

Abstract
In this review, we examine the multiple roles of ROS in the pathogenesis of melanoma, focusing on signal transduction and regulation of gene expression. In recent years, different studies have analyzed the dual role of ROS in regulating the redox system, with both negative and positive consequences on human health, depending on cell concentration of these agents. High ROS levels can result from an altered balance between oxidant generation and intracellular antioxidant activity and can produce harmful effects. In contrast, low amounts of ROS are considered beneficial, since they trigger signaling pathways involved in physiological activities and programmed cell death, with protective effects against melanoma. Here, we examine these beneficial roles, which could have interesting implications in melanoma treatment.
1. Introduction
It has become increasingly evident in recent years that the production of reactive oxygen species (ROS) is involved in the regulation of normal cell functions and that its dysregulation can be responsible for the onset of harmful events [1]. ROS are intermediate products in reduction-oxidation (redox) reactions during conversion of O2 to H2O. They include free oxygen radicals and nonradical oxidants, as reported in Table 1 that shows the main oxygen and nitrogen reactive species. Oxygen radicals include superoxide anion (O2·−), hydroxyl (·OH), peroxyl (ROO·) and alkoxyl radicals (RO·), nitric oxide (NO·), organic radicals (R·), thiyl radicals (RS·), sulfonyl radicals (ROS·), thiylperoxyl radicals (RSOO·), and disulfides (RSSR). Nonradical oxidants comprise hydrogen peroxide (H2O2), ozone (O3), singlet oxygen (1O2), organic hydroperoxides (ROOH), hypochloride (HOCl), peroxynitrite (ONO−), nitrosoperoxycarbonate anion (ONOOCO2−), nitrocarbonate anion (O2NOCO2−), dinitrogen dioxide (N2O2), and nitronium (NO2+), as well as highly reactive lipid- or carbohydrate-derived carbonyl compounds [2, 3]. It should be noted that H2O2 is an oxidizing agent that is classified as ROS since it generates the hydroxyl radical ·OH.
Table 1.
Main oxygen and nitrogen reactive species involved in cell function.
| Name | Formula | Characteristics |
|---|---|---|
| Hyperoxide/superoxide | ·O2− | Highly unstable, signaling function, synaptic plasticity |
| Hydrogen peroxide | H2O2 | Cell toxicity, signaling function, generation of other ROS |
| Hydroxyl radical | ·OH | Free radical, highly unstable, very reactive reagent |
| Alkoxyl radical | RO· | Free radical, reaction production of lipids |
| Peroxyl radical | ROO· | Free radical, reaction production of lipids |
| Hypochlorite anion | OCl− | Reactive oxygen species, reactive chlorine species, enzymatically generated by mieloperoxidase |
| Singlet oxygen | 1O2 | Induced/excited oxygen molecule, radical and nonradical form |
| Ozone | O3 | Environmental toxin |
| Nitric oxide | ·NO | Environmental toxin, endogenous signal molecule |
| Peroxynitrite | ONOO− | Highly reactive reaction intermediate of ·O2 and ·NO |
| Nitrogen dioxide | ·NO2 | Highly reactive radical, environmental toxin |
| Nitrogen oxides | NOx | Environmental toxins, including NO and ·NO2, derived from the combustion process |
RNS: reactive nitrogen species; ROS: reactive oxygen species.
1.1. Intracellular ROS Production
ROS may be produced by either nonenzymatic or enzymatic pathways and may originate from reactions involving organic compounds or ionizing radiations. Superoxide anion generates hydroxyl radical by interacting with hydrogen peroxide in the Haber-Weiss reaction. On the other hand, hydroxyl radical can be generated from H2O2 in the nonenzymatic Fenton reaction, in which Fe++or Cu++ act as single-electron donors [4]. Intracellular ROS can be produced at the level of mitochondria and plasma membranes. Production on membranes involves NAPH oxidase and 5-lipoxygenase enzymes (Figure 1). Multiple enzyme systems differently dislocated in the cell are involved in ROS production. Some of them are cytosolic enzymes, such as cyclooxygenases, myeloperoxidase, nitric oxide synthase (NOS), lipoxygenases, and microsomal cytochrome p450-dependent oxygenases [5, 6]. Others are restricted to various cell compartments, such as the mitochondria and cell membranes, as mentioned above.
Figure 1.
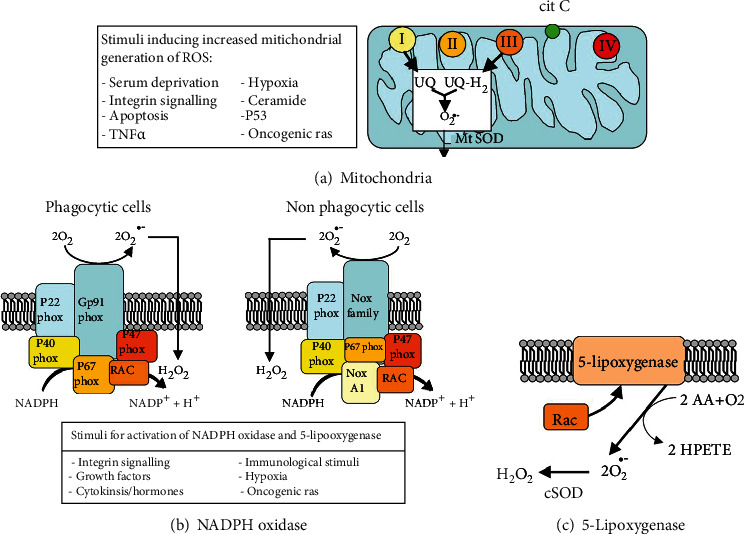
Main cellular sources of ROS. From Novo, E., Parola, M. “Redox Mechanisms in Hepatic Chronic Wound Healing and Fibrogenesis,” Fibrogenesis Tissue Repair 2008; 1: 5. doi: 10.1016/j.canlet.2008.02.035.
1.2. Plasma Membrane
It has been known for a long time that phagocytes can produce ROS at the plasma membrane level through the activation of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH) oxidase, as part of a host defense mechanism. However, it is now recognized that NADPH oxidase is also expressed in nonphagocytic cells. It is composed of membrane proteins, namely, the cytochrome b558, consisting of gp91phox or Nox2 and p22phox, and the small G protein Rap1A, as well as the cytosolic proteins p40phox, p47phox, p67phox, and G Rac2 (Figure 1). Upon activation, the cytosolic proteins are phosphorylated and recruited to the plasma membrane [7–11]. In this way, the transfer of electrons leads to the direct reduction of free oxygen. NADPH oxidases homologous to Nox2 have been classified into the following 3 groups, on the basis of their evolutionary relationships: (1) the group closest to Nox2, which includes Nox1, Nox3, and Nox4; (2) the dual oxidase groups, Duox-1 and Duox-2; and (3) Nox5 [12, 13]. Nox enzymes can be classified also into the following groups, based on their regulation: (a) the group including Nox1, Nox2, and Nox3, which need cytosolic regulator proteins to exert their function; (b) Nox4, which is constitutively activated; and (c) Nox5, Duox1, and Duox2, which bind calcium through their EF-hands and are controlled by intracellular calcium levels [13].
1.3. Mitochondria
Mitochondria represent the main site of cellular ROS generation (about 90%). In particular, O2·− is produced in the electron transport chain [1, 5] through the transferring of a single electron to oxygen by reduced coenzymes or prosthetic groups or xenobiotics reduced by specific enzymes [14]. Superoxide anion is released from complexes I and III at low levels into the mitochondrial matrix and in higher quantities into both sides of the inner mitochondrial membrane to easily reach their targets in the cytoplasm (Figure 1; [15]). Mitochondrial biogenesis and proper mitochondrial function are also essential to maintain physiological ROS levels and prevent the detrimental effects deriving from excessive ROS production. Such activity has great relevance in cell biology, since mitochondrial DNA is exposed to unbalanced ROS generation from both environmental and nutritive factors and is highly sensitive to oxidative damage. Initially, the mitochondrion was considered as an O2 sensor capable of responding to hypoxic conditions by ROS-mediated regulation of gene transcription [16]. However, mitochondria are involved in the transcriptional activity of the cell in several other ways. One of the most important for cell survival is mediated by the activation of the serine/threonine protein kinase D1 (PKD1) by mitochondrial ROS, leading to the induction of nuclear factor kB (NF-κB) [17]. This represents a novel mitochondria-to-nucleus signaling pathway involved in the activation of genes expressing antioxidant proteins [18, 19]. Indeed, this can be viewed as a homeostatic mechanism, since it results in PKD-mediated detoxification from mitochondrial reactive oxygen species.
1.4. Peroxisomes
Peroxisomes play an important role in the dynamic spin of ROS generation and scavenging. Indeed, these organelles are involved in a respiratory pathway uncoupled from oxidative phosphorylation, in which H2O2 is generated from O2 reduction [20]. Moreover, peroxisomes contain several types of ROS-generating enzymes such as xanthine oxidase that produces superoxide anion in the peroxisomal matrix and membranes [21, 22], NO synthase (NOS) that is responsible of formation of NO·, as well as enzymes involved in lipid metabolism, such as Acyl-CoA oxidases that generate H2O2 [23]. H2O2 and NO· pass through the peroxisomal membranes to take part in cellular signaling pathways.
1.5. Cell Detoxification from ROS
ROS homeostasis is maintained through the balance between production and scavenging by antioxidant mechanisms used by cells to prevent excessive ROS production and accumulation. Indeed, a persistent increase of ROS levels exceeding antioxidant cell defenses, or a deficient antioxidant system, can result in a state of “oxidative stress” that provokes severe modifications to the functions of cell components (Figure 2). Moreover, diverse types of ROS are heterogeneously distributed at different concentrations in the same tissue or in different tissues [24]. Detoxification from different types of ROS is obtained by an antioxidant system including either enzymes that are contained in different cell compartments, and selectively eliminate specific ROS species, or nonenzymatic molecules [25]. Structure and functions of antioxidant enzymes may be regulated by posttranslational changes such as phosphorylation, acetylation, and oxidation of cysteine residues [26]. In particular, oxidative posttranslational modifications (Ox-PTM) represent a switch able to change protein structure and function, including the catalytic properties of enzymes [27, 28]. In this regard, the thiol (RSH) functional group of cysteine residues within proteins represents the major cellular ROS target, especially of H2O2 and superoxide anion [29]. Therefore, cysteine residues and their intermolecular disulfide bond are considered to be “sensors” that are capable to monitor the redox homeostasis in cells [30–32]. H2O2 produced in response to growth factors oxidizes the thiol functional group of the cysteine residues contained in various signaling pathway proteins playing a role in modulation of their activities [29, 32, 33]. In fact, cystein oxidation may reversibly modulate the functions of some proteins, as shown in the following examples: (i) the inactivation of the catalytic site of protein tyrosine phosphatase SHP-2 induced by high concentrations of H2O2 allows PDGF to activate MAPK signaling [33–35], (ii) the p38 mitogen-activated protein kinase (MAPK) signaling pathway is activated in association with conformational modifications of the disulfide reductase thioredoxin (Trx) that leads to the release of the complexed protein apoptosis stimulating kinase 1 (ASK1) [36, 37], (iii) HOCl regulates the activity of matrix metalloprotease 7 (MMP-7) by oxidation of cystein residues in proenzyme metalloprotease 7 in vitro [38], and (iv) cystein oxidation increases the activity of the cell redox sensor Ca2+-release channels (ryanodinereceptors) in heart and skeletal muscle in vitro [39, 40].
Figure 2.
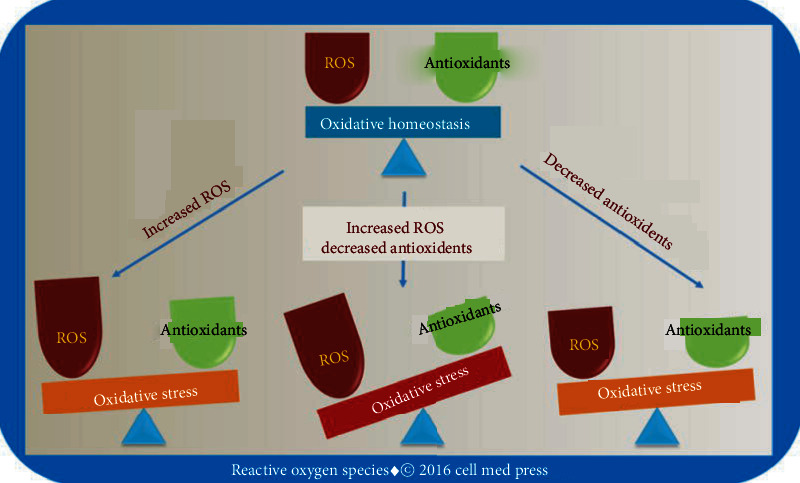
Oxidative homeostasis involves the balancing actions of ROS and antioxidants. Oxidative stress can result from excessive ROS production, decreased antioxidant activity, or both. From Li, R., Jia, Z., and Trush, M.A. “Defining ROS in Biology and Medicine,” React Oxyg Species (Apex), 2016; 1(1): 9–21. doi: 10.20455/ros.2016.803.
