

Abstract
The aorta is highly heterogeneous, containing many different types of cells that perform sophisticated functions to maintain aortic homeostasis. Recently, single-cell RNA sequencing (scRNA-seq) studies have provided substantial new insight into the heterogeneity of vascular cell types, the comprehensive molecular features of each cell type, and the phenotypic interrelationship between these cell populations. This new information has significantly improved our understanding of aortic biology and aneurysms at the molecular and cellular level. Here, we summarize these findings, with a focus on what scRNA-seq analysis has revealed about cellular heterogeneity, cellular transitions, communications among cell populations, and critical transcription factors in the vascular wall. We also review the information learned from scRNA-seq that has contributed to our understanding of the pathogenesis of vascular disease, such as the identification of cell types in which aneurysm-related genes and genetic variants function. Finally, we discuss the challenges and future directions of scRNA-seq applications in studies of aortic biology and diseases.
Keywords: Vascular disease, scRNA-seq
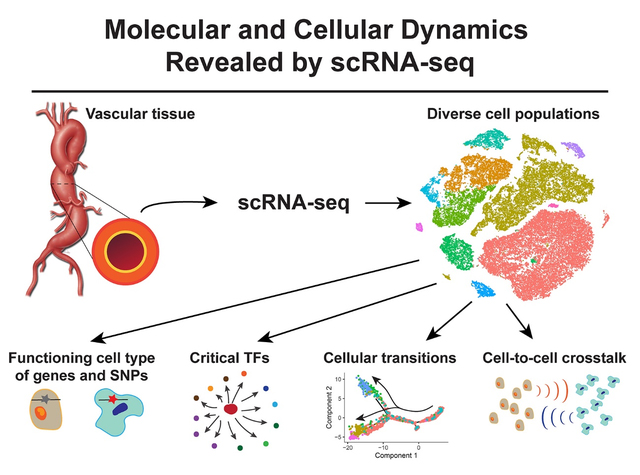
Graphical Abstract
INTRODUCTION
The aortic wall is highly heterogeneous, containing many different types of cells that perform sophisticated functions to maintain aortic homeostasis. Under stress, vascular cells undergo alterations in gene expression that change the structure and function of the aortic wall. Each type of cell responds to stress and contributes to vascular remodeling in a unique way. Therefore, identifying the genes and pathways that drive the cell type–specific response is critically important for understanding the vascular remodeling process.
Recent advances in single-cell sequencing technologies have allowed us to elucidate the epigenetic and gene expression profiles of diverse cell populations at single-cell resolution. Single-cell RNA sequencing (scRNA-seq) addresses the limitations of previous bulk mRNA methodologies by determining the genome-wide mRNA abundance of each individual captured cell in a given sample. ScRNA-seq has been increasingly applied to studies of the aorta and aortic aneurysms and has provided a wealth of information that has improved our understanding of aortic aneurysms at the molecular and cellular level. Here, we summarize these findings and discuss the limitations and future directions of single-cell technologies in aortic aneurysm research (Figure).
Figure. The application and findings of single-cell RNA sequencing (scRNA-seq ) analysis in aortic aneurysms.
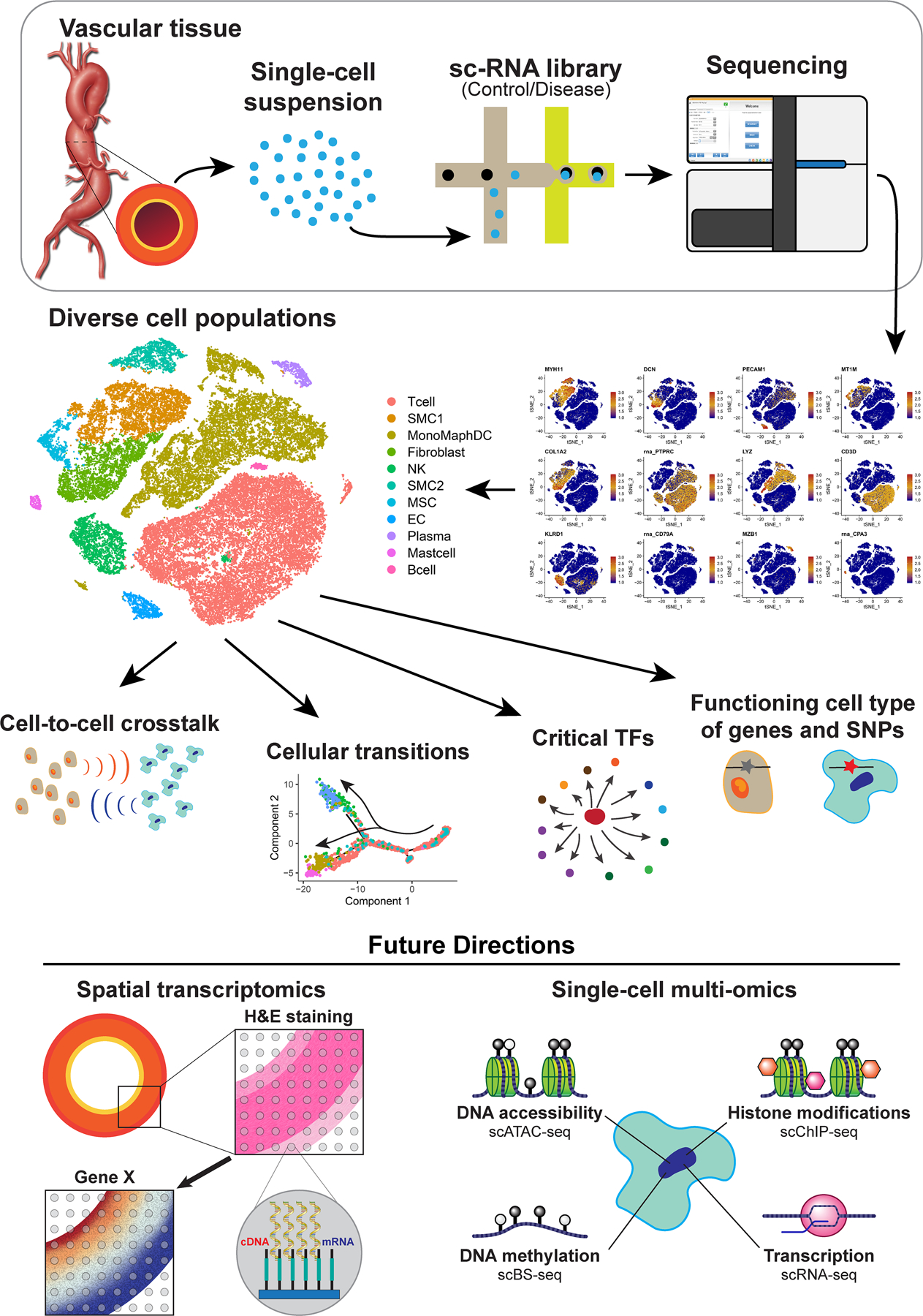
ScRNA-seq starts with the processing of aortic tissue into a single-cell suspension that is submitted for library preparation (the 10X Genomics platform is shown as an example) and then sequencing. ScRNA-seq analysis has revealed the cellular heterogeneity, cellular transitions, communications among cell populations, and critical transcription factors in the aortic wall. ScRNA-seq has also contributed to our understanding of the pathogenesis of aortic aneurysms through the identification of the functioning cell type of aneurysm-related genes and genetic variants. Future directions of scRNA-seq applications in studies of aortic aneurysms include the integration of scRNA-seq with spatial transcriptomics and single-cell multiomics. ScATAC-seq, single-cell sequencing assay for transposase-accessible chromatin; scBS-seq, single-cell bisulfite sequencing; scChIP-seq, single-cell chromatin immunoprecipitation sequencing; SNPs, single-nucleotide polymorphisms; TFs, transcription factors.
1. ScRNA-seq technologies applied in aortic aneurysm studies
