

Abstract
Immune homeostasis is maintained by a precise balance between effector immune cells and regulatory immune cells. Chronic deviations from immune homeostasis, driven by a greater ratio of effector to regulatory cues, can promote the development and propagation of inflammatory diseases/conditions (i.e., autoimmune diseases, transplant rejection, etc.). Current methods to treat chronic inflammation rely upon systemic administration of non-specific small molecules, resulting in broad immunosuppression with unwanted side effects. Consequently, recent studies have developed more localized and specific immunomodulatory approaches to treat inflammation through the use of local biomaterial-based delivery systems. In particular, this review focuses on (1) local biomaterial-based delivery systems, (2) common materials used for polymeric-delivery systems and (3) emerging immunomodulatory trends used to treat inflammation with increased specificity.
Keywords: hydrogels, microparticles, scaffolds, inflammation, immune homeostasis, autoimmunity, transplant rejection, controlled release
Graphical Abstract
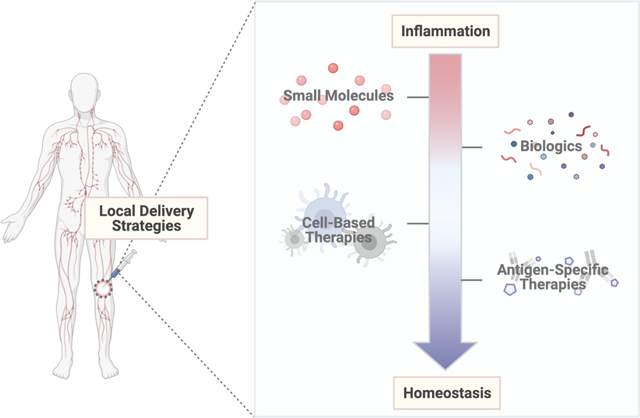
1. Introduction
Immune homeostasis, a state of immunological equilibrium, is maintained by a balance of effector immune cells (i.e., effector T cells, inflammatory macrophages, etc.) and regulatory immune cells (i.e., regulatory T cells, tolerogenic/immature dendritic cells, etc.) (Figure 1A). These cells work to maintain immune homeostasis through (1) eliminating disease and pathogens (i.e., cancer) by serving an immunostimulatory role or (2) preventing excess inflammation by serving a regulatory role. Chronic perturbations from immune homeostasis, either due to escape mechanisms (i.e., cancer) or failures in regulatory mechanisms (i.e., autoimmunity), can lead to improper balances of anti-inflammatory to inflammatory cues. In the case of cancer, anti-inflammatory cues outpace inflammatory cues, leading to disease propagation and metastasis (Figure 1B). On the other hand, failures in immune regulatory mechanisms, including the discrimination between non-self-antigens and self-antigens, as well as the elimination of autoreactive immune cells, can drive autoimmune diseases and transplant rejection (Figure 1C) [1]. This review will focus on methods to address immune imbalances in the latter scenario, during which a greater ratio of inflammatory to anti-inflammatory cues, as well as a lapse in inherent immunosuppressant mechanisms, results in improper immune activation, excessive inflammation, local tissue destruction and chronic disease.
Fig.1.

Maintenance of Immune Homeostasis. (A) When inflammatory cues (i.e., effector cells, mature dendritic cells, inflammatory cytokines) are balanced by anti-inflammatory cues (i.e., regulatory T cells, immature dendritic cells, anti-inflammatory cytokines, etc.), the immune system is in a state of immunological equilibrium. (B) If there is a greater ratio of anti-inflammatory to inflammatory signals, diseases such as cancer can develop and propagate. (C) If there is a greater ratio of inflammatory to anti-inflammatory signals, conditions such as autoimmune diseases and transplant rejection can develop and propagate.
Chronic perturbations in immune homeostasis, which result in excess inflammatory cues when compared to anti-inflammatory cues, can lead to the development and propagation of autoimmune diseases and the rejection of transplanted tissues. Autoimmune diseases, which affect roughly 7 to 9% of the general population, can be initiated because of environmental triggers (i.e., infections), genetic predispositions and signaling pathway mutations [2,3]. Once disease is initiated, it becomes difficult to control. Self-antigens, which are now driving the inflammatory reaction, cannot be eliminated [3]. For instance, in type 1 diabetes (T1D), destructive immune cells target self-antigens on insulin-producing cells known as beta-cells, leading to improper insulin production and a build-up of glucose within the bloodstream (hyperglycemia). Similarly, in multiple sclerosis (MS), recognition of self-antigens leads to an immunological attack on the myelin sheath, resulting in clinical symptoms of numbness and paralysis [4]. Although the anatomical sites impacted by these diseases are notably different, the underlying mechanism driving disease propagation is the same: an increasing accumulation of effector immune cells relative to regulatory immune cells [3]. A similar mechanism drives the rejection of transplanted cells, tissues and organs. Recognition of either intact non-self-antigens on donor antigen presenting cells (APCs) (direct recognition) or processed peptides on recipient APCs (indirect recognition) drives the activation and recruitment of destructive immune cells, leading to graft rejection affecting anywhere between 10% (liver) and 85% (vascularized composite) of transplanted tissues [5–7].
Approaches to mediate and treat these disorders rely upon the use of anti-inflammatory agents, such as corticosteroids, anti-metabolites and calcineurin inhibitors [8–10]. Overall, efficacy of these options is often limited by their specificity and method of administration. Many anti-inflammatory agents broadly suppress inflammation and are administered systemically in high dose concentrations, resulting in various side effects, including immunodeficiency toxicities (viral and bacterial infections, etc.) and systemic toxicities (nephrotoxicity, hepatotoxicity, etc.) [4,10–12]. In addition, systemic administration of agents is limited by renal/hepatic clearance and places the drug at risk for reduced pharmacological activity due to various physiological factors (i.e., pH, temperature, etc.) [13,14]. To minimize these side effects and maintain pharmacological activity of anti-inflammatory agents, biomaterial-based drug delivery systems have been explored as a method to deliver immunomodulatory treatments.
Biomaterial-based delivery systems provide several advantages over traditional administration methods, including the ability to enhance accumulation at the site of interest, reduce clearance, prevent undesired immune activation and achieve controlled drug release [13,14]. One of the more popular biomaterial strategies used to achieve these goals is nano-based delivery systems. There are many benefits to the use of nanotechnology for drug delivery. For one, nano-based systems can be designed to facilitate controlled drug delivery over time, reducing dose frequency and concentration of traditional anti-inflammatory agents [14]. In addition, nano-based approaches are very small in size, ranging from 10 to 1000 nanometers in diameter, allowing for enhanced tissue penetration and accumulation, as well as uptake by target cells [15,16]. As a result, nano-based delivery systems have been found to significantly increase drug concentration at specific sites, such as tumors, due to their localization/targeting ability [17]. Although drug concentration at a specific site can be increased using nano-based systems, it is worth noting that >95% of systemically administered nano-based delivery systems still accumulate elsewhere within the body [17]. In addition, nano-based delivery systems are often susceptible to rapid clearance by the reticuloendothelial system, further limiting their therapeutic effect [14,17–19].
Other biomaterial-based delivery systems, such as microparticles, hydrogels and in some cases, polymeric scaffolds, have emerged as viable candidates to address the limitations of nano-based delivery systems (i.e., off-target accumulation and effects). Local delivery approaches (i.e., microparticles, hydrogels, scaffolds) present a unique opportunity to further localize therapeutic effects, as these systems can be easily injected/implanted and engineered to avoid clearance by the immune system [4,20–23]. In addition, microparticles, hydrogels and polymeric scaffolds can facilitate controlled release, addressing the concern of high dose frequency and concentration associated with traditional anti-inflammatory agents [14]. Microparticles, particulate carriers on the order of microns (μm), are larger than nanoparticles [24,25]. As a result, microparticles typically act at the site of injection, as they are limited in their ability to cross biological barriers [24,26]. A similar trend is observed for hydrogels, which can also be classified based on size [27]. Macroscopic hydrogels are the largest class of hydrogels, ranging in size from millimeters to centimeters [27]. On the other hand, microgels, another class of hydrogels, have diameters on the order of microns [27]. Polymeric scaffolds, the final local delivery system discussed, are different from hydrogels and microparticles in that they are typically implanted to release or localize immunomodulatory cargo [22]. While implantable materials could be considered less desirable due to the potential immune response that develops because of implantation (i.e., foreign body response, protein adsorption, etc.), it is still important to analyze their ability to facilitate local immunomodulation through delivery of small molecules, biologics or antigen-specific therapeutics [28]. Altogether, microparticles, hydrogels and polymeric scaffolds are suitable platforms for local therapeutic administration, as their size limits clearance and/or migration and they can enhance therapeutic retention at the site of interest.
Several studies have illustrated the importance of local delivery strategies (microparticles, hydrogels, scaffolds) in inflammatory diseases (inflammatory arthritis, type 1 diabetes, inflammatory bowel disease, multiple sclerosis, etc.) and transplant rejection [29–44]. In the case of transplant rejection, intra-graft injections of various therapeutic payloads are capable of prolonging graft survival in vascularized composite allotransplantation, a specific type of transplant involving transplantation of multiple functional blocks of tissues (i.e., hand, face, etc.) [29,30,44]. In these studies, contralateral injections into the non-transplanted limb failed to prolong graft survival. In the case of inflammatory arthritis, direct administration of soluble drugs can result in escape from the joint cavity, prompting repeated injections to achieve a therapeutic effect [32–34,45]. To combat this effect, immunomodulatory drugs have been incorporated into various local drug delivery strategies for local intra-articular administration [32–37,46]. Together, these studies illustrate the importance of local delivery, as it can significantly improve treatment outcomes.
This review will focus on the use of microparticles, hydrogels and scaffolds as the most common local delivery strategies used to restore immune homeostasis in inflammatory disease models (i.e., transplant rejection, rheumatoid arthritis, multiple sclerosis, type 1 diabetes, periodontitis, etc.). We will first briefly review the most common polymeric materials, both synthetic and natural, that are used for local delivery systems. We will then summarize and analyze the immunological approaches most used to restore immune homeostasis from inflammation with local drug delivery strategies. Finally, we will highlight future directions of the field, as well as the path to clinical translation for local delivery strategies. Although this review focuses on microparticles, hydrogels and scaffolds as delivery methods, other strategies, such as nanocarriers and microneedle arrays, have applications in a variety of autoimmune disorders, as well as transplant rejection, and are discussed thoroughly elsewhere [47–50].
2. Polymeric materials used for local delivery systems
As previously mentioned, this review will discuss the use of microparticles, hydrogels and scaffolds as local drug delivery systems. Microparticles, hydrogels and scaffolds are engineered using a variety of fabrication techniques, including solvent evaporation, chemical crosslinking and solvent casting, respectively. For a more in-depth analysis of fabrication methods, as well as important parameters to consider during fabrication (i.e., size, shape, desired controlled release profile, etc.), common techniques and engineering parameters are discussed and analyzed in detail elsewhere [23,51–56].
The most common materials used for fabrication of these delivery systems are synthetic and natural polymers. Synthetic polymers are materials made of macromolecules that are typically chemically synthesized out of monomers [57,58]. Natural polymers, on the other hand, are materials that exist inherently in nature, usually as polysaccharides (alginate, chitosan, etc.) or proteins (gelatin, fibrin, collagen, etc.) [57,58]. This section will briefly highlight examples of both natural and synthetic polymers which are most used for local delivery systems for inflammatory diseases. For a more comprehensive analysis, the use of both natural and synthetic polymers in drug delivery, as well as their properties (i.e., biocompatibility, degradation, synthesis, etc.) have been extensively reviewed elsewhere [59,60].
2.1. Synthetic Polymers
Synthetic polymers can be easily synthesized and modified to achieve desired properties for individual applications (i.e., controlled release, enzymatic degradation, surface functionalization, etc.) [59]. Nonetheless, there are still some concerns in regard to the immunostimulatory potential of certain synthetic polymers [61–63]. Therefore, when choosing polymers for the development of delivery systems to treat inflammation, it is important to consider their immunomodulatory capacity. However, due to their tunable design parameters and ease of reproducibility, synthetic polymers are the most common material of choice for the development of drug delivery carriers. Thus, this section will briefly highlight the different classes of synthetic polymers, including polyesters, polyethers, poly (N-acrylamides), poloxamers and poly (amino acids), as well as discuss examples of their use as platforms to restore immune homeostasis from inflammation (Table 1).
Table 1.
Commonly used synthetic polymers for the development of local delivery strategies to restore immune homeostasis from inflammation.
| Polymer | Structure | Local Delivery Strategies | Ref(s) |
|---|---|---|---|
| Polyesters | |||
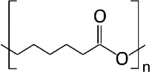
| Poly (caprolactone) (PCL) |
|
PEG-PCL-PEG MPs | [64] |
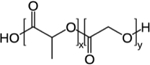
| Poly (lactic-co-glycolic acid) (PLGA) |
|
PLGA MPs | [59,65,66] |
| Polyethers | |||
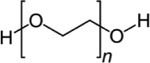
| Poly (ethylene glycol) (PEG) |
|
PEG-PCL-PEG MPs | [46,59,67,68] |
| Poly (ethylene glycol) methacrylate (PEGMA) |
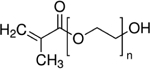
|
PEGMA hydrogel | [59,70,74] |
| Poly (N-acrylamides) | |||
| Poly (N-isopropylacrylamide) (pNIPAM) |
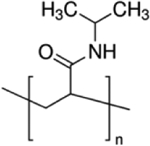
|
PLGA MPs/pNIPPAM hydrogel; pNIPAm-chitosan-hylauronic acid hydrogel | [59,76,79] |
| Poloxamers | |||
| Pluronic® F127 (F127) |
|
F127/F68 hydrogel | [33,37,80] |
| Pluronic® F68 (F68) |
|
F127/F68 hydrogel | [33,37,80] |
| Poly (amino acids) | |||
| Poly (lysine) (Lys) |
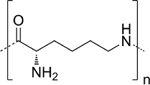
|
P-Lys-Ala-PLX hydrogel | [75,83] |
| Poly (alanine) (Ala) |
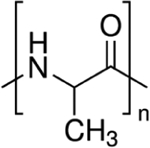
|
P-Lys-Ala-PLX hydrogel | [75,83] |
2.1.1. Polyesters
Polyesters, polymers most used for particulate systems, contain the ester functional group in their polymeric backbone [59]. Among polyesters, the most common examples for drug delivery applications are poly (caprolactone) (PCL), poly (lactic acid) (PLA) and poly (glycolic acid)) (PLG), as their degradation can facilitate controlled release. PCL is a hydrophobic, semi-crystalline polymer with a very slow hydrolysis rate [64]. PLA also has a relatively slow degradation rate, especially when compared to PLG, due to the presence of an extra methyl group on its’ polymeric backbone. The combination of PLA and PLG forms poly (lactic-co-glycolic acid) (PLGA), a block co-polymer that can be tuned to produce a desired degradation rate and material properties. In addition, the degradation products of PLA, PLG and PLGA are lactic and glycolic acid, which are further metabolized in vivo to carbon dioxide and water, which contributes to these materials being generally recognized as safe (GRAS) [59]. For these reasons, polyesters are the most common materials for local drug delivery strategies.
In addition to exhibiting desirable material properties, polyesters have also been shown to lack immunostimulatory effects and, in some cases, even exhibit immunosuppressive qualities [65,66]. When incubated with primary bone marrow-derived dendritic cells (DCs), PLGA microparticles failed to upregulate co-stimulatory molecules (CD80, CD86) when compared to PLGA/poly-β amino ester microparticles [65]. Furthermore, low molecular weight PLGA has been found to reduce the composite maturation index of costimulatory molecules (CD80, CD86, MHC II) on the surface of dendritic cells [66]. In addition, low molecular weight PLGA has been shown to induce T cell anergy, a key regulatory mechanism that renders effector T cells inactive [66]. Together, these studies suggest that, at least under certain circumstances, there is an immunomodulatory effect associated with polyester-based drug delivery systems.
2.1.2. Polyethers
Polyethers are also commonly used in drug delivery systems. For instance, poly (ethylene glycol) (PEG) and poly (glycerol) (PPG), are known to increase the circulation time of payloads by serving as a “stealth-agent” [59]. Both PEG and PPG can increase circulation time by (amongst other things) increasing the molar mass of the drug, resulting in decreased renal filtration [59]. “PEGylation,” which refers to the concept of coating matrices in PEG, can also protect materials from triggering immune responses by reducing protein adsorption and cell attachment [46,59,67,68]. For example, Erdemeli et al. highlighted the impact of PEG on protein adsorption when used in conjunction with PCL microparticles in a model of rheumatoid arthritis [46]. Protein adsorption on PEG-PCL-PEG microparticles was reduced when compared to PCL microparticles lacking PEG, primarily due to the steric hinderance generated from the presence of hydrophilic PEG in the matrix [46]. PEGlyation has also impacted the graft survival of transplanted islets. Giraldo et al. found that the combination of PEG with short-course immunotherapy decreased rejection when compared to short-course immunotherapy alone, highlighting the potential benefit of PEGlyation to existing treatments [67]. Although beneficial in these studies, some reports have expressed concern for the use of PEG, as it could potentially trigger complement activation as well as hypersensitivity reactions [59,69]. Nonetheless, reactions such as these are rare, and PEG is still considered a gold-standard for drug administration due to its’ widely proven benefits [59].
Other variations of PEG, such as poly (ethylene glycol) methacrylate (PEGMA) and poly (ethylene glycol) norbornene (PEGNB), have also been used as delivery systems in inflammation [70,71]. In the case of PEG-based hydrogels, incorporating methacrylate and norbornene as functional linkers enables more control over polymerization, as crosslinking can be controlled directly by photopolymerization (extensively reviewed by Lin et al.) [72,73]. Important for the context of this review, incorporation of methacrylate and norbornene results in ester linkages between the functional linkers and the PEG backbone [58,59]. As a result, functionalized-PEG hydrogels can hydrolytically degrade when present in an aqueous environment, a characteristic not exhibited by PEG itself [72,73].
PEGMA and PEGNB are also desirable materials due to their stimuli-responsive behaviors. As a thermoresponsive material, PEGMA can undergo phase transitions in response to temperature changes. Most thermoresponsive polymers have a lower critical solution temperature (LCST), below which polymer chains are hydrated and remain water soluble due to hydrogen bonding with the surrounding aqueous environment [74]. When temperatures reach above the LCST, hydrogen bonding is reduced and hydrophobic interactions dominate, prompting phase separation and aggregation that forms gels [70,74–78]. Abou-ElNour et al. demonstrated the thermoresponsive characteristics of PEGMA when used as a carrier for triamcinolone acetonide (small molecule drug) [70]. Notably, it was found that lower concentrations (2–10% w/v) of PEGMA, in conjunction with microparticles, resulted in irreversible aggregates when exposed to increasing temperatures, most likely due to hydrophobic interactions between the microparticles and the polymeric backbone [70]. These results are significant to consider when developing both thermoresponsive and composite release systems (discussed previously), as it could alter degradation, cargo release, and biocompatibility. While PEGMA is thermoresponsive, PEGNB is pH responsive. In slightly acidic conditions (pH = 6.0), PEGNB hydrogels exhibited limited mass swelling [73]. However, in basic conditions, swelling increased over time [73]. Due to their responsive nature, PEGMA and PEGNB can be utilized to fabricate “smart” delivery systems which can enable more precise control over controlled drug delivery.
2.1.3. Poly (N-acrylamides)
Aside from polyesters and polyethers, poly (N-acrylamides) have also been used for local biomaterial-based drug delivery systems. One of the most common poly (N-acrylamides) is poly (N-isopropylacrylamide) (PNIPAm), which is often utilized for its’ thermoresponsive behavior [59]. PNIPAm has an LCST of roughly 33°C which is very similar to body temperature (37°C), making it an attractive material for drug delivery [59]. Similar to PEGMA, described above, PNIPPAm exists as an aqueous solution below 33°C. Above this temperature, PNIPPAm becomes water insoluble and undergoes gelation.
Hou et al. explored the use of PNIPAm, in combination with chitosan and hyaluronic acid, as a hydrogel matrix in islet transplantation [76]. In this study, PNIPAm-chitosan-hyaluronic acid hydrogels were used to graft CTLA4-Ig, an immunomodulatory fusion protein to provide local immunosuppression for transplanted islets [76]. Notably, grafting CTLA4-Ig to the hydrogel decreased the LCST of the matrix to 28°C [76]. While still above room temperature, the resulting LCST is significantly below body temperature. Schilling et al. also developed a local biomaterial-based delivery system using PNIPAm in combination with PLGA MPs for local delivery of a corticosteroid to the sinuses in a model of chronic rhinosinusitis [79]. The resulting combined composite system (PNIPAm/PLGA MPs) exhibited a sol-gel transition at 35°C, which importantly, was the same sol-gel temperature demonstrated by PNIPPAm alone [79]. Although one of these systems was used to deliver a biologic (CTLA-Ig) and the other a small molecule (corticosteroid), they draw attention to the impact that drug incorporation (and the manner in which it occurs) can have on sol-gel temperatures. This could be an important parameter to consider when developing thermoresponsive biomaterial-based delivery systems, as the most ideal system would have a sol-gel transition temperature similar to body temperature.
2.1.4. Poloxamers
Poloxamers are another class of synthetic polymers commonly used in drug delivery. Poloxamers, also known as Pluronics®, are block copolymers consisting of both poly (ethylene oxide) (PEO) and poly (propylene oxide) (PPO) [80]. These polymers are desirable in drug delivery applications due to their thermoresponsive behaviors and ability to self-assemble into micelles (gel-like aggregates) in solution [80]. More specifically, poloxamers have critical micelle temperatures (CMT) and critical micelle concentrations (CMC), above which they form micelles [80]. On the other hand, when below the CMT/CMC, poloxamers exist as an aqueous solution.
Due to this reversible behavior, poloxamers have been used as drug delivery systems in inflammation [33,34,75,81,82]. Yin et al. explored the effect of Pluronic F127 (F127) and Pluronic F68 concentrations on various outcome measures (i.e., gelation temperature, gelation time, viscosity, etc.) [33]. Increasing F127 concentrations decreased the gelation temperature of the hydrogels. F127 concentrations exhibited a direct relationship with viscosity, where increased concentrations lead to increased solution viscosity at room temperature. This is an important parameter to consider in formulating injectable hydrogels, as increased viscosity could hinder injectability. Several more parameters for developing poloxamer thermoresponsive systems were explored by Wu et al. [37]. Wu et al. compared gelation temperatures of Soluplus, a commercially available block co-polymer, and Pluronic F127 (otherwise known as poloxamer P407) to select the best hydrogel for tacrolimus release. Due to its’ increased hydrophobicity, the sol-gel temperature of Soluplus was higher (range from 32.8° to 43.2°C) than that of P407 (range from 22.3°C to 28.3°C) [37]. Aside from hydrophobicity, polymer and additive (i.e., salt) concentration impacted gelation temperature [37]. More specifically, increasing concentrations of polymer and/or salt resulted in decreasing gelation temperatures. Since the sol-gel temperature of Soluplus was higher than that of P407, and within the range of normal body temperatures, it was concluded to be a more ideal thermoresponsive system. Observations from these studies highlight several variables to consider when formulating poloxamer thermoresponsive systems in inflammation, including viscosity, hydrophobicity, and polymer concentration.
2.1.5. Poly (amino acids)
Poly (amino acids) combine components of both natural and synthetic polymers. They are based on the same peptide bonds present in natural proteins but are prepared through synthetic processes [59]. Three of the most common poly (amino acids) include poly (glutamic acid), poly (lysine) and poly (aspartic acid) [59].
In drug delivery, poly (amino acids) are most commonly utilized in combination with other polymers, such as PEG and poloxamers, to form amphiphilic block co-polymers capable of self-assembly into micelles/hydrogels [75,83]. For instance, Lin et al. developed block co-polymer poloxamer-poly (amino acid) hydrogels to encapsulate and provide controlled release of tacrolimus in a model of murine skin allotransplantation [75]. PPG-PEG-PPG was used as the poloxamer and poly(alanine-lysine) was used as the poly (amino acid) in this study. Notably, the gelation temperature of the hydrogel decreased as the concentration of co-polymers increased, with solutions composed of less than 3 wt% unable to form gels. In addition, the hydrogels unexpectedly exhibited extremely low release rates of tacrolimus, resulting in transplant rejection due to low drug concentrations [75]. To address this issue, the authors added Pluronic 127 (F127), another poloxamer block copolymer consisting of poly (propylene oxide) (PPO) and poly (ethylene oxide) (PEO) (discussed briefly in 2.1.4.). F127 exhibits fast erosion kinetics in aqueous environments due to contact with solvent that prompts the hydrogel to fall below the critical gelation concentration, resulting in a loss of gel-like structure [84]. As a result, addition of F127 to the tacrolimus-eluting amino acid hydrogel resulted in higher release rates that corresponded with hydrogel degradation [75]. Altogether, these results highlight important design considerations, such as polymer concentration and matrix interactions, when developing poly (amino acid) delivery systems for controlled release in inflammation.
2.2. Natural Polymers
Although the above materials are readily implemented in drug delivery systems, synthetic polymers could also possess features that may limit their use. In some cases, synthetic materials, such as polyesters, have been found to induce an undesirable inflammatory response that skews the immunological reaction away from homeostasis [41]. Natural polymers could be utilized as an alternative or even incorporated in conjunction with synthetic polymers to capitalize on the benefits of both approaches (i.e., functionalization, controlled release, biomimicry, etc.). Natural polymers include polysaccharides (alginate, chitosan, etc.) or proteins (gelatin, fibrin, collagen, etc.) (Table 2). This section gives a brief overview of these materials and their corresponding characteristics that make them desirable for drug delivery carriers in inflammation. Several recent reviews have also explored these materials in more depth [85–88].
Table 2.
Commonly used natural polymers for the development of local delivery strategies to restore immune homeostasis from inflammation.
| Polymer | Structure | Local Delivery Strategies | Ref(s) |
|---|---|---|---|
| Polysaccharides | |||
| Alginate |

|
Alginate hydrogel; alginate/GelMA hydrogel; alginate capsules; alginate/PEI hydrogel; alginate MPs; alginate scaffold | [41,89–99] |
| Chitosan |
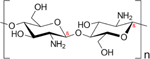
|
Chitosan/pNIPAm/hyaluronic acid hydrogels; alginate-MPs/chitosan hydrogels | [57,76,86, 92] |
| Hyaluronic acid |
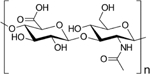
|
Hyaluronic acid hydrogel; hyaluronic acid/tyramine hydrogel; hyaluronic acid/collagen hydrogel | [34,36,104] |
| Agarose |
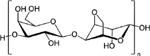
|
Agarose hydrogel; lipid microtubes/agarose hydrogel; gelatin-MPs/agarose scaffold | [108–110] |
| Protein-based polymers | |||
| Collagen |
|
Collagen scaffolds; hyaluronic acid/collagen hydrogels | [58,88,112] |
| Silk fibroin |
|
Silk hydrogels | [111,113,114] |
| Gelatin |
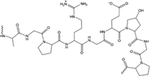
|
Gelatin hydrogels; gelatin-MPs/agarose scaffold | [110,115–118] |
2.2.1. Polysaccharides
Polysaccharides are naturally existing polymers composed of carbohydrates connected through glycosidic bonds [87]. Polysaccharides are attractive materials for drug delivery carriers due to their limited immunogenicity, functionalization (i.e., PEGylation) and biodegradability [87]. Common polysaccharides used for delivery matrices in treating inflammation include alginate, chitosan, hyaluronic acid and agarose.
Alginate is a polysaccharide isolated from brown algae. Due to its’ non-toxic profile, alginate delivery systems have been used for a variety of applications, including cell transplantation (i.e., islets, regulatory t cells, hybridoma cells, mesenchymal stem cells, etc.) and biologic delivery (i.e., antigen, cytokines, etc.) [41,89–99]. However, in the context of inflammation, it is important to consider the potential immune response associated with alginate. Since it is a natural polymer, there are numerous impurities (i.e., polyphenols, endotoxins, protein, etc.) that can be left during its’ isolation and processing [100]. One example of an immunostimulatory impurity in alginate is pathogen-associated molecular patterns (PAMPs), which can signal through immune receptors to trigger inflammatory responses [100]. Thus, it is important to produce or use alginate (or other natural polymers) which lack immunostimulatory impurities, such as PAMPs, in the development of delivery systems.
Nonetheless, alginate matrices are attractive carriers for local delivery strategies, specifically for stimuli-responsive applications, due to their sensitivity to both temperature and pH [101]. In general, natural polymers are known to have a quick sol-gel transition [74]. However, the quick sol-gel transition, which is exhibited by alginate, can limit its’ ability to be manipulated and injected for drug delivery [89]. Espona-Noguera et al. aimed to address this limitation by incorporating phosphate salt into alginate hydrogels for islet transplantation [89]. Increasing concentrations of phosphate salt resulted in increased gelation times. However, increasing concentrations also significantly reduce the elastic properties of the alginate hydrogels. While increased gelation times can be beneficial in the fact that there is a longer window available for manipulation prior to gelation, reduced elasticity could result in increased deformation when implemented in vivo. Therefore, it is important to consider these parameters when attempting to manipulate drug delivery matrices.
Chitosan, a polysaccharide derived from chitin, is another polysaccharide of interest in local drug delivery. In treating inflammation, it is often used for strategies aiming to achieve controlled release of immunomodulatory factors [57,76,86,92]. In addition to its’ ability to facilitate controlled release, chitosan is considered relatively non-toxic and can be incorporated within matrices to limit potential toxicity by other delivery polymers, making it an attractive material for inflammatory applications [76,87]. For instance, Hou et al. found that the addition of chitosan to PNIPAAm hydrogels also reduced inflammatory effects seen by the synthetic hydrogel alone [76]. Chitosan has also been demonstrated to have thermosensitive behaviors when combined with alginate microparticles to form composite hydrogels for local delivery of diclofenac sodium [32]. Although chitosan possesses several advantages that result in its’ use as a drug delivery material, chitosan could still be limited by its’ potential antigenicity. More specifically, chitosan has shown some promise as an adjuvant, as it can activate inflammatory signaling pathways [102]. However, a recent study has shown that high molecular weight chitosan could promote anti-inflammatory responses, whereas low molecular weight chitosan could promote inflammatory responses [103]. Therefore, it may be worthwhile to explore multiple molecular weights of chitosan in the development of delivery systems in order to avoid unanticipated immune responses.
Hyaluronic acid, composed of repeating units of N-acetyl-D-glucosamine and b-glucuronic acid, has also been used in local delivery systems, specifically for treating autoimmune diabetes (through islet transplantation) as well as rheumatoid arthritis [34,36,104]. As a hydrophilic polymer, hyaluronic acid has been used to improve the solubility of hydrophobic drugs and limit fibrotic growth sometimes associated with other polymers, such as alginate [34,88,104]. Of note, hyaluronic acid is known to interact with the immune system, specifically through CD44, a transmembrane receptor expressed on the surface of various immune cells [105]. Many studies have shown its’ inflammatory role, as hyaluronic acid has been demonstrated to increase inflammatory cytokine secretion through CD44 signaling [105]. However, other studies have highlighted that the inflammatory capacity of hyaluronic acid depends on its’ molecular weight [106]. High molecular weight hyaluronic acid has been shown to display immunosuppressive properties, whereas low molecular weight hyaluronic acid can be considered inflammatory [106]. Therefore, studies using hyaluronic acid to develop polymeric drug delivery systems should consider the molecular weight as a factor that could influence outcomes.
