

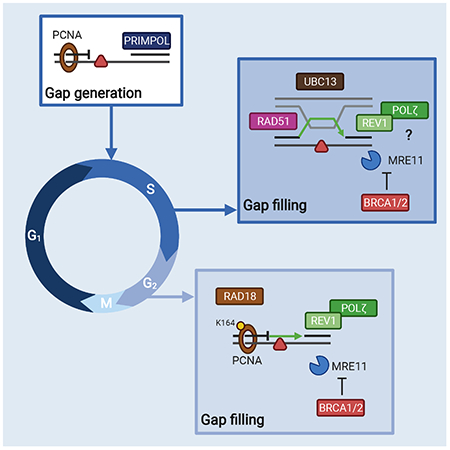
Summary
PRIMPOL repriming allows DNA replication to skip DNA lesions, leading to ssDNA gaps. These gaps must be filled to preserve genome stability. Using a DNA fiber approach to directly monitor gap filling, we studied the post-replicative mechanisms that fill the ssDNA gaps generated in cisplatin-treated cells upon increased PRIMPOL expression or when replication fork reversal is defective because of SMARCAL1 inactivation or PARP inhibition. We found that a mechanism dependent on the E3 ubiquitin ligase RAD18, PCNA monoubiquitination, and the REV1 and POLζ translesion synthesis polymerases promotes gap filling in G2. The E2 conjugating enzyme UBC13, the RAD51 recombinase, and REV1-POLζ are instead responsible for gap filling in S, suggesting that temporally distinct pathways of gap filling operate throughout the cell cycle. Furthermore, we found that BRCA1 and BRCA2 promote gap filling by limiting MRE11 activity and that simultaneously targeting fork reversal and gap filling enhances chemosensitivity in BRCA-deficient cells.
eTOC Blurb:
PRIMPOL generates ssDNA gaps, which must be filled to maintain genome stability. Tirman et al. show that two distinct pathways fill ssDNA gaps throughout the cell cycle in human cells and that BRCA proteins promote gap filling by limiting MRE11 activity. Disruption of these pathways enhances genome instability and chemosensitivity.
Graphical Abstract
Introduction
Single-stranded DNA (ssDNA) discontinuities (or gaps) are potentially genome destabilizing structures that require prompt repair (or filling) to prevent DNA breakage and genome instability. Different studies showed that ssDNA gaps are frequently present both on leading and lagging strands of DNA replication forks after treatment with a wide range of DNA-damaging agents (Diamant et al., 2012; Elvers et al., 2011; Jansen et al., 2009; Lehmann, 1972; Lopes et al., 2006; Meneghini, 1976; Quinet et al., 2014). Generation of gaps in the lagging strand can be explained by the discontinuous nature of Okazaki fragment synthesis. Conversely, leading strand gaps form when DNA synthesis resumes downstream of a replication-blocking lesion in a process termed fork repriming, which leaves unreplicated ssDNA gaps to be filled post-replicatively. Repriming is mediated by the DnaG primase in bacteria (Heller and Marians, 2006), the Polymerase α (Polα)/Primase complex and Ctf4 in yeast (Fumasoni et al., 2015), and by human Primase and DNA-directed Polymerase (PRIMPOL) in mammalian cells (Bianchi et al., 2013; García-Gómez et al., 2013; Mourón et al., 2013; Wan et al., 2013).
In recent years, there has been a surge of papers highlighting the role of PRIMPOL repriming during the replication stress response (Bai et al., 2020; Calvo et al., 2019; Gonzalez-Acosta et al., 2021; Keen et al., 2014; Kobayashi et al., 2016; Piberger et al., 2020; Quinet et al., 2021; Quinet et al., 2020; Schiavone et al., 2016; Švikoviç et al., 2019; Tirman et al., 2020). Replication fork reversal is another replication stress tolerance mechanism that promotes damage bypass or replication-coupled repair by remodeling the replication fork into a four-way junction structure (Neelsen and Lopes, 2015). PRIMPOL repriming rescues stalled forks when replication fork reversal is inactivated upon loss of the SMARCAL1 or HLTF translocases (Bai et al., 2020; Quinet et al., 2020), or when fork reversal is reduced by the loss of the PARP stimulating factor CARM1 (Genois et al., 2020). Although fork reversal helps replication forks deal with stress and lesions, it can also lead to pathological fork degradation when the regressed arms of the reversed forks are not adequately protected by BRCA proteins (Kolinjivadi et al., 2017; Lemaçon et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017). Recent studies showed that treatment with multiple cisplatin doses increases PRIMPOL expression and promotes PRIMPOL-mediated repriming in BRCA-deficient cells as an alternative strategy to cope with cisplatin-induced lesions and prevent pathological reversed fork degradation (Quinet et al., 2020).
The ssDNA gaps that accumulate behind the replication fork following repriming are filled post-replicatively by a process termed gap filling or post-replication repair (PRR). ssDNA gap filling is thought to be mediated by two main DNA damage tolerance (DDT) pathways: template switching (TS) and translesion DNA synthesis (TLS). While TS appears to be the main pathway of gap filling in E. coli (Berdichevsky et al., 2002; Laureti et al., 2015), early work suggested that TLS predominantly mediates gap filling in budding yeast (Daigaku et al., 2010; Gallo et al., 2019; Karras and Jentsch, 2010). However, later studies challenged this model by showing that the ssDNA gaps generated upon repriming by the Polα-primase complex are filled by TS or by an alternative homologous recombination (HR) salvage pathway in S. cerevisiae (Fumasoni et al., 2015; Gonzalez-Huici et al., 2014; Karras et al., 2013). While salvage HR is PCNA polyubiquitination-independent, gap filling by TS is facilitated by PCNA polyubiquitination and occurs early in the S phase in budding yeast (Branzei and Szakal, 2016; Gonzalez-Huici et al., 2014; Karras et al., 2013). This data led to the proposal that TS pathways act first during the S phase, whereas the more error-prone mechanisms, such as salvage HR and TLS, preferentially act later in the S phase or in G2 if the gaps cannot be properly filled by TS (Branzei and Szakal, 2016).
Different TLS enzymes have been proposed to be involved in gap filling in human cells (Diamant et al., 2012; Elvers et al., 2011; Quinet et al., 2016). In addition, an alternative HR pathway mediated by RAD51 was shown to efficiently fill gaps opposite of synthetic abasic sites or bulky adducts in mammalian cells (Adar et al., 2009; Piberger et al., 2020). However, how the different cell cycle phases affect the balance between TLS, TS, and alternative HR during post-replicative repair in the human genome remains largely unknown. Moreover, the relative contribution of PCNA mono- versus polyubiquitination to the PRR pathway choice in human cells has not been elucidated (Masuda and Masutani, 2019). Of note, the accumulation of ssDNA gaps is rapidly emerging as a key predictor of genome stability and chemotherapy response, especially in BRCA-deficient tumors, in which failure to properly repair ssDNA gaps has been associated with increased chemosensitivity (Simoneau et al., 2021; Cong et al., 2021; Lim et al., 2018; Nayak et al., 2020; Panzarino et al., 2020). However, how the choice between different PRR pathways or defective gap filling affects cell survival and genome stability remains poorly understood.
A major hurdle in studying mechanisms of gap filling has been the lack of an approach to directly visualize gap filling in human cells. In this study, we used a modified single-molecule DNA fiber technique to directly monitor gap filling in human cells and define the factors involved in PRR as a function of the different cell cycle phases. We found that a TLS pathway involving RAD18, PCNA monoubiquitination, and the REV1 and POLζ polymerases mediates gap filling in G2. Conversely, UBC13, a factor that promotes PCNA polyubiquitination, and the RAD51 recombinase promote gap filling in S, but not in G2, suggesting temporally distinct mechanisms of PRR in human cells. Furthermore, we found that BRCA proteins promote gap filling in both S and G2 phases by limiting MRE11 activity. Finally, we show that defective gap filling leads to DNA damage and genomic instability, as well as increased sensitivity to DNA-damaging chemotherapy in BRCA-deficient cancer cells.
Results
Suppression of fork reversal and increased PRIMPOL expression promote ssDNA gap accumulation.
PRIMPOL repriming enables replication fork progression in the presence of different replication obstacles, leading to accumulation of ssDNA gaps (Bai et al., 2020; Genois et al., 2020; Piberger et al., 2020; Quinet et al., 2020). Here, we demonstrate that PRIMPOL-dependent ssDNA gaps accumulate following treatment with cisplatin, a platinum-based compound that mainly generates intra-strand crosslinks (Helm and States, 2009). We monitored fork progression by DNA fiber assay, in which we pulse-labeled cells with the thymidine analog, IdU (red), for 20 minutes followed by labelling with the second thymidine analog, CldU (green), for 1 hour and concomitant treatment with 150 μM cisplatin. Next, we used the ssDNA-specific S1 nuclease to determine whether the CldU-labeled tracts contained ssDNA gaps (see scheme Figure 1A). The shorter DNA fiber tracts generated by S1 cleavage were used as a readout for the presence of ssDNA gaps (Quinet et al., 2017; Quinet et al., 2016). First, we used human osteosarcoma U2OS cells overexpressing a V5-tagged version of wild-type (WT) or primase-dead (C419G/H426Y, CH variant) PRIMPOL (Figure S1A) (Mourón et al., 2013). We found that cisplatin treatment led to ssDNA gap accumulation in U2OS cells overexpressing WT-PRIMPOL, but not in cells expressing CH-PRIMPOL, suggesting that the primase activity of PRIMPOL is required for ssDNA gap formation (Figures 1A and S1B).
Figure 1. Suppression of fork reversal and overexpression PRIMPOL lead to ssDNA gaps.
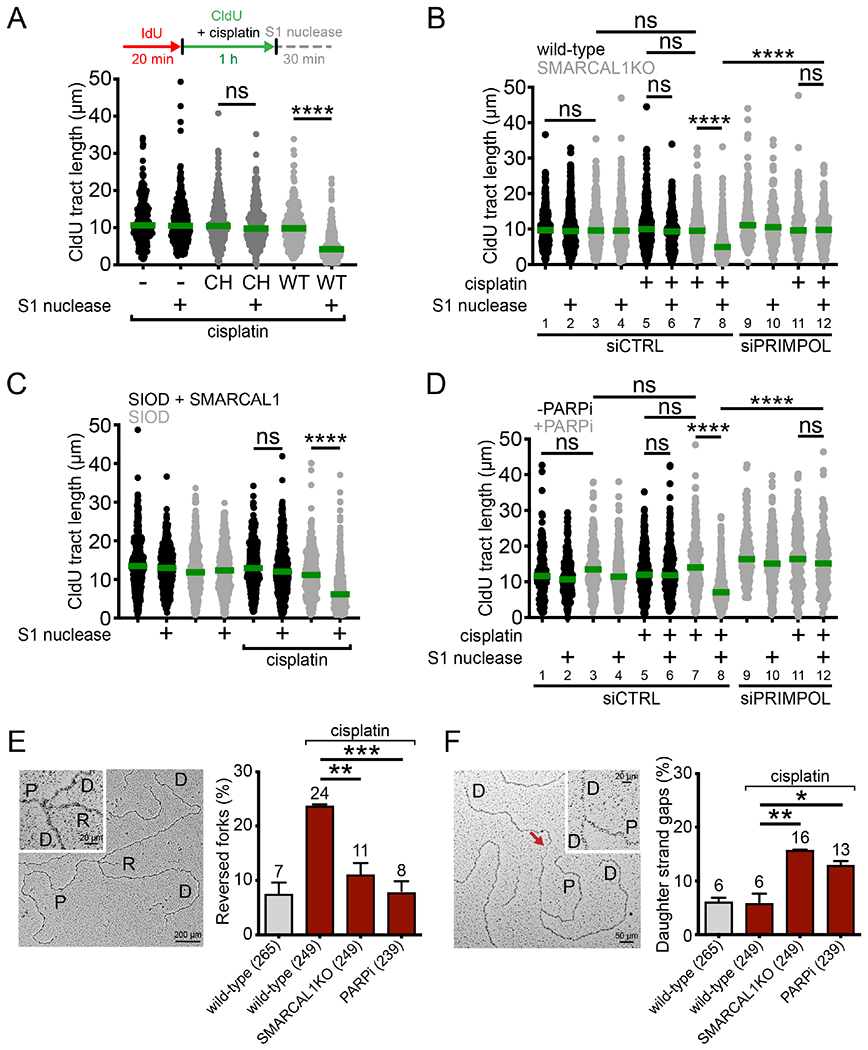
(A) Top, schematic of the DNA fiber assay with the S1 nuclease. Bottom, dot plot and median of CldU tract lengths in U2OS cells overexpressing WT- or CH-PRIMPOL + 150 μM cisplatin ± S1 nuclease (n=3). ns, non-significant, ****p < 0.0001. (B) Dot plot and median of CldU tract lengths in wild-type or SMARCAL1KO U2OS cells ± 150 μM cisplatin ± siCTRL (control siRNA) or siPRIMPOL ± S1 nuclease (n=3). ns, non-significant, ****p < 0.0001. (C) Dot plot and median of CldU tract lengths in SIOD cells or SIOD cells complemented with wild-type SMARCAL1 ± 150 μM cisplatin ± S1 nuclease (n=3). ns, non-significant, ****p < 0.0001. (D) Dot plot and median of CldU tract lengths in U2OS cells ± PARPi ± 150 μM cisplatin ± siCTRL or siPRIMPOL ± S1 nuclease (n=3). ns, non-significant, ****p < 0.0001. (E) Left, representative electron micrograph of a reversed replication fork. Right, percentage of reversed replication forks in wild-type, SMARCAL1KO, and PARPi-treated U2OS cells ± 150 μM cisplatin (n=3). Statistics: unpaired t test; **p < 0.01, ***p < 0.001. (F) Left, representative electron micrograph of a replication fork with an internal ssDNA gap behind the fork (daughter strand gap), indicated by the red arrow. Right, percentage of replication forks with daughter strand gaps in wild-type, SMARCAL1KO, and PARPi-treated U2OS cells ± 150 μM cisplatin (n=3). P: parental strand, D: daughter strand, R: reversed arm. Statistics: unpaired t test; *p < 0.05, **p < 0.01.
See also Figure S1.
Next, we found that inactivating the fork reversal enzyme SMARCAL1 using CRISPR-Cas9 gene editing also led to ssDNA gap accumulation (Figures 1B and S1C). Moreover, S1-dependent tract shortening was rescued by PRIMPOL depletion, confirming that PRIMPOL activity is required for ssDNA gap formation (Figure 1B, compare lanes 8 and 12). The same results were confirmed using patient Schimke Immunoosseous Dysplasia (SIOD) lymphocytes containing a splicing mutation in SMARCAL1 that compromises protein expression (Figures 1C and S1D) (Carroll et al., 2015), indicating that the observed effect is not cell type-specific. Importantly, loss of SMARCAL1 also led to PRIMPOL-dependent ssDNA gap accumulation in cells treated with a low dose (50 μM) of hydroxyurea (HU) or exposed to ultraviolet light (UV-C, 15 J/m2), suggesting that PRIMPOL repriming is more generally activated in response to different kinds of replication challenges (Figures S1E and S1F). Finally, we inhibited PARP activity with Olaparib (PARPi) as an alternative strategy to decrease fork reversal (Berti et al., 2013; Ray Chaudhuri et al., 2012) and found that treatment with PARPi also leads to PRIMPOL-dependent ssDNA gap accumulation (Figure 1D, lanes 8 and 12). These data collectively suggest that PRIMPOL-dependent ssDNA gaps accumulate when: 1) PRIMPOL expression is increased; 2) reversed fork formation is inhibited; and 3) accumulation of reversed forks is prevented by PARP inhibition.
To directly visualize PRIMPOL gaps generated as a consequence of impaired fork reversal, we combined in vivo psoralen crosslinking with electron microscopy (EM) and analyzed replication intermediates of wild-type, SMARCAL1KO, and PARPi-treated U2OS cells upon cisplatin treatment. As predicted, loss of SMARCAL1 or PARP inhibition decreased the frequency of reversed forks in U2OS cells treated with cisplatin (Figure 1E and Table S1A) and led to a concomitant increase in replication intermediates with daughter-strand ssDNA gaps (Figure 1F and Table S1B).
ssDNA gaps are repaired in S and G2 phase.