Since the first scRNA-seq method was reported in 2009,1 many new scRNA-seq technologies have been subsequently developed on the basis of different platforms (extensively reviewed elsewhere).2–4 Single-cell isolation technologies include low-throughput approaches such as pipette dilution, micromanipulation, and laser capture microdissection, whereas high-throughput large-scale technologies include traditional fluorescence-activated cell sorting (FACS) and systems that are microwell based (e.g., ICELL8), microfluidic based (e.g., Fluidigm C1), or droplet microfluidic based (e.g., InDrop, Drop-seq, and 10X Genomics). Several cDNA library preparation technologies, including Smart-seq, Smart-seq2, MARS-seq, and CEL-seq, have been used for low-throughput platforms and some large-scale (such as ICELL8, Fluidigm C1) single-cell isolation platforms. Most scRNA-seq studies of aortic aneurysm are performed by using the commercially available 10X Genomics system, which provides the high-throughput analysis of sequenced cells at a relatively low cost per cell and requires minimal laboratory skills. In some studies of aortic aneurysm, FACS or Fluidigm C1 have been used to process the cells, followed by library preparation with Smart-seq or Smart-seq2 (Table 1), which capture the full-length of mRNA and more genes per cell than the 10X Genomics system. The choice of scRNA-seq platforms depends on the aim of the study. For example, 10X Genomics may be used for capturing a heterogeneous cell population and rare cell types because it provides a large number of cells per sample, whereas Smart-seq and Smart-seq2 are better choices when advanced information such as alternative splicing and allele-specific expression is needed.
Table 1.
Studies of the Aorta or Aortic Aneurysm That Applied Single-cell RNA Sequencing
| Tissues | Technique | Analysis pipeline | # of cells/ # of datasets | Data deposit accession number | Main findings | Reference |
|---|---|---|---|---|---|---|
| Mouse infrarenal aorta, normal control and CaCl2-induced AAA (n=4 per group, pooled) | 10X Genomics | Seurat | 3896/2 | GSE164678 | Cell heterogeneity in early-stage murine AAA | 14 |
| Mouse infrarenal aorta, normal control and elastase-induced AAA (n=3 per group, pooled) | 10X Genomics | Seurat | 4642/3 | GSE152583 | AAA-relevant transcriptional signatures of vascular cells | 10 |
| Mouse aorta, AngII- and PCSK9-induced AAA (n=3, pooled) | 10X Genomics | CellRanger, Loupe Cell Browser | NA/1 | GSE118237 | Netrin-1 plays a role in AAA via macrophages | 30 |
| Mouse thoracic aorta, WT and Sting-deficient mice unchallenged or challenged with HFD/AngII (n=3 per group, pooled) | 10X Genomics | Seurat | NA/4 | NA | Potential mechanism of Sting in SMCs and macrophages in AAD | 31 |
| Mouse whole aorta, mice fed a chow or Western diet (n=2 per group) | 10X Genomics | Seurat | >10,000/4 | Broad Institute Single Cell Portal: https://portals.broadinstitute.org/single_cell/study/SCP289/single-cell-analysis-of-the-normal-mouse-aorta-reveals-functionally-distinct-endothelial-cell-populations | Identified 3 distinct EC populations in mouse aorta | 17 |
| Mouse whole aorta; mouse VSMC-lineage cells | 10X Genomics | Seurat | 2846/1; 3314/1 |
GSE117963 GSE79436 |
Detected Sca1+ VSMC-lineage cells | 9 |
| Mouse aortic media | Fluidigm C1 followed by Smart-seq | Customized | 143/4 | |||
| Myh11-derived Sca1+ cells from mouse aortic media | FACS followed by Smart-seq2 | Customized | 155/3 | |||
| Intact mouse aorta and four aortic segments (ascending, arch, thoracic, and abdominal aortas), WT mice fed with a chow diet, high-salt diet, or high-fat diet; mice with high plasma glucose | 10X Genomics | CellRanger, Seurat | 216 612/20 | NA | Elucidated the nature and range of aortic cell diversity, with implications for the treatment of metabolic pathologies | 13 |
| Mouse aortic root and ascending aorta, healthy controls and Fbn1C1041G/+ mice (n=3 or 4 per group, pooled) | 10X Genomics | Seurat | 17,083/4 | GSE153534 | SMC phenotype modulation in aneurysm of patient with MFS | 11 |
| Human aortic root, MFS patient (n=1) | 10X Genomics | Seurat | NA/1 | GSE153534 | ||
| Human ascending aorta, control (n=3) and sporadic ATAA (n=8) | 10X Genomics | Seurat | 48,128/11 | GSE155468 | Single-cell landscape of human ATAA | 12 |
AAA indicates abdominal aortic aneurysm; AAD, aortic aneurysm and dissection; AngII, angiotensin II; ATAA, ascending thoracic aortic aneurysm; EC, endothelial cell; FACS, fluorescence-activated cell sorting; HFD, high-fat diet; MFS, Marfan syndrome; NA, not available; VSMC, vascular smooth muscle cell; WT, wild-type.
