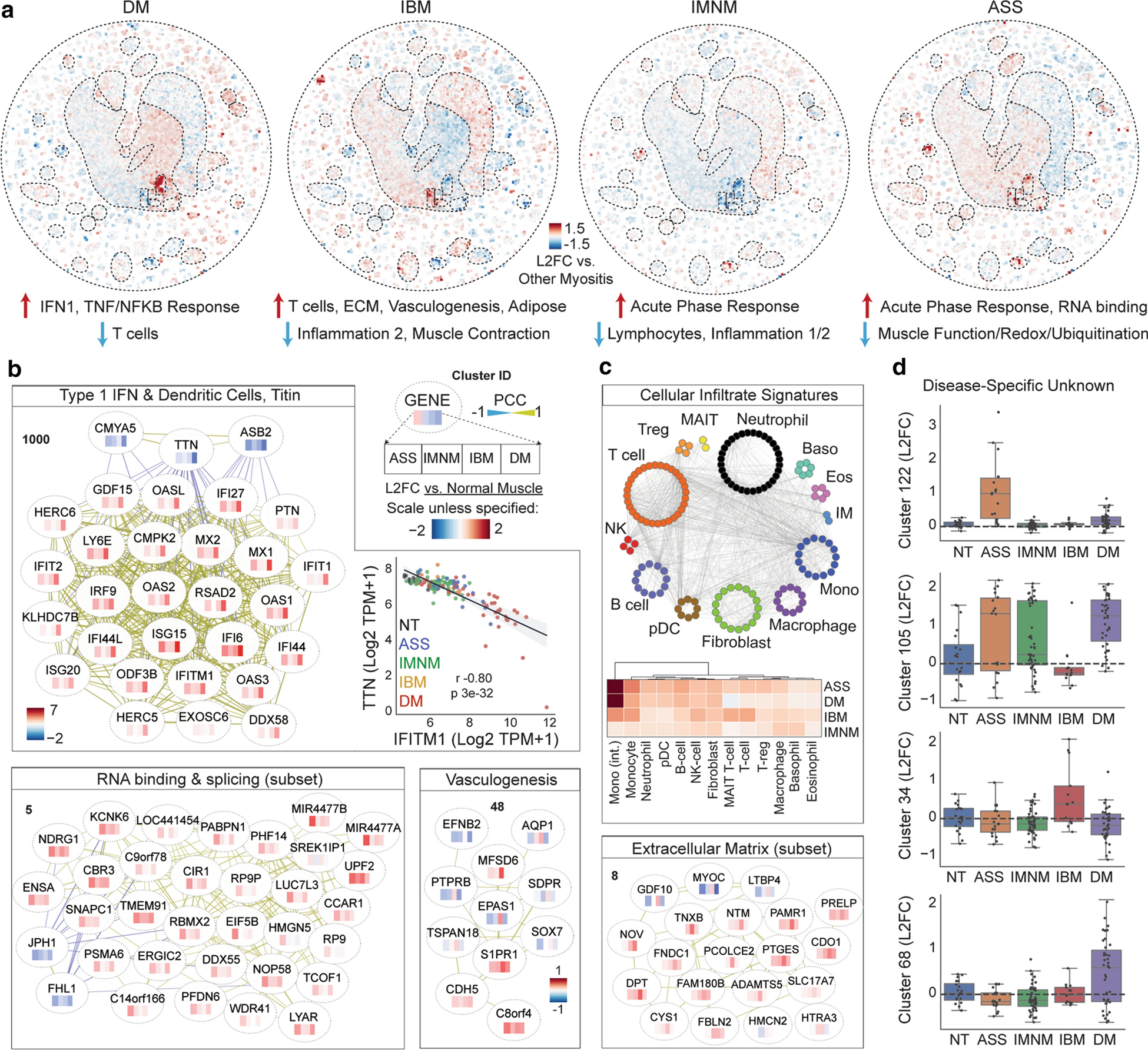
(a) Relative gene expression in each clinical subset of myositis as compared with all other myositis patients. Example clusters with differentially expressed genes are highlighted. (b) Clusters with expression changes specific to or enriched in one clinical subset of myositis patients. Heatmap inside node shows expression relative to normal controls; see key. Ancillary analyses corresponding to individual clusters are located within that cluster’s box; e.g. the top anticorrelation of TTN with IFITM1 is depicted in the IFN1 section.
The most striking difference between clinical subtypes is, perhaps unsurprisingly, the first described subtype-specific transcriptomic alteration in myositis: activation of type 1 interferon (IFN1)-inducible genes in DM [10, 11, 26] (Fig. 3b). A key IFN1 module gene is ISG15, a ubiquitin-like modifier conjugated to proteins in DM – particularly in regions of small, abnormal myofibers near the myofiber bundle periphery (perifascicular atrophy; PFA). This has led to hypotheses that aberrant ISG15 conjugation damages proteins necessary for muscle function, such as Titin (TTN), a key structural protein which is nearly absent at the protein level in micro-dissected PFA fibers [29]. Intriguingly, however, our network identified TTN as the strongest transcriptional anticorrelate with IFN1 genes (r=−0.8, p=3e-32; Fig. 3b). The marked magnitude of TTN transcript loss – approaching no detectable transcript in the most IFN1-high biopsies – strongly suggests that TTN loss in PFA regions is likely transcriptional rather than post-translational.