Next, we investigated the cell cycle phase during which the PRIMPOL-dependent ssDNA gaps are repaired by post-replication repair (PRR). We performed a kinetic S1 DNA fiber assay in PRIMPOL overexpressing (PRIMPOL-OE) U2OS cells treated with cisplatin. We digested the nuclei with the S1 nuclease every 4 hours over the course of 24 hours after CldU labeling and confirmed that PRIMPOL overexpression was maintained over the duration of the experiment (Figure S1A). As expected, cisplatin treatment led to significant tract shortening upon S1 cleavage at time 0, indicating the presence of gaps (Figure 2A). However, as gaps were filled, the CldU tract length gradually increased as a function of the timing of S1 nuclease treatment. In particular, the CldU tract length reached the same length observed in untreated cells when the S1 nuclease was added 16 hours after CldU labeling, suggesting that the ssDNA gaps are filled by this time. To determine the cell cycle phase corresponding with PRR, we labeled replicating PRIMPOL-OE cells with the thymidine analog EdU and monitored the progression of EdU-positive cells throughout the cell cycle every 4 hours over the course of 24 hours (see scheme Figure 2B, S2B). In agreement with previous studies (Harder and Rosenberg, 1970), cells treated with cisplatin progressed more slowly through S phase, reaching G2 phase between 12 and 16 hours, as opposed to 8 hours for untreated cells (Figures 2B, S2C and S2D). These results suggest that the ssDNA gaps generated by PRIMPOL overexpression start being repaired post-replicatively in S phase and are completely filled when cells have reached G2.
Figure 2. PRIMPOL-dependent gaps are repaired in S and G2 phase.
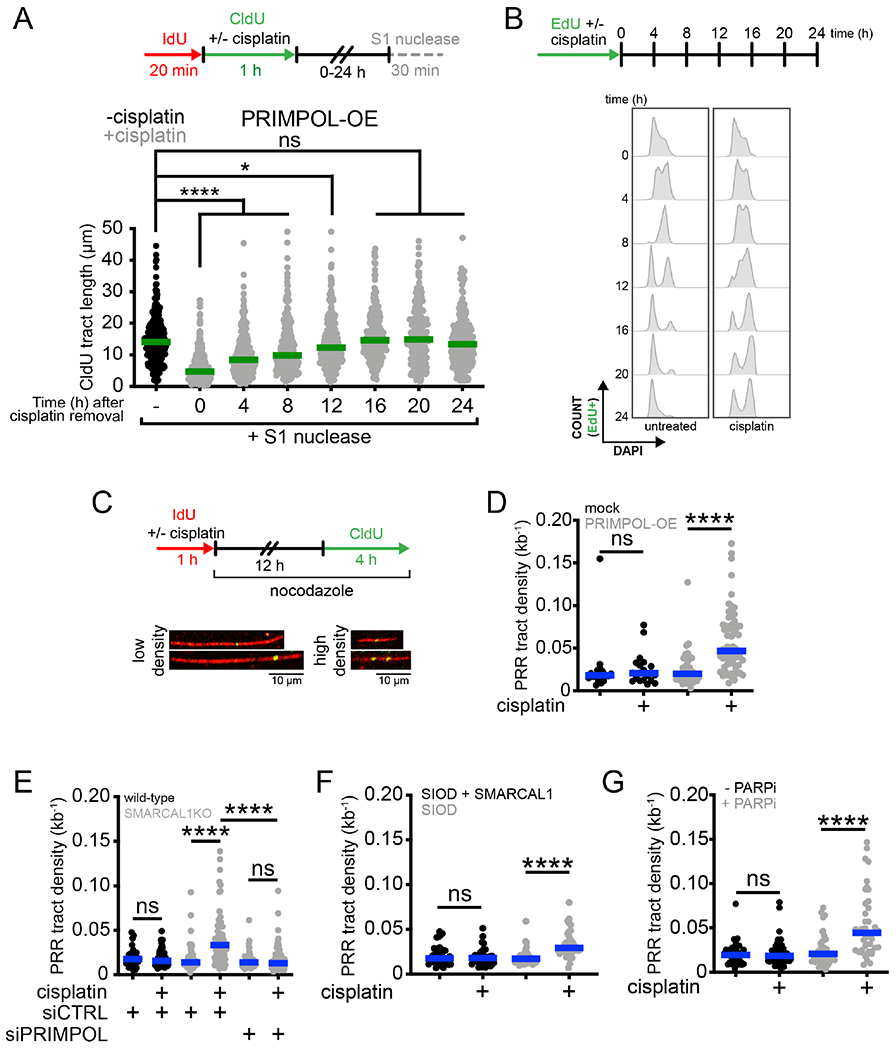
(A) Top, schematic of the DNA fiber assay with the S1 nuclease to determine the kinetics of ssDNA gap repair. Bottom, dot plot and median of CldU tract lengths in PRIMPOL-OE (overexpressing) U2OS cells ± 150 μM cisplatin treated with the S1 nuclease at the indicated times (n=3). ns, non-significant, *p < 0.05, ****p < 0.0001. (B) Top, schematic for kinetic flow cytometry analysis of replicating cells ± 150 μM cisplatin. Bottom, cell cycle histograms (stained with DAPI to assess DNA content) of EdU-positive PRIMPOL-OE U2OS cells ± 150 μM cisplatin, collected at the indicated times. (C) Top, schematic for post-replication repair (PRR) assay. Bottom, representative images of high-density and low-density PRR tracts. (D) Dot plot and median of PRR densities in mock-transfected or PRIMPOL-OE U2OS cells ± 150 μM cisplatin (n=3). ns, non-significant, ****p < 0.0001. (E) Dot plot and median of PRR densities in wild-type or SMARCAL1KO U2OS cells ± 150 μM cisplatin ± siCTRL or siPRIMPOL (n=3). ns, non-significant, ****p < 0.0001. (F) Dot plot and median of PRR densities in SIOD cells or SIOD cells complemented with SMARCAL1 ± 150 μM cisplatin (n=3). ns, non-significant, ****p < 0.0001. (G) Dot plot and median of PRR densities in U2OS cells ± PARPi ± 150 μM cisplatin (n=3). ns, non-significant, ****p < 0.0001.
See also Figure S2.
To directly visualize and quantify gap-filling events, we designed a modified version of the DNA fiber technique, termed PRR assay (Daigaku et al., 2010; Quinet et al., 2017; Quinet et al., 2016). We labeled nascent DNA with IdU and concomitantly treated the PRIMPOL-OE cells with 150 μM cisplatin for 1 hour to induce ssDNA gaps (Figure 1). Next, we added nocodazole to arrest cells in G2/M and avoid entry into the next cell cycle (Figure S2D). During the last 4 hours of nocodazole treatment, we added CldU to the cell culture media so that it could be incorporated during gap filling (Figure 2C). Since PRIMPOL-OE cells treated with cisplatin reached G2 after 12 hours (Figure 2B), we added CldU between 12 and 16 hours. IdU tracts that contained at least one CldU dot were scored as gap-filling events (Figure 2C). Results were expressed as density of PRR tracts per kilobase of DNA, which corresponds to the total number of CldU dots identified on a single tract divided by the total length of the tract expressed in kilobases. We found that the median PRR density of cisplatin-treated PRIMPOL-OE cells was over 2-fold higher than the mock transfected control at 16 hours (Figure 2D), in agreement with our finding that ssDNA gaps are effectively repaired in G2 (Figures 2A and 2B). We then confirmed that gaps are efficiently filled at this time point in SMARCAL1KO cells, SIOD lymphocytes, and PARPi-treated cells (Figures 2E-G). Collectively, these studies demonstrate that ssDNA gaps generated by PRIMPOL are efficiently filled post-replicatively in S and G2 phases of the cell cycle.
RAD18 and REV1-POLζ mediate gap filling in the G2 phase.
Next, we sought to elucidate the molecular mechanism of gap filling in G2. Monoubiquitinated PCNA recruits TLS enzymes and allows for direct bypass of DNA damage via polymerase switching (Kannouche et al., 2004), while polyubiquitinated PCNA promotes TS mechanisms of damage bypass (Hoege et al., 2002; Xiao et al., 2000). To identify the pathway responsible for gap filling, we knocked down RAD18, an E3 ubiquitin ligase that monoubiquitinates PCNA at K164 (Bailly et al., 1994; Hoege et al., 2002; Watanabe et al., 2004), and UBC13, an E2 conjugating enzyme that can further polyubiquitinate PCNA (Eddins et al., 2006; Hofmann and Pickart, 1999; Parker and Ulrich, 2009) in PRIMPOL-OE cells (Figure S3A). Loss of RAD18 reduced gap-filling density to control levels, whereas UBC13 knockdown did not affect gap-filling proficiency (Figure 3A). Importantly, we confirmed that RAD18-depleted cells did not display cell cycle defects, and that gaps were still generated in these cells, demonstrating that the lack of gap filling was not a consequence of defective cell cycle progression or impaired gap formation (Figures S3B and S3C). The effect of RAD18 depletion on PRR proficiency was validated in SMARCAL1KO U2OS cells (Figures 3B and S3D). To further define the role of RAD18 in gap filling, we used HEK293T cells expressing a K164R mutated version of PCNA, which cannot be ubiquitinated by RAD18 (Thakar et al., 2020) (Figure S3E). While gaps were still efficiently generated in the K164R mutant HEK293T cells, gap filling was significantly impaired (Figures 3C and S3F), suggesting that PCNA ubiquitination, along with RAD18, is required for PRR in G2.
Figure 3. Gap filling in G2 is dependent on RAD18, PCNA monoubiquitination and REV1-POLζ.
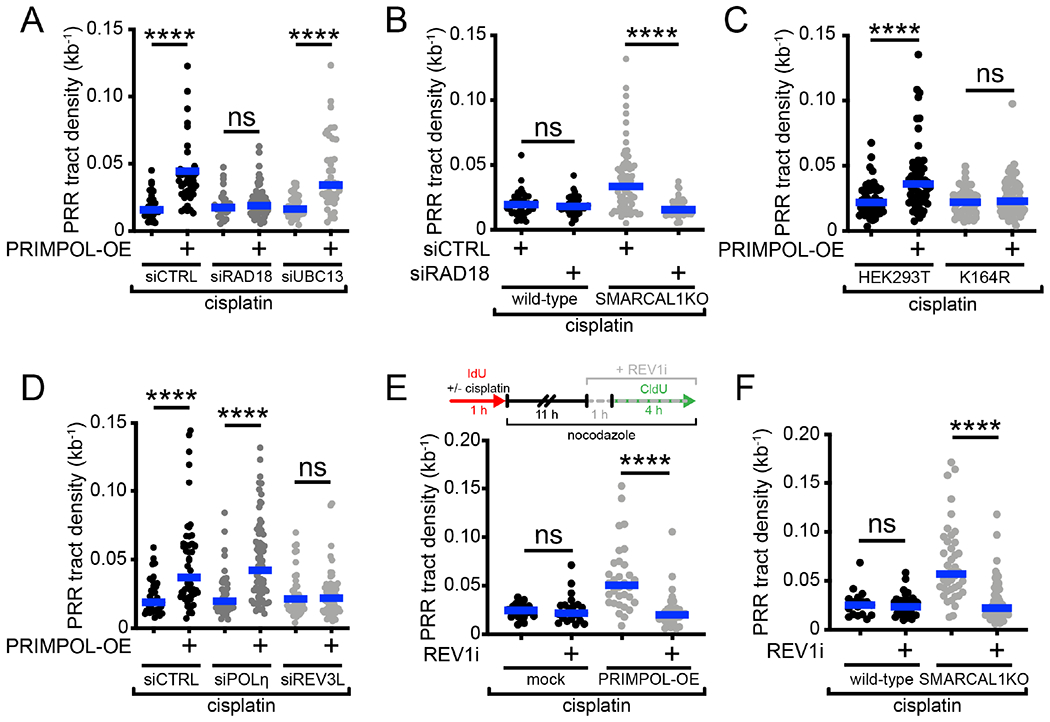
(A) Dot plot and median of PRR densities in wild-type or PRIMPOL-OE U2OS cells + 150 μM cisplatin ± siCTRL or siRAD18 or siUBC13 (n=3). ns, non-significant, ****p < 0.0001. (B) Dot plot and median of PRR densities in wild-type or SMARCAL1KO U2OS cells + 150 μM cisplatin ± siCTRL or siRAD18 (n=3). ns, non-significant, ****p < 0.0001. (C) Dot plot and median of PRR densities in wild-type HEK293T cells or HEK293T cells expressing the PCNA K164R mutant ± PRIMPOL-OE + 150 μM cisplatin (n=3). ns, non-significant, ****p < 0.0001. (D) Dot plot and median of PRR densities in wild-type or PRIMPOL-OE U2OS cells + 150 μM cisplatin ± siCTRL or siPOLηi or siREV3L (n=3). ns, non-significant, ****p < 0.0001. (E) Top, schematic of PRR assay treated with REV1 inhibitor (REVi, JH-RE-06). Bottom, dot plot and median of PRR densities in wild-type or PRIMPOL-OE U2OS cells + 150 μM cisplatin ± 2 μM REV1i (n=3). ns, non-significant, ****p < 0.0001. (F) Dot plot and median of PRR densities in wild-type or SMARCAL1KO U2OS + 150 μM cisplatin ± 2 μM REV1i (n=3). ns, non-significant, ****p < 0.0001.
See also Figure S3.
Next, we asked which TLS enzyme(s) is required for gap filling in G2 downstream of PCNA monoubiquitination. To this end, we tested two TLS polymerases, POLη and POLζ, previously shown to bypass platinum lesions (Hicks et al., 2010). POLη is a Y-family TLS polymerase (Prakash et al., 2005), whereas POLζ is a B-family TLS polymerase composed of a catalytic subunit (REV3L) and an accessory subunit (REV7), which can interact with monoubiquitinated PCNA (Lawrence and Hinkle, 1996). Gap filling was impaired by depletion of REV3L, but not POLη (Figures 3D, S3G and S3H). Again, we confirmed that this was not a consequence of defective cell cycle progression or impaired gap generation (Figures S3I and S3J). We next investigated the role of the REV1-POLζ interaction in this process. REV1 is a Y-family TLS polymerase that acts as a scaffold polymerase, binding to PCNA, DNA, and other TLS polymerases including POLζ (Ohmori et al., 2009; Vaisman and Woodgate, 2017). Treatment with the REV1 inhibitor, JH-RE-06, that disrupts the interaction between REV1 and POLζ (Wojtaszek et al., 2019) significantly abrogated gap filling in PRIMPOL-OE and SMARCAL1KO cells treated with cisplatin (Figures 3E and 3F). Collectively, our data suggest that PCNA monoubiquitination by RAD18 promotes ssDNA gaps filling by REV1-POLζ in G2 phase. Of note, we found that RAD18 is not required for gap filling following HU treatment, suggesting that cells utilize different gap filling pathways depending on the type of replication challenging agent (Figure S3K).
To study the recruitment of these factors to ssDNA gaps during gap filling, we employed a modified in situ proximity ligation assay (PLA) (Petruk et al., 2012; Soderberg et al., 2006). We treated wild-type and SMARCAL1KO U2OS cells with cisplatin for 1 hour, then placed them in nocodazole for 24 hours to arrest cells in G2/M. For the final hour of nocodazole treatment, EdU was added to the cell culture media to label gap filling events (Figures 4A and S4A). To limit our analysis to PRR events in G2, we only considered PLA events in cells positive for cyclin B1, a G2 marker (Hunter and Pines, 1994; Pines and Hunter, 1989). As a control, we found that preventing gap filling by RAD18 depletion or REV1 inhibition decreased EdU foci in G2-arrested SMARCAL1KO cells, confirming that EdU signal at this time point is an adequate proxy for gap filling (Figure S4B and S4C). Next, we monitored the interaction between RAD18 and EdU-labeled PRR events by PLA. As expected, we found an increased EdU-RAD18 PLA signal in SMARCAL1KO cells treated with cisplatin (Figures 4A and 4B). We obtained similar results in PLA experiments using antibodies against REV1 and EdU (Figures 4C and S4D), indicating that both RAD18 and REV1 are present at gaps during gap-filling events in G2. However, we did not observe a decrease in EdU foci following UBC13 depletion (Figures S4E and S4F) or an increase in PLA signal when probing for an interaction between UBC13 and EdU in cyclin B1-positive cells (Figures 4D and S4G), supporting our finding that UBC13 is not required for gap filling in G2.
