

Abstract
Kirsten Rat Sarcoma (KRAS) is a master oncogene involved in cellular proliferation and survival and is the most commonly mutated oncogene in all cancers. Activating KRAS mutations are present in over 90% of pancreatic ductal adenocarcinoma (PDAC) cases and are implicated in tumor initiation and progression. Although KRAS is a critical oncogene, and therefore an important therapeutic target, its therapeutic inhibition has been very challenging, and only recently specific mutant KRAS inhibitors have been discovered. In this review, we discuss the activation of KRAS signaling and the role of mutant KRAS in PDAC development. KRAS has long been considered undruggable, and many drug discovery efforts which focused on indirect targeting have been unsuccessful. We discuss the various efforts for therapeutic targeting of KRAS. Further, we explore the reasons behind these obstacles, novel successful approaches to target mutant KRAS including G12C mutation as well as the mechanisms of resistance.
Keywords: KRAS, Pancreatic cancer, KRASG12C, KRAS inhibitor, KRASG12C inhibitors, PROTAC, KRAS vaccine
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer. It is the 4th most common cause of cancer-related mortality in the USA with over 45,000 estimated deaths each year, and a consistently rising incidence rate [1–3]. PDAC remains one of the most recalcitrant malignancies with an overall 5-year survival rate of only 10%, which is the lowest among all cancers [3]. Diagnosis at a late stage, when the tumor had already metastasized, and the limited therapeutic options contribute to this poor prognosis [2].
The four most common genetic alterations in PDAC are activating mutations in KRAS and inactivating mutations in CDKN2A, TP53, and SMAD4 [4–6]. In 2016, Bailey et al. performed integrated genomic analysis of 456 PDAC samples and found 4 molecular subtypes including squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine exocrine (ADEX). The ADEX PDAC was shown to activate KRAS. Overall, the recurrently mutated genes were found to aggregate in several pathways including KRAS, G1-S cell cycle checkpoint, TGF-β, and WNT [7]. KRAS is the most commonly mutated oncogene in pancreatic cancer, found in over 95% of cases, and in approximately 90% of PDAC cases in particular, and is considered a master oncogene that regulates several cellular proliferation and survival pathways [4, 7]. Therefore, KRAS has been considered as the ultimate target for PDAC therapy. To date, several attempts have been made to target KRAS, most of which have been unsuccessful, which highlighted that it is notoriously difficult to target, and it was considered undruggable for a long time [8, 9]. There are several reasons behind this difficulty in drugging KRAS. First of all, GTP competitive inhibitors could not be developed due to the high affinity of KRAS for GTP in the picomolar range, whilst GTP is ubiquitously present in the cell in the sub-millimolar range, making competitive inhibition thermodynamically unfavorable [10, 11]. Other than the nucleotide binding pocket, no allosteric regulatory pocket and no other deep hydrophobic pockets were identified that were suitable for small molecule inhibitor development [12]. Nonetheless, the mutant KRAS G12C was successfully targeted recently, using mutant-specific covalent inhibitors that bind a novel allosteric pocket [13], which resulted in the FDA approval of KRAS G12C specific inhibitor AMG-510 (sotorasib). Although still challenging, perhaps it is now possible to view KRAS as a potentially druggable target.
In this review, we describe the different research efforts and the various strategies to target KRAS in PDAC. We discuss KRAS signaling and the role of KRAS in pancreatic cancer. Then, we discuss challenges and previous failed efforts to develop therapies to target KRAS and its pathways. Next, we examine some of the new and emerging therapeutics that target KRAS both directly and indirectly, including the new mutant specific KRAS G12C inhibitors, and recent efforts to target other KRAS mutants that are of interest for pancreatic cancer. We discuss several new therapeutics both in clinical and preclinical development, and promising emerging targeted therapy combinations. Finally, we discuss mechanisms of resistance that are associated with inhibiting KRAS signaling.
2. KRAS mutations in pancreatic cancer
2.1. KRAS signaling
The rat sarcoma (RAS) family of genes, comprising Kirsten RAS (KRAS), Harvey RAS (HRAS), and neuroblastoma RAS (NRAS), are frequently mutated in cancer [14]. The KRAS gene encodes a small GTPase protein that functions as a molecular switch for critical intracellular signaling pathways. KRAS cycles between an inactive GDP-bound state and an active GTP-bound state. In quiescent cells, KRAS is predominantly found in the inactive GDP-bound state. Upon activation of cellular transmembrane receptors such as EGFR by an extracellular signal, GDP is exchanged for GTP, resulting in KRAS activation and propagation of the signal through downstream effector pathways (Fig. 1). This exchange is facilitated by guanine nucleotide exchange factors (GEF)s, whereas intrinsic KRAS GTP hydrolysis is accelerated by GTPase activating proteins (GAPs). GEFs and GAPs tightly control KRAS cycling in normal cells, where GEFs ensure GTP exchange only in the presence of extracellular stimuli and GAPs ensure fast GTP hydrolysis and return to the inactive state [15, 16]. GTP-bound KRAS can activate a myriad of downstream effector pathways, including RAF/MAPK and PI3k/AKT signaling pathways, which results in cellular growth, proliferation, and survival.
Fig. 1. Schematic illustration of KRAS pathway and targeted preclinical and clinical agents.
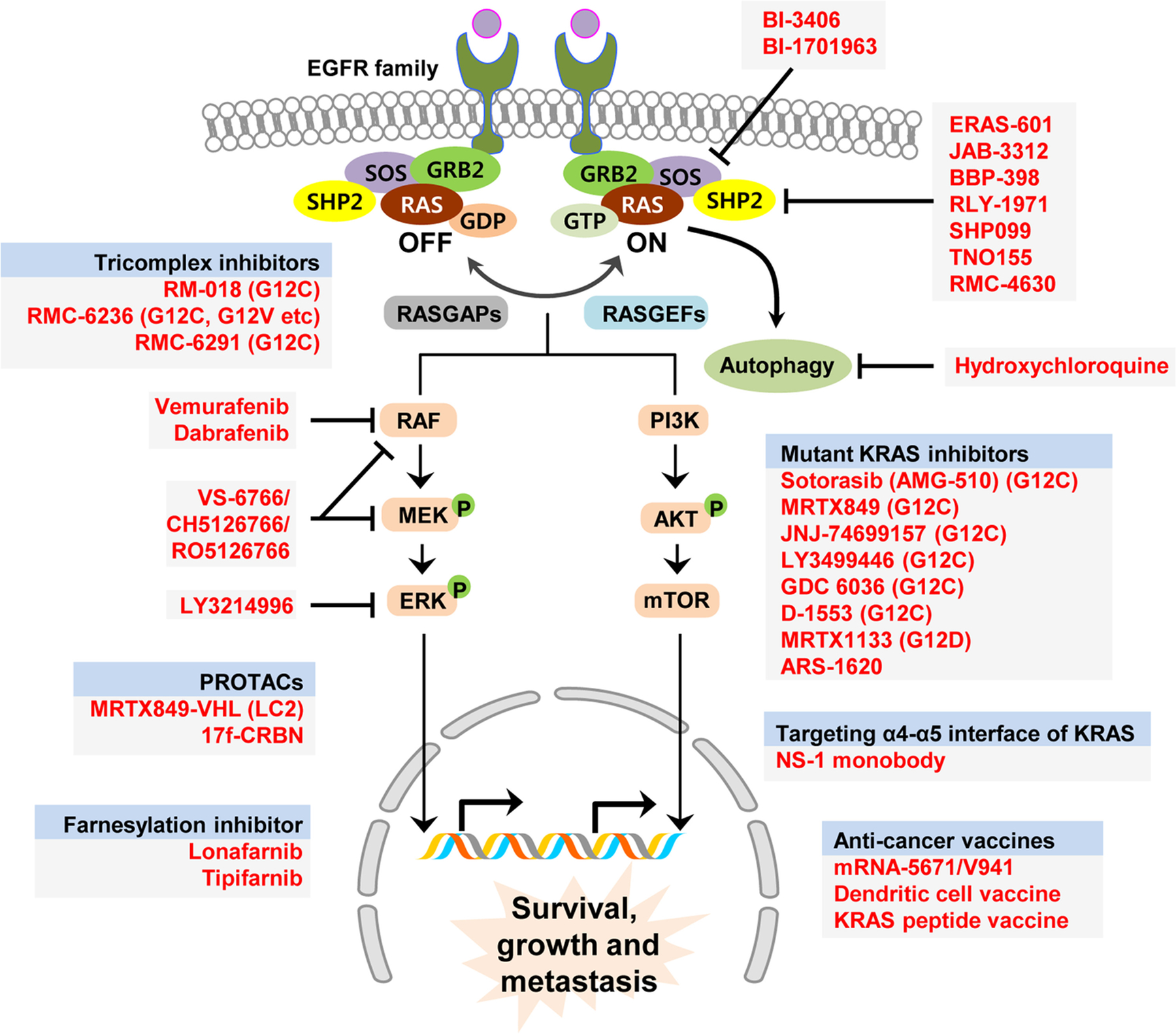
Receptors in the EGFR family upstream of KRAS transmit extracellular signals to activate KRAS. KRAS is activated via GDP to GTP exchange catalyzed by GEFs such as SOS, whereas GAPs catalyze the GTPase activity of KRAS. Downstream signaling pathways include the RAF/MEK/ERK and PI3K/AKT/mTOR pathways, which result in the expression of genes that promote survival, growth, and metastasis. Inhibitors or other targeted agents in the upstream or downstream of KRAS signaling have been depicted. RASGAPs RAS GTPase activating proteins, RASGEFs RAS guanine exchange factors, CRBN cereblon, PROTACs proteolysis targeting chimeras, VHL Von Hippel–Lindau tumor suppressor
2.2. KRAS mutations
Mutant forms of KRAS have an impaired ability to hydrolyze GTP, resulting in an aberrant hyperactive state that activates downstream signaling pathways leading to increased cellular proliferation [17]. Activating KRAS mutations are considered a major driver of pancreatic ductal adenocarcinoma (PDAC). A point mutation in codon 12 of the KRAS oncogene is the most frequent mutation and is found in the majority of PDAC cases [5, 14]. This mutation results in a single amino acid substitution of glycine by aspartic acid in G12D mutations (45%), valine in G12V mutations (35%), arginine in G12R mutations (17%), alanine in G12A mutations, or cysteine is G12C mutations [18]. These missense mutations inhibit the ability of GAPs to accelerate GTP hydrolysis, resulting in excessive activation of KRAS [19]. Less frequent mutations include G13, Q61, K117, and A146. Despite progress in targeting KRAS G12C, these mutations represent a minority of KRAS mutations in PDAC. The major driver mutations in PDAC are G12D and G12V which collectively account for approximately 80–90% of KRAS driver mutations in PDAC [20]. Therefore, targeting these mutations has the potential of having a significant impact on PDAC therapeutics.