Another difference between myositis subtypes was found at leukocyte-specific clusters. Consistent with histopathological observations, IBM patients had overexpression of T cell genes and a plasma cell marker (JCHAIN), ASS and DM patients had more modest differences and slightly higher expression of intermediate monocyte genes, and IMNM patients had minimal evidence of lymphocytic inflammation (Fig. 3c). Other expression programs which distinguished myositis subtypes included upregulation of RNA binding and translation genes specifically in ASS patients (eg, EIF5B; Fig. 3b). This connection to translation is intriguing, considering that autoantibodies to aminoacyl-tRNA synthetase proteins are the defining feature of ASS. Like the TTN-IFN1 anticorrelation, two essential muscle proteins mutated in congenital myopathies (JPH1 and FHL1) anticorrelated with this cluster, suggesting a potential link between this gene signature and pathogenesis. Elsewhere, the vasculogenesis cluster centered by Hypoxia-Inducible Factor 2α (EPAS1) was suppressed in most myositis subsets but overexpressed in IBM. Similarly, IBM specifically featured upregulation of certain extracellular matrix (ECM) genes including TNXB, PCOLCE2, and ADAMTS5. These gene sets may relate to “degenerative” features classically ascribed to IBM versus other myositis classes [5]. As with conserved clusters, many subtype-specific clusters emerged which have no known biological function (Fig. 3c).
Next, we investigated whether MSA-defined subsets of clinical groups in myositis would have divergent gene expression programs (Fig. 4a–b). Indeed, several clusters discriminated clinical groups by their MSA status. For example, cytoskeleton genes were induced in all forms of DM except anti-MDA5 DM (patient-level cluster signature scores, Fig. 4c). Moreover, induction of acute phase response, TNF response subset 1, and muscle regeneration genes were attenuated (respectively) in anti-TIF1, -NXP2, and -MDA5 subsets of DM. We also identify a cluster containing many genes expressed only in anti-Mi2 muscle, such as IFITM5, PRR35, DHRS2, CYP21A2, and the previously described MADCAM1 [25]. In IMNM, the contractile filaments subcluster was more intensely suppressed in anti-SRP patients. Several unknown clusters were also preferentially induced in one MSA subset. Overall, we identify and refine gene expression programs which distinguish myositis subtypes and hint at divergent pathophysiological mechanisms.
Figure 4: Transcriptomic divergence in serological subsets of clinical myositis diagnoses.
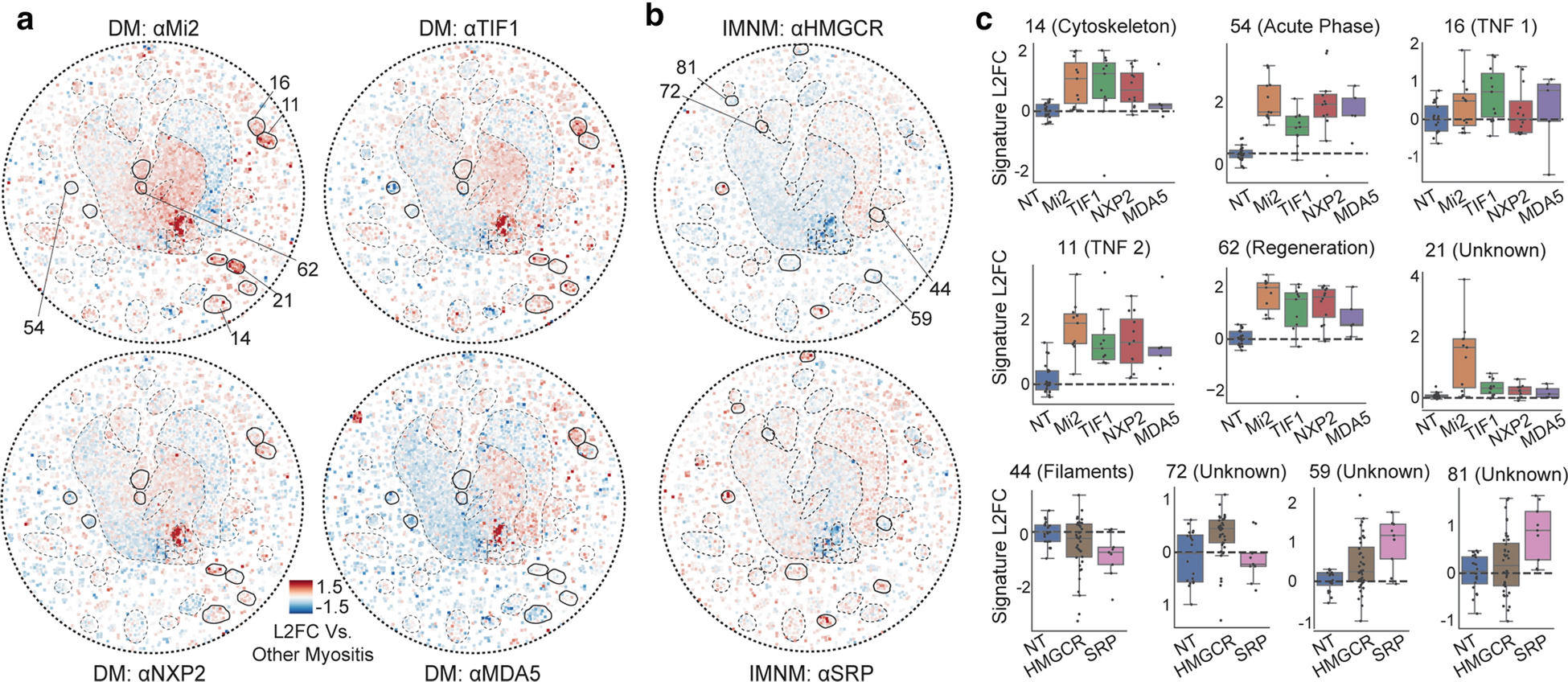
(a-b) Median gene expression of patients with the indicated DM-specific autoantibody in A or IMNM-specific autoantibody in B. Circles indicate a region of differential expression. (c) Gene expression signatures – mean log 2 fold change of cluster genes, where the cluster is indicated and highlighted in A or B – for individual patients across the indicated subsets. For more information on individual clusters (numbered), see Online Resource Supplementary Table 2.