Figure 4. REV1-POLζ, UBC13, and RAD51 mediate gap filling in S phase.
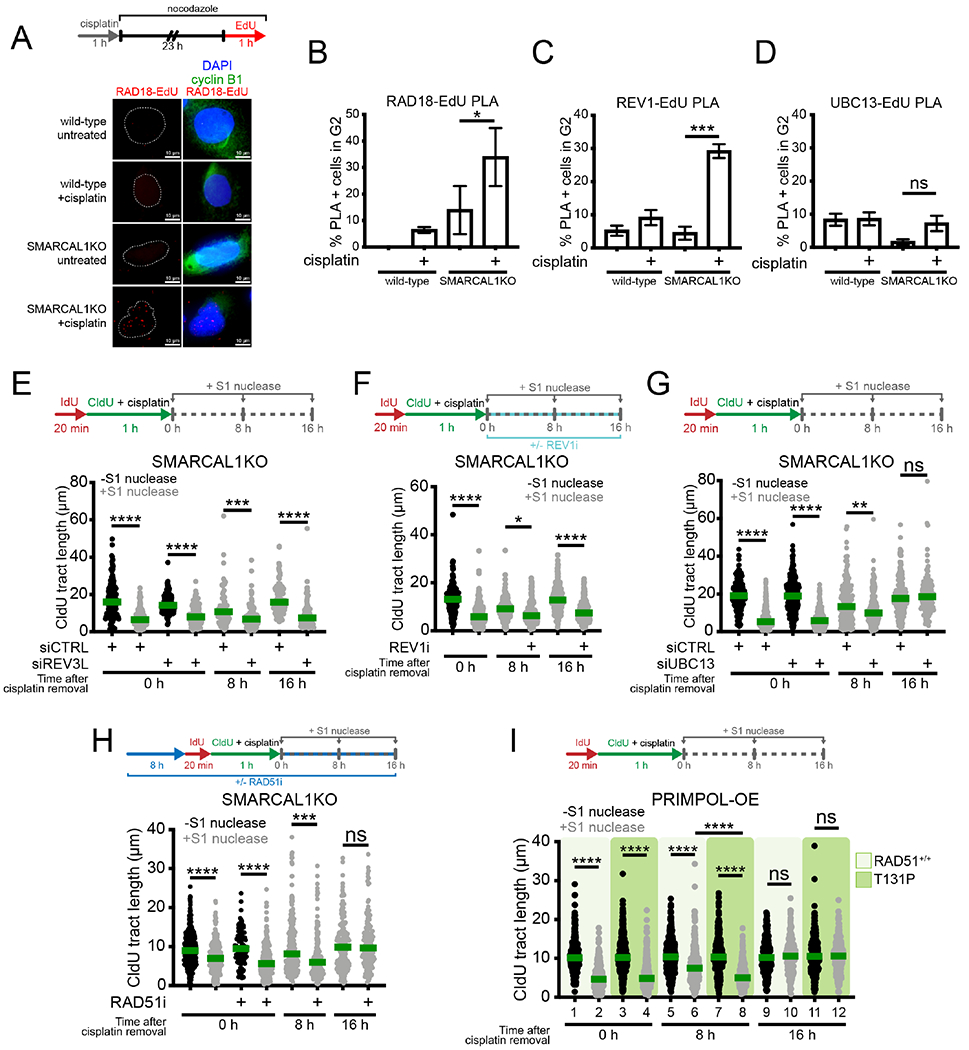
(A) Top, schematic of PLA assay. Bottom, representative images of RAD18-EdU PLA foci (red) wild-type and SMARCAL1KO U2OS cells ± 150 μM cisplatin. Cyclin B1 is used as a marker for G2. White dashes outline nuclear contours as detected by DAPI staining. (B) Percentage of cells in G2 (cyclin B1+) with >3 RAD18-EdU PLA foci (mean ± SEM) in wild-type and SMARCAL1KO U2OS cells (n=5). *p < 0.05. (C) Percentage of cells in G2 with >3 REV1-EdU PLA foci (mean ± SEM) in wild-type and SMARCAL1KO U2OS cells (n=3). ***p < 0.001. (D) Percentage of cells in G2 with >3 UBC13-EdU PLA foci (mean ± SEM) in wild-type and SMARCAL1KO U2OS cells (n=3). ns, non-significant. (E) Top, schematic of DNA fiber with S1 nuclease assay at the indicated times after cisplatin treatment. Bottom, dot plot and median of CldU tract lengths in SMARCAL1KO U2PS cells ± siCTRL or siREV3L, ± S1 nuclease at 0 hours or + S1 at 8 and 16 hours post a 1 hour treatment with 150 μM cisplatin (n=2). ***p < 0.001, ****p < 0.0001. (F) Top, schematic of DNA fiber with S1 nuclease assay with REV1 inhibitor (REV1i, JH-RE-06). Bottom, dot plot and median of CldU tract lengths in SMARCAL1KO U2OS cells ± 2 μM REV1i, ± S1 nuclease at 0 hours or + S1 at 8 and 16 hours post a 1 hour treatment with 150 μM cisplatin (n=2). *p < 0.05, ****p < 0.0001. (G) Top, schematic of DNA fiber with S1 nuclease assay at the indicated times after cisplatin treatment. Bottom, dot plot and median of CldU tract lengths in SMARCAL1 KO U2OS cells ± siCTRL or siUBC13, ± S1 nuclease at 0 hours or + S1 at 8 and 16 hours post a 1 hour treatment with 150 μM cisplatin (n=3). ns, non-significant, **p < 0.01, ****p < 0.0001. (H) Top, schematic of DNA fiber with S1 nuclease assay with RAD51 inhibitor (RAD51i, B02). Bottom, dot plot and median of CldU tract lengths in SMARCAL1KO U2OS cells ± 27 μM RAD51i, ± S1 nuclease at 0 hours or + S1 at 8 and 16 hours post a 1 hour treatment with 150 μM cisplatin (n=2). ns, non-significant, ***p < 0.001, ****p < 0.0001. (I) Top, schematic of DNA fiber with S1 nuclease assay at the indicated times after cisplatin treatment. Bottom, dot plot and median of CldU tract lengths in PRIMPOL-OE RAD51+/+ and RAD51 T131P cells ± S1 nuclease at 0, 8 and 16 hours post a 1 hour treatment with 150 μM cisplatin (n=3). ns, non-significant, ****p < 0.0001.
See also Figure S4.
UBC13, RAD51 and REV1-POLζ mediate gap filling in the S phase.
The PRR assay cannot be applied to study gap filling in S phase because the CldU signal originating from gap filling cannot be distinguished from CldU incorporation on replicating DNA. As an alternative strategy to study gap-filling activity in S phase, we employed the S1 DNA fiber assay and reasoned that depletion of factors involved in gap filling should lead to tract shortening upon S1 cleavage. Thus, we repeated our kinetic DNA fiber assay by adding the S1 nuclease at 0, 8, and 16 hours after cisplatin treatment. Based on our cell cycle progression data (Figures 2A and 2B), gap filling in S phase is measurable at 8 hours after cisplatin treatment via S1 nuclease DNA fiber. We found that REV3L depletion and REV1 inhibition induced S1-dependent tract shortening at both 8 and 16 hours (Figures 4E and 4F). These results further confirm that REV1-POLζ fills PRIMPOL-generated gaps in G2 (16 hours) and indicate that the same polymerases might also play a role in PRR during the S phase (8 hours). On the other hand, we observed that depletion of UBC13 or inactivation of RAD51 with the B02 inhibitor (Huang et al., 2011) led to tract shortening only at the 8 hour time point (Figures 4G and 4H). Similar results were obtained using patient fibroblasts expressing the RAD51-T131P mutant that destabilizes RAD51 nucleofilament formation (Figures 4I and S4I) (Wang et al., 2015). This suggests that an additional mechanism dependent on UBC13 and RAD51 contributes to gap filling in S, but not in G2. This conclusion is also supported by the PRR results showing that loss of UBC13 or inactivation of RAD51 does not affect gap filling in G2 (Figures 3A and S4H).
BRCA proteins promote gap filling by limiting MRE11 activity.
On the basis of studies suggesting that loss of BRCA1 promotes ssDNA gap accumulation (Cong et al., 2021; Lim et al., 2018; Panzarino et al., 2020), we sought to investigate the function of BRCA proteins in gap filling. We found that BRCA1 depletion prevents PRR in PRIMPOL-OE U2OS cells, even when using high cisplatin concentrations (Figures 5A and S5A). Importantly, BRCA1 depletion did not affect cell cycle progression to G2 (Figure S5B). The same results were recapitulated in the BRCA1-null ovarian cancer cell line (UWB1.289; named UW here) overexpressing PRIMPOL or lacking SMARCAL1 (Figures 5B and 5C). Of note, we did not detect any defect in gap generation using the S1 DNA fiber assay because S1 treatment led to tract shortening in both BRCA1-depleted PRIMPOL-OE cells (Figure S5C) and SMARCAL1-depleted UW cells (Figure S5D). Moreover, we obtained similar results in BRCA2-null ovarian cancer cells (PEO1), suggesting that loss of either BRCA1 or BRCA2 compromises gap filling in G2 (Figures S5E-S5G).
Figure 5. BRCA1 promotes gap filling by limiting MRE11 activity.
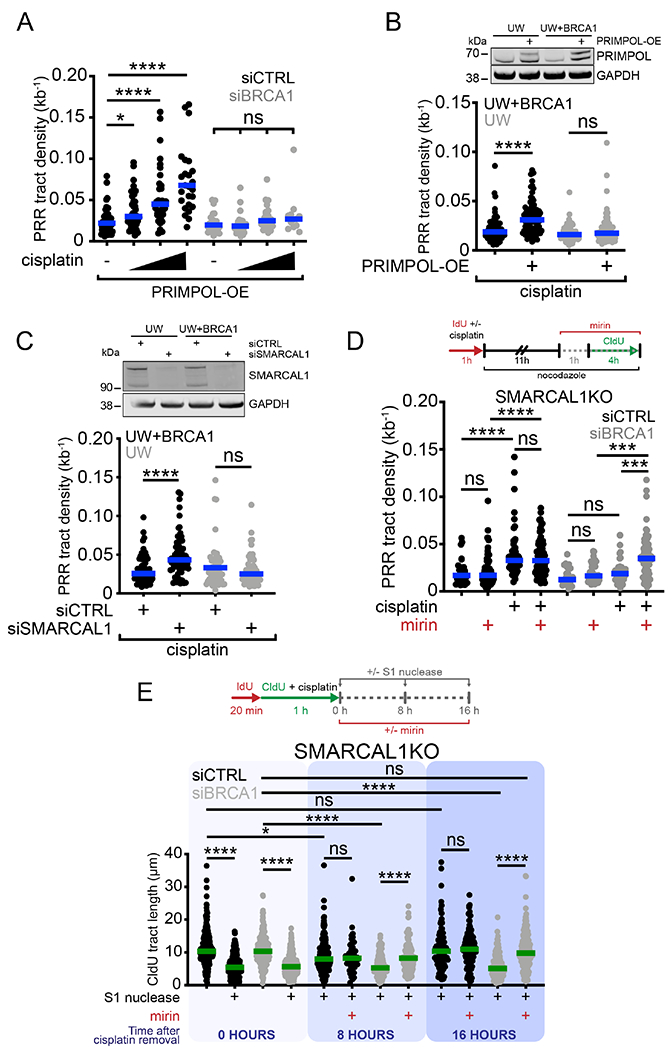
(A) Dot plot and median of PRR densities in PRIMPOL-OE U2OS cells treated with increasing concentrations of cisplatin (150, 300, and 600 μM) ± siCTRL or siBRCA1 (n=3). ns, non-significant, *p < 0.05, ****p < 0.0001. (B) Top, expression of PRIMPOL after transfection of V5-tagged PRIMPOL in UW and UW+BRCA1 cells. Bottom, dot plot and median of PRR densities in UW and UW+BRCA1 cells ± PRIMPOL-OE + 150 μM cisplatin (n=3). ns, non-significant, ****p < 0.0001. (C) Top, expression of SMARCAL1 after siRNA (siCTRL or siSMARCAL1) knockdown in UW and UW+BRCA1 cells. Bottom, dot plot and median of PRR densities in UW and UW+BRCA1 cells ± siCTRL or siSMARCAL1 + 150 μM cisplatin (n=3). ns, non-significant, ****p < 0.0001. (D) Top, schematic of PRR assay with MRE11 inhibitor (mirin). Bottom, dot plot and median of PRR densities in SMARCAL1KO U2OS cells ± 150 μM cisplatin ± siCTRL or siBRCA1 ± 50 μM mirin (n=3). ns, non-significant, ***p < 0.001, ****p < 0.0001. (E) Top, schematic of kinetic fiber with S1 nuclease and MRE11 inhibitor (mirin). Bottom, dot plot and median of CldU tract lengths in SMARCAL1KO cells ± siCTRL or siBRCA1 ± 50 μM mirin ± S1 nuclease at 0 or + S1 at 8 and 16 hours post a 1 hour treatment with 150 μM cisplatin. ns, non-significant, *p < 0.05, ****p < 0.0001.
See also Figure S5.
Homologous recombination factors have well-established roles in protecting reversed replication forks from MRE11-dependent degradation (Kolinjivadi et al., 2017; Lemaçon et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017). We hypothesized that BRCA proteins might be also required to protect ssDNA gaps from aberrant MRE11 processing. To test this hypothesis, we repeated the PRR assay by adding mirin, an MRE11 inhibitor (Dupre et al., 2008), when gaps are filled with CldU in G2 (see scheme Figure 5D). MRE11 inhibition rescued the defect in gap filling observed in BRCA1-depleted SMARCAL1KO cells (Figures 5D, S5H and S5I). Analogous results were obtained in BRCA2-depleted SMARCAL1KO cells (Figure S5J and S5K). This suggests that BRCA1 and BRCA2 limit MRE11 activity and that, in their absence, extensive MRE11-dependent degradation of ssDNA gaps might prevent efficient gap filling in G2.
To test whether BRCA proteins are also required for gap filling in S phase, we treated SMARCAL1KO BRCA1- or BRCA2-depleted cells with the S1 nuclease at 0, 8 or 16 hours after CldU incorporation. First, we confirmed the presence of ssDNA gaps in BRCA-proficient and – deficient cells at 0 hours by adding the S1 nuclease immediately after CldU labeling (Figures 5E and S5L). Next, we found that S1 treatment at 8 and 16 hours after CldU labeling still generated shorter tracts in BRCA-deficient cells relative to BRCA-proficient cells (Figures 5E and S5L), suggesting that BRCA1 and BRCA2 facilitate gap filling in both S and G2 phases of the cell cycle. Addition of mirin at 8 and 16 hours after gap generation rescued tract shortening, suggesting that BRCA1 and BRCA2 promote gap filling by limiting MRE11 activity both in S and G2 (Figures 5E and S5L).
Preventing gap filling promotes genome instability and affects cell survival.
Given the emerging impact of ssDNA gap accumulation on genome integrity and response to chemotherapy (Bai et al., 2020; Cong et al., 2021; Lim et al., 2018; Panzarino et al., 2020), we aimed to determine the consequences of impaired gap filling on genome stability. To prevent gap filling, we depleted REV3L, BRCA1, or BRCA2. We found that depletion of REV3L leads to a more pronounced accumulation of DSBs and G1-specific 53BP1 nuclear bodies 48 hours after cisplatin treatment (5 μM) in SMARCAL1KO compared to wild-type U2OS cells (Figures 6A-D). BRCA1 depletion also led to a significant accumulation of DSBs and 53BP1 nuclear bodies, and this effect was more marked when gap filling was prevented by BRCA1 depletion in SMARCAL1KO cells (Figures 6E and 6F). Similar results were obtained by depleting BRCA2 (Figure S6A). Collectively, these results show that preventing gap filling by either inactivating the REV1-POLζ pathway or by depleting BRCA1 or BRCA2 leads to increased DNA damage and genome instability. Furthermore, combination of BRCA1 deficiency with the REV1 inhibitor, JH-RE-06, further increased G1-specific 53BP1 nuclear bodies in SMARCAL1KO U2OS cells (Figures 6G and S6B).