1.6. Antioxidant Enzymatic Systems
1.6.1. Superoxide Dismutases (SODs)
Among the antioxidant enzymes, the most representative is SODs that block the excessive activity of O2·− at the production sites [41] through the conversion of superoxide anion to H2O2 (Figure 1). SODs show high antioxidative activity although they are present at low levels in the cell. Indeed, ROS concentration is balanced by the regulation of epigenetic and posttranscriptional expression of three genes encoding in mammals for three different isoforms of SODs: copper-zinc superoxide dismutase (Cu/ZnSOD, SOD1) encoded by the sod1 gene, manganese superoxide dismutase (MnSOD, SOD2) encoded by sod2, and extracellular superoxide dismutase (EC-SOD, SOD3) encoded by sod3. SODs differ on the basis of their molecular weight, homodimer or homotetramer forms, metal cofactors, location into the cell, or the extracellular environment [42–44]. The distribution of SOD1 activity within the cell is related with the sites of superoxide anion production, thus confining the antioxidant activity in cytoplasm [42, 45], nuclei, and lysosomes [41], as well as in the matrix of peroxisomes [46]. In particular, peroxisomes use SOD1 to directly scavenge the superoxide anion O2·−produced as an additional by-product of xanthine oxidase. A small fraction of SOD1 in the mitochondrial intermembrane space [47] has a minor role as antioxidant [41]. The superoxide anion undergoes different degradation pathways, according to the site of its production and its inability to cross the lipid barrier of membranes [48]. O2·−generated in the inner membranes is dismuted to H2O2 by SOD1, while it becomes the substrate for SOD2 in the mitochondrial matrix [3, 49–51]. Extracellular SOD3 is the only specific antioxidant enzyme that eliminates superoxide in the extracellular environment. It is present in extracellular fluids, such as plasma, lymph, and synovial fluid [52, 53], although it is also strongly expressed in a cell- and tissue-specific way in various types of connective tissues and in the walls of blood vessels at the level of the extracellular matrix and on cell membranes [54, 55], in the lung [56, 57]. Binding of SOD3 to the extracellular matrix and to cell surfaces is mediated by heparin sulfate proteoglycans [58]. Cellular up-take of SOD3 and its subsequent nuclear translocation, induced by vitamin C, defend the DNA from ROS activity and/or transcription of redox-sensitive genes [59, 60]. The protective effect of SOD3 against oxidative DNA damage was demonstrated in 17β-estradiol- (E2-) induced breast tumors in female rats and in MCF-10 breast epithelial cells that significantly express SOD3 after treatment with the antioxidant butylated hydroxyanisole (BHA) or vitamin C [60]. Indeed, extracellular SOD3 can translocate to the nucleus [59]. In the extracellular matrix SOD3 regulates the bioavailability of nitric oxide in vessel walls and transforms nitric oxide into peroxynitrite, a potent oxidizing agent [61]. Similarly to SOD1, the activity of SOD3 is associated with reduction and reoxidation of the copper ion at the catalytic site. The transfer of copper to specific cellular targets occurs by the transport protein Antioxidant-1 (Atox1) that could play a role in SOD3 upregulation [62].
1.6.2. Catalase (CAT)
This enzyme is encoded by CAT gene that is involved in the synthesis of four subunits bound to heme group. CAT is located in peroxisomes and in the cytosol and acts as scavenger of hydrogen peroxide derived from SOD activity, through its conversion to water and molecular oxygen. This transformation occurs at a high rate of turnover, since 6 million molecules of H2O2 are converted per minute [63]. The reaction catalyzed by CAT includes two steps. After interaction with H2O2, CAT is transformed into an oxidized intermediate named compound I and returns in its reduced state by reacting with a second molecule of H2O2. In presence of low H2O2 concentrations and donors of one electron, compound I is transformed into an intermediate called compound II [64]. Since H2O2 is removed by different cellular enzymatic systems, catalase antioxidant activity may be different in various tissues because of its diverse expression and concentration levels [65].
1.6.3. Glutathione (GSH) System
An important detoxifying mechanism occurring in the conversion of H2O2 to O2 is the glutathione (L-γ-glutamyl-L-cysteinyl-glycine) multienzyme system that includes reduced glutathione (GSH), glutathione reductase (GR), glutathione peroxidases (GPx), and glutathione S-transferases (GST). The importance of glutathione is also linked with different activities, such as transport of amino acids across the plasma membranes, regulation of transcription factors, including NFkB, or ability to act as a cofactor of numerous ROS detoxifying enzymes. GSH is an electron-donor substrate [66] occurring in the reduction of protein disulfide bonds to cysteine in oxidative stress conditions. In this way, GSH is oxidized to glutathione disulfide (GSSG) in a cytosolic coupled reaction in which GPX catalyzes a H2O2 transformation to H2O and O2 [63]. Next, a constitutive activity of GR converts GSSG to GSH in the mitochondrial matrix. GSH can also be produced by de novo synthesis or extracellular uptake. The GSH/GSSG ratio is an indicator of cell oxidative stress [67]. GPXs can be considered as an enzyme family originating from a common gene ancestor. They are located in the cytosol and in mithocondria and play an important role as scavengers of lipid peroxides. Expression of the GPX isoforms differs in relation to the type of tissue. Gpx1, Gpx2, and Gpx6 genes are involved in glutathione-mediated age-related detoxification and redox mechanisms and are probably coregulated by an antioxidant response element (ARE) in the promoter region [68]. Consistent with the presence of the rare amino acid selenocysteine (SeCys) residue at the active site, GPXs are distinct in selenocysteine containing GPXs (GPX1-4, GPX6) and nonselenocysteine containing GPXs (GPX5, GPX7, GPX8), that include a Cys residue rather than SeCys [69, 70]. Among the selenocysteine-containing GPXs, GPX1 is located in various cell compartments such as cytosol, nucleus, and mitochondria and is ubiquitously expressed in all mammalian tissues, whereas GPX2 is expressed in breast and intestinal epithelial cells [71], but its substrates are unknown [69]. GPX3 is mostly synthesized in kidney cells, but it is considered as a plasma component, since it is secreted extracellularly as a result of lack of a retention signal for the endoplasmic reticulum [69]. From the blood, GPX3 binds to the basement membranes of the gastrointestinal tract, epididymis, and lung epithelia [72]. GPX3 uses various secreted thiols as reductants, such as thioredoxin and glutaredoxin, in addition to GSH as an electron donor. GPX4 (PHGPX) acts with GSH to reduce phospholipid hydroperoxides contained in membranes and lipoproteins [73]. It is present in three different isoforms in cytoplasm, mitochondria, and sperm nucleus. The cytosolic form is ubiquitously expressed, while the mitochondrial and nuclear forms are mainly expressed in testis [69]. Among GPXs that do not contain seleno-cysteine, GPX5 plays a role as a phospholipid hydroperoxidase, and it is mostly and differentially expressed in three different transcripts in mouse epididymis, where it prevents cell oxidative stress and DNA mutations [69, 74–76]. GPX7 is a nonselenocysteine-containing phospholipid hydroperoxide glutathione peroxidase (NPGPX) that is expressed in many tissues as a mitochondrial, as well as a nonmitochondrial form in the cytosol [77]. GPX7 protects against singlet oxygen-generated lipid peroxidation by removing lipid hydroperoxides from cell membranes [78, 79]. In addition, GPX7 plays an important role in oxidative stress as an endoplasmic reticulum- (ER-) resident sensor/transducer of signals in pathways involved in the regulation of ROS levels, through its interaction with target proteins [70, 80]. Furthermore, hydroperoxide reduction by GPX7, in association with ER oxidoreductin1 (Ero1) flavoproteins and GRP78 chaperone, is correlated with formation or rearrangement of disulfide bonds in oxidative protein folding mediated by protein disulfide isomerase (PDI) [81]. Indeed, GSH and PDI are both considered alternative substrates of GPX7, since the active site of GPX7 oxidizes PDI when GSH concentration is low [82]. GPX8 is a transmembrane protein located in the ER, like GPX7, with which it shares similarity in amino acid sequences and domains, the association with Ero1, and PDI peroxidase activity in disulfide bond formation with the production H2O2 [83], which is quickly eliminated by GPX8 to protect cells from oxidative stress [70, 84]. Moreover, GPX8 has been shown to be a cellular substrate of hepatitis C virus NS3-4 protease [85]. GSTs include eight classes of isoenzymes (Alpha, Mu, Pi, Theta, Kappa, Sigma, Zeta, and Omega). In addition to their detoxification role, consisting in the reduction of hydroperoxides with the formation of GSSG, or conjugation of GSH with nonsubstrate ligands in the metabolism of exogenous compounds, such as chemical carcinogens or drugs, they also play a noncatalytic activity, such as membrane transport [86, 87]. GSTs are mainly located in cytosol, but they were also detected in nucleus and in mitochondria. In particular, glutathione S-transferase Kappa 1 (GSTK1) is present in peroxisomes, where it plays a fundamental role in the elimination of lipid peroxides [88]. It shows a TRX-like domain analogous to that of GPX [89].
1.6.4. Thioredoxin (Trx)
Trx is a cellular redox system, including Trx and Trx reductase. Trxs include three isoforms coupled with GSH/GSSG system in redox regulation: Trx1 that is located in the cytoplasm and moves into the nucleus in oxidative stress situations, where it interacts with transcription factors, whereas Trx2 is present in mitochondria, and Sp-Trx3 is spermatid-specific [90, 91]. NADPH-dependent Trx reductase catalyzes the reduction of disulfide bonds of two cysteine residues contained in the catalytic site of Trx [3]. Trx reductase is protected by calcium from nitrosoureas deactivation in metastatic melanotic and amelanotic melanomas [92].
1.6.5. Peroxiredoxins (PRDXs)
PRDXs are thioredoxin peroxidases that catalyze the reduction of organic hydroperoxides, H2O2, and peroxynitrite [3, 93]. In mammalian cells, PRDXs include six isoforms that are grouped into three classes, namely, the 1-Cys (Prx VI) atypical 2-Cys (Prx V) and 2-Cys subgroups (Prxs I–IV), along with the recycle of sulfenic acid with formation of a disulfide linkage between two cysteine residues in their active site [94]. PRDXs are distributed in the cytosol, mitochondria, peroxisomes, plasma membranes, nucleus, and endoplasmic reticulum and are involved in programmed cell death and cell proliferation [2, 23, 66, 90, 94–96]. Among PRDXs, the function of PRDX1 as an antioxidant enzyme is currently debated, since it has been found to be extremely susceptible to oxidative stress [97]. In contrast, under oxidative stress conditions, an important role seems to be played by the ER-resident protein PRDXIV, which acts as H2O2 sensor for protein folding [98, 99]. However, PRDXs show a low catalytic activity compared to the efficiency of other antioxidant enzymes, such as GPXs or catalase. Despite this, PRDXs are the main scavengers of signaling ROS, and their activity is regulated by phosphorylation and sulfenylation.
1.6.6. Sulfiredoxins (SRXs)
These make up a family of proteins containing a conserved cysteine residue and catalyze the reduction of sulfenylated PRDX, which reactivate them.
1.6.7. Glutaredoxins (GRXs)
GRXs reduce cysteine residues in disulphide substrates and are reduced, in turn, by NADPH-dependent glutathione reductase [100].
1.7. Antioxidant Nonenzymatic Systems
Nonenzymatic antioxidants also play a critical role in neutralizing free radicals and oxidants inside cells, since they modify ROS to form less reactive species. They include water-soluble antioxidants that are present in cytosol, such as ascorbate, reduced GSH, which plays a role as a spontaneous antioxidant, and low molecular-weight scavengers such as coenzyme Q-10, lipoic acid, and lipid-soluble antioxidants, such as a-tocopherols, carotenoids, flavonoids, and omega-3 acids, as well as trace metals, such as selenium and zinc that can be externally supplied [101]. In addition to its role as spontaneous antioxidant, GSH can function as an electron-donor substrate in enzymatic reactions for ROS detoxification [66]. Cancer cells have developed the ability to specifically upregulate antioxidant mechanisms, in order to elude the negative effects of the high ROS levels induced by genetic changes and altered metabolism. Therefore, these homeostatic systems give cancer cells a selective advantage for their survival. This is considered an apparent paradox, since high production of ROS is associated with elevated antioxidant levels in cancer [102, 103]. Several lines of evidence indicate that ROS may act as a double-edged sword, since they function either as harmful agents or as beneficial signaling molecules. Thus, they can function as “bad” or “good” molecules depending on type, quantity, duration, and localization of specific ROS production in cell physiology [103]. On the “huggly” side, one should consider the aptitude of ROS to induce chronic diseases, including cancer [104], which is linked to the ability of excessive ROS production to produce cell or tissue damage. Alternatively, ROS produced by nonphagocytic cells may trigger a physiological response to many extracellular stimuli, whereas ROS produced by phagocytic cells defend the host against microorganisms.
1.8. ROS as Harmful Molecules
ROS can induce deregulation of redox-sensitive pathways and considerable damage to macromolecules, such as oxidation of proteins with enzyme inhibition, peroxidation of lipids at plasma membranes with production of aldehyde derivatives, DNA single- and double-strand breaks, base substitutions, formation of adducts [105], and chromosomal aberrations [106], as well as genomic instability, epigenetic, and gene expression modifications involving tumor suppressor genes and oncogenes [63, 107]. Moreover, ROS can stimulate signaling pathways, such as Ca2+-signaling, protein phosphorylation, and transcription factor activation or act as second messengers in the activation of pathways triggered by ligand-receptor interaction [108].
1.9. ROS as Beneficial Molecules
ROS are not only damaging for cells but are also considered critical signaling molecules in the regulation of various pathways. In fact, ROS can act as beneficial molecules, since they affect several cell functions. It is known that ROS determine a positive or negative cell response on proliferation in relation to their concentration. In breast cancer MCF-7 cells, low levels of hydrogen peroxide and increased superoxide anion concentrations are linked to reduce activity of MnSOD, leading to increased cell proliferation, while the overexpression of MnSOD leads to augmented production of H2O2 and suppression of the malignant phenotype, with inhibition of hypoxic accumulation of hypoxia-inducible factor-1 (HIF-1) and vascular endothelial growth factor (VEGF) [109]. It was reported that ROS play diverse roles on lung carcinoma H460 cells, depending on the type and quantity of specific ROS produced [110]. In particular, ·OH promotes cell motility, while O2·− and H2O2 play an inhibitory role in cell migration and invasion. Cell migration is promoted by treatment with catalase, whereas it is inhibited by hydrogen peroxide [110].