Oncogenic KRAS activation is influenced by the specific mutation and the presence of extracellular stimuli. Studies have shown that mutant KRAS undergoes rapid nucleotide cycling [21, 22] and requires activation by upstream signals for prolonged activation of KRAS and enhanced downstream signaling [21, 23, 24], supporting that KRAS is not locked in a static active state, and its activation is modulated by upstream signaling. Interestingly, biochemical profiling of the different mutant KRAS forms revealed intrinsic differences in the kinetics and biochemical properties between them [25]. Intrinsic GTP hydrolysis activity in the mutant forms differs according to the specific point mutation. Among G12 mutants, G12C, G12D, G13D mutant forms have high intrinsic GTPase activity, whereas G12A, G12R, G12V mutants have a low intrinsic GTP hydrolysis rate [25]. Mutants also differ in their sensitivity to GAP-mediated GTP hydrolysis and affinity for RAF kinase. Moreover, studies have shown that different KRAS mutations result in biological differences in downstream pathway activation in the tumor and in response to therapy [26–28]. Studies have also demonstrated that different KRAS mutations could be associated with prognosis and different disease outcomes in pancreatic cancer [28–31]. This implies that the specific KRAS mutations possibly have a variable impact on tumor biology. These biochemical and physiological differences may have implications on which approach would be more suitable to target the various KRAS mutant forms.
Several critical downstream effector pathways are activated by oncogenic KRAS signaling. The three major effector pathways are the RAF-MEK-ERK MAPK pathway, PI3K-AKT-mTOR pathway, and the Ral guanine nucleotide exchange factor (RalGEF) pathway. These pathways have been the target of intense drug development research for various types of cancer, as they are commonly deregulated in various malignancies either by upstream mutations such as RAS or receptor tyrosine kinases (RTKs), or activating mutations of proteins within these pathways [32–34]. KRAS and downstream effector pathways regulate several cellular processes in pancreatic cancer. There is a plethora of evidence that mutant KRAS signaling is involved in promoting cellular growth, proliferation, survival, and metastasis. More recently, accumulating evidence is showing that KRAS mutations are associated with the regulation of diverse cellular processes in pancreatic cancer including autophagy, macropinocytosis, metabolic perturbations, and modulation of the tumor microenvironment [35–40].
3. KRAS in pancreatic cancer
3.1. Role of KRAS in pancreatic cancer development and subsistence
PDAC is thought to progress through a multistage process, where premalignant lesions accumulate mutations as they progress into malignant invasive carcinoma [41]. These lesions are known as pancreatic intraepithelial neoplasia (PanINs) and are classified according to the degree of cellular dysplasia from PanIN1 to PanIN3. KRAS mutations are found at the earliest stage of PDAC development (PanIN1) [42], suggesting that it is important for tumor initiation, and additional mutations in other genes are required for tumor progression. Several transgenic and genetically engineered mouse models (GEMMs) have been used to elucidate the role of mutant KRAS in PDAC initiation and progression [43–45]. The multistage development of PanIN lesions into invasive cancer has been recapitulated in the transgenic KRAS G12D mouse model [43, 44]. In those studies, all of the transgenic mice develop PanIN lesions, but only a subset of the animals develop invasive disease. These results show that mutant KRAS drives cancer initiation, but acquisition of further mutations is required for progression. Studies in GEMMs also explored whether mutant KRAS signaling is still required at later stages of tumor development, well after initiation [46–48]. An inducible mutant KRAS model has been developed, where KRAS G12D expression can be reversibly induced in the pancreata of mice. In this model, turning off oncogenic KRAS expression in established pancreatic and metastatic tumors resulted in tumor regression [46–48]. Collectively, these studies have shown that KRAS oncogenic signaling is critical for both initiation and maintenance of pancreatic cancer; therefore, it is the ideal target for therapy.
3.2. Role of KRAS in pancreatic microenvironment and metastasis
Pancreatic cancer has a dense fibrotic stroma which is composed of various different components including the extra cellular matrix (ECM), cancer associated fibroblasts (CAFs), the vasculature, and immune cells and has been shown to have a dichotomous role that can be pro-tumorigenic or anti-tumorigenic [49–51]. Oncogenic KRAS is implicated in the regulation of the tumor microenvironment (TME) and the recruitment of pro-tumorigenic cells resulting in tumor invasion and metastasis [40, 52]. CAFs are the major cell type within the pancreatic cancer stroma, and evidence shows they have the capacity to promote tumor growth and progression. On the other hand, pancreatic tumor cells secrete a variety of factors and proteins that can activate and alter the function of CAFs and other cells in the TME. The hedgehog signaling pathway has been identified as a critical regulator of pancreatic cancer initiation and maintenance [53]. Pancreatic tumor cells secrete hedgehog ligand, which acts on nearby CAFs promoting a desmoplastic response [54, 55]. The role of hedgehog pathway in pancreatic cancer remains controversial and has been shown to have a dichotomous dose-dependent response that could either promote or inhibit tumor growth [56]. Recently, mutant KRAS has been shown to be involved in the activation of paracrine hedgehog signaling, and the promotion of reciprocal signaling between pancreatic tumor cells and the stroma [57]. In a proteomic and phosphoproteomic approach, it was found that KRAS G12D mutated tumor cells secrete sonic hedgehog, which activates pancreatic stellate cells, which are a subtype of CAFs, resulting in widespread alterations in the stellate cells, which in turn secrete growth factors that promote the desmoplastic response and also signal back to the tumor cells [57].
In addition to governing this crosstalk between tumor cells and CAFs, oncogenic KRAS has been implicated in regulating the immune microenvironment by promoting inflammation and immune evasion [40, 52]. KRAS mutations have been correlated with low immune cell infiltration in PDAC which is associated with a poorer prognosis [58]. Mechanisms controlling this immune evasion are not completely understood, but are likely to involve crosstalk between multiple cells including KRAS-driven tumor cells, CAFs, myeloid derived tumor suppressor cells, and tumor associated macrophages [59]. In a study using a pancreatic cancer syngeneic mouse model, KRAS knockout (KO) or KRAS G12D pancreatic tumor cells were injected into the pancreata of mice and monitored for tumor growth. Mice injected with KRAS KO cells rejected the cells or only grew tumors after a long latency period and were highly infiltrated by B and T lymphocytes, whereas cells with oncogenic KRAS grew rapidly, were able to evade immune regulation, and lacked B and T cell infiltration [60], suggesting a direct role for KRAS activation in promoting an immune-evasive microenvironment. Additionally, direct targeting of KRAS using G12C specific inhibitors has been shown to alter the immune microenvironment by increasing the infiltration of immune cells including CD8+ T-cells, increasing cytokine signaling, and antigen presentation [61, 62]. Taken together, these data demonstrate the essential role of KRAS in maintaining an immunosuppressive TME and an opportunity to combine KRAS targeting with immunotherapy for sustained responses to therapy.
4. Challenges in Kras targeted therapy in PDAC
4.1. Targeting KRAS directly
KRAS is a critical oncogene in over 80–90% of PDAC cases; therefore, it is a prime target in these oncogene addicted tumors. Significant research efforts have been focused on targeting KRAS both directly and indirectly (Fig. 1). Despite this, KRAS remains an elusive target in pancreatic cancer as various targeted therapies have failed to elicit sufficient clinical benefit. As previously mentioned, the structure of the KRAS protein lacking deep hydrophobic pockets, and its high affinity to GTP, prevented the development of effective KRAS inhibitors. Recently, mutant KRAS inhibitors have been developed which will be discussed later in this review.
Due to difficulties in drugging KRAS directly, researchers investigated various methods of indirect targeting (Table 1). These efforts have focused on abrogating KRAS signaling through inhibiting its activation, or its downstream pathways. Post-translational prenylation of KRAS, which is catalyzed by farnesyl transferases (FTases), is required for its membrane localization and consequently signal transduction [63, 64]. FTase inhibitors (FTIs) have been designed to target KRAS farnesylation aiming to prevent its membrane localization, thereby preventing its activation, and some have been clinically investigated, including drugs such as lonafarnib and tipifarnib [65–67]. Despite initial excitement about FTase inhibitors for KRAS mutant PDAC, some of which progressed to phase III clinical trials for various cancer types, results in PDAC have been disappointing eventually [68–70]. In the presence of FTIs, KRAS was alternatively prenylated by geranylgeranyl transferase 1 (GGTase1) and targeted to the plasma membrane [71–73]. This showed that KRAS mutant tumors have the intrinsic ability to resist FTI therapy by alternative prenylation. A potential approach to combat this resistance is the dual inhibition of FTase and GGTase1 activity [74]. However, these approaches have a limited therapeutic window as GGTase 1 inhibitors have wide ranging targets.
Table 1.
Unsuccessful studies in targeting KRAS in pancreatic cancer
| Agent | Rationale | Mechanism of resistance | Reference |
|---|---|---|---|
| Farnesyl transferase inhibitors (FTIs) | Prevention of KRAS plasma membrane localization | Alternative prenylation by geranyl geranyl transferases, which allows KRAS localization at the plasma membrane | [68–70] |
| RAF inhibitors (e.g., vemurafenib) | Inhibition of KRAS mediated RAF activation and downstream signaling | RAF inhibitors bound to wild type RAF in KRAS mutant cells, resulted in the formation of RAF dimers that activated downstream signaling | [77–80] |
| mTOR inhibitors (e.g., everolimus) | Inhibition of PI3K pathway downstream of KRAS | Activation of alternative downstream pathways; Paradoxical AKT phosphorylation and cyclin D1 activation leading to increased proliferation. | [85, 86] |
4.2. Targeting KRAS downstream effector pathways
The RAF-MEK-ERK MAPK pathway is one of the best characterized pathways downstream from KRAS, and it has been intensively studied as a therapeutic target for KRAS mutant tumors [75]. Active KRAS induces the phosphorylation and dimerization of RAF, thereby activating it. Consequently, RAF phosphorylates MEK1 and MEK2, which in turn phosphorylate ERK1 and ERK2 [76]. ERK subsequently activates transcription factors that promote cellular proliferation. The activation of this MAPK pathway downstream of KRAS is critical for its oncogenic signaling. Several RAF, MEK, and ERK inhibitors have been developed, and some have been clinically studied in PDAC. RAF inhibitors vemurafenib and dabrafenib have been successful for the treatment of BRAF mutated tumors but failed in KRAS-mutant cancers. Both vemurafenib and dabrafenib resulted in the paradoxical transactivation of RAF dimers in BRAF-wild type KRAS-mutant cancers and therefore resulted in the propagation rather than the suppression of downstream signaling [77–80]. Pan-RAF inhibitors that target both monomers and dimers of RAF could potentially overcome its paradoxical activation [81].