An example where hyaluronic acid was successfully used to resolve inflammation is the use of its’ crosslinked form for [34]-articular delivery of methotrexate in inflammatory arthritis [34]. In vivo studies demonstrated that crosslinked hyaluronic acid loaded with methotrexate led to increased articular residence time which enabled reversal of rheumatoid arthritis [34].
A less frequently used polysaccharide of interest in the development of delivery systems in inflammation is agarose. Agarose, a polysaccharide derived from seaweed, is a desirable material due to its’ slow degradation rate, self-gelling ability and ease of manipulability (i.e., porosity, functionalization) [85]. Furthermore, agarose-treated dendritic cells have been shown to induce regulatory immune cells (at comparable levels to untreated immature dendritic cells), as well as expression of immunomodulatory proteins (both anti-inflammatory and inflammatory) [107]. Therefore, agarose-based biomaterials themselves could potentially be used to modulate the immune response.
Agarose, either as a hydrogel or a scaffold, has been used to facilitate local delivery of immunomodulators as well as cells in models of inflammation [108–110]. When utilizing agarose as a scaffold, one parameter to consider is its’ potential porosity due to the fabrication process (i.e., lyophilization). While high porosity can be beneficial for cell-based delivery approaches by allowing an exchange of nutrients, high porosity can also facilitate faster release of cargo. Accordingly, Srinivasan et al. incorporated gelatin microparticles within an agarose scaffold to provide an extra barrier to cargo diffusion, which can impact release (Figure 2A, 2B) [110]. Interestingly, for this combination approach, it was found that release kinetics were primarily associated with the number of microparticles dispersed within the platform [110]. Lower amounts of microparticles (<1010) resulted in reduced burst release when compared to higher quantities of microparticles (Figure 2C).
Fig.2.
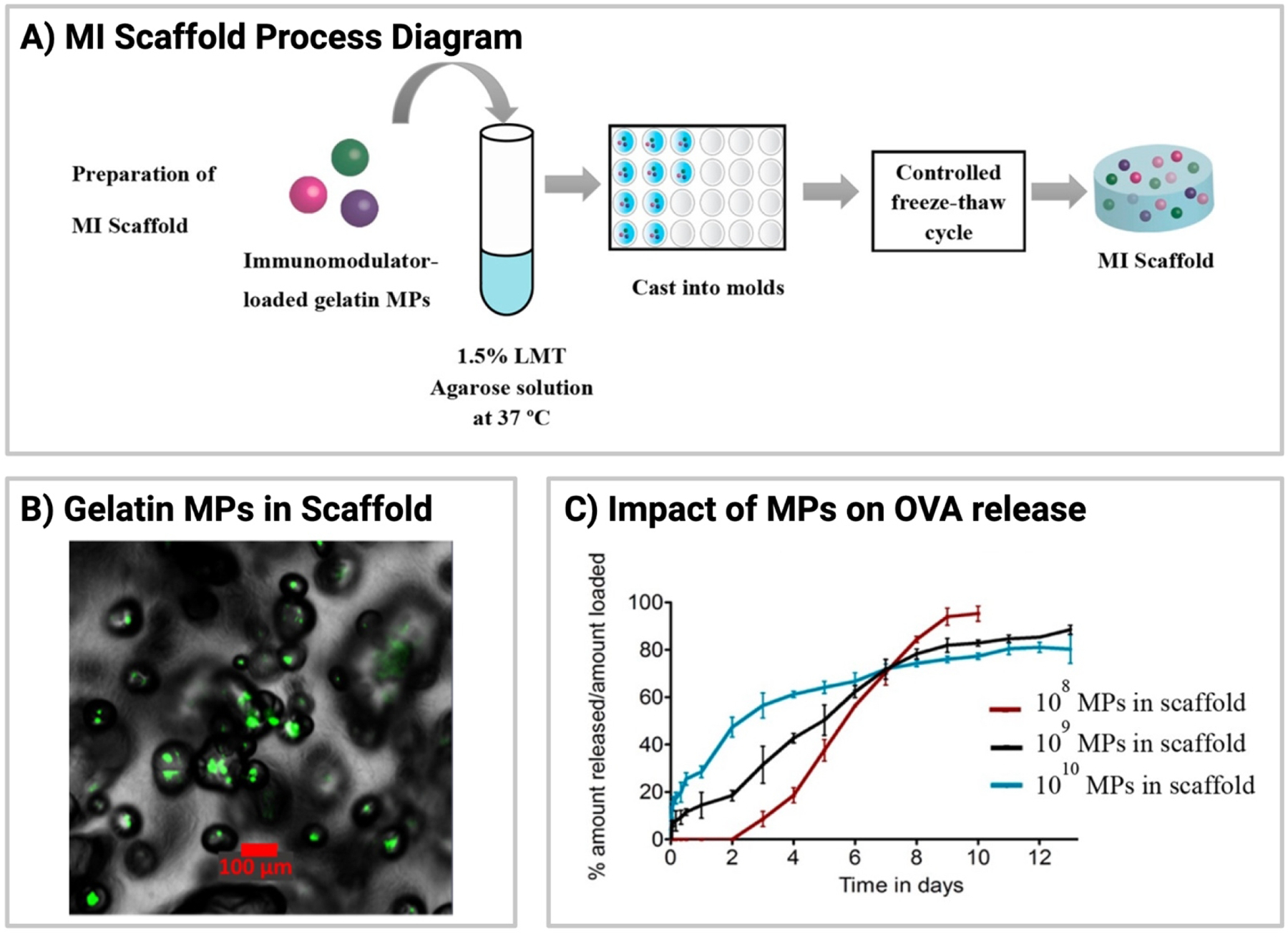
Gelatin-MPs embedded in agarose scaffold. (A) Process diagram demonstrating the fabrication of scaffolds containing gelatin-MPs loaded with immunomodulatory signals. (B) Confocal microscopy image of gelatin-MPs (green) within an agarose scaffold. (C) Effect of the number density of gelatin-MPs on ovalbumin release, where ovalbumin is a model protein. All images/data in this figure reprinted with permission from S. Srinivasan, J.E. Babensee, Controlled Delivery of Immunomodulators from a Biomaterial Scaffold Niche to Induce a Tolerogenic Phenotype in Human Dendritic Cells, Acs Biomater Sci Eng. 6 (2020) 4062–4076. https://doi.org/10.1021/acsbiomaterials.0c00439. Copyright 2021. American Chemical Society.
2.2.1. Proteins
Aside from polysaccharides, proteins such as collagen and silk fibroin, have been used for both islet transplantation and controlled release of biologics in autoimmunity [111–114]. Collagen, one of the main components of the extracellular matrix, is a biodegradable material with low antigenicity [58,88]. Griffin et al. used collagen scaffolds to locally deliver high doses of antigen in a murine model of experimental autoimmune encephalomyelitis (EAE) [112]. Similar to collagen, silk fibroin, isolated from silkworm cocoons, is also desirable for local delivery systems due to its’ inherent slow degradation rate and self-assembling nature [88,111]. Silk fibroin contains several crystalline domains, in addition to amorphous domains, which can self-assemble into micelles [88]. Under the presence of external stimuli, the micelles can further aggregate to form a hydrogel-like network [88]. In addition to its’ property of self-assembly, silk fibroin contains numerous available groups on side chains for functionalization, which can be used to tune its’ degradation kinetics [88]. Kumar et al. explored the use of silk hydrogels, specifically a blended composition of mulberry silk and non-mulberry silk, for islet encapsulation and controlled release of immunomodulatory factors [114]. Notably, silk hydrogels were able to facilitate controlled release of two immunomodulatory factors, interleukin-4 and dexamethasone, over two weeks.
Another protein-based natural polymer is gelatin, which is derived from collagen [115]. Gelatin has been used alone, as well as in conjunction with other release systems, to facilitate controlled release of biologics in models of inflammation [110,116]. These studies demonstrated the feasibility of gelatin-based microparticles to controllably deliver biologics over a short timeframe (~10 days). Nonetheless, gelatin-based delivery carriers can be limited by their rapid disintegration in aqueous environments, resulting in burst release of encapsulated cargo [110,115]. To combat this, gelatin crosslinking can be increased to reduce the degradation rate [110,115]. The addition of methacryloyl groups to gelatin, resulting in gelatin methacryloyl hydrogels (GelMA), is a common approach used to crosslink the material through photopolymerization [117]. GelMA hydrogels have also been used in models of inflammation to facilitate controlled release of biologics and provide immunoisolation of transplanted cells [90,118]. Aside from limitations regarding degradation and release, gelatin-based materials are considered biocompatible due to their low antigenicity, and in the case of GelMA, their similarity to the extracellular matrix [115,117]. As a result of their ability to facilitate controlled release and low antigenicity, gelatin has become a desirable carrier for drug delivery applications in inflammation.
3. Applying local delivery methods to restore immune homeostasis
The biomaterials discussed above are the most common materials used for the development of local delivery systems (i.e., microparticles, hydrogels, scaffolds). These delivery systems can then be administered locally through a variety of delivery routes (i.e., subcutaneous injection, intra-articular injection, implantation) in an effort to promote immune homeostasis in a diverse range of pathologies. One important consideration is that biomaterials themselves have the potential to spark an immune response (either anti-inflammatory or inflammatory). Multiple recent reviews have discussed the impact of certain materials, as well as their properties (i.e., size, hydrophobicity, molecular weight, etc.), on the immune response [119–121]. In addition, the delivery of these materials (i.e., injection, implantation, etc.) can disturb the local immunological milieu and risk triggering immune reactions (i.e., foreign body response, complement activation, platelet activation, etc.) [28,122,123]. For a more comprehensive analysis, the interaction between common materials used for local drug delivery systems and their delivery routes with the immune system is extensively reviewed elsewhere [14,28,119–124].
Although materials themselves can be used to modulate the immune system, this review will focus on the use of biomaterials as local delivery strategies for cargo delivery. Local delivery of four main immunological approaches (i.e., small molecules, biologics, cells or antigen-specific therapeutics) has shown to be beneficial in a wide variety of disease indications, ranging from transplant rejection to autoimmunity. The following section will discuss these approaches in order of complexity, beginning with the delivery of small molecules and biologics, and finishing with antigen-specific approaches.
3.1. Delivery of small molecules
Approaches to restore immune homeostasis rely upon the use of systemic anti-inflammatory agents, such as non-steroidal anti-inflammatory agents, corticosteroids, anti-metabolites, mTOR inhibitors and calcineurin inhibitors [8–10]. Although using different mechanisms of action, these agents are similar in their lack of specificity, resulting in widespread immunosuppression accompanied by systemic side effects (i.e., infection, nephrotoxicity, hepatotoxicity, etc.) [8–10]. To address these issues, local drug delivery strategies can be used to repurpose immunosuppressant drugs to minimize side effects and restrict immunomodulation to the site of interest.
3.1.1. Non-steroidal anti-inflammatory agents (NSAIDs)
Non-steroidal anti-inflammatory agents are the broadest class of anti-inflammatory drugs in terms of their mechanism of action. NSAIDs block the production of prostaglandins, which play a role in driving acute inflammation, from almost all nucleated cells by inhibiting cyclooxygenase enzymes [125,126]. While blocking cyclooxygenases (COX) enzymes in sites of inflammation can be beneficial, COX inhibition in normal tissues can lead to side effects such as excess bleeding and ulcers [125]. Therefore, local drug delivery strategies have been used to localize the effects of certain NSAIDs (Table 3) [32,33,127,128].
Table 3.
Local delivery strategies used for small molecule delivery to restore immune homeostasis from inflammation.
| Small Molecules | Local Delivery Strategy | Disease Model | Ref(s) |
|---|---|---|---|
| Non-steroidal anti-inflammatory agents | |||
| Diclofenac sodium | Alginate MPs/chitosan hydrogel | Inflammatory arthritis | [32] |
| Indomethacin | PEI NPs/pluronic hydrogel | - | [33] |
| Celecoxib | PLGA/cyclodextrin MPs | - | [127] |
| PLGA-silica MPs/PLLA scaffold | Periodontal disease | [128] | |
| Corticosteroids | |||
| Dexamethasone | PDMS scaffold | Islet transplantation | [132] |
| Ascorbyl palmitate hydrogel | Inflammatory bowel disease | [134] | |
| PLGA micelles | Islet transplantation | [38] | |
| Triamcinolone acetonide | TGMS hydrogel | Inflammatory arthritis | [133] |
| PLA/mPEG-PDL MPs | Inflammatory arthritis | [138] | |
| PLA/mPEG-PDL MPs/ PEGMA hydrogel | Inflammatory arthritis | [70] | |
| Betamethasone | Gellan gum hydrogel | Inflammatory arthritis | [136] |
| Prednisolone | PLGA MPs | Inflammatory arthritis | [135] |
| Mometasone furoate | PLGA MPS/PNIPPAm hydrogel | Chronic rhinosinusitis | [79] |
| Anti-Metabolites | |||
| Methotrexate | Alginate MPs/methylcellulose hydrogel | Inflammatory arthritis | [149] |
| PEI-NPs/pluronic hydrogel | Inflammatory arthritis | [33] | |
| PFK15 inhibitor | paKG MPs | Inflammatory arthritis | [152] |
| mTOR inhibitors | |||
| Rapamycin | PLGA/PCL MPs | Islet transplantation | [157] |
| Elastin-like polypeptide depots | Sjögren’s Syndrome | [158] | |
| PLGA MPs | Ex vivo autoimmunity | [156] | |
| Everolimus/Temsirolimus | PLGA MPs | Ex vivo autoimmunity | [156] |
| Calcineurin Inhibitors | |||
| Cyclosporin A | PLGA MPs/alginate hydrogel | Cell transplantation | [162] |
| Tacrolimus | TGMS hydrogel | Vascularized composite allotransplantation | [144] |
| TGMS hydrogel | Vascularized composite allotransplantation | [31] | |
| TGMS hydrogel | Vascularized composite allotransplantation | [142] | |
| Poly(amino acid)/pluronic hydrogel | Skin allotransplantation | [75] | |
| Supramolecular hydrogel | Liver transplantation | [164] | |
| PLGA MPs | Islet transplantation | ||
| PLGA NPs/RADA16 hydrogel | Stem cell transplantation | [165] | |
| TGMS hydrogel | Vascularized composite allotransplantation | [143] | |
| Soluplus/pluronic hydrogel | Inflammatory arthritis | [37] | |
| PLGA MPs/fibrin gel | - | [170], [169] | |
Many NSAIDs, such as diclofenac sodium and indomethacin, inhibit both isoforms of cyclooxygenase enzymes (COX-1 and COX-2). To restrict their action to a specific site of interest, local delivery of both diclofenac sodium and indomethacin has been explored [32,33]. Qi et al. developed a composite system composed of alginate microspheres and chitosan hydrogels to facilitate short-term controlled release of diclofenac sodium following intra-articular injection [32]. In a model of rheumatoid arthritis, intra-articular injection of the composite system reduced joint swelling [32]. While joint swelling was reduced following treatment, weekly intra-articular administration over three weeks failed to prevent disease progression. While diclofenac sodium can relieve symptoms associated with rheumatoid arthritis, it is likely ineffective at halting disease progression. Therefore, it could be worthwhile to explore other biologics for a more specific and robust mechanism of action that can significantly inhibit disease progression. Local delivery of indomethacin has also been explored using a composite system composed of polyethyleneimine nanoparticles within a Pluronic hydrogel [33]. In this study, Yin et al. locally delivered indomethacin alongside methotrexate, an anti-metabolite (discussed in subsequent sections) intra-articularly in a model of rheumatoid arthritis [33]. It was found that indomethacin release was significantly reduced, and controlled release prolonged, when released from nanoparticle-containing hydrogels (D-NGel) when compared to nanoparticles alone. This study demonstrates the ability to develop a local, controlled release system for indomethacin, while highlighting the ability to modulate release through addition of a secondary barrier that can impact diffusion.
While many NSAIDs, such as those mentioned above, inhibit both COX isoforms, few NSAIDs inhibit COX-2 only, slightly increasing their specificity [129]. An example of a COX-2 inhibitor is celecoxib, which has been reformulated using multiple drug delivery strategies [127,128]. Cannavà et al. reformulated celecoxib using PLGA microparticles and tested its’ efficacy in vitro on human chondrocytes [127]. In this study, the authors sought to increase release of celecoxib from PLGA MPs, as celecoxib’s high lipophilicity can greatly delay release [127]. It was found that the addition of dimethyl-β-cyclodextrin during fabrication had a dose-dependent effect on cargo release, as high concentrations increased burst effects. As a result of increased release, PLGA microparticles with dimethyl-β-cyclodextrin significantly decreased in vitro nitric oxide synthase expression when compared to microparticles lacking cyclodextrin [127]. In this case, the authors showed increased therapeutic efficacy when burst release was enhanced. However, these results are limited by the timescale of the study, as human chondrocytes were stimulated and treated for only 72 hours. Therefore, it could be important to conduct a longer study to determine the impact of this approach over time.
3.1.2. Corticosteroids
Corticosteroids, steroid hormones that can be produced by the body, also have an expansive mechanism of action. More specifically, corticosteroids regulate inflammatory responses by altering gene transcription in leukocytes, leading to reduced pro-inflammatory cytokine secretion and immune cell activation [130]. Due to their non-specificity, corticosteroids are used in a variety of inflammatory disorders/conditions, including allergy, asthma, inflammatory bowel disease, islet transplant rejection, rheumatoid arthritis and chronic rhinosinusitis [131]. Several of these corticosteroids, including dexamethasone, triamcinolone acetonide, prednisolone, betamethasone and mometasone furoate have been reformulated using local drug delivery systems to treat inflammatory bowel disease, islet transplant rejection, rheumatoid arthritis and chronic rhinosinusitis (Table 3) [38,70,79,132–138].
One of the earliest synthesized corticosteroids, dexamethasone, has been reformulated using biomaterial-based drug delivery strategies in multiple studies. Dexamethasone (DEX) exerts its’ anti-inflammatory effects through multiple mechanisms, including decreased immune cell migration and proliferation [139]. In addition, dexamethasone is capable of modulating macrophage phenotype over time and even skew polarization towards an anti-inflammatory phenotype (M2) [132,140]. Jiang et al. explored the effects of local delivery of dexamethasone on islet transplantation with the goal of enhancing graft viability and glycemic control [132]. When co-encapsulated with islet cells in a porous polydimethylsiloxane (PDMS) scaffold, low concentrations of dexamethasone significantly improved islet engraftment [132]. Further examination of the mechanisms responsible showed that localized dexamethasone-loaded scaffolds fostered macrophage polarization towards an M2 phenotype, characterized by the expression of CXC3CR1. Notably, dexamethasone-loaded scaffolds did not impact cell infiltration into the scaffold, and only cell phenotype was altered [132]. This study demonstrates the promise of delivering low concentrations of immunosuppressants, facilitated using local delivery systems, in reducing inflammation and promoting cell survival. Similar effects were observed by Kuppan et al. when islets were co-localized with DEX-PLGA micelles under the kidney capsule in allogeneic islet transplantation [38]. In this study, co-localization of DEX-micelles in combination with monoclonal antibody therapy (DEX-micelle + mAb) was compared against empty micelles with monoclonal antibody therapy (empty micelle + mAb). Contralateral administration of empty micelles + mAb therapy failed to promote allogenic islet transplant survival when compared to islet co-localization with DEX-micelles, highlighting the importance of local delivery. Together, these studies illustrate the enhanced efficacy of dexamethasone when administered at low doses and localized to the site of interest, which is made possible with local biomaterial-based delivery systems.
Triamcinolone acetonide (TA), an alternative corticosteroid to dexamethasone, functions similarly to dexamethasone. Trimcinoclone acetonide also inhibits leukocyte and macrophage migration to inflammatory sites [141]. TA has been reformulated using drug delivery strategies to facilitate local delivery in rheumatoid arthritis (RA) following intra-articular injection [133,137,138]. Joshi et al. developed an enzyme-responsive hydrogel using triglycerol monostearate (TGMS/TG-18) to deliver TA in arthritis joints [133]. TGMS is a common enzyme-response material taken from the Generally Recognized as Safe (GRAS) list provided by the U.S. Food and Drug Administration. Due to its’ ability to degrade in response to external stimuli (i.e., MMPs, esterases), similar to what happens to native tissues in the body (ECM), TGMS has been used multiple times as a local drug delivery carrier in inflammatory disorders (Figure 3A) [142–145]. In this case, TGMS was used to controllably deliver TA in response to the presence of degradative enzymes upregulated in synovial fluid from arthritic joints [133]. In vitro release kinetics demonstrated stimuli-responsive release of TA in the presence of both MMPs and synovial fluid from human rheumatoid arthritis joins (SF-RA) (Figure 3B, 3C). When locally administered to hind paws in a model of inflammatory arthritis, TA-loaded hydrogels reduced severity, paw thickness and clinical score in arthritic joints when compared to empty hydrogels [133]. Importantly, local administration of TA-loaded TGMS also performed significantly better than soluble TA at reducing arthritis severity, potentially due to rapid joint clearance of soluble TA.
Fig.3.
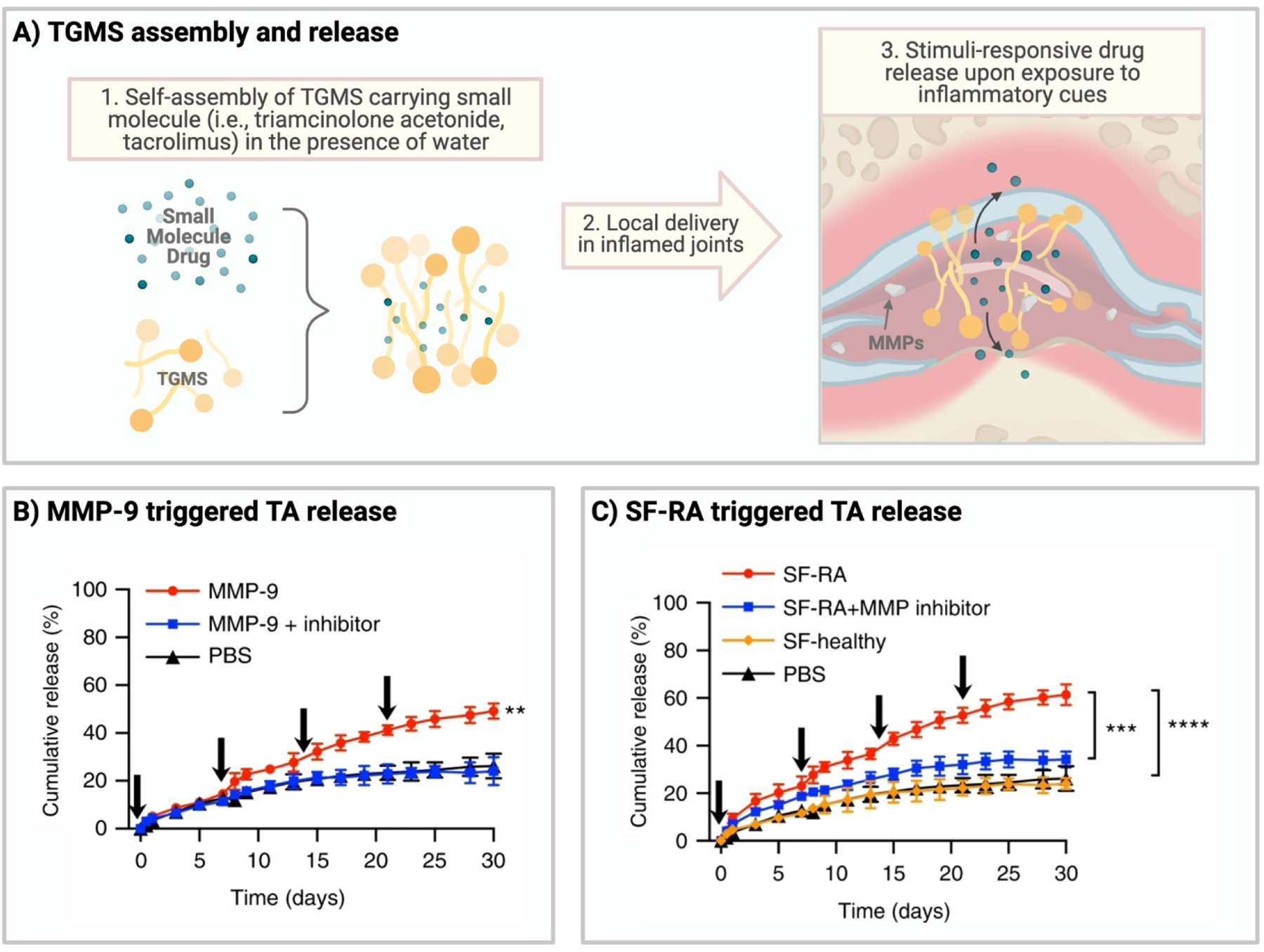
TGMS assembly and release of TA. (A) Schematic illustrating fabrication and application of TGMS in inflammation. (B) Effect of matrix mellatoproteinase 9 (MMP-9) on cumulative release of TA. (C) Effect of synovial fluid from arthritic joints (SF-RA) on cumulative release of TA. Data in Figures 3B and 3C reprinted from N. Joshi, J. Yan, S. Levy, S. Bhagchandani, K.V. Slaughter, N.E. Sherman, J. Amirault, Y. Wang, L. Riegel, X. He, T.S. Rui, M. Valic, P.K. Vemula, O.R. Miranda, O. Levy, E.M. Gravallese, A.O. Aliprantis, J. Ermann, J.M. Karp, Towards an arthritis flare-responsive drug delivery system, Nat Commun. 9 (2018) 1275. https://doi.org/10.1038/s41467-018-03691-1. Creative Commons license https://creativecommons.org/licenses/by/4.0/. No changes were made to the material.
Abou-ElNour et al. also reformulated TA for intra-articular injection using poly-lactic acid (PLA) and PEG-poly-δ-decalactone (PEG-PDL) microparticles, which were found to provide short-term controlled release [138]. Notably, PEG-PDL polymer was unable to form microparticles alone. Therefore, PLA was incorporated to form the delivery matrix. Characterization of the resulting particles demonstrated the formation of drug-polymer crystals following incorporation of PEG-PDL. When implemented in a model of inflammatory arthritis, PLA/PEG-PDL microparticles inhibited inflammation for a longer period when compared to microparticles lacking PEG-PDL [138]. It is possible that crystal formation led to increased drug retention over time, allowing for a prolonged effect. Results from these studies suggest the importance of and the ability to increase therapeutic retention/availability of TA through the use of local biomaterial-based delivery systems.
Although these studies suggest that local administration of corticosteroids could be beneficial to enhance their effects, it is worth noting that long-term use of corticosteroids is often associated with drug resistance, limiting their effectiveness [131,146]. Therefore, it would be worth exploring the use of alternative small molecules, discussed below, or even more specific immunomodulators, discussed in the subsequent sections, to maintain immune homeostasis long-term.
3.1.3. Anti-metabolites
Anti-metabolites, another class of anti-inflammatory drugs, are like corticosteroids in that they exert effects on multiple immune cell populations. However, anti-metabolites impact cell metabolism to generate immunomodulatory effects. More specifically, effector cells (i.e., effector T cells, proinflammatory macrophages, etc.) require a switch from oxidative phosphorylation to glycolysis to support proliferation and proinflammatory molecule expression, a concept known as the Warburg effect [147]. The metabolic switch enables immune cells to function in inflammatory states, which typically exhibit hypoxic conditions. Therefore, by restricting or inhibiting processes involved in the switch from oxidative phosphorylation to glycolysis, or glycolysis in general, anti-metabolites can promote anti-inflammatory effects [147].
One of the most common anti-metabolites is methotrexate (MTX), which appears to act through different mechanisms of action based on dosage. At high doses, methotrexate targets the folate pathway where it inhibits nucleotide synthesis and damages DNA replication [148]. At lose doses, it is likely that methotrexate targets amido-imidazole-carbox-amido-ribonucleotide, leading to the release of anti-inflammatory adenosine [148]. Adenosine can exert anti-inflammatory effects by signaling through its’ receptor A2A to inhibit T cell proliferation and activation [148]. Required dosage can be minimized using local drug delivery strategies, which can constrain drug to a specific site of interest and avoid high doses associated with systemic administration.
In the case of rheumatoid arthritis, local drug delivery strategies have been used to constrain the action of methotrexate to the joint to limit systemic exposure (Table 3) [149,150]. For example, Dhanka et al. studied the impact of local methotrexate delivery using alginate microparticles embedded in a sodium hyaluronate-methylcellulose hydrogel (MTX-MPs-H) in a model of inflammatory arthritis [149]. The combination microparticle-hydrogel delivery system provided short-term controlled release over several days. These results were a stark contrast to hydrogel delivery of methotrexate alone, which only released methotrexate over a few hours. When intra-articularly administered in a model of inflammatory arthritis, MTX-MPs-H significantly decreased paw thickness and swelling when compared to soluble methotrexate and methotrexate-loaded microparticles alone, suggesting a reduction in inflammation [149]. These results suggest that intra-articular administration of MTX-MPs-H increased therapeutic retention within the joint when compared to soluble administration or methotrexate-microparticles alone. It is possible that the increased retention provided by the local drug delivery strategy then enabled a significant reduction in inflammation. Yin et al. also developed a composite system, composed of polyethyleneimine nanoparticles within a Pluronic hydrogel matrix (D-NGel), to facilitate controlled release of methotrexate and indomethacin intra-articularly [33]. Similar to Dhanka et al., Yin et al. found that controlled release of methotrexate was significantly improved when nanoparticles were used in conjunction with the hydrogel, suggesting the benefit of adding an additional barrier to diffusion when developing controlled release systems. In a collagen-induced arthritis model, intra-articular treatment with D-NGel reduced levels of TNF-α and IL-1β, two inflammatory cytokines responsive for synovial damage in RA [33]. Importantly, levels of TNF-α were slightly more reduced in the knee joint than the ankle joint, likely attributed to direct intra-articular administration to the knee joint. In addition, combination treatment with both methotrexate and indomethacin significantly improved arthritis symptoms when compared to methotrexate or indomethacin in hydrogels alone [33]. Therefore, it may be advantageous to treat disease with multiple small molecules that have different mechanisms of action to improve disease outcomes. In addition, results from these two studies demonstrate the ability to tune controlled release by providing additional barriers to diffusion.