Analysis pipeline indicates the analysis from gene count per cell to cluster identification.
2. Diverse cell populations in normal and aneurysmal aortic wall
The aortic wall consists of many types of cells, including vascular smooth muscle cells (SMCs), fibroblasts, endothelial cells, mesenchymal stem cells, pericytes, and immune cells. Although the major cell types of the aortic wall are known, scRNA-seq has revealed heterogeneity within the major cell types, providing a higher-resolution view of the cell populations within the aortic wall. In turn, this heterogeneity compels us to reconsider the roles of each cell type in the aortic wall, especially in the scenario of aortic aneurysm, and may be a factor in the mechanism of aneurysm formation.
SMC clusters
SMCs are one of the most important constituents of the aortic wall. Located in the medial layer, SMCs are responsible for the contraction of the aortic wall. SMCs possess remarkable plasticity, allowing them to withstand the dynamics of blood flow and stress on the aortic wall and respond to vascular injury and remodeling. The change in SMC gene expression profiles is the molecular basis of SMC phenotype switching. Previously, SMCs were believed to have two phenotypes: contractile and synthetic.5–8 Contractile SMCs were characterized by the high expression of contractile genes, whereas synthetic SMCs were considered to have a dedifferentiated state characterized by the decreased expression of contractile genes, as well as increased proliferation, migration, and extracellular matrix (ECM) production. However, single-cell transcriptomic studies have revealed that SMC populations are even more dynamic than previously thought and have significantly improved our understanding of SMC phenotype changes.
By combining single-cell transcriptomics with lineage tracing in the cells of healthy mouse vessels, Dobnikar et al.9 detected a rare population of SMC lineage cells that express the multipotent progenitor marker stem cell antigen 1 (Sca1). These Sca1+ cells showed the downregulation of contractile vascular SMC genes and the upregulation of genes associated with the SMC response to inflammation and growth factors. In the aneurysmal infrarenal abdominal aortas of mice, Zhao et al.10 identified four SMC subpopulations: quiescent-contractile SMCs, proliferative-contractile SMCs, dedifferentiated SMCs, and inflammatory-like SMCs. In adult Fbn1C1041G/+ mice with Marfan syndrome, a distinct cluster of transcriptomically modulated SMCs was identified in aortic aneurysm tissue through single-cell transcriptomic data analysis.11 In another single-cell transcriptome study of human aneurysmal ascending aortic tissue, five types of SMCs were identified, including contractile SMCs, stressed SMCs, two types of proliferating SMCs, and fibromyocytes (modulated SMCs),12 supporting the plasticity and multi-phenotypic nature of SMCs. Compared with contractile SMCs, the two types of proliferating SMCs exhibited a gene expression profile indicative of high proliferation but with no sign of increased migration or ECM production.12 Interestingly, one of the proliferating SMC clusters (proliferating SMC1) showed the highest expression of contraction-related genes. In fibromyocytes, ECM production was increased, but migration and proliferation were not.12 These studies, which are summarized in Table 2, support that multiple types of SMCs exist in aortic tissue. In aneurysm, phenotypes of SMCs may switch from contraction towards higher proliferation, inflammation, and fibroblast transition.
Table 2.
Studies of SMC Phenotypes in the Normal and Aneurysmal Aorta Identified by Using Single-cell RNA Sequencing
| Species | Disease (Model) | Tissue | Phenotype of SMCs | Reference |
|---|---|---|---|---|
| Mouse (C57BL/6) | Healthy (Myh11-CreERt2/Confetti ) | Ascending and descending aorta | Ly6a/Sca1+ SMC, with downregulated expression of contractile genes and upregulated expression of inflammation-related genes and growth factors | 9 |
| Mouse (C57BL/6J) | Aneurysmal (mice treated with elastase for 7 or 14 days) | Infrarenal aorta | Quiescent-contractile SMCs, proliferative-contractile SMCs, dedifferentiated SMCs, and inflammatory-like SMCs | 10 |
| Mouse (C57BL/6J) | Aneurysmal (MFS; Fbn1C1041G/+) | Aortic root and ascending aorta | One SMC and one fibromyocyte cluster | 11 |
| Human | Aneurysmal (MFS) | Aortic root | One SMC and one fibromyocyte cluster | 11 |
| Human | Normal control and sporadic ATAA | Ascending aorta | Contractile SMC, stressed SMC, proliferating SMCs (two types), and fibromyocytes | 12 |
ATAA indicates ascending thoracic aortic aneurysm; MFS, Marfan syndrome; SMC, smooth muscle cell.