The PI3K-AKT-mTOR pathway, which can be activated by KRAS signaling, has also been the center of multiple drug development efforts as it is involved in various cancers [33]. There are three classes of PI3Ks in the human cell. GTP-bound KRAS activates Class I PI3Ks, which in turn recruit AKT to the plasma membrane, resulting in the activation of mTOR. Several AKT substrates are involved in the regulation of diverse functions such as cellular growth, proliferation, survival, and metabolism [82]. The PI3K-AKT-mTOR pathway is upregulated in pancreatic neuroendocrine tumors (PNETs) [83]. The mTOR inhibitor everolimus showed benefit for patients with PNETs in a phase III clinical trial, resulting in its FDA approval for advanced PNETs [84]. However, PI3K pathway inhibitors failed as single agents in PDAC [85, 86], possibly because of interconnected signaling pathways downstream of KRAS which could be alternatively activated upon the inhibition of one pathway. Therefore, targeting both PI3K and MAPK pathways has emerged as a potential therapeutic strategy [87, 88]. Preclinical results have shown strong synergy between inhibitors targeting the two pathways. Clinical trials however have not been successful in translating those results so far.
5. New and emerging strategies for targeting KRAS
5.1. Mutation-specific direct targeting of KRAS
Efforts to target KRAS directly have resulted in the development of KRAS G12C mutant-selective small molecule inhibitors [8, 13, 21, 22, 61, 89–91]. Using X-ray crystallography and mass spectrometry, Shokat and colleagues identified the druggable switch-II pocket in KRAS G12C where covalent small molecule inhibitors can bind [13]. They developed a series of compounds that irreversibly bind this pocket resulting in the disruption of switch-I and switch-II regions of KRAS making the GDP-bound state more favorable. These inhibitors effectively lock KRAS in the inactive GDP-bound state, preventing its binding to RAF and the activation of downstream signaling pathways. The reactivity of the cysteine found in KRAS G12C, which is a result of the missense mutation in codon 12, allows it to be exploited for the development of covalent small molecule inhibitors [92]. Since these inhibitors specifically bind the cysteine in the mutant form, they are mutant-specific and are expected to spare wild-type cells that lack this mutation, thereby reducing off-target toxicities.
Subsequent drug discovery efforts by multiple groups resulted in the development of several more potent covalent inhibitors of KRAS (G12C), including sotorasib, adagrasib (MRTX849), JNJ-74699157, and LY3499446, which rapidly moved into clinical investigation. Sotorasib has shown promising results in a clinical trial (NCT03600883) and has been given accelerated approval by the FDA in May 2021 for non-small cell lung cancer (NSCLC) [93]. Results from a phase II clinical trial of sotorasib in previously treated NSCLC (NCT03600883) have been recently reported [94]. The results are promising with 37.1% of the patients achieving an overall objective response (ORR), disease control (DCR) achieved in 80.6% of patients, and a median progression free survival of 6.8 months. A phase III trial is currently underway (NCT04303780). However, sotorasib has not been extensively examined in patients with pancreatic cancer.
MRTX849 is currently in clinical trials for patients with KRAS G12C mutant cancers, including pancreatic (NCT03785249). In phase I and II clinical trials, NSCLC patients achieved 45% objective response rate (ORR) and 96% disease control rate (DCR). The compound is currently in phase III clinical trial for NSCLC (NCT04685135) and colorectal cancer patients (NCT04793958). In preclinical studies, MRTX849 showed potent inhibition of cellular proliferation in a pancreatic cancer cell line [95]. One patient with pancreatic cancer has been reported to have a confirmed partial response in the phase I/Ib cohort (NCT03785249).
Another G12C inhibitor, LY3499446, was being investigated in a phase I clinical trial (NCT04165031). Despite promising preclinical results, the clinical trial has been terminated due to toxicities. Additional KRAS G12C inhibitors are in clinical trials including JNJ-74699157, GDC 6036, and D-1553. Details about these inhibitors have not been published, and data from ongoing clinical trials have not been reported.
KRAS G12C is the predominant KRAS mutation in NSCLC and is associated with tobacco mutation signatures [96]. However, G12D is the most prevalent KRAS mutation in PDAC, and only a small subset of patients harbors the G12C mutation. Therefore, most PDAC patients cannot derive benefit from these new inhibitors. Nonetheless, efforts to target KRAS G12D are underway. Various groups are working on developing new approaches and lead compounds to target GTP-bound KRAS G12D. A cyclic peptide, KD2, can selectively target GTP-bound KRAS G12D and has been shown to have the ability to access and bind the switch-II groove in mutant KRAS [97]. This shows that it is possible to drug GTP-bound KRAS G12D, but these cyclic peptides do not readily enter the cell, and further development of more drug-like compounds is required [98]. A bicyclic peptide called KS-58 showed activity against KRAS G12D mutated pancreatic cancer both in vitro and in vivo [99]. These studies provide proof-of-concept evidence that KRAS G12D mutation could be potentially targeted, which would benefit a larger number of PDAC patients. A small molecule inhibitor of mutant KRAS G12D called MRTX1133 is reported to selectively target the G12D mutant form of KRAS and result in regression of tumors harboring the mutation [100]. This drug candidate is in preclinical development, and details have not been published.
A new class of KRAS small molecule inhibitors, termed tricomplex inhibitors, that target the GTP-bound form of specific KRAS mutants is being developed (Fig. 2). These inhibitors bind in a novel pocket that is formed between the target mutant KRAS and a chaperone protein, cyclophilin A [101], thereby overcoming the challenge of finding a suitable pocket for inhibitors in KRAS. When tricomplex inhibitors enter the cell, they associate with Cyclophilin A, which is ubiquitous in the cell, forming a bi-complex, which then binds to KRAS via protein-protein surface interaction, resulting in the tricomplex [101]. The association of the chaperone with KRAS physically prevents it from binding to downstream effector proteins, thus blocking further signal transduction. The inhibitor RM-018, which is a prototype of G12C tricomplex inhibitors, is active against KRAS G12C mutated cancer cells that are resistant to other mutant-specific inhibitors [101]. A closely related tricomplex inhibitor of the GTP-bound active KRAS G12C, RMC-6291, is in preclinical development and on track to go to clinical trials. Additionally, a tricomplex multi-RAS inhibitor, RMC-6236, which can target multiple different mutations of KRAS, including KRAS G12V and different forms of RAS is also being developed [98]. Details of these inhibitors and their activity have not been published.
Fig. 2. Various novel approaches to target mutant KRAS.
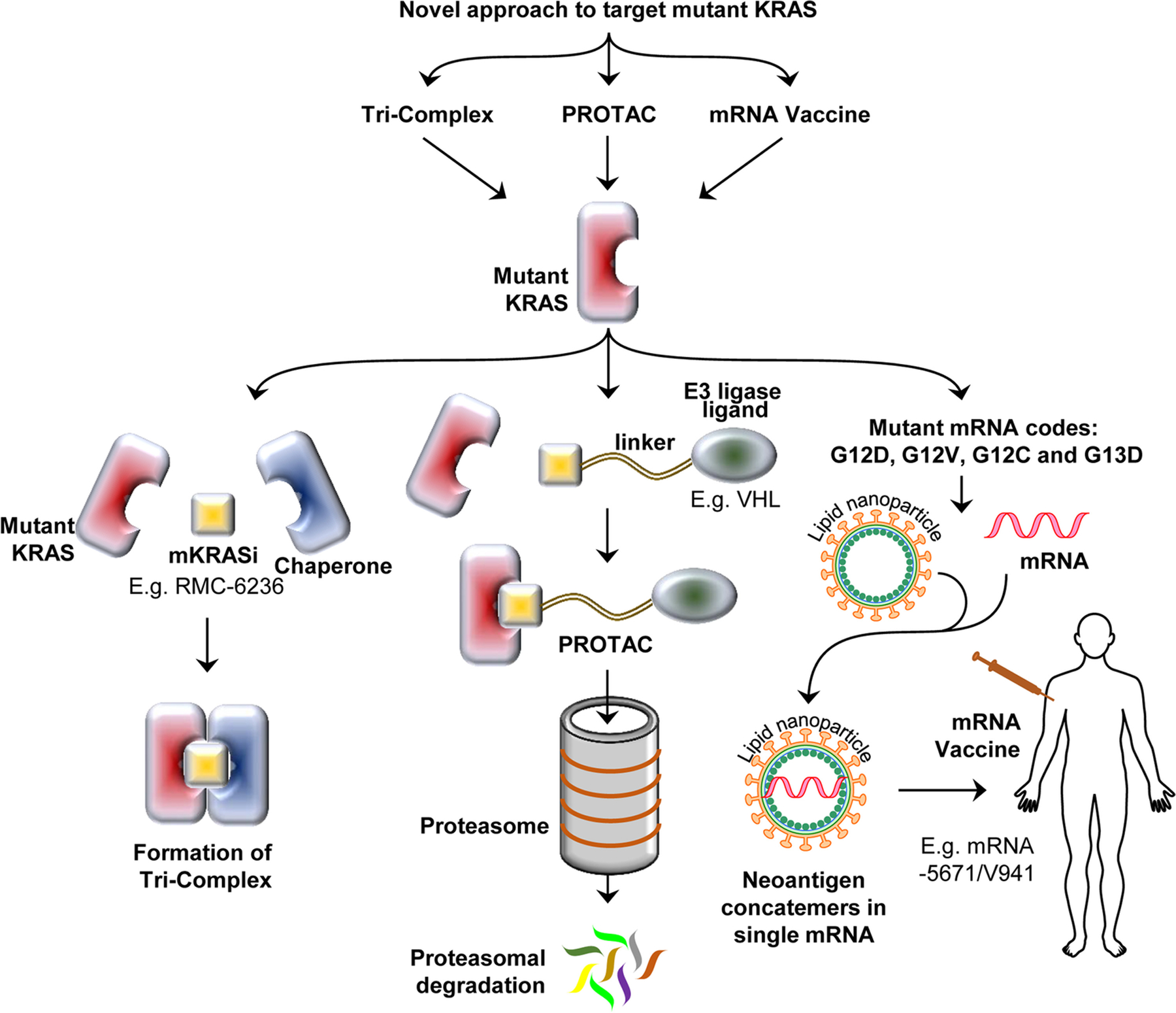
Basic concepts of targeting mutated KRAS using tricomplex inhibitors, PROTACs, and mRNA vaccine have been illustrated in the figure. Tricomplex inhibitors utilize an endogenous chaperone protein to create a novel pocket for small molecule KRAS inhibitors. PROTACs recruit an endogenous E3 Ligase to mutant KRAS, thus targeting it for proteasomal-mediated degradation. mRNA vaccines utilize the mRNA code of mutant KRAS, encapsulated in a lipid nanoparticle to stimulate the immune response against tumor-associated neoantigens. mKRASi mutant KRAS inhibitor, PROTACs proteolysis targeting chimeras, VHL Von Hippel–Lindau tumor suppressor
5.2. Non-mutation specific inhibitors
5.2.1. SOS1 interaction inhibition
Son of Sevenless (SOS1) is a guanine nucleotide exchange factor (GEF) that acts as an activator of KRAS[16]. SOS1 binds GDP-bound KRAS and catalyzes the exchange of GDP for GTP. SOS1 is critical for the regulation of KRAS signaling, and its depletion results in reduced cellular proliferation [102]. Therefore, the binding of SOS1 to KRAS emerged as a potential therapeutic strategy to target KRAS. Several molecules that block the interaction between SOS1 and KRAS have been developed [103, 104]. BI-3406 is a potent SOS1 inhibitor that is orally bioavailable, with anti-proliferative activity against cells harboring various KRAS mutations, but not on KRAS wild-type cells [105]. BI-3406 was shown to inhibit cellular proliferation and KRAS downstream signaling, and was synergistic with MEK inhibitors [105]. BI-1701963 is a SOS1 inhibitor derived from BI-3406, which is currently being evaluated in a phase I clinical trial (NCT04111458) as a single agent or in combination with trametinib (MEK inhibitor) in patients with KRAS mutated cancers. Results have not been published.