Aside from methotrexate, one emerging small molecule that could be classified as an anti-metabolite is PFK15. PFK15 is a small molecule inhibitor of PFKFB3, an enzyme that controls the rate of glycolysis [151,152]. In the context of autoimmunity, PFKFB3 has been found to be upregulated in a variety of cancers, as well as in synovial tissue and fibroblast-like synoviocytes from rheumatoid arthritis patients [151,153]. Treatment with PFK15 has shown anti-glycolytic effects in models of gastric cancer and collagen-induced arthritis [151,153]. To enhance the efficacy of PFK15, Mangal et al. explored the subcutaneous delivery of PFK15, alongside disease-associated antigen, using alpha-ketoglutarate and diol-based microparticles (paKG-MPs) (Figure 4A) [152]. paKG-MPs, synthesized from aKG,, an immunosuppressive Kreb’s cycle intermediate, have been shown to controllably delivery aKG following particle degradation, leading to anti-inflammatory effects (i.e., reduced glycolysis, reduced CD4+ T cell responses, etc.) [154]. Delivery of PRK15 using immunosuppressive paKG-MPs was found to modulate dendritic cell glycolysis and metabolic pathways (Figure 4B) [152]. As this study delivered PFK15 alongside antigen, it will be further discussed in Section 3.4.2. Nonetheless, these studies highlight the potential benefit of choosing immunosuppressive materials to deliver immunomodulatory signals, as it could further modulate the immune response to enable the desired response.
Fig.4.
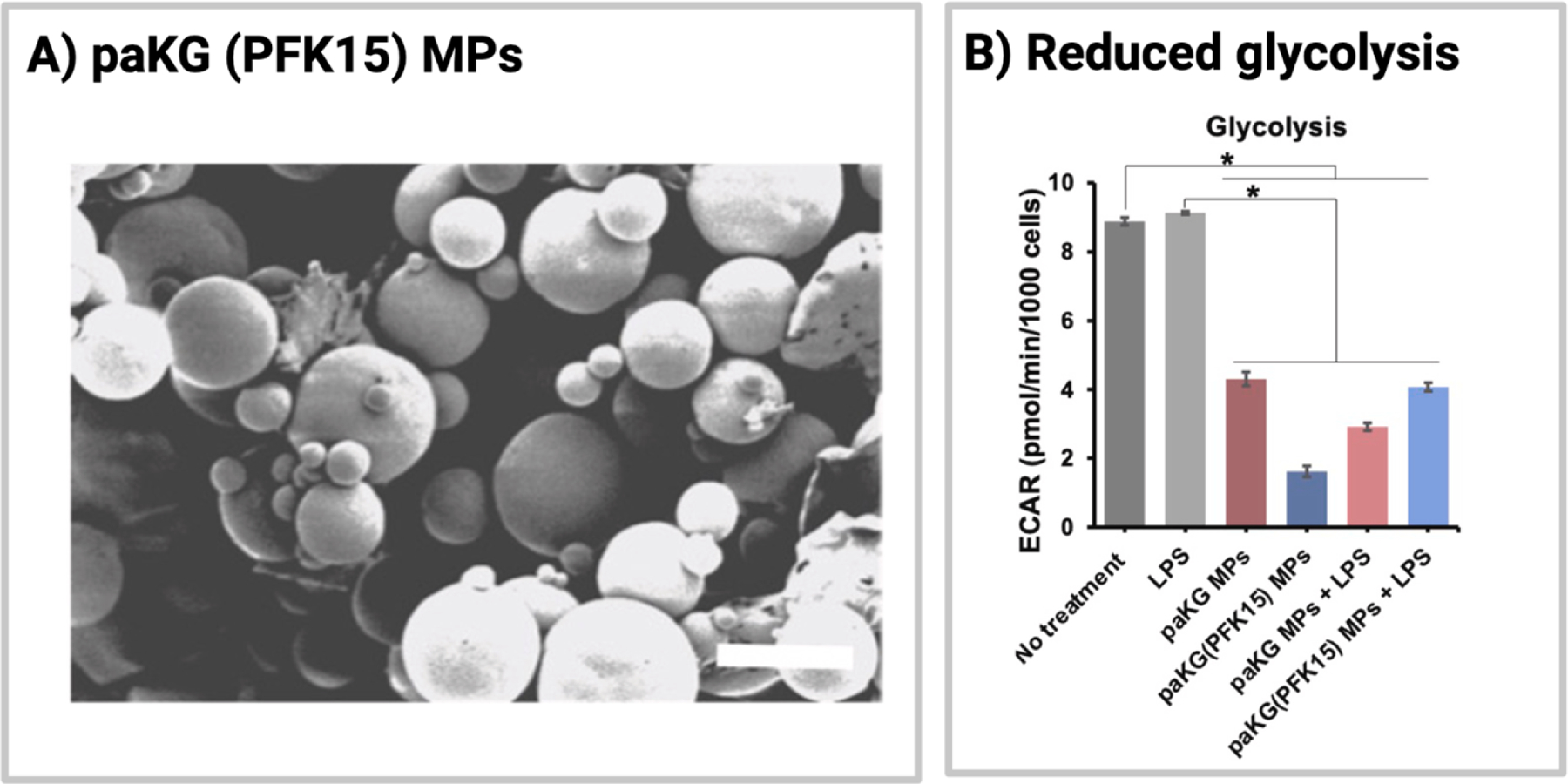
Structure and function of paKG(PFK15)-MPs. (A) Scanning electron microscopy image of paKG(PFK15)-MPs. (B) Effect of paKG- and paKG(PFK15)-MPs on glycolysis (ECAR) in bone-marrow dendritic cells. Reprinted from Biomaterials, Volume 277, Mangal et al., “Inhibition of glycolysis in the presence of antigen generates suppressive antigen-specific responses and restrains rheumatoid arthritis in mice,” Copyright 2021 with permission from Elsevier.
3.1.4. mTOR inhibitors
Rapamycin, another type of anti-metabolite, can be further classified as an mTOR inhibitor. mTOR exists in two structurally different complexes (mTORC1 and mTORC2) which serve different functions in growth and metabolism. mTORC1 activation leads to the translation of various metabolic enzymes that drive cell proliferation, whereas mTORC2 enhances metabolism by signaling through the Akt pathway [155]. Together, mTORC1 and mTORC2 can regulate immune cell differentiation, generating both effector and regulatory immune cells [155,156]. Activation of the mTORC complex (mTORC1/2) results in increased glycolysis that allows for effector cell differentiation and proliferation. Inhibition of mTOR, on the other hand, impacts glycolysis to skew immune cell subtypes towards a memory cell phenotype [148]. Rapamycin (Rapa), which targets mTORC1, has been shown to promote Treg differentiation from CD4+ T cells, potentially restoring immune homeostasis in inflammatory disorders by changing the balance between effector and regulatory subtypes [155,156]. To skew phenotypes away from inflammation, rapamycin has been incorporated in local delivery strategies to foster local immunosuppression [156–158].
Microparticles and polypeptide-forming depots, specifically, have been utilized to facilitate local delivery of rapamycin (Table 3) [156–158]. For instance, Fan et al. developed PLGA microparticles, in conjunction with PCL microparticles, to facilitate local, controlled release of rapamycin to enhance islet graft transplantation in the anterior chamber of the eye [157]. Islet transplantation in the anterior chamber of the eye is a suitable alternative to the typical method of administration into the hepatic portal system, as the hepatic portal system exposes islets to low oxygen levels and shear stress that can promote islet graft failure [157]. Nonetheless, islets in the anterior chamber of the eye are still prone to immunological rejection after vascularization. Fan et al. found that co-transplantation of islets with Rapa-MPs significantly extended islet survival when compared to blank microparticles [157]. Importantly, when the ratio of microparticles to islets was decreased, islet rejection rate was not impacted [157]. These results suggest that reduced concentrations of Rapa-MPs could be used to prolong islet survival, potentially due to the ability of local drug delivery to reduce required dose concentrations. However, it has been found that rapamycin could be toxic to pancreatic β-cells, reducing cell proliferation, viability and insulin secretion [159]. Therefore, it may be of use to explore alternative immunosuppressants for islet transplantation.
Rapamycin has also been locally delivered using elastin-like polypeptides in a murine model of Sjögren’s syndrome, a systemic autoimmune disease that targets the lacrimal glands resulting in clinical symptoms of dry eyes and mouth [158,160]. In this study, intralacrimal injection of elastin-like polypeptides carrying rapamycin formed a polymeric drug depot at the site of injection and significantly suppressed lymphocytic infiltration [158]. In addition, local delivery of rapamycin significantly improved tear production [158]. Importantly, the authors found that lacrimal injections of rapamycin containing elastin-like polypeptides resulted in less systemic toxicity (i.e., bodyweight loss, high blood glucose levels, etc.) when compared to subcutaneous injections [158]. Together, these results demonstrate the importance of local delivery in reducing inflammation and administration-associated toxicities.
Due to its’ poor water solubility and broad immunosuppression, Rapa, as well as newer generations of Rapa (i.e., everolimus, temsirolimus, etc.) have been incorporated into particulate systems to skew local immune responses in both ex vivo and in vivo models of inflammation (Table 3) [156,157]. For instance, Gosselin et al. individually encapsulated Rapa, everolimus and temsirolimus using PLGA microparticles to examine their immunosuppressive effects in an ex vivo model of autoimmunity [156]. In this study, co-incubation of dendritic cells (DCs) with MPs reduced DC activation, characterized by reduced levels of CD80, CD86 and CD40, and decreased T cell proliferation and inflammatory cytokine expression. While demonstrated ex vivo, co-incubation or localization of PLGA MPs with dendritic cells clearly exerted anti-inflammatory effects. Future studies using this approach should examine the efficacy of this system in vivo, specifically through local administration in models of autoimmunity.
3.1.5. Calcineurin Inhibitors
A more specific class of small molecule drugs are calcineurin inhibitors (CNIs), which are a class of immunosuppressive drugs that inhibit T cell activation. CNIs bind to intracellular proteins that inhibit the activity of calcineurin, an intracellular protein which, in tandem with nuclear factor of activated T cells (NFAT), upregulates cytokine and costimulatory molecule expression [161]. By inhibiting this protein, full T cell activation cannot occur.
Biomaterial-based drug delivery strategies have been used in numerous instances to locally deliver two CNIs, cyclosporin A and tacrolimus, to promote local immunomodulation and limit systemic side effects (Table 3) [31,37,40,75,142–144,162–166]. Cyclosporin A (CsA), the first immunosuppressant used to treat transplant patients, is often associated with chronic nephrotoxicity that limits its’ efficacy for long-term graft survival [167]. Thus, local biomaterial-based drug delivery strategies could be used to localize immunosuppression and limit systemic nephrotoxicity. For instance, Song et al. explored the effects of local delivery of CsA on xenogeneic cell transplant through use of PLGA microparticles embedded in an alginate hydrogel [162]. The resulting composite system, which provided controlled release of CsA over several weeks, was then incorporated into a 3D printed PCL/PLGA scaffold to enhance load-bearing capacity to reduce breakage and corresponding unintended drug release [162]. Co-localization of xenogeneic cells with the 3D printed-CsA-composite system reduced graft infiltration of CD4+ and CD8+ T cells while increasing graft survival [162]. While this study did not compare the local 3D printed delivery system against systemic CsA, it demonstrates the potential of such a strategy to restore immune homeostasis.
Given the nephrotoxicity of CsA, immunosuppressant protocols have shifted to the use of tacrolimus (TAC). TAC exerts’ its effects through a similar mechanism of action to CsA, specifically by inhibiting the function of calcineurin. TAC also results in a similar rate of graft loss to that of CsA; however, renal function and cardiovascular risk profiles are greatly improved by its’ use [168]. Nonetheless, TAC is still administered systemically and places the patient at risk for other complications. As a result, TAC has been incorporated in numerous delivery systems for local delivery [31,37,40,75,142–144,164–166,169]. Several of these studies have explored the use of triglycerol monostearate (TGMS) (discussed previously) to facilitate local, enzyme-responsive release of TAC. TAC-TGMS was shown to release TAC in response to various local inflammatory stimuli (lipase, MMPs, activated macrophage supernatant, etc.) and enhance hindlimb graft survival in both rodent and porcine models of vascularized composite transplantation [31,142–144]. For instance, in an in vivo model of lipopolysaccharide-induced inflammation, TAC was successfully released from TGMS in response to LPS, an acute inflammatory signal [144]. However, TAC was also released under non-inflammatory conditions, potentially explained for by the unintended inflammatory effect cause by hydrogel injection itself. In addition, after incorporating near-infrared dye (NIRD) to monitor TAC release, it was found that NIRDs were not released in response to degradable enzymes [144]. NIRDs are hydrophilic molecules; therefore, it is possible that its’ release is governed by passive diffusion, rather than TAC, which preferentially remains in the TGMS hydrogel. Therefore, it may be important to consider cargo choice when using enzyme-responsive systems.
In these studies, the benefits of local administration of TAC through biomaterial-based drug delivery were also demonstrated. Local delivery of TAC reduced systemic TAC concentrations when compared to soluble TAC administration, decreasing the risk of systemic side effects [31,164]. In addition, Fries et al. observed that both low doses and high doses of TAC hydrogels prolonged graft survival [143]. Therefore, biomaterial-based local drug delivery systems may make it possible to use a lower treatment dose and achieve the same survival outcome. In addition, local, intra-graft administration of TAC hydrogels prolonged graft survival when compared to both soluble and contralateral administration of TAC [142]. Together, these results highlight the importance of local biomaterial-based drug delivery strategies, as they can result in superior survival outcomes and reduce the required dose concentration to achieve a therapeutic effect.
3.2. Delivery of Biologics
Aside from small molecule drugs, various biologics, such as antibodies, cytokines, and chemokines, have been delivered to manipulate local immunological responses (Figure 5, Table 4). Delivery of biologics could be considered a more specific and effective approach for accomplishing a specific task, as many of the biologics discussed below are innately expressed and utilized by the body to promote immune homeostasis. Thus, lower dosages could be used, and side effects could be minimized. As compared to systemic administration (or even local, soluble administration) of immunosuppressive agents, this difference could be significant.
Fig. 5.
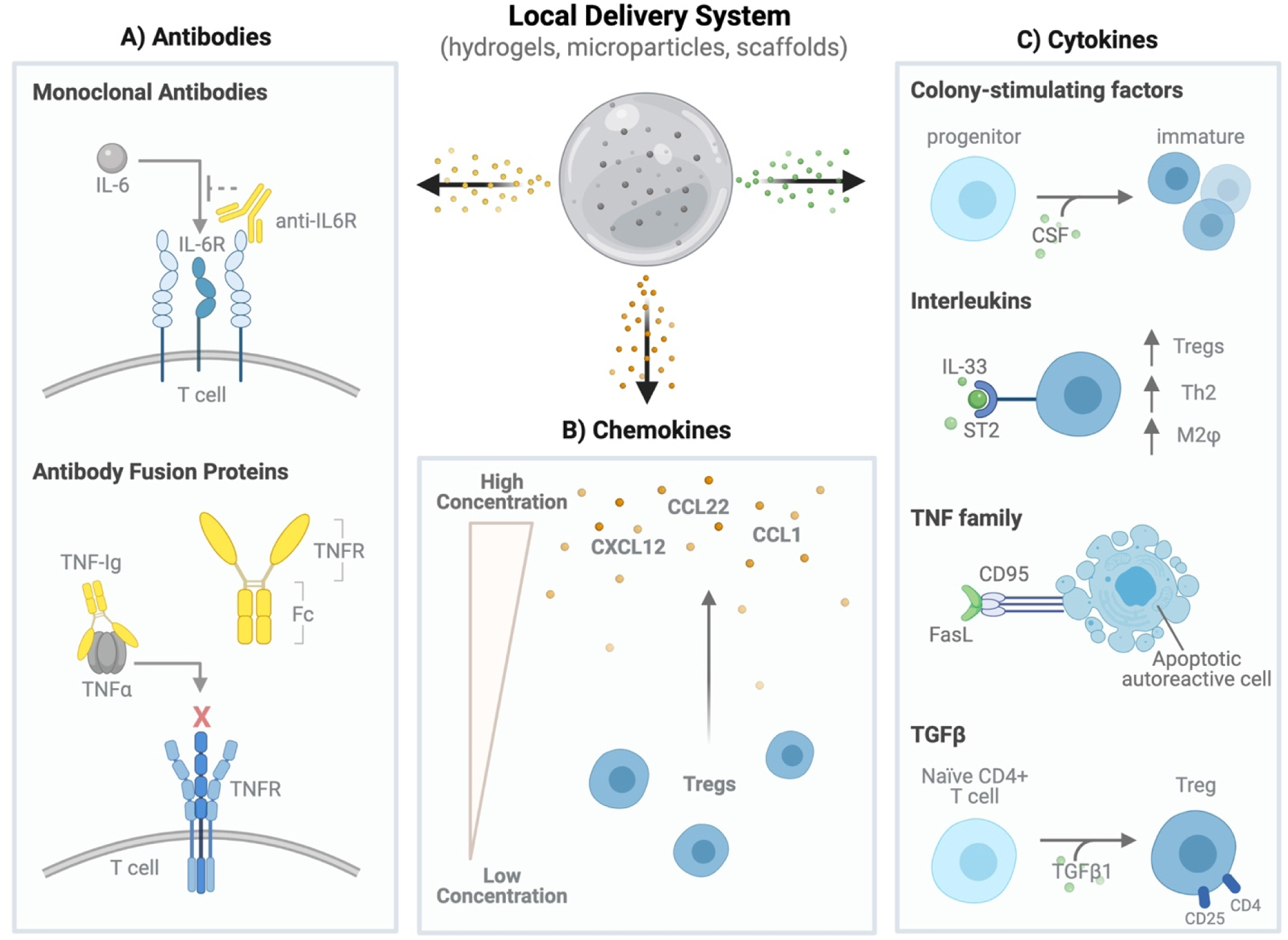
Classes of biologics delivered by local biomaterial-based delivery strategies. (A) Antibody-based treatments are used to interfere with signaling of inflammatory cytokines such as IL-6 and TNFα. (B) Chemokines, such as CCL22, CXCL12 and CCL1, can be delivered to generate a chemotactic gradient capable of recruiting regulatory T cells to a specific site of interest. (C) Various cytokines can be delivered to drive differentiation of immature or tolerogenic dendritic cell subtypes (colony-stimulating factors), increase regulatory immune responses (interleukins), induce apoptosis of autoreactive immune cells (TNF family) or generate regulatory T cells (TGFβ1).
Table 4.
Local delivery strategies used for biologic delivery to restore immune homeostasis from inflammation.
| Biologics | Delivery Strategy | Disease Model | Ref(s) |
|---|---|---|---|
| Antibodies | |||
| Anti-IL-6R mAb | GelMA hydrogel | Skin transplantation | [118] |
| Anti-TNFa mAb | PLGA MPs | In vitro neutralization | [179] |
| Self-assembled nanofiber hydrogel | In vitro neutralization | [180] | |
| CTLA4-Ig | Acellular dermal matrices | Islet transplantation | [39] |
| pNIPAAM-chitosan-hyaluronic hydrogel | Islet transplantation | [76] | |
| TNF-Ig | MPEG-PCL-MPEG MPs | - | [46] |
| Cytokines | |||
| GM-CSF | Puramatrix™ hydrogel | Inflammatory diabetes | [186] |
| Interleukin-2 | Alginate-GelMA hydrogel | Islet transplantation | [90] |
| Interleukin-33 | PLG scaffold | Islet transplantation | [197] |
| Interleukin-1Ra | Gelatin MPs | Islet transplantation | [76] |
| Pluronic F127 hydrogel | - | [81] | |
| b-TCP MP | - | [199] | |
| FasL | Agarose hydrogel-lipid microtubes | Cell transplantation | [109] |
| PEG hydrogel MPs (microgels) | Islet transplantation | [201] | |
| TGF-β1 | Alginate scaffold | Allogenic cell transplantation | [99] |
| PLGA MPs | Vascularized composite allotransplantation | [30] | |
| PEG-PLGA MPs | Inflammatory arthritis | [190] | |
| Chemokines | |||
| CCL22 | PLGA MPs | Allogeneic cell transplantation | [210] |
| PLGA MPs | Vascularized composite allotransplantation | [44] | |
| CXCL12 | Alginate MPs | Islet transplantation | [98] |
| CCL1 | Alginate-GelMA hydrogel | Islet transplantation | [90] |
3.2.1. Antibodies
Antibodies are secreted by plasma B cells and form the foundation of humoral immunity. There are two main types of antibodies: monoclonal antibodies and polyclonal antibodies. Monoclonal antibodies are produced by one pool of B lymphocytes, resulting in a homogeneous and consistent population of antibodies that recognize one specific antigen [171]. Polyclonal antibodies, on the other hand, are heterogenous and respond to a variety of antigens, making them less specific [171]. Due to their specificity, monoclonal antibodies are one of the earliest examples of biologic-based therapies [172]. In treating inflammation, monoclonal antibodies have been used to either prompt the destruction of cytotoxic lymphocytes (depleting antibodies) or block the function of inflammatory proteins (non-depleting antibodies) [173]. Aside from monoclonal antibodies, fusion proteins, constructed from antibody fragments and domains of native transmembrane proteins, can be constructed to inhibit downstream effects of certain ligands [174]. This subsection will focus the use of both monoclonal antibodies and fusion proteins in local delivery systems used for inflammatory disorders (Figure 5A, Table 4).
3.2.1.1. Monoclonal Antibodies
Monoclonal antibodies (mAb) are biologic treatments with a broad range of applications (i.e., cancer, inflammation, etc.). Systemic administration of mAbs is limited by their short half-life, large size and susceptibility to enzymatic degradation, thus limiting circulation time and tissue permeability, two variables needed to achieve a therapeutic effect [175]. Biomaterial-based delivery strategies have been used to address these limitations; however, early studies have focused on biomaterial-based mAb modifications (i.e., PEG) or using nanoparticle carriers [175–177]. Focus has been on these approaches because most FDA approved antibody therapies are approved to treat systemic or metastatic diseases (i.e., leukemia, lymphoma, sickle cell disease) in hard-to-reach anatomical sites (i.e., brain). Therefore, reformulation strategies have concentrated on reducing clearance and improving barrier penetration of antibody therapies using stealth-agents such as PEG and nanoparticulate carriers [175,176]. Nonetheless, local delivery of mAb could also be used to induce immunosuppression in inflammatory diseases where disease is constrained, such as rheumatoid arthritis and transplant rejection (i.e., VCA transplant, islet transplant, etc.).
One example of a monoclonal antibody that has been locally delivered to induce immunosuppression in graft rejection is anti-interleukin-6R (anti-IL-6R). Anti-IL-6R is a mAb against the receptor of IL-6, a cytokine that can inhibit TGFB1-mediated differentiation of Tregs from naïve CD4+ T cells [118,178]. Anti-IL-6R has been locally delivered using GelMA hydrogels in a model of allogeneic skin transplantation [118]. Subcutaneous administration of anti-IL-6R directly under the skin graft resulted in increased levels of CD4+CD25+Foxp3+ Tregs in the draining lymph nodes, without affecting levels of CD4+ and CD8+ T cells in the spleen, suggesting the benefit of local immunomodulation [118]. Moreover, GelMA/Anti-IL-6R significantly prolonged alloskin graft survival when compared to systemic anti-IL-6R and GelMA only treatment groups [118]. Together, these results highlight both the efficacy of anti-IL-6R in rejection and the ability of subcutaneous, local drug delivery to limit systemic immunosuppression.
Another target of monoclonal antibody therapy is tumor necrosis factor alpha (TNFα). TNFα is a potent apoptotic cytokine produced by macrophages and monocytes that can promote chronic inflammation. Anti-TNFα mAb can inhibit inflammatory responses sparked by TNFα by preventing binding between TNFα and its’ receptor (TNFR) [179,180]. Local delivery of anti-TNFα mAbs, through both the use of PLGA microparticles and self-assembled nanofiber hydrogels, has been explored in vitro [179,180]. Use of both PLGA microparticles and self-assembled nanofiber hydrogels enabled controlled release of anti-TNFα, which resulted in TNFα neutralization and increased fibroblast viability [179,180]. Although initial results yielded from these studies are promising, additional studies exploring local delivery in clinically relevant models of inflammation in vivo would further validate the efficacy of these systems.
3.2.1.2. Antibody Fusion Proteins
Fusion proteins consist of fragments of antibodies (otherwise known as immunoglobulins (Ig)) that include extracellular domains of other molecules, typically transmembrane proteins. Fusion proteins provide specificity, offered by the binding domain, and stability, given by the Fc region of the antibody. One common fusion protein used in inflammatory disorders is CTLA4-Ig, which combines the extracellular domain of CTLA4, a checkpoint molecule that downregulates immune responses, with the Fc portion of IgG. The resulting protein can interfere with CD28-CD80 and CD28-CD86 interactions, which enable T cell activation [76,181]. More specifically, CD80 and CD86, which are expressed on the surface of antigen presenting cells, interact with CD28 expressed on the surface of naïve T cells to trigger their activation and differentiation [76,181]. CTLA4-Ig can interrupt this interaction by binding to both CD80 and CD86, preventing interaction with CD28. As a result, T cell differentiation is blocked, leading to anergy and cell death [181].
Local delivery of CTLA4-Ig has been facilitated using PNIPAAm-chitosan-hyaluronic acid hydrogels and acellular dermal matrices to enhance survival of transplanted allogeneic islets in T1D [39,76]. Zhang et al. grafted CTLA4-Ig to human-derived acellular dermal matrices to provide local immunosuppression in islet transplantation [39]. Localization of CTLA4-Ig reduced immune cell infiltration and significantly prolonged islet allograft survival when compared to contralaterally implanted CTAL4-Ig and WT mice [39]. Hou et al. also studied the impact of grafted and locally delivered CTLA4-Ig on islet transplantation by grafting CTLA4-Ig to PNIPAAm-chitosan-hyaluronic acid hydrogels [76]. Using these hydrogels, CTLA4-Ig was delivered alongside IL-1Ra, an interleukin-1 receptor antagonist, to study their effect on islet survival [76]. While the effects of CTLA4-Ig alone were not examined in this study, CTLA4-Ig and IL1Ra releasing hydrogels limited immunological infiltration when compared to blank controls [76]. Further studies exploring the effects of CTLA4-Ig alone could provide insights into the mechanisms responsible. Nonetheless, these results demonstrate the potential of both local administration and CTLA4-Ig in restoring immune homeostasis.
Another fusion protein of interest is tumor necrosis factor alpha immunoglobulin (TNF-Ig), which combines the TNF receptor with the Fc region of IgG. [46]. As discussed above, TNFα is a potent, apoptotic cytokine that can promote inflammation through inducing expression of other proinflammatory cytokines [182]. Its’ role in various inflammatory diseases, such as rheumatoid arthritis, has been extensively studied [46,183]. An example of a TNF-Ig is etanercept (ETN), which functions as a soluble decoy receptor for TNFα. More specifically, etanercept binds to soluble TNFα, preventing its’ binding to TNFR and rendering TNFα biologically inactive [182]. ETN administration typically occurs systemically, leading to potentially toxic side effects. Therefore, Erdemli et al. explored using MPEG-PCL-MPEG microspheres to encapsulate ETN for local delivery [46]. In vitro studies using this system exhibited prolonged therapeutic release over at least 60 days with limited protein adsorption due to the presence of hydrophilic PEG chains [46]. These results demonstrate the ability to develop controllable release systems for ETN. However, as mentioned above, the efficacy of such a system is worth exploring in vivo.
3.2.2. Cytokines
Another common class of biologics used for immunomodulation are cytokines, which are proteins secreted by immune cells that skew immune responses through binding to specific receptors. Cytokines can be further classified into several subtypes, including colony-stimulating factors, interleukins and TNF family proteins [173]. Among the colony-stimulating factors, granulocyte-monocyte colony-stimulating factor (GM-CSF), which is secreted by T cells (Th1, Th2 and Th17), spurs the production and differentiation of granulocytes, macrophages and dendritic cells [173]. Previous work has shown that low doses of GM-CSF can promote the production of immature dendritic cells, characterized by low expression of several costimulatory molecules (MHCII, CD80, CD86) [184,185]. However, Yoon et al. demonstrated that local hydrogel delivery of GM-CSF alone, without the addition of other tolerogenic factors, did not significantly delay the onset of inflammatory diabetes when compared to the no treatment group [186]. As a result, GM-CSF has been used in conjunction with other biologics (antigen, cytokines, etc.) to further ensure the development of regulatory phenotypes (Figure 5C, Table 4) [41,185–189].
Interleukins (IL), cytokines first identified as being secreted by leukocytes, have also been incorporated in local delivery systems in an effort to skew immunological responses. Interleukin-2 (IL-2), specifically, is a common cytokine of interest, as it is critical for the differentiation and maintenance of regulatory T cells (Table 4) [30,90,173,190]. In clinical trials for graft-versus-host disease and type 1 diabetes, daily low dose subcutaneous injections of IL-2 showed promise in reducing disease incidence by expanding populations of CD4+CD25+Foxp3+CD127- regulatory T cells [191–193]. Nonetheless, soluble IL-2 has several limitations. IL-2 has a short half-life and high doses have been shown to expand effector T cells (Th1), potentially tipping the homeostasis balance towards inflammation [192,194]. Incorporation of IL-2 into local delivery systems can address both issues, as it can improve drug half-life and reduce the required dosage, reinforcing the development of Th2 (i.e., anti-inflammatory cytokine secretion, Treg proliferation, etc.) rather than Th1 responses. For instance, Kim et al. encapsulated IL-2 within alginate-GelMA hydrogels alongside Tregs to provide localized immunosuppression for transplanted islets [90]. Local delivery of IL-2 improved induced Treg suppressive function, characterized by enhanced expression of CD25 and Foxp3, two markers central to the development of Tregs [90]. These results suggest that the localized dosage of IL-2 in this instance fostered immunosuppressive responses, rather than effector responses. Nonetheless, it is important to restrict the differentiation of CD8+ effector T cells, which could be induced through delivery of IL-2. To address this potential limitation, IL-2 has been incorporated in multiple other local delivery strategies that release combinations of factors in an effort to further skew the immune response away from inflammation [30,96,190].
A specific family of interleukins that are of interest for local delivery are interleukin-1 cytokines, which include IL-33 and IL-1 receptor antagonist (IL-1Ra). IL-33 has been shown to have both inflammatory and anti-inflammatory properties [195,196]. In terms of its’ anti-inflammatory actions, IL-33 can promote Th2 responses and skew macrophage polarization to an M2 phenotype through interaction with its’ receptor (ST2) expressed on the surface of Th2 cells, Tregs and group 2 innate lymphoid cells (ILC2) [195,196]. The local anti-inflammatory and immunosuppressive effects of IL-33 were recently studied by Liu et al. in a model of islet transplantation, where a PLG scaffold was used to locally release IL-33 into a cell transplant microenvironment (Table 4) [197]. Incorporation of IL-33 resulted in a local expansion of Foxp3+ Tregs and a decrease in CD8+ T cells [197]. Moreover, local delivery of IL-33 induced a Th2 response, characterized by enhanced expression of Th2 cytokines IL-4, IL-5 and IL-13, as well as expanded populations of ST2+ ILC2 [197]. The enhanced anti-inflammatory effect resulted in a significant increase in islet allograft survival when compared to blank controls [197]. Observations from this study highlight the potential of IL-33 in mediating inflammation.