1.10. Factors Affecting the Biological Functions of ROS
It is now well established that the concentration of ROS within cells plays a crucial role on cell functions [4, 102]. Low ROS levels have protective effects on normal physiological activities, serving as effectors in the immunological defense against pathogens, mediators in signaling pathways involved in various cellular processes, such as proliferation, differentiation, survival, and critical intermediates in vascular tone regulation, control of ventilation, and erythropoietin production. At subtoxic concentrations, H2O2 acts as an intracellular messenger in pathways regulated by different growth factors, such as PDGF and EGF. Increased concentrations of H2O2 inactivate protein tyrosine phosphatases (PTPs) and the lipid phosphatase PTEN, leading to accumulation of phosphorylated proteins capable of activating various signaling pathways [111]. On the contrary, at moderate or high concentrations, ROS stimulate the expression of genes reactive to stress and can produce harmful oxidative DNA damage. However, the effects of high levels of ROS depend on the cell type, since they can generate harmful consequences, such as oxidative stress, when they act on normal cells, or host-beneficial effects when the act on cancer cells, by causing their growth suppression through cell cycle inhibition and apoptosis. Therefore, in addition to the concentration of ROS, also the cell type influences the response to oxidative stress. A complex interplay occurs between ROS and various cell systems, and this must be considered in evaluating the effects of ROS on cell functions. Many factors come into play in the balance between advantageous and detrimental effects. In fact, in addition to the amount of ROS and the cell type, various conditions, including the length of oxidant production, the kind of reactive species produced, and the source of production (plasma membranes, cytosol, nucleus, mitochondria, peroxisome, endoplasmic reticulum), may contribute to trigger a physiological response or cell/tissue damage. Thus, both the type of ROS and their local concentration collectively determine whether redox signaling or oxidative stress-induced damage occurs. The location of the molecular target(s) and the associated physiological or pathological conditions should also be considered [26, 112]. These are only the most cited factors determining the cell fate. As the imbalance in ROS generation and removal is linked to the development or exacerbation of many pathological conditions, like cancer, chronic inflammation, metabolic diseases, aging, and neurodegenerative disorders, it has become increasingly clear that ROS metabolism must be kept under strict control, both in terms of quantity and quality. Therefore, a thorough understanding of such issues may help in the prevention and management of several diseases and in reverting the harmful effects of ROS in living systems.
1.11. Role of ROS as Messengers
A persistent increase of ROS causes modifications in gene expression and activation of cell death pathways, such as apoptosis or necrosis, thereby significantly contributing to disease onset and progression. Specificity in ROS signaling pathways is linked to sensors that are sensitive to variations in ROS cell concentration and regulate the expression of scavengers to maintain a basal ROS level [100]. As mentioned above, elevated concentrations of different kinds of ROS are responsible for cell damage, whereas moderate levels of ROS act as regulatory mediators in the activation of redox-sensitive signaling pathways in response to the action of various extracellular stimuli. These include cytokines, such as transforming growth factor β (TGF-β) [113, 114], TNF-alpha [115], and interleukin-1 b [116], as well as growth factors [116–118], such as platelet-derived growth factor (PDGF) [119] and epidermal growth factor (EGF) that trigger H2O2 generation [120]. These extracellular stimuli are in turn associated with transcriptional cell changes related to oxidative stress [121]. The specificity of ROS on diverse signaling pathways also depends on the pKa of signaling molecules, the redox status around the signaling pathway, and the total redox status in the cell [122]. In this context, both extracellular ROS, as hydrogen peroxide, or oxidants produced within the cells, as superoxide anion, may act as second messengers in signaling transduction systems and in regulating pathways sensitive to the redox status [5, 123]. Such pathways involve signaling molecules, including p38/mitogen activated protein kinases (MAPKs), phosphatidyl inositol-3 kinase (PI3K)/Akt [124, 125], various protein kinase C (PKC) family members, and c-Jun N-terminal kinase (JNK) [126, 127], that are modulated by NADPH oxidase and pathway-specific transcription factors such as nuclear factor-κB (NF-κB) [128], activator protein 1 (AP-1), hypoxia inducible factor (HIF), and p53. Only few signaling molecules and pathways have been found to be activated by mROS increase. In particular, proapoptotic signaling molecules, such as c-Jun N terminal kinase (JNK) and p38, in response to high levels of mROS induce apoptosis through caspase activation [129]. On the contrary, Akt, activated in response to exogenous ROS or growth factors, protects cells from apoptosis mediated by mROS through downregulation of Bcl-2 family members. In T lymphocytes, ROS activate two distinct pathways: superoxide anion triggers the proapoptotic FasL signal pathway, and H2O2 production regulates the proliferative signal by ERK activation [130, 131].
1.12. ROS and Cancer
Compared to normal cells, cancer cells are exposed to high levels of ROS, which may represent a response to increased metabolism, elevated production of growth factors and activation of cell signaling pathways [120], altered mitochondrial or peroxisomal functions, increased activity of oxidant enzymes, or increased generation of ROS in inflammation [132, 133]. On the other hand, ROS in cancer cells are involved in the regulation of signaling molecules during cell cycle progression and proliferation [134], survival, apoptosis [135], intercellular adhesion, cell motility, and angiogenesis [3]. This effect may be of particular interest, since ROS regulate signal transduction pathways during apoptosis, and antioxidants such as N-acetylcysteine, or overexpression of MnSOD can block or suspend apoptosis. An example is offered by SOD3 which regulates cell proliferation in a dose-dependent manner. In anaplastic thyroid cancer cells, high SOD3 levels are correlated with cell growth arrest through p53-p21 signaling, while decreased SOD3 mRNA expression correlates with increased malignant cell proliferation mediated by the Ras oncogene signal pathway [136, 137]. Indeed, ROS affect with different signals the transduction pathways and the expression of genes involved in cell proliferation. In this way, superoxide plays a role of second messenger in the Ras and Rac signaling pathway and is involved in uncontrolled cell transformation and proliferation [138], despite H2O2 best meets the criteria required of a second messenger such as enzymatic production and degradation [139]. The main features of cancer cells, along with melanoma and nonmelanoma skin tumors, are the generation of extracellular superoxide anion and the so-called “H2O2-catabolizing phenotype” [140–145]. The catalase activity at the membrane level prevents transformed cells from receiving signals promoting apoptosis. In contrast, interestingly, catalase inhibition activates apoptosis only in cancer cells and not in normal ones [146]. When Nox1 is silenced, the proapoptotic effect of catalase inhibition is abolished. Therefore, the malignancy is held by anion superoxide generation and H2O2 elimination, such indicating the beneficial role for H2O2 in specifically eliminating cancer cells through apoptosis. The role of the hydrogen peroxide in inducing apoptosis is currently deemed to be of considerable importance so as to promote therapeutic plans that provide for the inhibition of catalase membrane [147]. Therefore, the promalignant effects of ROS are not to be assigned to hydrogen peroxide, which activates apoptosis, but rather to the production of superoxide ion. More recent studies have highlighted the role of redox imbalance in skin carcinogenesis and its complexity, due to the multiple and interrelated pathways involved. In the skin, low levels of ROS induce physiological responses, such as wound healing and skin repair, besides dermal angiogenesis [148]. If ROS accumulation occurs in response to external stimuli or in pathological conditions, DNA damage may result in carcinogenesis and development of melanoma, spinous cell carcinoma, and basal cell carcinoma. However, if toxic levels of ROS are reached, cell death can disrupt tumor development. ROS act in several ways in inducing skin cancer: they can activate protooncogenes, such as BRAF, NRas, or suppress antioncogenes, such as p53, or modify gene expression through epigenetic alterations, or affect the related signal pathways [149]. ROS affect a series of transcription factors that are involved in carcinogenesis, such as those regulating proliferation, metastasis, angiogenesis, and inflammation. Therefore, ROS has become a privileged target in cancer therapy [150].
1.13. ROS in Signal Transduction with Negative Effects in Melanoma
Although ROS have been considered just as harmful agents in the past, recent evidence supports their crucial role as regulator molecules in various signaling pathways, whose effects are context-dependent. An altered redox status occurs in melanoma development through the activation of different signal transduction pathways mediated by NADPH oxidase family members (including NOX 1-5 and DUOX1/2) and other ROS-producing molecules. Figure 3 depicts the balance between ROS production and detoxification in melanocytes. Different studies report changes in expression levels of proteins involved in oxidant mechanisms, but the exact molecular mechanisms responsible for ROS-dependent promotion of melanoma development have not yet been fully elucidated. NOXs/DUOXs are responsible for maintaining optimal cellular levels of ROS. Tumor cells may produce high levels of ROS that are the result of deregulation of NOXs present either in melanoma (NOX1 and NOX4) or in various other types of tumors, such as prostate cancer (NOX1 and NOX5) and glioblastoma (NOX4) [151]. Nox1 and Nox4 are overexpressed in melanoma cell lines, including metastatic cells. In particular, in the melanoma cell line Wm3211 the overexpression of Nox1 correlated with increased cell invasion. Nox4 mediates melanoma proliferation by regulating G2-M cell cycle progression, and modifications in Nox4 expression are present in the early stages of melanoma development [152]. Interestingly, expression profiles for Nox4 show that proliferating normal epithelial melanocytes highly express only Nox4 and its associated subunit p22phox, while gp91phox (Nox2) is only slightly expressed. In contrast, some human melanoma cells strongly express the NADPH oxidase components p22phox and gp91phox in plasma membrane and the cytosolic p67phox, whereas p47phox expression level is low [153]. In B16 mouse melanoma cells, Nox4 is expressed in large quantities compared to other isoforms. Nox4 and ROS production are increased by the α-melanocyte-stimulating hormone (α-MSH), via the microphthalmia-associated transcription factor (MITF), which induces Nox4 gene expression [154]. Since melanin synthesis induces the silencing of Nox4 gene by increasing tyrosinase gene expression, ROS generation could be also inversely correlated with melanin formation via a negative feedback mechanism of regulation that may be altered in skin pathologies [154]. Upregulation of Nox1 is an early event in melanoma transformation [155] and Nox1 overexpression regulates melanoma invasion through upregulation of matrix metalloproteinase-2 [156]. Inhibition of Nox1 activity blocks migration of melanoma cells [157]. NADPH oxidases are responsible for the costitutive activation of the transcription factor NFkB by malignant melanoma cells which in turn results in increased cell proliferation. NADPH oxidase inhibitors reduce proliferation. In particular, the NADPH oxidase inhibitor diphenylene iodonium (DPI) inhibits the costitutive DNA binding of transcription factors to NFkB and cAMP-response elements, thus suggesting a crucial role of NADPH oxidase in melanoma proliferation [153] and of antioxidant therapy for the interruption of oxidant signaling in melanoma [158]. This assumption is confirmed by more recent studies showing that some enzymes involved in the regulation of redox status, such as peroxiredoxins (Prx) I and Prx II, are downregulated in melanoma as compared to dysplastic and benign naevi. Moreover, sulfiredoxin and Prx IV expression apparently showed a protective role in melanoma and was associated with a better prognosis [159]. The complex interplay between UV radiation and oxidative stress leading to melanoma is shown in Figure 4.
Figure 3.
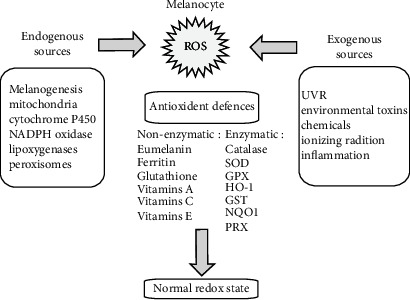
Listed are various endogenous and exogenous sources of ROS in melanocytes. ROS levels are decreased by the action of a number of antioxidants. From Denat L. et al. “Melanocytes as Instigators and Victims of Oxidative Stress,” Journal of Investigative Dermatology, 2014 Jun; 134(6): 1512-1518. doi: 10.1038/jid.2014.65.
Figure 4.
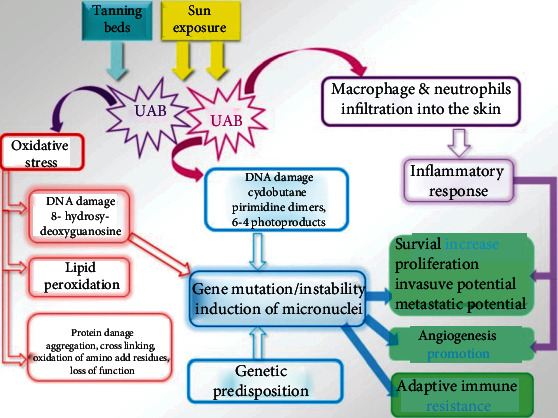
UV radiation and oxidative stress promote melanoma formation. Both sun exposure and tanning beds are sources of UV. Among these, UVA is mostly responsible for oxidative stress, causing DNA and protein damage as well as lipid peroxidation. DNA damage leads to gene instability and mutagenesis, impacting the main cellular processes involved in cancerogenesis, cancer growth, metastasis, and adaptive immune resistance. Likewise, UVB modulate those processes by the activation of inflammatory response. From Wróbel, S., Przybyło, M and Stępień, E. “The Clinical Trial Landscape for Melanoma Therapies,” J. Clin. Med. 2019, 8(3): 368. doi: 10.3390/jcm8030368.
