

Abstract
Introduction.
The clinical implications of abnormal chromosomal microarray (CMA) remain unclear for children less than 1 year of age with critical heart disease. Our objective was to determine whether abnormal CMA was related to surgical severity scores or to pre-determined clinical outcomes, including cardiac arrest.
Methods.
Retrospective review of children under 1 year of age admitted to a pediatric cardiac intensive care unit from December, 2014 to September, 2017. Associations between CMA result and cardiac arrest, syndromic abnormalities, and extracardiac anomalies were evaluated. A simple and multivariable logistic regression model was used to analyze associations between STAT mortality category and CMA result.
Results.
The overall prevalence of abnormal microarray was 48/168 (29%), with peak prevalence in AV septal defects and left-sided obstructive lesions. There was no statistical association between surgical severity scores and abnormal CMA (STAT 1/2 vs. 3+, odds ratio 0.56, p=0.196). Abnormal CMA was associated with a higher prevalence of cardiac arrest (5/48 abnormal CMA vs. 2/120 normal CMA, p=0.02). Abnormal CMA was associated with a higher frequency of syndromic abnormalities (18/48 abnormal CMA vs. 13/120 normal CMA, p<0.001).
Conclusions.
There was a high prevalence of abnormal CMA findings in the pediatric cardiac population less than 1 year of age (29%), associated with cardiac arrest, but not associated with surgical risk score. The absence of a standardized protocol for ordering a CMA in the setting of congenital heart disease results in a highly variable prevalence data.
Keywords: pediatric, congenital heart disease, chromosomal microarray, cardiac arrest, surgical risk score
INTRODUCTION
Congenital heart disease is the leading cause of death related to birth defects and has been associated with a chromosomal abnormality in 12-30% of cases.[1-4] There is no consensus on which clinical presentations are associated with the highest prevalence of abnormal chromosomal microarray (CMA). For example, some studies show that septal defects have the highest diagnostic yields.[5-7] Others have found that right ventricular outflow tract obstructions (RVOTO) have the highest diagnostic yield.[8] Few studies have linked CMA abnormalities to cardiac clinical outcomes in the first year of life. Most existing research on the prevalence of abnormal microarray genetic testing in congenital heart disease is based on the developmental biology of cardiac malformations. However, the clinical severity of a disease is not easily determined by the developmental biology categorization, so correlating CMA with clinical outcomes research has been challenging. For example, the clinical severity of a small primum atrial septal defect is usually mild compared to an unbalanced atrioventricular canal, even though both arise from endocardial cushion defects.
The goal of this retrospective study was to link abnormal CMA and clinical cardiac outcomes. We used cardiac surgical severity scores as a classification system to test this hypothesis as well as clearly defined cardiac outcomes, such as a cardiac arrest during admission. We included only children admitted at less than 1 year of life because the surgical options are most predictable in the first year of life.
METHODS
All patients less than 1 year of age admitted to our tertiary, pediatric cardiac intensive care unit (CICU) from 12/1/14 to 9/1/17 were identified from the Pediatric Cardiac Critical Care Consortium (PC4) database for this retrospective review study. Only the first admission was counted for each patient so that each patient was only included once. The presence of syndromic abnormalities (a characteristic group of signs and symptoms) was determined by the attending physician or subspecialist on service, as recorded in the PC4 database. Information on clinical outcomes was obtained from the PC4 database and by direct review of electronic medical records (EPIC, Verona, WI). The Society of Thoracic Surgeons-European Association for Cardio-Thoracic Surgery (STAT) mortality category, the Risk Adjustment for Congenital Heart Surgery (RACHS) risk category, and data on surgical complications were obtained from the Society of Thoracic Surgeons (STS) database. In patients with multiple scores from different surgeries during the same admission, the surgery with the highest score was recorded. Results from chromosomal microarray (CMA) tests were obtained from medical records. Microarray results are stable genetic results (not age-dependent). Therefore, all CMA results were included in the analysis, independent of the date of CMA collection. All data were centralized in the REDCap (Research Electronic Data Capture) system hosted at Northwestern University Clinical and Translational Sciences Institute. Appropriate human subjects approval was obtained from our Institutional Review Board.
Race was self-reported by the parent or guardian at the time of registration. Congenital heart disease diagnoses were grouped based on the National Birth Defect Prevention Study classification system with additional categories for arrhythmia and cardiomyopathy.[9] Hypoplastic left heart syndrome (HLHS) with single ventricle physiology was classified separately from left ventricular disease with biventricular physiology.