Deep learning classification of patients by their network profiles
Because disease course and therapy response are highly variable in myositis, a central goal is to identify molecular subtypes which may predict outcomes and guide prospective trials. An emerging approach to subclassify high dimension data profiles, such as biopsy transcriptomes, is to convert them to images suitable for analysis by deep learning algorithms. Such algorithms have outperformed conventional machine learning in genomics classification problems [20, 33]. We reasoned that our network architecture discriminates pathways and disease signatures and may be a useful image framework to compare patient transcriptomes. Thus, we created an image of each patient’s transcriptome mapped onto our network, trained a convolutional neural network (CNN) to stratify each image into clinical groups, and then extracted the model’s features for unsupervised hierarchical clustering.
Clustering by CNN-derived features yielded a concise hierarchy of patient subgroups (Fig. 5a). One distinct cluster represented a subset of DM patients with extreme activation of pro-inflammatory (e.g., IFN1) pathways and suppression of muscle function genes – these patients were considered to have a “severe” transcriptional profile, in line with a previous study linking interferon pathway upregulation with more severe muscle pathology [26]. At the individual gene level, severe DM patients were distinguished from the “mild” DM cluster by upregulation of genes such as KLHDC7B, FGF21, and TMEM63C (severe vs. mild DM FPKM, 5.25 vs. 0.22, 3.6 vs. 0, and 2.58 vs. 0.16, respectively; all P<0.0001). Of note, transcriptome-scale comparisons for each subset described here are available in Online Resource Supplementary Table 4. Beyond DM, patients in each other group tended to form one primary cluster (dendrogram colors, Fig. 5a). However, seven patients had network more aligned with other clinical groups. For example, one DM patient – featuring greater lymphocyte and acute phase response induction, less TNF and IFN1 induction, and worse suppression of muscle function and apoptosis suppression genes than is typical for DM – was grouped with ASS patients (Fig. 5a–b). Elsewhere, two IBM and two ASS patients clustered with the NT control samples, suggesting these muscles may have had low disease activity when assayed (Fig. 5c).
Figure 5: Deep learning organization of patients by their transcriptional network features.
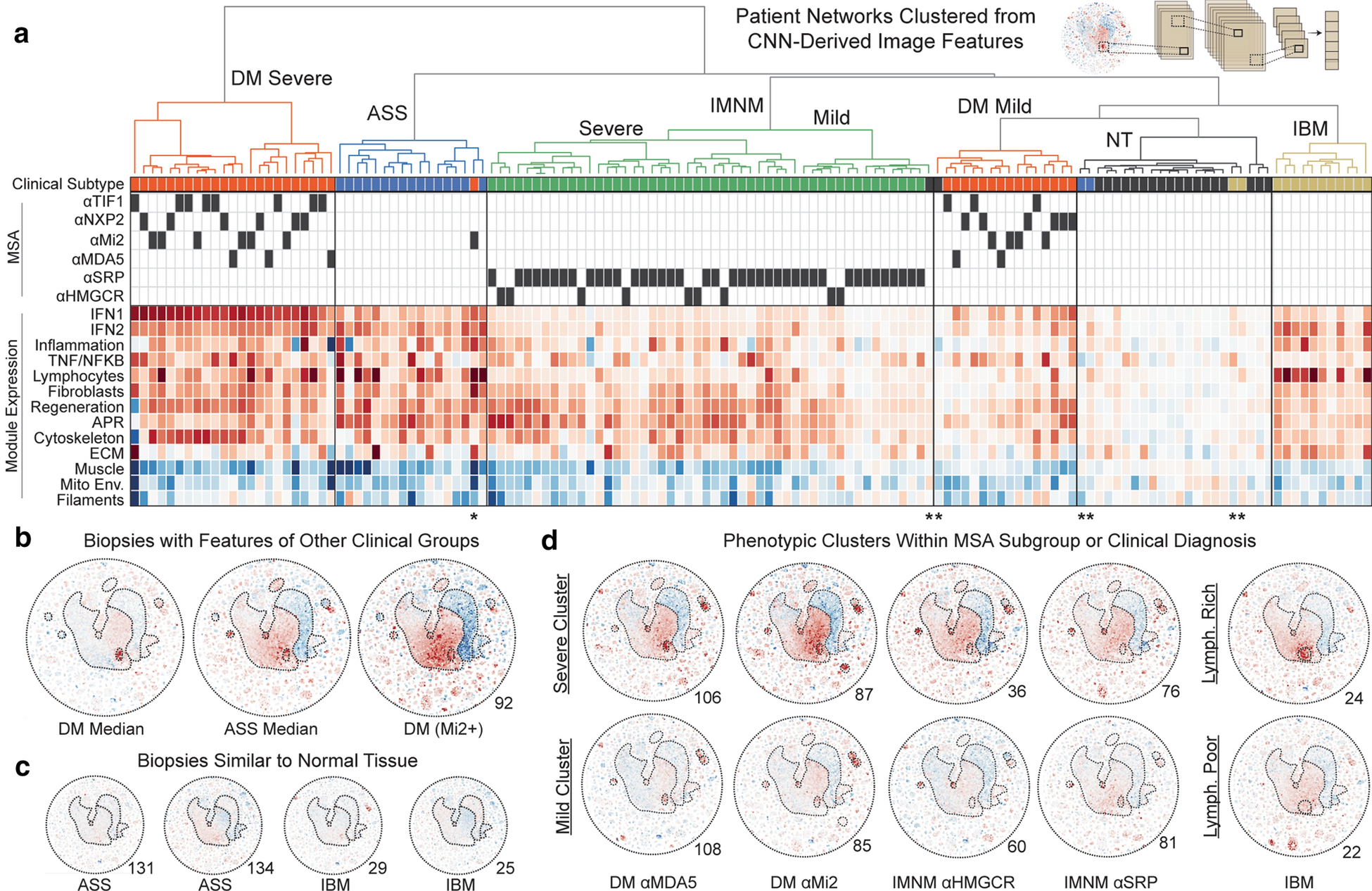
(a) Features, extracted from a convolutional neural network (CNN) trained to classify patients by their network image, were used to hierarchically cluster patients into groups with the least variance. Clinical features and signature scores for dynamic clusters corresponding to known biological processes are shown below the dendrogram. Dark boxes indicate MSA presence. Red/blue indicate overexpression and suppression as in prior figures. (b) An example biopsy from a DM patient which had many features enriched in ASS patients. (c) Four biopsies which had minimal global changes and clustered with normal tissue biopsies. (d) Example phenotypic subsets within clinical or MSA-defined subgroups include patients with delineations by overall severity of transcriptomic rewiring as well as a lymphocyte-rich subset of IBM.
