

Abstract
Nitric oxide (NO) relaxes vascular smooth muscle cells (SMCs) and dilates blood vessels by increasing intracellular levels of cyclic guanosine monophosphate (cGMP), which stimulates the activity of cGMP-dependent protein kinase (PKG). However, the vasodilator mechanisms downstream of PKG remain incompletely understood. Here, we found that transient receptor potential melastatin 4 (TRPM4) cation channels, which are activated by Ca2+ released from the sarcoplasmic reticulum (SR) through inositol triphosphate receptors (IP3Rs) under native conditions, are essential for SMC membrane depolarization and vasoconstriction. We hypothesized that signaling via the NO/cGMP/PKG pathway causes vasodilation by inhibiting TRPM4. We found that TRPM4 currents activated by stretching the plasma membrane or directly activating IP3Rs were suppressed by exogenous NO or a membrane-permeable cGMP analog, the latter of which also impaired IP3R-mediated release of Ca2+ from the SR. The effects of NO on TRPM4 activity were blocked by inhibition of soluble guanylyl cyclase or PKG. Notably, upon phosphorylation by PKG, IRAG (IP3R-associated PKG substrate) inhibited IP3R-mediated Ca2+ release, and knockdown of IRAG expression diminished NO-mediated inhibition of TRPM4 activity and vasodilation. Using superresolution microscopy, we found that IRAG, PKG, and IP3Rs form a nanoscale signaling complex on the SR of SMCs. We conclude that NO/cGMP/PKG signaling through IRAG inhibits IP3R-dependent activation of TRPM4 channels in SMCs to dilate arteries.
Significance Statement
Nitric oxide is a gaseous vasodilator produced by endothelial cells that is essential for cardiovascular function. Although NO-mediated signaling pathways have been intensively studied, the mechanisms by which they relax SMCs to dilate blood vessels remain incompletely understood. In this study, we show that NO causes vasodilation by inhibiting the activity of Ca2+-dependent TRPM4 cation channels. Probing further, we found that NO does not act directly on TRPM4 but instead initiates a signaling cascade that inhibits its activation by blocking the release of Ca2+ from the SR. Thus, our findings reveal the essential molecular pathways of NO-induced vasodilation—a fundamental unresolved concept in cardiovascular physiology.
Keywords: nitric oxide, TRP channels, PKG, IP3Rs
Graphical Abstract
Graphical Abstract.
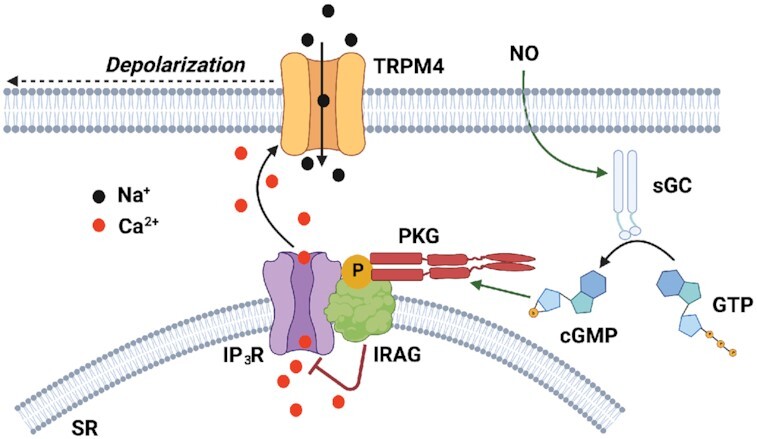
Introduction
Inward cation currents conducted by the melastatin (M) transient receptor potential (TRP) channel, TRPM4, depolarize the plasma membrane of vascular smooth muscle cells (SMCs) to constrict cerebral arteries 1,2. This process is essential for cerebral blood flow autoregulation during increases in intraluminal pressure 2,3. TRPM4 channels in SMCs are activated by Ca2+ released from the sarcoplasmic reticulum (SR) through inositol trisphosphate receptors (IP3Rs) 4 following mechanical- or agonist-induced activation of type1 angiotensin II receptors (AT1Rs) 5,6 and other Gq-protein coupled receptors (GqPCRs) 7. Although the signaling cascades that open TRPM4 channels to cause vasoconstriction have been described, nothing is currently known about potential endogenous mechanisms that inhibit TRPM4 activity to elicit vasodilation.