5.2.2. SHP2 inhibitors|
The protein SHP2 is a tyrosine phosphatase encoded by the gene PTPN11 that is involved in signal transduction in various signaling pathways including KRAS MAPK signaling. Its role in KRAS signaling is still incompletely understood, but it is thought to recruit other critical proteins such as SOS1 and GRB2 [106–108]. Although SHP2 is a phosphatase, it has a positive activating role in KRAS signaling and is considered a critical protein in oncogenic signaling. SHP2 plays an important role in cancer development in KRAS-mutated PDAC and NSCLC models, and its deletion delays progression of established PDAC and NSCLC xenografts in mice [109]. Targeting SHP2 showed synergy with MEK inhibition and resulted in tumor growth inhibition in PDAC and NSCLC models in vivo [109]. Moreover, it was shown recently that SHP2 is essential for the remodeling of the vasculature in melanoma and colon cancer tumor models, and its inhibition impairs the tumor vasculature, thus reducing tumor growth [110]. Whether SHP2 plays a similar role in PDAC remains to be seen.
SHP2 has an intrinsic regulatory mechanism, as it is found in a closed conformation due to intramolecular domain-domain interactions that prevents access of its active site [111]. This autoinhibitory state was the basis for a screen to find a molecule that would lock SHP2 in its inactive autoinhibited state [112]. The compound SHP099 was discovered, and it was able to suppress MAPK signaling and reduce tumor growth in vivo [112]. This discovery catalyzed drug development efforts to target SHP2, which resulted in multiple clinical candidates that are in various stages of clinical testing [113]. RMC-4630 is currently in phase I/Ib clinical trials as monotherapy or in combination with an ERK inhibitor (NCT03634982, NCT04916236). RMC-4630 is based on the molecule RMC-4550 which was shown preclinically to inhibit proliferation of tumor cells harboring certain KRAS mutations [114]. Another SHP2 inhibitor, TNO155, is currently in several phase I and II clinical trials as a monotherapy or in combination with various targeted therapies (NCT04000529, NCT04330664, NCT03114319). TNO155 was discovered through structural and property-based design of a lead molecule discovered in a high throughput screening approach to find an allosteric inhibitor of SHP2. TNO155 was able to inhibit MAPK signaling and reduce tumor growth in vivo [115], and was found to be synergistic with several other drugs which are being tested in clinical trials [116]. Several other SHP2 inhibitors are also in clinical trials, including ERAS-601 (NCT04670679), JAB-3312 (NCT04121286), BBP-398 (NCT04528836), and RLY-1971 (NCT04252339). Little detail is available about these inhibitors, and results from the trials are not yet available.
5.2.3. Cancer vaccination
Cancer vaccination has recently emerged as a potential immunological therapeutic strategy. Tumors have neoantigens, and they can be utilized to vaccinate the patient to stimulate the immune system to attack cancer cells that present those antigens. This approach has been mostly studied as a personalized medicine approach, with each patient vaccinated with antigens specific to their tumor [117–120]. Recently, a new mRNA vaccine has entered a phase I clinical trial. mRNA-5671/V941 encodes mutant KRAS and is being investigated in patients with KRAS-mutant tumors (NCT03948763) (Fig. 2). In preclinical models, strong T-cell responses were elicited after vaccination in mice with two different (unspecified) KRAS mutations [110]. The clinical trial is testing the vaccine as a single agent or in combination with the monoclonal anti PD-1 antibody, pembrolizumab. Other types of vaccination for KRAS mutated patients are also in clinical trials, including dendritic cell vaccines (NCT03592888) and peptide vaccines (NCT04117087).
5.2.4. Adoptive cell therapies
Adoptive cell transfer is an immunotherapeutic approach where the patient’s lymphocytes, which either have anti-tumor specificity or are manipulated to target tumor antigens, are expanded ex vivo and then infused back into the patient [121]. Identifying a safe and efficient antigenic target is critical when manipulating the specificity of the lymphocytes [122]. Tumor neoantigens are the most suitable target since they are virtually foreign antigens where central tolerance was not induced, thus increasing efficiency and minimizing toxicities [123]. RAS oncogene has long been known to be immunogenic. Recently, a patient with metastatic colorectal cancer was found to have KRAS G12D reactive T-cells within the tumor-infiltrating lymphocytes (TILs) [124]. The patient was treated with a transfer of an expanded population of TILs with 4 different specificities to mutant KRAS G12D, and all seven of the patient’s metastatic lesions regressed [124]. This showed that adoptive transfer of T-cells with mutant KRAS specificity is a valid therapeutic approach that could potentially benefit patients with these mutations. Two clinical trials were started to investigate a therapeutic approach that uses the patient’s peripheral lymphocytes which are transduced with murine T-cell receptors that target mutant KRAS G12D (NCT03745326) or G12V (NCT03190941). These therapies are HLA-restricted, which is required for the recognition of the tumor cells by the mutant KRAS specific T cells. Although these therapies were promising, the trials have since been suspended, and no results have been reported. It remains to be seen whether any other cell-based immunologic therapies would be developed and would benefit pancreatic cancer patients.
5.3. Emerging strategies in preclinical development
5.3.1. PROTAC
Proteolysis targeting chimeras (PROTACs) are an emerging therapeutic approach to target proteins of interest for degradation, and it is an especially attractive area of research for protein targets that are considered undruggable using small molecule inhibitors (Fig. 2) [125]. PROTACs are composed of two peptides that are linked together: one is a ligand of the target protein, and the other recruits an E3 ubiquitin ligase to ubiquitinate the target protein and mark it for proteasomal degradation. A PROTAC called LC-2 that targets mutant KRAS G12C has been developed which is composed of MRTX849 linked to a VHL recruiter peptide [126]. MRTX849 binds covalently to mutant KRAS, and then VHL E3 ligase is recruited and mutant KRAS is destroyed by ubiquitin-mediated proteasomal degradation [126]. LC-2 resulted in sustained degradation of KRAS and inhibition of MAPK signaling [126]. Another PROTAC was developed to target PDEδ, which is critical for KRAS diffusion in the cytosol and its plasma membrane localization [127]. The compound 17f was able to degrade PDEδ, diminish KRAS signaling, and inhibit cellular proliferation and tumor growth in a colorectal cancer model with mutated KRAS [128]. For implementation in the clinic, these compounds need further development to enhance the efficacy and potency. These preclinical studies represent proof of concept for a promising novel therapeutic approach for KRAS.
5.3.2. α4–α5 interface inhibitors
A monobody termed NS-1 has been developed as a biologic that targets the α4–α5 interface of KRAS [129–131]. This inhibitor prevents the dimerization of RAS, thus blocking RAF activation and downstream signaling [131]. Intracellular expression of NS-1 was able to prevent the formation and progression of pancreatic cancer in mice [132]. This shows that targeting this new site is a potential strategy for KRAS inhibition. However, cellular permeability of large molecules such as NS-1 is an obstacle, and thus further drug development is required to target the α4–α5 interface.
6. Novel combinations
KRAS signaling pathways are highly diverse and interconnected; therefore, abrogating KRAS signaling is not a trivial task. We highlight here some of the novel targeted therapy combinations that are in clinical and preclinical testing (Tables 2 and 3). Combinations have the advantage of potentially targeting multiple aspects of KRAS signaling at the same time, which could result in more efficacious attenuation of signal transduction. Recently, genomic studies have revealed co-occurring mutations in KRAS mutated PDAC which can be utilized to develop potential therapeutic combinations that target the genes and the pathways that are involved in PDAC [7, 133]. These pathways include MAPK and PI3K-AKT-mTOR pathways downstream of KRAS, as well as EGFR overexpression upstream of KRAS [134, 135]. Additionally, DNA damage repair genes have been implicated in PDAC tumorigenesis, as deficiencies in BRCA and mismatch repair genes (MMR) have been identified as major mutational mechanisms [7], and therefore represent targetable therapeutic vulnerabilities. Development of combinatorial targeting strategies is an important approach to combat resistance against single agent therapy. One approach is vertical combinations, where multiple proteins of the same pathway are targeted, for instance targeting multiple effectors of the KRAS-RAF-MEK-ERK signaling pathway. The other approach is horizontal targeting, where targets across different KRAS downstream pathways are inhibited at the same time.
Table 2.