Whole genome CMAs were used to detect copy number variants. The CMA result was a dichotomous variable classified as either abnormal or normal. Abnormal CMA results occurred when one or more missing or extra chromosomal segments were present.[10] The Genetic Testing Registry (GTR) was used to identify genes that occurred in abnormal CMA regions.[11] Copy number imbalances deemed to be benign or likely benign by current guidelines were excluded from analysis.[12]
CMAs were obtained during clinical care and were performed in a CLIA-certified lab. The lab performed array comparative genomic hybridization analysis with an oligonucleotide array designed to detect copy number imbalances (losses or gains) of specific chromosomal regions and across the genome. Quality indicators and sex chromosome internal controls were reviewed in each case to ensure successful hybridization. Only 5/48 abnormal CMA results were classified as variants of uncertain significance (VUS). These were classified as abnormal for the main analysis. As a sensitivity analysis, VUS were also dichotomized as “normal”. Statistical conclusions were unchanged with the alternative classification, including no effect on the cardiac arrest analysis. All statistical analysis was performed in the statistical software R version 3.5.1 (R Core Team, 2018), and an alpha level of 0.05 was considered for all hypothesis testing.
Primary analysis considered an association between STAT score and CMA result. The analytic cohort for the primary analysis included all patients in the PC4 database with CMA results and a STAT mortality category. The cohort for the secondary analyses included all patients in the PC4 database with CMA results (STAT score was not required for secondary analyses). Descriptive statistics summarized demographic and clinical variables overall and by CMA result. Associations between demographics and clinical outcomes with CMA results as well as associations of cardiac arrest with CMA results and history of cardiac surgery were tested using chi-squared tests or a Fisher’s exact test, in the case of low cell counts. For the primary analysis, a logistic regression model was used to assess the association between STAT mortality category and CMA result. Specifically, we considered a dichotomized version of the STAT mortality category due to low cell counts at individual score levels. Sensitivity and specificity were estimated at each possible cut-point for STAT mortality category, and the optimal threshold for dichotomization was determined to be less than or equal to 2 via Youden’s index. A multivariable logistic regression model was constructed to control for sex and race as biologically plausible confounders in the multivariable model. A Hosmer-Lemeshow test was used to assess the goodness of fit. Similar methods were employed for the second independent variable of interest, RACHS risk category, where the cutpoint was also found to be less than or equal to 2.
RESULTS
Between December 2014 and September 2017, 540 patients less than 1 year of age were admitted to the CICU. Of those, chromosomal microarray (CMA) was performed in 168 (Figure 1). An abnormal CMA was present in 48/168 patients (28.6%). The median age at admission was 1 day of life (interquartile range 0–18 days) for those who had a CMA. There was no difference in the prevalence of abnormal CMA results by gender (24/95 male vs. 24/73 female, p=0.28) or by dichotomized self-report of race (26/87 white vs. 22/80 non-white, p=0.73, Table 1). All abnormal microarray results from this study are reported in Online Resource 1.
Fig 1.

Frequency of microarray testing in the cardiac intensive care unit. All patients admitted to the CICU under 1 year of age were evaluated. Microarray testing was at the discretion of the attending intensivist. STAT mortality category is a risk score developed for congenital heart surgery and was assigned based on the highest STAT mortality category assigned during the hospitalization. CICU = cardiac intensive care unit; STAT = The Society of Thoracic Surgeons-European Association for Cardio-Thoracic Surgery
Table 1:
Demographics and primary outcome comparison
| Variable | Total=168 | Abnormal CMA (48) |
Normal CMA (120) |
P-Value |
|---|---|---|---|---|
| Admission Age in Days, median (IQR) | 1.0 (0.0-17.5) | 2.0 (0.0-97.5) | 1.0 (0.0-10.5) | 0.14 |
| Sex | ||||
| Male | 95 | 24 | 71 | 0.28 |
| Female | 73 | 24 | 49 | |
| Race (%) | ||||
| Caucasian | 90 (53.6) | 27 (56.3) | 63 (52.5) | |
| Hispanic/Latino | 46 (27.4) | 9 (18.8) | 37 (30.8) | 0.34 |
| African American | 19 (11.3) | 7 (14.6) | 12 (10.0) | |
| Asian | 9 (5.4) | 4 (8.3) | 5 (4.2) | |
| Other | 53 (31.5) | 12 (25.0) | 41 (34.2) | |
| CICU Length of Stay in Days, median (IQR) | 8 (3-17) | 7 (2-16) | 9 (3-17) | |
The presence of syndromic abnormalities was defined on clinical grounds by the treating clinician. An abnormal CMA was associated with a higher frequency of syndromic abnormalities. Of those with an abnormal CMA, 18/48 (37.5%) had a syndromic abnormality while 13/120 (10.8%) patients with a normal CMA had a syndromic abnormality (p<0.001).