Figure 6. Inhibition of gap filling leads to DNA double-stranded breaks and G1-specifc 53BP1 nuclear bodies.
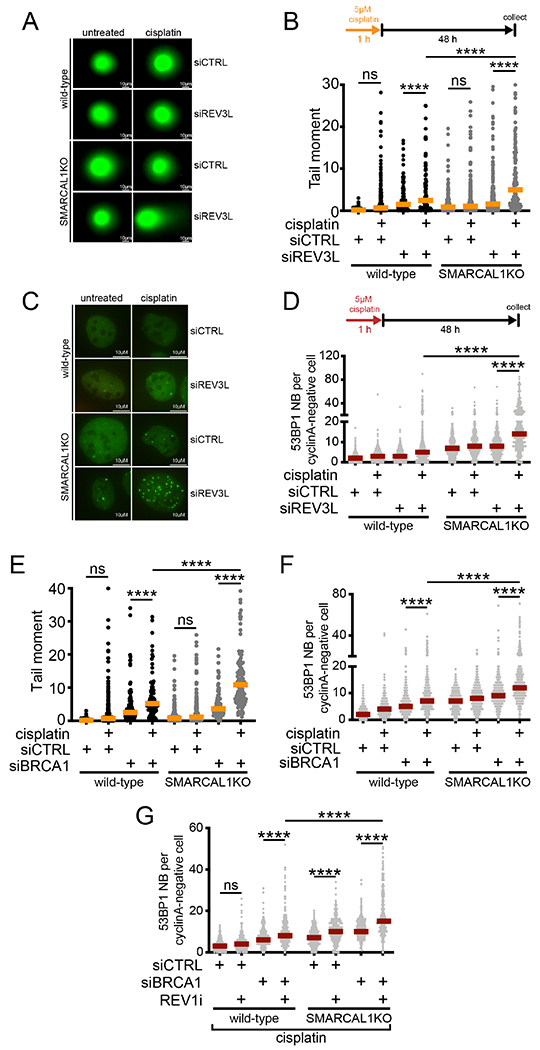
(A) Representative images of comets in wild-type and SMARCAL1KO U2OS cells ± siCTRL or siREV3L ± 5 μM cisplatin. (B) Top, schematic of comet assay. Bottom, dot plot and median of tail moments in wild-type and SMARCAL1KO U2OS cells ± siCTRL or siREV3L ± 5 μM cisplatin (n=3). ns, non-significant, ****p < 0.0001. (C) Representative images of 53BP1 nuclear bodies in G1 in wild-type and SMARCAL1KO U2OS cells ± siCTRL or siREV3L ± 5 μM cisplatin. (D) Top, schematic of experiment. Bottom, dot plot and median of G1-specific 53BP1 bodies in wild-type and SMARCAL1KO U2OS cells ± siCTRL or siREV3L ± 5 μM cisplatin (n=3). ****p < 0.0001. (E) Dot plot and median of tail moments in wild-type and SMARCAL1KO U2OS cells ± siCTRL or siBRCA1 ± 5 μM cisplatin (n=2). ns, non-significant, ****p<0.0001. (F) Dot plot and median of G1-specific 53BP1 bodies in wild-type and SMARCAL1 KO U2OS cells ± siCTRL or siBRCA1 ± 5 μM cisplatin (n=3). ****p < 0.0001. (G) Dot plot and median of G1-specific 53BP1 bodies in wild-type and SMARCAL1KO U2OS cells ± siCTRL or siBRCA1 ± 2 μM REV1i + 5 μM cisplatin (n=2). ns, non-significant, ****p < 0.0001.
See also Figure S6.
Next, we investigated the effect of impaired gap filling on cell survival. We found that depleting BRCA1 or REV3L sensitizes U2OS cells to cisplatin treatment, and that this effect is more marked in SMARCAL1KO cells, mirroring the results obtained from the 53BP1 body analysis (Figures 7A and 7B). To validate these findings, we extended our analysis to the BRCA1-mutant UW ovarian cancer cells treated with either PARPi, REV1i, or cisplatin alone or using different combinations of these compounds (Figure 7C). In agreement with previous findings, the BRCA1-mutant UW cells are more sensitive to treatment with PARPi or cisplatin alone compared to the UW+BRCA1 cells expressing wild-type BRCA1 (lanes 1, 3, 4 and 9, 11, 12). The UW cells were also moderately sensitive to the REV1 inhibitor JH-RE-06 alone (lanes 1, 2 and 9, 10), suggesting that these cells require a functional TLS pathway. Interestingly, a combinatorial treatment with PARP and REV1 inhibitors further compromised cell survival in the UW, but not in the UW+BRCA1 cells (lanes 2, 5 and 10, 13). This effect was exacerbated by the addition of cisplatin to a greater extent in the UW cells relative to the UW+BRCA1 cells (lanes 4, 8 and 12, 16). Similar results were obtained using the BRCA2-mutant PEO1 ovarian cancer cells, even though these cells were not sensitive to treatment with the JH-RE-06 inhibitor alone, at least at the concentration used in this study (Figure S6C). Collectively, these results suggest that inactivating fork reversal and concomitantly preventing ssDNA gap repair leads to increased cellular sensitivity to DNA-damaging drugs in different cellular models.
Figure 7. Inhibition of gap filling negatively affects cell survival.
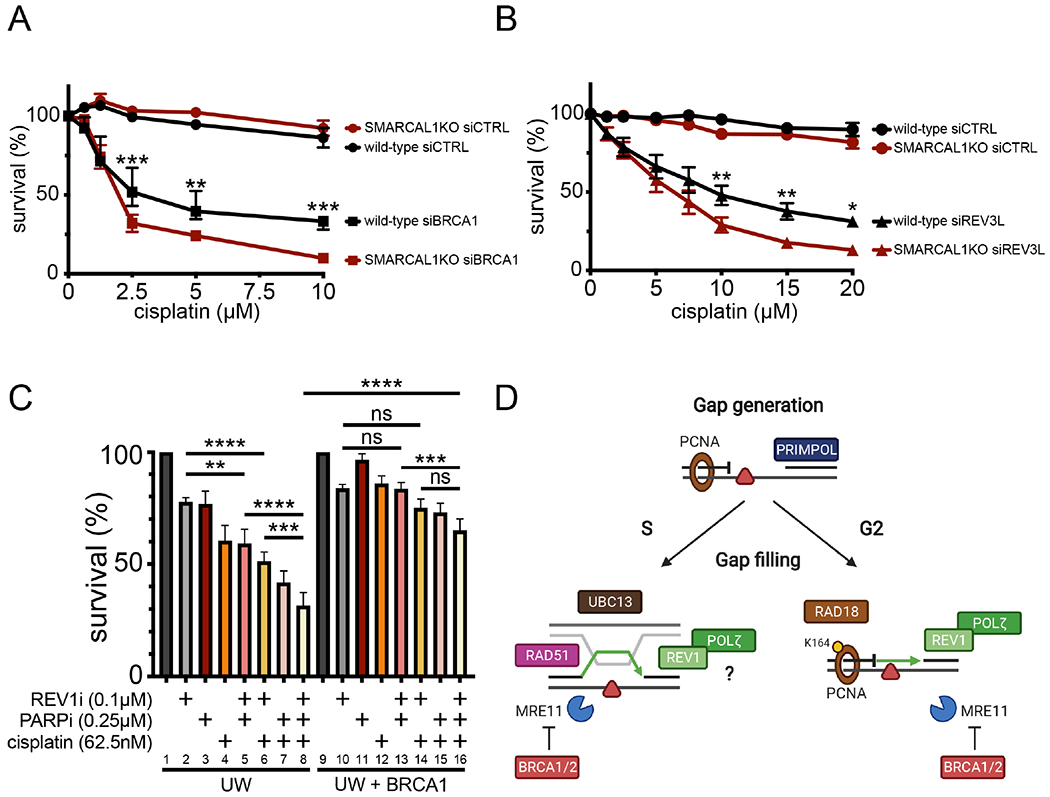
(A) Cell survival of wild-type and SMARCAL1KO U2OS cells ± siCTRL or siBRCA1 6 days after acute (1 hour) treatment with the indicated doses of cisplatin (means ± SEM) (n=3). **p < 0.01, ***p < 0.001. (B) Cell survival of wild-type and SMARCAL1KO U2OS cells ± siCTRL or siREV3L 6 days after acute (1 hour) treatment with the indicated doses of cisplatin (means ± SEM) (n=5). *p < 0.05, **p < 0.01. (C) Cell survival of UW and UW+BRCA1 cells ± indicated chronic doses (6 days) of PARPi (Olaparib), cisplatin, and REV1i (JH-RE-06) (n=3). ns, non-significant, **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) Working model for the mechanisms of gap filling following PRIMPOL repriming. In G2, RAD18 promotes PCNA monoubiquitination at K164, which, in turn, recruits the REV1-POLζ complex to fill gaps by TLS. UBC13 and RAD51 are instead required for gap filling in S suggesting that these factors mediate a TS mechanism to fill gaps in S. REV1 and POLζ are also required for gap filling in S phase, suggesting that they either participate in the same pathway or that TLS is active both in S and G2 phase. BRCA1 and BRCA2 protect ssDNA gaps from aberrant MRE11 activity in both S and G2 phase.
See also Figure S6.
Discussion
ssDNA gaps are frequent structures that accumulate on newly synthesized DNA under conditions of replication stress. These gaps need to be adequately processed and filled to preserve genome stability. Here, we implemented an approach to directly monitor gap filling under three different conditions that promote ssDNA gap accumulation upon cisplatin treatment: 1) PRIMPOL overexpression; 2) SMARCAL1 inactivation; and 3) PARP inhibition. Our findings indicate that human cells primarily employ a TLS mechanism dependent on the E3 ubiquitin ligase RAD18, PCNA monoubiquitination, and the REV1-POLζ polymerases to fill gaps in G2 under these conditions. An alternative pathway dependent on the E2 conjugating enzyme UBC13, the RAD51 recombinase, and REV1-POLζ support gap filling in S but not in G2, suggesting that distinct pathways of PRR operate throughout the cell cycle. Moreover, efficient gap filling requires the breast cancer susceptibility proteins BRCA1 and BRCA2 to limit MRE11 activity.
Repriming is emerging as a central mechanism that allows replication forks to skip DNA lesions with a minimal effect on fork elongation (Bai et al., 2020; Mourón et al., 2013; Quinet et al., 2020). How cells choose between repriming and alternative DNA replication stress response pathways is still a subject of investigation. We found that repriming is activated in response to different kinds of replication challenges under conditions where PRIMPOL expression is increased, replication fork reversal is inactivated upon loss of the SMARCAL1 translocase, or the accumulation of reversed replication forks is compromised because of PARP inhibition. Our finding that repriming is activated upon treatment with cisplatin, UV-C, and low doses of HU, which does not induce DNA damage, suggest that repriming is a general mechanism to deal with replication stress even when replication forks do not face DNA lesions. Along the same line, a recent study showed that loss of another fork remodeler HLTF suppresses fork reversal and promotes PRIMPOL repriming upon treatment with HU (Bai et al., 2020). Mutations in SMARCAL1 can lead to SIOD, a multi-system disorder characterized by growth defects, immune deficiencies, renal failure and other complex phenotypes (Boerkoel et al., 2000). Our finding that repriming is activated in SIOD lymphocytes containing a splicing mutation in SMARCAL1 suggests that the replication-associated DNA damage previously detected in these cells (Bansbach et al., 2010) could be related to the hyperactivation of repriming. Moreover, our finding that PRIMPOL repriming becomes a predominant pathway to deal with DNA lesions upon PARP inhibition has important therapeutic implications as PARP inhibitors are widely used for cancer treatment (Ashworth and Lord, 2018; D’Andrea, 2018; Pilie et al., 2019).
The impact of employing repriming versus other replication stress response pathways on genome stability and cell survival largely relies on the ability of cells to repair the PRIMPOL-dependent ssDNA gaps. Coupling the S1 nuclease assay to cell cycle analysis we found that the PRIMPOL-dependent ssDNA gaps start to be repaired in S and that repair is completed in G2. Next, we used an ad hoc PRR assay to define the molecular steps that promote gap filling in G2 in human cells. Our findings show that human cells utilize a TLS pathway dependent on the E3 ubiquitin ligase RAD18, PCNA monoubiquitination, and the REV1-POLζ polymerases to fill gaps formed upon cisplatin treatment in G2. Previous studies identified that REV1, POLη, and POLζ are required to bypass platinum lesions at replication forks (Hicks et al., 2010). Our data show that cisplatin lesions can be skipped by repriming and that REV1 and POLζ, but not POLη, fill the ssDNA gaps containing cisplatin monoadducts behind replication forks. These results agree with previous findings suggesting that cells might utilize different TLS polymerases at the replication fork versus behind the fork to deal with the same type of lesion (Quinet et al., 2016). PCNA ubiquitination was shown to be required for post-replicative ssDNA gap repair in budding yeast (Daigaku et al., 2010; Karras and Jentsch, 2010), but its role in gap filling in higher eukaryotes remained controversial. In particular, expression of the ubiquitination-deficient K164R-PCNA mutant seems to compromise gap filling in avian DT-40 cells, but not in mouse embryonic fibroblasts in response to UV-C irradiation (Edmunds et al., 2008; Temviriyanukul et al., 2012). In our study, we used human HEK293T cells harboring the K164R-PCNA mutation and found that PCNA ubiquitination is required for gap filling, at least in G2. Moreover, our finding that the E3 ubiquitin ligase RAD18, which promotes PCNA monoubiquitination, but not the E2 conjugating enzyme UBC13, which promotes PCNA polyubiquitination, is required for gap filling in G2 suggests that this process is mediated by PCNA monoubiquitination in the G2 phase.
We found that UBC13 and RAD51 are instead required for gap filling in the S phase of the cell cycle. On the basis of these findings, we propose that a TS pathway mediated by UBC13 and RAD51 promotes gap filling in the S phase. The concept that the cell cycle phase defines the choice between different gap-filling pathways is supported by studies in yeast showing that loss of PCNA polyubiquitination by depletion of UBC13 reduces the formation of sister chromatid junctions, which are associated with TS-mediated repair of DNA damage in S phase (Karras et al., 2013). In this context, RAD51 would facilitate template switching between sister chromatids in S phase (Liberi et al., 2005; Vanoli et al., 2010). This model is supported by a recent report describing a RAD51-dependent HR pathway to repair PRIMPOL-dependent ssDNA gaps in human cells (Piberger et al., 2020). Moreover, an earlier study suggested that RAD51 is required for HR-dependent gap filling using a gapped plasmid repair assay to directly assess gap filling in mouse and human cells (Adar et al., 2009). However, defining the role of RAD51 in gap filling is complicated by the lack of direct methodologies to investigate homology-mediated mechanisms that do not necessarily involve strand transfer and by the multiple functions of RAD51 during the replication stress response. For example, RAD51 is required for both reversed fork formation and protection (Hashimoto et al., 2010; Kolinjivadi et al., 2017; Malacaria et al., 2019; Zellweger et al., 2015), and might have a mechanistically-ill-defined role during break-induced replication (Kramara et al., 2018). A proper analysis of the function of RAD51 in gap filling would require the development of separation of function mutants that selectively target its other replication stress response functions without affecting its role in gap filling, as well as of new methodologies to directly monitor homology-mediated mechanisms in S phase. In addition to UBC13 and RAD51, we found that REV1 and POLζ are required for gap filling in S phase. This finding suggests either that TLS is active both in the S and G2 phases, or that REV1-POLζ is also required for the UBC13-mediated pathway in S phase. Indeed, earlier studies showed that the REV1 and POLζ polymerases are required for HR-repair (Okada et al., 2005; Sharma et al., 2012; Sonoda et al., 2003), suggesting that these polymerases might have additional functions in TS or HR-mediated gap filling and that there might be a cross-talk between factors involved in TLS and TS/HR.
Recent studies suggested that loss of BRCA proteins promotes ssDNA gap accumulation upon PARP inhibition or HU treatment (Cong et al., 2021; Lim et al., 2018; Panzarino et al., 2020). Moreover, we showed that the gaps that form in BRCA1-deficient cells challenged with platinum-based compounds are PRIMPOL-dependent (Quinet et al., 2020). However, the mechanistic basis for the PRIMPOL-dependent ssDNA gap accumulation in BRCA-deficient cells remained unclear. Here, we found that inhibiting MRE11 nuclease activity restores gap filling in BRCA-deficient cells. On the basis of these findings, we propose that MRE11 activity plays a crucial role in gap processing and that deregulated resection by MRE11 in BRCA-deficient cells promotes accumulation of unresolved ssDNA gaps.