Fibroblast clusters
Fibroblasts are composed of dynamic cell populations that play a key role in vascular remodeling. Therefore, it is important to identify the diverse fibroblast subclusters and understand each of their specific features and functions. In a study of aortic tissues from mice (a) under normal conditions, (b) with high blood glucose levels, (c) with high dietary salt, or (d) with high-fat intake, three fibroblast clusters were identified: CD34high, SERPINF1high, and cartilage-like fibroblasts.13 According to results of gene ontology analysis, CD34high fibroblasts were enriched in the expression of genes that regulate SMC hypertrophy (IGFBP5, PI16 and HN1), angiogenesis (ANXA1, KLF4, and TGFBR2), and ECM organization (FN1, LAMC1, TNXB, ADAMTS5, TIMP2, FBN1, and COL14A1). Cartilage-like fibroblasts expressed genes associated with cartilage development (OTOR, ACAN, UCMA, and COMP), suggesting that they may modulate the tensile strength and vascular resilience of the aorta. In early-stage abdominal aortic aneurysm (AAA) tissue from mice, two clusters of fibroblasts were identified.14 One cluster (Fibro-1) was characterized by the expression of proteoglycans (e.g. Dcn and Lum) and Sfrp4, whereas the other fibroblast cluster (Fibro-2)—most likely proliferative fibroblasts—showed the increased expression of Pclaf and Mki67. In our previous study,12 we identified two clusters of fibroblasts (Fibroblast1 and Fibroblast2) in human aneurysmal and control ascending aorta. Fibroblast1 cells highly expressed elastin (ELN), whereas Fibroblast2 cells highly expressed fibrillin-1 (FBN1) and proteoglycans (e.g. LUM and DCN). In addition, the Fibroblast1 cluster showed higher cell-cell and cell-ECM junction scores than the Fibroblast2 cluster did.
Endothelial cell clusters
In the aorta, endothelial cells form the inner cellular lining and play important roles in hemostatic balance, permeability, and blood cell trafficking.15 Thus, uncovering the dynamics and epigenetic regulation of the endothelial cell transcriptome at the single-cell level during aortic aneurysm development may deepen our understanding of the potential contributions of endothelial dysfunction to disease formation. In a single-cell transcriptomic study of endothelial cells from 11 mouse tissues, transcriptomes were found to be similar among endothelial cells from different vascular beds (e.g., arteries, veins, capillaries, and lymphatics) within the same tissue type, although endothelial cell heterogeneity varied by tissue type.16 This suggested that the different roles of endothelial cells in particular microenvironments contribute substantially to the heterogeneous transcriptome profiles of endothelial cells.
Although the relationship between the heterogeneous transcriptome of aortic endothelial cells and aortic aneurysm is not fully understood, a few studies have focused on the scRNA-seq analysis of aortic endothelial cells and have revealed significant findings. Kalluri et al.17 identified three mouse aortic endothelial cell populations with distinct gene expression profiles, suggesting the functional specialization of endothelial cell subpopulations in (a) ECM production, (b) lipid handling and angiogenesis, and (c) lymphatic function. Lukowski et al.18 identified two mouse aortic endothelial cell populations: endovascular progenitors and differentiated endothelial cells. In another study of whole mouse aorta, three endothelial cell populations were identified: CD34high, THY1high, and activated endothelial cell clusters.13 On the basis of their expression profiles, CD34high and THY1high endothelial cells may respond to paracrine signals to proliferate and promote vasculogenesis. However, understanding the functions of and differences between the CD34high and THY1high subpopulations requires further exploration.
Immune cell clusters
Immune cells are also an important part of the aortic wall. The accumulation and activation of immune cells in the aorta are a main feature of aortic diseases, including aortic aneurysms. ScRNA-seq analysis of human and mouse aortic tissues showed the presence of macrophages, T lymphocytes, B lymphocytes, natural killer cells, and mast cells. In our study,12 we showed that macrophages and T lymphocytes are abundant and heterogeneous in the human aortic wall and that their populations are increased in aneurysmal aortic tissue. However, many questions remain. Further efforts are needed to address how the diverse immune cell population is related to aortic aneurysm and what the differences are in the immune cell population between aortic aneurysm and other aortic diseases such as atherosclerosis. A scRNA-seq–based meta-analysis of leukocyte diversity in mouse models of atherosclerosis19 is recommended for a comprehensive review of immune cells in the aorta. With the increasing availability of additional scRNA-seq data, we anticipate that a meta-analysis on the human aorta will be forthcoming and may provide valuable information about the heterogeneity of aortic cells.
Variance of cell subpopulations among studies
ScRNA-seq–based studies have suggested the existence of heterogeneity among the major cell types in aneurysmal and control aortic tissues. However, the features/classifications and the subpopulation percentages are not always consistent among different studies. In addition to sample variation, several technical reasons may be attributable for these differences. First, variations may arise from sample preparation. Different tissue processing methods applied in these studies most likely lead to different recovery rates of various types of cells. For example, in studies by Kalluri et al.17 and He et al.,13 three endothelial cell clusters were identified, whereas in another study of mouse aneurysmal infrarenal abdominal aorta, the endothelial cell population was small and subclusters were not identified.10 Second, variations may arise from data analysis tools. ScRNA-seq data are processed and manipulated in multiple steps to reduce the dimensions and integrate data before cluster identification. Even though a similar pipeline of analysis is applied among different studies, small parameters such as the normalization method and clustering resolution may affect the clustering outcomes. Finally, variations may arise from data interpretation. After clustering, the researchers’ method of annotating the clusters may be influenced by their research background. Integrative analysis of all the datasets from these studies may help to gain a comprehensive understanding of current single-cell studies on aortic aneurysm and eliminate some technical variance among studies.