A first mechanism by which excessive ROS production can promote melanoma formation and development is simply via direct induction of DNA damage and mutagenesis. ROS, however, may also favor the development of melanoma and other tumors by modulating the activation of signaling pathways and transcription factors, such as NF-κΒ. For example, ROS-induced oxidation of LC8, a multifunctional protein of the dynein motor complex, can increase NF-κΒ activation. This is linked to the ability of reduced LC8 to bind to the NF-κB component I-κBα in a redox-dependent manner, blocking its phosphorylation by IKK. ROS-oxidized LC8 dissociates from I-κBα, leading to NF-κB activation [160]. NF-κB activation, in turn, could favor melanoma progression through its antiapoptotic effects and by creating an inflammatory microenvironment [161]. ROS, however, may also inhibit NF-κB activation via oxidation of thioredoxin, a protein that, when present in the nucleus in a reduced state, increases NF-κB activity by promoting its binding to DNA [160]. Moreover, prolonged oxidative stress may lead to direct oxidation of NF-κB heterodimers and reduced DNA binding. Thus, while acute oxidative stress leads to increased NF-κB activation, sustained ROS production may have more complex effects.
1.14. ROS as Beneficial Molecules in Melanoma
ROS can be considered beneficial molecules in melanoma as they may, under certain conditions, activate programmed cell death. In association with selected inhibitors of cell growth, ROS may be mediators in signal pathways leading to apoptosis. The proapoptotic antitumor antibiotic DC-81-enediyne induces death of human melanoma A375 cells by the involvement of ROS, caspase-3 activation, PARP degradation, and activation of the p38/MAPK and AP-1 signaling pathways [162]. Moreover, curcumin activates apoptosis in the same cell line [163]. Treatment with Parthenolide too induces apoptosis through ROS generation, leading to depletion of proteinthiols and glutathione (GSH) and dissipation of the mitochondrial membrane potential (Dcm), with condensation and fragmentation of chromatin and activation of caspase-independent and AIF-mediated apoptosis in melanoma cells [164]. Cytokine melanoma differentiation associated gene 7- (mda-7-) induced apoptosis in melanoma cells is mediated by ROS that induce significant decrease in both BCL-2 and BCL-XL and upregulation of BAX and BAK [165]. The generation of ROS is the signal pathway triggered by Benzofuroxan N-Br and N-I derivatives to induce cytoxicity and inhibition of AKT activation in melanoma B16F10-Nex2 cells [166]. A similar mechanism is employed by Spatane diterpinoids isolated from the brown marine algae Stoechospermum marginatum that they were shown to induce apoptosis in B16F10 melanoma cells in a concentration-dependent manner through ROS generation. The resulting oxidative stress induced an imbalance in Bax/Bcl-2 ratio that disrupted the inner mitochondrial transmembrane potential (ΔΨm) resulting in cytochrome c redistribution to the cytoplasm and activation of caspase-mediated apoptotic pathway. Moreover, apoptosis was reached also through another signaling pathway involving the deregulation of PI3K/AKT. Such effects were also shown in C57BL/6 mice bearing B16F10 melanoma. Spatane diterpinoid from the brown algae, Stoechospermum marginatum, induces apoptosis via ROS-induced mitochondrial-mediated caspase dependent pathway in murine B16F10 melanoma cells [167]. Another mechanism employed by ROS as antimelanoma agents was shown by studies reporting that Simvastatin, a prooxidant agent responsible for an increased amount of intracellular ROS and overexpression of catalase and peroxiredoxin-1, is able to induce senescence in human melanoma cells by activation of p53/p21 pathway [168]. A similar effect is exerted by Nexrutine that increases the constitutively elevated oxidative stress in melanoma cells to inhibit their survival mediated by PI3K/AKT/mTOR [169]. Photodynamic therapy-induced ROS increase has been shown to significantly reduce melanoma proliferation through cell autophagy mechanism, such supporting the notion that the oxidative stress is responsible of melanoma cell damaging [170]. Several compounds are able to induce apoptosis in melanoma cells through ROS generation by either mitochondria-dependent [171] or mitochondria-independent pathways [172]. The former mechanism is engaged by cerium (Ce) oxide nanoparticles (CNP; nanoceria) which selectively kills A375 melanoma cells through the increase of ROS concentration, prevalently hydrogen peroxide, at mitochondrial level. Such event occurs concomitantly to mitochondrial thiol oxidation and is followed by modifications in mitochondrial bioenergetics, dynamics, and cristae morphology, and ultimately by mitochondrial dysfunction-induced cell death [173]. Moreover, increased concentrations of ROS can reduce melanoma development through activation of cell cycle regulators and arrest of cell cycle in G2/M phase by the inhibition of Cdc25c and cyclin A [171]. Recently, several small molecule ROS inducers have been employed with the aim to pharmacologically elevate intracellular levels of ROS through various mechanisms and to target and disrupt tumor cells in a selective way [174]. For example, atmospheric gas plasmas (AGP) are able to upregulate intracellular ROS and to induce apoptosis in melanoma, but not in normal melanocyte, cells by oxidative stress-induced activation of the TNF-ASK1-JNK/p38–caspase-3/7 apoptotic pathway [175]. More recently, it has been shown that VB1, a compound purified from the seed of the Chinese herb Vitex negundo, inhibits melanoma cell proliferation and promotes apoptosis by increasing ROS levels, thereby causing DNA damage and cell death. The effect was selective for melanoma cells, including BRAF inhibitor-resistant cells [176]. Similar effects were exerted by the novel calchone derivative lj-1-59 that, by inducing ROS elevation, blocks melanoma cell proliferation at the G2/M phase and triggers different apoptosis pathways [177].
Treatment of melanoma through the combined effects of two different ROS inducers has been successfully achieved employing low-dose UVA irradiation and brusatol (BR), a quassinoid isolated from Brucea javanica plant. Both cause ROS accumulation, the former one by endogenous photosensitization [178], the latter one by deregulating nuclear factor E2-related factor 2 (Nrf2), a transcription factor belonging to the cap “n”collar family of leucine-zipper (b-ZIP) proteins. Following oxidative stress, Nrf2 is activated and induces the expression of genes, such as heme oxygenase 1 (HO-1), NAD(P)H:quinone oxidoreductase-1 (NQO1), and glutathione S-transferase (GST) drug transporters to restore homoeostasis [179]. On the contrary, it was clearly shown that inhibition of the Nrf2-mediated antioxidant defense system sensitizes cancer cell to therapy [180]. Indeed, it was reported that BR is a potent inhibitor of Nrf2 activation, and that in such a way, it can reduce tumor proliferation and cancer chemoresistance [181]. More recently, it was shown that, in A375 melanoma cells, cotreatment with (UVA + BR) either inhibited melanoma cell growth and proliferation both in vitro and in vivo and/or activated cell apoptosis. The UVA-induced increase in of ROS was further enhanced by the BR-mediated caused reduction of Nrf2 expression with a consequent inhibition of AKT signaling. Therefore, cotreatment of UVA and BR reduced melanoma development by blocking AKT-Nrf2 cascades [182]. These data have promoted studies showing that the excess of antioxidants is detrimental in melanoma treatment as it causes melanoma cell metastasis (Figure 5).
Figure 5.
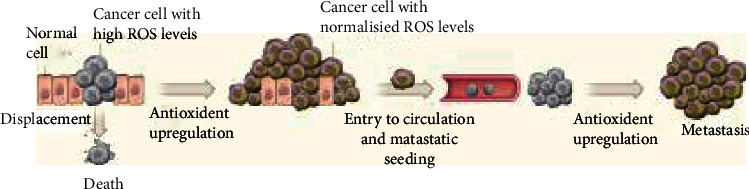
High metabolic activity can lead to increased ROS levels and cell death in melanoma and other cancer cells. Conversely, effective antioxidant responses can result in cell growth and metastasis. From Harris, I., Brugge, J. “The Enemy of My Enemy Is My Friend”. Nature 2015 12 (11); 527 (7577): 170-1. doi:10.1038/nature15644, commentary to Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. “Oxidative Stress Inhibits Distant Metastasis by Human Melanoma Cells.” Nature. 2015 Nov 12; 527 (7577): 186-91. doi:10.1038/nature15726. Epub 2015 Oct 14.
1.15. ROS and Epigenetic Pathways in Melanoma
In the last decades, several evidences pointed to the role of epigenetics in cancer onset and progression as key factor involved in tumorigenesis, even though cancer was generally considered to be the resultant of genetic mutation accumulation [183]. Really, it has been clearly shown that also the genetic view regards changes in gene expression prevalent in cancer [183–185]. Closer studies of the epigenetic regulatory mechanisms showed that a gene expression state is determined not only at the transcriptional level but even more at posttranscriptional level. A complex scenario emerged where the simple genetic concept that a mutation in oncogenes or suppressor genes led to cancer by an abnormal or reduced expression, respectively, is now substituted by a variety of mechanisms involving either gene chemical modifications or an intricate network of regulatory RNAs.
The genetic path to cancer is relatively straightforward: mutation of tumor suppressors and/or oncogenes causes either loss or gain of function and abnormal expression. The epigenetic pathway to cancer is more complex and is determined by changes in chromatin structure, including DNA methylation, histone variants and histone modifications, and nucleosome remodeling, as well as small noncoding regulatory RNAs [186]. During tumor initiation and progression, the epigenome goes through multiple alterations, including a genome-wide loss of DNA methylation (hypomethylation), frequent increases in promoter methylation of CpG islands, changes in nucleosome occupancy, and modification profiles. It is now believed that oxidative stress may affect the so-called epigenetic machinery, since an emerging role of ROS as inducers of epigenetic changes has been established. To understand the molecular mechanisms underlying the redox balance, the use of epi-drugs that have been available in the last few years proved to be crucial. They can take part in epigenetic processes by the reactions of nucleophilic substitution.
ROS are active intermediaries in either DNA methylation or histone chemical modifications, but they have been also implicated in the regulation of microRNA (miRNA) pathways, by altering mRNA stability and their transport inside the cytosol. ROS may induce an aberrant hypermethylation by increasing the expression of DNA methyltrasferases (DNMTs). The methylation of CpG islands in the promoter of oncosuppressor genes leads to gene silencing and tumor onset [187]. On the other hands, ROS are also able to induce a global hypomethylation of the genome [149] and, intriguingly, the hypomethylation of histone H3K9 leads to melanoma epigenetic instability [188]. ROS may interfere with gene expression by affecting also the histone acetylation/deacetylation level by their activity on acetyltransferase (HAT) or histone deacetylase (HDAC). Gene expression alterations caused by ROS-induced histone modification depend either on the amino acid residues involved or the level of histone acetylation [189]. It is generally believed that histone deacetylase (HDACi) inhibitors may trigger cancer cell death through ROS generation [190, 191]. In this context, the histone deacetylase inhibitor vorinostat, which is well known to induce 8-oxo-G, a marker of oxidative DNA damage [192], was shown to dramatically increase ROS levels only in BRAF (V600E) mutant melanomas, which acquired resistance to MAPK inhibitors. This type of melanoma already displayed elevated levels of ROS that were further increased by vorinostat treatment. In such a way, a very significant tumor regression was observed, as consequence of the toxic elevation of cell ROS. Interestingly, the vorinostat effect was BRAF (V600E) mutant and MAPK inhibitors-resistant melanoma specific [193]. Among the HDACs involved in cancer development, HDAC6 has caught the attention of many scholars, and recently, it has been shown to be related to melanoma onset [194]. Previous studies have shown that histone deacetylase 6 (HDAC6) plays critical roles in many cellular processes related to cancer. However, its biological roles in the development of melanoma remain unexplored. Our aim was to investigate whether HDAC6 has a biological role in human melanoma development and to understand its underlying mechanism. In the present study, HDAC6 expression was upregulated in melanoma tissues and cell lines. Knockdown of HDAC6 significantly inhibited the proliferation and colony formation ability of A375.S2 cells, promoted cell arrest at G0/G1 phase and apoptosis. Additionally, western blotting analysis showed that HDAC6 silencing suppressed Bcl-2 and enhanced Bax levels, activated caspase-9 and caspase-3, further activated the release of cytochrome c from mitochondria into the cytoplasm, and finally induced apoptosis involving the mitochondrial pathway. Knockdown of HDAC6 triggered a significant generation of ROS and disruption of mitochondrial membrane potential (MMP). Furthermore, the ROS inhibitor NAC reduced HDAC6 siRNA-induced ROS production and blocked HDAC6 siRNA-induced loss of MMP and apoptosis. NAC also significantly blocked HDAC6 siRNA-induced mtDNA copy number decrease and mitochondrial biogenesis and degradation imbalance. In conclusion, the results showed that knockdown of HDAC6 induced apoptosis in human melanoma A375.S2 cells through a ROS-dependent mitochondrial pathway. Interplay between ROS signaling and miRNA pathway was described as possible cause of cancer, since both are dysregulated in this pathology [195]. But not only that, in several types of cancer, including skin cancer, there is a reciprocal relationship between ROS and miRNA profiles. Indeed, miRNAs may be regulated, induced, or repressed, by ROS as well as by hypoxia, or in turn are themselves capable of inducing ROS increase [196]. miRNAs are greatly implicated in the regulation of oxidative stress by interacting with the nuclear factor erythroid 2-related factor 2 (Nrf2), a transcription factor that controls the expression of several genes involved in the response to oxidative stress. As some miRNAs modify the expression of genes responsible either for ROS production or antioxidant response, they have been defined “redoximiRs” [197]. Recent studies have evidenced the double way in which ROS and miRNA interact and the underlying mechanisms [196]. A microRNA profiling analysis revealed that exposure to H2O2 modifies the set of microRNA contents through epigenetic modifications, such as alteration of the methylation status of miRNA genes or biogenesis. In such a way, the oxidative stress may interfere with carcinogenesis not only by DNA damage-induced mutations but also through epigenetic alterations of miRNA genes. Similarly, miRNAs are capable of regulating intracellular ROS levels by targeting enzyme involved in ROS generation and elimination [198]. Dysregulation of miR-125b following ROS exposure has been involved in skin carcinogenesis via interfering with the expression of genes involved in cell proliferation [198]. Many other miRNAs silencing the expression of oncogenes able to induce cell proliferation have been shown to be hypermethylated by the oxidative stress in melanoma, such as miR-34b, miR-34c, miR-148, and miR-9. Thereby, their targets MYC and cyclin-dependent kinase 6 resulted to be overexpressed leading to cell transformation [199]. Recently, special attention was paid to the complex network of long noncoding RNAs (lncRNA), miRNAs, and mRNAS, named “competing endogenous RNA (ceRNA)” that regulates gene expression in cancer cells by posttranscriptional mechanisms, as firstly described by Salmena et al. [200]. lncRNAs are longer than 200 nucleotides and are unable to exert protein-coding activity. They function as sponges towards miRNA, preventing them from blocking gene expression [201]. According to the hypothesis of Salmena et al., lncRNAs, by affecting mRNA expression through the interaction with miRNA response elements, widen genetic information, and if an alteration occurs in the network equilibrium, it may have great implications in cancer pathogenesis. Recently, therapeutic interventions based on lncRNA analysis improved prognosis and quality of life in melanoma patients in early stages of disease or with Breslow thickness less than 2 mm [202]. Six lncRNAs (AL050303, LINC00707, LINC01324, RP11-85G21, RP4-794I6.4, and RP5-855F16) have been shown to affect MAPK pathways, immune and inflammatory responses, and focal adhesion pathways, thereby suggesting that they may be significant in the prognosis of patients with melanoma [203]. More recently, a signature of seven lncRNAs has been implicated in melanoma metastasis, as they are differentially expressed in primary and metastatic melanoma and are independently related to overall survival [204]. Among the identified seven lncRNAs, myocardial infarction associated transcript (MIAT) aroused great interest and significance as prognostic marker in melanoma. It was shown to be expressed in patients with a better prognosis, and its potential role in improving immune response was evidenced in light of a greater sensitivity to immunotherapy. Interestingly, H19 that was found to be dysregulated in metastatic melanoma, as compared with primary melanoma, and to be associated with a poor diagnosis constitutes an integral part of the hypoxia, p53, and cancer pathway [205]. Several lncRNAs have been correlated with an increase of RO and shown to be regulators of cardiovascular diseases related to hypoxia, cardiotoxicity, and ischemia-reperfusion [206]. Therefore, ROS act not only in signal transduction pathways but also interfere with the competing endogenous RNA network in regulating gene expression. In this context, many efforts are aimed at employing lncRNA-based therapies in ROS-related diseases. For example, knockdown of the lcnRNA growth arrest-specific transcript 5 (GAS5) increases the levels of superoxide anion and oxidized glutathiones by altering the redox balance in melanoma cells. In such a way, reduced GAS5 expression is linked to progression of melanoma [207]. It is now well established that ROS are crucial mediators of signaling cascades regulating cell proliferation, invasion, migration, and apoptosis and that they are able to activate or suppress important cell functions. A fine interplay exists between ROS and noncoding RNAs, in which the former are regulated by noncoding RNAs and, in turn, the expression of the latter is modified by ROS-induced alterations of proteins responsible for noncoding RNA transcription and maturation [208]. However, although considerable evidence points to the involvement of ROS in the expression of coding and noncoding RNA in cancer, much remains to be known about this in melanoma.