We next investigated subclusters within clinical groups. Like DM, IMNM segregated into two branches reflecting the severity of muscle regeneration, inflammation, and muscle function gene alterations. This delineation corresponded with strength levels (median strength in severe vs. mild IMNM, 6.5/10 vs. 9.0/10, P<0.01). Intriguingly, severity-delimited subgroups in DM and IMNM had a proportionate representation of MSAs. For example, anti-MDA5+ DM patients generally have mild muscle disease clinically [13], but certain anti-MDA5+ patients had robust muscle transcriptomic alterations (Fig. 5d). On the other hand, while anti-Mi2+ patients have severe muscle disease clinically [27] and had extreme transcriptional changes on average, several anti-Mi2+ patients better aligned with the mild transcriptomic cluster. Beyond groups reflecting the magnitude of transcriptomic changes, other phenotypic subclusters emerged. For example, several IBM patients had markedly attenuated changes in lymphocyte gene expression (e.g., CD3G) and may be considered “lymphocyte-poor” (Fig. 5d). Further analysis revealed that this lymphocyte-poor subset, which was not delineated by clinical features such as time to biopsy, had reduced expression of genes enriched for inflammatory responses (e.g., OAS2, OAS3, IFIT1) and elevated expression of genes which regulate protein folding (e.g., HSF1, HSF4, HSPA1B, HSPB1). Altogether, our data indicate that the global transcriptional network of each biopsy provides information not encompassed by clinical subtype, MSA, or single pathway alterations. Such information may be relevant for predicting patient course or therapy response.
Myositis network topology hints at pathway membership and hierarchy
As we better understand the pathways dysregulated in myositis muscle, priorities will shift toward understanding which pathway members are most influential, and how we may manipulate them therapeutically. We first sought to investigate critical drivers of myositis altered pathways. Thus, we calculated the relative degree centrality of each gene – a measure of how connected that gene is within each pathway and often indicative of importance in that pathway [38]. This approach successfully identified hubs with overarching control of their correlated process, such as the master regulators MYOG (myogenesis [4]) and SOCS3 (NFKB [8]) (Fig. 6a). We also identified putative hubs with little existing literature linking them to their module, such as RGS19 (inflammation [15]) and KDM5A (nuclear body [28]). These genes may be high priority targets for basic inquiry. Last, we sought to nominate novel pathway members. On our network, we marked understudied genes as well as ncRNAs, a traditionally understudied class of genes (Fig. 6b). Illustrating a potential for biological discovery, we highlight the myogenesis cluster, containing an unknown gene (C11orf1), a ncRNA (LINCMD1), and several genes with no known role in muscle regeneration (Fig. 6c). To determine whether these genes are part of a true muscle regeneration program versus indirect correlates of muscle damage, we leveraged two models of myogenesis: human skeletal muscle myoblast (HSMM) differentiation and cardiotoxin mouse muscle injury. In both systems, cluster genes were robustly altered in a temporal fashion analogous to known myogenesis factors (Fig. 6d), including the markedly induced HN1/JPT-1 and suppressed LMOD1 which previously had no connection to muscle regeneration (Fig. 6e). Thus, the myositis co-expression network is a resource to generate or refine hypotheses about novel genes with potentially important roles in myositis-relevant pathways.