Clinical development of strategies to target KRAS in cancer
| Agent | Target | Clinical trial number (Clinicaltrials.gov) | Remarks |
|---|---|---|---|
| Sotorasib (AMG-510) | Direct mutant KRAS G12C covalent inhibitor |
NCT03600883
NCT04185883 NCT04933695 NCT04303780 |
Sotorasib obtained accelerated FDA approval. Ongoing phase Ib, II, and III trials for patients with G12C mutations. |
| MRTX849 | Direct mutant KRAS G12C covalent inhibitor |
NCT04330664, NCT03785249 NCT04685135 NCT04793958 NCT04613596 |
KRYSTAL trials: Phase I/II trials for patients with G12C mutations. Phases II and III trials for NSCLC and CRC patients with G12C mutations. |
| LY3499446 | Direct mutant KRAS G12C covalent inhibitor | NCT04165031 | Trial terminated due to unanticipated toxicities. |
| JNJ-74699157 | Direct mutant KRAS G12C inhibitor | NCT04006301 | Trial completed, no results reported |
| GDC 6036 | Direct mutant KRAS G12C inhibitor | NCT04449874 | Phase I trial for tumors with G12C mutations. |
| D-1553 | Direct mutant KRAS G12C inhibitor | NCT04585035 | Phase I/II trial for tumors with G12C mutations. |
| BI-1701963 | SOS1 inhibitor | NCT04111458 | Phase I trial for solid KRAS mutated tumors. |
| RMC-4630 | SHP2 inhibitor |
NCT03634982
NCT04916236 |
Phase I/Ib trial for tumors with MAPK pathway hyperactivation, including KRAS mutated. |
| TNO155 | SHP2 inhibitor |
NCT04000529
NCT03114319 NCT04330664 |
Phase I and II trials |
| ERAS-601 | SHP2 inhibitor | NCT04670679 | FLAGSHP-1 phase I/Ib trial in advanced or metastatic solid tumors as a single agent or in combination with MEK inhibitor |
| JAB-3312 | SHP2 inhibitor |
NCT04045496
NCT04720976 NCT04121286 |
Phase I/IIa clinical trials for patients with solid tumors |
| BBP-398 | SHP2 inhibitor | NCT04528836 | Phase I trial for patients with advanced solid tumors |
| RLY-1971 | SHP2 inhibitor | NCT04252339 | Phase I trial for patients with advanced solid tumors. However, it excludes patients with KRAS G12D, G12V, G13X, and Q61X mutations. |
| mRNA-5671/V941 | Mutant KRAS mRNA vaccine | NCT03948763 | Phase I trial for KRAS mutated solid tumors |
| KRAS peptide vaccine | Mutant KRAS long peptide vaccine | NCT04117087 | Phase I trial in CRC and PDAC |
| mDC3/8-KRAS Vaccine | Dendritic cell vaccine | NCT03592888 | Phase I trial in KRAS mutated resectable PDAC |
| Anti-KRAS G12D/G12V mTCR PBL | Adoptive cell transfer |
NCT03745326
NCT03190941 |
Trials have been suspended. |
CRC colorectal carcinoma, FDA food and drug administration, mTCR PBL murine T-cell receptor transduced peripheral blood lymphocytes, PDAC pancreatic ductal adenocarcinoma
Table 3.
Preclinical development of novel strategies to target KRAS in pancreatic cancer
| Agent | Target | Mechanism of action | Reference |
|---|---|---|---|
| Tricomplex inhibitors (RMC-6291, RMC-6236) | Direct mutant KRAS inhibitors | Small molecule binds a chaperone protein forming a complex that binds to KRAS in a tricomplex that prevents KRAS-mediated signaling. | [98] |
| KD2 cyclic peptide | Direct mutant KRAS inhibitor | Binds and inhibits the GTP-bound form of mutant KRAS G12D in the switch II groove | [97] |
| KS-58 bicyclic peptide | Direct mutant KRAS inhibitor | Cyclic peptide with unnatural amino acids that binds mutant KRAS G12D | [99] |
| MRTX1133 | Direct mutant KRAS inhibitor | Binds mutant KRAS G12D | [100] |
| NS-1 | α4–α5 interface inhibitor | Monobody that binds KRAS surface preventing dimerization. | [129, 130, 132] |
| LC-2 | PROTAC targeting mutant KRAS | PROTAC composed of MRTX849 linked to a VHL recruiter, resulting in ubiquitin-mediated proteasomal degradation of KRAS G12C | [126] |
| 17f | PROTAC targeting PDEδ | Degradation of PDEδ prevents KRAS localization at plasma membrane, thereby preventing signaling. | [128] |
Novel therapeutic vertical combinations include SHP2 inhibitor combinations with novel KRAS G12C inhibitors, with ERK inhibitors, or with MEK inhibitors. Combining SHP2 inhibitor with KRAS G12C inhibitors ARS-1620 and AMG 510 in a panel of pancreatic, colon, and lung cancer cell lines harboring G12C mutation resulted in sustained KRAS signaling attenuation and prevented feedback reactivation of RAS signaling compared to G12C single agent inhibition [136]. Similar combinations are being investigated clinically; a combination of MRTX849 with TNO155 (SHP2 inhibitor) is being evaluated for solid tumors in a phase I/II clinical trial (NCT04330664), and another trial is being conducted to evaluate the combination of JDQ443, a KRAS G12C inhibitor, with TNO155 or spartalizumab, a PD-1 inhibitor (NCT04699188). These trials are currently recruiting, and results have not yet been reported.
Evidence suggests that SHP2 inhibition is a potential strategy to limit adaptive resistance to ERK pathway inhibition in KRAS mutant cancers [137, 138]. A new phase I/Ib clinical trial is going to study the combination of a SHP2 inhibitor, RMC-4630, with an ERK inhibitor, LY3214996, in patients with metastatic pancreatic, colorectal cancer, and NSCLC which harbor KRAS mutations (NCT04916236). Additionally, combinations of various SHP2 inhibitors with MEK inhibitors are being evaluated in clinical trials (NCT04670679, NCT03989115).
Additionally, KRAS G12C inhibitors are in combinatorial trials with various targeted and immunological therapies. Although checkpoint inhibitors have had great success in various cancers, most trials in PDAC did not show significant benefit [139]. Trials are investigating combinations of G12C inhibitors with checkpoint inhibitors for possible synergy (NCT04185883, NCT04613596) [61]. Synergy between G12C inhibitors and checkpoint inhibitors would provide proof of concept for future combinations with any novel G12D or G12V inhibitors, which would benefit a greater proportion of pancreatic cancer patients. Other investigational combinations with G12C inhibitors include MEK inhibitors, EGFR inhibitors, mTOR inhibitors, CDK 4/6 inhibitors, and VEGF inhibitors.
A dual RAF-MEK inhibitor, VS-6766/ CH5126766/ RO5126766, that targets the kinase activity of RAF and MEK simultaneously has been tested clinically in certain malignancies [140–142]. In preclinical evaluation, VS-6766 was able to suppress RAF and MEK kinase activity in a panel of cancer cell lines including KRAS mutated PDAC cells, was able to reduce tumor growth in NSCLC and colorectal carcinoma (CRC) mouse models, and showed synergy with G12C inhibitors in NSCLC and CRC cells [143–145]. The combination of VS-6766 with defactinib has been recently granted breakthrough therapy designation by the FDA, which came after encouraging results from the phase II FRAME trial (NCT04625270) for ovarian cancer irrespective of KRAS mutation status [146]. Nonetheless, KRAS mutant patients had a higher ORR (70%) than patient with wild-type KRAS (52%) [146]. A phase II clinical trial is underway to evaluate the combination in KRAS mutated solid tumors (NCT04620330).
Another combinatorial approach is targeting multiple arms of KRAS signaling. Co-targeting of the MAPK and PI3K pathways has been explored as a strategy to prevent adaptive resistance that results from the activation of one of those pathways upon the inhibition of the other. Clinical trials have investigated combinations targeting both MAPK and PI3K pathways, including combinations of PI3K and MEK inhibitors, AKT and MEK inhibitors, and mTOR and MEK inhibitors. Although these combinations showed synergistic activity in preclinical models, there were issues of tolerability and rate-limiting toxicities in clinical trials.
Finally, some combinations aim to target KRAS pathways in addition to cellular processes that are known to be deregulated in pancreatic cancer, such as autophagy. Autophagy is upregulated in PDAC cells [35] and is thought to be implicated in chemotherapy resistance [147]. Thus, autophagy has been investigated as a potential target for PDAC. Hydroxychloroquine is an anti-malaria medication that inhibits autophagy and has been studied in a phase II clinical trial as monotherapy for PDAC without much clinical benefit [148]. Combinations of chloroquine or hydroxychloroquine with gemcitabine have also been tested in clinical trials but failed to show any therapeutic benefit over gemcitabine alone [149, 150]. Interest remains in autophagy inhibition in combination with targeted therapeutics to treat KRAS mutant PDAC. Preclinical studies have shown that inhibition of autophagy was synergistic with MAPK pathway inhibitors in PDAC models. Combinations of MAPK pathway inhibition (ERK or MEK inhibitors) with hydroxychloroquine are currently being investigated in multiple clinical trials for malignancies with KRAS mutations (NCT04145297, NCT03825289, NCT04132505, and NCT04214418).
7. Mechanisms and strategies for overcoming resistance
Although novel inhibitors of mutant KRAS and its downstream targets showed promising results in preclinical and clinical investigations, studies to understand the mechanism of resistance to mutant KRAS or downstream pathway inhibitors are very limited. Recent investigations in a cohort of 38 NSCLC patients treated with KRAS G12C inhibitors revealed resistance development in 45% of the patients. Notable acquired genetic changes in KRAS were G12D/R/V/W, G13D, Q61H, R68S, H95D/Q/R, Y96C, and KRAS G12C allele amplification [151]. Such mutations may elevate the levels of GTP-bound active KRAS protein and can prevent binding to drugs [91]. Amplification of MET; mutational activation of NRAS, BRAF, MAP2K1, and RET; oncogene fusion including ALK, RET, BRAF, RAF1, and FGFR3; and loss-of-function mutations in NF1 and PTEN were also shown to play a significant role in the development of resistance [151]. It has been demonstrated that KRAS inhibition mediated by mutant KRAS inhibitors, including KRAS G12C inhibitor sotorasib, or MEK inhibitors, such as trametinib, leads to adaptive resistance in PDAC [152]. Such inhibition causes integrin-linked kinase (ILK) mediated activation of the mTORC2 molecule RICTOR and phosphorylation of AKT at Ser-473 in several PDAC mouse models and human tumors [152]. It is evident that RICTOR expression is correlated with poor prognosis in PDAC patients [153], and both RICTOR/mTORC2 activations are associated with PDAC progression [154]. Inhibition of mTORC2 alone stimulates phosphorylation of ERK [152, 155] resulting in increased cell survival. However, in combination with mutant KRAS or MEK inhibitor, it promotes cell death in PDAC cells [152], suggesting an important role of mTORC2 signaling in the resistance of PDAC.