Our results provide new insight into the function of BRCA1 and BRCA2 in the DNA replication stress response by suggesting that BRCA proteins regulate resection of ssDNA gaps, in addition to protecting reversed forks (Kolinjivadi et al., 2017; Lemaçon et al., 2017; Mijic et al., 2017; Taglialatela et al., 2017). We show that BRCA proteins are required for efficient gap filling both in S and G2. Conversely, RAD51 inactivation suppresses gap filling in S but not in G2. This finding opens the intriguing scenario that the mechanism by which BRCA proteins protect gaps from resection in G2 is different from the mechanism by which they protect replication intermediates in S phase, which was previously suggested to require RAD51 binding (Hashimoto et al., 2010; Kolinjivadi et al., 2017; Mijic et al., 2017). A possible model is that BRCA2 facilitates BRCA1 recruitment, which in turn directly inhibits MRE11 activity, as suggested by in vitro studies (Paull et al., 2001). Moreover, MRE11 interacts with the BRCA1-CtIP complex after DNA damage (Greenberg et al., 2006), and this complex is enriched in G2 (Chen et al., 2008; Yu and Chen, 2004). Further work would be necessary to prove that this interaction is indeed important to limit MRE11 activity in G2.
The idea that ssDNA gaps become toxic intermediates if they are not repaired in a timely fashion is supported by recent studies suggesting that ssDNA gap accumulation correlates with chemotherapy response in BRCA-deficient cells (Cong et al., 2021; Lim et al., 2018; Panzarino et al., 2020). In agreement with this data, we found that preventing gap filling leads to increased cell sensitivity to cisplatin and genomic instability. Our findings provide further support to the model that, in addition to the previously reported HR and fork protection defects, ssDNA gap accumulation contributes to increased sensitivity of BRCA-deficient cells to treatment with Olaparib or cisplatin, relative to BRCA-proficient cells. Moreover, our data suggest that inactivating fork reversal by SMARCAL1 depletion or PARP inhibition and concomitantly preventing ssDNA gap repair by loss of BRCA1 (or BRCA2) and REV1 inhibition leads to significantly increased sensitivity to DNA-damaging drugs. These findings open new avenues toward the design of novel combinatorial treatments for BRCA-deficient tumors that simultaneously target the fork reversal and gap-filling pathways.
Limitations of Study
Finally, this study has limitations. First, depletion of SMARCAL1 or treatment with PARPi could have other effects beyond suppressing replication fork reversal and promoting ssDNA gap accumulation. Second, the PRR assay cannot be applied to directly monitor gap filling in S phase because the signal originating from ssDNA gap filling cannot be distinguished from the signal originating from incorporation of the thymidine analogs on replicating DNA. Moreover, gap filling could be mediated by other polymerases or by different mechanisms in other cell types or when cells are challenged with agents different from cisplatin, as suggested by our experiments with HU. Regarding the role of MRE11 in gap processing, our observation that inhibition of MRE11 activity restores gap filling in BRCA1-deficient cells does not rule out the possibility that other nucleases contribute to gap processing in addition to MRE11. Rigorous studies would be required to establish how the size of the ssDNA gaps changes upon inhibition of different nucleases and how this change affects the efficiency of gap filling. Finally, further studies would be required to determine the impact of employing TS versus TLS pathways of gap filling on mutagenesis and genome stability.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Alessandro Vindigni (avindigni@wustl.edu).
Materials availability:
No new plasmids or cell lines were generated in this study. All plasmids and cell lines generated in the Vindigni laboratory are available upon request. Any plasmids or cell lines obtained from other sources should be requested from those investigators.
Data and code availability:
All data is available in the main text, supplemental materials, or via Mendeley at https://dx.doi.org/10.17632/2sc5bwyw6s.1.
The paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and cell lines
The human osteosarcoma U2OS cells (female) (American Type Culture Collection), the SMARCAL1 knocked-out (KO) U2OS cells (Liu et al., 2020) were grown in DMEM media supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C, 5% CO2. The patient Schimke Immunoosseous Dysplasia (SIOD) lymphocytes (female) transfected with an empty plasmid (pLPCX) and their complemented counterpart expressing wild-type SMARCAL1 (pDC1072) (provided by Dr. David Cortez, Vanderbilt University School of Medicine) (Carroll et al., 2015) were grown in DMEM media supplemented with 15% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 1.5 μg/mL puromycin at 37°C, 5% CO2. The RAD51+/+ and T131P-RAD51 mutant patient fibroblasts (female) (provided by Agata Smogorzewska, Rockefeller University) (Wang et al., 2015) were grown in DMEM media supplemented with 15% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, non-essential amino acids, and glutamax at 37°C in 5% O2. The BRCA1-mutant ovarian cancer cells UWB1.289 (female) (named UW for simplicity) and their complemented derivative expressing wild-type BRCA1, UWB1.289+BRCA1 (named UW+BRCA1 for simplicity) (provided by Dr. Lee Zou, Harvard Medical School) (Lemaçon et al., 2017; Yazinski et al., 2017), were cultivated in 50% RPMI media (ATCC, 30-2001), 50% MEGM bullet kit (Lonza CC-3150) supplemented with 3% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin (+ 400 μg/mL G418 (G8168, Millipore Sigma) in the case of UW+BRCA1) at 37°C, 5% CO2. The human embryonic kidney 293 cells (HEK293T) (American Type Culture Collection) and the HEK293T (female) expressing a K164R mutant of PCNA (provided by Dr. George-Lucian Moldovan, Penn State Cancer Institute) (Thakar et al., 2020) were grown in DMEM media supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C, 5% CO2. The human ovarian adenocarcinoma BRCA2-deficient PEO1 cells (female) and the BRCA2 proficient reversion cell line, C4-2 (provided by Dr. Sharon Cantor, University of Massachusetts Medical School) (Panzarino et al., 2020) were grown in DMEM media supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C, 5% CO2.
METHOD DETAILS
Gene silencing with RNAi and transfection of PRIMPOL constructs
Transient gene depletions were carried out using the Lipofectamine RNAiMax transfection reagent (13778-150, Thermo Fisher Scientific), according to the manufacturer’s instruction and the following siRNA at a final concentration of 50 nM: custom-made 5’-GAG GAA ACC GUU GUC CUC AGU GUA U-3’ (Dharmacon) for PRIMPOL (Vallerga et al. 2015; Quinet et al. 2020), custom-made 5’-GCC GGA UCU GAA AAA UAA C-3’ (Dharmacon) for RAD18 (Yoon et al., 2012), custom-made 5’-CCA GAU GAU CCA UUA GCA A-3’ (Dharmacon) for UBC13 (Zhao et al., 2007), SMARTpool siRNA for REV3L (M-006302-01, Dharmacon) (Quinet et al., 2016). POLη was depleted with 25nM SMARTpool siRNA (L-006454-01, Dharmacon) (Shachar et al., 2009), SMARCAL1 with 20 nM siGENOME individual siRNA (D-013058-04-02, Dharmacon) (Carvajal-Maldonado et al., 2019; Taglialatela et al., 2017), BRCA1 with 10 nM SMARTpool siRNA (L-003461-00, Dharmacon) (Tian et al. 2013; Lemaçon et al. 2017; Quinet et al 2020), and BRCA2 with 10 nM SMARTpool siRNA (L-003462-00, Dharmacon) (Carvajal-Maldonado et al., 2019; Lemaçon et al., 2017). Silencer select negative control #1 siRNA (4390843, Ambion) was used as control siRNA at the same concentration of the most concentrated siRNA used in the same experiment (Quinet et al., 2020).
Wild-type (WT) and primase-dead (CH) PRIMPOL constructs (Mourón et al., 2013) were transiently over-expressed in U2OS, UW/UW+BRCA1, and RAD51+/+ and T131P-RAD51 cells following transfection of appropriate plasmids using Transit-LT1 Transfection reagent (MIR 2304, Mirus) following the manufacturer’s instruction. Experiments overexpressing PRIMPOL were performed between 24- and 72-hours post-transfection.
Drugs and cell treatments
Cisplatin (P4394, Millipore Sigma) was dissolved in 10X PBS at a 5 mM concentration stock and stored at −20°C. Aliquots were warmed at 60°C for 10 minutes then diluted in cell growth media to the indicated final concentrations for experimental use. Cells were treated for indicated times at 37°C, 5% CO2. Aliquots were frozen and thawed no more than 3 times before disposal. The PARP inhibitor Olaparib (PARPi, AZD2281, Selleckchem) was dissolved at DMSO at a 10 mM concentration stock and immediately dissolved in cell growth media to the indicated final concentrations for experimental use at 37°C, 5% CO2. Hydroxyurea (HU, H8627, Millipore Sigma) was dissolved in water at a 1 M concentration stock and stored at −20°C. HU was dissolved in cell growth media to a final concentration of 50 mM and cells were treated for 1 hour at 37°C, 5% CO2. For UV-C irradiation, cells were washed with warmed (37°C) PBS and then exposed to a UV-C lamp (XX-15S, Bench lamp, 254 nm, 95-0042-05, UVP) at a rate of 1 J/m2/s as monitored by a UV radiometer and UV-C sensor (97-0015-02 and 97-0016-01, UVP) for 15 seconds for a final dose of 15 J/m2. Nocodazole (M1404, Millipore Sigma) was dissolved in DMSO at a 2 mg/mL stock concentration and diluted in cell growth media to a final concentration of 200 ng/mL. Cells were treated for indicated times at 37°C, 5% CO2. The REV1 inhibitor JH-RE-06 (REV1i, GLXC-21219, AOBIOUS Laboratories) (Wojtaszek et al., 2019) was dissolved in DMSO to a stock concentration of 10 mM and diluted in growth media to the indicated final concentrations for experimental use at 37°C, 5% CO2. REV1i in solution was stored at −20°C for no longer than a month. The MRE11 inhibitor mirin (M9948, Millipore Sigma) was dissolved in DMSO at a 50 mM stock concentration and diluted in cell growth media to a final concentration of 50 μM at 37°C, 5% CO2. The RAD51 inhibitor B02 (S8434, Selleck Chem) (Huang et al., 2011) was dissolved in DMSO at a 50 mM stock concentration and diluted in cell growth media to a final concentration of 27 μM at 37°C, 5% CO2.
S1 nuclease DNA fiber assay
Cells were pulse-labeled with 20 μM 5-lodo-2’-deoxyuridine (IdU, Millipore Sigma) for 20 minutes, washed twice with PBS, then pulse-labeled with 200 μM 5-Chloro-2’-deoxyuridine (CldU, Millipore Sigma) for 1 hour, followed by two washes with PBS. In the case of cisplatin or hydroxyurea treatment, cells were treated with drug concurrently with CldU for 1 hour. In the case of UV-C irradiation, cells were irradiated immediately before addition of CldU. For experiments with the PARP inhibitor, Olaparib, 10 μM Olaparib was added 2 hours prior to analog incorporation and maintained in the cell media during the entire labeling period with and without cisplatin, up until permeabilization. For experiments with the REV1 inhibitor (REV1i), 2 μM REV1i was added immediately after CldU incorporation for indicated times, up until permeabilization. For experiments with the RAD51 inhibitor, B02, 27 μM B02 was added 8 hours prior to analog incorporation and maintained in the cell media during the entire labeling period with and without cisplatin, up until permeabilization. For experiments with the MRE11 inhibitor, mirin, 50 μM mirin was added immediately after CldU incorporation for indicated times up until permeabilization. After CldU incorporation, cells were permeabilized with CSK100 (100 mM NaCl, 10 mM MOPS pH 7, 3 mM MgCl2, 300 mM sucrose and 0.5% Triton-X100 in water) for 5 minutes at room temperature (RT), washed once in S1 buffer (30 mM sodium acetate pH 4.6, 10 mM zinc acetate, 5% glycerol, 50 mM NaCl in water), then treated with the S1 nuclease (18001-016, Thermo Fisher Scientific) at 20 U/mL in S1 buffer for 30 minutes at 37°C, and collected in PBS + 0.1% BSA with cell scraper. Nuclei were then pelleted at −4500 × g for 5 min at 4°C, then resuspended in PBS to a final concentration of −1500 nuclei/mL (nuclei cannot be quantified, so initial number of cells plated and pellet size should be considered here) (Quinet et al., 2017). To spread DNA into fibers, 2 μL of cells was added to a positively charged glass slide and lysed with 5.5 μl of lysis buffer (200 mM Tris-HCl pH 7.5, 50 mM EDTA, 0.5% SDS in water) by gently pipetting up and down. Slides were kept at RT for 5 min, then tilted at a 20 - 45° angle to spread the fibers at a constant, low speed. Slides were then dried at RT in a dark chamber for 10-15 minutes. DNA was then fixed with a freshly prepared solution of 3:1 methanol:glacial acetic acid for 5 minutes, dried at RT in the dark, then stored at 4°C overnight. To immunostain, DNA was rehydrated in PBS twice for 5 minutes, then denatured with 2.5 M HCl for 45 minutes at RT. Slides were then washed with PBS four times for 3 minutes and blocked with pre-warmed 5% BSA in PBS at 37°C for 1 hour. Fibers were then immuno-stained with rat anti-BrdU (1/200, Ab6326, Abeam) and mouse anti-BrdU (1/40, 347580, BD Biosciences) for 1 hour 30 minutes at RT in a dark, humid chamber. After primary antibody incubation, slides were washed four times with PBS + 0.1%Tween-20 for 3 minutes with gentle shaking for the final 30 seconds, washed once with PBS, then incubated with anti-rat Alexa Fluor 488 and anti-mouse Alexa Fluor 568 (1/150, A21470 and A21124, respectively, Thermo Fisher Scientific) for 1 hour at RT. Slides were again washed four times with PBS + 0.1%Tween-20 for 3 minutes with gentle shaking, washed once with PBS, then mounted with Prolong Gold Antifade Reagent (P36930, Thermo Fisher Scientific) (Quinet et al., 2017). Images were acquired with LAS AF software using a Leica DMi8 confocal microscope with either a 63x/1.4 or 40x/1.15 oil immersion objective. For all the DNA fiber experiments, tracts were measured only on forks characterized by contiguous IdU-CldU signals (i.e. progressing replication forks). Only fiber tracts where the beginning and end of each color was unambiguously defined were considered in the analysis. The length of each tract was measured manually using the segmented line tool on ImageJ software (National Institute of Health) (Schneider et al., 2012). The pixel values were converted into μm using the scale bar generated by the microscope software. Size distribution of tract lengths or ratios from individual DNA fibers were plotted as scatter dot plot with a line representing the median. Data were pooled from independent experiments. At least 150 individual tracts were scored for each data set.
Post-replication repair (PRR) assay
Asynchronous cells were pulse-labeled with 20 μM IdU for 1 hour. For treated samples, indicated concentrations of cisplatin were added to growth media concurrently with IdU. After 1 hour, cells were washed twice with previously warmed (37 °C) PBS and placed in growth media with 200 ng/mL nocodazole for 16 to 24 hours. During the final 4 hours of nocodazole incubation, CldU was added to the growth media at a final concentration of 20 μM to be incorporated during PRR (gap filling). In the experiments with the RAD51 inhibitor, B02, 27 μM B02 was added immediately after IdU incorporation with nocodazole and for the duration of the CldU incorporation. In the experiments with mirin and the REV1 inhibitor, JH-RE-06, cells were pre-treated with the indicated inhibitor (50 μM mirin or 2 μM REV1i) concurrently with nocodazole for 1 hour prior to CldU incorporation and during the entire duration of CldU incorporation, for a total of 5 hours. In the PRR experiments with Olaparib, cells were pre-treated with 10 μM Olaparib for 2 hours prior to the IdU treatment and during the entire duration of the experiment. After CldU incorporation, cells were washed carefully with previously warmed PBS, trypsinized, and collected on ice. Cells were then pelleted at 350 × g for 5 min at 4°C. Supernatant was aspirated and cells were resuspended in cold PBS to a final concentration of −1500 cells/μL. Cells are immediately lysed, spread into fibers, and immuno-stained as specified in S1 nuclease DNA fiber assay (Quinet et al., 2017; Quinet et al., 2016). A minimum of 10-15 PRR events were scored per sample, unless there were less than 10 total events. Only IdU tracts with at least 1 CldU dot were considered PRR events. Tracts with CldU dots that were not centered on the IdU tract or covering the entire width of the fiber were not scored. The level of background of each image was taken into consideration in order to accurately distinguish CldU dots from background. Analysis of pictures with high levels of background was avoided. The length of each IdU tract with at least one CldU dot was measured manually using the segmented line tool on ImageJ software (National Institute of Health) (Schneider et al., 2012) and pixel values were converted into μm using the scale bar generated by the microscope software. Lengths were further converted into kilobases (1 μm = 2.59 kilobases) (Jackson and Pombo, 1998) and PRR density was calculated by dividing the total number of CldU dots on an IdU tract by the length of that IdU tract in kilobases (Daigaku et al., 2010; Quinet et al., 2016). Per-kilobase densities were plotted as scatter dot plots with the line representing the median. Data were pooled from at least two independent experiments.