2. Concluding Remarks
One of the main causes of melanoma onset is exposure to ultraviolet rays, which induce the production of significant concentrations of ROS. However, in this neoplastic condition, ROS represent much more than secondary products of the redox processes induced by ultraviolet rays. The concentration, the production site, and the specific type of ROS determine their activities and roles, influence their involvement in almost all cellular processes, and outline their benefits and harms. It is now well known that ROS are a double-edged blade or two sides of the same coin, since in certain conditions they can be detrimental to the cell, while in others they are an integral and necessary part in signal conduction pathways. Moreover, recent evidence indicates that ROS also play an increasingly important role in the regulation of gene expression. They interfere not only with the processes of DNA methylation and histone acetylation/deacetylation but also with the increasingly complex RNA network, including mRNA, miRNA, and ceRNA, although in melanoma the relationships between ROS and ceRNA have not yet been defined. Such crucial involvement of ROS in key mechanisms of cellular functions suggests that they will represent useful targets for therapeutic approaches in melanoma.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
Isabella Venza and Mario Venza have to be considered as first name.
References
- 1.Balaban R. S., Nemoto S., Finkel T. Mitochondria, oxidants, and aging. Cell . 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Davies M. J. The oxidative environment and protein damage. Biochimica et Biophysica Acta . 2005;1703(2):93–109. doi: 10.1016/j.bbapap.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Liou G. Y., Storz P. Reactive oxygen species in cancer. Free Radical Research . 2010;44(5):479–496. doi: 10.3109/10715761003667554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pham-Huy L. A., He H., Pham-Huy C. Free radicals, antioxidants in disease and health. International journal of biomedical science: IJBS . 2008;4(2):89–96. [PMC free article] [PubMed] [Google Scholar]
- 5.Finkel T. Signal transduction by reactive oxygen species. Journal of Cell Biology . 2011;194(1):7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zangar R. C., Bollinger N., Weber T. J., Tan R. M., Markillie L. M., Karin N. J. Reactive oxygen species alter autocrine and paracrine signaling. Free Radical Biology and Medicine . 2011;51(11):2041–2047. doi: 10.1016/j.freeradbiomed.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Babior B. M. NADPH oxidase: an update. Blood . 1999;93(5):1464–1476. doi: 10.1182/blood.V93.5.1464. [DOI] [PubMed] [Google Scholar]
- 8.Chanock S. J., el Benna J., Smith R. M., Babior B. M. The respiratory burst oxidase. Journal of Biological Chemistry . 1994;269(40):24519–24522. doi: 10.1016/S0021-9258(17)31418-7. [DOI] [PubMed] [Google Scholar]
- 9.Clark R. A. Activation of the neutrophil respiratory burst oxidase. The Journal of infectious diseases . 1999;179(Supplement 2):S309–S317. doi: 10.1086/513849. [DOI] [PubMed] [Google Scholar]
- 10.DeLeo F. R., Quinn M. T. Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. Journal of leukocyte biology . 1996;60(6):677–691. doi: 10.1002/jlb.60.6.677. [DOI] [PubMed] [Google Scholar]
- 11.Wientjes F. B., Segal A. W., Hartwig J. H. Immunoelectron microscopy shows a clustered distribution of NADPH oxidase components in the human neutrophil plasma membrane. Journal of leukocyte biology . 1997;61(3):303–312. doi: 10.1002/jlb.61.3.303. [DOI] [PubMed] [Google Scholar]
- 12.Bedard K., Krause K. H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological reviews . 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 13.Brandes R. P., Weissmann N., Schröder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radical Biology and Medicine . 2014;76:208–226. doi: 10.1016/j.freeradbiomed.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 14.Turrens J. F. Mitochondrial formation of reactive oxygen species. The Journal of physiology . 2003;552(Part 2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muller F. L., Liu Y., van Remmen H. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. Journal of Biological Chemistry . 2004;279(47):49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 16.Chandel N. S., Maltepe E., Goldwasser E., Mathieu C. E., Simon M. C., Schumacker P. T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proceedings of the National Academy of Sciences . 1998;95(20):11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storz P., Döppler H., Toker A. Protein kinase Cδ selectively regulates protein kinase D-dependent activation of NF-κB in oxidative stress signaling. Molecular and cellular biology . 2004;24(7):2614–2626. doi: 10.1128/MCB.24.7.2614-2626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storz P., Döppler H., Ferran C., Grey S. T., Toker A. Functional dichotomy of A20 in apoptotic and necrotic cell death. Biochemical Journal . 2005;387(Part 1):47–55. doi: 10.1042/BJ20041443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Storz P., Döppler H., Toker A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Molecular and cellular biology . 2005;25(19):8520–8530. doi: 10.1128/MCB.25.19.8520-8530.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Duve C., Baudhuin P. Peroxisomes (microbodies and related particles) Physiological reviews . 1966;46(2):323–357. doi: 10.1152/physrev.1966.46.2.323. [DOI] [PubMed] [Google Scholar]
- 21.Bonekamp N. A., Völkl A., Fahimi H. D., Schrader M. Reactive oxygen species and peroxisomes: struggling for balance. Biofactors . 2009;35(4):346–355. doi: 10.1002/biof.48. [DOI] [PubMed] [Google Scholar]
- 22.del Río L., Sandalio L. M., Palma J. M., Bueno P., Corpas F. J. Metabolism of oxygen radicals in peroxisomes and cellular implications. Free Radical Biology and Medicine . 1992;13(5):557–580. doi: 10.1016/0891-5849(92)90150-f. [DOI] [PubMed] [Google Scholar]
- 23.Schrader M., Fahimi H. D. Peroxisomes and oxidative stress. Biochimica et Biophysica Acta . 2006;1763(12):1755–1766. doi: 10.1016/j.bbamcr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 24.Zorov D. B., Juhaszova M., Sollott S. J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiological reviews . 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birben E., Sahiner U. M., Sackesen C., Erzurum S., Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organization Journal . 2012;5(1):9–19. doi: 10.1097/WOX.0b013e3182439613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy M. P., Holmgren A., Larsson N. G., et al. Unraveling the biological roles of reactive oxygen species. Cell metabolism . 2011;13(4):361–366. doi: 10.1016/j.cmet.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klomsiri C., Karplus P. A., Poole L. B. Cysteine-based redox switches in enzymes. Antioxidants & redox signaling . 2011;14(6):1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marino S. M., Gladyshev V. N. Analysis and Functional Prediction of Reactive Cysteine Residues. Journal of Biological Chemistry . 2012;287(7):4419–4425. doi: 10.1074/jbc.R111.275578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winterbourn C. C., Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radical Biology and Medicine . 1999;27(3-4):322–328. doi: 10.1016/S0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 30.Chung H. S., Wang S. B., Venkatraman V., Murray C. I., van Eyk J. E. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circulation research . 2013;112(2):382–392. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagahara N. Intermolecular disulfide bond to modulate protein function as a redox-sensing switch. Amino Acids . 2011;41(1):59–72. doi: 10.1007/s00726-010-0508-4. [DOI] [PubMed] [Google Scholar]
- 32.Paulsen C. E., Carroll K. S. Orchestrating redox signaling networks through regulatory cysteine switches. ACS chemical biology . 2010;5(1):47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bartosz G. Reactive oxygen species: destroyers or messengers? Biochemical pharmacology . 2009;77(8):1303–1315. doi: 10.1016/j.bcp.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 34.Meng T. C., Fukada T., Tonks N. K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Molecular cell . 2002;9(2):387–399. doi: 10.1016/S1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 35.Xu D., Rovira I. I., Finkel T. Oxidants painting the cysteine chapel: redox regulation of PTPs. Developmental cell . 2002;2(3):251–252. doi: 10.1016/S1534-5807(02)00132-6. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh C. C., Papaconstantinou J. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. The FASEB Journal . 2006;20(2):259–268. doi: 10.1096/fj.05-4376com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Janssen-Heininger Y. M., Mossman B. T., Heintz N. H., et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radical Biology and Medicine . 2008;45(1):1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fu X., Kassim S. Y., Parks W. C., Heinecke J. W. Hypochlorous Acid Oxygenates the Cysteine Switch Domain of Pro-matrilysin (MMP-7): Journal of Biological Chemistry . 2001;276(44):41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 39.Aracena-Parks P., Goonasekera S. A., Gilman C. P., Dirksen R. T., Hidalgo C., Hamilton S. L. Identification of Cysteines Involved in S-Nitrosylation, S-Glutathionylation, and Oxidation to Disulfides in Ryanodine Receptor Type 1. Journal of Biological Chemistry . 2006;281(52):40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- 40.Mi T., Xiao Z., Guo W., et al. Role of Cys3602 in the function and regulation of the cardiac ryanodine receptor. Biochemical Journal . 2015;467(1):177–190. doi: 10.1042/BJ20141263. [DOI] [PubMed] [Google Scholar]
- 41.Chang L. Y., Slot J. W., Geuze H. J., Crapo J. D. Molecular immunocytochemistry of the CuZn superoxide dismutase in rat hepatocytes. The Journal of cell biology . 1988;107(6 Part 1):2169–2179. doi: 10.1083/jcb.107.6.2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johnson F., Giulivi C. Superoxide dismutases and their impact upon human health. Molecular aspects of medicine . 2005;26(4-5):340–352. doi: 10.1016/j.mam.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Miao L., Clair D. K. Regulation of superoxide dismutase genes: implications in disease. Free Radical Biology and Medicine . 2009;47(4):344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zelko I. N., Mariani T. J., Folz R. J. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radical Biology and Medicine . 2002;33(3):337–349. doi: 10.1016/S0891-5849(02)00905-X. [DOI] [PubMed] [Google Scholar]
- 45.Crapo J. D., Oury T., Rabouille C., Slot J. W., Chang L. Y. Copper, zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proceedings of the National Academy of Sciences . 1992;89(21):10405–10409. doi: 10.1073/pnas.89.21.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh I. Mammalian peroxisomes: metabolism of oxygen and reactive oxygen species. Annals of the New York Academy of Sciences . 1996;804(1 Peroxisomes):612–627. doi: 10.1111/j.1749-6632.1996.tb18648.x. [DOI] [PubMed] [Google Scholar]
- 47.Sturtz L. A., Diekert K., Jensen L. T., Lill R., Culotta V. C. A Fraction of Yeast Cu,Zn-Superoxide Dismutase and Its Metallochaperone, CCS, Localize to the Intermembrane Space of Mitochondria: A physiological role for SOD1 in guarding against mitochondrial oxidative damage. Journal of Biological Chemistry . 2001;276(41):38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 48.Bak D. W., Weerapana E. Cysteine-mediated redox signalling in the mitochondria. Molecular BioSystems . 2015;11(3):678–697. doi: 10.1039/c4mb00571f. [DOI] [PubMed] [Google Scholar]
- 49.Fukai T., Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxidants & redox signaling . 2011;15(6):1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Han D., Williams E., Cadenas E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochemical Journal . 2001;353(Part 2):411–416. doi: 10.1042/bj3530411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Storz P. Mitochondrial ROS - radical detoxification, mediated by protein kinase D. Trends in cell biology . 2007;17(1):13–18. doi: 10.1016/j.tcb.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 52.Marklund S. L. Human copper-containing superoxide dismutase of high molecular weight. Proceedings of the National Academy of Sciences . 1982;79(24):7634–7638. doi: 10.1073/pnas.79.24.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marklund S. L., Holme E., Hellner L. Superoxide dismutase in extracellular fluids. Clinica chimica acta . 1982;126(1):41–51. doi: 10.1016/0009-8981(82)90360-6. [DOI] [PubMed] [Google Scholar]