3. Dynamic cellular transitions and communications in the aortic wall
Analyzing cellular transitions by using trajectory analysis
In addition to its usefulness in identifying cell populations, another important application of scRNA-seq technology at the cellular level is in trajectory analysis. Trajectory analysis is used to illustrate cell-cell relationships and transitions by characterizing the differences in cells that result from dynamic evolution processes. Rather than dividing cells into discrete clusters, cells are placed along a continuous path that represents the evolution of the process,20 enabling the study of how cells transition from one state to another. In the aortic root/ascending aorta of Fbn1C1041G/+ mice (Marfan syndrome mice) the pseudotime analysis of scRNA-seq data by using Monocle3 revealed a continuous cell state transition from SMCs to fibromyocytes at the transcriptome level.11 This transition was driven by the Tcf21 gene, which was previously shown to be protective against coronary artery disease (CAD) in a scRNA-seq study of atherosclerotic plaque from the aortic root of Myh11-CreERT2; ROSAtdT/+; ApoE−/− lineage-tracing mice.21 By applying R package Monocle, we12 showed that SMCs have the potential to transdifferentiate into four other clusters that fall into the categories of macrophage-like nonimmune cells/remodeling macrophages or T cell–like non-immune cells/phenotype-switched T cells.
Uncovering cell-to-cell crosstalk with computational predictions
ScRNA-seq can also be used to study intercellular communications. When identifying crosstalk between interacting cells, a communication score is typically calculated for each pair of interacting proteins by using the expression value of corresponding genes as input.22 The following four core scoring functions can be used to calculate the communication score: expression threshold, expression product, expression correlation, and differential combinations.22 We estimated the cell-cell junction and cell-ECM junction score by using the expression product method,12 and the results of our analysis suggested that modulated SMCs (fibromyocytes) exhibit higher cell junction than do other type of SMCs. In addition to the core scoring functions, many other tools that employ advanced statistical methods can be used to identify intercellular communication,22 such as CellPhoneDB,23,24 CellChat,25 and ICELLNET.26 He and colleages13 constructed intercellular networks of mouse aortic cells by using CellPhoneDB. Strong interactions were observed for the stromal cell–EC, stromal cell–SMC, and EC–SMC networks.13 Compared with the healthy aorta, the at-risk aorta (from mice with high salt, high fat, or high glucose) exhibited extensively decreased interactions between SMCs and ECs and SMCs and stromal cells. Furthermore, these three types of cells showed diminished interactions within their own subtypes.13 To date, cell-to-cell crosstalk analyses of the aorta and aortic aneurysm have provided evidence of potentially critical intercellular crosstalk, as well as a new avenue for estimating the features of cell clusters identified by scRNA-seq studies. However, further investigation is required to understand how this crosstalk maintains aortic homeostasis and protection and what specific ligand-receptor pairs are responsible for aortic dysfunction and aneurysm development.
4. Understanding the control of gene expression profiles at single-cell resolution
ScRNA-seq data provide genome-wide differential gene expression profiles among different cell types and under different conditions. However, it is important to identify the key transcription factors that control these differential gene expression profiles. Several tools have been developed for gene regulatory network analysis,27 which can be used to identify the potential transcription factors responsible for (or associated with) differential gene expression. Pan et al.28 analyzed mouse scRNA-seq data of SMC lineages by using Virtual Inference of Protein activity by Enriched Regulon (VIPER) and identified the significant activation or repression of multiple master regulators (e.g., transforming growth factor beta β signaling, retinoic acid [RA] signaling) in SMC-derived intermediate cells versus SMCs. Using scRNA-seq data from aortic and coronary artery tissues of young and old cynomolgus monkeys, Zhang et al.29 constructed the gene regulatory networks of transcription factors and their target genes with SCENIC workflow. They identified six transcription factors (i.e., FOXO3A, TCF3, GLISI, NFIX, TGIF1, and RBPJ) that may serve as master regulators of vascular aging. Further experimental studies have verified that FOXO3A deficiency is a key driver of arterial endothelial aging. Although there have been no reports of transcription factor identification by using the gene regulatory network analysis of scRNA-seq data in aortic aneurysms, this type of analysis is expected to be applied in aortic aneurysm studies in the near future and will illustrate the potentially critical transcription factors for aortic aneurysm formation and development.