In their landmark study, Furchgott and Zawadski demonstrated that nitric oxide (NO) released from the endothelium in response to the muscarinic receptor agonist acetylcholine relaxed precontracted arteries 8. Nitric oxide is a highly soluble gas generated within endothelial cells during the conversion of L-arginine to L-citrulline by the Ca2+-regulated enzyme, endothelial nitric oxide synthase (eNOS). Nitric oxide diffuses to overlying SMCs and stimulates soluble guanylyl cyclase (sGC), an enzyme that catalyzes the conversion of guanosine-5’-triphosphate (GTP) to the second messenger, cyclic guanosine monophosphate (cGMP). Elevated levels of intracellular cGMP stimulate the activity of a family of cGMP-dependent protein kinases (PKGs) 9, which then phosphorylate target proteins, including IP3R-associated cGMP-kinase substrate (IRAG) 10. IRAG, encoded by Mrvi1, is a 125-kDa protein secured to the SR/endoplasmic reticulum (ER) membrane by a C-terminal anchoring domain 10,11. IRAG forms a stable complex with IP3Rs in SMCs 12 that, when phosphorylated at Ser696 by the PKG1β isoform, suppresses the release of Ca2+ from the SR/ER 10,13. Hence, IRAG is an NO/cGMP/PKG-dependent negative regulator of IP3R-mediated Ca2+ release. IRAG has been linked to NO-mediated vasodilation 12,14, but the mechanisms responsible for SMC relaxation are not currently known.
In this study, we investigated the effects of the endothelium-dependent vasodilator NO on TRPM4 channels in native SMCs and cerebral resistance arteries, testing the hypothesis that NO dilates arteries by stimulating phosphorylation of IRAG to inhibit IP3R-driven TRPM4 channel activity. Our findings show that administration of NO or a membrane-permeable cGMP analog inhibits the activity of TRPM4 channels in native SMCs from cerebral arteries, a response that is dependent on PKG. NO/cGMP/PKG does not act directly on TRPM4, but instead inhibits TRPM4 activation by Ca2+ released from the SR through IP3Rs. Using superresolution microscopy, we also show that IP3Rs, IRAG, and PKG form a nanoscale complex on the SR of SMCs from cerebral arteries. We report that NO-mediated inhibition of TRPM4 and dilation of cerebral arteries is dependent on the expression of IRAG. We conclude that NO causes vasodilation by stimulating PKG-mediated phosphorylation of IRAG, thereby inhibiting the release of Ca2+ from the SR through IP3Rs and diminishing Ca2+-dependent TRPM4 channel activity.
Results
NO Inhibits TRPM4 Through cGMP and PKG
TRPM4 currents in cerebral artery SMCs were recorded by patch-clamp electrophysiology using the perforated-patch configuration, which allows control of membrane potential but minimizes disruption of intracellular Ca2+ dynamics. After seal formation and perforation, transient inward cation currents (TICCs) conducted by TRPM4 channels were recorded in cells voltage-clamped at −70 mV. As previously shown 5,6, TICCs were increased upon stretching of the plasma membrane by application of negative pressure via the patch pipette and were inhibited by the TRPM4 blocker, 9-phenanthrol (Figure 1A and B). Ion substitution was used to verify that TICCs are indeed cation currents. In a prior study, TICC activity was eliminated upon replacement of Na+ with the non-permeable cation N-methyl-D-glucamine (NMDG) 15, indicating that extracellular Na+ is necessary for these currents. Further control studies show that the substitution of NaCl in the bath solution with Na-gluconate had no effect on TICCs activity (Figure S1), providing additional evidence that the current is carried by Na+ and not Cl– ions. To test a possible role for NO in stretch-induced, TRPM4-mediated TICCs, we applied different concentrations of the exogenous NO donor and S-nitroso-N-acetylpenicillamine (SNAP), chosen because of its high stability and potential for long-term NO release 16,17, and measured TICC activity. SNAP, applied to the bath after activation of TICCs with negative pressure, induced a concentration-dependent reduction in TICC activity with an IC50 (50% inhibitory concentration) of 3.5 µM (Figure 1C and D), indicating that NO inhibits TRPM4 channels.
Figure 1.

NO inhibits TRPM4 channel activity by activating the NO/cGMP/PKG signaling cascade. (A) Representative trace from a perforated patch-clamp experiment demonstrating that TICC activity, induced by applying negative pressure (−20 mmHg) through the patch pipette to stretch the plasma membrane, is blocked by the selective TRPM4 inhibitor 9-phenanthrol (30 µM). (B) Summary data demonstrating that TICC activity is inhibited by 9-phenanthrol (*P < .05; n = 6 cells from four animals). (C) Representative trace from a perforated patch-clamp experiment showing the effects of SNAP (100 µM) on TICCs induced by application of negative pressure (−20 mmHg) through the patch pipette. (D) Concentration–response curve for the inhibition of TICCs by SNAP (n = 5 cells from two animals per concentration). (E) Diagram depicting the NO/cGMP/PKG signaling cascade and pharmacological interventions. (F) Representative trace from a perforated patch-clamp experiment demonstrating that TICC activity induced by negative pressure (−20 mmHg) is significantly reduced by dibutyryl-cGMP (0.5 µM). (G) Summary data showing the significant reduction in TICC activity in response to dibutyryl-cGMP (*P < .05; n = 7 cells from four animals). (H) Representative trace showing that SNAP (30 µM) does not block TICC activity in the presence of the selective sGC inhibitor NS2028 (1 µM). (I) Summary data showing that NS2028 blocks the inhibitory effects of SNAP (n = 7 cells from five animals). (J) Representative trace showing that SNAP (30 µM) does not block TICC activity in the presence of the selective PKG inhibitor KT5823 (1 µM). (K) Summary data showing that KT5823 blocks the inhibitory effects of SNAP (n = 6 cells from four animals).