Cell cycle analysis
For cell cycle analysis of cells in the S phase, asynchronous cells were treated with 10 μm 5-ethynyl-2’-deoxyuridine (EdU, E10187, Thermo Fisher Scientific) for 1 hour, washed twice with pre-warmed PBS, treated with and without 150 μM cisplatin for 1 hour, washed twice with pre-warmed PBS, then allowed to recover for the indicated times in fresh culture media at 37°C, 5% CO2. Cells were harvested between 0 and 24 hours and fixed in 2% paraformaldehyde for 15 min at RT. After fixation, samples were blocked with 1% BSA/PBS for 10 min, then permeabilized in 1% BSA, 0.5% saponin (8047-15-2, Millipore Sigma) in the dark for 30 min. Permeabilized cells were then incubated with the Click-iT EdU Alexa Fluor 488 Imaging Kit (C10337, Thermo Fisher Scientific) in the dark for 30 min before staining with DAPI (1% BSA, 0.1 mg/mL RNase A, 2 μg/mL DAPI) for 20 min in the dark at RT. Samples were run through flow cytometry (FACSCanto II, BD Biosciences), and data were analyzed with FlowJo. At least 10,000 events were analyzed per sample. Cells were gated for EdU positivity to follow the cell cycle progression of cells that were in the S phase at the time of cisplatin treatment.
RT-qPCR
Total RNA was extracted using the PureLink RNA mini Kit (12183018A, Thermo Fisher Scientific), cDNA was synthesized by M-MLV Reverse Transcriptase (28025013, Thermo Fisher Scientific), and PCR was performed using iQTM SYBR Green supermix (1708880, Biorad) by the CFX96 Real Time PCR Detection System (Biorad), according to the manufacturers’ instructions. The following primers were used: BRCA1 (Quinet et al., 2020): forward AGAAACCACCAAGGTCCAAAG, reverse GGGCCCATAGCAACAGATTT; PRIMPOL (Vallerga et al., 2015): forward TGTGGCTTTGGAGGTTACTGA, reverse TTCTACTGAAGTGCCGATACTGT; POLη (Christmann et al., 2016): forward ATCTTCTACTGGCACAAG, reverse ACATTATCTCCATCACTTCA; REV3L: forward TCATGAGAAGGAAAGACACTTTATG, reverse GCTGTAGGAGGTAGGGAATATG (Quinet et al., 2020); ACTIN: forward CTCGCCTTTGCCGATCC, reverse ATGCCGGAGCCGTTGTC was used as an endogenous control. The results were calculated according to the 2-ΔΔCt methodology and are shown as relative expressions to the correspondent control.
Western blot
Proteins were extracted with lysis buffer (50 mM Tris-HCl pH 7.5, 20 mM NaCl, 1 mM MgCl2, 0.1% SDS, 1X protease inhibitor) and benzonase (71206, Novagen) at 250 U/mL for 20 min on ice. Total protein concentration was measured using Pierce BCA protein assay kit (23227, Thermo Fisher Scientific) according to the manufacturer’s instructions. 1X NuPAGE LDS sample buffer (NP0007, Thermo Fisher Scientific) and 200 mM DTT were added and proteins were denatured at 95°C for 5 min. 10-25 μg proteins were loaded onto a NuPAGE Novex 4%–12% Bis-Tris Gel (NP0322BOX, Thermo Fisher Scientific) and run with 1X NuPAGE MES SDS Running buffer (NP0002, Thermo Fisher Scientific). Proteins were transferred onto a 0.45 μm pore nitrocellulose membrane (10600002, GE Healthcare Life Sciences) by cold wet-transfer in 1X Tris/Glycine Buffer (1610734, Biorad) and 20% Methanol at constant 400 mA for 45-60 min. Membranes were blocked with 5% milk (170-6404, Biorad) in TBS-0.1% Tween-20 or PBS-0.1% Tween-20 for total proteins or with 5% BSA in PBS-0.1% Tween-20 for ubiquitinated proteins for 1 hour at RT. The following primary antibodies were used in 5% milk or BSA in TBS-0.1% Tween-20 or PBS-0.1% Tween-20: Rabbit anti-PRIMPOL (1/1,000; custom-made), Mouse anti-V5 (1/5,000, R96025, Thermo Fisher Scientific), Mouse anti-SMARCAL1 (1/1,000, (A-2) sc-376377, Santa Cruz), Rabbit anti-RAD18 (1/1,000, 9040S, Cell Signaling), Mouse anti-UBC13 (1/500, sc376470, Santa Cruz), Mouse anti-PCNA (1/2,000, PC10, sc-56, Santa Cruz), Rabbit anti-PCNA-Ub Lys164 (1/1,000, 13439, Cell Signaling), Rabbit anti-GAPDH (1/20,000, ab181602, Abeam), Rabbit anti-Vinculin (1/5,000, ab129002, Abeam). IRDye Infrared secondary antibodies from LI-COR were used, proteins were detected by Odyssey CLx (1/20,000, LI-COR) and images were prepared with Image Studio Lite (LI-COR) (Quinet et al., 2020).
Electron microscopy
For the EM analysis of replication intermediates, 5-10×106 wild-type, SMARCAL1KO, or PARPi-treated U2OS cells were harvested immediately after treatment with 150 μM cisplatin for 1 hour. For experiments with the PARP inhibitor Olaparib, 10 μM Olaparib was added for 2 hours prior to cisplatin treatment, and for the duration of cisplatin treatment for a total of 3 hours. Untreated cells were also included. DNA was cross-linked by incubating with 10 μg/mL 4,5’,8-trimethylpsoralen (T6137, Millipore Sigma) followed by a 3-minute exposure to 366 nm UV light on a precooled metal block, for a total of three rounds. Cells were lysed and genomic DNA was isolated from the nuclei by proteinase K digestion (25530-015, Life Technologies) and chloroform-isoamyl alcohol extraction. Genomic DNA was purified by isopropanol precipitation and digested with PvuII HF (R3151S, New England Biolabs) with the appropriate buffer for 4 hours at 37°C. Replication intermediates were enriched on a benzoylated naphthoylated DEAE-cellulose (B6385, Millipore Sigma) column. Samples were prepared for EM visualization by spreading the purified, concentrated DNA on a carbon-coated grid in the presence of benzyl-dimethyl-alkylammonium chloride (B6295, Millipore Sigma), followed by platinum rotary shadowing. Images were obtained on a JEOL JEM-1400 electron microscope using a bottom mounted AMT XR401 camera. Analysis was performed using ImageJ software (National Institute of Health) (Schneider et al., 2012). EM analysis allows distinguishing duplexed DNA, which is expected to appear as a 10 nm thick fiber after the platinum/carbon coating step necessary for EM visualization, from ssDNA, which has a reduced thickness of 5-7 nm. Criteria used for the assignment of a three-way junction, indicative of a replication fork, include the joining of three DNA fibers into a single junction, with two symmetrical daughter strands and single parental strand. Reversed replication forks consist of four DNA fibers joined at a single junction, consisting of two symmetrical daughter strands, one parental strand and the addition of a typically shorter fourth strand, representative of the reversed arm. The length of the two daughter strands corresponding to the newly replicated duplex should be equal (b=c), whereas the length of the parental arm and the regressed arm can vary (a ≠ b = c ≠ d). Conversely, canonical Holliday junction structures will be characterized by arms of equal length (a = b, c = d). Particular attention is paid to the junction of the reversed replication fork in order to observe the presence of a bubble structure, indicating that the junction is opened up and that it is simply not the result of the occasional crossover of two DNA molecules. These four-way junctions of reversed replication forks may also be collapsed and other indicators, such as daughter strand symmetry, presence of single-stranded DNA at the junction, or the entire structure itself, are all considered during analysis (Neelsen et al., 2014). The frequency of reversed forks and daughter strand ssDNA gaps in a sample is computed using the Prism software.
Proximity Ligation Assay
PLA immunofluorescence was performed according to manufacturer’s protocol (Duolink, DUO82040, Millipore Sigma) with minor modifications adapted from Taglialatela et. al 2017. Cells were plated on coverslips at 80% confluency and allowed to attach for 24 hours. Cells were then treated with 150 μM cisplatin for 1 hour, washed twice with PBS before incubation with 200 ng/mL nocodazole for 24 hours. Next, 2 μM REV1 inhibitor (JH-RE-06, GLXC-21219, AOBIOUS Laboratories) was added to the media for 1 hour before and during incubation with 10 μM 5-ethynyl-2’-deoxyuridine (EdU, E10187, Thermo Fisher Scientific) for 1 hour, for a total of 2 hours. Cells were then fixed in 4% paraformaldehyde for 10 minutes at RT. and washed with PBS. Fixed slides were then permeabilized in PBS with 0.5% Triton X-100 for 10 minutes at RT and blocked in FBS for 30 min at 37°C. Coverslips were then incubated with a Click-iT reaction cocktail (10 μM Biotin Azide [B10184, Thermo Fisher Scientific], 10 mM (+) Sodium L-Ascorbate [A4034, Sigma], 10mM Copper (II) sulfate pentahydrate (CuSO4, C489, Thermo Fisher Scientific]) for 30 min at RT. Primary antibodies were diluted in PBS with 5% BSA and incubated with slides for 1 hour at 37°C. The primary antibodies used were anti-cyclin B1 (AF6000, Novus, 1:1000), Rabbit anti-RAD18 (9040S, Cell Signaling, 1:1000), Mouse anti-REV1 (SC-393022, Santa Cruz, 1:1000), Mouse anti-UBC13 (SC-376470, Santa Cruz, 1:1000), Mouse anti-Biotin (200-002-211, Jackson, 1:1000), Rabbit anti-Biotin (A150-109A, Bethyl, 1:3000). Alexa Fluor 488 donkey anti-goat secondary (A11055, Like Tech, 1:1000) was used to probe for cyclin B1. PLA reactions were then performed according to manufacturer instructions using the Duolink kit. Images were acquired using a Leica DMi8 confocal microscope with a 63x/1.4 oil immersion objective. Negative controls were not incubated with EdU. Positive controls were incubated with Mouse anti-biotin and Rabbit anti-biotin together.
Cell survival
Cell survival assay was performed using Cell Proliferation Kit II (XTT, 11465015001, Millipore Sigma) by seeding 1.3 × 104 cells per well in a 24-well plate in duplicate the day prior to treatment. Cells were then treated for 1 hour with the indicated doses of cisplatin and cell survival was assessed after 6 days of treatment. The absorbance was measured at 450 nm with a reference wavelength at 692 nm. Results were expressed as percentage of the corresponding untreated control. For CellTiter 96 Aqueous Non-Radioactive Cell proliferation assay (MTS, G5430, Promega), 1.5 × 103 wild-type and SMARCAL1KO U2OS cells transfected with siCTRL or siREV3L were seeded in a 96-well plate in quadruplicate the day prior to treatment, then they were treated with the indicated doses of cisplatin for 1 hour and survival was assessed 6 days after treatment. For PEO1 and C4-2 cells survival assay, 1.5 × 103 cells were seeded in 96-well plate in quadruplicate the day prior to treatment. Cells were then treated for 6 days with or without 0.5 μM Olaparib, 0.5 μM cisplatin, and 0.1 μM REV1i. The absorbance was measured at 490 nm and the results were expressed as percentage of the corresponding untreated sample.
Plates were scanned using the Infinite 200Pro Reader (Tecan) with Tecan i-control software. For the CellTiter Glo experiments, 800 UW or UW+BRCA1 cells were seeded in flat-bottom black/clear 96-well plates in triplicate the day prior to treatment. Cells were then treated chronically with or without 0.25 μM PARPi, 62.5 nM cisplatin, and 0.1 μM REV1i. After 6 days, plates were cooled to room temperature for 30 minutes and 50 μL of CellTiter Glo mix (Promega, G7571) was added to each well. Plates were incubated at room temperature for 15 minutes and scanned using an EnVision 2103 Multilabel Reader (Perkin Elmer) with Envision Manager software version 1.13.3009.1409. Viability was calculated as the ratio of the average signal of three technical replicates in drug-treated conditions compared to untreated control.
G1-specific 53BP1 nuclear bodies
48 hours after chronic treatment with 5 μM cisplatin with and without 2 μM JH-RE-06, wild-type and SMARCAL1KO U2OS cells were fixed with 4% PFA for 10 min, washed three times in PBS, permeabilized in 0.5% Triton X-100 for 10 min at RT, and blocked in BSA-T (5% BSA, 0.05% Tween 20) for at least 1 hour at RT. Immunostaining of the 53BP1 nuclear bodies was performed with rabbit anti-53BP1 antibody (1:1,000; Novus Biologicals, NM100-304S) and mouse anti-cyclin A antibody (1:100; Santa Cruz, SC-271682) in a humid chamber at 4°C overnight. Slides were then washed three times in PBS. Secondary antibodies anti-mouse Alexa Fluor 594 (1:1,000; Thermo Fisher Scientific, A11005) and anti-rabbit Alexa Fluor 488 (1:1,000; Thermo Fisher Scientific, A11034) were incubated with cells for 1 hour at RT in the dark. Slides were again washed three times in PBS. Nuclei were stained with 0.05 μg/mL DAPI for 10 min at RT. The slides were mounted with Prolong Gold Antifade reagent (Invitrogen, P36930). Images were captured with 40x objective, and at least 150 cyclin A-negative cells were analyzed per condition (Wood et al., 2020).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Prism 8 (GraphPad Software). Details of the individual statistical tests are indicated in the figure legends and results. In all cases: ns, non-significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. All experiments were repeated at least three times unless otherwise noted. Statistical differences in DNA fiber tract lengths, PRR densities, comet tail moments, and G1 specific 53BP1 bodies were all determined by Kruskal-Wallis followed by Dunn’s multiple comparisons test. Statistical differences for the electron microscopy experiments were determined by an unpaired t test. Statistical differences in PLA+ cells in G2, EdU+ cells in G2, and cell survival of the UW ± BRCA1 and PEO1/C4-2 cells were determined by one-way ANOVA followed by Tukey’s multiple comparison test. Statistical differences in cell survival of wild-type and SMARCAL1KO U2OS cells were determined by one-way ANOVA followed by Bonferroni test comparing wild-type siBRCA1 vs SMARCAL1KO siBRCA1 or wild-type siREV3L vs SMARCAL1KO siREV3L.