We examined associations between microarray result and clinical outcomes (Table 2). Cardiac arrest occurred in 5/48 patients with an abnormal CMA (10.4%); whereas cardiac arrest occurred in only 2/120 patients with normal microarray (1.7%, p=0.02). Cardiac arrest was not associated with history of cardiac surgery before or during the first hospital admission at our institution (p=0.97) or availability of CMA results (p=0.39). Table 3 lists the abnormal CMA results in patients with cardiac arrest. We analyzed each CMA abnormality to determine if the abnormality encompassed genes associated with structural cardiac abnormalities, cardiomyopathy or rhythm disorders. None of the genes in regions of CMA abnormalities were associated with known genotype-phenotype correlations for cardiac disease or arrhythmia.
Table 2:
Associations Between Clinical Outcomes and Microarray Result
| level | Abnormal Microarray |
Normal Microarray |
p-value a | |
|---|---|---|---|---|
| n | 48 | 120 | ||
| Mortality (%) | No | 43 ( 89.6) | 108 (90.0) | 0.999 |
| Yes | 5 ( 10.4) | 12 (10.0) | ||
| Cardiac Cath (%) | No | 36 ( 75.0) | 82 (68.3) | 0.601 |
| Yes | 12 (25.0) | 37 (30.8) | ||
| Missing | 0 ( 0.0) | 1 (0.8) | ||
| Readmission within 30 days of discharge (%) | No | 34 ( 70.8) | 90 (75.0) | 0.464 |
| Yes | 8 ( 16.7) | 22 (18.3) | ||
| Missing | 6 ( 12.5) | 8 (6.7) | ||
| Cardiac Arrest (%) | No | 43 ( 89.6) | 117 (97.5) | 0.021 |
| Yes | 5 ( 10.4) | 2 (1.7) | ||
| Missing | 0 ( 0.0) | 1 (0.8) | ||
| Extracardiac Anomaly (%) | No | 33 ( 68.8) | 87 (72.5) | 0.766 |
| Yes | 15 (31.2) | 33 (27.5) | ||
| Syndromic Abnormality (%) | No | 30 (62.5) | 107 (89.2) | <0.001 |
| Yes | 18 (37.5) | 13 (10.8) |
P-value from Chi-square or Fisher’s exact test, as appropriate
Table 3.
Abnormal microarray results in children less than 1 with cardiac arrest
| Diagnosis | Microarray Result |
|---|---|
| Hypoplastic Left Heart Syndrome | arr[hg19] 7q11.23(72,688,285-74,161,142)x3 |
| Pulmonary Atresia | arr[hg19] Xq26.2(131,942,553-132,603,622)x3 |
| Hypoplastic Left Heart Syndrome | arr[hg19] 15q11.2(22,628,639-23,178,762)x1 |
| Conotruncal Defect | arr[hg19] 16p11.2(28,497,756-28,498,146)x1 |
| Pulmonary Atresia | arr[GRCh37] 11q24.2q25(124,723,867-135,006,516)x3,14q32.33(104,133,013-107,349,540)x1 |
arr = microarray
hg19/GRCh37 = type of microarray test performed
STAT mortality category was available in 111/168 patients with CMA results. There was no trend in the prevalence of abnormal CMA across STAT classifications (Figure 2). An unadjusted model found no statistical association between STAT mortality category (dichotomized as 1, 2 vs. 3, 4, 5) and abnormal CMA (odds ratio 0.57, CI95 0.23-1.36, p=0.2). A multivariable model including race and sex as possible confounders confirmed this finding (odds ratio 0.56, CI95 0.23-1.35, p=0.2). As a sensitivity analysis, the same tests were performed using RACHS risk category (dichotomized as 1,2 vs. 3, 4, 5) as the independent variable of interest. Similar results were found in the 107/168 patients who had both a RACHS risk category and a CMA (odds ratio 0.65, CI95 0.26-1.69 p=0.37, Online Resource 2).
Fig 2.