5. Understanding the mechanism of aortic aneurysm–related genes and genetic variants
Decoding the functioning cell type of aortic aneurysm–related genes
Many genes have been shown to be important for aortic degeneration and may play a role in aneurysm formation. However, it is important to identify the cells in which these genes exert their adverse effects and to examine the cell-specific functions of these genes in the aortic wall. By profiling the single-cell transcriptomic landscape of murine AAA, Hadi et al.30 found that 83% of the cells expressing Ntn1 were macrophages.30 Targeted deletion of the gene encoding netrin-1 in hematopoietic lineage cells protected mice from developing AAA, supporting a causative role of macrophage-derived netrin-1 in AAA development.30 We have recently shown that Sting, which encodes an important pro-inflammatory cytosolic DNA sensing signaling molecule, was highly expressed in two SMC clusters, three fibroblast clusters, and one macrophage cluster31 in the aortic wall, and its expression in these cells was induced by aortic stress. Sting-deficient mice showed significant reductions in the challenge-induced DNA damage response, inflammatory response, dedifferentiation, and cell death in one SMC cluster, as well as reductions in the inflammatory response and matrix metalloproteinase expression in the macrophage cluster. ScRNA-seq analysis has increased our understanding of the comprehensive roles that STING plays in multiple cells that are responsible for its contribution to aortic enlargement, dissection, and rupture in both the thoracic and abdominal aorta in mice with sporadic aortic aneurysm and dissection.31
Integration of scRNA-seq with genome-wide association studies (GWAS)
During the past decade, GWAS have provided valuable information regarding human genetic polymorphisms and have been used to identify single-nucleotide polymorphisms (SNPs) that are potentially associated with aneurysms. However, a huge gap in knowledge exists regarding germline polymorphisms and the initiation and development of aneurysms. It remains unclear how these polymorphisms may affect cell structure and function in a cell-type–specific way and contribute to disease formation. Answering these questions is critical for understanding the pathogenesis of aneurysms. By overlapping the GWAS results with the differentially expressed genes identified in the scRNA-seq analysis of human carotid atherosclerotic plaques, researchers found that cardiovascular disease susceptibility genes were particularly enriched in the macrophages, endothelial cells, and SMCs of aortic lesions.32 We combined the cell-type–specific differentially expressed genes identified by scRNA-seq , the SNPs identified by GWAS, and the chromatin interactions identified by promoter capture Hi-C to infer the cell types in which those genetic variants and their associated genes function to cause or worsen aortic aneurysm. We identified 13 genes affected by aneurysm-related SNPs and showed the cell types in which those genes function. Particularly, we found that the transcription factor ERG may be critical for aortic function in SMCs, fibroblasts, and endothelial cells.12
Additionally, integration with GWAS data could lead to new findings. Pan et al.28 used sc-RNAseq data to analyze regulatory networks and showed that RA signaling may serve as a prominent modulator of SMC transition during atherosclerosis. When they sought additional clues from GWAS data about the role of RA signaling in SMC phenotype switching and atherosclerosis, they found enriched signals for CAD in the loci of multiple RA signaling target genes. Furthermore, CAD-associated SNPs were eQTLs (expression quantitative trait loci) for some RA signaling genes in CAD-relevant tissues, and the CAD risk alleles were associated with decreased expression of RA-regulated genes.
6. Challenges and future directions
Challenges in vascular tissue processing and data interpretation
Several challenges in sample processing and single-cell sample preparation should be considered in the interpretation of data. First, tissue digestion and processing can be harmful to vascular cells. The recovery rate varies significantly among different types of cells and in various regions with different types of vascular remodeling.33 For example, inflammatory cells are relatively easier to recover than SMCs. Cells in highly fibrotic regions are harder to disassociate than those in nonfibrotic regions. In addition, SMCs in damaged and dissected areas are more vulnerable to harsh tissue digestion. These issues are particularly problematic in diseased human aortic tissues and lead to biased recovery rates among different types of cells. Furthermore, the percentile of each type of cell identified by using scRNA-seq may not represent the actual cell proportion in the tissue. This limits our ability to determine the changes in cell populations under different conditions. Additionally, tissue processing itself also triggers a stress response. We12 and others9 have identified a SMC cluster with the high expression of stress-related genes, which may result from both the disease process and tissue processing. Therefore, precautions should be taken during tissue processing and data interpretation. In the situation that the tissue may not be applicable for scRNA-seq, such as frozen tissue, single-nuclear sequencing may serve as an alternative approach. Despite that the design of the single-cell approach is to capture one cell per reaction system, two or more cells may fall into one drop or well. Thus, during the quality control step of data processing, cells with extremely high gene counts are suspected as doublets and are excluded. Similarly, when a newly identified cell type shows features of two cell types, a doublet is suspected. In these situations, further experimental validation such as immunostaining should be performed. Finally, although scRNA-seq provides a genome-wide gene expression profile, the mRNAs captured and detected per cell are actually a small fraction of the transcriptome. The dropout of scRNA-seq data, which also exists in other types of single-cell sequencing, makes data integration challenging.
Future directions in spatial transcriptomics
Single-cell technology is a quickly advancing area. Several new techniques have been developed that further empower our tools for analysis. Spatially resolved transcriptomics reveal the spatial patterns of gene expression, which would greatly expand our knowledge of the complex, multicellular vascular wall. One spatial transcriptomic method is based on an array of spots, which are printed with a spatial barcode and reverse transcription primer.34 The tissue is fixed, stained, imaged, and permeabilized on the array. Reverse transcription occurs while the tissue is still in place, generating a cDNA library that incorporates the spatial barcodes and preserves the spatial information. Integration of scRNA-seq and spatial transcriptomics has been used for the in situ detection of gene expression profiles in human heart tissue.35 Spatial single-cell transcriptomics analysis provides profound information, particularly with respect to the spatial signatures of gene expression that are influenced by the local microenvironment. With this technology, we are able to pinpoint exactly where a new subpopulation or cell type identified by using scRNA-seq is located in the aortic wall. This information, together with the gene expression profile data, is expected to provide a more sophisticated functional landscape of the aorta.