In another study, Chen et al. observed that murine PDAC cells can tolerate acute and sustained KRAS silencing by activating a reversible cell state [156]. The study used an RNAi-based inducible system for conditional silencing of KRAS at both alleles. The reversible state was characterized by morphological changes, proliferative kinetics, and the capacity for tumor initiation. Although no mutational or transcriptional alterations were found during the KRAS-silenced state, global phosphoproteomic analysis revealed the involvement of a number of cellular signaling events including the activation of the focal adhesion pathway. Such findings indicate that focal adhesion signaling is a possible resistance mechanism to KRAS inhibition. Identification and targeting of key molecules of this pathway may overcome resistance to KRAS inhibitors. Although targeting of focal adhesion kinase (FAK) or SRC was not sufficient to induce cell death in KRAS-inhibited cells selectively, other molecules of this pathway could be potentially targeted which warrants further investigation [156]. Moreover, alterations of multiple RTK-RAS-MAPK pathways also contribute to the resistance to the KRAS G12C inhibitor adagrasib resistance, and clinical trials combining RTK or SHP2 inhibitors with mutant KRAS inhibitors are underway (NCT04330664, NCT04185883) [151]. Additionally, alternative prenylation against FTI therapy as described above plays a role in the development of resistance. It is important to understand the mechanism of resistance against novel KRAS targeted inhibitors to ensure sustained and durable remissions. In-depth understanding of resistance mechanisms is crucial for optimal therapeutic targeting of PDAC even when mutation specific KRAS inhibitors are unavailable.
7.1. The role of tumor microenvironment in resistance
The TME of PDAC plays an important role in the development of resistance [157]. CAFs have been implicated in driving the desmoplastic response in PDAC [59, 158], which ultimately contributes to the resistance to various therapies [159] including KRAS targeting agents. A mechanistic study revealed that CAF’s expressed vitamin D receptor is involved in the development of resistance, and calcipotriol, a vitamin D receptor agonist, can revert CAFs to their original state in a mouse model [160]. Such targeting of tumor stroma could potentially enhance the delivery of KRAS specific inhibitors or other targeting agents. Recently in 2020, Hou et al. observed an upregulation of HDAC5 in a gain-of-function study in the inducible KRASG12D/Trp53−/− PDAC mouse model [161]. The epigenetic regulation mediated by HDAC5 enhanced the recruitment of macrophages into the TME, which promotes resistance to KRAS targeting. An increase in CCL2, due to the suppression of its negative regulator SOCS3, helps in the recruitment of macrophages. These tumor-associated macrophages (TAMs) can support cancer cells by bypassing KRAS signaling through the activation of TGF-β in a SMAD-4 dependent manner [161]. Therapy resistance in PDAC could also result from autophagic degradation of CAFs which releases different metabolites that are utilized by the cancer cells [159, 162–164]. The secretion of metabolites with structural similarity to drugs by CAFs, such as the metabolite deoxycytidine which is similar to gemcitabine, enhances the tumor’s capability of drug resistance [165, 166]. Besides CAFs, neurons present in the TME can support PDAC growth by releasing amino acid serine [162].
KRAS mutated TME includes numerous types of cells. Targeting cell types that significantly contribute to PDAC growth along with KRAS pathway targeting agents could potentially be a sustainable strategy to overcome resistance. Targeting autophagy of both tumor cells and CAFs is a promising approach to inhibit tumor growth in in vivo [167] and can be combined with KRAS targeted agents. Additionally, targeting TME recruited neurons in PDAC using TRK inhibitor [168] along with KRAS targeted inhibitors could be a novel approach. Research showed convincing findings of simultaneous targeting of TGF-β and oncogenic KRAS using antifibrotic fraxinellone-loaded CGKRK-modified nanoparticles (Frax-NP- CGKRK) and siRNA-loaded lipid-coated calcium phosphate (LCP) biomimetic nanoparticles (siKras-LCP-ApoE3) respectively in a pancreatic cancer mouse model [169]. Additionally, combining a SHP2 inhibitor with KRAS G12C inhibitor successfully abrogated RTK feedback signaling and adaptive resistance to G12C inhibitor in in vitro, xenograft, and syngeneic KRAS G12C-mutant PDAC model [170]. This combination was shown to remodel the tumor immune microenvironment by decreasing myeloid suppressor cells and increasing CD8+ T cells [113, 170].
8. Conclusion
Despite decades of intensive research, pancreatic cancer remains a highly lethal malignancy with an extremely poor prognosis, and cytotoxic therapeutics provide minimal benefit for pancreatic cancer patients. The newly approved and emerging KRAS G12C inhibitors could only benefit a small subset of pancreatic cancer patients, whereas there are no approved drugs to target KRAS G12D and G12V, the major mutations in PDAC. Nonetheless, there is now renewed hope that KRAS could be a targetable oncogene, and several promising advances have been made in the field including direct and indirect targeting strategies. Combinations of various targeted therapeutics and immunotherapy seem to be the direction that holds most promise for the future for PDAC patients. Clinical trials are actively investigating the therapeutic targeting of multiple KRAS-related pathways and molecules. Results from these trials would inform future research directions and clinical trials on tailoring the correct combinations according to the molecular alterations for each patient’s cancer, as well as elucidating the mechanisms of resistance to tackle it when it arises. In light of the successes of G12C inhibitors, ongoing work to develop other specific mutant KRAS inhibitors could potentially result in promising new therapeutic approaches to target the major driver mutations in PDAC. Finally, research into combining KRAS targeted drugs with pancreatic tumor microenvironment modulating agents for enhanced drug delivery and efficacy will be promising avenues of future therapeutic strategies.
Funding
Work in the lab of Azmi AS is supported by R37CA215427, R01CA24060701A1, and SKY Foundation Inc.
Conflict of interest
ASA received funding from Karyopharm Therapeutics, Janssen, Rhizen, and EISAI. ASA serves as a consultant for GLG and Guidepoint.
Footnotes
Publisher's Disclaimer: This AM is a PDF file of the manuscript accepted for publication after peer review, when applicable, but does not reflect post-acceptance improvements, or any corrections. Use of this AM is subject to the publisher’s embargo period and AM terms of use. Under no circumstances may this AM be shared or distributed under a Creative Commons or other form of open access license, nor may it be reformatted or enhanced, whether by the Author or third parties. See here for Springer Nature’s terms of use for AM versions of subscription articles: https://www.springernature.com/gp/open-research/policies/accepted-manuscript-terms
References
- 1.da Costa WL Jr., Oluyomi AO, and Thrift AP, Trends in the incidence of pancreatic adenocarcinoma in all 50 United States examined through an age-period-cohort analysis. JNCI Cancer Spectr, 2020. 4(4): p. pkaa033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGuigan A, et al. , Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol, 2018. 24(43): p. 4846–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, et al. , Cancer statistics, 2021. CA Cancer J Clin, 2021. 71(1): p. 7–33. [DOI] [PubMed] [Google Scholar]
- 4.Waddell N, et al. , Whole genomes redefine the mutational landscape of pancreatic cancer. Nature, 2015. 518(7540): p. 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witkiewicz AK, et al. , Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun, 2015. 6: p. 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network. Electronic address, a.a.d.h.e. and N. Cancer Genome Atlas Research, Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell, 2017. 32(2): p. 185–203 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailey P, et al. , Genomic analyses identify molecular subtypes of pancreatic cancer. Nature, 2016. 531(7592): p. 47–52. [DOI] [PubMed] [Google Scholar]
- 8.Nagasaka M, et al. , KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat Rev, 2020. 84: p. 101974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takashima A and Faller DV, Targeting the RAS oncogene. Expert Opin Ther Targets, 2013. 17(5): p. 507–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gysin S, et al. , Therapeutic strategies for targeting ras proteins. Genes & cancer, 2011. 2(3): p. 359–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Müller MP, et al. , Nucleotide based covalent inhibitors of KRas can only be efficient in vivo if they bind reversibly with GTP-like affinity. Sci Rep, 2017. 7(1): p. 3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong LA, et al. , Crystal structures at 2.2 A resolution of the catalytic domains of normal ras protein and an oncogenic mutant complexed with GDP. J Mol Biol, 1991. 217(3): p. 503–16. [DOI] [PubMed] [Google Scholar]
- 13.Ostrem JM, et al. , K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature, 2013. 503(7477): p. 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prior IA, Lewis PD, and Mattos C, A comprehensive survey of Ras mutations in cancer. Cancer Res, 2012. 72(10): p. 2457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bos JL, Rehmann H, and Wittinghofer A, GEFs and GAPs: Critical elements in the control of small G proteins. Cell, 2007. 129(5): p. 865–877. [DOI] [PubMed] [Google Scholar]
- 16.Hennig A, et al. , Ras activation revisited: Role of GEF and GAP systems. Biol Chem, 2015. 396(8): p. 831–48. [DOI] [PubMed] [Google Scholar]
- 17.Mann KM, et al. , KRAS-related proteins in pancreatic cancer. Pharmacol Ther, 2016. 168: p. 29–42. [DOI] [PubMed] [Google Scholar]
- 18.Moore AR, et al. , RAS-targeted therapies: Is the undruggable drugged? Nat Rev Drug Discov, 2020. 19(8): p. 533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Logsdon CD and Lu W, The significance of Ras activity in pancreatic cancer initiation. Int J Biol Sci, 2016. 12(3): p. 338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lennerz JK and Stenzinger A, Allelic ratio of KRAS mutations in pancreatic cancer. Oncologist, 2015. 20(4): p. e8–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patricelli MP, et al. , Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov, 2016. 6(3): p. 316–29. [DOI] [PubMed] [Google Scholar]
- 22.Janes MR, et al. , Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell, 2018. 172(3): p. 578–589.e17. [DOI] [PubMed] [Google Scholar]
- 23.Daniluk J, et al. , An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest, 2012. 122(4): p. 1519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H, et al. , Oncogenic K-Ras requires activation for enhanced activity. Oncogene, 2014. 33(4): p. 532–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter JC, et al. , Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res, 2015. 13(9): p. 1325–35. [DOI] [PubMed] [Google Scholar]
- 26.Shao T, et al. , Recombinant expression of different mutant K-ras gene in pancreatic cancer Bxpc-3 cells and its effects on chemotherapy sensitivity. Sci China Life Sci, 2014. 57(10): p. 1011–7. [DOI] [PubMed] [Google Scholar]
- 27.Cayron C and Guillermet-Guibert J, The type of KRAS mutation drives PI3Kα/γ signalling dependency: Implication for the choice of targeted therapy in pancreatic adenocarcinoma patients. Clinics and Research in Hepatology and Gastroenterology, 2021. 45(1): p. 101473. [DOI] [PubMed] [Google Scholar]