IL-1 receptor antagonist (IL-1Ra), another member of the interleukin-1 cytokine, also exerts anti-inflammatory effects by inhibiting interaction of interleukin-1 (inflammatory cytokine) with its’ cognate receptor. IL-1Ra is made endogenously in response to inflammatory stimuli and its’ administration has been found beneficial to restore immune homeostasis in several models of inflammation (reviewed by Arend et al.) [198]. Due to its’ short biological half-life, IL-1Ra has been incorporated in several local delivery systems for immunomodulation (Table 4) [76,81,199]. Hou et al. developed gelatin microparticles capable of providing short-term IL-1Ra release [76]. When applied in vitro in a model of IFNg-induced cell death, treatment with IL-1Ra microparticles significantly enhanced beta-cell viability when compared to controls [76]. These results suggest the potential benefit of utilizing IL-1Ra local delivery strategies in both islet transplantation and autoimmune diabetes. Clements et al. also explored the impact of local delivery of IL-1Ra in a healing rat medial collateral ligament model [199]. Local delivery of IL-1Ra through conjugation to b-tricalcium phosphate beads increased local concentrations of IL-1Ra in MCLs when compared to soluble IL-1Ra injections [199]. Together, these studies highlight the immunomodulatory potential of IL-1Ra and benefit of local delivery systems.
A third subtype of cytokine is TNF family cytokines, which can function in both inflammatory and anti-inflammatory roles. An example of a TNF family cytokine is Fas Ligand (FasL), which triggers apoptosis through interaction with its’ transmembrane receptor Fas (CD95) [200]. It is responsible for maintaining immune homeostasis through the death of T cells and autoreactive B cells, which can promote unwanted inflammation. Due to its’ immunosuppressive role, FasL has been incorporated in local delivery systems to serve immunomodulatory purposes in cell transplantation (Table 4) [109,201]. Alvarado-Velez et al. developed agarose hydrogels containing lipid microtubes to locally release FasL to enhance the transplantation of mesenchymal stem cells (MSCs) in a model of traumatic brain injury [109]. Local delivery of FasL reduced the percentage of CD8+ cytotoxic T cells at the site of injury. In addition, co-encapsulation of FasL and MSCs within agarose hydrogels enhanced transplant survival of MSCs.
Although results from Alvarado-Velez et al. suggest the potential benefits of soluble FasL, some studies have discussed limitations pertaining to its’ use in this form. For instance, FasL may be more effective at inducing apoptosis in its membrane-bound (rather than soluble) form [202,203]. Therefore, multiple studies have explored the induction of apoptosis by externally conjugating FasL to delivery systems or cells [201–203]. For example, Headen et al. explored the efficacy of local FasL delivery through external conjugation of FasL to microgels composed of PEG [201]. In this study, direct conjugation of streptavidin-FasL to microgels significantly hindered the apoptotic activity of the ligand [201]. To address this limitation, the authors then developed biotinylated hydrogel microparticles (microgels) as a method to display streptavidin-FasL (FasL-microgels) [201]. This approach takes advantage of the natural, strong affinity of streptavidin for biotin to provide conjugation rather than relying on direct, chemical conjugation. When applied to lymphoma cells in vitro, FasL microgels resulted in dose-dependent apoptotic effects [201]. In a model of islet transplantation, FasL-microgels, in combination with short-course rapamycin, trended towards increasing the ratio of CD4+CD25+Foxp3+ Tregs to CD8+CD44hiCD62Llo effector T cells (Teff) [201]. Notably, there was no statistically significant difference in this ratio (Tregs:Teff) between control microgels (lacking FasL) and control microgels in combination with rapamycin, demonstrating the immunomodulatory benefit of including FasL. Overall, this study demonstrates the potential of FasL while highlighting significant biomaterial design considerations (i.e., external conjugation vs. soluble, conjugation method, etc.) to improve efficacy.
One cytokine that falls outside the classifications of colony-stimulating factors, interleukins and TNF family proteins is transforming growth factor beta (TGF-β), which also exerts a potent immunosuppressive effect. More specifically, TGF-β promotes naïve CD4+ T cell differentiation into induced Tregs in the absence of other proinflammatory cues (i.e., IL-6) [173,204]. In addition, TGF-β can inhibit Th1 differentiation, proliferation and function [173,204]. As a result of its’ important role in maintaining the balance of immune homeostasis, TGF-β has been incorporated in several local biomaterial systems to induce immunosuppressive effects (Table 4) [30,99,190,205–208]. For instance, Orr et al. explored functionalization of TGF-β to alginate scaffolds as a method to promote local Treg differentiation [99]. It was found that membrane-bound TGF-β was superior to soluble TGF-β in promoting the generation of FoxP3+ Tregs, potentially due to enhanced biologic retention and signal transduction supported by presentation of TGF-β to immune cells [99]. When affinity-bound TGF-β alginate scaffolds were co-transplanted with allogeneic fibroblasts, fibroblast viability was significantly increased when compared to biomaterial scaffolds lacking TGF-β [99]. Yang et al. also explored surface functionalization of TGF-β [207]. In this study, TGF-β was engineered to express 1-methyl-2-diphenylphosphino-terethalate (MDT) through a reaction with free amines in NHS-PEG. Amide bonds were then formed between surfaces of interest (i.e., glass beads, antigen presenting cells, etc.) and MDT-TGF-β. Surface-bound TGF-β, on both glass beads and antigen presenting cells, significantly enhanced expression of FoxP3+ Tregs when compared to controls lacking TGF-β [207]. Unlike the previous study, high doses of surface-bound TGF-β did not yield greater Treg populations when compared to soluble TGF-β [207]. While functionalized TGF-β did not appear to enhance the immunosuppressive effect in this study, these results show that surface functionalization does not appear to inhibit or reduce the effect of TGF-β. In addition, these studies demonstrate the ability to functionalize TGF-β1 through multiple approaches, which could be further optimized based on the delivery material of choice.
Aside from surface functionalization, TGF-β has also been encapsulated within local delivery systems to provide local soluble delivery of the protein, specifically in models of allotransplantation and rheumatoid arthritis [30,190]. In these studies, PLGA microparticles or PEG-PLGA microparticles were utilized to facilitate local delivery of soluble TGF-β, which enabled controlled release and enhanced biologic availability at the site of interest [30,190]. Interestingly, Fisher et al. found that individual local administration of TGF-β-MPs in a model of vascularized composite allotransplantation failed to provide long-term allograft survival [30]. When administered alongside two other microparticle formulations (releasing IL-2 and rapamycin), graft survival was significantly prolonged [30]. Therefore, it could be beneficial to incorporate TGF-β alongside other immunosuppressive cues to enhance the effect of the cytokine. This phenomenon is especially important when considering the plasticity of Tregs. While TGF-β can promote the development of Tregs, it is also involved in the differentiation of Th9 cells and Th17 cells if in the presence of additional cytokines, such as IL-2/IL-4 and IL-6/IL-21/IL-23, respectively. Therefore, it may be very important to control the local microenvironment through a combination of multiple other factors that provide the appropriate immunological context, which will be discussed further in subsequent sections.
3.2.3. Chemokines
Another approach to generate regulatory microenvironments is to release chemokines, a specific class of cytokine responsible for directing cell migration. Delivery systems that release chemokines in inflammatory disorders can be used to recruit various immune cells to the site of inflammation, where they can then inhibit effector mechanisms and promote immunological homeostasis (Figure 5B, Table 4).
One example of a chemokine of recent interest is CCL22, which can attract regulatory T cells through interaction with CCR4. In the context of cancer, CCL22 has been shown to drive the active recruitment of Tregs to promote tumor evasion [209]. To capitalize on this innate mechanism, Jhunjhunwala et al. developed PLGA microparticle formulations which mimic this effect by facilitating local, controlled release of CCL22 in order to enrich Tregs at a site of interest [210]. In vivo subcutaneous injection of CCL22-MPs resulted in preferential recruitment of intravenously administered alloactivated Tregs to the site of administration [210]. These formulations were also able to restore the immune-evasion ability of lung carcinoma cells (which do not secrete CCL22) as compared to bolus CCL22 injections at the site of the tumor [210]. These results suggest the importance of local, controlled release of CCL22, which promoted Treg migration and allogenic cell survival. Fisher et al. then implemented CCL22-MPs to recruit regulatory T cells in a model of vascularized composite allotransplantation (VCA) [44]. Subcutaneous injection of CCL22-MPs in VCA reduced the local expression of several proinflammatory mediators (TNF-a, IFNg, IL-17A, Perforin-1, etc.) and increased the proportion of Foxp3+ Tregs (% FoxP3+ of total CD4+ or CD8+ T cells) in allografted skin [44]. As a result, treatment with CCL22-MPs significantly prolonged hindlimb allotransplant when compared to allografts treated with Blank MP, soluble CCL22 or rapamycin. Notably, when long-term surviving animals (>200 days) treated with CCL22-MPs were challenged with syngeneic, donor and third-party skin grafts, animals accepted syngeneic and donor grafts but rejected third-party grafts (in the complete absence of systemic immunosuppressants), suggesting that CCL22-MPs are capable of inducing donor-specific tolerance [44]. It is likely that donor-specific tolerance was induced due to the initial presence of endogenous antigen prior to CCL22-MP administration. However, this effect may not be seen in cases where the endogenous antigens) of interest are not present, potentially requiring the administration of additional exogenous antigen alongside the biologic treatment (discussed in subsequent sections). Nonetheless, results pertaining to the use of CCL22-MPs demonstrate the regulatory role of CCL22 and the importance of controlled release, which was able to generate a chemotactic gradient capable of recruiting regulatory T cells to a specific site of interest.
Two other chemokines, CXCL12 and CCL1, have also been examined for their capacity to recruit regulatory T cells (Table 4). Early studies investigating the role of CXCL12 with respect to regulatory T cells showed that bone marrow expression of CXCL12 drives Treg migration and local accumulation through interaction with its’ receptor, CXCR4 [211]. Chen et al. found that coating pancreatic islets with CXCL12 promotes the accumulation of Foxp3+ T cells within and surrounding transplanted islets in vivo [98]. Local delivery of CXCL12-coated islets in a non-obese diabetic (NOD) mouse model of autoimmune diabetes failed to prevent rejection of transplanted islets, potentially due to the lack of a physical barrier that offers protection from pre-generated humoral antibodies [98]. To address this concern, islets and CXCL12 were then incorporated within alginate microparticles, which led to enhanced function and survival of both allo- and xenoislets when assessed in NOD mice [98]. In studying the mechanism that provided this effect, it was found that NOD mice isolated CD4+CD25+ Treg cells migrated towards a chemotactic gradient of CXCL12, potentially due to their high expression of CXCR4 [98]. This study supports the ability of local chemotactic gradients to attract regulatory cells. However, in the case of cell transplantation, it may be beneficial to design a more complex delivery system that enhances their immunoisolation.
Similar to CXCL12, CCL1 also functions as a chemokine for Tregs through interaction with its’ receptor CCR8, which is expressed on the surface Tregs [90]. Kim et al. studied the effects of local CCL1 delivery, alongside delivery of IL-2 (previously described), through the use of an alginate-GelMA hydrogel [90]. Alginate-GelMA hydrogels were used to encapsulate induced Tregs and natural Tregs (expanded upon in section 3.3.1) with CCL1 and IL-2 in order to enhance viability of co-encapsulated islets [90]. Incorporation of CCL1 within the delivery matrix resulted in local Treg migration [90]. However, migration was significantly reduced when compared to unencapsulated CCL1. These results could be due to the structure of the delivery matrix, which was engineered with high porosity to maintain encapsulated cell viability by allowing proper nutrient exchange [90]. Therefore, it is possible that excess CCL1 diffusion reduced the strength of the chemotactic gradient, ultimately reducing Treg migration. While this study highlights the ability of CCL1 to attract regulatory T cells, it also draws attention to design parameters (i.e., the properties of the delivery system) to consider when designing local chemokine delivery systems.
3.3. Strategies for cell-based immunomodulation
Although local delivery of both small molecules and biologics has shown promise in restoring immune homeostasis, it may be more efficient and effective to directly deliver immunosuppressive or tolerogenic immune cells to skew the immune response. Cell-based immunomodulation can be achieved through two main approaches: (1) ex vivo isolation and delivery or (2) in situ induction and expansion of these subtypes (Table 5).
Table 5.
Local delivery strategies for cell-based approaches to restore immune homeostasis from inflammation.
| Cell-Based Approaches | Delivery Strategy | Disease Model | Ref(s) | |
|---|---|---|---|---|
| Ex vivo isolation/delivery of regulatory subtypes | ||||
| Dendritic cells | PEG hydrogels | Multiple sclerosis | [42] | |
| Regulatory T cells | Alginate-GelMA hydrogels | Islet transplantation | [90] | |
| PEGNB hydrogels | Peripheral nerve allotransplantation | [71] | ||
| Mesenchymal stem cells | Dextran-PEG hydrogels | Islet transplantation | [221] | |
| Silk hydrogels | Islet transplantation | [111] | ||
| PEO/Alginate MPs | Systemic lupus erythematosus | [222] | ||
| In situ engineering of endogenous subtypes | ||||
| Dendritic Cells | GM-CSF, dexamethasone, peptidoglycan | Gelatin MPs | Ex vivo autoimmunity | [110] |
| Tacrolimus, clondronate liposomes | PLGA MPs/Matrigel | Islet transplantation | [227] | |
| Rapamycin, retinoic acid, IL-10, TGF-β1 | PLGA MPs | Ex vivo autoimmunity | [229] | |
| Macrophages | CCL2 | PLGA MPs | Periodontitis | [232] |
| Regulatory T cells | Anti-CD3/CD28, TGF-β1 | PLGA/PBAE MPs | - | [233] |
| TGF-β1, rapamycin, IL-2 | PLGA MPs | - | [234] | |
| TGF-β1, rapamycin, IL-2 | PLGA MPs | Contact dermatitis | [235] | |
| TGF-β1, rapamycin, IL-2 | PLGA/PEG-PLGA MPs | Dry eye disease | [236] | |
| TGF-β1, rapamycin, IL-2 | PLGA MPS | Vascularized composite allotransplantation | [30] | |
| TGF-β1, rapamycin, IL-2 | PLGA/PEG-PLGA MPs | Inflammatory arthritis | [190] | |
3.3.1. Ex vivo isolation and delivery of regulatory subtypes
Regulatory subtypes of interest for the treatment of inflammatory disorders include immature/tolerogenic dendritic cells, regulatory T cells and mesenchymal stem cells (Figure 6). Dendritic cells (DCs) can be either inflammatory or anti-inflammatory depending on their maturation. Mature DCs have an immunogenic phenotype, characterized by elevated expression of costimulatory molecules CD40, CD80 and CD86 [212]. Tolerogenic DCs, or immature DCs (tolDCs), are characterized by low expression of CD80 and CD86, as well as expression of negative co-stimulatory molecules (PD-1 and CD70) (Figure 6A) [212,213]. tolDCs exert immunosuppressive effects through multiple mechanisms. For one, tolDCs secrete the cytokine IL-10, which induces the generation of regulatory T cells [212,213]. In addition, interaction of co-inhibitory marker PD-1 on tolDCs and its’ ligand, PD-L1, on the surface of T cells reduces T cell activation [213]. Due to these immunosuppressive functions, tolDCs are a target for studies that are aiming to induce tolerance or restore homeostasis in inflammation.
Fig.6.
Classification and function of regulatory immune cell subtypes. Key surface markers and immunological functions for (A) tolerogenic dendritic cells, (B) regulatory T cells and (C) mesenchymal stem cells. Information adapted from references [212–219].
Studies utilizing tolDCs to exert immunosuppressive effects in inflammation typically must first induce their generation from naïve cells through interactions with signaling molecules, such as IL-10, dexamethasone, vitamin D, rapamycin and TGF-β1 [213]. The immunosuppressive effects of IL-10 treated-tolDCs (DC10s) were recently studied in a murine model of autoimmune encephalomyelitis [42]. To ensure local immunomodulation, Thomas et al. delivered cells using PEG hydrogels [42]. Local delivery of DC10s utilizing PEG hydrogels reduced levels of T cells, B cells and DCs at site of injection [42]. In addition, local delivery of DC10s reduced maturation markers (CD86, CD54) of endogenous DCs (from the host) at the hydrogel injection site. As a result of these outcomes, local delivery of DC10s led to increased survival and attenuated paralysis in a murine model of MS [42]. Together, these results highlight the capability of DCs manipulated ex vivo to reduce the inflammatory characteristics of multiple sclerosis to restore immune homeostasis.
Another regulatory subtype of interest for local delivery is regulatory T cells. Typically characterized by high expression of CD25, the IL-2 receptor, and Foxp3, a transcription factor central to their development and stability, Tregs are central mediators of homeostasis and tolerance (Figure 6B) [212,214]. There are multiple types of Tregs, the most prominent two being “natural” Tregs and induced Tregs. Natural Tregs, otherwise known as thymic Tregs, originate in the thymus where they express Foxp3 [90,212,214,215]. Induced Tregs, on the other hand, differentiate from naïve CD4+ T cells and acquire Foxp3 expression in the periphery after encountering antigen [90,214]. Altogether, Tregs enforce suppression through multiple mechanisms, including contact-dependent mechanisms and non-contact dependent mechanisms, such as secretion of immunosuppressive cytokines (i.e., IL-10, TGF-β1) [212,215].
Both subtypes of Tregs, nTregs and iTregs, were studied by Kim et. al. in an effort to generate a local immunosuppressive environment that supports the transplantation of islets [90]. To do so, murine islets were co-encapsulated with either nTregs or iTregs in alginate-GelMA hydrogels. In the presence of human peripheral blood mononuclear cells, islet viability was significantly enhanced when compared to islets without Tregs, illustrating the immunoprotective effect of Tregs [90]. Roballo et al. also explored the use of Tregs to induce short-term immunosuppression for peripheral nerve (PN) allografts [71]. To enhance local accumulation and immunosuppression, the authors developed PEGNB hydrogels to deliver Tregs proximal to the allograft [71]. In vitro studies demonstrated that Tregs could be released from the hydrogel over a 14-day window. When implemented in vivo in a model of transplantation, local release of Tregs reduced the local accumulation of host derived CD4+ cells [71]. The overall effect of local Treg delivery on allograft survival was not assessed. However, allografts in the presence of Treg hydrogels regenerated to similar magnitudes of autografts [71]. Results of both studies demonstrate the feasibility of locally released Tregs to induce immunosuppressive effects that could promote transplant viability; however, a study assessing overall allograft survival could be beneficial.
In addition to DCs and Tregs, mesenchymal stem cells (MSCs) are a common cell subtype of interest used to induce regulatory effects following local delivery. MSCs are multipotent progenitor cells, positive for CD73, CD105 and CD90 markers, that can differentiate into various cell types (i.e., osteocytes, chondrocytes, adipocytes) (Figure 6C) [216]. More importantly, MSCs have also been shown to induce immunosuppressive and tolerogenic effects through a variety of mechanisms, such as the induction of TGF-β1 and IL-10 secretion [217–219]. In addition, MSCs can shift the balance between effector and regulatory cells by promoting monocyte differentiation towards an M2 phenotype (anti-inflammatory phenotype), reducing DC maturation, and expanding populations of Tregs [217,220]. Therefore, MSCs hold promise as a cell-therapy strategy used to restore homeostasis.
Cell therapy approaches utilizing MSCs typically involve their systemic administration, thus relying on the homing ability of MSCs to reach the targeted site [221]. To eliminate this variable and potentially improve their effect, MSCs have been incorporated in multiple local delivery systems to skew immune responses away from inflammation (Table 5) [111,221,222]. For instance, Ayenehdeh et al. used dextran-PEG hydrogels to co-embed adipose-tissue derived MSCs (AT-MSCs) with allogenic islets to assess AT-MSC effects in a murine model of streptozotocin induced diabetes [221]. Local delivery of AT-MSCs alongside islets reduced graft infiltration of immune cells and inflammatory cytokine production (IL-17, IFNγ) [221]. Similarly, Davis et al. developed silk hydrogels for co-encapsulation of islets and MSCs [111]. It was found that incorporation of MSCs suppressed splenocyte proliferation in a mixed lymphocyte reaction, illustrating their immunomodulatory effect [111]. In addition, islet response to glucose stimulation was enhanced by the local presence of MSCs provided by silk hydrogels [111]. Together, these studies highlight the ability of locally placed MSCs to generate anti-inflammatory responses that could skew immune responses away from inflammation.
3.3.2. In situ engineering of endogenous regulatory subtypes
Although cell therapy approaches have promising effects, there are numerous obstacles that they must overcome for implementation. Ex vivo manipulation and reinfusion of cell therapies rely upon initial isolation of a patient’s cells from blood. Tregs, which can be isolated from either peripheral blood or umbilical cord blood, represent 2–10% of circulating CD4+ T cells [223,224]. However, within that subset, circulating CD4+CD25+Foxp3+ Tregs represent less than 2%, making isolation difficult [224]. In addition to challenges related to their isolation, Tregs can be unstable under inflammatory conditions, leading to the loss of their Foxp3+ phenotype and corresponding suppressive function [224,225] Many of the same issues (function, isolation, processing, scale-up, etc.) exist for MSCs and DCs [212,218,220,226]. MSCs can differ in their differentiation and immunomodulatory capacity based on their source, and similarly to DCs, can acquire diverse subtypes based on the local microenvironment and timing of infusion [218,220,226]. Based on their plasticity, these cell therapy approaches risk generating proinflammatory subtypes with a high financial burden based on the manufacturing steps involved.
Each of the cell therapy-based approaches discussed above take advantage of the ability of live cells to exert dynamic and sophisticated modes of regulation yet suffer from the associated regulatory and processing complications. But, if it were possible to direct or coordinate the trafficking and/or activation of endogenous cells in their microenvironment using engineered systems, it may be feasible to take advantage of the promise of cell therapies while avoiding the significant roadblocks of ex vivo therapies, as described above. As a result, several groups have explored the in situ induction and expansion of tolerogenic subtypes, specifically antigen-presenting cells and regulatory T cells, through local delivery of soluble factors and/or biologics (Table 5).
3.3.2.1. In Situ Manipulation of Antigen-Presenting Cells
Antigen presenting cells (APCs), specifically dendritic cells and macrophages, have been targeted in situ to develop regulatory subtypes. APCs link innate immunity to adaptive immunity, resulting in the activation and induction of effector responses that could promote inflammation. However, dendritic cells and macrophages can also function in a regulatory role that have the potential to restore immune homeostasis. In the case of dendritic cells, their immature or tolerogenic phenotype (discussed above) could achieve this goal. Srinivasan et al. induced this phenotype in human cells in vitro through controlled release of GM-CSF, dexamethasone (DEX) and peptidoglycan (PGN) [110]. GM-CSF was chosen for its’ ability to serve as a growth factor that promotes differentiation of progenitors into DCs [110]. DEX and PGN were chosen to serve as immunosuppressive and maturation signals, respectively [110]. Using gelatin-MPs, the encapsulated immunomodulators were released sequentially to follow the stages of dendritic cell proliferation and maturation [110]. More specifically, MPs were engineered to deliver GM-CSF over days 0 to 3, DEX over days 3 to 5 and PGN during day 6. When incubated with human peripheral blood mononuclear cells in vitro, immunomodulatory MPs induced tolerogenic DCs [110]. Tolerogenic DCs expressed low levels of maturation surface markers (i.e., CD80, CD86 and MHC), but exhibited increased expression of immunoglobulin-like transcript 3 (ILT-3), a marker of the regulatory DC phenotype. Furthermore, co-incubation of MP-treated DCs with allogeneic T cells resulted in dose-dependent decreased in T-cell proliferation [110]. These results demonstrate the potential of a new method to induce tolerogenic DCs, which was made possible through the intentional engineering of local delivery systems to sequentially release immunomodulatory biologics.
Tolerogenic DCs have also been induced through release of tacrolimus from locally placed PLGA microparticles in conjunction with clodronate liposomes [227]. Past and present studies involving liposomal clodronate illustrated its’ ability to deplete inflammatory macrophages [227,228]. In this study, Pathak et al. explored the local co-delivery of liposomal clodronate and tacrolimus using MPs embedded in Matrigel to induce tolerogenic DCs and protect transplanted xenogeneic islets from rejection [227]. A single, subcutaneous dose of the local delivery system decreased expression of DC costimulatory markers (i.e., CD40, CD80, CD86 and MHCII) in both the graft-tissue and draining lymph nodes [227]. This phenotype is characteristic of tolerogenic DCs, as previously described. As a result of tolerogenic DC induction, and their subsequent secretion of immunosuppressive factors (TGF-β1, IL-10), populations of CD4+CD25+Foxp3+ Tregs were also significantly increased in the spleen and draining lymph nodes [227]. The induction of regulatory subtypes, initially spurred by the production of tolerogenic dendritic cells, led to indefinite graft survival of transplanted islets [227]. Importantly, a single dose of the controlled release system was sufficient to induce regulatory effects [227]. Therefore, it is possible that high doses and/or dose frequency of individual immunomodulatory factors could be avoided using a local, controlled release system.
Lewis et al. also induced tolerogenic DCs through local delivery of immunomodulators from two distinct sizes of PLGA microparticles [229]. PLGA microparticles encapsulating either rapamycin or retinoic acid were engineered to be 2 μm in diameter, with the intention of being phagocytosed by antigen presenting cells for intracellular processing [229]. IL-10 or TGF-β1, on the other hand, were encapsulated in larger microparticles (30 μm) to prevent phagocytosis, resulting in extracellular release [229]. Single and pairwise combinations of MPs were then studied for their effects on DC maturation. The combination of rapamycin/TGF-β1 microparticles excelled at reducing the expression of costimulatory molecules (MHC II, CD80, CD86) [229]. However, the combination of retinoic acid/IL-10 resulted in the least T-cell proliferation [229]. Although different combinations of payloads excelled in fostering diverse immunosuppressant outcomes, it was apparent that combination microparticles were superior to soluble or single MP treatment, demonstrating the synergistic or combinatorial effect of using multiple immunosuppressant or tolerogenic signals.
Local delivery strategies have also been used to target macrophages to induce their anti-inflammatory, otherwise known as alternatively activated macrophages (M2). Unlike M1 macrophages, which are generally considered inflammatory, M2 macrophages secrete anti-inflammatory cytokines (i.e., IL-10, IL-1Ra, TGF-β1) and promote tissue remodeling [230,231]. Induction of M2 macrophages in situ has been achieved through the local delivery of CCL2, a macrophage chemokine and polarizing factor, using PLGA microparticles [232]. In vitro studies demonstrated the ability of CCL2 to induce M2 macrophages from bone marrow derived macrophages. The efficacy of this system was then explored in vivo in a murine model of periodontitis, a chronic inflammatory tooth disorder [232]. Local delivery of CCL2-MPs increased the ratio of M2:M1 macrophages, enhanced anti-inflammatory cytokine secretion and prevented periodontitis-induced bone loss [232]. Together, these studies show the potential in targeting multiple subsets of antigen presenting cells to restore immune homeostasis.
3.3.2.2. In Situ Manipulation of Regulatory T Cells
Regulatory T cells are also of interest for in situ induction and manipulation. Rhodes et al. explored the use of PLGA/PBAE microparticles, functionalized with anti-CD3/CD28 and encapsulating TGF-β1, to generate Foxp3+ Tregs [233]. Anti-CD3/CD28 were included within the matrix to induce T cell activation, which in the presence of TGF-β1, can result in the development of Tregs. To enhance surface conjugation of anti-CD3/CD28, PBAE was incorporated with PLGA to form the local delivery matrix. The use of PLGA/PBAE MPs induced greater proportions of CD4+CD25+Foxp3+ Tregs when compared to PLGA MPs alone, as higher proportions of anti-CD3/CD28 were conjugated to the surface [233]. PLGA/PBAE MPs were also superior at suppressing CD4+ T cell proliferation in vitro when compared to PLGA MPs. In vivo studies were then conducted to assess the development of Tregs following intravenous administration of PLGA/PBAE MPs [233]. While administration of PLGA/PBAE MPs trended towards the development of a regulatory phenotype in lymph nodes, these results were not obtained in a model of inflammation. Therefore, the efficacy of this approach could be further validated in a model of inflammation where other endogenous stimuli may prevent the development of regulatory subtypes. Furthermore, the results observed in this study may be enhanced if the delivery system were to be administered directly to the site of interest (i.e., lymphatics, subcutaneous, etc.), as the size of the fabricated particles (~3 μm) could facilitate local immunomodulation. In addition, this study highlighted an additional design consideration (i.e., material choice) to enhance protein conjugation, which has been proven beneficial in inducing regulatory effects.
Treg differentiation has also been facilitated by local delivery of three, specific immunomodulatory factors, TGF-β1, rapamycin, and IL-2 (TRI-MP) (Figure 7B) [30,190,234–236]. Induced Tregs, discussed previously, have been shown to differentiate from naïve CD4+ T cells in response to the presence of TGF-β1 [237]. TGF-β1, however, is also crucial for the development of Th17 cells, effector cells implicated in the pathogenesis of autoimmunity [237]. Therefore, additional factors, such as IL-2 and rapamycin, can be used limit plasticity and enhance stability of iTregs [237,238]. Early studies performed by Jhunjhunwala et al. demonstrated that TRI-MP were as effective as soluble factors at inducing Tregs with an immunosuppressive phenotype in vitro [234]. When prophylactically and subcutaneously administered during hapten sensitization by a contact allergen, TRI-MP significantly increased the ratio of Tregs:Teff and decreased expression of several pro-inflammatory cytokines (i.e., IL-1β, TNFα, IFNγ) [235]. Notably, prophylactic treatment of TRI-MPs suppressed immune responses in an allergen-specific manner when re-challenged with antigen following initial sensitization [235]. These results suggest the ability of the system to induce antigen-specific immune responses that promote tolerance and restore immune homeostasis. Furthermore, this study elucidated the benefit of both controlled release and local delivery. While TRI-MPs significantly enhanced local Treg populations, administration of soluble TRI factors (bolus administration) failed to provide the same effect [235]. In addition, administration of TRI-MP at a site distal to the site of initial sensitization did not impact the ratio of Tregs:Teff. Altogether, these results suggest the benefit of local, controlled delivery as well as the potential to induce antigen-specific responses through the delivery of several biologics.
Fig.7.