- 54.Stralin P., Karlsson K., Johansson B. O., Marklund S. L. The interstitium of the human arterial wall contains very large amounts of extracellular superoxide dismutase. Arteriosclerosis, thrombosis, and vascular biology . 1995;15(11):2032–2036. doi: 10.1161/01.ATV.15.11.2032. [DOI] [PubMed] [Google Scholar]
- 55.Stralin P., Marklund S. L. Vasoactive factors and growth factors alter vascular smooth muscle cell EC-SOD expression. American Journal of Physiology-Heart and Circulatory Physiology . 2001;281(4):H1621–H1629. doi: 10.1152/ajpheart.2001.281.4.H1621. [DOI] [PubMed] [Google Scholar]
- 56.Loenders B., van Mechelen E., Nicoläı S., et al. Localization of extracellular superoxide dismutase in rat lung: neutrophils and macrophages as carriers of the enzyme. Free Radical Biology and Medicine . 1998;24(7-8):1097–1106. doi: 10.1016/S0891-5849(97)00434-6. [DOI] [PubMed] [Google Scholar]
- 57.Oury T. D., Chang L. Y., Marklund S. L., Day B. J., Crapo J. D. Immunocytochemical localization of extracellular superoxide dismutase in human lung. Laboratory investigation . 1994;70(6):889–898. [PubMed] [Google Scholar]
- 58.Sandstrom J., Carlsson L., Marklund S. L., Edlund T. The heparin-binding domain of extracellular superoxide dismutase C and formation of variants with reduced heparin affinity. Journal of Biological Chemistry . 1992;267(25):18205–18209. doi: 10.1016/S0021-9258(19)37173-X. [DOI] [PubMed] [Google Scholar]
- 59.Ookawara T., Kizaki T., Takayama E., et al. Nuclear translocation of extracellular superoxide dismutase. Biochemical and biophysical research communications . 2002;296(1):54–61. doi: 10.1016/S0006-291X(02)00804-5. [DOI] [PubMed] [Google Scholar]
- 60.Singh B., Bhat H. K. Superoxide dismutase 3 is induced by antioxidants, inhibits oxidative DNA damage and is associated with inhibition of estrogen-induced breast cancer. Carcinogenesis . 2012;33(12):2601–2610. doi: 10.1093/carcin/bgs300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nozik-Grayck E., Suliman H. B., Piantadosi C. A. Extracellular superoxide dismutase. The international journal of biochemistry & cell biology . 2005;37(12):2466–2471. doi: 10.1016/j.biocel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 62.Itoh S., Ozumi K., Kim H., et al. Novel mechanism for regulation of extracellular SOD transcription and activity by copper: role of antioxidant-1. Free Radical Biology and Medicine . 2009;46(1):95–104. doi: 10.1016/j.freeradbiomed.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valko M., Rhodes C. J., Moncol J., Izakovic M., Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-biological interactions . 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 64.Rovira C. Structure, protonation state and dynamics of catalase compound II. ChemPhysChem . 2005;6(9):1820–1826. doi: 10.1002/cphc.200400633. [DOI] [PubMed] [Google Scholar]
- 65.Ho Y. S., Xiong Y., Ma W., Spector A., Ho D. S. Mice Lacking Catalase Develop Normally but Show Differential Sensitivity to Oxidant Tissue Injury. Journal of Biological Chemistry . 2004;279(31):32804–32812. doi: 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- 66.Andreyev A. Y., Kushnareva Y. E., Starkov A. A. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Moscow) . 2005;70(2):200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 67.Schafer F. Q., Buettner G. R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free radical biology and medicine . 2001;30(11):1191–1212. doi: 10.1016/S0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka C., Coling D. E., Manohar S., et al. Expression pattern of oxidative stress and antioxidant defense-related genes in the aging Fischer 344/NHsd rat cochlea. Neurobiology of aging . 2001;33(8):e1814–e1841. doi: 10.1016/j.neurobiolaging.2011.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brigelius-Flohé R., Maiorino M. Glutathione peroxidases. Biochimica et Biophysica Acta . 2013;1830(5):3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 70.Chen Y. I., Wei P. C., Hsu J. L., Su F. Y., Lee W. H. NPGPx (GPx7): a novel oxidative stress sensor/transmitter with multiple roles in redox homeostasis. American journal of translational research . 2016;8(4):1626–1640. [PMC free article] [PubMed] [Google Scholar]
- 71.Chu F. F., Esworthy R. S., Lee L., Wilczynski S. Retinoic acid induces Gpx 2 gene expression in MCF-7 human breast cancer cells. The Journal of nutrition . 1999;129(10):1846–1854. doi: 10.1093/jn/129.10.1846. [DOI] [PubMed] [Google Scholar]
- 72.Burk R. F., Olson G. E., Winfrey V. P., Hill K. E., Yin D. Glutathione peroxidase-3 produced by the kidney binds to a population of basement membranes in the gastrointestinal tract and in other tissues. American Journal of Physiology-Gastrointestinal and Liver Physiology . 2011;301(1):G32–G38. doi: 10.1152/ajpgi.00064.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trachootham D., Lu W., Ogasawara M. A., Valle N. R. D., Huang P. Redox regulation of cell survival. Antioxidants & redox signaling . 2008;10(8):1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hall L., Williams K., Perry A. C. F., Frayne J., Jury J. A. The majority of human glutathione peroxidase type 5 (GPX5) transcripts are incorrectly spliced: implications for the role of GPX5 in the male reproductive tract. Biochemical Journal . 1998;333(1):5–9. doi: 10.1042/bj3330005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Taylor A., Robson A., Houghton B. C., Jepson C. A., Ford W. C., Frayne J. Epididymal specific, selenium-independent GPX5 protects cells from oxidative stress-induced lipid peroxidation and DNA mutation. Human Reproduction . 2013;28(9):2332–2342. doi: 10.1093/humrep/det237. [DOI] [PubMed] [Google Scholar]
- 76.Zhang T., Chabory E., Britan A., et al. GPX5, the selenium-independent glutathione peroxidase-encoding single copy gene is differentially expressed in mouse epididymis. Reproduction, Fertility and Development . 2008;20(5):615–625. doi: 10.1071/RD08008. [DOI] [PubMed] [Google Scholar]
- 77.Wang H. P., Qian S. Y., Schafer F. Q., Domann F. E., Oberley L. W., Buettner G. R. Phospholipid hydroperoxide glutathione peroxidase protects against singlet oxygen-induced cell damage of photodynamic therapy. Free radical biology and medicine . 2001;30(8):825–835. doi: 10.1016/S0891-5849(01)00469-5. [DOI] [PubMed] [Google Scholar]
- 78.Margis R., Dunand C., Teixeira F. K., Margis-Pinheiro M. Glutathione peroxidase family - an evolutionary overview. The FEBS journal . 2008;275(15):3959–3970. doi: 10.1111/j.1742-4658.2008.06542.x. [DOI] [PubMed] [Google Scholar]
- 79.Utomo A., Jiang X., Furuta S., et al. Identification of a Novel Putative Non-selenocysteine Containing Phospholipid Hydroperoxide Glutathione Peroxidase (NPGPx) Essential for Alleviating Oxidative Stress Generated from Polyunsaturated Fatty Acids in Breast Cancer Cells. Journal of Biological Chemistry . 2004;279(42):43522–43529. doi: 10.1074/jbc.M407141200. [DOI] [PubMed] [Google Scholar]
- 80.Wei P. C., Hsieh Y. H., Su M. I., et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Molecular cell . 2012;48(5):747–759. doi: 10.1016/j.molcel.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang L., Zhang L., Niu Y., Sitia R., Wang C. C. Glutathione peroxidase 7 utilizes hydrogen peroxide generated by Ero1α to promote oxidative protein folding. Antioxidants & redox signaling . 2014;20(4):545–556. doi: 10.1089/ars.2013.5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bosello-Travain V., Conrad M., Cozza G., et al. Protein disulfide isomerase and glutathione are alternative substrates in the one Cys catalytic cycle of glutathione peroxidase 7. Biochimica et Biophysica Acta . 2013;1830(6):3846–3857. doi: 10.1016/j.bbagen.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 83.Nguyen V. D., Saaranen M. J., Karala A. R., et al. Two endoplasmic reticulum PDI peroxidases increase the efficiency of the use of peroxide during disulfide bond formation. Journal of molecular biology . 2011;406(3):503–515. doi: 10.1016/j.jmb.2010.12.039. [DOI] [PubMed] [Google Scholar]
- 84.Ramming T., Hansen H. G., Nagata K., Ellgaard L., Appenzeller-Herzog C. GPx8 peroxidase prevents leakage of H2O2 from the endoplasmic reticulum. Free Radical Biology and Medicine . 2014;70:106–116. doi: 10.1016/j.freeradbiomed.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 85.Morikawa K., Gouttenoire J., Hernandez C., et al. Quantitative proteomics identifies the membrane-associated peroxidase GPx8 as a cellular substrate of the hepatitis C virus NS3-4A protease. Hepatology . 2014;59(2):423–433. doi: 10.1002/hep.26671. [DOI] [PubMed] [Google Scholar]
- 86.Lushchak V. I. Glutathione homeostasis and functions: potential targets for medical interventions. Journal of Amino Acids . 2012;2012:26. doi: 10.1155/2012/736837.736837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sheehan D., Meade G., Foley V. M., Dowd C. A. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochemical journal . 2001;360(1):1–16. doi: 10.1042/bj3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morel F., Rauch C., Petit E., et al. Gene and Protein Characterization of the Human Glutathione _S_ -Transferase Kappa and Evidence for a Peroxisomal Localization. Journal of biological chemistry . 2004;279(16):16246–16253. doi: 10.1074/jbc.M313357200. [DOI] [PubMed] [Google Scholar]
- 89.Li J., Xia Z., Ding J. Thioredoxin-like domain of human kappa class glutathione transferase reveals sequence homology and structure similarity to the theta class enzyme. Protein science . 2005;14(9):2361–2369. doi: 10.1110/ps.051463905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Circu M. L., Aw T. Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Biology and Medicine . 2010;48(6):749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matsuzawa A. Thioredoxin and redox signaling: roles of the thioredoxin system in control of cell fate. Archives of biochemistry and biophysics . 2017;617:101–105. doi: 10.1016/j.abb.2016.09.011. [DOI] [PubMed] [Google Scholar]
- 92.Schallreuter K. U., Gleason F. K., Wood J. M. The mechanism of action of the nitrosourea anti-tumor drugs on thioredoxin reductase, glutathione reductase and ribonucleotide reductase. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research . 1990;1054(1):14–20. doi: 10.1016/0167-4889(90)90199-N. [DOI] [PubMed] [Google Scholar]
- 93.Poynton R. A., Hampton M. B. Peroxiredoxins as biomarkers of oxidative stress. Biochimica et Biophysica Acta . 2014;1840(2):906–912. doi: 10.1016/j.bbagen.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 94.Wood Z. A., Schroder E., Robin Harris J., Poole L. B. Structure, mechanism and regulation of peroxiredoxins. Trends in biochemical sciences . 2003;28(1):32–40. doi: 10.1016/S0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 95.Immenschuh S., Baumgart-Vogt E. Peroxiredoxins, oxidative stress, and cell proliferation. Antioxidants & redox signaling . 2005;7(5-6):768–777. doi: 10.1089/ars.2005.7.768. [DOI] [PubMed] [Google Scholar]
- 96.Perkins A., Nelson K. J., Parsonage D., Poole L. B., Karplus P. A. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends in biochemical sciences . 2015;40(8):435–445. doi: 10.1016/j.tibs.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ding C., Fan X., Wu G. Peroxiredoxin 1- an antioxidant enzyme in cancer. Journal of cellular and molecular medicine . 2017;21(1):193–202. doi: 10.1111/jcmm.12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rhee S. G., Woo H. A., Kil I. S., Bae S. H. Peroxiredoxin Functions as a Peroxidase and a Regulator and Sensor of Local Peroxides. Journal of Biological Chemistry . 2012;287(7):4403–4410. doi: 10.1074/jbc.R111.283432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zito E., Melo E. P., Yang Y., Wahlander A., Neubert T. A., Ron D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Molecular cell . 2010;40(5):787–797. doi: 10.1016/j.molcel.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.D'Autreaux B., Toledano M. B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nature reviews Molecular cell biology . 2007;8(10):813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 101.Lobo V., Patil A., Phatak A., Chandra N. Free radicals, antioxidants and functional foods: impact on human health. Pharmacognosy reviews . 2010;4(8):118–126. doi: 10.4103/0973-7847.70902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cairns R. A., Harris I. S., Mak T. W. Regulation of cancer cell metabolism. Nature Reviews Cancer . 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 103.Panieri E., Santoro M. M. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell death & disease . 2016;7(6, article e2253) doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zuo L., Zhou T., Pannell B. K., Ziegler A. C., Best T. M. Biological and physiological role of reactive oxygen species--the good, the bad and the ugly. Acta physiologica . 2015;214(3):329–348. doi: 10.1111/apha.12515. [DOI] [PubMed] [Google Scholar]