Figure 6: Predicting pathway members and hierarchy from the myositis network topology.
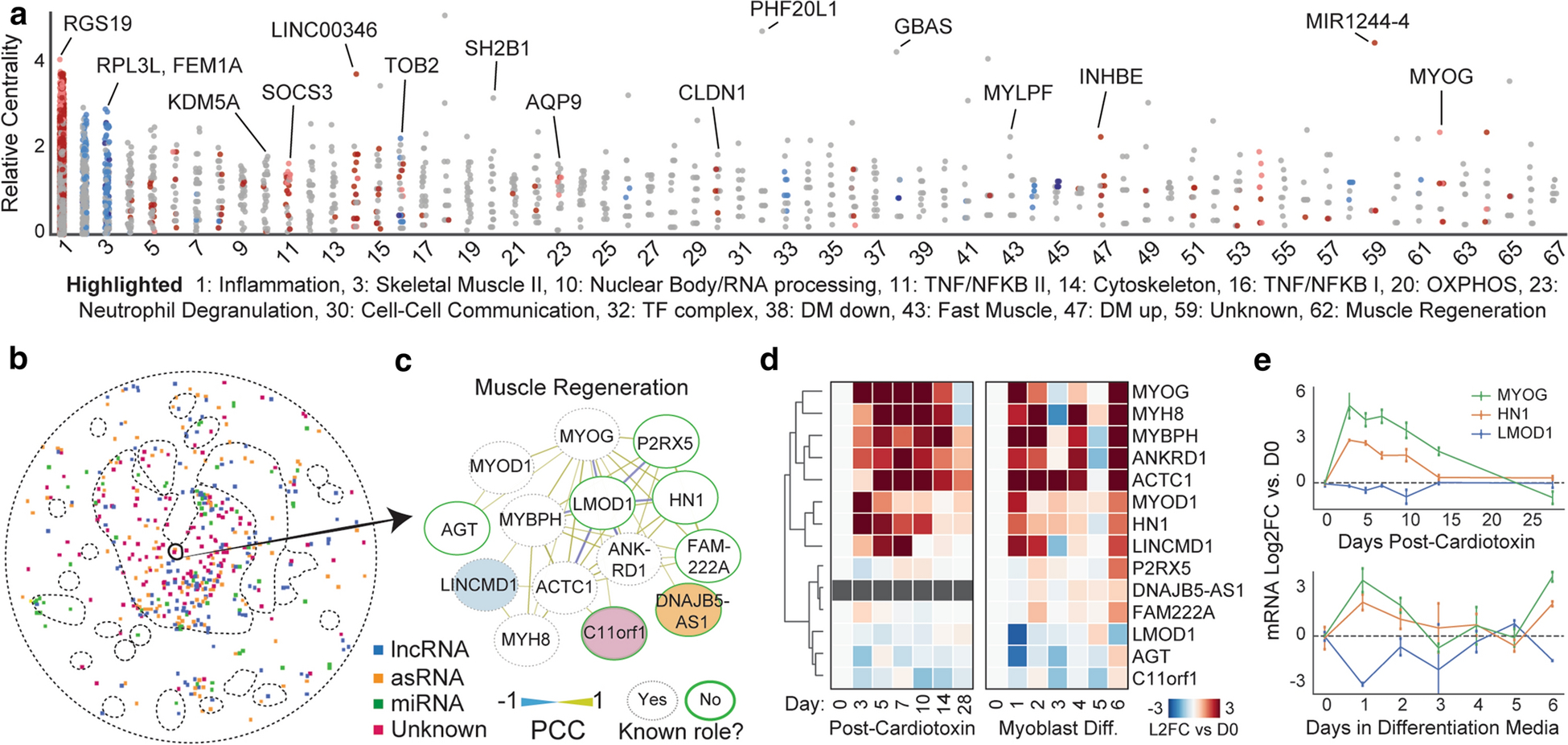
(a) A measure of the importance of a given gene in its cluster, the intra-cluster degree centrality, expressed relative to the average gene in the cluster. For more information on individual clusters (numbered), see Online Resource Supplementary Table 2. (b) Noncoding transcripts and unknown genes are present in most clusters. (c) The muscle regeneration cluster 62, which contains noncoding transcripts, unknown genes, and genes with no known connection to myogenesis. The layout is hierarchical based on degree. (d) Expression of regeneration cluster genes in two models of muscle regeneration/myogenesis, mouse cardiotoxin injury and human skeletal muscle myoblast differentiation. (e) Temporal transcriptional regulation of the master myogenesis regulator MYOG and two example genes without known roles in muscle differentiation, HN1 (induced, positive correlate) and LMOD1 (suppressed, negative correlate).