Supplementary Material
Key resource table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-BrdU (B44) | BD Biosciences | Cat# 347580; RRID: AB_400326 |
| Rat monoclonal anti-BrdU [BU1/75 (ICR1)] | Abcam | Cat# ab6326; RRID: AB_305426 |
| Rabbit polyclonal anti-PRIMPOL | This study | AB_2892623; RRID: AB_2892623 |
| Mouse monoclonal IgG2a anti-V5 | Thermo Fisher Scientific | Cat# R960-25; RRID: AB_2556564 |
| Rabbit monoclonal anti-RAD18 (D2B8) | Cell Signaling Technology | Cat# 9040; RRID: AB_2756446 |
| Mouse monoclonal anti-REV1 | Santa Cruz Biotechnology | Cat# SC-393022; RRID: AB_2885169 |
| Mouse Monoclonal anti-UBC13 | Santa Cruz Biotechnology | Cat# SC-376470; RRID: AB_11150503 |
| Mouse monoclonal anti-PCNA [PC10] | Santa Cruz Biotechnology | Cat# sc-56; RRID: AB_62810 |
| Rabbit Monoclonal anti-Ubiquityl-PCNA (Lys164) | Cell Signaling Technology | Cat#: 13439; RRID: AB_2798219 |
| Mouse polyclonal anti-Biotin | Jackson Labs | Cat# 200-002-211; RRID: AB_2339006 |
| Rabbit polyclonal anti-Biotin | Bethyl | Cat# A150-109A; RRID: AB_67327 |
| Mouse Monoclonal anti-SMARCAL1 (A-2) | Santa Cruz Biotechnology | Cat#: SC-376377; RRID: AB_20987841 |
| Rabbit polyclonal anti-53BP1 | Novus Biologicals | Cat# NB100-304; RRID: AB_10003037 |
| Goat polyclonal anti-Cyclin B1 | Novus Biologicals | Cat# AF6000; RRID: AB_10003037 |
| Mouse polyclonal anti-Cyclin A | Santa Cruz Biotechnology | Cat# SC-271682; RRID: AB_1964555 |
| Rabbit monoclonal anti-GAPDH [EPR1689] | Abcam | Cat# Ab181602; RRID: AB_2630358 |
| Mouse monoclonal anti-TUBULIN | Millipore Sigma | Cat# T5168; RRID: AB_477579 |
| Rabbit Monoclonal anti-Vinculin | Abcam | Cat# Ab129002; RRID: AB_11144129 |
| Alexa Fluor 488 Chicken anti-Rat IgG (H+L) | Thermo Fisher Scientific | Cat# A21470; RRID: AB_2535873 |
| Alexa Fluor 546 Goat anti-Mouse IgG1 | Thermo Fisher Scientific | Cat# A21123; RRID: AB_2535765 |
| Alexa Fluor 594 Goat anti-Mouse IgG (H+L) | Thermo Fisher Scientific | Cat# A11005; RRID: AB_2534073 |
| Alexa Fluor 488 Goat anti-Rabbit IgG (H+L) | Thermo Fisher Scientific | Cat# A11034; RRID: AB_2576217 |
| Alexa Flour 488 Donkey polyclonal anti-Goat | Like Tech | Cat# A11055; RRID: AB_2534102 |
| IRDye 800CW Goat anti-Rabbit IgG (H+L) | LI-COR Biosciences | Cat# 925-32211; RRID: AB_2651127 |
| IRDye 680RD Goat anti-Rabbit IgG (H+L) | LI-COR Biosciences | Cat# 925-68071; RRID: AB_2721181 |
| IRDye 800CW Goat anti-Mouse IgG (H+L) | LI-COR Biosciences | Cat# 925-32210; RRID:AB_2687825 |
| IRDye 680RD Goat anti-Mouse IgG (H+L) | LI-COR Biosciences | Cat# 925-68070; RRID: AB_2651128 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cisplatin | Millipore Sigma | Cat# P4394-250MG |
| Hydroxyurea | Millipore Sigma | Cat# H8627-5G |
| Olaparib | Selleck Chemicals | Cat# S1060 |
| Mirin | Millipore Sigma | Cat# M9948-5MG |
| JH-RE-06 (REV1i) | AOBIOUS Laboratories | Cat# AOB13138 |
| B02 (RAD51i) | Selleck Chem | Cat# S8434 |
| DMSO (Dimethyl Sulfoxide) | Thermo Fisher Scientific | Cat# BP231 |
| IdU (5-Iodo-2′-deoxyuridine) | Millipore Sigma | Cat# I7125-5G |
| CldU (5-Chloro-2′-deoxyuridine) | Millipore Sigma | Cat# C6891-100MG |
| EdU (5-Ethynyl-2′-deoxyuridine) | Thermo Fisher Scientific | Cat# A10044 |
| Prolong Gold Antifade reagent | Invitrogen | Cat# P36930 |
| Nocodazole | Millipore Sigma | Cat# M1404 |
| DAPI (4’,6-Diamidino-2-phenylindole dihydrochloride) | Millipore Sigma | Cat# D9542 |
| Triton X-100 | Fisher Scientific | Cat# BP151-500 |
| RNAimax transfection reagent | Thermo Fisher Scientific | Cat# 13778-150 |
| TransIT LT1 transfection reagent | Mirus | Cat# MIR 2300 |
| S1 nuclease | Thermo Fisher Scientific | Cat# 18001-016 |
| Biotin Azide | Thermo Fisher Scientific | Cat# B10184 |
| (+) Sodium L-Ascorbate | Sigma | Cat# A4034 |
| Copper (II) Sulfate Petahydrate (CuSO4) | Thermo Fisher Scientific | Cat# C489 |
| FBS | Millipore Sigma | Cat# F0926 |
| BSA | Gold Biotechnology | Cat# A-420-250 |
| Saponin | Millipore Sigma | Cat# 8047-15-2 |
| Paraformaldehyde | Thermo Fisher | Cat# AAJ19943K2 |
| PureLink RNAse A (20 mg/mL) | Thermo Fisher Scientific | Cat# 12091039 |
| Benzonase | Novagen | Cat# 71206 |
| Pierce Protease inhibitor tablets, EDTA-free | Thermo Fisher Scientific | Cat# A32965 |
| G 418 disulfate salt solution | Millipore Sigma | Cat# G8168 |
| TMP (4,5’,8-Trimethylpsoralen) | Millipore Sigma | Cat# T6137-100MG |
| Proteinase K | Life Technologies | Cat# 25530-015 |
| PvuII HF | New England Biolabs | Cat# R3151S |
| Benzoylated Naphthoylated DEAE-Cellulose | Millipore Sigma | Cat# B6385-25G |
| Benzalkonium chloride (BAC) | Millipore Sigma | Cat# B6295 |
| Uranyl Acetate | Electron Microscopy Sciences | Cat# 541-09-3 |
| Critical Commercial Assays | ||
| Click-It EdU Alexa Fluor 488 Imaging Kit | Thermo Fisher Scientific | Cat# C10337 |
| CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay (MTS) | Promega | Cat# G5440 |
| Cell proliferation Kit II (XTT) | Millipore Sigma | Cat# 11465015001 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7571 |
| PureLink RNA mini Kit | Thermo Fisher Scientific | Cat# 12183018A |
| M-MLV Reverse Transcriptase | Thermo Fisher Scientific | Cat# 28025013 |
| iQTM SYBR Green supermix | Biorad | Cat# 41708880 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Duolink | Sigma-Aldrich | Cat# DUO92101 |
| Deposited Data | ||
| Unprocessed microscopy images, gels, and blots | This paper, Mendeley data | http://dx.doi.org/10.17632/2sc5bwyw6s.1 |
| Experimental Models: Cell Lines | ||
| Human: wild-type U2OS | ATCC | HTB-96 |
| Human: SMARCAL1KO U2OS | David Cortez lab ; Liu et al., 2020 | N/A |
| Human: Schimke Immunoosseous Dysplasia lymphocytes (SIOD) | David Cortez lab; Carroll et al., 2015 | N/A |
| Human: SIOD+SMARCAL1 | David Cortez lab; Carroll et al., 2015 | N/A |
| Human: HEK293T | ATCC | N/A |
| Human: HEK293T K164R | George-Lucian Moldovan lab; Thakar et al., 2020 | N/A |
| Human: UWB1.289 | Lee Zou lab; Yazinski et al., 2017 | CRL-2945 |
| Human: UWB1.289 + BRCA1 | Lee Zou lab; Yazinski et al., 2017 | CRL-2946 |
| Human: C4-2 | Sharon Cantor Lab; Panzarino et al., 2021 | N/A |
| Human: PEO1 | Sharon Cantor Lab; Panzarino et al., 2021 | N/A |
| Human: RAD51+/+ CRISPR corrected (RA2630) | Agata Smogorzewska lab; Wang et al., 2015 | N/A |
| Human: T131P RAD51 mutant human fibroblasts | Agata Smogorzewska lab; Wang et al., 2015 | N/A |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| BRCA1 RT-qPCR primer F: | Lemacon et al., 2017 | AGAAACCACCAAGGTCCAAAG |
| BRCA1 RT-qPCR primer R: | Lemacon et al., 2017 | GGGCCCATAGCAACAGATTT |
| BRCA2 RT-qPCR primer F: | Lemacon et al., 2017 | AGGACTTGCCCCTTTCGTCTA |
| BRCA2 RT-qPCR primer R: | Lemacon et al., 2017 | TGCAGCAATTAACATATGAGG |
| REV3L RT-qPCR primer F: | Quinet et al., 2020 | TCATGAGAAGGAAAGACACTTTATG |
| REV3L RT-qPCR primer R: | Quinet et al., 2020 | GCTGTAGGAGGTAGGGAATATG |
| POLH RT-qPCR primer F: | Christmann et al., 2016 | ATCTTCTACTGGCACAAG |
| POLH RT-qPCR primer R: | Christmann et al., 2016 | ACATTATCTCCATCACTTCA |
| PRIMPOL RT-qPCR primer F: | Vallerga et al., 2015 | TGTGGCTTTGGAGGTTACTGA |
| PRIMPOL RT-qPCR primer R: | Vallerga et al., 2015 | TTCTACTGAAGTGCCGATACTGT |
| Beta-Actin RT-qPCR primer F: | Quinet et al., 2020 | CTCGCCTTTGCCGATCC |
| Beta-Actin RT-qPCR primer R: | Quinet et al., 2020 | ATGCCGGAGCCGTTGTC |
| Silencer Select Negative control #1 siRNA (siCTRL) | Ambion | Cat# 4390843 |
| siRNA PRIMPOL | Dharmacon (custom siRNA) | GAGGAAACCGUUGUCCUCAGUGUAU |
| siRNA RAD18 | Dharmacon (custom siRNA) | GCCGGAUCUGAAAAAUAAC |
| siRNA UBC13 | Dharmacon (custom siRNA) | CCAGAUGAUCCAUUAGCAA |
| siRNA POLH (POLη) | Dharmacon | Cat# L-006454 |
| siRNA REV3L | Dharmacon | Cat# L-006302 |
| siRNA BRCA1 | Dharmacon | Cat# L-007287 |
| siRNA BRCA2 | Dharmacon | Cat# L-003462 |
| siRNA SMARCAL1 | Dharmacon | Cat# D-013058-04-0002 |
| Recombinant DNA | ||
| Plasmid: pcDNA3.1_nV5-DEST V5-WT-PRIMPOL (V5-WT-PRIMPOL) | Juan Méndez lab; Mourón et al. 2013 | N/A |
| Plasmid: pcDNA3.1_nV5-DEST V5-CH-PRIMPOL (V5-CH-PRIMPOL) | Juan Méndez lab; Mourón et al. 2013 | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ RRID: SCR_003070 |
| GraphPad Prism | GraphPad software | http://www.graphpad.com RRID:SCR_002798 |
| FlowJo | FlowJo LLC | https://www.flowjo.com/ RRID: SCR_008520 |
| ImageStudioLite2 | LiCOR Odyssey | https://www.licor.com/bio/image-studio-lite/ RRID: SCR_013715 |
| LAS (Leica Application Suite) AF software | Leica | https://www.leica-microsystems.com/products/microscope-software/ |
| Envision Manager software | Perkin Elmer | http://envision.bioe.orst.edu/Downloads.aspx RRID:SCR_016681 |
| Other | ||
| MEGM Mammary Epithelial Cell Growth Medium BulletKit | Lonza | Cat# CC-3150 |
| JEOL 1200 EX Electron Microscope | JEOL | N/A |
| AMTXR41 Camera | AMT | N/A |
| Leica EM ACE600 Coater | Leica | N/A |
| Leica DMi8 confocal microscope | Leica | N/A |
| Odyssey CLx Imaging System | LI-COR Biosciences | N/A |
| CFX96 Real Time PCR Detection System | Biorad | N/A |
| S1000 Thermal Cycler | Biorad | N/A |
| FACSCanto II | BD Biosciences | N/A |
| UVP CL-1000L Crosslinker | Fisher Scientific | Cat# UVP95017401 |
| UVC Bulb ( UVP XX-15S, Bench lamp, 254 nm) | MidSci | Cat# UVP95004205 |
| UVP UV Radiometer | MidSci | Cat# UVP97001502 |
| UVP UVC Sensor (254 nm) | MidSci | Cat# UVP97001601 |
| EnVision 2103 Multilabel Reader | Perkin Elmer | N/A |
| SpectraMax 340 Microplate Reader | Molecular Devices | N/A |
Highlights:
Two temporally distinct pathways fill ssDNA gaps in human cells
RAD18, PCNA monoubiquitination, and REV1-POLζ promote gap filling in G2.
UBC13, RAD51, and REV1-POLζ promote gap filling in S.
BRCA1 and BRCA2 promote gap filling by limiting MRE11 activity in S and G2.