- 105.Wauchope O. R., Beavers W. N., Galligan J. J., Mitchener M. M., Kingsley P. J., Marnett L. J. Nuclear oxidation of a major peroxidation DNA adduct, M1dG, in the genome. Chemical research in toxicology . 2015;28(12):2334–2342. doi: 10.1021/acs.chemrestox.5b00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Limoli C. L., Giedzinski E. Induction of chromosomal instability by chronic oxidative stress. Neoplasia . 2003;5(4):339–346. doi: 10.1016/S1476-5586(03)80027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fruehauf J. P., Meyskens F. L., Jr. Reactive oxygen species: a breath of life or death? Clinical Cancer Research . 2007;13(3):789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- 108.Suzuki Y. J., Forman H. J., Sevanian A. Oxidants as stimulators of signal transduction. Free Radical Biology and Medicine . 1997;22(1-2):269–285. doi: 10.1016/S0891-5849(96)00275-4. [DOI] [PubMed] [Google Scholar]
- 109.Wang M., Kirk J. S., Venkataraman S., et al. Manganese superoxide dismutase suppresses hypoxic induction of hypoxia- inducible factor-1 _α_ and vascular endothelial growth factor. Oncogene . 2005;24(55):8154–8166. doi: 10.1038/sj.onc.1208986. [DOI] [PubMed] [Google Scholar]
- 110.Luanpitpong S., Talbott S. J., Rojanasakul Y., et al. Regulation of Lung Cancer Cell Migration and Invasion by Reactive Oxygen Species and Caveolin-1. Journal of Biological Chemistry . 2010;285(50):38832–38840. doi: 10.1074/jbc.M110.124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rhee S. G., Kang S. W., Jeong W., Chang T. S., Yang K. S., Woo H. A. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Current opinion in cell biology . 2005;17(2):183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 112.Storz P. Reactive oxygen species in tumor progression. Frontiers in Bioscience . 2005;10(1-3):1881–1896. doi: 10.2741/1667. [DOI] [PubMed] [Google Scholar]
- 113.Abrigo J., Rivera J. C., Simon F., Cabrera D., Cabello-Verrugio C. Transforming growth factor type beta (TGF-β) requires reactive oxygen species to induce skeletal muscle atrophy. Cellular Signalling . 2016;28(5):366–376. doi: 10.1016/j.cellsig.2016.01.010. [DOI] [PubMed] [Google Scholar]
- 114.Das S. J., Lovicu F. J., Collinson E. J. Nox4 plays a role in TGF-β–Dependent lens epithelial to mesenchymal transition. Investigative ophthalmology & visual science . 2016;57(8):3665–3673. doi: 10.1167/iovs.16-19114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kim J. J., Lee S. B., Park J. K., Yoo Y. D. TNF- _α_ -induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-XL. Cell Death & Differentiation . 2010;17(9):1420–1434. doi: 10.1038/cdd.2010.19. [DOI] [PubMed] [Google Scholar]
- 116.Yang D., Elner S. G., Bian Z. M., Till G. O., Petty H. R., Elner V. M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Experimental eye research . 2007;85(4):462–472. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Holmstrom K. M., Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature reviews Molecular cell biology . 2014;15(6):411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 118.Rhee S. G., Chang T. S., Bae Y. S., Lee S. R., Kang S. W. Cellular regulation by hydrogen peroxide. Journal of the American Society of Nephrology . 2003;14(suppl 3):S211–S215. doi: 10.1097/01.ASN.0000077404.45564.7E. [DOI] [PubMed] [Google Scholar]
- 119.Bae Y. S., Sung J. Y., Kim O. S., et al. Platelet-derived Growth Factor-induced H2O2 Production Requires the Activation of Phosphatidylinositol 3-Kinase. Journal of Biological Chemistry . 2000;275(14):10527–10531. doi: 10.1074/jbc.275.14.10527. [DOI] [PubMed] [Google Scholar]
- 120.Bae Y. S., Kang S. W., Seo M. S., et al. Epidermal Growth Factor (EGF)-induced Generation of Hydrogen Peroxide: Journal of Biological Chemistry . 1997;272(1):217–221. doi: 10.1074/jbc.272.1.217. [DOI] [PubMed] [Google Scholar]
- 121.Liu H., Colavitti R., Rovira I. I., Finkel T. Redox-dependent transcriptional regulation. Circulation research . 2005;97(10):967–974. doi: 10.1161/01.RES.0000188210.72062.10. [DOI] [PubMed] [Google Scholar]
- 122.al Ghouleh I., Khoo N. K., Knaus U. G., et al. Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radical Biology and Medicine . 2011;51(7):1271–1288. doi: 10.1016/j.freeradbiomed.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rhee S. G. Redox signaling: hydrogen peroxide as intracellular messenger. Experimental & molecular medicine . 1999;31(2):53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- 124.Jin S. Y., Lee H. S., Kim E. K., Ha J. M., Kim Y. W., Bae S. Reactive oxygen species and PI3K/Akt signaling in cancer. Free Radical Biology and Medicine . 2014;75(Supplement 1):S34–S35. doi: 10.1016/j.freeradbiomed.2014.10.773. [DOI] [PubMed] [Google Scholar]
- 125.Usatyuk P. V., Fu P., Mohan V., et al. Role of c-Met/Phosphatidylinositol 3-Kinase (PI3k)/Akt Signaling in Hepatocyte Growth Factor (HGF)-mediated Lamellipodia Formation, Reactive Oxygen Species (ROS) Generation, and Motility of Lung Endothelial Cells. Journal of Biological Chemistry . 2014;289(19):13476–13491. doi: 10.1074/jbc.M113.527556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Poljsak B., Suput D., Milisav I. Achieving the balance between ROS and antioxidants: when to use the synthetic antioxidants. Oxidative Medicine and Cellular Longevity . 2013;2013:11. doi: 10.1155/2013/956792.956792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shen H. M., Liu Z. G. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radical Biology and Medicine . 2006;40(6):928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 128.Tobar N., Villar V., Santibanez J. F. ROS-NFkappaB mediates TGF-beta1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Molecular and cellular biochemistry . 2010;340(1-2):195–202. doi: 10.1007/s11010-010-0418-5. [DOI] [PubMed] [Google Scholar]
- 129.Pearl-Yafe M., Halperin D., Scheuerman O., Fabian I. The p38 pathway partially mediates caspase-3 activation induced by reactive oxygen species in Fanconi anemia C cells. Biochemical pharmacology . 2004;67(3):539–546. doi: 10.1016/j.bcp.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 130.Calvani M., Comito G., Giannoni E., Chiarugi P. Time-dependent stabilization of hypoxia inducible Factor-1α by different intracellular sources of reactive oxygen species. PLoS One . 2012;7(10, article e38388) doi: 10.1371/journal.pone.0038388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Devadas S., Zaritskaya L., Rhee S. G., Oberley L., Williams M. S. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. The Journal of experimental medicine . 2002;195(1):59–70. doi: 10.1084/jem.20010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bulua A. C., Simon A., Maddipati R., et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) Journal of Experimental Medicine . 2011;208(3):519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Naik E., Dixit V. M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. Journal of Experimental Medicine . 2011;208(3):417–420. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cai T., Kuang Y., Zhang C., et al. Glucose-6-phosphate dehydrogenase and NADPH oxidase 4 control STAT3 activity in melanoma cells through a pathway involving reactive oxygen species, c-SRC and SHP2. American journal of cancer research . 2015;5(5):1610–1620. [PMC free article] [PubMed] [Google Scholar]
- 135.Lee M. H., Hong S. H., Park C., et al. Hwang-Heuk-San induces apoptosis in HCT116 human colorectal cancer cells through the ROS-mediated activation of caspases and the inactivation of the PI3K/Akt signaling pathway. Oncology reports . 2016;36(1):205–214. doi: 10.3892/or.2016.4812. [DOI] [PubMed] [Google Scholar]
- 136.Castellone M. D., Langella A., Cantara S., et al. Extracellular superoxide dismutase induces mouse embryonic fibroblast proliferative burst, growth arrest, immortalization, and consequent in vivo tumorigenesis. Antioxidants & redox signaling . 2014;21(10):1460–1474. doi: 10.1089/ars.2013.5475. [DOI] [PubMed] [Google Scholar]
- 137.Laukkanen M. O. Extracellular superoxide dismutase: growth promoter or tumor suppressor? Oxidative medicine and cellular longevity . 2016;2016:9. doi: 10.1155/2016/3612589.3612589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Irani K., Goldschmidt-Clermont P. J. Ras, superoxide and signal transduction. Biochemical pharmacology . 1998;55(9):1339–1346. doi: 10.1016/s0006-2952(97)00616-3. [DOI] [PubMed] [Google Scholar]
- 139.Forman H. J., Maiorino M., Ursini F. Signaling functions of reactive oxygen species. Biochemistry . 2010;49(5):835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bauer G. Targeting extracellular ROS signaling of tumor cells. Anticancer research . 2014;34(4):1467–1482. [PubMed] [Google Scholar]
- 141.Deichman G. I. Natural selection and early changes of phenotype of tumor cells in vivo: acquisition of new defense mechanisms. Biochemistry (Mosc) . 2000;65(1):78–94. [PubMed] [Google Scholar]
- 142.Deichman G. I. Early phenotypic changes of in vitro transformed cells during in vivo progression: possible role of the host innate immunity. Seminars in cancer biology . 2002;12(4):317–326. doi: 10.1016/S1044-579X(02)00018-4. [DOI] [PubMed] [Google Scholar]
- 143.Deichman G. I., Kluchareva T. E., Matveeva V. A., Kushlinsky N. E., Bassalyk L. S., Vendrov E. L. Clustering of discrete cell properties essential for tumorigenicity and metastasis. I. Studies of Syrian hamster embryo fibroblasts spontaneously transformed in vitro. International journal of cancer . 1989;44(5):904–907. doi: 10.1002/ijc.2910440526. [DOI] [PubMed] [Google Scholar]
- 144.Deichman G. I., Vendrov E. L. Characteristics of in vitro transformed cells essential for their in vivo survival, selection and metastasizing activity. International journal of cancer . 1986;37(3):401–409. doi: 10.1002/ijc.2910370312. [DOI] [PubMed] [Google Scholar]
- 145.Deichman G. J., Matveeva V. A., Kashkina L. M., et al. Cell transforming genes and tumor progression: in vivo unified secondary phenotypic cell changes. International journal of cancer . 1998;75(2):277–283. doi: 10.1002/(SICI)1097-0215(19980119)75:2<277::AID-IJC17>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 146.Heinzelmann S., Bauer G. Multiple protective functions of catalase against intercellular apoptosis-inducing ROS signaling of human tumor cells. Biological Chemistry . 2010;391(6):675–693. doi: 10.1515/BC.2010.068. [DOI] [PubMed] [Google Scholar]
- 147.Bauer G. Tumor cell-protective catalase as a novel target for rational therapeutic approaches based on specific intercellular ROS signaling. Anticancer research . 2012;32(7):2599–2624. [PubMed] [Google Scholar]
- 148.Rudolf J., Raad H., Taieb A., Rezvani H. R. NADPH oxidases and their roles in skin homeostasis and carcinogenesis. Antioxidants & redox signaling . 2018;28(13):1238–1261. doi: 10.1089/ars.2017.7282. [DOI] [PubMed] [Google Scholar]
- 149.Xian D., Lai R., Song J., Xiong X., Zhong J. Emerging perspective: role of increased ROS and redox imbalance in skin carcinogenesis. Oxidative Medicine and Cellular Longevity . 2019;2019:11. doi: 10.1155/2019/8127362.8127362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Aggarwal V., Tuli H. S., Varol A., et al. Role of reactive oxygen species in cancer progression: molecular mechanisms and recent advancements. Biomolecules . 2019;9(11):p. 735. doi: 10.3390/biom9110735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Weyemi U., Redon C. E., Parekh P. R., Dupuy C., Bonner W. M. NADPH oxidases NOXs and DUOXs as putative targets for cancer therapy. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents) . 2013;13(3):502–514. doi: 10.2174/1871520611313030013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yamaura M., Mitsushita J., Furuta S., et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer research . 2009;69(6):2647–2654. doi: 10.1158/0008-5472.CAN-08-3745. [DOI] [PubMed] [Google Scholar]
- 153.Brar S. S., Kennedy T. P., Sturrock A. B., et al. An NAD(P) H oxidase regulates growth and transcription in melanoma cells. American Journal of Physiology-Cell Physiology . 2002;282(6):C1212–C1224. doi: 10.1152/ajpcell.00496.2001. [DOI] [PubMed] [Google Scholar]
- 154.Liu G. S., Peshavariya H., Higuchi M., et al. Microphthalmia-associated transcription factor modulates expression of NADPH oxidase type 4: a negative regulator of melanogenesis. Free Radical Biology and Medicine . 2012;52(9):1835–1843. doi: 10.1016/j.freeradbiomed.2012.02.040. [DOI] [PubMed] [Google Scholar]
- 155.Govindarajan B., Sligh J. E., Vincent B. J., et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. The Journal of clinical investigation . 2007;117(3):719–729. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Liu F., Gomez Garcia A. M., Meyskens F. L., Jr. NADPH oxidase 1 overexpression enhances invasion via matrix metalloproteinase-2 and epithelial-mesenchymal transition in melanoma cells. Journal of investigative dermatology . 2012;132(8):2033–2041. doi: 10.1038/jid.2012.119. [DOI] [PubMed] [Google Scholar]