DISCUSSION
Understanding the molecular landscape of clinically heterogeneous disorders should improve our ability to diagnose and classify patients, prioritize new therapeutic targets, and predict which patients will respond to a drug targeting a given pathway. Because myositis patients are relatively rare, obtaining sufficient samples to compare the activation of dynamically regulated pathways across clinical and serological subtypes has remained challenging. Here, by leveraging a relatively large number of biopsies and employing an analysis strategy agnostic to pre-defined gene sets or patient groupings, we report a map of dynamic gene expression programs in myositis.
Among the core and subtype-enriched pathways we identify, several are previously undescribed – such as the conserved suppression of the stress-resilience linked humanin isoforms and the preferential upregulation of RNA processing genes in ASS – and may serve as the basis for future studies. Our network also refines programs previously connected to myositis, such as the subset of universally down-regulated myofilaments and specific markers of neutrophil degranulation in myositis muscle. Importantly, the understudied genes present in most clusters may also serve as a launching point for basic studies into fundamental processes altered in myositis. Many of these understudied genes are strikingly differentially expressed or have high intra-cluster connectivity, and are likely unknown due to funding practices and historical technical constraints rather than lack of importance [35]. Robust transcriptional anticorrelations, such as that between IFN1-induced genes and TTN, may also serve as the basis for mechanistic studies of myositis-dysregulated processes. Specifically, the loss of TTN transcription in IFN1-high muscle (observed here at the biopsy level) and protein (at the level of individual dissected PFA fibers [29]) suggests that IFN1 provokes a signaling change – whether epigenetic or at the level of a single transcription factor – to abrogate transcription of a key muscle structural protein. In vitro studies of the interplay between IFN1 and the factors controlling TTN transcription may thus reveal a therapeutic strategy to restore integrity of affected fibers in DM.
With regards to patient classification, we provide proof of concept for the utility of treating myositis patient transcriptomics profiles as images suitable for deep learning. The classification performance of our model suggests the potential for diagnostic applications of image-based transcriptome recognition algorithms in myositis, though validation in an external cohort of patients will be required to assess the true performance characteristics of this approach. Such studies may use our source code and model weights to further test and improve our algorithm, which can be accessed through an interactive notebook (github.com/mendillolab). Future studies should also explore less defined or overlap myositis cases, which will likely have distinct network characteristics relevant to classification and upstream aspects of pathogenesis. Beyond classification into classic disease groups, we were intrigued to find phenotypic subclusters within clinical groupings, such as severe vs. mild DM and lymphocyte rich vs. poor IBM. That transcriptomic subgroups spanned MSAs suggests that while certain expression programs can reliably delineate patients by their MSA, the overall transcriptional state of these patients may still be strikingly similar. Thus, the global transcriptional state – as viewed in network form or by reduction to a set of CNN-derived features – may be an ideal surrogate for certain aspects of patient phenotyping (e.g., disease activity), whereas specific cluster signatures or MSAs may be better in others (e.g., therapy response or extra-muscular manifestations, or in subtle diagnostic distinctions between highly similar groups).
Finally, we emphasize that our network may serve as a resource for future studies. For each cluster, the constituent genes, expression across myositis subsets, and degree of correlation with strength and serum creatine kinase are included in Online Resource Supplementary Tables 2 and 3. Additionally, individuals studying genes or pathways relevant to myositis – or other similar disorders – may benefit from directly identifying their connections in our interactive network (see Online Resource). Our network also allows imposition of external gene expression profiles to rapidly visualize and contextualize alteration of consequential pathways, with or without neural network analysis, both of which may be useful as clinical transcription profiling in myositis becomes more common. More broadly, the methodology used in this study should be applicable to any number of heterogeneous conditions where patient subgrouping, pathogenic mechanisms, and therapeutic response predictors are incompletely understood.
Supplementary Material
Online Resource Supplementary Table 1: Epidemiologic and muscle biopsy features of the patients.
Online Resource Supplementary Table 2: Expression levels, constituent genes, and clinical correlates of myositis network modules. Attached as Excel file due to size.
Online Resource Supplementary Table 3: Gene-level expression values and module centrality estimates. Attached as Excel file due to size
Online Resource Supplementary Table 4: Transcriptomic differences between phenotypic subclusters nominated by the deep learning algorithm. Attached as Excel file due to size
Acknowledgements:
This research was supported in part by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health. We are also grateful to Dr. Peter Buck, whose generous support made this work possible. D.R.A. was supported by the National Institutes of Health (F30CA264513 and T32GM008152). M.L.M is supported by the Susan G. Komen Foundation (CCR17488145). M.L.M was also supported by Kimmel Scholar (SKF-16-135) and Lynn Sage Scholar awards. The authors have no conflicts of interest to disclose.