Acknowledgments
We would like to thank Juan Mendez (CNIO, Madrid, Spain) for providing the wild-type (WT-PRIMPOL) and primase-dead PRIMPOL constructs (CH-PRIMPOL), David Cortez (Vanderbilt, TN, USA) for providing the SMARCAL1KO U2OS and the SMARCAL1 mutant SIOD cells, George-Lucian Moldovan (Pennsylvania State, PA, USA) for the PCNA K164R mutant cells, and Agata Smogorzewska (Rockefeller, NY, USA) for providing the Q16 RAD51-T131P patient fibroblasts. This work was supported by the NCI under grant numbers R01CA237263 to A.V., R01CA248526 to A.V. and L.Z., and F30CA254215 to E.C. Figures were created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Adar S, Izhar L, Hendel A, Geacintov N, and Livneh Z (2009). Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res 37, 5737–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth A, and Lord CJ (2018). Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat Rev Clin Oncol 15, 564–576. [DOI] [PubMed] [Google Scholar]
- Bai G, Kermi C, Stoy H, Schiltz CJ, Bacal J, Zaino AM, Hadden MK, Eichman BF, Lopes M, and Cimprich KA (2020). HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol Cell 78, 1237–1251 e1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly V, Lamb J, Sung P, Prakash S, and Prakash L (1994). Specific complex formation between yeast RAD6 and RAD18 proteins: a potential mechanism for targeting RAD6 ubiquitin-conjugating activity to DNA damage sites. Genes Dev 8, 811–820. [DOI] [PubMed] [Google Scholar]
- Bansbach CE, Boerkoel CF, and Cortez D (2010). SMARCAL1 and replication stress: an explanation for SIOD? Nucleus 1, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky A, Izhar L, and Livneh Z (2002). Error-free recombinational repair predominates over mutagenic translesion replication in E. coli. Mol Cell 10, 917–924. [DOI] [PubMed] [Google Scholar]
- Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. (2013). Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nature Structural & Molecular Biology 20, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V, Taylor E, Stevanovic I, Green AJ, Stracker TH, Lindsay HD, et al. (2013). PrimPol Bypasses UV Photoproducts during Eukaryotic Chromosomal DNA Replication. Molecular Cell 52, 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerkoel CF, O’Neill S, Andre JL, Benke PJ, Bogdanovic R, Bulla M, Burguet A, Cockfield S, Cordeiro I, Ehrich JH, et al. (2000). Manifestations and treatment of Schimke immuno-osseous dysplasia: 14 new cases and a review of the literature. Eur J Pediatr 159, 1–7. [DOI] [PubMed] [Google Scholar]
- Branzei D, and Szakal B (2016). DNA damage tolerance by recombination: Molecular pathways and DNA structures. DNA Repair (Amst) 44, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo PA, Sastre-Moreno G, Perpiñá C, Guerra S, Martínez-Jiménez MI, and Blanco L (2019). The invariant glutamate of human PrimPol DxE motif is critical for its Mn2+-dependent distinctive activities. DNA Repair 77, 65–75. [DOI] [PubMed] [Google Scholar]
- Carroll C, Hunley TE, Guo Y, and Cortez D (2015). A novel splice site mutation in SMARCAL1 results in aberrant exon definition in a child with Schimke immunoosseous dysplasia. Am J Med Genet A 167A, 2260–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Maldonado D, Byrum AK, Jackson J, Wessel S, Lemacon D, Guitton-Sert L, Quinet A, Tirman S, Graziano S, Masson JY, et al. (2019). Perturbing cohesin dynamics drives MRE11 nuclease-dependent replication fork slowing. Nucleic Acids Res 47, 1294–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Nievera CJ, Lee AY, and Wu X (2008). Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem 283, 7713–7720. [DOI] [PubMed] [Google Scholar]
- Christmann M, Boisseau C, Kitzinger R, Berac C, Allmann S, Sommer T, Aasland D, Kaina B, and Tomicic MT (2016). Adaptive upregulation of DNA repair genes following benzo(a)pyrene diol epoxide protects against cell death at the expense of mutations. Nucleic Acids Res 44, 10727–10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S, Krais J, VanderVere-Carozza PS, Pawelczak KS, Calvo J, et al. (2021). Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Andrea AD (2018). Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 71, 172–176. [DOI] [PubMed] [Google Scholar]
- Daigaku Y, Davies AA, and Ulrich HD (2010). Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 465, 951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamant N, Hendel A, Vered I, Carell T, Reissner T, de Wind N, Geacinov N, and Livneh Z (2012). DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res 40, 170–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, et al. (2008). A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat Chem Biol 4, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddins MJ, Carlile CM, Gomez KM, Pickart CM, and Wolberger C (2006). Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nat Struct Mol Biol 13, 915–920. [DOI] [PubMed] [Google Scholar]
- Edmunds CE, Simpson LJ, and Sale JE (2008). PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 30, 519–529. [DOI] [PubMed] [Google Scholar]
- Elvers I, Johansson F, Groth P, Erixon K, and Helleday T (2011). UV stalled replication forks restart by re-priming in human fibroblasts. Nucleic Acids Res 39, 7049–7057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumasoni M, Zwicky K, Vanoli F, Lopes M, and Branzei D (2015). Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polalpha/Primase/Ctf4 Complex. Mol Cell 57, 812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo D, Kim T, Szakal B, Saayman X, Narula A, Park Y, Branzei D, Zhang Z, and Brown GW (2019). Rad5 Recruits Error-Prone DNA Polymerases for Mutagenic Repair of ssDNA Gaps on Undamaged Templates. Mol Cell 73, 900–914 e909. [DOI] [PubMed] [Google Scholar]
- García-Gómez S, Reyes A, Martínez-Jiménez MI, Chocrón ES, Mourón S, Terrados G, Powell C, Salido E, Méndez J, Holt IJ, et al. (2013). PrimPol, an Archaic Primase/Polymerase Operating in Human Cells. Molecular Cell 52, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genois MM, Gagne JP, Yasuhara T, Jackson J, Saxena S, Langelier MF, Ahel I, Bedford MT, Pascal JM, Vindigni A, et al. (2020). CARM1 regulates replication fork speed and stress response by stimulating PARP1. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Acosta D, Blanco-Romero E, Ubieto-Capella P, Mutreja K, Miguez S, Llanos S, Garcia F, Munoz J, Blanco L, Lopes M, et al. (2021). PrimPol-mediated repriming facilitates replication traverse of DNA interstrand crosslinks. EMBO J, e106355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Huici V, Szakal B, Urulangodi M, Psakhye I, Castellucci F, Menolfi D, Rajakumara E, Fumasoni M, Bermejo R, Jentsch S, et al. (2014). DNA bending facilitates the error-free DNA damage tolerance pathway and upholds genome integrity. EMBO J 33, 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, and Livingston DM (2006). Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev 20, 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder HC, and Rosenberg B (1970). Inhibitory effects of anti-tumor platinum compounds on DNA, RNA and protein syntheses in mammalian cells in virtro. Int J Cancer 6, 207–216. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Chaudhuri AR, Lopes M, and Costanzo V (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nature Structural & Molecular Biology 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, and Marians KJ (2006). Replication fork reactivation downstream of a blocked nascent leading strand. Nature 439, 557–562. [DOI] [PubMed] [Google Scholar]
- Helm CW, and States JC (2009). Enhancing the efficacy of cisplatin in ovarian cancer treatment - could arsenic have a role. J Ovarian Res 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks JK, Chute CL, Paulsen MT, Ragland RL, Howlett NG, Gueranger Q, Glover TW, and Canman CE (2010). Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links. Mol Cell Biol 30, 1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, and Jentsch S (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141. [DOI] [PubMed] [Google Scholar]
- Hofmann RM, and Pickart CM (1999). Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell 96, 645–653. [DOI] [PubMed] [Google Scholar]
- Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, and Mazin AV (2011). Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol 6, 628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, and Pines J (1994). Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell 79, 573–582. [DOI] [PubMed] [Google Scholar]
- Jackson DA, and Pombo A (1998). Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 140, 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen JG, Tsaalbi-Shtylik A, Hendriks G, Verspuy J, Gali H, Haracska L, and de Wind N (2009). Mammalian polymerase zeta is essential for post-replication repair of UV-induced DNA lesions. DNA Repair (Amst) 8, 1444–1451. [DOI] [PubMed] [Google Scholar]
- Kannouche PL, Wing J, and Lehmann AR (2004). Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14, 491–500. [DOI] [PubMed] [Google Scholar]
- Karras GI, Fumasoni M, Sienski G, Vanoli F, Branzei D, and Jentsch S (2013). Noncanonical role of the 9-1-1 clamp in the error-free DNA damage tolerance pathway. Mol Cell 49, 536–546. [DOI] [PubMed] [Google Scholar]
- Karras GI, and Jentsch S (2010). The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141, 255–267. [DOI] [PubMed] [Google Scholar]
- Keen BA, Bailey LJ, Jozwiakowski SK, and Doherty AJ (2014). Human PrimPol mutation associated with high myopia has a DNA replication defect. Nucleic acids research 42, 12102–12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Guilliam TA, Tsuda M, Yamamoto J, Bailey LJ, Iwai S, Takeda S, Doherty AJ, and Hirota K (2016). Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle 15, 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Molecular Cell 67, 867–881.e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramara J, Osia B, and Malkova A (2018). Break-Induced Replication: The Where, The Why, and The How. Trends Genet 34, 518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laureti L, Demol J, Fuchs RP, and Pages V (2015). Bacterial Proliferation: Keep Dividing and Don’t Mind the Gap. PLoS Genet 11, e1005757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CW, and Hinkle DC (1996). DNA polymerase zeta and the control of DNA damage induced mutagenesis in eukaryotes. Cancer Surv 28, 21–31. [PubMed] [Google Scholar]
- Lehmann AR (1972). Post-replication repair of DNA in ultraviolet-irradiated mammalian cells. No gaps in DNA synthesized late after ultraviolet irradiation. Eur J Biochem 31, 438–445. [DOI] [PubMed] [Google Scholar]
- Lemaçon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nature Communications 8, 860–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta-Ramusino C, Lopes M, Pellicioli A, Haber JE, and Foiani M (2005). Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19, 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KS, Li H, Roberts EA, Gaudiano EF, Clairmont C, Sambel LA, Ponnienselvan K, Liu JC, Yang C, Kozono D, et al. (2018). USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol Cell 72, 925–941 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Krishnamoorthy A, Zhao R, and Cortez D (2020). Two replication fork remodeling pathways generate nuclease substrates for distinct fork protection factors. Sci Adv 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Foiani M, and Sogo JM (2006). Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21, 15–27. [DOI] [PubMed] [Google Scholar]
- Malacaria E, Pugliese GM, Honda M, Marabitti V, Aiello FA, Spies M, Franchitto A, and Pichierri P (2019). Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat Commun 10, 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda Y, and Masutani C (2019). Spatiotemporal regulation of PCNA ubiquitination in damage tolerance pathways. Crit Rev Biochem Mol Biol 54, 418–442. [DOI] [PubMed] [Google Scholar]
- Meneghini R (1976). Gaps in DNA synthesized by ultraviolet light-irradiated WI38 human cells. Biochim Biophys Acta 425, 419–427. [DOI] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, et al. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nature Communications 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourón S, Rodriguez-Acebes S, Martínez-Jiménez MI, García-Gómez S, Chocrón S, Blanco L, and Méndez J (2013). Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nature Structural & Molecular Biology 20, 1383–1389. [DOI] [PubMed] [Google Scholar]
- Nayak S, Calvo JA, Cong K, Peng M, Berthiaume E, Jackson J, Zaino AM, Vindigni A, Hadden MK, and Cantor SB (2020). Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci Adv 6, eaaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Chaudhuri AR, Follonier C, Herrador R, and Lopes M (2014). Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods Mol Biol 1094, 177–208. [DOI] [PubMed] [Google Scholar]
- Neelsen KJ, and Lopes M (2015). Replication fork reversal in eukaryotes: from dead end to dynamic response. Nature Reviews Molecular Cell Biology 16, 207–220. [DOI] [PubMed] [Google Scholar]
- Ohmori H, Hanafusa T, Ohashi E, and Vaziri C (2009). Separate roles of structured and unstructured regions of Y-family DNA polymerases. Adv Protein Chem Struct Biol 78, 99–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Sonoda E, Yoshimura M, Kawano Y, Saya H, Kohzaki M, and Takeda S (2005). Multiple roles of vertebrate REV genes in DNA repair and recombination. Mol Cell Biol 25, 6103–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzarino NJ, Krais JJ, Cong K, Peng M, Mosqueda M, Nayak SU, Bond SM, Calvo JA, Doshi MB, Bere M, et al. (2020). Replication Gaps Underlie BRCA-deficiency and Therapy Response. Cancer Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JL, and Ulrich HD (2009). Mechanistic analysis of PCNA poly-ubiquitylation by the ubiquitin protein ligases Rad18 and Rad5. EMBO J 28, 3657–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Cortez D, Bowers B, Elledge SJ, and Gellert M (2001). Direct DNA binding by Brca1. Proc Natl Acad Sci U S A 98, 6086–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petruk S, Sedkov Y, Johnston DM, Hodgson JW, Black KL, Kovermann SK, Beck S, Canaani E, Brock HW, and Mazo A (2012). TrxG and PcG proteins but not methylated histones remain associated with DNA through replication. Cell 150, 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piberger AL, Bowry A, Kelly RDW, Walker AK, Gonzalez-Acosta D, Bailey LJ, Doherty AJ, Mendez J, Morris JR, Bryant HE, et al. (2020). PrimPol-dependent single-stranded gap formation mediates homologous recombination at bulky DNA adducts. Nat Commun 11, 5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilie PG, Gay CM, Byers LA, O’Connor MJ, and Yap TA (2019). PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin Cancer Res 25, 3759–3771. [DOI] [PubMed] [Google Scholar]
- Pines J, and Hunter T (1989). Isolation of a human cyclin cDNA: evidence for cyclin mRNA and protein regulation in the cell cycle and for interaction with p34cdc2. Cell 58, 833–846. [DOI] [PubMed] [Google Scholar]
- Prakash S, Johnson RE, and Prakash L (2005). Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 74, 317–353. [DOI] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017). DNA Fiber Analysis: Mind the Gap! Methods Enzymol 591, 55–82. [DOI] [PubMed] [Google Scholar]
- Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, and Menck CFM (2016). Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Research 44, 5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Cybulla E, Meroni A, and Vindigni A (2021). To skip or not to skip: choosing repriming to tolerate DNA damage. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Šviković S, Lemaçon D, Carvajal-Maldonado D, González-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. (2020). PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Molecular Cell 77, 461–474.e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Vessoni AT, Rocha CR, Gottifredi V, Biard D, Sarasin A, Menck CF, and Stary A (2014). Gap-filling and bypass at the replication fork are both active mechanisms for tolerance of low-dose ultraviolet-induced DNA damage in the human genome. DNA Repair (Amst) 14, 27–38. [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, and Lopes M (2012). Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol 19, 417–423. [DOI] [PubMed] [Google Scholar]
- Schiavone D, Jozwiakowski SK, Romanello M, Guilbaud G, Guilliam TA, Bailey LJ, Sale JE, and Doherty AJ (2016). PrimPol Is Required for Replicative Tolerance of G Quadruplexes in Vertebrate Cells. Molecular Cell 61, 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J, Reissner T, Chaney S, Friedberg EC, Wang Z, Carell T, et al. (2009). Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J 28, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Hicks JK, Chute CL, Brennan JR, Ahn JY, Glover TW, and Canman CE (2012). REV1 and polymerase zeta facilitate homologous recombination repair. Nucleic Acids Res 40, 682–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoneau A, Xiong R, Zou L, 2021. The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes Dev. 35, 1271–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, et al. (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- Sonoda E, Okada T, Zhao GY, Tateishi S, Araki K, Yamaizumi M, Yagi T, Verkaik NS, van Gent DC, Takata M, et al. (2003). Multiple roles of Rev3, the catalytic subunit of polzeta in maintaining genome stability in vertebrates. EMBO J 22, 3188–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šviković S, Crisp A, Tan-Wong SM, Guilliam TA, Doherty AJ, Proudfoot NJ, Guilbaud G, and Sale JE (2019). R-loop formation during S phase is restricted by PrimPol-mediated repriming. The EMBO Journal 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang J-W, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Molecular Cell 68, 414–430.e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temviriyanukul P, van Hees-Stuivenberg S, Delbos F, Jacobs H, de Wind N, and Jansen JG (2012). Temporally distinct translesion synthesis pathways for ultraviolet light-induced photoproducts in the mammalian genome. DNA Repair (Amst) 11, 550–558. [DOI] [PubMed] [Google Scholar]
- Thakar T, Leung W, Nicolae CM, Clements KE, Shen B, Bielinsky AK, and Moldovan GL (2020). Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nat Commun 11, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F, Sharma S, Zou J, Lin SY, Wang B, Rezvani K, Wang H, Parvin JD, Ludwig T, Canman CE, Zhang D, 2013. BRCA1 promotes the ubiquitination of PCNA and recruitment of translesion polymerases in response to replication blockade. Proc Natl Acad Sci. USA 110, 13558–13563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirman S, Cybulla E, Quinet A, Meroni A, and Vindigni A (2020). PRIMPOL ready, set, reprime! Crit Rev Biochem Mol Biol, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisman A, and Woodgate R (2017). Translesion DNA polymerases in eukaryotes: what makes them tick? Crit Rev Biochem Mol Biol 52, 274–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallerga MB, Mansilla SF, Federico MB, Bertolin AP, and Gottifredi V (2015). Rad51 recombinase prevents Mre11 nuclease-dependent degradation and excessive PrimPol-mediated elongation of nascent DNA after UV irradiation. Proc Natl Acad Sci U S A 112, E6624–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoli F, Fumasoni M, Szakal B, Maloisel L, and Branzei D (2010). Replication and recombination factors contributing to recombination-dependent bypass of DNA lesions by template switch. PLoS Genet 6, e1001205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Lou J, Xia Y, Su B, Liu T, Cui J, Sun Y, Lou H, and Huang J (2013). hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO reports 14, 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AT, Kim T, Wagner JE, Conti BA, Lach FP, Huang AL, Molina H, Sanborn EM, Zierhut H, Cornes BK, et al. (2015). A Dominant Mutation in Human RAD51 Reveals Its Function in DNA Interstrand Crosslink Repair Independent of Homologous Recombination. Mol Cell 59, 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, and Yamaizumi M (2004). Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 23, 3886–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszek JL, Chatterjee N, Najeeb J, Ramos A, Lee M, Bian K, Xue JY, Fenton BA, Park H, Li D, et al. (2019). A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 178, 152–159 e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M, Quinet A, Lin YL, Davis AA, Pasero P, Ayala YM, and Vindigni A (2020). TDP-43 dysfunction results in R-loop accumulation and DNA replication defects. J Cell Sci 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Chow BL, Broomfield S, and Hanna M (2000). The Saccharomyces cerevisiae RAD6 group is composed of an error-prone and two error-free postreplication repair pathways. Genetics 155, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A, et al. (2017). ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 31, 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Prakash S, and Prakash L (2012). Requirement of Rad18 protein for replication through DNA lesions in mouse and human cells. Proc Natl Acad Sci U S A 109, 7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, and Chen J (2004). DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol 24, 9478–9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, and Lopes M (2015). Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao GY, Sonoda E, Barber LJ, Oka H, Murakawa Y, Yamada K, Ikura T, Wang X, Kobayashi M, Yamamoto K, et al. (2007). A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol Cell 25, 663–675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data is available in the main text, supplemental materials, or via Mendeley at https://dx.doi.org/10.17632/2sc5bwyw6s.1.
The paper does not report any original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.