- 157.Prasad R., Kappes J. C., Katiyar S. K. Inhibition of NADPH oxidase 1 activity and blocking the binding of cytosolic and membrane-bound proteins by honokiol inhibit migratory potential of melanoma cells. Oncotarget . 2016;7(7):7899–7912. doi: 10.18632/oncotarget.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Brar S. S., Kennedy T. P., Whorton A. R., et al. Reactive oxygen species from NAD(P)H:quinone oxidoreductase constitutively activate NF-kappaB in malignant melanoma cells. American Journal of Physiology-Cell Physiology . 2001;280(3):C659–C676. doi: 10.1152/ajpcell.2001.280.3.C659. [DOI] [PubMed] [Google Scholar]
- 159.Hintsala H. R., Soini Y., Haapasaari K. M., Karihtala P. Dysregulation of redox-state-regulating enzymes in melanocytic skin tumours and the surrounding microenvironment. Histopathology . 2015;67(3):348–357. doi: 10.1111/his.12659. [DOI] [PubMed] [Google Scholar]
- 160.Lingappan K. NF-κB in oxidative stress. Current opinion in toxicology . 2018;7:81–86. doi: 10.1016/j.cotox.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dolcet X., Llobet D., Pallares J., Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows archiv . 2005;446(5):475–482. doi: 10.1007/s00428-005-1264-9. [DOI] [PubMed] [Google Scholar]
- 162.Chen C. Y., Chen Y. K., Wang J. J., et al. DC-81-enediyne induces apoptosis of human melanoma A375 cells: involvement of the ROS, p38 MAPK, and AP-1 signaling pathways. Cell biology and toxicology . 2013;29(2):85–99. doi: 10.1007/s10565-012-9238-6. [DOI] [PubMed] [Google Scholar]
- 163.Chen J., Li L., Su J., Li B., Chen T., Wong Y. S. Synergistic apoptosis-inducing effects on A375 human melanoma cells of natural borneol and curcumin. PLoS One . 2014;9(6, article e101277) doi: 10.1371/journal.pone.0101277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.D'Anneo A., Carlisi D., Lauricella M., et al. Parthenolide induces caspase-independent and AIF-mediated cell death in human osteosarcoma and melanoma cells. Journal of cellular physiology . 2013;228(5):952–967. doi: 10.1002/jcp.24131. [DOI] [PubMed] [Google Scholar]
- 165.Dent P., Yacoub A., Hamed H. A., et al. The development of MDA-7/IL-24 as a cancer therapeutic. Pharmacology & therapeutics . 2010;128(2):375–384. doi: 10.1016/j.pharmthera.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Farias C. F., Massaoka M. H., Girola N., et al. Benzofuroxan derivatives N-Br and N-I induce intrinsic apoptosis in melanoma cells by regulating AKT/BIM signaling and display anti metastatic activity in vivo. BMC Cancer . 2015;15(1):p. 807. doi: 10.1186/s12885-015-1835-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Velatooru L. R., Baggu C. B., Janapala V. R. Spatane diterpinoid from the brown algae, Stoechospermum marginatum induces apoptosis via ROS induced mitochondrial mediated caspase dependent pathway in murine B16F10 melanoma cells. Molecular carcinogenesis . 2016;55(12):2222–2235. doi: 10.1002/mc.22463. [DOI] [PubMed] [Google Scholar]
- 168.Guterres F. A., Martinez G. R., Rocha M. E. M., Winnischofer S. M. B. Simvastatin rises reactive oxygen species levels and induces senescence in human melanoma cells by activation of p53/p21 pathway. Experimental cell research . 2013;319(19):2977–2988. doi: 10.1016/j.yexcr.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 169.Hambright H. G., Meng P., Kumar A. P., Ghosh R. Inhibition of PI3K/AKT/mTOR axis disrupts oxidative stress-mediated survival of melanoma cells. Oncotarget . 2015;6(9):7195–7208. doi: 10.18632/oncotarget.3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Santos G. M. P., Oliveira S., Monteiro J. C. S., et al. ROS-induced autophagy reduces B16F10 melanoma cell proliferative activity. Lasers in medical science . 2018;33(6):1335–1340. doi: 10.1007/s10103-018-2489-6. [DOI] [PubMed] [Google Scholar]
- 171.Hsiao Y. P., Tsai C. H., Wu P. P., et al. Cantharidin induces G2/M phase arrest by inhibition of Cdc25c and cyclin A and triggers apoptosis through reactive oxygen species and the mitochondria-dependent pathways of A375.S2 human melanoma cells. International journal of oncology . 2014;45(6):2393–2402. doi: 10.3892/ijo.2014.2689. [DOI] [PubMed] [Google Scholar]
- 172.Kwon S. J., Lee J. H., Moon K. D., et al. Induction of apoptosis by isoegomaketone from Perilla frutescens L. in B16 melanoma cells is mediated through ROS generation and mitochondrial-dependent, -independent pathway. Food and chemical toxicology . 2014;65:97–104. doi: 10.1016/j.fct.2013.12.031. [DOI] [PubMed] [Google Scholar]
- 173.Aplak E., von Montfort C., Haasler L., et al. CNP mediated selective toxicity on melanoma cells is accompanied by mitochondrial dysfunction. PLoS One . 2020;15(1, article e0227926) doi: 10.1371/journal.pone.0227926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hong Y., Sengupta S., Hur W., Sim T. Identification of novel ROS inducers: quinone derivatives tethered to long hydrocarbon chains. Journal of medicinal chemistry . 2015;58(9):3739–3750. doi: 10.1021/jm501846y. [DOI] [PubMed] [Google Scholar]
- 175.Ishaq M., Kumar S., Varinli H., et al. Atmospheric gas plasma-induced ROS production activates TNF-ASK1 pathway for the induction of melanoma cancer cell apoptosis. Molecular biology of the cell . 2014;25(9):1523–1531. doi: 10.1091/mbc.e13-10-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liu N., Wang K. S., Qi M., et al. Vitexin compound 1, a novel extraction from a Chinese herb, suppresses melanoma cell growth through DNA damage by increasing ROS levels. Journal of Experimental & Clinical Cancer Research . 2018;37(1):p. 269. doi: 10.1186/s13046-018-0897-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Li K., Zhao S., Long J., et al. A novel chalcone derivative has antitumor activity in melanoma by inducing DNA damage through the upregulation of ROS products. Cancer cell international . 2020;20(1):p. 36. doi: 10.1186/s12935-020-1114-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Svobodova A., Walterova D., Vostalova J. Ultraviolet light induced alteration to the skin. Biomedical Papers-Palacky University in Olomouc . 2006;150(1):25–38. doi: 10.5507/bp.2006.003. [DOI] [PubMed] [Google Scholar]
- 179.Suzuki T., Motohashi H., Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends in pharmacological sciences . 2013;34(6):340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 180.Lee S., Lim M. J., Kim M. H., et al. An effective strategy for increasing the radiosensitivity of human lung cancer cells by blocking Nrf2-dependent antioxidant responses. Free Radical Biology and Medicine . 2012;53(4):807–816. doi: 10.1016/j.freeradbiomed.2012.05.038. [DOI] [PubMed] [Google Scholar]
- 181.Ren D., Villeneuve N. F., Jiang T., et al. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proceedings of the National Academy of Sciences . 2011;108(4):1433–1438. doi: 10.1073/pnas.1014275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wang M., Shi G., Bian C., et al. UVA irradiation enhances brusatol-mediated inhibition of melanoma growth by downregulation of the Nrf2-mediated antioxidant response. Oxidative medicine and cellular longevity . 2018;2018:15. doi: 10.1155/2018/9742154.9742154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hanahan D., Weinberg R. A. Hallmarks of cancer: the next generation. Cell . 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 184.Baylin S. B., Jones P. A. A decade of exploring the cancer epigenome -- biological and translational implications. Nature Reviews Cancer . 2011;11(10):726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sandoval J., Esteller M. Cancer epigenomics: beyond genomics. Current opinion in genetics & development . 2012;22(1):50–55. doi: 10.1016/j.gde.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 186.Sharma S., Kelly T. K., Jones P. A. Epigenetics in cancer. Carcinogenesis . 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wu Q., Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Current drug targets . 2015;16(1):13–19. doi: 10.2174/1389450116666150113121054. [DOI] [PubMed] [Google Scholar]
- 188.Sang Y., Deng Y. Current insights into the epigenetic mechanisms of skin cancer. Dermatologic therapy . 2019;32(4, article e12964) doi: 10.1111/dth.12964. [DOI] [PubMed] [Google Scholar]
- 189.RM S. K., Wang Y., Zhang X., et al. Redox components: key regulators of epigenetic modifications in plants. International journal of molecular sciences . 2020;21(4):p. 1419. doi: 10.3390/ijms21041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Chen C. S., Wang Y. C., Yang H. C., et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer research . 2007;67(11):5318–5327. doi: 10.1158/0008-5472.CAN-06-3996. [DOI] [PubMed] [Google Scholar]
- 191.Eot-Houllier G., Fulcrand G., Magnaghi-Jaulin L., Jaulin C. Histone deacetylase inhibitors and genomic instability. Cancer letters . 2009;274(2):169–176. doi: 10.1016/j.canlet.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 192.Petruccelli L. A., Dupéré-Richer D., Pettersson F., Retrouvey H., Skoulikas S., Miller W. H. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One . 2011;6(6, article e20987) doi: 10.1371/journal.pone.0020987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wang L., Leite de Oliveira R., Huijberts S., et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell . 2018;173(6):1413–1425.e14. doi: 10.1016/j.cell.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 194.Bai J., Lei Y., An G. L., He L. Down-regulation of deacetylase HDAC6 inhibits the melanoma cell line A375.S2 growth through ROS-dependent mitochondrial pathway. PLoS One . 2015;10(3, article e0121247) doi: 10.1371/journal.pone.0121247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Droge W. Free radicals in the physiological control of cell function. Physiological reviews . 2002;82(1):47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 196.Lin Y. H. MicroRNA networks modulate oxidative stress in cancer. International journal of molecular sciences . 2019;20(18):p. 4497. doi: 10.3390/ijms20184497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Fierro-Fernández M., Miguel V., Lamas S. Role of redoximiRs in fibrogenesis. Redox Biology . 2016;7:58–67. doi: 10.1016/j.redox.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.He J., Jiang B. H. Interplay between reactive oxygen species and microRNAs in cancer. Current pharmacology reports . 2016;2(2):82–90. doi: 10.1007/s40495-016-0051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Lujambio A., Calin G. A., Villanueva A., et al. A microRNA DNA methylation signature for human cancer metastasis. Proceedings of the National Academy of Sciences . 2008;105(36):13556–13561. doi: 10.1073/pnas.0803055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P. P. A ceRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell . 2011;146(3):353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Perkel J. M. Visiting "noncodarnia". Biotechniques . 2013;54(6):301–304. doi: 10.2144/000114037. [DOI] [PubMed] [Google Scholar]
- 202.Chen X., Guo W., Xu X. J., et al. Melanoma long non-coding RNA signature predicts prognostic survival and directs clinical risk-specific treatments. Journal of dermatological science . 2017;85(3):226–234. doi: 10.1016/j.jdermsci.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 203.Yang S., Xu J., Zeng X. A six-long non-coding RNA signature predicts prognosis in melanoma patients. International journal of oncology . 2018;52(4):1178–1188. doi: 10.3892/ijo.2018.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Liu N., Liu Z., Liu X., Chen H. Comprehensive analysis of a competing endogenous RNA network identifies seven-lncRNA signature as a prognostic biomarker for melanoma. Frontiers in oncology . 2019;9:p. 935. doi: 10.3389/fonc.2019.00935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Matouk I. J., Mezan S., Mizrahi A., et al. The oncofetal H19 RNA connection: hypoxia, p53 and cancer. Biochimica et Biophysica Acta . 2010;1803(4):443–451. doi: 10.1016/j.bbamcr.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 206.Dong Y., Xu W., Liu C., Liu P., Li P., Wang K. Reactive oxygen species related noncoding RNAs as regulators of cardiovascular diseases. International journal of biological sciences . 2019;15(3):680–687. doi: 10.7150/ijbs.30464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chen L., Yang H., Yi Z., et al. LncRNA GAS5 regulates redox balance and dysregulates the cell cycle and apoptosis in malignant melanoma cells. Journal of cancer research and clinical oncology . 2019;145(3):637–652. doi: 10.1007/s00432-018-2820-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Babu K. R., Tay Y. The Yin-Yang regulation of reactive oxygen species and microRNAs in cancer. International journal of molecular sciences . 2019;20(21):p. 5335. doi: 10.3390/ijms20215335. [DOI] [PMC free article] [PubMed] [Google Scholar]