Future directions in single-cell multiomics
Single-cell sequencing assay for transposase-accessible chromatin (scATAC-seq) provides information regarding the accessibility of chromatin, which can be further analyzed for enriched and dynamic motifs and accessibility of cis-elements at the single-cell level. The motifs and cis-elements can be further interpreted to identify the transcription factors and genes that are involved in the regulation of certain types of cells or that are regulated by treatments. By integrating scRNA-seq and scATAC-seq datasets, Andueza et al.36 identified several potential transcription factors (e.g., RELA, AP1, STAT1, and TEAD1) that mediate flow disturbance–induced endothelial cell reprograming and phenotype changes in the carotid arteries of mice. This approach has also been used in a study that identified Isl1 and Nkx2–5 as critical transcription factors in controlling cardiac progenitor cell fate transitions.37 In addition, several methods of single-cell DNA methylation profiling38 and single-cell chromatin immunoprecipitation sequencing (ChIP-seq) analysis39,40 have been developed. Single-cell DNA methylation profiling allows us to study how genes are regulated by DNA methylation at the single-cell level. Single-cell ChIP-seq analysis of histone modifications provides information about functional genomic elements and their regulation of the genome at the single-cell level. The application of these techniques, which have not yet been used in the field of cardiovascular research, would shed light on the diverse aortic cell population and provide mechanistic details underlying aortic aneurysm formation such as how the gene program is epigenetically regulated and which transcription factors are involved.
Gene expression is regulated at multiple levels, including DNA polymorphisms, mutations, methylation, histone modification, chromatin remodeling, and posttranscriptional regulation. Future developments in integrated single-cell multiomics will allow us to uncover multiple molecular profiles in each individual cell that will, in turn, greatly enhance our understanding of the epigenetic control of dynamic gene expression, cell reprogramming, and phenotypic changes in aortic aneurysm formation and development.
CONCLUSIONS
The application of scRNA-seq in the field of aortic aneurysm research has allowed us to characterize the cellular heterogeneity of the aortic wall, especially with respect to cell phenotype switching under aneurysm conditions. In addition, by providing information about the transcription factors that drive vascular disease, as well as the functioning cell type of disease-associated genes and genetic variants, scRNA-seq can reveal valuable information about the molecular mechanisms underlying aortic aneurysms. Integrating scRNA-seq with other types of omics data, including spatial transcriptomics and sc-ATACseq data, will greatly expand our knowledge of vascular biology in a systematic way. However, the application of scRNA-seq in aortic aneurysm studies is still in its early stages; thus, the data should be interpreted with caution, and new experimental findings should be validated.
Highlights.
Single-cell transcriptomic studies have revealed that cell populations in the vascular wall are more dynamic than previously thought and have significantly improved our understanding of smooth muscle phenotype changes.
Furthermore, the identification of transcription factors and the functioning cell type of aneurysm-related genes and genetic variants at single-cell resolution has improved our understanding of the pathogenesis of aortic aneurysm.
The integration of single-cell RNA sequencing data with other types of omics data allows us to uncover multiple molecular profiles in each individual cell and has the potential to greatly enhance our understanding of aortic aneurysm formation and development.
Acknowledgments
The authors thank Nicole Stancel, PhD, ELS(D), of the Department of Scientific Publications at the Texas Heart Institute, for providing editorial support, and Scott Weldon, MA, at the Division of Cardiothoracic Surgery, Baylor College of Medicine, for editing of the Figure.
Sources of Funding
Our research is supported by grants from the American Heart Association (AHA) Vascular Diseases Strategically Focused Research Networks (SFRN) (AHA18SFRN33960114), and NIH HL131980, HL143359 and HL127111. Dr. Li is supported by the Victor A. McKusick Fellowship Grant from The Marfan Foundation. Dr. LeMaire’s work is supported in part by the Jimmy and Roberta Howell Professorship in Cardiovascular Surgery at Baylor College of Medicine.
Nonstandard Abbreviations and Acronyms
- AAA
abdominal aortic aneurysm
- CAD
coronary artery disease
- ChIP-seq
chromatin immunoprecipitation sequencing
- ECM
extracellular matrix
- FACS
fluorescence-activated cell sorting
- GWAS
genome-wide association studies
- RA
retinoic acid
- scATAC-seq
single-cell sequencing assay for transposase-accessible chromatin
- scRNA-seq
single-cell RNA sequencing
- SMC
smooth muscle cells
- SNP
single-nucleotide polymorphism
Footnotes
Disclosures
The authors report no conflicts of interest. Dr. LeMaire serves as a consultant for Terumo Aortic and Baxter Healthcare; serves as a principal investigator for clinical studies sponsored by Terumo Aortic and CytoSorbants; and serves as a co-investigator for clinical studies sponsored by W.L. Gore & Associates.