Footnotes
Publisher's Disclaimer: This AM is a PDF file of the manuscript accepted for publication after peer review, when applicable, but does not reflect post-acceptance improvements, or any corrections. Use of this AM is subject to the publisher’s embargo period and AM terms of use. Under no circumstances may this AM be shared or distributed under a Creative Commons or other form of open access license, nor may it be reformatted or enhanced, whether by the Author or third parties. See here for Springer Nature’s terms of use for AM versions of subscription articles: https://www.springernature.com/gp/open-research/policies/accepted-manuscript-terms
Compliance with Ethical Standards: The authors have no conflicts of interest to disclose. All patient participants provided written informed consent as outlined in IRB-approved protocols. Mouse experiments followed established IACUC protocols. All non-identifying data and code are made available in supplemental files to the manuscript and at github.com/mendillolab.
REFERENCES
- 1.Amici DR, Jackson JM, Truica MI, Smith RS, Abdulkadir SA, Mendillo ML (2021) FIREWORKS: a bottom-up approach to integrative coessentiality network analysis. Life Sci Alliance 4: Doi 10.26508/lsa.202000882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amici DR, Pinal-Fernandez I, Mazala DA, Lloyd TE, Corse AM, Christopher-Stine L, Mammen AL, Chin ER (2017) Calcium dysregulation, functional calpainopathy, and endoplasmic reticulum stress in sporadic inclusion body myositis. Acta Neuropathol Commun 5: 24 Doi 10.1186/s40478-017-0427-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amici DR, Pinal-Fernandez I, Pagkatipunan R, Mears A, de Lorenzo R, Tiniakou E, Albayda J, Paik JJ, Lloyd TE, Christopher-Stine L et al. (2019) Muscle endurance deficits in myositis patients despite normal manual muscle testing scores. Muscle Nerve 59: 70–75 Doi 10.1002/mus.26307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bentzinger CF, Wang YX, Rudnicki MA (2012) Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 4: Doi 10.1101/cshperspect.a008342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benveniste O, Stenzel W, Hilton-Jones D, Sandri M, Boyer O, van Engelen BG (2015) Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the degenerative chicken. Acta Neuropathol 129: 611–624 Doi 10.1007/s00401-015-1384-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bodzioch M, Lapicka-Bodzioch K, Zapala B, Kamysz W, Kiec-Wilk B, Dembinska-Kiec A (2009) Evidence for potential functionality of nuclearly-encoded humanin isoforms. Genomics 94: 247–256 Doi 10.1016/j.ygeno.2009.05.006 [DOI] [PubMed] [Google Scholar]
- 7.Boufenzer A, Carrasco K, Jolly L, Brustolin B, Di-Pillo E, Derive M, Gibot S (2021) Potentiation of NETs release is novel characteristic of TREM-1 activation and the pharmacological inhibition of TREM-1 could prevent from the deleterious consequences of NETs release in sepsis. Cellular & Molecular Immunology: Doi 10.1038/s41423-020-00591-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carow B, Rottenberg ME (2014) SOCS3, a Major Regulator of Infection and Inflammation. Front Immunol 5: 58 Doi 10.3389/fimmu.2014.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I (2011) A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147: 358–369 Doi 10.1016/j.cell.2011.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, Barohn RJ, Saperstein DS, Briemberg HR, Ericsson M et al. (2005) Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 57: 664–678 Doi 10.1002/ana.20464 [DOI] [PubMed] [Google Scholar]
- 11.Greenberg SA, Sanoudou D, Haslett JN, Kohane IS, Kunkel LM, Beggs AH, Amato AA (2002) Molecular profiles of inflammatory myopathies. Neurology 59: 1170–1182 Doi 10.1212/wnl.59.8.1170 [DOI] [PubMed] [Google Scholar]
- 12.Halim D, Wilson MP, Oliver D, Brosens E, Verheij JB, Han Y, Nanda V, Lyu Q, Doukas M, Stoop H et al. (2017) Loss of LMOD1 impairs smooth muscle cytocontractility and causes megacystis microcolon intestinal hypoperistalsis syndrome in humans and mice. Proc Natl Acad Sci U S A 114: E2739–E2747 Doi 10.1073/pnas.1620507114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall JC, Casciola-Rosen L, Samedy LA, Werner J, Owoyemi K, Danoff SK, Christopher-Stine L (2013) Anti-melanoma differentiation-associated protein 5-associated dermatomyositis: expanding the clinical spectrum. Arthritis Care Res (Hoboken) 65: 1307–1315 Doi 10.1002/acr.21992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill C, Flyvbjerg A, Rasch R, Bak M, Logan A (2001) Transforming growth factor-beta2 antibody attenuates fibrosis in the experimental diabetic rat kidney. J Endocrinol 170: 647–651 Doi 10.1677/joe.0.1700647 [DOI] [PubMed] [Google Scholar]
- 15.Kehrl JH (2016) The impact of RGS and other G-protein regulatory proteins on Galphai-mediated signaling in immunity. Biochem Pharmacol 114: 40–52 Doi 10.1016/j.bcp.2016.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kendall FP, McCreary EK, Provance PG (1993) Muscles, testing and function : with Posture and pain. Williams & Wilkins, City [Google Scholar]
- 17.Lacy P (2006) Mechanisms of degranulation in neutrophils. Allergy Asthma Clin Immunol 2: 98–108 Doi 10.1186/1710-1492-2-3-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P (2015) The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425 Doi 10.1016/j.cels.2015.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA (2014) Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology 83: 426–433 Doi 10.1212/WNL.0000000000000642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyu B, Haque A (2018) Deep Learning Based Tumor Type Classification Using Gene Expression Data. bioRxiv: 364323 Doi 10.1101/364323 [DOI] [Google Scholar]