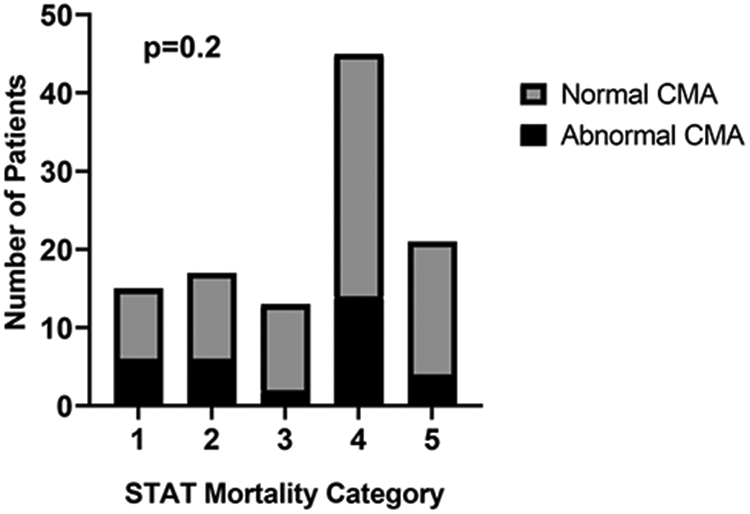
Distribution of chromosomal microarray by STAT mortality category. Results of microarray analysis are presented across STAT mortality category (1 = lowest severity; 5 = maximum severity). The dark shading represents the frequency of abnormal CMA results (range 15-40% of cases within mortality category). The distribution of abnormal CMA tests did not significantly change across STAT mortality category (p=0.2).
The frequency of abnormal CMA results varied by diagnosis (Table 4). The prevalence of abnormal CMA was over 25% in only 5 classes of congenital heart disease: right and left outflow tract obstruction, pulmonary venous anomalies, conotruncal defects, and atrioventricular septal defects (AVSD), although analysis was limited by small cell counts in AVSD.
Table 4:
Distribution of abnormal microarray among diagnoses
| Diagnosis | Number of Cases | Abnormal CMA (% of cases) |
Prior Reports of Abnormal CMA frequency a |
|---|---|---|---|
| Atrioventricular septal defect | 2 | 1 (50) | 0%[8], 25%[6], 33%[7], 53%[5] |
| Left ventricular outflow tract obstruction b | 25 | 12 (48) | 17%[7], 23%[6], 24%[5], 36%[8] |
| Anomalous pulmonary vein origin, connection, or other venous anomaly | 5 | 2 (40) | 0%[7], 0%[6], 40%[5] |
| Right ventricular outflow tract obstruction c | 24 | 9 (38) | 6%[7], 8%[5], 25%[6], 46%[8] |
| Conotruncal defect d | 48 | 14 (29) | 11%[7], 13%[8], 19%[5], 23%[6] |
| Hypoplastic left heart syndrome | 26 | 6 (23) | |
| Septal defect e | 10 | 2 (20) | 23%[8], 33%[7], 48%[6], 56%[5] |
| Cardiomyopathy | 5 | 1 (20) | |
| Other f | 6 | 1 (17) | 21%[6], 25%[7], 31%[5] |
| Single ventricle, other than HLHS | 8 | 0 (0) | 0%[8] |
| Patent ductus arteriosus | 3 | 0 (0) | 33%[8] |
| Arrhythmia | 5 | 0 (0) | |
| Normal heart | 1 | 0 (0) | |
| Total | 168 | 48/168 (29%) | 14% [7], 24% [6], 28% [8] |
CMA, Chromosomal microarray; HLHS, hypoplastic left heart syndrome
Citations numbers from References section
Coarctation of the aorta, aortic stenosis, bicuspid aortic valve, interrupted aortic arch, aortic arch hypoplasia, Shone complex.
Pulmonary stenosis or atresia, discontinuous pulmonary arteries, Ebstein anomaly of the tricuspid valve.
Tetralogy of Fallot, transposition of the great arteries, double-outlet right ventricle, truncus arteriosus.
Ventricular septal defect, atrial septal defect.
Aortic aneurysm, high output cardiac failure due to vascular shunt, myocarditis, coronary anomalies, primary valve disease, pulmonary hypertension, vascular ring.
DISCUSSION
In our primary analysis, increased clinical severity of surgical disease was not associated with an increased prevalence of CMA abnormalities. The lack of an association between STAT mortality category and abnormal CMA among children less than 1 year of age who require surgery continues to reinforce the hypothesis that in the circulatory system, developmental biology provides a stronger framework for understanding chromosomal abnormalities than the surgical severity of the lesion.