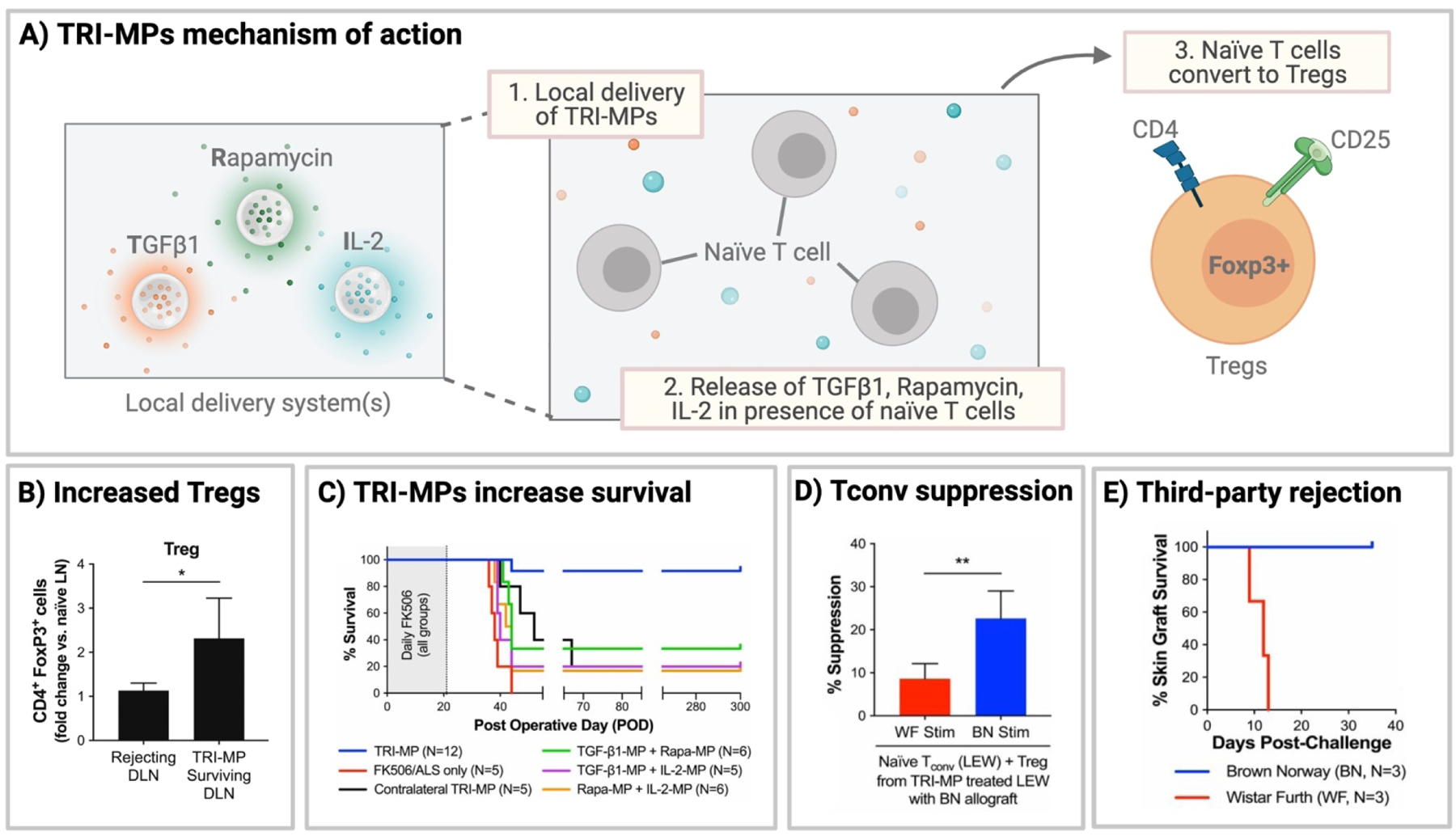
TRI-MPs induce regulatory T cells and donor-specific tolerance in VCA. (A) Schematic illustrating the mechanism of action for Treg-inducing microspheres, where local release of TGFβ1, rapamycin and IL-2 induce Treg conversion from naïve T cells. (B) Effect of TRI-MP treatment on Treg populations in allograft draining inguinal lymph nodes. (C) Impact of TRI-MPs on hindlimb graft survival in a model of vascularized composite allotransplantation (VCA). (D) Increased suppression of TRI-MP treated Tregs on conventional T cell proliferation when stimulated with donor splenocytes when compared to third-party splenocytes. (E) Secondary skin graft challenge demonstrates acceptance of donor skin grafts, but rejection of third-party skin grafts, illustrating the induction of donor-specific tolerance. Data in figures 7B, 7C, 7D and 7E reprinted from J.D. Fisher, S.C. Balmert, W. Zhang, R. Schweizer, J.T. Schnider, C. Komatsu, L. Dong, V.E. Erbas, J.V. Unadkat, A.M. Aral, A.P. Acharya, Y. Kulahci, H.R. Turnquist, A.W. Thomson, M.G. Solari, V.S. Gorantla, S.R. Little, Treg-inducing microparticles promote donor-specific tolerance in experimental vascularized composite allotransplantation, Proc National Acad Sci. 116 (2019) 25784–25789. https://doi.org/10.1073/pnas.1910701116. Copyright (2021) National Academy of Sciences.
Several other studies explored the impact of TRI-MP and resulting Treg induction, but in models of dry eye disease, vascularized composite allotransplantation and rheumatoid arthritis [30,190,236]. Fisher et al. found that subcutaneous administration of TRI-MP in VCA allografts increased the presence of suppressive cytokines (i.e., IL-10, IL-35, TGF-β1) and cell populations (CD4+Foxp3+ Tregs) in graft tissue and allograft draining lymph nodes, respectively (Figure 7B) [30]. In a model of vascularized composite allotransplantation, local injections of TRI-MPs significantly prolonged hindlimb allograft survival (>200 days) (Figure 7C) [30]. These results were not observed for pairwise iterations of TRI-MP or when TRI-MP was injected contralaterally, demonstrating the importance of multi-factor delivery and site-specific immunomodulation. Importantly, conventional T cell stimulation, specifically in the presence of TRI-MP-treated Tregs and donor splenocytes, was significantly suppressed when compared to stimulation with TRI-MP-treated Tregs and third-party splenocytes (Figure 7D) [30]. As a result, third-party skin grafts were rejected, whereas donor skin grafts were accepted (Figure 7E). These results suggest that TRI-MPs were able to induce donor-specific tolerance.[30] Bassin et al. then tested TRI-MP in a model of collagen-induced arthritis [190]. Subcutaneous injection of TRI-MP significantly reduced levels of arthritic autoantibodies, inflammatory cytokine expression and immune cell infiltration [190]. Overall, these studies highlight the impact of in situ Treg generation on restoring immune homeostasis and the potential for local biologic release systems to induce donor-specific tolerance.
3.4. Delivery of antigen-specific therapeutics
In some inflammatory conditions such as T1D, MS and RA, self-antigens (Ag) are recognized by the immune system, prompting an unwanted immune response against normal tissue. Therefore, antigen-specific tolerance is particularly important in these disease models to maintain or restore immune homeostasis. While the above studies (Fisher et al., Balmert et al., etc.) demonstrates the ability to induce donor-specific tolerance through delivery of biologics alone, the mechanisms of this tolerance are not entirely understood. It is possible that the presence of endogenous antigens in an inflammatory context in the individual local sites (i.e., grafted limb, transdermal to ear skin) contributed to the antigen-specific tolerance observed in these studies. In the absence of this context, or in cases where the antigen of interest is known, this antigen itself could be delivered, alone or alongside other factors, to induce antigen-specific tolerance.
Antigen-specific strategies have been developed to induce tolerance or facilitate immunosuppressive responses against specific disease-associated antigens (i.e., myelin oligodendrocyte glycoprotein (multiple sclerosis), myelin proteolipid protein (multiple sclerosis), insulin (diabetes), etc.). However, there are still concerns in regard to their method of administration, as systemic administration can propagate disease and result in anaphylaxis rather than resolution [41,239–241]. More specifically, systemic peptide administration generates a profound response by enabling extensive immune cell activation throughout the body [239]. Local antigen administration, on the other hand, results in uptake by a limited population of antigen-presenting cells, which then migrate to secondary lymphatic organs to induce a restricted immune response. To facilitate this response, local delivery strategies can be used to constrain antigen delivery to specific anatomical sites of interest, prompting immune responses in local draining lymph nodes. The two most employed strategies for the development of such systems are to deliver (1) antigen alone or (2) antigen in combination with other immunosuppressive/tolerogenic signals (i.e., small molecules, biologics, etc.) to induce a more robust, specific response (Table 6). The next section of this review will discuss examples of antigen-specific therapeutics and highlight their potential to restore immune homeostasis.
Table 6.
Local delivery strategies for antigen or antigen and small molecules/biologics used to restore immune homeostasis from inflammation.
| Antigen-Specific Approach | Delivery Strategy | Disease Model | Ref(s) | |
|---|---|---|---|---|
| Antigen | ||||
| PLP | PLGA MPs | Multiple sclerosis | [240,241] | |
| Collagen scaffold | Multiple sclerosis | [112] | ||
| Ins29–23 | PLGA MPs | Inflammatory diabetes | [245] | |
| Antigen and small molecules/biologics | ||||
| MOG | Rapamycin | PLGA MPs | Multiple sclerosis | [43] |
| Dexamethasone | Acetalated dextran MPs | Multiple sclerosis | [246] | |
| Vitamin D3, TGF-β1, GM-CSF | PLGA MPs | Multiple sclerosis | [185] | |
| Insulin B | Vitamin D3, TGF-β1, GM-CSF | PLGA MPs | Inflammatory diabetes | [187] |
| Denatured insulin | Vitamin D3, TGF-β1, GM-CSF | PLGA MPs | Inflammatory diabetes | [188] |
| Collagen II | Vitamin D3, TGF-β1, GM-CSF | PLGA MPs | Inflammatory arthritis | [248] |
| PFK15 inhibitor | Poly-aKG MPs | Inflammatory arthritis | [152] | |
3.4.1. Delivery of antigen alone
Delivery of antigen alone can be capable of restoring mechanisms of central and peripheral tolerance, such as T cell anergy, T cell exhaustion, negative selection and Treg expansion, which often fail during inflammatory disorders (Figure 8). T cell anergy is a state of unresponsiveness, promoted by improper engagement of all signals required for naïve T cell activation. Naïve T cell activation is a result of three signals: (1) engagement of the T cell receptor (TCR) by peptide-bound major histocompatibility complexes expressed on antigen-presenting cells (APCs), (2) co-stimulation and (3) cytokine secretion/signaling [239,240,242]. Signal 2, co-stimulation, also involves the binding of adhesion molecules expressed on the surface of T cells (LFA-1) and APCs (ICAM) [239,240,242]. Interaction between these adhesion molecules is necessary for efficient T cell activation. In the case of incomplete co-stimulation, T cells are not fully activated and thus enter a state of anergy, during which the cell lives but remains insensitive to stimuli. To induce this state, Zhao et al. developed PLGA microparticles to both locally and controllably deliver a bifunctional peptide inhibitor (BPI) following subcutaneous administration [240,241]. The BPI contains a peptide epitope for myelin proteolipid protein (PLP), an antigen implicated in multiple sclerosis, a peptide linker and LABL, a peptide for ICAM. With an affinity for both MHC and ICAM, the BPI limits T cell activation by blocking the interaction of LFA-1 and ICAM (Figure 8A) [240]. Importantly, the authors of this study compared routes of administration to enhance the efficacy of PLP-BPI. It was found that intravenous and subcutaneous injections of soluble PLP-BPI effectively induced suppression in a model of EAE [240]. However, these methods of administration lead to increased toxicity. PLP-BPI microparticles, on the other hand, induced suppression of EAE without the toxicity seen by soluble administration [240]. Together, these results highlight the ability to interrupt T cell activation to reduce inflammation while demonstrating the importance of local, controllable release systems in limiting side effects.
Fig. 8.
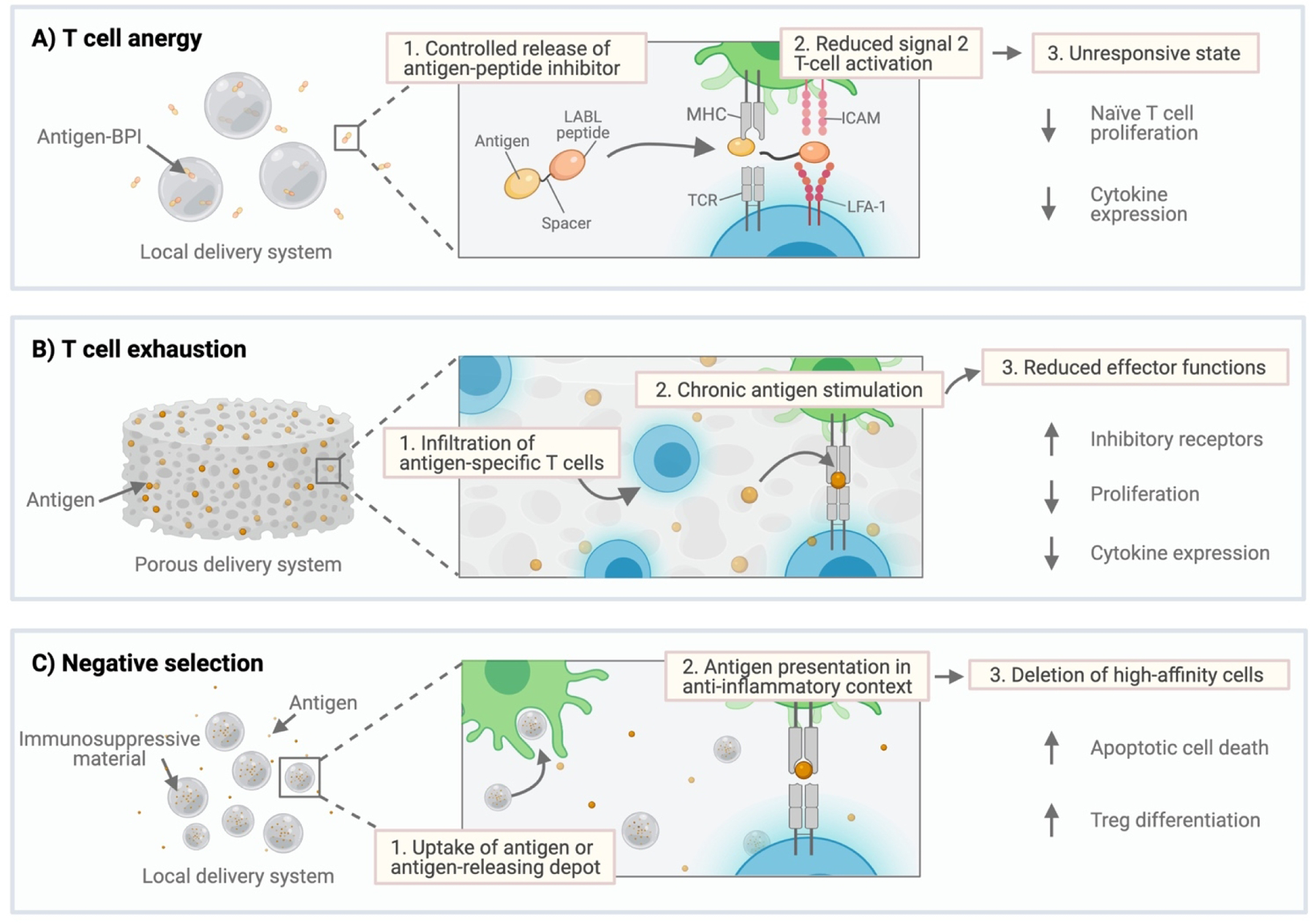
Strategies used to restore immune homeostasis through local delivery of antigen alone. A) Local delivery of bifunctional peptide inhibitor (BPI) engineered to limit signal 2 of T cell activation which occurs between APCs (green) and T cells (blue). Improper activation can then lead to an anergic state characterized by reduced naïve T cell proliferation and decreased cytokine expression. B) Local delivery of high concentrations of antigen in a restricted area can induce T cell exhaustion due to chronic antigen stimulation. T cell exhaustion is characterized by increased expression of inhibitory receptors, decreased proliferation, and cytokine expression. C) Negative selection, which results in the deletion of autoreactive cells, can be induced through delivery of antigen in an anti-inflammatory context provided by the delivery system itself.
Another antigen-specific mechanism used to induce tolerance and restore immune homeostasis is T cell exhaustion, which results from exposure to constant and/or excess concentrations of antigen [243,244]. T cell exhaustion typically occurs during chronic infection and it is characterized by a loss of effector functions (i.e., cytokine secretion, degranulation) over time [243,244]. In addition, this state of unresponsiveness is often exhibited by T cells in tumors, where impaired T cell proliferation, reduced cytokine secretion and increased inhibitory receptor expression promote tumor cell survival [244]. In the case of autoimmunity, induction of T cell exhaustion could promote a return to immune homeostasis. Such an approach has been explored in a model of multiple sclerosis, where Griffin et al. developed porous collagen depots to provide a high local concentration of PLP to infiltrating autoreactive immune cells (Figure 8B) [112]. Upon infiltration, autoreactive cells were then overstimulated with PLP, inducing apoptosis due to chronic exposure [112]. Subcutaneous implantation of the construct in a murine model of EAE significantly reduced disease incidence, with zero of the treated mice developing disease [112]. Observations from this study establish the ability to reduce systemic disease incidence through local immunomodulation, providing a potentially safer method of treatment.
Aside from generating states of anergy and exhaustion, antigen delivery has been used to induce negative selection while expanding populations of Tregs. Negative selection involves the deletion of self-reactive cells after exposure to antigen within a tolerogenic context [244]. Local delivery of Ins29–23, a known autoantigen in T1D, through use of PLGA microparticles has been explored as a method to induce negative selection [245]. In this approach, Liu et al. used PLGA microparticles as both the delivery system and the tolerogenic signal, as PLGA has been shown to induce an immunosuppressant immune cell phenotype (Figure 8C) [66,245]. The resulting microparticles were 2–4 μm in diameter, making them small enough for uptake by antigen presenting cells [24,245]. Controlled release of Ins29–23 using PLGA microparticles completely prevented the development of T1D, potentially due to reduced splenic populations of CD4+ T cells from negative selection and increased early populations of CD4+C25+Foxp3+ Tregs [245]. Results from this study highlight the potential capacity to choose an immunosuppressant material, rather than an additional biologic, to serve as a tolerogenic signal. Nonetheless, although proving to be immunosuppressive in this setting, PLGA microparticles have also induced inflammatory responses in other situations. Therefore, this study could be further enhanced by the inclusion of additional tolerogenic factors (discussed in the next section).
3.4.2. Delivery of antigen and small molecules or biologics
As mentioned previously, mechanisms such as anergy and exhaustion can promote cell survival by limiting effector mechanisms, a common approach exploited by cancers to facilitate immune evasion [244]. However, anergic and exhausted cells can be “rescued” from their states, resulting in immune cells which can now respond to antigen again. Although beneficial in the context of cancer, “rescue” of autoreactive cells could be detrimental in the context of autoimmunity. To prevent mechanisms of rescue from occurring, it may be beneficial to deliver antigen alongside additional tolerogenic or immunosuppressive signals, such as (1) standard pharmaceuticals or (2) biologics, to generate antigen-specific cell populations that are not immunosuppressant due to their anergic or exhausted state (Table 6).
Such an approach has been explored to induce antigen-specific immune cells in models of multiple sclerosis, type 1 diabetes, and rheumatoid arthritis (Figure 6) [43,152,185–189,229,246–248]. Among standard pharmaceuticals, three small molecules that have been delivered locally with disease-associated antigen are rapamycin, dexamethasone and PFK15 (discussed previously) [43,246]. Tostanoski et al. locally delivered rapamycin, alongside myelin oligodendrocyte glycoprotein (MOG), using PLGA microparticles to induce antigen-specific tolerance in multiple sclerosis [43]. When studied in a murine model of MS, intranodal administration of MOG/Rapamycin-MPs greatly expanded populations of Tregs, resulting in improved clinical scores and reduced MS disease incidence when compared to controls [43]. More so, a single dose of MOG/Rapamycin-MPs in established disease was capable of reversing disease incidence and promoting weight gain [43]. The antigen-specific action of MOG/Rapamycin-MPs was confirmed ex vivo, during which isolated cells were restimulated with MOG. Following restimulation, cells isolated from animals treated with MOG/Rapamycin-MPs exhibited reduced levels of inflammatory cytokine production (IFNg, IL-17) [43]. This study demonstrates the immunomodulatory potential of inducing antigen-specific immune responses during disease progression, rather than prophylactically, in autoimmunity.
Local delivery of MOG using acetylated dextran microparticles, as opposed to PLGA MPs (described above), has also been explored as a method to induce antigen-specific immune responses in multiple sclerosis [246]. In this study, Peine et al. subcutaneously administered microparticles co-encapsulating MOG and dexamethasone following symptom onset. Codelivery of MOG/dexamethasone-MPs led to a significant reduction in MS clinical scores [246]. Notably, delivery of MOG/dexamethasone-MPs was superior in ameliorating disease progression when compared to dexamethasone-MPs alone [246]. These results suggest the potential benefit of including disease-specific antigens in local delivery strategies to enhance efficacy.
The last small molecule that has been locally delivered simultaneous to antigen is PFK15, a PFKFB3 inhibitor shown to induce anti-inflammatory effects by inducing metabolic changes [151–153]. In this study, Mangal et al. explored the local delivery of PFK15 alongside collagen type II, a self-antigen implicated in inflammatory arthritis, using paKG-MPs (known as paKG(PFK15+bc2)-MPs ) [152]. As mentioned previously, delivery of PRK15 can alter the metabolic programming and corresponding function of dendritic cells, as shown by reduced glycolysis and altered pro- and anti-inflammatory gene expression [152]. In a model of collagen-induced arthritis, subcutaneous injection of paKG(PFK15+bc2)-MPs to arthritic limbs (back paws) reduced arthritic scores by the end of the study (day 65) when compared to the no treatment group [152]. Importantly, local delivery of paKG(PFK15+bc2)-MPs induced both local and systemic antigen-specific responses, as demonstrated by increased populations of proliferating regulatory T cells, as well as decreased populations of Th1 and Th17 cells in both the popliteal (local measurement) and cervical lymph nodes (systemic measurement) [152]. Together, these results highlight the potential to induce antigen-specific anti-inflammatory responses by altering metabolic pathways within dendritic cells in the presence of disease-associated antigen.
Many studies have also explored the delivery of antigen alongside a variety of biologics (i.e., cytokines) to enhance the specificity of the responses generated. For instance, Lewis et al. expanded on their previous work (discussed in section 3.3.2.1) to induce antigen-specific tolerogenic dendritic cells in autoimmune diabetes (Figure 9A) [187,229]. In the subsequent study, the four factors selected for encapsulation in PLGA MPs were vitamin D3, insulin B, TGF-β1 and GM-CSF [187,229]. Vitamin D3 (VD3) was chosen based on its’ ability to prevent inflammatory DC maturation [187]. Insulin B was used as the peptide, as it is a key anti-islet autoantigen involved in the development of autoimmune diabetes [249]. Finally, TGF-β1 and GM-CSF were included due to their role in the expansion of Tregs and chemotaxis of monocytes and lymphocytes, respectively [186,187]. VD3 and insulin B were loaded in phagocytosable PLGA MPs, whereas TGF-β1 and GM-CSF were loaded in larger MPs incapable of migrating elsewhere or being phagocytosed [187]. When applied in a prevention study of autoimmune diabetes in 4-week-old mice, subcutaneous administration of all four types of MPs significantly delayed the onset of diabetes and resulted in increased survival [187]. Importantly, all four factors (vitamin D3, insulin B, TGF-β1, GM-CSF) were required to significantly prevent diabetes onset, demonstrating the efficacy of delivering multiple biologic signals to induce tolerance.
Fig. 9.

Local delivery of immunomodulatory factors to induce antigen-specific tolerogenic dendritic cells. (A) GM-CSF can drive dendritic cell maturation from progenitors, while Vitamin D3 and TGFβ1 provide immunosuppressive signals that skew dendritic cell maturation away from an inflammatory phenotype and towards an anti-inflammatory phenotype (tolerogenic/immature DCs). The resulting DCs exhibit low expression of MHC II and CD80/86, but high expression of ILT-3. (B) Combination microparticle treatment of bone-marrow derived macrophages reduced the composite maturation index of the cells. (C) Microparticle injection (REGvac, indicated by solid black arrows) reversed the progression of collagen-induced arthritis in mice with moderately established disease (clinical score = 5). Data from Figures 6B and 6C reprinted with permission from: R. Allen, S. Chizari, J.A. Ma, S. Raychaudhuri, J.S. Lewis, Combinatorial, Microparticle-Based Delivery of Immune Modulators Reprograms the Dendritic Cell Phenotype and Promotes Remission of Collagen-Induced Arthritis in Mice, Acs Appl Bio Mater. 2 (2019) 2388–2404. https://doi.org/10.1021/acsabm.9b00092. Copyright 2021 American Chemical Society.
To improve this platform and ensure its’ clinical relevance, Stewart et al. explored the immunomodulatory microparticles developed above in a late-stage diabetes prevention model [188,247]. Four-week-old mice, used in the previous study, do not display typical symptoms (i.e., leukocyte infiltration) exhibited by patients at the time of T1D diagnosis [188,247]. Aside from changing models, denatured insulin was encapsulated instead of insulin B to expand the peptide library being processed and presented for tolerization [188]. In addition, microparticles were engineered to release higher doses of GM-CSF and TGF-β1 [188]. In vitro studies established the ability of dMPs (all four factors) to alter DC phenotype by reducing expressing of maturation markers [188]. In a late-stage diabetes model, subcutaneous administration of dMPs significantly prevented diabetes onset and reversed recent-onset diabetes for a short period of time [188]. These results demonstrate the ability of local codelivery of multiple biologics to generate desirable immune responses in a more clinically applicable model. When implemented in models of MS and RA, local delivery of antigen-, vitamin D3-, TGF-β1- and GM-CSF- MPs yielded similar results [185,248]. In both cases, disease-specific peptides, such as myelin oligodendrocyte glycoprotein (MOG) for MS, and collagen II for RA, were substituted for denatured insulin. In a murine model of MS, dMP treatment suppressed EAE in an antigen-dependent manner [185]. As for its’ application in RA, initial in vitro experiments demonstrated that the combination of VD3-MPs and TGFβ1-MPs was required to reduce the composite maturation index (expression of CD80/CD86/MHCII) of bone-marrow derived macrophages, emphasizing the importance of multi-factor delivery (Figure 9B) [248]. In a model of collagen-induced arthritis, dMP administration (otherwise known as REGvac) reversed disease progression (Figure 9C) [248]. RA progression was also reversed in an antigen-specific manner, as isolated cells from dMP-treated mice did not proliferate or produce inflammatory cytokines when rechallenged with antigen [248]. Together, these studies demonstrate the ability of dMPs to restore or maintain immune homeostasis in several clinically relevant models of inflammation.
To further demonstrate the clinical translatability of the approach discussed above, Brusko et al. assessed the effect of dMPs on human cells, specifically human-derived monocytes/macrophages, DCs and T cells [189]. Treatment of monocytes and human-derived DCs resulted in a suppressive cell phenotype, characterized by resistance to LPS-induced maturation and enhanced expression anti-inflammatory mediators (i.e., PD-L1, IDO, etc.) [189]. To expand upon this study and based on its’ success in both 4 and 8-week-old NOD mice, RA, MS, dMPs were then used in combination with low-dose anti-CD3 immunotherapy, a T cell depleting treatment, to assess its’ ability to prevent diabetes in 12-week-old mice [247]. Although previous results were promising, the combination treatment failed to prevent T1D [247]. It is possible that the effects of both dMPs and anti-CD3 immunotherapy were insufficient to overcome disease progression in 12-week-old mice. Nonetheless, a suppressive phenotype of both dendritic cells and CD4+ T cells developed in the draining lymph nodes [247]. In the case of dendritic cells, dMP treatment reduced the expression of CD86 and MHCII. CD4+ T cells, on the other hand, exhibited increased expression of PD-1. These effects were not observed in distal lymphoid organs, illustrating the importance of local delivery.
Altogether, these studies illustrate the ability of local delivery of antigen in combination with other factors to induce antigen-specific immune responses in various models of inflammation (multiple sclerosis, inflammatory diabetes, rheumatoid arthritis). In addition, these results highlight the importance of tuning delivery systems (i.e., loading, timing of administration, controlled release) to the disease in question, as disease progression can limit the effectiveness of administered treatments. Nonetheless, these studies demonstrate the ability to restore immune homeostasis using local, antigen-specific delivery strategies given both prior to symptom onset and during pathogenesis, enhancing the clinical relevance of the approaches.
4. Future directions and clinical translation
Local delivery strategies are emerging as viable platforms to restore immune homeostasis in the context of inflammation, specifically through delivery of small molecules, biologics (i.e., antibodies, cytokines, chemokines), cells and antigen-specific approaches. Nonetheless, there is still areas where the field could benefit from further exploration. In this section, we will highlight future directions in the field, as well as discuss clinical translation of these technologies.
4.1. Future directions for local delivery strategies in inflammation
Over the past few decades, significant advancements have been made to develop novel approaches which treat inflammation with increased efficacy. Current standard immunosuppressant regimens are limited by their (1) systemic administration, which promotes off-target effects and dosing toxicities, and (2) lack of specificity in regard to their mechanism of action. As mentioned previously, the manner in which agents are administered has a direct impact on the efficacy of the approach [14]. For instance, conventional small molecules are typically administered systemically, requiring high dose concentrations to reach the intended anatomical target. In addition, systemically administered drugs are limited by their clearance by the reticuloendothelial system. To address these limitations, local drug delivery strategies (i.e., microparticles, hydrogels, scaffolds), which sequester immunosuppressant or anti-inflammatory cargo at a specific site of interest for immunomodulation, are emerging as viable platforms to address these limitations.
The most common approach for localized drug delivery is to repurpose FDA-approved and clinically implemented pharmaceuticals (discussed in section 3.1.). While this approach can improve pharmaceutical retention following administration (i.e., subcutaneous, intra-articular, co-localization, etc.), it fails to address one of the main limitations associated with the use of small molecules: non-specific, broad mechanisms of action. Standard immunosuppressants typically broadly suppression inflammation by altering immune cell function (i.e., metabolism, activation, cytokine production, etc.), regardless of immune cell subtype and the disease in question. On the other hand, biologic-based treatments (i.e., antibodies, cytokines, chemokines, antigens, etc.) have the potential to restore immune homeostasis in a more specific or disease-dependent manner. More specifically, local biologic-based delivery systems can release particular cargo (i.e., CCL22, antigen) known to impact specific immune cells which have been found critical to the restoration of immune homeostasis in a certain disease state. Since such an approach, local delivery of biologic-based approaches, enables more precise control over the resulting immune response, we predict that research in this area will continue to grow.
Among local biologic-based delivery strategies, delivery of antibody-based treatments, including monoclonal antibodies and fusion proteins, is less commonly explored. It is possible that these strategies have been studied less due to their inherently longer half-life (monoclonal antibodies—days to weeks, fusion proteins—days), as drug delivery is often used to improve half-life [250]. In addition, local delivery system fabrication processes could damage antibody stability, potentially limiting bioactivity and effectiveness [175,250]. Nonetheless, local delivery systems could still be used to increase antibody-based treatment retention at a specific site of interest, which could enhance overall efficacy and limit off-target effects [250]. Therefore, this is an area which could benefit from more exploration. However, it will be important to consider formulation parameters (i.e., stabilizers, temperature, mechanical stress, freeze/thaw stress, pH, etc.) which can negatively impact antibody folding and resulting function (extensively reviewed by Awwad et al.) [175].