- 21.Mammen AL, Casciola-Rosen LA, Hall JC, Christopher-Stine L, Corse AM, Rosen A (2009) Expression of the dermatomyositis autoantigen Mi-2 in regenerating muscle. Arthritis Rheum 60: 3784–3793 Doi 10.1002/art.24977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR, Casciola-Rosen LA (2011) Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 63: 713–721 Doi 10.1002/art.30156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET et al. (2016) A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351: 271–275 Doi 10.1126/science.aad4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinal-Fernandez I, Amici DR, Parks CA, Derfoul A, Casal-Dominguez M, Pak K, Yeker R, Plotz P, Milisenda JC, Grau-Junyent JM et al. (2019) Myositis Autoantigen Expression Correlates With Muscle Regeneration but Not Autoantibody Specificity. Arthritis Rheumatol 71: 1371–1376 Doi 10.1002/art.40883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, Pak K, Miller FW, Milisenda JC, Grau-Junyent JM, Selva-O’Callaghan A, Carrion-Ribas C, Paik JJ et al. (2020) Machine learning algorithms reveal unique gene expression profiles in muscle biopsies from patients with different types of myositis. Ann Rheum Dis 79: 1234–1242 Doi 10.1136/annrheumdis-2019-216599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinal-Fernandez I, Casal-Dominguez M, Derfoul A, Pak K, Plotz P, Miller FW, Milisenda JC, Grau-Junyent JM, Selva-O’Callaghan A, Paik J et al. (2019) Identification of distinctive interferon gene signatures in different types of myositis. Neurology 93: e1193–e1204 Doi 10.1212/WNL.0000000000008128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pinal-Fernandez I, Mecoli CA, Casal-Dominguez M, Pak K, Hosono Y, Huapaya J, Huang W, Albayda J, Tiniakou E, Paik JJ et al. (2019) More prominent muscle involvement in patients with dermatomyositis with anti-Mi2 autoantibodies. Neurology 93: e1768–e1777 Doi 10.1212/WNL.0000000000008443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, Bodenmiller B, Campbell P, Carninci P, Clatworthy M et al. (2017) The Human Cell Atlas. Elife 6: Doi 10.7554/eLife.27041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salajegheh M, Kong SW, Pinkus JL, Walsh RJ, Liao A, Nazareno R, Amato AA, Krastins B, Morehouse C, Higgs BW et al. (2010) Interferon-stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann Neurol 67: 53–63 Doi 10.1002/ana.21805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schepetkin IA, Khlebnikov AI, Giovannoni MP, Kirpotina LN, Cilibrizzi A, Quinn MT (2014) Development of small molecule non-peptide formyl peptide receptor (FPR) ligands and molecular modeling of their recognition. Curr Med Chem 21: 1478–1504 Doi 10.2174/0929867321666131218095521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seto N, Torres-Ruiz JJ, Carmona-Rivera C, Pinal-Fernandez I, Pak K, Purmalek MM, Hosono Y, Fernandes-Cerqueira C, Gowda P, Arnett N et al. (2020) Neutrophil dysregulation is pathogenic in idiopathic inflammatory myopathies. JCI Insight 5: Doi 10.1172/jci.insight.134189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 Doi 10.1101/gr.1239303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharma A, Vans E, Shigemizu D, Boroevich KA, Tsunoda T (2019) DeepInsight: A methodology to transform a non-image data to an image for convolution neural network architecture. Sci Rep 9: 11399 Doi 10.1038/s41598-019-47765-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Starnes T, Broxmeyer HE, Robertson MJ, Hromas R (2002) Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J Immunol 169: 642–646 Doi 10.4049/jimmunol.169.2.642 [DOI] [PubMed] [Google Scholar]
- 35.Stoeger T, Gerlach M, Morimoto RI, Nunes Amaral LA (2018) Large-scale investigation of the reasons why potentially important genes are ignored. PLoS Biol 16: e2006643 Doi 10.1371/journal.pbio.2006643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550 Doi 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tjondrokoesoemo A, Schips T, Kanisicak O, Sargent MA, Molkentin JD (2016) Genetic overexpression of Serpina3n attenuates muscular dystrophy in mice. Hum Mol Genet 25: 1192–1202 Doi 10.1093/hmg/ddw005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Dam S, Vosa U, van der Graaf A, Franke L, de Magalhaes JP (2018) Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform 19: 575–592 Doi 10.1093/bib/bbw139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, Mitch WE (2009) IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol 20: 604–612 Doi 10.1681/ASN.2008060628 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Resource Supplementary Table 1: Epidemiologic and muscle biopsy features of the patients.
Online Resource Supplementary Table 2: Expression levels, constituent genes, and clinical correlates of myositis network modules. Attached as Excel file due to size.
Online Resource Supplementary Table 3: Gene-level expression values and module centrality estimates. Attached as Excel file due to size
Online Resource Supplementary Table 4: Transcriptomic differences between phenotypic subclusters nominated by the deep learning algorithm. Attached as Excel file due to size