In our retrospective analysis of patients in whom a CMA was obtained, abnormal CMA was associated with a higher risk of cardiac arrest during hospitalization. This supports existing data by Alten and colleagues, who found an odds ratio of 1.36 in favor of cardiac arrest in children with “any chromosomal abnormality” using registry methods.[13] The current study provides new data by analyzing the specific genes associated with each CMA finding. In this cohort, cardiac arrest was not associated with disruptions in known genes contributing to structural heart disease, cardiomyopathy or channelopathy. The absolute numbers of cardiac arrests in our study are low; however, our data support the important hypothesis that genetic abnormalities with low individual effect sizes contribute an incremental risk for cardiac arrest.
Our data also support a large genotype-based study in children with congenital heart disease. A recent study of 2,517 patients with congenital heart disease and whole exome sequencing identified copy number variants at 15q11.2 as a potential association with reduced transplant-free survival.[14] While limited to a single observation, CMA in our study revealed a 15q11.2 deletion in one patient with cardiac arrest.
In summary, the association between abnormal CMA and risk for cardiac arrest is important because a rapid and cost-effective test such as CMA may improve clinical outcomes during inpatient care if patients at increased risk of cardiac arrest can be identified early in the patient’s CICU care.
Finally, we tabulated the distribution of abnormal CMA results among structural congenital heart diagnoses. We provide detailed relationships between diagnoses and abnormal CMA in this cohort. For example, the prevalence of abnormal CMA in the setting of pulmonary venous anomalies was 40% in our cohort, matching the data in Ahrens et al, who also found a prevalence of 40% in a cohort of 347 cardiac patients. However, the prevalence in other studies has been lower.[6,7] The wide variation in reported results has inhibited the development of a single CMA protocol in cardiac intensive care units. The next step should be a coordinated, prospective effort to evaluate CMA in the setting of children less than 1 who present to cardiac intensive care units with close evaluation of clinically-relevant outcome measurements.
The growing number of small, single-center studies evaluating CMA in the CICU suggests that the time is ripe to interrogate a larger network to make a definitive, prospective conclusion about the value of CMA in the CICU. This is especially important because some centers have begun to order CMA routinely. While it is not yet standard of care in every CICU, CMA has begun to be labelled as standard of care and can replace karyotype in many settings.[2,15,16] The data in the current study are a step toward establishing the clinical utility for CMA in neonatal congenital heart disease. We reinforce a preliminary association between abnormal CMA and cardiac arrest during hospitalization. We demonstrate that surgical risk scores were not statistically linked with abnormal CMA and provide additional preliminary data about lesion-specific prevalence.
The generalizability of these data are limited by the methods of a single-center retrospective study. While this study adds new genotype analysis in cardiac arrest, our data are potentially influenced by the lack of a single ordering protocol for CMA in our cardiac intensive care unit during this period. Thus, differences in practice patterns among individual providers may have influenced our retrospective analysis.
CONCLUSIONS
There was a high prevalence of abnormal CMA findings in the pediatric cardiac population less than 1 year of age (29%), linked to cardiac arrest outcomes but not associated with surgical risk score. The absence of a standardized protocol for ordering a CMA in the setting of congenital heart disease results in highly variable prevalence data.
Supplementary Material
Funding
Research reported in this publication was supported, in part, by the National Institutes of Health, National Heart, Lung and Blood Institute, grant number K23HL130554, the American Heart Association Mentored Clinical and Population Research Award (Dallas, TX). REDCap access was provided by Northwestern University Clinical and Translational Sciences Institute, funded in part by NIH UL1TR001422.
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to report.