- 28.Ihle NT, et al. , Effect of KRAS oncogene substitutions on protein behavior: Implications for signaling and clinical outcome. J Natl Cancer Inst, 2012. 104(3): p. 228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bournet B, et al. , KRAS G12D mutation subtype is a prognostic factor for advanced pancreatic adenocarcinoma. Clin Transl Gastroenterol, 2016. 7(3): p. e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian ZR, et al. , Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncology, 2018. 4(3): p. e173420–e173420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ogura T, et al. , Prognostic value of K-ras mutation status and subtypes in endoscopic ultrasound-guided fine-needle aspiration specimens from patients with unresectable pancreatic cancer. Journal of Gastroenterology, 2013. 48(5): p. 640–646. [DOI] [PubMed] [Google Scholar]
- 32.Dhillon AS, et al. , MAP kinase signalling pathways in cancer. Oncogene, 2007. 26(22): p. 3279–90. [DOI] [PubMed] [Google Scholar]
- 33.Fruman DA, et al. , The PI3K pathway in human disease. Cell, 2017. 170(4): p. 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gentry LR, et al. , Ral small GTPase signaling and oncogenesis: More than just 15minutes of fame. Biochim Biophys Acta, 2014. 1843(12): p. 2976–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang S, et al. , Pancreatic cancers require autophagy for tumor growth. Genes Dev, 2011. 25(7): p. 717–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hobbs GA, et al. , Atypical KRAS(G12R) mutant is impaired in PI3K signaling and macropinocytosis in pancreatic cancer. Cancer Discov, 2020. 10(1): p. 104–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki T, et al. , Mutant KRAS drives metabolic reprogramming and autophagic flux in premalignant pancreatic cells. Cancer Gene Ther, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dey P, et al. , Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discov, 2020. 10(4): p. 608–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pupo E, et al. , KRAS-driven metabolic rewiring reveals novel actionable targets in cancer. Front Oncol, 2019. 9: p. 848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dias Carvalho P, et al. , KRAS oncogenic signaling extends beyond cancer cells to orchestrate the microenvironment. Cancer Res, 2018. 78(1): p. 7–14. [DOI] [PubMed] [Google Scholar]
- 41.Bryant KL, et al. , KRAS: Feeding pancreatic cancer proliferation. Trends in Biochemical Sciences, 2014. 39(2): p. 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanda M, et al. , Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology, 2012. 142(4): p. 730–733.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hingorani SR, et al. , Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell, 2003. 4(6): p. 437–50. [DOI] [PubMed] [Google Scholar]
- 44.Hingorani SR, et al. , Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell, 2005. 7(5): p. 469–83. [DOI] [PubMed] [Google Scholar]
- 45.Aguirre AJ, et al. , Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev, 2003. 17(24): p. 3112–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Collins MA, et al. , Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One, 2012. 7(12): p. e49707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collins MA, et al. , Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest, 2012. 122(2): p. 639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ying H, et al. , Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell, 2012. 149(3): p. 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rhim AD, et al. , Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell, 2014. 25(6): p. 735–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhattacharjee S, et al. , Tumor restriction by type I collagen opposes tumor-promoting effects of cancer-associated fibroblasts. J Clin Invest, 2021. 131(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iwamoto C, et al. , Bone marrow-derived macrophages converted into cancer-associated fibroblast-like cells promote pancreatic cancer progression. Cancer Letters, 2021. 512: p. 15–27. [DOI] [PubMed] [Google Scholar]
- 52.Hamarsheh S.a., et al. , Immune modulatory effects of oncogenic KRAS in cancer. Nature communications, 2020. 11(1): p. 5439–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thayer SP, et al. , Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature, 2003. 425(6960): p. 851–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian H, et al. , Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A, 2009. 106(11): p. 4254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walter K, et al. , Overexpression of smoothened activates the sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clin Cancer Res, 2010. 16(6): p. 1781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mathew E, et al. , Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep, 2014. 9(2): p. 484–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tape Christopher J., et al. , Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell, 2016. 165(4): p. 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pu N, et al. , Genetic landscape of prognostic value in pancreatic ductal adenocarcinoma microenvironment. Ann Transl Med, 2019. 7(22): p. 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorchs L and Kaipe H, Interactions between cancer-associated fibroblasts and T cells in the pancreatic tumor microenvironment and the role of chemokines. Cancers (Basel), 2021. 13(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ischenko I, et al. , KRAS drives immune evasion in a genetic model of pancreatic cancer. Nature Communications, 2021. 12(1): p. 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Canon J, et al. , The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature, 2019. 575(7781): p. 217–223. [DOI] [PubMed] [Google Scholar]
- 62.Briere DM, et al. , The KRAS(G12C) inhibitor MRTX849 reconditions the tumor immune microenvironment and sensitizes tumors to checkpoint inhibitor therapy. Mol Cancer Ther, 2021. 20(6): p. 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kato K, et al. , Isoprenoid addition to Ras protein is the critical modification for its membrane association and transforming activity. Proc Natl Acad Sci U S A, 1992. 89(14): p. 6403–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ahearn IM, et al. , Regulating the regulator: post-translational modification of RAS. Nature Reviews Molecular Cell Biology, 2012. 13(1): p. 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berndt N, Hamilton AD, and Sebti SM, Targeting protein prenylation for cancer therapy. Nat Rev Cancer, 2011. 11(11): p. 775–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang W. h., et al. , Post-translational modification of KRAS: potential targets for cancer therapy. Acta Pharmacologica Sinica, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brock EJ, et al. , How to target activated Ras proteins: Direct inhibition vs. induced mislocalization. Mini Rev Med Chem, 2016. 16(5): p. 358–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brunner TB, et al. , Farnesyltransferase inhibitors: An overview of the results of preclinical and clinical investigations. Cancer Res, 2003. 63(18): p. 5656–68. [PubMed] [Google Scholar]
- 69.Rao S, et al. , Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol, 2004. 22(19): p. 3950–7. [DOI] [PubMed] [Google Scholar]
- 70.Van Cutsem E, et al. , Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol, 2004. 22(8): p. 1430–8. [DOI] [PubMed] [Google Scholar]
- 71.Whyte DB, et al. , K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem, 1997. 272(22): p. 14459–64. [DOI] [PubMed] [Google Scholar]
- 72.Basso AD, Kirschmeier P, and Bishop WR, Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res, 2006. 47(1): p. 15–31. [DOI] [PubMed] [Google Scholar]
- 73.Rowell CA, et al. , Direct demonstration of geranylgeranylation and farnesylation of Ki-Ras in vivo. J Biol Chem, 1997. 272(22): p. 14093–7. [DOI] [PubMed] [Google Scholar]
- 74.Kazi A, et al. , Dual farnesyl and geranylgeranyl transferase inhibitor thwarts mutant KRAS-driven patient-derived pancreatic tumors. Clin Cancer Res, 2019. 25(19): p. 5984–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Drosten M and Barbacid M, Targeting the MAPK pathway in KRAS-driven tumors. Cancer Cell, 2020. 37(4): p. 543–550. [DOI] [PubMed] [Google Scholar]
- 76.Terrell EM and Morrison DK, Ras-mediated activation of the Raf family kinases. Cold Spring Harb Perspect Med, 2019. 9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Poulikakos PI, et al. , RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature, 2010. 464(7287): p. 427–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hatzivassiliou G, et al. , RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature, 2010. 464(7287): p. 431–5. [DOI] [PubMed] [Google Scholar]
- 79.Heidorn SJ, et al. , Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell, 2010. 140(2): p. 209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanchez-Laorden B, et al. , BRAF inhibitors induce metastasis in RAS mutant or inhibitor-resistant melanoma cells by reactivating MEK and ERK signaling. Sci Signal, 2014. 7(318): p. ra30. [DOI] [PubMed] [Google Scholar]
- 81.Peng S-B, et al. , Inhibition of RAF isoforms and active dimers by LY3009120 leads to anti-tumor activities in RAS or BRAF mutant cancers. Cancer Cell, 2015. 28(3): p. 384–398. [DOI] [PubMed] [Google Scholar]
- 82.Hoxhaj G and Manning BD, The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer, 2020. 20(2): p. 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Missiaglia E, et al. , Pancreatic endocrine tumors: Expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol, 2010. 28(2): p. 245–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yao JC, et al. , Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med, 2011. 364(6): p. 514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wolpin BM, et al. , Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology, 2009. 27(2): p. 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Javle MM, et al. , Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: Results of two phase II studies. BMC Cancer, 2010. 10: p. 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Halilovic E, et al. , PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res, 2010. 70(17): p. 6804–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Williams TM, et al. , Cotargeting MAPK and PI3K signaling with concurrent radiotherapy as a strategy for the treatment of pancreatic cancer. Molecular cancer therapeutics, 2012. 11(5): p. 1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fell JB, et al. , Identification of the clinical development candidate MRTX849, a covalent KRAS(G12C) inhibitor for the treatment of cancer. J Med Chem, 2020. 63(13): p. 6679–6693. [DOI] [PubMed] [Google Scholar]
- 90.Ostrem JM and Shokat KM, Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat Rev Drug Discov, 2016. 15(11): p. 771–785. [DOI] [PubMed] [Google Scholar]
- 91.Lito P, et al. , Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science, 2016. 351(6273): p. 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jackson PA, et al. , Covalent modifiers: A chemical perspective on the reactivity of α,β-unsaturated carbonyls with thiols via hetero-Michael addition reactions. Journal of Medicinal Chemistry, 2017. 60(3): p. 839–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.FDA approves first KRAS inhibitor: Sotorasib. Cancer Discov, 2021. [DOI] [PubMed] [Google Scholar]
- 94.Skoulidis F, et al. , Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hallin J, et al. , The KRAS(G12C) inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer discovery, 2020. 10(1): p. 54–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dogan S, et al. , Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-mutant cancers. Clin Cancer Res, 2012. 18(22): p. 6169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang Z, et al. , GTP-state-selective cyclic peptide ligands of K-Ras(G12D) block its interaction with Raf. ACS Cent Sci, 2020. 6(10): p. 1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dougherty PG, Sahni A, and Pei D, Understanding cell penetration of cyclic peptides. Chem Rev, 2019. 119(17): p. 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sakamoto K, Masutani T, and Hirokawa T, Generation of KS-58 as the first KRas(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Scientific reports, 2020. 10(1): p. 21671–21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.https://www.mirati.com/science/programs/kras-inhibitors/kras-g12d-inhibitor/ in Mirati Therapeutics Website. Accessed on: 07/07/2021.