Aside from delivery of individual biologics, cell-based approaches and antigen-specific therapeutics have the potential to act in extremely specific manners to restore immune homeostasis without many of the side effects seen by traditional pharmaceuticals. Cell-based approaches, specifically, have advantages over administration of biologics. Cells can sense and dynamically respond to their local environment in a very controlled and precise manner (i.e., only responding if provided with the biological cues to do so) [251]. Small molecule drugs and biologics, on the other hand, produce an effect that is only dependent upon its’ presence (and concentration) in a local environment. Nonetheless, there are numerous limitations associated with cell-based therapies (i.e., isolation process, monetary burden, variability, etc.). In situ induction of these subtypes represent a potentially more elegant way to take advantage of the intricate sensing and immunomodulatory effects of cell-based therapies while avoiding limitations associated with traditional cell-based therapies. This has been demonstrated by locally presenting specific biologics in combination, which can enrich regulatory immune cells (i.e., regulatory T cells, immature dendritic cells, M2 macrophages, etc.) at a specific site of interest. In several cases, local delivery of these biologics in the presence of endogenous antigen is even capable of creating antigen-specific or donor-specific tolerance to restore homeostasis. Further investigation is necessary to understand this phenomenon. Overall, in situ induction of regulatory subtypes could address some limitations associated with traditional cell-based therapies; however, there are still numerous obstacles to clinical translation, summarized in Section 4.2.
Local delivery strategies have also recently incorporated exogeneous antigen alongside biologics to achieve antigen-specific tolerance. Nevertheless, there are still many limitations with this approach. One of the main limitations with antigen-specific therapeutics is the identification of disease-associated peptides, which vary between diseases [252,253]. While many autoantigens that trigger the initial immune response have been discovered, many have not. In addition, antigen epitopes can change over time due to epitope spreading [252]. Even more so, autoantigens can differ from patient to patient, with certain antigens dominating disease in one case but not the other. Altogether, the heterogeneity of autoantigens can significantly limit the application of antigen-specific therapeutics. Nonetheless, given the success of preclinical antigen-specific therapeutic approaches, further research in this area is merited and could benefit from the identification of additional autoantigens.
4.2. Clinical translation of local delivery strategies
To fully benefit human health, the strategies discussed throughout this review must be translated to the clinic. However, there are numerous barriers to overcome for successful clinical translation, including initial manufacturing and scale-up concerns. Some of these obstacles are highlighted below (Table 7); however, a more comprehensive analysis of these obstacles can be found elsewhere [254–258].
Table 7.
Key obstacles in the clinical translation of local delivery strategies.
| Obstacle | Description of Challenges | Ref(s) |
|---|---|---|
| Formulation Development | ||
| Scale-up | Complex formulations challenging to scale | [254] |
| Specifications needed to limit batch-to-batch variability | [254,257] | |
| Sterilization | Aseptic vs. terminal sterilization decision | [258] |
| Can damage polymeric chains | [257] | |
| Can damage biologic-based cargo | [256] | |
| Preclinical Evaluation | ||
| Dose scaling | Pre-existing dose scaling guidance based on systemic exposure | [259,260] |
| Biologics distribute differently than small molecules | [259,260] | |
| In vitro to In vivo models | Need more emphasis on pharmacokinetic (i.e., absorption) and pharmacodynamic (i.e., toxicity) studies | [254] |
| In vitro to in vivo models do not always correlate | [256,262] | |
| Cost | ||
| Overall Cost | Between $161 million to $2 billion USD | [261] |
| Clinical Trials (average cost) | Phase I: $6.6 million USD; Phase II: $16 million USD; Phase III: $11.9 million USD | [261] |
One of the first translational barriers to overcome is large-scale manufacturing of the formulation/product. During initial formulation development, many of these delivery systems are manufactured in small quantities for initial proof-of-concept animal models. However, translation into the clinic requires producing much larger quantities of the product for both large-scale animal models (typically non-human primate) and eventually, human clinical trials. Large-scale manufacturing is susceptible to variability, potentially producing a product that does not perform based on initial studies. Therefore, specifications for formulations (i.e., delivery system size, stability, morphology, cargo encapsulation and release, etc.) should be well-documented and reproducible prior to pursuing large-scale manufacturing to ensure limited batch-to-batch variability and correct biologic activity of the product [254,257].
Another barrier to translation for local delivery strategies, such as those discussed here, is dose scaling for human clinical trials. The Food and Drug Administration (FDA) has clear guidance regarding the calculation of human equivalent doses (HED) for new drugs and biological therapeutics; however, it only pertains to products with systemic exposure. In this instance, the “no-observed adverse effect level (NOAEL),” or the highest concentration of a drug (determined by previous studies) that causes no adverse effects, is scaled to a human equivalent dose by accounting for body surface area [259]. The technologies discussed here, however, are not expected to facilitate whole-body distribution of a drug due to their local placement and reduced dose frequency/concentration. Therefore, it may be more applicable to scale local delivery systems in a manner similar to vaccines, which also rely upon inducing immunological effects in local sites (i.e., site of injection, draining lymph nodes) [260]. This approach is further supported in the case of biologic-based local delivery approaches (i.e., cytokines, chemokines, antibodies), as biologics, unlike small molecules, cannot pass through biological membranes easily and can be degraded by proteolytic enzymes found within the body [259]. Thus, biologics are also expected to have smaller volume distributions when compared to small molecules. For this reason, the FDA suggests scaling biologic-based approaches on a mg·kg−1 basis, rather than the traditional approach based on surface area [259]. Overall, dose scaling of local delivery systems should likely not be based off allometric scaling, and will need to account for new variables, including the site of administration, local distribution and pharmacokinetic/dynamic data. Further research into this topic is merited in order to develop a standardized approach for dose scaling of localized immunomodulatory approaches.
Translation of local delivery strategies can also be limited by product sterilization, which is required for human clinical trials. Sterile products can either be achieved by aseptic processing or terminal sterilization [258]. Aseptic processing requires all materials (i.e., polymer, containers, drugs/proteins, etc.) to be sterilized separately. These materials are then utilized during a manufacturing process, which also maintains sterility, to produce the final product. Terminal sterilization, on the other hand, consists of sterilizing the product in its’ final form. Due to the intricate steps and environments required by aseptic processing, as well as the potential for microbial contamination, terminal sterilization is typically preferred [258]. Nonetheless, polymeric delivery systems, as well as the active pharmaceutical ingredients (API) being delivered, can be directly impacted by the sterilization process. In the case of PLGA-based delivery systems, sterilization can alter polymeric chains, which can easily impact other formulation characteristics (i.e., controlled release, degradation, drug encapsulation, etc.) [257]. For protein-based local delivery systems, terminal sterilization methods (i.e., steam, dry heat and ethylene oxide) can damage the incorporated protein [256]. Overall, careful consideration should go into selecting the correct sterilization method, and activity testing should be performed following sterilization to ensure proper function.
Perhaps the most inhibitory obstacle to clinical translation is cost, as bringing a drug to market has been estimated to cost between $161 million to $2 billion USD [261]. Specific to the field of immunomodulatory drugs, Phase I studies cost an average of $6.6. million USD. Phase II and III trials average $16 million and $11.9 million USD, respectively [261]. To assist with the financial burden associated with clinical translation of these products, several agencies (private and public) have established funding programs (i.e., National Institute of Health Small Business Research (SBIR), Coulter Foundation, Michigan—Pittsburgh—Wyss Interdisciplinary Translational Project, etc.). While these programs assist tremendously, the funds provided are insufficient to bring a product through to Phase III trials, resulting on reliance on private investors.
Although there are numerous obstacles associated with clinical translation, multiple local delivery strategies have successfully advanced to clinical trials or to the market (Table 8). Many of these strategies (i.e., Ozurdex®, Dextenza®, Zilretta®, etc.) have been engineered to provide local delivery of small molecules (i.e., dexamethasone, triamcinolone acetonide, etc.), as discussed in section 3.1. One unique product is Arestin®, which is intended for the treatment of periodontal disease. Periodontal disease begins as an initial inflammatory response to bacterial biofilms. In some cases, inflammation is never resolved, and chronic inflammation becomes the main pathology of the disease [263]. To treat this disease, Arestin®, composed of polymeric microparticles, has been engineered to locally release minocycline in the periodontal pocket. While minocycline is primarily thought of as an antibiotic, recent studies have demonstrated its’ anti-inflammatory effects in various models, including periodontitis [264].
Table 8.
Local delivery strategies which are FDA-approved, in clinical trials or are in preclinical development used to restore immune homeostasis.
| Cargo Class | Local Delivery System Name (Company/Institution), description |
Stage of Development (ClinicalTrials.gov Identifier) |
Disease Indication |
|---|---|---|---|
| Small molecule | |||
| Dexamethasone | Ozurdex® (Allergan); polymeric implant | FDA-approved | Diabetic macular edema; noninfectious uveitis (inflammation) |
| Dextenza® (Ocular Therapeutics); polymeric hydrogel | FDA-approved | Allergic conjunctivitis; postsurgical ocular inflammation | |
| OTX-DED (Ocular Therapeutics) | Phase II Trials (NCT04747977) | Dry Eye Disease | |
| Triamcinolone acetonide | Zilretta® (Flexion Therapeutics); polymeric microparticles | FDA-approved | Osteoarthritis |
| Mometasome furoate | Propel® (Intersect ENT); dissolvable stent | FDA-approved | Chronic sinusitis |
| Sinuva® (Intersect ENT); dissolvable stent | FDA-approved | Chronic sinusitis | |
| Diclofenac sodium | Voltaren® Gel (GSK); topical gel | FDA-approved | Inflammatory arthritis |
| Minocycline | Arestin® (OraPharma); polymeric microparticles | FDA-approved | Periodontitis |
| Protein | |||
| Recombinant human glutamic acid dehydrogenase (rhGAD65) | GAD-alum (Diamyd); aluminum hydrogel | Phase II Trials (NCT04262479) | Latent Autoimmune Diabetes |
| Interleukin-10 | Prevascar (Prevascar); polymeric hydrogel | FDA-approved | Wound healing |
| Antigen-specific therapeutics | |||
| Protein immunomodulators, disease-associated antigen | Inspira-01 (Inspira Therapeutics); dual-sized polymeric microspheres | Pre-clinical | Type 1 Diabetes |
| Other | |||
| Undisclosed small molecules, biologics, nucleic acids | Alivio™ (PureTech); polymeric hydrogels | Pre-clinical | Autoimmune diseases |
Aside from small molecule delivery systems, protein-based and antigen-specific treatment approaches have begun to emerge on the market. One example of an antigen-specific treatment is Inspira-01, a dual-sized biodegradable polymeric microparticle platform in preclinical development for the treatment of autoimmune diabetes. By releasing multiple immune modifiers alongside disease-associated antigen, Inspira-01 can promote a regulatory phenotype in antigen presenting cells to prevent and delay T1D.
5. Conclusions
Overall, significant progress has been made in the past few decades to develop new methods that restore immune homeostasis in the context of inflammation. While the use of standard pharmaceuticals to treat inflammation is unlikely to end in the immediate future, shifting to locally delivered, biologic-based (i.e., protein, cell, antigen, etc.) approaches could represent a way to capitalize on pre-existing biological signals that the immune system already uses to produce more targeted regulatory effects (both anatomically and mechanistically). This is especially the case for combinatorial factor delivery, during which multiple signals are presented in a local context to precisely direct the resulting regulatory response. Combinatorial delivery is particularly important when considering the plasticity of many immune cells (i.e., regulatory T cells, Th17 cells, dendritic cells, macrophages, etc.), where the presence or lack of one single biological cue can change drastically change the immunological outcome. Indeed, cells are naturally presented with several signals in their native environment, with each one contributing to the overall message (or instruction) communicated to that cell.
Local delivery of these immunomodulatory cues presents a unique opportunity to address the limitations of systemic treatments, including rapid clearance and off-target effects. Furthermore, since local delivery strategies can be administered in a specific site of interest (i.e., intra-articular space), these approaches can reduce the dose frequency and concentration required to achieve a therapeutic effect. As a result, local delivery strategies can reduce the whole-body distribution of immunosuppressant or anti-inflammatory agents. Although nanoparticulate carriers can be engineered to address several of these limitations, nano-based approaches still rely on trafficking to specific sites of interest and can result in accumulation in undesired anatomical sites. While nano-based systems will continue to advance and one day overcome these issues, local drug delivery strategies, such as those discussed throughout this review, already have the ability to overcome these limitations. Therefore, we anticipate that in the future, treatments which (1) mimic pre-existing biological cues to more precisely control the resulting immune response and (2) are administered locally, rather than systemically, will begin to emerge as front runners in the development of futuristic immunomodulatory strategies intended to treat inflammation and immune rejection for a vast array of diseases where there still is no clinical solution.
Highlights.
Microparticles, hydrogels and scaffolds as local delivery strategies.
Local delivery strategies offer advantages over systemic or bolus administration.
Shift from small molecule treatments to antigen-specific treatments to restore immune homeostasis in the context of inflammation.
5. Acknowledgements
E. Bentley is supported by the National Institutes of Health [grant number T32 AI 74490; NIH/NIAID].
All graphics were created with Biorender.com.
Abbreviations
- Ab
Antibody
- Ag
Antigen
- APC
Antigen presenting cell
- CCL1
C-C Motif Chemokine 1
- CCL22
C-C Motif Chemokine 22
- CD
Cluster of differentiation
- CMC
Critical micelle concentration
- CMT
Critical micelle temperature
- CNI
Calcineurin inhibitor
- COX
Cyclooxygenases
- CsA
Cyclosporin A
- CSF
Colony stimulating factor
- CXCL12
C-X-C Motif Chemokine 12
- DC
Dendritic cell
- DEX
Dexamethasone
- EAE
Experimental autoimmune encephalomyelitis
- ETN
Etanercept
- FasL
Fas ligand
- Foxp3
Forkhead Box P3
- GelMA
Gelatin methacryloyl
- GM-CSF
Granulocyte-macrophage colony-stimulating factor
- Ig
Immunoglobulin
- IL
Interleukin
- IL-1Ra
Interleukin-1 receptor antagonist
- IL-2
Interleukin-2
- IL-33
Interleukin-33
- IL-6
Interleukin-6
- iTreg
Induced regulatory T cell
- LCST
Lower critical solution temperature
- M1
Anti-inflammatory macrophage
- M2
Inflammatory macrophage
- mAb
Monoclonal antibody
- MHC
Major histocompatibility complex
- MMP
Matrix mellatoproteinase
- MOG
Oligodendrocyte glycoprotein
- MP
Microparticle
- MS
Multiple Sclerosis
- MSC
Mesenchymal stem cell
- MTX
Methotrexate
- NSAID
Non-steroidal anti-inflammatory agent
- nTreg
Natural regulatory T cell
- PBAE
Poly-β amino ester
- PCL
Polycaprolactone
- PDL
Poly-δ-decalactone
- PDMS
Polydimethylsiloxane
- PEG
Polyethylene glycol
- PEGMA
Polyethylene glycol methacrylate
- PEGNB
Polyethylene glycol norbornene
- PEO
Polyethylene oxide
- PLA
Polylactic acid
- PLG
Polyglycolic acid
- PLGA
Polylactic-co-glycolic acid
- PLP
Myelin proteolipid protein
- PNIPPAm
Poly-N-isopropylacrylamide
- PPG
Polyglycerol
- PPO
Polypropylene oxide
- RA
Rheumatoid arthritis
- Rapa
Rapamycin
- T1D
Type 1 Diabetes
- TA
Triamcinolone acetonide
- TAC
Tacrolimus
- TGFβ1
Transforming growth factor beta 1
- TGMS
Triglycerol monostearate
- TNFα
Tumor necrosis factor alpha
- TNFR
Tumor necrosis factor receptor
- tolDC
Tolerogenic dendritic cell
- Treg
Regulatory T cell
- VCA
Vascularized composite allotransplantation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- [1].Khan TA, Reddy ST, Immunological principles regulating immunomodulation with biomaterials, Acta Biomater. 10 (2014) 1720–1727. 10.1016/j.actbio.2013.12.011. [DOI] [PubMed] [Google Scholar]
- [2].Theofilopoulos AN, Kono DH, Baccala R, The multiple pathways to autoimmunity, Nat Immunol. 18 (2017) 716–724. 10.1038/ni.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rosenblum MD, Remedios KA, Abbas AK, Mechanisms of human autoimmunity, J Clin Invest. 125 (2015) 2228–2233. 10.1172/jci78088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kwiatkowski AJ, Stewart JM, Cho JJ, Avram D, Keselowsky BG, Nano and Microparticle Emerging Strategies for Treatment of Autoimmune Diseases: Multiple Sclerosis and Type 1 Diabetes, Adv Healthc Mater. 9 (2020) 2000164. 10.1002/adhm.202000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Benichou G, Thomson AW, Direct versus Indirect Allorecognition Pathways: On the Right Track, Am J Transplant. 9 (2009) 655–656. 10.1111/j.1600-6143.2009.02572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Moreau A, Varey E, Anegon I, Cuturi M-C, Effector Mechanisms of Rejection, Csh Perspect Med. 3 (2013) a015461. 10.1101/cshperspect.a015461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Uluer MC, Brazio PS, Woodall JD, Nam AJ, Bartlett ST, Barth RN, Vascularized Composite Allotransplantation: Medical Complications, Curr Transplant Reports. 3 (2016) 395–403. 10.1007/s40472-016-0113-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J, Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies, Bone Res. 6 (2018) 15. 10.1038/s41413-018-0016-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dandel M, Lehmkuhl HB, Knosalla C, Hetzer R, Impact of different long-term maintenance immunosuppressive therapy strategies on patients’ outcome after heart transplantation, Transpl Immunol. 23 (2010) 93–103. 10.1016/j.trim.2010.04.007. [DOI] [PubMed] [Google Scholar]
- [10].Black CK, Termanini KM, Aguirre O, Hawksworth JS, Sosin M, Solid organ transplantation in the 21 st century, Ann Transl Medicine. 6 (2018) 409–409. 10.21037/atm.2018.09.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Leventhal JR, Mathew JM, Outstanding questions in transplantation: Tolerance, Am J Transplant. 20 (2020) 348–354. 10.1111/ajt.15680. [DOI] [PubMed] [Google Scholar]
- [12].Chandrasekharan D, Issa F, Wood KJ, Achieving operational tolerance in transplantation: how can lessons from the clinic inform research directions?, Transplant Int. 26 (2013) 576–589. 10.1111/tri.12081. [DOI] [PubMed] [Google Scholar]
- [13].Fenton OS, Olafson KN, Pillai PS, Mitchell MJ, Langer R, Advances in Biomaterials for Drug Delivery, Adv Mater. 30 (2018) 1705328. 10.1002/adma.201705328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tibbitt MW, Dahlman JE, Langer R, Emerging Frontiers in Drug Delivery, J Am Chem Soc. 138 (2016) 704–717. 10.1021/jacs.5b09974. [DOI] [PubMed] [Google Scholar]
- [15].Jong WHD, Borm PJA, Drug delivery and nanoparticles: Applications and hazards, Int J Nanomed. Volume 3 (2008) 133–149. 10.2147/ijn.s596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].McNeil SE, Characterization of Nanoparticles Intended for Drug Delivery, Methods Mol Biology. 697 (2010) 3–8. 10.1007/978-1-60327-198-1_1. [DOI] [Google Scholar]
- [17].Park K, Facing the Truth about Nanotechnology in Drug Delivery, Acs Nano. 7 (2013) 7442–7447. 10.1021/nn404501g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mitchell MJ, Billingsley MM, Haley RM, Wechsler ME, Peppas NA, Langer R, Engineering precision nanoparticles for drug delivery, Nat Rev Drug Discov. 20 (2021) 101–124. 10.1038/s41573-020-0090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chenthamara D, Subramaniam S, Ramakrishnan SG, Krishnaswamy S, Essa MM, Lin F-H, Qoronfleh MW, Therapeutic efficacy of nanoparticles and routes of administration, Biomaterials Res. 23 (2019) 20. 10.1186/s40824-019-0166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bracho Sanchez E, Xia CQ, Clare-Salzler MJ, Keselowsky BG, Micro and Nano Material Carriers for Immunomodulation, Am J Transplant. 16 (2016) 3362–3370. 10.1111/ajt.13878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gammon JM, Jewell CM, Engineering Immune Tolerance with Biomaterials, Adv Healthc Mater. 8 (2019) 1801419. 10.1002/adhm.201801419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Calori IR, Braga G, da C.C. de Jesus P, Bi H, Tedesco AC, Polymer Scaffolds as Drug Delivery Systems, Eur Polym J. 129 (2020) 109621. 10.1016/j.eurpolymj.2020.109621. [DOI] [Google Scholar]
- [23].Balmert SC, Little SR, Biomimetic Delivery with Micro- and Nanoparticles, Adv Mater. 24 (2012) 3757–3778. 10.1002/adma.201200224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fisher JD, Acharya AP, Little SR, Micro and nanoparticle drug delivery systems for preventing allotransplant rejection, Clin Immunol. 160 (2015) 24–35. 10.1016/j.clim.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Danhier F, Ansorena E, Silva JM, Coco R, Breton AL, Préat V, PLGA-based nanoparticles: An overview of biomedical applications, J Control Release. 161 (2012) 505–522. 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- [26].Kohane DS, Microparticles and nanoparticles for drug delivery, Biotechnol Bioeng. 96 (2007) 203–209. 10.1002/bit.21301. [DOI] [PubMed] [Google Scholar]
- [27].Li J, Mooney DJ, Designing hydrogels for controlled drug delivery, Nat Rev Mater. 1 (2016) 16071. 10.1038/natrevmats.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gardner AB, Lee SKC, Woods EC, Acharya AP, Biomaterials-Based Modulation of the Immune System, Biomed Res Int. 2013 (2013) 1–7. 10.1155/2013/732182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang S, Xiong Y, Wang Y, Chen J, Yang J, Sun B, Evaluation of PLGA microspheres with triple regimen on long-term survival of vascularized composite allograft – an experimental study, Transplant Int. 33 (2020) 450–461. 10.1111/tri.13574. [DOI] [PubMed] [Google Scholar]
- [30].Fisher JD, Balmert SC, Zhang W, Schweizer R, Schnider JT, Komatsu C, Dong L, Erbas VE, Unadkat JV, Aral AM, Acharya AP, Kulahci Y, Turnquist HR, Thomson AW, Solari MG, Gorantla VS, Little SR, Treg-inducing microparticles promote donor-specific tolerance in experimental vascularized composite allotransplantation, Proc National Acad Sci. 116 (2019) 25784–25789. 10.1073/pnas.1910701116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dzhonova DV, Olariu R, Leckenby J, Banz Y, Prost J-C, Dhayani A, Vemula PK, Voegelin E, Taddeo A, Rieben R, Local Injections of Tacrolimus-loaded Hydrogel Reduce Systemic Immunosuppression-related Toxicity in Vascularized Composite Allotransplantation, Transplantation. 102 (2018) 1684–1694. 10.1097/tp.0000000000002283. [DOI] [PubMed] [Google Scholar]
- [32].Qi X, Qin X, Yang R, Qin J, Li W, Luan K, Wu Z, Song L, Intra-articular Administration of Chitosan Thermosensitive In Situ Hydrogels Combined With Diclofenac Sodium–Loaded Alginate Microspheres, J Pharm Sci. 105 (2016) 122–130. 10.1016/j.xphs.2015.11.019. [DOI] [PubMed] [Google Scholar]
- [33].Yin N, Guo X, Sun R, Liu H, Tang L, Gou J, Yin T, He H, Zhang Y, Tang X, Intra-articular injection of indomethacin–methotrexate in situ hydrogel for the synergistic treatment of rheumatoid arthritis, J Mater Chem B. 8 (2019) 993–1007. 10.1039/c9tb01795j. [DOI] [PubMed] [Google Scholar]
- [34].Seo J, Park SH, Kim MJ, Ju HJ, Yin XY, Min BH, Kim MS, Injectable Click-Crosslinked Hyaluronic Acid Depot To Prolong Therapeutic Activity in Articular Joints Affected by Rheumatoid Arthritis, Acs Appl Mater Inter. 11 (2019) 24984–24998. 10.1021/acsami.9b04979. [DOI] [PubMed] [Google Scholar]
- [35].Présumey J, Salzano G, Courties G, Shires M, Ponchel F, Jorgensen C, Apparailly F, Rosa GD, PLGA microspheres encapsulating siRNA anti-TNFalpha: Efficient RNAi-mediated treatment of arthritic joints, Eur J Pharm Biopharm. 82 (2012) 457–464. 10.1016/j.ejpb.2012.07.021. [DOI] [PubMed] [Google Scholar]
- [36].Kim KS, Park SJ, Yang J-A, Jeon J-H, Bhang SH, Kim B-S, Hahn SK, Injectable hyaluronic acid–tyramine hydrogels for the treatment of rheumatoid arthritis, Acta Biomater. 7 (2011) 666–674. 10.1016/j.actbio.2010.09.030. [DOI] [PubMed] [Google Scholar]
- [37].Wu H, Wang K, Wang H, Chen F, Huang W, Chen Y, Chen J, Tao J, Wen X, Xiong S, Novel self-assembled tacrolimus nanoparticles cross-linking thermosensitive hydrogels for local rheumatoid arthritis therapy, Colloids Surfaces B Biointerfaces. 149 (2017) 97–104. 10.1016/j.colsurfb.2016.10.013. [DOI] [PubMed] [Google Scholar]
- [38].Kuppan P, Kelly S, Polishevska K, Hojanepesov O, Seeberger K, Korbutt GS, Pepper AR, Co-localized immune protection using dexamethasone-eluting micelles in a murine islet allograft model, Am J Transplant. 20 (2020) 714–725. 10.1111/ajt.15662. [DOI] [PubMed] [Google Scholar]
- [39].Zhang W, Gorantla VS, Campbell PG, Li Y, Yang Y, Komatsu C, Weiss LE, Zheng XX, Solari MG, Biopatterned CTLA4/Fc Matrices Facilitate Local Immunomodulation, Engraftment, and Glucose Homeostasis After Pancreatic Islet Transplantation, Diabetes. 65 (2016) 3660–3666. 10.2337/db16-0320. [DOI] [PubMed] [Google Scholar]
- [40].Nguyen TT, Pham TT, Nguyen HT, Nepal MR, Phung CD, You Z, Katila N, Pun NT, Jeong TC, Choi D-Y, Park P-H, Yong CS, Kim JO, Yook S, Jeong J-H, Engineering “cell-particle hybrids” of pancreatic islets and bioadhesive FK506-loaded polymeric microspheres for local immunomodulation in xenogeneic islet transplantation, Biomaterials. 221 (2019) 119415. 10.1016/j.biomaterials.2019.119415. [DOI] [PubMed] [Google Scholar]
- [41].Verbeke CS, Gordo S, Schubert DA, Lewin SA, Desai RM, Dobbins J, Wucherpfennig KW, Mooney DJ, Multicomponent Injectable Hydrogels for Antigen-Specific Tolerogenic Immune Modulation, Adv Healthc Mater. 6 (2017) 1600773. 10.1002/adhm.201600773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Thomas AM, Beskid NM, Blanchfield JL, Rosado AM, García AJ, Evavold BD, Babensee JE, Localized hydrogel delivery of dendritic cells for attenuation of multiple sclerosis in a murine model, J Biomed Mater Res A. (2020). 10.1002/jbm.a.37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tostanoski LH, Chiu Y-C, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell CM, Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific, Cell Reports. 16 (2016) 2940–2952. 10.1016/j.celrep.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fisher JD, Zhang W, Balmert SC, Aral AM, Acharya AP, Kulahci Y, Li J, Turnquist HR, Thomson AW, Solari MG, Gorantla VS, Little SR, In situ recruitment of regulatory T cells promotes donor-specific tolerance in vascularized composite allotransplantation, Sci Adv. 6 (2020) eaax8429. 10.1126/sciadv.aax8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Garg NK, Singh B, Tyagi RK, Sharma G, Katare OP, Effective transdermal delivery of methotrexate through nanostructured lipid carriers in an experimentally induced arthritis model, Colloids Surfaces B Biointerfaces. 147 (2016) 17–24. 10.1016/j.colsurfb.2016.07.046. [DOI] [PubMed] [Google Scholar]