Availability of data and material
Not applicable
Code availability
Not applicable
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
REFERENCES
- 1.Hartman RJ, Rasmussen SA, Botto LD, Riehle-Colarusso T, Martin CL, Cragan JD, Shin M, Correa A (2011) The contribution of chromosomal abnormalities to congenital heart defects: a population-based study. Pediatr Cardiol 32 (8): 1147–1157. doi: 10.1007/s00246-011-0034-5 [DOI] [PubMed] [Google Scholar]
- 2.van Nisselrooij AEL, Lugthart MA, Clur SA, Linskens IH, Pajkrt E, Rammeloo LA, Rozendaal L, Blom NA, van Lith JMM, Knegt AC, Hoffer MJV, Aten E, Santen GWE, Haak MC (2020) The prevalence of genetic diagnoses in fetuses with severe congenital heart defects. Genet Med 22 (7): 1206–1214. doi: 10.1038/s41436-020-0791-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nees SN, Chung WK (2020) Genetic Basis of Human Congenital Heart Disease. Cold Spring Harb Perspect Biol 12 (9). doi: 10.1101/cshperspect.a036749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meberg A, Hals J, Thaulow E (2007) Congenital heart defects--chromosomal anomalies, syndromes and extracardiac malformations. Acta Paediatr 96 (8):1142–1145. doi: 10.1111/j.1651-2227.2007.00381.x [DOI] [PubMed] [Google Scholar]
- 5.Ahrens-Nicklas RC, Khan S, Garbarini J, Woyciechowski S, D'Alessandro L, Zackai EH, Deardorff MA, Goldmuntz E (2016) Utility of genetic evaluation in infants with congenital heart defects admitted to the cardiac intensive care unit. Am J Med Genet A 170 (12):3090–3097. doi: 10.1002/ajmg.a.37891 [DOI] [PubMed] [Google Scholar]
- 6.Geddes GC, Basel D, Frommelt P, Kinney A, Earing M (2017) Genetic Testing Protocol Reduces Costs and Increases Rate of Genetic Diagnosis in Infants with Congenital Heart Disease. Pediatr Cardiol 38 (7):1465–1470. doi: 10.1007/s00246-017-1685-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buckley JR, Kavarana MN, Chowdhury SM, Scheurer MA (2015) Current Practice and Utility of Chromosome Microarray Analysis in Infants Undergoing Cardiac Surgery. Congenit Heart Dis 10 (3):E131–138. doi: 10.1111/chd.12241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu XL, Li R, Fu F, Pan M, Han J, Yang X, Zhang YL, Li FT, Liao C (2017) Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr 17 (1):117. doi: 10.1186/s12887-017-0863-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Botto LD, Lin AE, Riehle-Colarusso T, Malik S, Correa A (2007) Seeking causes: Classifying and evaluating congenital heart defects in etiologic studies. Birth Defects Res A Clin Mol Teratol 79 (10):714–727. doi: 10.1002/bdra.20403 [DOI] [PubMed] [Google Scholar]
- 10.Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST (2011) American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13 (7):680–685. doi: 10.1097/GIM.0b013e3182217a3a [DOI] [PubMed] [Google Scholar]
- 11.Information NCfB (2019) GTR: Genetic Testing Registry. U.S. National Library of Medicine. https://www.ncbi.nlm.nih.gov/gtr/. Accessed 3/1/2019 2019 [Google Scholar]
- 12.Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, Raca G, Ritter DI, South ST, Thorland EC, Pineda-Alvarez D, Aradhya S, Martin CL (2020) Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 22 (2):245–257. doi: 10.1038/s41436-019-0686-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alten JA, Klugman D, Raymond TT, Cooper DS, Donohue JE, Zhang W, Pasquali SK, Gaies MG (2017) Epidemiology and Outcomes of Cardiac Arrest in Pediatric Cardiac ICUs. Pediatr Crit Care Med 18 (10):935–943. doi: 10.1097/pcc.0000000000001273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boskovski MT, Homsy J, Nathan M, Sleeper LA, Morton S, Manheimer KB, Tai A, Gorham J, Lewis M, Swartz M, Alfieris GM, Bacha EA, Karimi M, Meyer D, Nguyen K, Bernstein D, Romano-Adesman A, Porter GA Jr., Goldmuntz E, Chung WK, Srivastava D, Kaltman JR, Tristani-Firouzi M, Lifton R, Roberts AE, Gaynor JW, Gelb BD, Kim R, Seidman JG, Brueckner M, Mayer JE Jr., Newburger JW, Seidman CE (2020) De Novo Damaging Variants, Clinical Phenotypes, and Post-Operative Outcomes in Congenital Heart Disease. Circ Genom Precis Med 13 (4):e002836. doi: 10.1161/CIRCGEN.119.002836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Helm BM, Freeze SL (2016) Genetic Evaluation and Use of Chromosome Microarray in Patients with Isolated Heart Defects: Benefits and Challenges of a New Model in Cardiovascular Care. Front Cardiovasc Med 3:19. doi: 10.3389/fcvm.2016.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Committee Opinion No.682: Microarrays and Next-Generation Sequencing Technology: The Use of Advanced Genetic Diagnostic Tools in Obstetrics and Gynecology (2016). Obstet Gynecol 128 (6):e262–e268. doi: 10.1097/aog.0000000000001817 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.