- 101.Tanaka N, et al. , Clinical acquired resistance to KRASG12C inhibition through a novel KRAS switch-II pocket mutation and polyclonal alterations converging on RAS-MAPK reactivation. Cancer Discov, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liceras-Boillos P, et al. , Sos1 disruption impairs cellular proliferation and viability through an increase in mitochondrial oxidative stress in primary MEFs. Oncogene, 2016. 35(50): p. 6389–6402. [DOI] [PubMed] [Google Scholar]
- 103.Hillig RC, et al. , Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS-SOS1 interaction. Proc Natl Acad Sci U S A, 2019. 116(7): p. 2551–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Burns MC, et al. , Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proceedings of the National Academy of Sciences, 2014. 111(9): p. 3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hofmann MH, et al. , BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov, 2021. 11(1): p. 142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li W, et al. , A new function for a phosphotyrosine phosphatase: Linking GRB2-Sos to a receptor tyrosine kinase. Mol Cell Biol, 1994. 14(1): p. 509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vogel W and Ullrich A, Multiple in vivo phosphorylated tyrosine phosphatase SHP2 engages binding to Grb2 via tyrosine 584. Cell Growth Differ, 1996. 7(12): p. 1589–97. [PubMed] [Google Scholar]
- 108.Dance M, et al. , The molecular functions of Shp2 in the Ras/mitogen-activated protein kinase (ERK1/2) pathway. Cell Signal, 2008. 20(3): p. 453–9. [DOI] [PubMed] [Google Scholar]
- 109.Ruess DA, et al. , Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat Med, 2018. 24(7): p. 954–960. [DOI] [PubMed] [Google Scholar]
- 110.Wang Y, et al. , Targeting the SHP2 phosphatase promotes vascular damage and inhibition of tumor growth. EMBO Molecular Medicine, 2021. n/a(n/a): p. e14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heppner DE and Eck MJ, A structural perspective on targeting the RTK/Ras/MAP kinase pathway in cancer. Protein Sci, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chen YN, et al. , Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature, 2016. 535(7610): p. 148–52. [DOI] [PubMed] [Google Scholar]
- 113.Kerr DL, Haderk F, and Bivona TG, Allosteric SHP2 inhibitors in cancer: Targeting the intersection of RAS, resistance, and the immune microenvironment. Curr Opin Chem Biol, 2021. 62: p. 1–12. [DOI] [PubMed] [Google Scholar]
- 114.Nichols RJ, et al. , RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol, 2018. 20(9): p. 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.LaMarche MJ, et al. , Identification of TNO155, an allosteric SHP2 inhibitor for the treatment of cancer. J Med Chem, 2020. 63(22): p. 13578–13594. [DOI] [PubMed] [Google Scholar]
- 116.Liu C, et al. , Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin Cancer Res, 2021. 27(1): p. 342–354. [DOI] [PubMed] [Google Scholar]
- 117.Carreno BM, et al. , Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science, 2015. 348(6236): p. 803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ott PA, et al. , An immunogenic personal neoantigen vaccine for patients with melanoma. Nature, 2017. 547(7662): p. 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cafri G, et al. , mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Invest, 2020. 130(11): p. 5976–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sahin U, et al. , Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature, 2017. 547(7662): p. 222–226. [DOI] [PubMed] [Google Scholar]
- 121.Wang Z and Cao YJ, Adoptive cell therapy targeting neoantigens: A frontier for cancer research. Front Immunol, 2020. 11: p. 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Roudko V, Greenbaum B, and Bhardwaj N, Computational prediction and validation of tumor-associated neoantigens. Front Immunol, 2020. 11: p. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schumacher TN, Scheper W, and Kvistborg P, Cancer neoantigens. Annual Review of Immunology, 2018. 37(1): p. 173–200. [DOI] [PubMed] [Google Scholar]
- 124.Tran E, et al. , T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med, 2016. 375(23): p. 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chamberlain PP and Hamann LG, Development of targeted protein degradation therapeutics. Nature Chemical Biology, 2019. 15(10): p. 937–944. [DOI] [PubMed] [Google Scholar]
- 126.Bond MJ, et al. , Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent Sci, 2020. 6(8): p. 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chandra A, et al. , The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol, 2011. 14(2): p. 148–58. [DOI] [PubMed] [Google Scholar]
- 128.Cheng J, et al. , Discovery of novel PDEδ degraders for the treatment of KRAS mutant colorectal cancer. J Med Chem, 2020. 63(14): p. 7892–7905. [DOI] [PubMed] [Google Scholar]
- 129.Khan I, Spencer-Smith R, and O’Bryan JP, Targeting the α4-α5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene, 2019. 38(16): p. 2984–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Spencer-Smith R, et al. , Targeting the α4-α5 interface of RAS results in multiple levels of inhibition. Small GTPases, 2019. 10(5): p. 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Spencer-Smith R, et al. , Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol, 2017. 13(1): p. 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Khan I, et al. , Targeting the KRAS α4-α5 allosteric interface inhibits pancreatic cancer tumorigenesis. Small GTPases, 2021: p. 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jones S, et al. , Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science, 2008. 321(5897): p. 1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ardito CM, et al. , EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell, 2012. 22(3): p. 304–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Navas C, et al. , EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell, 2012. 22(3): p. 318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ryan MB, et al. , Vertical pathway inhibition overcomes adaptive feedback resistance to KRAS(G12C) inhibition. Clin Cancer Res, 2020. 26(7): p. 1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ahmed TA, et al. , SHP2 drives adaptive resistance to ERK signaling inhibition in molecularly defined subsets of ERK-Dependent Tumors. Cell Rep, 2019. 26(1): p. 65–78.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fedele C, et al. , SHP2 inhibition prevents adaptive resistance to MEK inhibitors in multiple cancer models. Cancer Discov, 2018. 8(10): p. 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schizas D, et al. , Immunotherapy for pancreatic cancer: A 2020 update. Cancer Treat Rev, 2020. 86: p. 102016. [DOI] [PubMed] [Google Scholar]
- 140.Dual RAF-MEK Inhibitor Assessed. Cancer Discov, 2021. 11(1): p. 5–6. [DOI] [PubMed] [Google Scholar]
- 141.Guo C, et al. , Intermittent schedules of the oral RAF-MEK inhibitor CH5126766/VS-6766 in patients with RAS/RAF-mutant solid tumours and multiple myeloma: A single-centre, open-label, phase 1 dose-escalation and basket dose-expansion study. Lancet Oncol, 2020. 21(11): p. 1478–1488. [DOI] [PubMed] [Google Scholar]
- 142.Guo C and Banerji U, Searching for treatments for non-G12C-KRAS mutant cancers. Br J Cancer, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ishii N, et al. , Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res, 2013. 73(13): p. 4050–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wada M, et al. , The dual RAF/MEK inhibitor CH5126766/RO5126766 may be a potential therapy for RAS-mutated tumor cells. PLoS One, 2014. 9(11): p. e113217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pachter, RAS targeted drug development meeting 2020. Verastem Oncology Website: https://www.verastem.com/research/raf-mek-inhibition/vs-6766/. Accessed on: 6/28/2021. [Google Scholar]
- 146.Verastem Oncology Receives Breakthrough Therapy Designation for VS-6766 with Defactinib in Recurrent Low-Grade Serous Ovarian Cancer. Verastem Oncology Press Release. https://investor.verastem.com/news-releases/news-release-details/verastem-oncology-receives-breakthrough-therapy-designation-vs. Accessed on: 06/28/2021, 2021. [Google Scholar]
- 147.Sui X, et al. , Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis, 2013. 4(10): p. e838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wolpin BM, et al. , Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist, 2014. 19(6): p. 637–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Karasic TB, et al. , Effect of gemcitabine and nab-paclitaxel with or without hydroxychloroquine on patients with advanced pancreatic cancer: A phase 2 randomized clinical trial. JAMA Oncol, 2019. 5(7): p. 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Samaras P, et al. , Phase I study of a chloroquine-gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother Pharmacol, 2017. 80(5): p. 1005–1012. [DOI] [PubMed] [Google Scholar]
- 151.Awad MM, et al. , Acquired resistance to KRAS(G12C) inhibition in cancer. N Engl J Med, 2021. 384(25): p. 2382–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Brown WS, et al. , Overcoming adaptive resistance to KRAS and MEK inhibitors by co-targeting mTORC1/2 complexes in pancreatic cancer. Cell Rep Med, 2020. 1(8): p. 100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schmidt KM, et al. , Inhibition of mTORC2 component RICTOR impairs tumor growth in pancreatic cancer models. Oncotarget, 2017. 8(15): p. 24491–24505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Driscoll DR, et al. , mTORC2 signaling drives the development and progression of pancreatic cancer. Cancer Res, 2016. 76(23): p. 6911–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Soares HP, et al. , Dual PI3K/mTOR inhibitors induce rapid overactivation of the MEK/ERK pathway in human pancreatic cancer cells through suppression of mTORC2. Mol Cancer Ther, 2015. 14(4): p. 1014–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen PY, et al. , Adaptive and reversible resistance to kras inhibition in pancreatic cancer cells. Cancer Res, 2018. 78(4): p. 985–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ryan DP, Hong TS, and Bardeesy N, Pancreatic adenocarcinoma. N Engl J Med, 2014. 371(11): p. 1039–49. [DOI] [PubMed] [Google Scholar]
- 158.Cirri P and Chiarugi P, Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev, 2012. 31(1–2): p. 195–208. [DOI] [PubMed] [Google Scholar]
- 159.Kerk SA, et al. , Metabolic networks in mutant KRAS-driven tumours: Tissue specificities and the microenvironment. Nat Rev Cancer, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sherman MH, et al. , Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell, 2014. 159(1): p. 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hou P, et al. , Tumor microenvironment remodeling enables bypass of oncogenic KRAS dependency in pancreatic cancer. Cancer Discov, 2020. 10(7): p. 1058–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chaudhri VK, et al. , Metabolic alterations in lung cancer-associated fibroblasts correlated with increased glycolytic metabolism of the tumor. Mol Cancer Res, 2013. 11(6): p. 579–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sousa CM, et al. , Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature, 2016. 536(7617): p. 479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhao H, et al. , Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife, 2016. 5: p. e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dalin S, et al. , Deoxycytidine release from pancreatic stellate cells promotes gemcitabine resistance. Cancer Res, 2019. 79(22): p. 5723–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Halbrook CJ, et al. , Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab, 2019. 29(6): p. 1390–1399 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yan Y, et al. , The effects and the mechanisms of autophagy on the cancer-associated fibroblasts in cancer. J Exp Clin Cancer Res, 2019. 38(1): p. 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Banh RS, et al. , Neurons release serine to support mRNA translation in pancreatic cancer. Cell, 2020. 183(5): p. 1202–1218 e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pei Y, et al. , Sequential targeting TGF-beta signaling and KRAS mutation increases therapeutic efficacy in Pancreatic Cancer. Small, 2019. 15(24): p. e1900631. [DOI] [PubMed] [Google Scholar]
- 170.Fedele C, et al. , SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J Exp Med, 2021. 218(1). [DOI] [PMC free article] [PubMed] [Google Scholar]