REFERENCES
- 1.Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, Lao K, Surani MA. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009:6:377–382. [DOI] [PubMed] [Google Scholar]
- 2.Chen G, Ning B, Shi T. Single-cell RNA-seq technologies and related computational data analysis. Front Genet. 2019:10:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valihrach L, Androvic P, Kubista M. Platforms for single-cell collection and analysis. Int J Mol Sci. 2018:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Li T, Liu F, Chen Y, Yao J, Li Z, Huang Y, Wang J. Comparative analysis of droplet-based ultra-high-throughput single-cell RNA-seq systems. Mol Cell. 2019:73:130–142 e135. [DOI] [PubMed] [Google Scholar]
- 5.Halayko AJ, Solway J. Invited Review: Molecular mechanisms of phenotypic plasticity in smooth muscle cells. Journal of Applied Physiology. 2001:90:358–368. [DOI] [PubMed] [Google Scholar]
- 6.Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J. 2007:15:100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fei J, Cui XB, Wang JN, Dong K, Chen SY. ADAR1-mediated RNA editing, a novel mechanism controlling phenotypic modulation of vascular smooth muscle cells. Circ Res. 2016:119:463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, Hwa J, Yu J, Martin KA. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. 2013:128:2047–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E, Dzierzak E, Bennett MR, Spivakov M, Jorgensen HF. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun. 2018:9:4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao G, Lu H, Chang Z, Zhao Y, Zhu T, Chang L, Guo Y, Garcia-Barrio MT, Chen YE, Zhang J. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc Res. 2021:117:1402–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedroza AJ, Tashima Y, Shad R, Cheng P, Wirka R, Churovich S, Nakamura K, Yokoyama N, Cui JZ, Iosef C, Hiesinger W, Quertermous T, Fischbein MP. Single-cell transcriptomic profiling of vascular smooth muscle cell phenotype modulation in Marfan syndrome aortic aneurysm. Arterioscler Thromb Vasc Biol. 2020:40:2195–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Ren P, Dawson A, et al. Single-cell transcriptome analysis reveals dynamic cell populations and differential gene expression patterns in control and aneurysmal human aortic tissue. Circulation. 2020:142:1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He D, Mao A, Zheng C-B, Kan H, Zhang K, Zhang Z, Feng L, Ma X. Aortic heterogeneity across segments and under high fat/salt/glucose conditions at the single-cell level. National Science Review. 2020:7:881–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang H, Zhou T, Stranz A, DeRoo E, Liu B. Single-cell RNA sequencing reveals heterogeneity of vascular cells in early stage murine abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2021:ATVBAHA120315607. [DOI] [PMC free article] [PubMed]
- 15.Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007:100:158–173. [DOI] [PubMed] [Google Scholar]
- 16.Kalucka J, de Rooij L, Goveia J, et al. Single-cell transcriptome atlas of murine endothelial cells. Cell. 2020:180:764–779 e720. [DOI] [PubMed] [Google Scholar]
- 17.Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, Regev A, Kathiresan S, Gupta RM. Single-cell analysis of the normal mouse aorta reveals functionally distinct endothelial cell populations. Circulation. 2019:140:147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukowski SW, Patel J, Andersen SB, Sim SL, Wong HY, Tay J, Winkler I, Powell JE, Khosrotehrani K. Single-cell transcriptional profiling of aortic endothelium identifies a hierarchy from endovascular progenitors to differentiated cells. Cell Rep. 2019:27:2748–2758 e2743. [DOI] [PubMed] [Google Scholar]
- 19.Zernecke A, Winkels H, Cochain C, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. 2020:127:402–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cannoodt R, Saelens W, Saeys Y. Computational methods for trajectory inference from single-cell transcriptomics. Eur J Immunol. 2016:46:2496–2506. [DOI] [PubMed] [Google Scholar]
- 21.Wirka RC, Wagh D, Paik DT, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. 2019:25:1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armingol E, Officer A, Harismendy O, Lewis NE. Deciphering cell-cell interactions and communication from gene expression. Nat Rev Genet. 2021:22:71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Efremova M, Vento-Tormo M, Teichmann SA, Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat Protoc. 2020:15:1484–1506. [DOI] [PubMed] [Google Scholar]
- 24.Vento-Tormo R, Efremova M, Botting RA, et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature. 2018:563:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat Commun. 2021:12:1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noel F, Massenet-Regad L, Carmi-Levy I, Cappuccio A, Grandclaudon M, Trichot C, Kieffer Y, Mechta-Grigoriou F, Soumelis V. Dissection of intercellular communication using the transcriptome-based framework ICELLNET. Nat Commun. 2021:12:1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen H, Tran D, Tran B, Pehlivan B, Nguyen T. A comprehensive survey of regulatory network inference methods using single-cell RNA sequencing data. Brief Bioinform. 2020. [DOI] [PMC free article] [PubMed]
- 28.Pan H, Xue C, Auerbach BJ, et al. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation. 2020:142:2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, Zhang S, Yan P, et al. A single-cell transcriptomic landscape of primate arterial aging. Nature Communications. 2020:11:2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hadi T, Boytard L, Silvestro M, et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat Commun. 2018:9:5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo W, Wang Y, Zhang L, et al. Critical role of cytosolic DNA and its sensing adaptor STING in aortic degeneration, dissection, and rupture. Circulation. 2020:141:42–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Depuydt MAC, Prange KHM, Slenders L, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. 2020:127:1437–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams JW, Winkels H, Durant CP, Zaitsev K, Ghosheh Y, Ley K. Single cell RNA sequencing in atherosclerosis research. Circ Res. 2020:126:1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asp M, Bergenstrahle J, Lundeberg J. Spatially resolved transcriptomes-next generation tools for tissue exploration. Bioessays. 2020:42:e1900221. [DOI] [PubMed] [Google Scholar]
- 35.Asp M, Giacomello S, Larsson L, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019:179:1647–1660 e1619. [DOI] [PubMed] [Google Scholar]
- 36.Andueza A, Kumar S, Kim J, Kang DW, Mumme HL, Perez JI, Villa-Roel N, Jo H. Endothelial reprogramming by disturbed flow revealed by single-cell RNA and chromatin accessibility study. Cell Rep. 2020:33:108491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia G, Preussner J, Chen X, Guenther S, Yuan X, Yekelchyk M, Kuenne C, Looso M, Zhou Y, Teichmann S, Braun T. Single cell RNA-seq and ATAC-seq analysis of cardiac progenitor cell transition states and lineage settlement. Nature Communications. 2018:9:4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karemaker ID, Vermeulen M. Single-cell DNA methylation profiling: Technologies and biological applications. Trends Biotechnol. 2018:36:952–965. [DOI] [PubMed] [Google Scholar]
- 39.Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, Bernstein BE. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015:33:1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grosselin K, Durand A, Marsolier J, et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat Genet. 2019:51:1060–1066. [DOI] [PubMed] [Google Scholar]