- [46].Erdemli Ö, Özen S, Keskin D, Usanmaz A, Batu ED, Atilla B, Tezcaner A, In vitro evaluation of effects of sustained anti-TNF release from MPEG-PCL-MPEG and PCL microspheres on human rheumatoid arthritis synoviocytes, J Biomater Appl. 29 (2014) 524–542. 10.1177/0885328214535958. [DOI] [PubMed] [Google Scholar]
- [47].Getts DR, Shea LD, Miller SD, King NJC, Harnessing nanoparticles for immune modulation, Trends Immunol. 36 (2015) 419–427. 10.1016/j.it.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kumar L, Verma S, Singh M, Chalotra T, Utreja P, Advanced Drug Delivery Systems for Transdermal Delivery of Non-Steroidal Anti-Inflammatory Drugs: A Review, Curr Drug Deliv. 15 (2018) 1087–1099. 10.2174/1567201815666180605114131. [DOI] [PubMed] [Google Scholar]
- [49].Brusini R, Varna M, Couvreur P, Advanced nanomedicines for the treatment of inflammatory diseases, Adv Drug Deliver Rev. 157 (2020) 161–178. 10.1016/j.addr.2020.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Jin K, Luo Z, Zhang B, Pang Z, Biomimetic nanoparticles for inflammation targeting, Acta Pharm Sinica B. 8 (2018) 23–33. 10.1016/j.apsb.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Han FY, Thurecht KJ, Whittaker AK, Smith MT, Bioerodable PLGA-Based Microparticles for Producing Sustained-Release Drug Formulations and Strategies for Improving Drug Loading, Front Pharmacol. 7 (2016) 185. 10.3389/fphar.2016.00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].O’Donnell PB, McGinity JW, Preparation of microspheres by the solvent evaporation technique, Adv Drug Deliver Rev. 28 (1997) 25–42. 10.1016/s0169-409x(97)00049-5. [DOI] [PubMed] [Google Scholar]
- [53].Dhandayuthapani B, Yoshida Y, Maekawa T, Kumar DS, Polymeric Scaffolds in Tissue Engineering Application: A Review, Int J Polym Sci. 2011 (2011) 1–19. 10.1155/2011/290602. [DOI] [Google Scholar]
- [54].Hoare TR, Kohane DS, Hydrogels in drug delivery: Progress and challenges, Polymer. 49 (2008) 1993–2007. 10.1016/j.polymer.2008.01.027. [DOI] [Google Scholar]
- [55].Ahmed EM, Hydrogel: Preparation, characterization, and applications: A review, J Adv Res. 6 (2015) 105–121. 10.1016/j.jare.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Rothstein SN, Federspiel WJ, Little SR, A unified mathematical model for the prediction of controlled release from surface and bulk eroding polymer matrices, Biomaterials. 30 (2009) 1657–1664. 10.1016/j.biomaterials.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sionkowska A, Current research on the blends of natural and synthetic polymers as new biomaterials: Review, Prog Polym Sci. 36 (2011) 1254–1276. 10.1016/j.progpolymsci.2011.05.003. [DOI] [Google Scholar]
- [58].Mogoşanu GD, Grumezescu AM, Natural and synthetic polymers for wounds and burns dressing, Int J Pharmaceut. 463 (2014) 127–136. 10.1016/j.ijpharm.2013.12.015. [DOI] [PubMed] [Google Scholar]
- [59].Englert C, Brendel JC, Majdanski TC, Yildirim T, Schubert S, Gottschaldt M, Windhab N, Schubert US, Pharmapolymers in the 21st Century: Synthetic Polymers in Drug Delivery Applications, Prog Polym Sci. 87 (2018) 107–164. 10.1016/j.progpolymsci.2018.07.005. [DOI] [Google Scholar]
- [60].Liechty WB, Kryscio DR, Slaughter BV, Peppas NA, Polymers for Drug Delivery Systems, Annu Rev Chem Biomol. 1 (2010) 149–173. 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pillai O, Panchagnula R, Polymers in drug delivery, Curr Opin Chem Biol. 5 (2001) 447–451. 10.1016/s1367-5931(00)00227-1. [DOI] [PubMed] [Google Scholar]
- [62].Skwarczynski M, Zhao G, Boer JC, Ozberk V, Azuar A, Cruz JG, Giddam AK, Khalil ZG, Pandey M, Shibu MA, Hussein WM, Nevagi RJ, Batzloff MR, Wells JW, Capon RJ, Plebanski M, Good MF, Toth I, Poly(amino acids) as a potent self-adjuvanting delivery system for peptide-based nanovaccines, Sci Adv. 6 (2020) eaax2285. 10.1126/sciadv.aax2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Shakya AK, Kumar A, Nandakumar KS, Adjuvant properties of a biocompatible thermo-responsive polymer of N-isopropylacrylamide in autoimmunity and arthritis, J Roy Soc Interface. 8 (2011) 1748–1759. 10.1098/rsif.2011.0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Woodruff MA, Hutmacher DW, The return of a forgotten polymer—Polycaprolactone in the 21st century, Prog Polym Sci. 35 (2010) 1217–1256. 10.1016/j.progpolymsci.2010.04.002. [DOI] [Google Scholar]
- [65].Little SR, Lynn DM, Ge Q, Anderson DG, Puram SV, Chen J, Eisen HN, Langer R, Poly-β amino ester-containing microparticles enhance the activity of nonviral genetic vaccines, P Natl Acad Sci Usa. 101 (2004) 9534–9539. 10.1073/pnas.0403549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Allen RP, Bolandparvaz A, Ma JA, Manickam VA, Lewis JS, Latent, Immunosuppressive Nature of Poly(lactic-co-glycolic acid) Microparticles, Acs Biomater Sci Eng. 4 (2018) 900–918. 10.1021/acsbiomaterials.7b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Giraldo JA, Molano RD, Rengifo HR, Fotino C, Gattás-Asfura KM, Pileggi A, Stabler CL, The impact of cell surface PEGylation and short-course immunotherapy on islet graft survival in an allogeneic murine model, Acta Biomater. 49 (2017) 272–283. 10.1016/j.actbio.2016.11.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Peppas NA, Keys KB, Torres-Lugo M, Lowman AM, Poly(ethylene glycol)-containing hydrogels in drug delivery, J Control Release. 62 (1999) 81–87. 10.1016/s0168-3659(99)00027-9. [DOI] [PubMed] [Google Scholar]
- [69].Sellaturay P, Nasser S, Ewan P, Polyethylene Glycol–Induced Systemic Allergic Reactions (Anaphylaxis), J Allergy Clin Immunol Pract. 9 (2021) 670–675. 10.1016/j.jaip.2020.09.029. [DOI] [PubMed] [Google Scholar]
- [70].Abou-ElNour M, Soliman ME, Skouras A, Casettari L, Geneidi AS, Ishak RAH, Microparticles-in-Thermoresponsive/Bioadhesive Hydrogels as a Novel Integrated Platform for Effective Intra-articular Delivery of Triamcinolone Acetonide, Mol Pharmaceut. 17 (2020) 1963–1978. 10.1021/acs.molpharmaceut.0c00126. [DOI] [PubMed] [Google Scholar]
- [71].Roballo KCS, Dhungana S, Jiang Z, Oakey J, Bushman JS, Localized delivery of immunosuppressive regulatory T cells to peripheral nerve allografts promotes regeneration of branched segmental defects, Biomaterials. 209 (2019) 1–9. 10.1016/j.biomaterials.2019.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lin C-C, Anseth KS, PEG Hydrogels for the Controlled Release of Biomolecules in Regenerative Medicine, Pharmaceut Res. 26 (2009) 631–643. 10.1007/s11095-008-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lin C, Ki CS, Shih H, Thiol–norbornene photoclick hydrogels for tissue engineering applications, J Appl Polym Sci. 132 (2015) n/a–n/a. 10.1002/app.41563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kim Y-J, Matsunaga YT, Thermo-responsive polymers and their application as smart biomaterials, J Mater Chem B. 5 (2017) 4307–4321. 10.1039/c7tb00157f. [DOI] [PubMed] [Google Scholar]
- [75].Lin H-C, Anggelia MR, Cheng C-C, Ku K-L, Cheng H-Y, Wen C-J, Wang AYL, Lin C-H, Chu I-M, A Mixed Thermosensitive Hydrogel System for Sustained Delivery of Tacrolimus for Immunosuppressive Therapy, Pharm. 11 (2019) 413. 10.3390/pharmaceutics11080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Hou H-Y, Fu S-H, Liu C-H, Chen J-P, Hsu BR-S, The graft survival protection of subcutaneous allogeneic islets with hydrogel grafting and encapsulated by CTLA4Ig and IL1ra, Polym J. 46 (2014) 136–144. 10.1038/pj.2013.71. [DOI] [Google Scholar]
- [77].Badeau BA, DeForest CA, Programming Stimuli-Responsive Behavior into Biomaterials, Annu Rev Biomed Eng. 21 (2019) 1–25. 10.1146/annurev-bioeng-060418-052324. [DOI] [PubMed] [Google Scholar]
- [78].Roy D, Brooks WLA, Sumerlin BS, New directions in thermoresponsive polymers, Chem Soc Rev. 42 (2013) 7214–7243. 10.1039/c3cs35499g. [DOI] [PubMed] [Google Scholar]
- [79].Schilling AL, Kulahci Y, Moore J, Wang EW, Lee SE, Little SR, A thermoresponsive hydrogel system for long-acting corticosteroid delivery into the paranasal sinuses, J Control Release. 330 (2021) 889–897. 10.1016/j.jconrel.2020.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Bodratti AM, Alexandridis P, Formulation of Poloxamers for Drug Delivery, J Funct Biomaterials. 9 (2018) 11. 10.3390/jfb9010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Akash MSH, Rehman K, Li N, Gao J-Q, Sun H, Chen S, Sustained Delivery of IL-1Ra from Pluronic F127-Based Thermosensitive Gel Prolongs its Therapeutic Potentials, Pharmaceut Res. 29 (2012) 3475–3485. 10.1007/s11095-012-0843-0. [DOI] [PubMed] [Google Scholar]
- [82].Almoshari Y, Ren R, Zhang H, Jia Z, Wei X, Chen N, Li G, Ryu S, Lele SM, Reinhardt RA, Wang D, GSK3 inhibitor-loaded osteotropic Pluronic hydrogel effectively mitigates periodontal tissue damage associated with experimental periodontitis, Biomaterials. 261 (2020) 120293. 10.1016/j.biomaterials.2020.120293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Bae Y, Kataoka K, Intelligent polymeric micelles from functional poly(ethylene glycol)-poly(amino acid) block copolymers, Adv Drug Deliver Rev. 61 (2009) 768–784. 10.1016/j.addr.2009.04.016. [DOI] [PubMed] [Google Scholar]
- [84].Kojarunchitt T, Hook S, Rizwan S, Rades T, Baldursdottir S, Development and characterisation of modified poloxamer 407 thermoresponsive depot systems containing cubosomes, Int J Pharmaceut. 408 (2011) 20–26. 10.1016/j.ijpharm.2011.01.037. [DOI] [PubMed] [Google Scholar]
- [85].Yazdi MK, Taghizadeh A, Taghizadeh M, Stadler FJ, Farokhi M, Mottaghitalab F, Zarrintaj P, Ramsey JD, Seidi F, Saeb MR, Mozafari M, Agarose-based biomaterials for advanced drug delivery, J Control Release. 326 (2020) 523–543. 10.1016/j.jconrel.2020.07.028. [DOI] [PubMed] [Google Scholar]
- [86].Bernkop-Schnürch A, Dünnhaupt S, Chitosan-based drug delivery systems, Eur J Pharm Biopharm. 81 (2012) 463–469. 10.1016/j.ejpb.2012.04.007. [DOI] [PubMed] [Google Scholar]
- [87].Miao T, Wang J, Zeng Y, Liu G, Chen X, Polysaccharide-Based Controlled Release Systems for Therapeutics Delivery and Tissue Engineering: From Bench to Bedside, Adv Sci. 5 (2018) 1700513. 10.1002/advs.201700513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Tong X, Pan W, Su T, Zhang M, Dong W, Qi X, Recent advances in natural polymer-based drug delivery systems, React Funct Polym. 148 (2020) 104501. 10.1016/j.reactfunctpolym.2020.104501. [DOI] [Google Scholar]
- [89].Espona-Noguera A, Ciriza J, Cañibano-Hernández A, Fernandez L, Ochoa I, del Burgo LS, Pedraz JL, Tunable injectable alginate-based hydrogel for cell therapy in Type 1 Diabetes Mellitus, Int J Biol Macromol. 107 (2018) 1261–1269. 10.1016/j.ijbiomac.2017.09.103. [DOI] [PubMed] [Google Scholar]
- [90].Kim J, Hope CM, Gantumur N, Perkins GB, Stead SO, Yue Z, Liu X, Asua AU, Kette FD, Penko D, Drogemuller CJ, Carroll RP, Barry SC, Wallace GG, Coates PT, Encapsulation of Human Natural and Induced Regulatory T-Cells in IL 2 and CCL1 Supplemented Alginate-GelMA Hydrogel for 3D Bioprinting, Adv Funct Mater. 30 (2020) 2000544. 10.1002/adfm.202000544. [DOI] [Google Scholar]
- [91].Weaver JD, Headen DM, Coronel MM, Hunckler MD, Shirwan H, García AJ, Synthetic poly(ethylene glycol)-based microfluidic islet encapsulation reduces graft volume for delivery to highly vascularized and retrievable transplant site, Am J Transplant. 19 (2019) 1315–1327. 10.1111/ajt.15168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Hillberg AL, Oudshoorn M, Lam JBB, Kathirgamanathan K, Encapsulation of porcine pancreatic islets within an immunoprotective capsule comprising methacrylated glycol chitosan and alginate, J Biomed Mater Res Part B Appl Biomaterials. 103 (2015) 503–518. 10.1002/jbm.b.33185. [DOI] [PubMed] [Google Scholar]
- [93].Ma M, Chiu A, Sahay G, Doloff JC, Dholakia N, Thakrar R, Cohen J, Vegas A, Chen D, Bratlie KM, Dang T, York RL, Hollister-Lock J, Weir GC, Anderson DG, Core–Shell Hydrogel Microcapsules for Improved Islets Encapsulation, Adv Healthc Mater. 2 (2013) 667–672. 10.1002/adhm.201200341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zhang J, Zhu Y, Song J, Xu T, Yang J, Du Y, Zhang L, Rapid and Long-Term Glycemic Regulation with a Balanced Charged Immune-Evasive Hydrogel in T1DM Mice, Adv Funct Mater. 29 (2019) 1900140. 10.1002/adfm.201900140. [DOI] [Google Scholar]
- [95].Cari L, Montanucci P, Basta G, Petrillo MG, Ricci E, Pescara T, Greco A, Cipriani S, Shimizu J, Migliorati G, Nocentini G, Calafiore R, Riccardi C, Microencapsulated G3C Hybridoma Cell Graft Delays the Onset of Spontaneous Diabetes in NOD Mice by an Expansion of Gitr + Treg Cells, Diabetes. 69 (2020) 965–980. 10.2337/db19-0087. [DOI] [PubMed] [Google Scholar]
- [96].Bollyky PL, Vernon RB, Falk BA, Preisinger A, Gooden MD, Nepom GT, Gebe JA, IL-10 Induction from Implants Delivering Pancreatic Islets and Hyaluronan, J Diabetes Res. 2013 (2013) 1–9. 10.1155/2013/342479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Iansante V, Dhawan A, Masmoudi F, Lee CA, Fernandez-Dacosta R, Walker S, Fitzpatrick E, Mitry RR, Filippi C, A New High Throughput Screening Platform for Cell Encapsulation in Alginate Hydrogel Shows Improved Hepatocyte Functions by Mesenchymal Stromal Cells Co-encapsulation, Frontiers Medicine. 5 (2018) 216. 10.3389/fmed.2018.00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Chen T, Yuan J, Duncanson S, Hibert ML, Kodish BC, Mylavaganam G, Maker M, Li H, Sremac M, Santosuosso M, Forbes B, Kashiwagi S, Cao J, Lei J, Thomas M, Hartono C, Sachs D, Markmann J, Sambanis A, Poznansky MC, Alginate Encapsulant Incorporating CXCL12 Supports Long‐Term Allo- and Xenoislet Transplantation Without Systemic Immune Suppression, Am J Transplant. 15 (2015) 618–627. 10.1111/ajt.13049. [DOI] [PubMed] [Google Scholar]
- [99].Orr S, Strominger I, Eremenko E, Vinogradov E, Ruvinov E, Monsonego A, Cohen S, TGF-β affinity-bound to a macroporous alginate scaffold generates local and peripheral immunotolerant responses and improves allocell transplantation, Acta Biomater. 45 (2016) 196–209. 10.1016/j.actbio.2016.08.015. [DOI] [PubMed] [Google Scholar]
- [100].Juárez GAP, Spasojevic M, Faas MM, de Vos P, Immunological and Technical Considerations in Application of Alginate-Based Microencapsulation Systems, Frontiers Bioeng Biotechnology. 2 (2014) 26. 10.3389/fbioe.2014.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Hariyadi DM, Islam N, Current Status of Alginate in Drug Delivery, Adv Pharmacol Pharm Sci. 2020 (2020) 1–16. 10.1155/2020/8886095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Komi DEA, Sharma L, Cruz CSD, Chitin and Its Effects on Inflammatory and Immune Responses, Clin Rev Allerg Immu. 54 (2018) 213–223. 10.1007/s12016-017-8600-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Chang S-H, Lin Y-Y, Wu G-J, Huang C-H, Tsai GJ, Effect of chitosan molecular weight on anti-inflammatory activity in the RAW 264.7 macrophage model, Int J Biol Macromol. 131 (2019) 167–175. 10.1016/j.ijbiomac.2019.02.066. [DOI] [PubMed] [Google Scholar]
- [104].Harrington S, Williams J, Rawal S, Ramachandran K, Stehno-Bittel L, Hyaluronic Acid/Collagen Hydrogel as an Alternative to Alginate for Long-Term Immunoprotected Islet Transplantation*, Tissue Eng Pt A. 23 (2017) 1088–1099. 10.1089/ten.tea.2016.0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lee-Sayer SSM, Dong Y, Arif AA, Olsson M, Brown KL, Johnson P, The Where, When, How, and Why of Hyaluronan Binding by Immune Cells, Front Immunol. 6 (2015) 150. 10.3389/fimmu.2015.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Litwiniuk M, Krejner A, Speyrer MS, Gauto AR, Grzela T, Hyaluronic Acid in Inflammation and Tissue Regeneration., Wounds Compend Clin Res Pract. 28 (2016) 78–88. [PubMed] [Google Scholar]
- [107].Park J, Gerber MH, Babensee JE, Phenotype and polarization of autologous T cells by biomaterial-treated dendritic cells, J Biomed Mater Res A. 103 (2015) 170–184. 10.1002/jbm.a.35150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Huang Z, Zhang Z, Zha Y, Liu J, Jiang Y, Yang Y, Shao J, Sun X, Cai X, Yin Y, Chen J, Dong L, Zhang J, The effect of targeted delivery of anti-TNF-α oligonucleotide into CD169+ macrophages on disease progression in lupus-prone MRL/lpr mice, Biomaterials. 33 (2012) 7605–7612. 10.1016/j.biomaterials.2012.06.074. [DOI] [PubMed] [Google Scholar]
- [109].Alvarado-Velez M, Enam SF, Mehta N, Lyon JG, LaPlaca MC, Bellamkonda RV, Immunosuppressive hydrogels enhance allogeneic MSC survival after transplantation in the injured brain, Biomaterials. 266 (2021) 120419. 10.1016/j.biomaterials.2020.120419. [DOI] [PubMed] [Google Scholar]
- [110].Srinivasan S, Babensee JE, Controlled Delivery of Immunomodulators from a Biomaterial Scaffold Niche to Induce a Tolerogenic Phenotype in Human Dendritic Cells, Acs Biomater Sci Eng. 6 (2020) 4062–4076. 10.1021/acsbiomaterials.0c00439. [DOI] [PubMed] [Google Scholar]
- [111].Davis NE, Beenken-Rothkopf LN, Mirsoian A, Kojic N, Kaplan DL, Barron AE, Fontaine MJ, Enhanced function of pancreatic islets co-encapsulated with ECM proteins and mesenchymal stromal cells in a silk hydrogel, Biomaterials. 33 (2012) 6691–6697. 10.1016/j.biomaterials.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Griffin JD, Song JY, Huang A, Sedlacek AR, Flannagan KL, Berkland CJ, Antigen-specific immune decoys intercept and exhaust autoimmunity to prevent disease, Biomaterials. 222 (2019) 119440. 10.1016/j.biomaterials.2019.119440. [DOI] [PubMed] [Google Scholar]
- [113].Hamilton DC, Shih HH, Schubert RA, Michie SA, Staats PN, Kaplan DL, Fontaine MJ, A silk-based encapsulation platform for pancreatic islet transplantation improves islet function in vivo, J Tissue Eng Regen M. 11 (2017) 887–895. 10.1002/term.1990. [DOI] [PubMed] [Google Scholar]
- [114].Kumar M, Gupta P, Bhattacharjee S, Nandi SK, Mandal BB, Immunomodulatory injectable silk hydrogels maintaining functional islets and promoting anti-inflammatory M2 macrophage polarization, Biomaterials. 187 (2018) 1–17. 10.1016/j.biomaterials.2018.09.037. [DOI] [PubMed] [Google Scholar]
- [115].Foox M, Zilberman M, Drug delivery from gelatin-based systems, Expert Opin Drug Del. 12 (2015) 1547–1563. 10.1517/17425247.2015.1037272. [DOI] [PubMed] [Google Scholar]
- [116].Okunishi K, Dohi M, Fujio K, Nakagome K, Tabata Y, Okasora T, Seki M, Shibuya M, Imamura M, Harada H, Tanaka R, Yamamoto K, Hepatocyte Growth Factor Significantly Suppresses Collagen-Induced Arthritis in Mice, J Immunol. 179 (2007) 5504–5513. 10.4049/jimmunol.179.8.5504. [DOI] [PubMed] [Google Scholar]
- [117].Yue K, Santiago GT, Alvarez MM, Tamayol A, Annabi N, Khademhosseini A, Synthesis, properties, and biomedical applications of gelatin methacryloyl (GelMA) hydrogels, Biomaterials. 73 (2015) 254–271. 10.1016/j.biomaterials.2015.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Uehara M, Li X, Sheikhi A, Zandi N, Walker B, Saleh B, Banouni N, Jiang L, Ordikhani F, Dai L, Yonar M, Vohra I, Kasinath V, Orgill DP, Khademhosseini A, Annabi N, Abdi R, Anti-IL-6 eluting immunomodulatory biomaterials prolong skin allograft survival, Sci Rep-Uk. 9 (2019) 6535. 10.1038/s41598-019-42349-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Andorko JI, Jewell CM, Designing biomaterials with immunomodulatory properties for tissue engineering and regenerative medicine, Bioeng Amp Transl Medicine. 2 (2017) 139–155. 10.1002/btm2.10063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Lewis JS, Roy K, Keselowsky BG, Materials that harness and modulate the immune system, Mrs Bull. 39 (2014) 25–34. 10.1557/mrs.2013.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Hubbell JA, Thomas SN, Swartz MA, Materials engineering for immunomodulation, Nature. 462 (2009) 449–460. 10.1038/nature08604. [DOI] [PubMed] [Google Scholar]
- [122].Jiskoot W, van Schie RMF, Carstens MG, Schellekens H, Immunological Risk of Injectable Drug Delivery Systems, Pharmaceut Res. 26 (2009) 1303–1314. 10.1007/s11095-009-9855-9. [DOI] [PubMed] [Google Scholar]
- [123].Jarvi NL, Balu-Iyer SV, Immunogenicity Challenges Associated with Subcutaneous Delivery of Therapeutic Proteins, Biodrugs. 35 (2021) 125–146. 10.1007/s40259-020-00465-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Evans CH, Kraus VB, Setton LA, Progress in intra-articular therapy, Nat Rev Rheumatol. 10 (2014) 11–22. 10.1038/nrrheum.2013.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Crofford LJ, Use of NSAIDs in treating patients with arthritis, Arthritis Res Ther. 15 (2013) S2. 10.1186/ar4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Ricciotti E, FitzGerald GA, Prostaglandins and Inflammation, Arteriosclerosis Thrombosis Vasc Biology. 31 (2011) 986–1000. 10.1161/atvbaha.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Cannavà C, Tommasini S, Stancanelli R, Cardile V, Cilurzo F, Giannone I, Puglisi G, Ventura CA, Celecoxib-loaded PLGA/cyclodextrin microspheres: Characterization and evaluation of anti-inflammatory activity on human chondrocyte cultures, Colloids Surfaces B Biointerfaces. 111 (2013) 289–296. 10.1016/j.colsurfb.2013.06.015. [DOI] [PubMed] [Google Scholar]
- [128].Hao Y, Tian R, Lv K, Liu Z, Ni J, Yuan P, Bai Y, Chen X, Stimuli responsive co-delivery of celecoxib and BMP2 from micro-scaffold for periodontal disease treatment, J Mater Sci Technol. 75 (2021) 216–224. 10.1016/j.jmst.2020.10.027. [DOI] [Google Scholar]
- [129].Gong L, Thorn CF, Bertagnolli MM, Grosser T, Altman RB, Klein TE, Celecoxib pathways, Pharmacogenet Genom. 22 (2012) 310–318. 10.1097/fpc.0b013e32834f94cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Cain DW, Cidlowski JA, Immune regulation by glucocorticoids, Nat Rev Immunol. 17 (2017) 233–247. 10.1038/nri.2017.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Ramamoorthy S, Cidlowski JA, Corticosteroids Mechanisms of Action in Health and Disease, Rheum Dis Clin N Am. 42 (2016) 15–31. 10.1016/j.rdc.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Jiang K, Weaver JD, Li Y, Chen X, Liang J, Stabler CL, Local release of dexamethasone from macroporous scaffolds accelerates islet transplant engraftment by promotion of anti-inflammatory M2 macrophages, Biomaterials. 114 (2017) 71–81. 10.1016/j.biomaterials.2016.11.004. [DOI] [PubMed] [Google Scholar]
- [133].Joshi N, Yan J, Levy S, Bhagchandani S, Slaughter KV, Sherman NE, Amirault J, Wang Y, Riegel L, He X, Rui TS, Valic M, Vemula PK, Miranda OR, Levy O, Gravallese EM, Aliprantis AO, Ermann J, Karp JM, Towards an arthritis flare-responsive drug delivery system, Nat Commun. 9 (2018) 1275. 10.1038/s41467-018-03691-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Zhang S, Ermann J, Succi MD, Zhou A, Hamilton MJ, Cao B, Korzenik JR, Glickman JN, Vemula PK, Glimcher LH, Traverso G, Langer R, Karp JM, An inflammation-targeting hydrogel for local drug delivery in inflammatory bowel disease, Sci Transl Med. 7 (2015) 300ra128–300ra128. 10.1126/scitranslmed.aaa5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Khaled KA, Sarhan HA, Ibrahim MA, Ali AH, Naguib YW, Prednisolone-Loaded PLGA Microspheres. In Vitro Characterization and In Vivo Application in Adjuvant-Induced Arthritis in Mice, Aaps Pharmscitech. 11 (2010) 859–869. 10.1208/s12249-010-9445-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Oliveira IM, Gonçalves C, Shin ME, Lee S, Reis RL, Khang G, Oliveira JM, Enzymatically crosslinked tyramine-gellan gum hydrogels as drug delivery system for rheumatoid arthritis treatment, Drug Deliv Transl Re. (2020) 1–13. 10.1007/s13346-020-00855-9. [DOI] [PubMed] [Google Scholar]
- [137].Rudnik-Jansen I, Schrijver K, Woike N, Tellegen A, Versteeg S, Emans P, Mihov G, Thies J, Eijkelkamp N, Tryfonidou M, Creemers L, Intra-articular injection of triamcinolone acetonide releasing biomaterial microspheres inhibits pain and inflammation in an acute arthritis model, Drug Deliv. 26 (2019) 226–236. 10.1080/10717544.2019.1568625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Abou-ElNour M, Ishak RAH, Tiboni M, Bonacucina G, Cespi M, Casettari L, Soliman ME, Geneidi AS, Triamcinolone acetonide-loaded PLA/PEG-PDL microparticles for effective intra-articular delivery: synthesis, optimization, in vitro and in vivo evaluation, J Control Release. 309 (2019) 125–144. 10.1016/j.jconrel.2019.07.030. [DOI] [PubMed] [Google Scholar]
- [139].Giles AJ, Hutchinson M-KND, Sonnemann HM, Jung J, Fecci PE, Ratnam NM, Zhang W, Song H, Bailey R, Davis D, Reid CM, Park DM, Gilbert MR, Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy, J Immunother Cancer. 6 (2018) 51. 10.1186/s40425-018-0371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Wofford KL, Cullen DK, Spiller KL, Modulation of macrophage phenotype via phagocytosis of drug-loaded microparticles, J Biomed Mater Res A. 107 (2019) 1213–1224. 10.1002/jbm.a.36617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].FRANCHIMONT D, Overview of the Actions of Glucocorticoids on the Immune Response: A Good Model to Characterize New Pathways of Immunosuppression for New Treatment Strategies, Ann Ny Acad Sci. 1024 (2004) 124–137. 10.1196/annals.1321.009. [DOI] [PubMed] [Google Scholar]
- [142].Gajanayake T, Olariu R, Leclère FM, Dhayani A, Yang Z, Bongoni AK, Banz Y, Constantinescu MA, Karp JM, Vemula PK, Rieben R, Vögelin E, A single localized dose of enzyme-responsive hydrogel improves long-term survival of a vascularized composite allograft, Sci Transl Med. 6 (2014) 249ra110–249ra110. 10.1126/scitranslmed.3008778. [DOI] [PubMed] [Google Scholar]
- [143].Fries CA, Lawson SD, Wang LC, Slaughter KV, Vemula PK, Dhayani A, Joshi N, Karp JM, Rickard RF, Gorantla VS, Davis MR, Graft-implanted, enzyme responsive, tacrolimus-eluting hydrogel enables long-term survival of orthotopic porcine limb vascularized composite allografts: A proof of concept study, Plos One. 14 (2019) e0210914. 10.1371/journal.pone.0210914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Dzhonova D, Olariu R, Leckenby J, Dhayani A, Vemula PK, Prost J-C, Banz Y, Taddeo A, Rieben R, Local release of tacrolimus from hydrogel-based drug delivery system is controlled by inflammatory enzymes in vivo and can be monitored non-invasively using in vivo imaging, Plos One. 13 (2018) e0203409. 10.1371/journal.pone.0203409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Tibbitt MW, Rodell CB, Burdick JA, Anseth KS, Progress in material design for biomedical applications, Proc National Acad Sci. 112 (2015) 14444–14451. 10.1073/pnas.1516247112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Barnes PJ, Mechanisms and resistance in glucocorticoid control of inflammation, J Steroid Biochem Mol Biology. 120 (2010) 76–85. 10.1016/j.jsbmb.2010.02.018. [DOI] [PubMed] [Google Scholar]
- [147].Gaber T, Strehl C, Buttgereit F, Metabolic regulation of inflammation, Nat Rev Rheumatol. 13 (2017) 267–279. 10.1038/nrrheum.2017.37. [DOI] [PubMed] [Google Scholar]
- [148].Pålsson-McDermott EM, O’Neill LAJ, Targeting immunometabolism as an anti-inflammatory strategy, Cell Res. 30 (2020) 300–314. 10.1038/s41422-020-0291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Dhanka M, Pawar V, Chauhan DS, Jain NK, R.S. P, Shetty C, Kumawat MK, Prasad R, Srivastava R, Synthesis and characterization of an injectable microparticles integrated hydrogel composite biomaterial: In-vivo biocompatibility and inflammatory arthritis treatment, Colloids Surfaces B Biointerfaces. 201 (2021) 111597. 10.1016/j.colsurfb.2021.111597. [DOI] [PubMed] [Google Scholar]
- [150].Yin N, Tan X, Liu H, He F, Ding N, Gou J, Yin T, He H, Zhang Y, Tang X, A novel indomethacin/methotrexate/MMP-9 siRNA in situ hydrogel with dual effects of anti-inflammatory activity and reversal of cartilage disruption for the synergistic treatment of rheumatoid arthritis, Nanoscale. 12 (2020) 8546–8562. 10.1039/d0nr00454e. [DOI] [PubMed] [Google Scholar]
- [151].Zou Y, Zeng S, Huang M, Qiu Q, Xiao Y, Shi M, Zhan Z, Liang L, Yang X, Xu H, Inhibition of 6-phosphofructo-2-kinase suppresses fibroblast-like synoviocytes-mediated synovial inflammation and joint destruction in rheumatoid arthritis, Brit J Pharmacol. 174 (2017) 893–908. 10.1111/bph.13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Mangal JL, Inamdar S, Le T, Shi X, Curtis M, Gu H, Acharya AP, Inhibition of glycolysis in the presence of antigen generates suppressive antigen-specific responses and restrains rheumatoid arthritis in mice, Biomaterials. (2021) 121079. 10.1016/j.biomaterials.2021.121079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Zhu W, Ye L, Zhang J, Yu P, Wang H, Ye Z, Tian J, PFK15, a Small Molecule Inhibitor of PFKFB3, Induces Cell Cycle Arrest, Apoptosis and Inhibits Invasion in Gastric Cancer, Plos One. 11 (2016) e0163768. 10.1371/journal.pone.0163768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Mangal JL, Inamdar S, Yang Y, Dutta S, Wankhede M, Shi X, Gu H, Green M, Rege K, Curtis M, Acharya AP, Metabolite releasing polymers control dendritic cell function by modulating their energy metabolism, J Mater Chem B. 8 (2020) 5195–5203. 10.1039/d0tb00790k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Jones RG, Pearce EJ, MenTORing Immunity: mTOR Signaling in the Development and Function of Tissue-Resident Immune Cells, Immunity. 46 (2017) 730–742. 10.1016/j.immuni.2017.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Gosselin EA, Tostanoski LH, Jewell CM, Controlled Release of Second Generation mTOR Inhibitors to Restrain Inflammation in Primary Immune Cells, Aaps J. 19 (2017) 1175–1185. 10.1208/s12248-017-0089-1. [DOI] [PubMed] [Google Scholar]
- [157].Fan Y, Zheng X, Ali Y, Berggren P-O, Loo SCJ, Local release of rapamycin by microparticles delays islet rejection within the anterior chamber of the eye, Sci Rep-Uk. 9 (2019) 3918. 10.1038/s41598-019-40404-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Ju Y, Edman MC, Guo H, Janga SR, Peddi S, Louie SG, Junge JA, MacKay JA, Hamm-Alvarez SF, Intralacrimal Sustained Delivery of Rapamycin Shows Therapeutic Effects without Systemic Toxicity in a Mouse Model of Autoimmune Dacryoadenitis Characteristic of Sjögren’s Syndrome, Biomacromolecules. 22 (2020) 1102–1114. 10.1021/acs.biomac.0c01468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Barlow AD, Nicholson ML, Herbert TP, Evidence for Rapamycin Toxicity in Pancreatic β-Cells and a Review of the Underlying Molecular Mechanisms, Diabetes. 62 (2013) 2674–2682. 10.2337/db13-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Vivino F, Bunya VY, Massaro-Giordano G, Johr CR, Giattino SL, Schorpion A, Shafer B, Peck A, Sivils K, Rasmussen A, Chiorini JA, He J, Ambrus JL, Sjogren’s syndrome: An update on disease pathogenesis, clinical manifestations and treatment, Clin Immunol. 203 (2019) 81–121. 10.1016/j.clim.2019.04.009. [DOI] [PubMed] [Google Scholar]
- [161].Azzi JR, Sayegh MH, Mallat SG, Calcineurin Inhibitors: 40 Years Later, Can’t Live Without …, J Immunol. 191 (2013) 5785–5791. 10.4049/jimmunol.1390055. [DOI] [PubMed] [Google Scholar]
- [162].Song T-H, Jang J, Choi Y-J, Shim J-H, Cho D-W, 3D-Printed Drug/Cell Carrier Enabling Effective Release of Cyclosporin A for Xenogeneic Cell-Based Therapy, Cell Transplant. 24 (2015) 2513–2525. . [DOI] [PubMed] [Google Scholar]
- [163].Unadkat JV, Schnider JT, Feturi FG, Tsuji W, Bliley JM, Venkataramanan R, Solari MG, Marra KG, Gorantla VS, Spiess AM, Single Implantable FK506 Disk Prevents Rejection in Vascularized Composite Allotransplantation, Plast Reconstr Surg. 139 (2017) 403e–414e. 10.1097/prs.0000000000002951. [DOI] [PubMed] [Google Scholar]
- [164].Wu J, Zheng Z, Chong Y, Li X, Pu L, Tang Q, Yang L, Wang X, Wang F, Liang G, Immune Responsive Release of Tacrolimus to Overcome Organ Transplant Rejection, Adv Mater. 30 (2018) 1805018. 10.1002/adma.201805018. [DOI] [PubMed] [Google Scholar]
- [165].Li R, Liang J, He Y, Qin J, He H, Lee S, Pang Z, Wang J, Sustained Release of Immunosuppressant by Nanoparticle-anchoring Hydrogel Scaffold Improved the Survival of Transplanted Stem Cells and Tissue Regeneration, Theranostics. 8 (2018) 878–893. 10.7150/thno.22072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Kojima R, Yoshida T, Tasaki H, Umejima H, Maeda M, Higashi Y, Watanabe S, Oku N, Release mechanisms of tacrolimus-loaded PLGA and PLA microspheres and immunosuppressive effects of the microspheres in a rat heart transplantation model, Int J Pharmaceut. 492 (2015) 20–27. 10.1016/j.ijpharm.2015.07.004. [DOI] [PubMed] [Google Scholar]
- [167].Tedesco D, Haragsim L, Cyclosporine: A Review, J Transplant. 2012 (2012) 230386. 10.1155/2012/230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Krämer BK, Montagnino G, del Castillo D, Margreiter R, Sperschneider H, Olbricht CJ, Krüger B, Ortuño J, Köhler H, Kunzendorf U, Stummvoll H-K, Tabernero JM, Mühlbacher F, Rivero M, Arias M, Efficacy and safety of tacrolimus compared with cyclosporin A microemulsion in renal transplantation: 2 year follow-up results, Nephrol Dial Transpl. 20 (2005) 968–973. 10.1093/ndt/gfh739. [DOI] [PubMed] [Google Scholar]
- [169].Tajdaran K, Shoichet MS, Gordon T, Borschel GH, A novel polymeric drug delivery system for localized and sustained release of tacrolimus (FK506), Biotechnol Bioeng. 112 (2015) 1948–1953. 10.1002/bit.25598. [DOI] [PubMed] [Google Scholar]
- [170].Zuo KJ, Shafa G, Chan K, Zhang J, Hawkins C, Tajdaran K, Gordon T, Borschel GH, Local FK506 drug delivery enhances nerve regeneration through fresh, unprocessed peripheral nerve allografts, Exp Neurol. 341 (2021) 113680. 10.1016/j.expneurol.2021.113680. [DOI] [PubMed] [Google Scholar]
- [171].Lipman NS, Jackson LR, Trudel LJ, Weis-Garcia F, Monoclonal Versus Polyclonal Antibodies: Distinguishing Characteristics, Applications, and Information Resources, Ilar J. 46 (2005) 258–268. 10.1093/ilar.46.3.258. [DOI] [PubMed] [Google Scholar]
- [172].Buss NA, Henderson SJ, McFarlane M, Shenton JM, de Haan L, Monoclonal antibody therapeutics: history and future, Curr Opin Pharmacol. 12 (2012) 615–622. 10.1016/j.coph.2012.08.001. [DOI] [PubMed] [Google Scholar]
- [173].Murphy K, Casey Weaver, Charles Janeway, Janeway’s immunobiology, 2017. [Google Scholar]
- [174].Murer P, Neri D, Antibody-cytokine fusion proteins: a novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation, New Biotechnol. 52 (2019) 42–53. 10.1016/j.nbt.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Awwad S, Angkawinitwong U, Overview of Antibody Drug Delivery, Pharm. 10 (2018) 83. 10.3390/pharmaceutics10030083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Ben-Akiva E, Witte SE, Meyer RA, Rhodes KR, Green JJ, Polymeric micro- and nanoparticles for immune modulation, Biomater Sci-Uk. 7 (2018) 14–30. 10.1039/c8bm01285g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Chapman AP, PEGylated antibodies and antibody fragments for improved therapy: a review, Adv Drug Deliver Rev. 54 (2002) 531–545. 10.1016/s0169-409x(02)00026-1. [DOI] [PubMed] [Google Scholar]
- [178].Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S, The pro- and anti-inflammatory properties of the cytokine interleukin-6, Biochimica Et Biophysica Acta Bba - Mol Cell Res. 1813 (2011) 878–888. 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- [179].Foong KS, Patel R, Forbes A, Day RM, Anti-Tumor Necrosis Factor-Alpha–Loaded Microspheres as a Prospective Novel Treatment for Crohn’s Disease Fistulae, Tissue Eng Part C Methods. 16 (2010) 855–864. 10.1089/ten.tec.2009.0599. [DOI] [PubMed] [Google Scholar]
- [180].Kaplan JA, Barthélémy P, Grinstaff MW, Self-assembled nanofiber hydrogels for mechanoresponsive therapeutic anti-TNFα antibody delivery, Chem Commun. 52 (2016) 5860–5863. 10.1039/c6cc02221a. [DOI] [PubMed] [Google Scholar]
- [181].Bluestone JA, St. Clair EW, Turka LA, CTLA4Ig: Bridging the Basic Immunology with Clinical Application, Immunity. 24 (2006) 233–238. 10.1016/j.immuni.2006.03.001. [DOI] [PubMed] [Google Scholar]
- [182].Esposito E, Cuzzocrea S, TNF-Alpha as a Therapeutic Target in Inflammatory Diseases, Ischemia-Reperfusion Injury and Trauma, Curr Med Chem. 16 (2009) 3152–3167. 10.2174/092986709788803024. [DOI] [PubMed] [Google Scholar]
- [183].Popa C, Netea MG, van Riel PLCM, van der Meer JWM, Stalenhoef AFH, The role of TNF-α in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk, J Lipid Res. 48 (2007) 751–762. 10.1194/jlr.r600021-jlr200. [DOI] [PubMed] [Google Scholar]
- [184].Lutz MB, Suri RM, Niimi M, Ogilvie ALJ, Kukutsch NA, Rößner S, Schuler G, Austyn JM, Immature dendritic cells generated with low doses of GM CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo, Eur J Immunol. 30 (2000) 1813–1822. . [DOI] [PubMed] [Google Scholar]
- [185].Cho JJ, Stewart JM, Drashansky TT, Brusko MA, Zuniga AN, Lorentsen KJ, Keselowsky BG, Avram D, An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis, Biomaterials. 143 (2017) 79–92. 10.1016/j.biomaterials.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Yoon YM, Lewis JS, Carstens MR, Campbell-Thompson M, Wasserfall CH, Atkinson MA, Keselowsky BG, A combination hydrogel microparticle-based vaccine prevents type 1 diabetes in non-obese diabetic mice, Sci Rep-Uk. 5 (2015) 13155. 10.1038/srep13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG, A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice, Clin Immunol. 160 (2015) 90–102. 10.1016/j.clim.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, Dolgova NV, Xia C, Brusko TM, Wasserfall CH, Clare-Salzler MJ, Atkinson MA, Keselowsky BG, Dual-Sized Microparticle System for Generating Suppressive Dendritic Cells Prevents and Reverses Type 1 Diabetes in the Nonobese Diabetic Mouse Model, Acs Biomater Sci Eng. 5 (2019) 2631–2646. 10.1021/acsbiomaterials.9b00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Brusko MA, Stewart JM, Posgai AL, Wasserfall CH, Atkinson MA, Brusko TM, Keselowsky BG, Immunomodulatory Dual-Sized Microparticle System Conditions Human Antigen Presenting Cells Into a Tolerogenic Phenotype In Vitro and Inhibits Type 1 Diabetes-Specific Autoreactive T Cell Responses, Front Immunol. 11 (2020) 574447. 10.3389/fimmu.2020.574447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Bassin EJ, Buckley AR, Piganelli JD, Little SR, TRI microparticles prevent inflammatory arthritis in a collagen-induced arthritis model, Plos One. 15 (2020) e0239396. 10.1371/journal.pone.0239396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, Fonfrede M, Rosenzwajg M, Bernard C, Klatzmann D, Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial, Lancet Diabetes Endocrinol. 1 (2013) 295–305. 10.1016/s2213-8587(13)70113-x. [DOI] [PubMed] [Google Scholar]
- [192].Tahvildari M, Dana R, Low-Dose IL-2 Therapy in Transplantation, Autoimmunity, and Inflammatory Diseases, J Immunol. 203 (2019) 2749–2755. 10.4049/jimmunol.1900733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, Armand P, Cutler C, Ho VT, Treister NS, Bienfang DC, Prasad S, Tzachanis D, Joyce RM, Avigan DE, Antin JH, Ritz J, Soiffer RJ, Interleukin-2 and Regulatory T Cells in Graft-versus-Host Disease, New Engl J Medicine. 365 (2011) 2055–2066. 10.1056/nejmoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Boyman O, Sprent J, The role of interleukin-2 during homeostasis and activation of the immune system, Nat Rev Immunol. 12 (2012) 180–190. 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- [195].Miller AM, Role of IL-33 in inflammation and disease, J Inflamm. 8 (2011) 22. 10.1186/1476-9255-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Liew FY, Girard J-P, Turnquist HR, Interleukin-33 in health and disease, Nat Rev Immunol. 16 (2016) 676–689. 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- [197].Liu JMH, Zhang X, Joe S, Luo X, Shea LD, Evaluation of biomaterial scaffold delivery of IL-33 as a localized immunomodulatory agent to support cell transplantation in adipose tissue, J Immunol Regen Medicine. 1 (2018) 1–12. 10.1016/j.regen.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Arend WP, The balance between IL-1 and IL-1Ra in disease, Cytokine Growth F R. 13 (2002) 323–340. 10.1016/s1359-6101(02)00020-5. [DOI] [PubMed] [Google Scholar]
- [199].Clements AEB, Groves ER, Chamberlain CS, Vanderby R, Murphy WL, Microparticles Locally Deliver Active Interleukin-1 Receptor Antagonist In Vivo, Adv Healthc Mater. 7 (2018) 1800263. 10.1002/adhm.201800263. [DOI] [PubMed] [Google Scholar]
- [200].Volpe E, Sambucci M, Battistini L, Borsellino G, Fas–Fas Ligand: Checkpoint of T Cell Functions in Multiple Sclerosis, Front Immunol. 7 (2016) 382. 10.3389/fimmu.2016.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Headen DM, Woodward KB, Coronel MM, Shrestha P, Weaver JD, Zhao H, Tan M, Hunckler MD, Bowen WS, Johnson CT, Shea L, Yolcu ES, García AJ, Shirwan H, Local immunomodulation with Fas ligand-engineered biomaterials achieves allogeneic islet graft acceptance, Nat Mater. 17 (2018) 732–739. 10.1038/s41563-018-0099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Yolcu ES, Zhao H, Bandura-Morgan L, Lacelle C, Woodward KB, Askenasy N, Shirwan H, Pancreatic Islets Engineered with SA-FasL Protein Establish Robust Localized Tolerance by Inducing Regulatory T Cells in Mice, J Immunol. 187 (2011) 5901–5909. 10.4049/jimmunol.1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Yolcu ES, Askenasy N, Singh NP, Cherradi S-EL, Shirwan H, Cell Membrane Modification for Rapid Display of Proteins as a Novel Means of Immunomodulation FasL-Decorated Cells Prevent Islet Graft Rejection, Immunity. 17 (2002) 795–808. 10.1016/s1074-7613(02)00482-x. [DOI] [PubMed] [Google Scholar]
- [204].Batlle E, Massagué J, Transforming Growth Factor-β Signaling in Immunity and Cancer, Immunity. 50 (2019) 924–940. 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Koprivica I, Gajic D, Saksida T, Cavalli E, Auci D, Despotovic S, Pejnovic N, Stosic-Grujicic S, Nicoletti F, Stojanovic I, Orally delivered all-trans-retinoic acid- and transforming growth factor-β-loaded microparticles ameliorate type 1 diabetes in mice, Eur J Pharmacol. 864 (2019) 172721. 10.1016/j.ejphar.2019.172721. [DOI] [PubMed] [Google Scholar]
- [206].Liu JMH, Zhang J, Zhang X, Hlavaty KA, Ricci CF, Leonard JN, Shea LD, Gower RM, Transforming growth factor-beta 1 delivery from microporous scaffolds decreases inflammation post-implant and enhances function of transplanted islets, Biomaterials. 80 (2016) 11–19. 10.1016/j.biomaterials.2015.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Yang EY, Kronenfeld JP, Gattás-Asfura KM, Bayer AL, Stabler CL, Engineering an “infectious” Treg biomimetic through chemoselective tethering of TGF-β1 to PEG brush surfaces, Biomaterials. 67 (2015) 20–31. 10.1016/j.biomaterials.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Shah NJ, Mao AS, Shih T-Y, Kerr MD, Sharda A, Raimondo TM, Weaver JC, Vrbanac VD, Deruaz M, Tager AM, Mooney DJ, Scadden DT, An injectable bone marrow–like scaffold enhances T cell immunity after hematopoietic stem cell transplantation, Nat Biotechnol. 37 (2019) 293–302. 10.1038/s41587-019-0017-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, Biota C, Doffin AC, Durand I, Olive D, Perez S, Pasqual N, Faure C, Ray-Coquard I, Puisieux A, Caux C, Blay J-Y, Ménétrier-Caux C, Regulatory T Cells Recruited through CCL22/CCR4 Are Selectively Activated in Lymphoid Infiltrates Surrounding Primary Breast Tumors and Lead to an Adverse Clinical Outcome, Cancer Res. 69 (2009) 2000–2009. 10.1158/0008-5472.can-08-2360. [DOI] [PubMed] [Google Scholar]
- [210].Jhunjhunwala S, Raimondi G, Glowacki AJ, Hall SJ, Maskarinec D, Thorne SH, Thomson AW, Little SR, Bioinspired Controlled Release of CCL22 Recruits Regulatory T Cells In Vivo, Adv Mater. 24 (2012) 4735–4738. 10.1002/adma.201202513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Zou L, Barnett B, Safah H, LaRussa VF, Evdemon-Hogan M, Mottram P, Wei S, David O, Curiel TJ, Zou W, Bone Marrow Is a Reservoir for CD4+CD25+ Regulatory T Cells that Traffic through CXCL12/CXCR4 Signals, Cancer Res. 64 (2004) 8451–8455. 10.1158/0008-5472.can-04-1987. [DOI] [PubMed] [Google Scholar]
- [212].Horwitz DA, Fahmy TM, Piccirillo CA, Cava AL, Rebalancing Immune Homeostasis to Treat Autoimmune Diseases, Trends Immunol. 40 (2019) 888–908. 10.1016/j.it.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Eslami-Kaliji F, Sarafbidabad M, Rajadas J, Mohammadi MR, Dendritic Cells as Targets for Biomaterial-Based Immunomodulation, Acs Biomater Sci Eng. 6 (2020) 2726–2739. 10.1021/acsbiomaterials.9b01987. [DOI] [PubMed] [Google Scholar]
- [214].Dominguez-Villar M, Hafler DA, Regulatory T cells in autoimmune disease, Nat Immunol. 19 (2018) 665–673. 10.1038/s41590-018-0120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Josefowicz SZ, Lu L-F, Rudensky AY, Regulatory T Cells: Mechanisms of Differentiation and Function, Immunology. 30 (2012) 531–564. 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Khan RS, Newsome PN, A Comparison of Phenotypic and Functional Properties of Mesenchymal Stromal Cells and Multipotent Adult Progenitor Cells, Front Immunol. 10 (2019) 1952. 10.3389/fimmu.2019.01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Lopez-Santalla M, Fernandez-Perez R, Garin MI, Mesenchymal Stem/Stromal Cells for Rheumatoid Arthritis Treatment: An Update on Clinical Applications, Cells. 9 (2020) 1852. 10.3390/cells9081852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Abedi M, Alavi-Moghadam S, Payab M, Goodarzi P, Mohamadi-jahani F, Sayahpour FA, Larijani B, Arjmand B, Mesenchymal stem cell as a novel approach to systemic sclerosis; current status and future perspectives, Cell Regen. 9 (2020) 20. 10.1186/s13619-020-00058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Heyes R, Iarocci A, Tchoukalova Y, Lott DG, Immunomodulatory Role of Mesenchymal Stem Cell Therapy in Vascularized Composite Allotransplantation, J Transplant. 2016 (2016) 1–10. 10.1155/2016/6951693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Podestà MA, Remuzzi G, Casiraghi F, Mesenchymal Stromal Cells for Transplant Tolerance, Front Immunol. 10 (2019) 1287. 10.3389/fimmu.2019.01287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Ayenehdeh JM, Niknam B, Rasouli S, Hashemi SM, Rahavi H, Rezaei N, Soleimani M, Liaeiha A, Niknam MH, Tajik N, Immunomodulatory and protective effects of adipose tissue-derived mesenchymal stem cells in an allograft islet composite transplantation for experimental autoimmune type 1 diabetes, Immunol Lett. 188 (2017) 21–31. 10.1016/j.imlet.2017.05.006. [DOI] [PubMed] [Google Scholar]
- [222].Nie M, Chen G, Zhao C, Gan J, Alip M, Zhao Y, Sun L, Bio-inspired adhesive porous particles with human MSCs encapsulation for systemic lupus erythematosus treatment, Bioact Mater. 6 (2021) 84–90. 10.1016/j.bioactmat.2020.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G, Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity, Front Immunol. 10 (2019) 43. 10.3389/fimmu.2019.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Yang J-H, Eun S-C, Therapeutic application of T regulatory cells in composite tissue allotransplantation, J Transl Med. 15 (2017) 218. 10.1186/s12967-017-1322-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Sawant DV, Vignali DAA, Once a Treg, always a Treg?, Immunol Rev. 259 (2014) 173–191. 10.1111/imr.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Ochando J, Ordikhani F, Jordan S, Boros P, Thomson AW, Tolerogenic dendritic cells in organ transplantation, Transplant Int. 33 (2020) 113–127. 10.1111/tri.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Pathak S, Acharya S, Regmi S, Shrestha P, You Z, Bae YK, Park MH, Yook S, Kim J, Park SY, Jeong D, Yong CS, Kim JO, Chang JH, Jeong J, Particulate-Based Single-Dose Local Immunosuppressive Regimen for Inducing Tolerogenic Dendritic Cells in Xenogeneic Islet Transplantation, Adv Healthc Mater. (2020) 2001157. 10.1002/adhm.202001157. [DOI] [PubMed] [Google Scholar]
- [228].Haque MR, Lee DY, Ahn C-H, Jeong J-H, Byun Y, Local Co-Delivery of Pancreatic Islets and Liposomal Clodronate Using Injectable Hydrogel to Prevent Acute Immune Reactions in a Type 1 Diabetes, Pharmaceut Res. 31 (2014) 2453–2462. 10.1007/s11095-014-1340-4. [DOI] [PubMed] [Google Scholar]
- [229].Lewis JS, Roche C, Zhang Y, Brusko TM, Wasserfall CH, Atkinson M, Clare-Salzler MJ, Keselowsky BG, Combinatorial delivery of immunosuppressive factors to dendritic cells using dual-sized microspheres, J Mater Chem B. 2 (2013) 2562–2574. 10.1039/c3tb21460e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Spiller KL, Koh TJ, Macrophage-based therapeutic strategies in regenerative medicine, Adv Drug Deliver Rev. 122 (2017) 74–83. 10.1016/j.addr.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Sridharan R, Cameron AR, Kelly DJ, Kearney CJ, O’Brien FJ, Biomaterial based modulation of macrophage polarization: a review and suggested design principles, Mater Today. 18 (2015) 313–325. 10.1016/j.mattod.2015.01.019. [DOI] [Google Scholar]
- [232].Zhuang Z, Yoshizawa-Smith S, Glowacki A, Maltos K, Pacheco C, Shehabeldin M, Mulkeen M, Myers N, Chong R, Verdelis K, Garlet GP, Little S, Sfeir C, Induction of M2 Macrophages Prevents Bone Loss in Murine Periodontitis Models, J Dent Res. 98 (2019) 200–208. 10.1177/0022034518805984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Rhodes KR, Meyer RA, Wang J, Tzeng SY, Green JJ, Biomimetic Tolerogenic Artificial Antigen Presenting Cells for Regulatory T Cell Induction, Acta Biomater. 112 (2020) 136–148. 10.1016/j.actbio.2020.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Jhunjhunwala S, Balmert SC, Raimondi G, Dons E, Nichols EE, Thomson AW, Little SR, Controlled release formulations of IL-2, TGF-β1 and rapamycin for the induction of regulatory T cells, J Control Release. 159 (2012) 78–84. 10.1016/j.jconrel.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Balmert SC, Donahue C, Vu JR, Erdos G, Falo LD, Little SR, In vivo induction of regulatory T cells promotes allergen tolerance and suppresses allergic contact dermatitis, J Control Release. 261 (2017) 223–233. 10.1016/j.jconrel.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Ratay ML, Balmert SC, Acharya AP, Greene AC, Meyyappan T, Little SR, TRI Microspheres prevent key signs of dry eye disease in a murine, inflammatory model, Sci Rep-Uk. 7 (2017) 17527. 10.1038/s41598-017-17869-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Eisenstein EM, Williams CB, The Treg/Th17 Cell Balance: A New Paradigm for Autoimmunity, Pediatr Res. 65 (2009) 26R–31R. 10.1203/pdr.0b013e31819e76c7. [DOI] [PubMed] [Google Scholar]
- [238].Kopf H, de la Rosa GM, Howard OMZ, Chen X, Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells, Int Immunopharmacol. 7 (2007) 1819–1824. 10.1016/j.intimp.2007.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Miller SD, Turley DM, Podojil JR, Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease, Nat Rev Immunol. 7 (2007) 665–677. 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
- [240].Zhao H, Kiptoo P, Williams TD, Siahaan TJ, Topp EM, Immune response to controlled release of immunomodulating peptides in a murine experimental autoimmune encephalomyelitis (EAE) model, J Control Release. 141 (2010) 145–152. 10.1016/j.jconrel.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Kobayashi N, Kobayashi H, Gu L, Malefyt T, Siahaan TJ, Antigen-Specific Suppression of Experimental Autoimmune Encephalomyelitis by a Novel Bifunctional Peptide Inhibitor, J Pharmacol Exp Ther. 322 (2007) 879–886. 10.1124/jpet.107.123257. [DOI] [PubMed] [Google Scholar]
- [242].Pearson RM, Casey LM, Hughes KR, Miller SD, Shea LD, In vivo reprogramming of immune cells: Technologies for induction of antigen-specific tolerance, Adv Drug Deliver Rev. 114 (2017) 240–255. 10.1016/j.addr.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Wherry EJ, T cell exhaustion, Nat Immunol. 12 (2011) 492–499. 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- [244].Schietinger A, Greenberg PD, Tolerance and exhaustion: defining mechanisms of T cell dysfunction, Trends Immunol. 35 (2014) 51–60. 10.1016/j.it.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Liu M, Feng D, Liang X, Li M, Yang J, Wang H, Pang L, Zhou Z, Yang Z, Kong D, Li C, Old Dog New Tricks: PLGA Microparticles as an Adjuvant for Insulin Peptide Fragment-Induced Immune Tolerance against Type 1 Diabetes, Mol Pharmaceut. 17 (2020) 3513–3525. 10.1021/acs.molpharmaceut.0c00525. [DOI] [PubMed] [Google Scholar]
- [246].Peine KJ, Guerau-de-Arellano M, Lee P, Kanthamneni N, Severin M, Probst GD, Peng H, Yang Y, Vangundy Z, Papenfuss TL, Lovett-Racke AE, Bachelder EM, Ainslie KM, Treatment of Experimental Autoimmune Encephalomyelitis by Codelivery of Disease Associated Peptide and Dexamethasone in Acetalated Dextran Microparticles, Mol Pharmaceut. 11 (2014) 828–835. 10.1021/mp4005172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Stewart JM, Posgai AL, Leon JJ, Haller MJ, Keselowsky BG, Combination Treatment with Antigen-Specific Dual-Sized Microparticle System Plus Anti-CD3 Immunotherapy Fails to Synergize to Improve Late-Stage Type 1 Diabetes Prevention in Nonobese Diabetic Mice, Acs Biomater Sci Eng. 6 (2020) 5941–5958. 10.1021/acsbiomaterials.0c01075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Allen R, Chizari S, Ma JA, Raychaudhuri S, Lewis JS, Combinatorial, Microparticle-Based Delivery of Immune Modulators Reprograms the Dendritic Cell Phenotype and Promotes Remission of Collagen-Induced Arthritis in Mice, Acs Appl Bio Mater. 2 (2019) 2388–2404. 10.1021/acsabm.9b00092. [DOI] [PubMed] [Google Scholar]
- [249].Nakayama M, Insulin as a key autoantigen in the development of type 1 diabetes, Diabetes Metabolism Res Rev. 27 (2011) 773–777. 10.1002/dmrr.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Schweizer D, Serno T, Goepferich A, Controlled release of therapeutic antibody formats, Eur J Pharm Biopharm. 88 (2014) 291–309. 10.1016/j.ejpb.2014.08.001. [DOI] [PubMed] [Google Scholar]
- [251].Fischbach MA, Bluestone JA, Lim WA, Cell-Based Therapeutics: The Next Pillar of Medicine, Sci Transl Med. 5 (2013) 179ps7–179ps7. 10.1126/scitranslmed.3005568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Pozsgay J, Szekanecz Z, Sármay G, Antigen-specific immunotherapies in rheumatic diseases, Nat Rev Rheumatol. 13 (2017) 525–537. 10.1038/nrrheum.2017.107. [DOI] [PubMed] [Google Scholar]
- [253].Serra P, Santamaria P, Antigen-specific therapeutic approaches for autoimmunity, Nat Biotechnol. 37 (2019) 238–251. 10.1038/s41587-019-0015-4. [DOI] [PubMed] [Google Scholar]
- [254].Hua S, de Matos MBC, Metselaar JM, Storm G, Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization, Front Pharmacol. 9 (2018) 790. 10.3389/fphar.2018.00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Mandal A, Clegg JR, Anselmo AC, Mitragotri S, Hydrogels in the clinic, Bioeng Transl Medicine. 5 (2020) e10158. 10.1002/btm2.10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Spiller KL, Vunjak-Novakovic G, Clinical translation of controlled protein delivery systems for tissue engineering, Drug Deliv Transl Re. 5 (2015) 101–115. 10.1007/s13346-013-0135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Operti MC, Bernhardt A, Grimm S, Engel A, Figdor CG, Tagit O, PLGA-based nanomedicines manufacturing: technologies overview and challenges in industrial scale-up, Int J Pharmaceut. 605 (2021) 120807. 10.1016/j.ijpharm.2021.120807. [DOI] [PubMed] [Google Scholar]
- [258].Galante R, Pinto TJA, Colaço R, Serro AP, Sterilization of hydrogels for biomedical applications: A review, J Biomed Mater Res Part B Appl Biomaterials. 106 (2018) 2472–2492. 10.1002/jbm.b.34048. [DOI] [PubMed] [Google Scholar]
- [259].Sharma V, McNeill JH, To scale or not to scale: the principles of dose extrapolation, Brit J Pharmacol. 157 (2009) 907–921. 10.1111/j.1476-5381.2009.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Davis HL, Novel vaccines and adjuvant systems: The utility of animal models for predicting immunogenicity in humans, Hum Vaccines. 4 (2008) 246–250. 10.4161/hv.4.3.5318. [DOI] [PubMed] [Google Scholar]
- [261].Sertkaya A, Wong H-H, Jessup A, Beleche T, Key cost drivers of pharmaceutical clinical trials in the United States, Clin Trials. 13 (2016) 117–126. 10.1177/1740774515625964. [DOI] [PubMed] [Google Scholar]
- [262].Anselmo AC, Mitragotri S, An overview of clinical and commercial impact of drug delivery systems, J Control Release. 190 (2014) 15–28. 10.1016/j.jconrel.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Cekici A, Kantarci A, Hasturk H, Dyke TEV, Inflammatory and immune pathways in the pathogenesis of periodontal disease, Periodontol 2000. 64 (2014) 57–80. 10.1111/prd.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Garrido-Mesa N, Zarzuelo A, Gálvez J, Minocycline: far beyond an antibiotic, Brit J Pharmacol. 169 (2013) 337–352. 10.1111/bph.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]