In the canonical signaling cascade, NO stimulates sGC to generate cGMP, which in turn increases the activity of PKGs (Figure 1E). To determine if the NO/cGMP/PKG signaling pathway is responsible for the inhibition of TRPM4, we treated cells with the membrane-permeable cGMP analog, dibutyryl-cGMP, and measured TICC activity by patch-clamping cells in the perforated-patch configuration. TICCs induced by applying negative pressure were inhibited by dibutyryl-cGMP, indicating that cGMP inhibits TRPM4 channel activity in SMCs (Figure 1F and G). In addition, SNAP had no effect on negative-pressure-induced TICC activity in the presence of the selective sGC blocker NS2028, implying that the inhibitory effects of NO require the generation of cGMP by sGC (Figure 1H and I). We further found that the selective PKG inhibitor KT5823 also abolished the TRPM4-inhibitory effects of SNAP, suggesting an essential role for PKG in this process (Figure 1J and K). These data collectively indicate that NO acts through increases in intracellular cGMP levels and subsequent stimulation of PKG activity to inhibit TRPM4 currents in SMCs.
We also used patch-clamp electrophysiology to investigate the effects of NO on large-conductance Ca2+-activated K+ (BK) activity in cerebral artery SMCs. Under physiological conditions, BK channels in SMCs are activated by transient subcellular Ca2+ rich domains generated by the release of Ca2+ ions into the cytosol from the SR through clusters of type 2 ryanodine receptors (RyR2s) 18–20. These Ca2+ signals, known as Ca2+ sparks, activate clusters of BK channels to generate large, outward K+ currents called spontaneous outward transient currents (STOCs). We recorded STOCs from cerebral artery SMCs using the perforated-patch-clamp configuration, and found that SNAP had no effect on amplitude and frequency (Figure S2). These findings suggest that BK channel activity, Ca2+ sparks, functional coupling of BK channels and RyR2s, and total SR Ca2+ store load were not acutely affected by NO.
NO Does Not Act Directly on TRPM4 Channels
To determine if the NO/cGMP/PKG pathway directly affects TRPM4 channels, we used patch-clamp electrophysiology. In the first experimental series, the Ca2+ ionophore ionomycin was added to SMCs to raise intracellular [Ca2+] and directly activate TRPM4 channels. As expected, TICC activity, measured in the perforated-patch configuration, was substantially increased by ionomycin (Figure 2A and B). Direct elevation of NO levels by co-application of SNAP had no effect on the stimulatory effects of ionomycin (Figure 2C and D), suggesting that NO/cGMP/PKG signaling does not directly block TRPM4 or diminish the sensitivity of the channel to Ca2+.
Figure 2.

The NO/cGMP/PKG pathway does not directly inhibit TRPM4. (A) Representative trace from a perforated patch-clamp experiment demonstrating that TICC activity is activated by the Ca2+ ionophore ionomycin (1 µM). (B) Summary data showing the increase in TICC activity in response to ionomycin (*P < .05; n = 8 cells from four animals). (C) Representative trace from a perforated patch-clamp experiment demonstrating that ionomycin (1 µM) reactivates SNAP (30 µM)-inhibited TICC activity. (D) Summary data showing reversal of the inhibitory effects of SNAP by ionomycin (*P < .05; n = 8 cells from four animals). (E) Time course of a representative conventional whole-cell patch-clamp recording showing that an outwardly rectifying cation current activated by applying voltage ramps from −100 to +100 mV during the first 100 s after breaking into the cell is abolished by the selective TRPM4 blocker 9-phenanthrol (30 µM). (F) I–V relationships for experiments shown in (E) in the presence and absence of 9-phenanthrol. Currents for I–V relationships were obtained 100 s after break-in (arrow). (G) Quantification of whole-cell TRPM4 currents at +100 mV (n = 6 cells from four animals). (H) Time course of a representative conventional whole-cell patch-clamp recording showing that whole-cell TRPM4 currents are unaffected by SNAP (100 µM) but are abolished by the selective TRPM4 blocker 9-phenanthrol (30 µM). (I) I–V relationships for experiments shown in (H) in the presence and absence of SNAP. Currents for I–V relationships were obtained 100 s after break-in (arrow). (J) Quantification of the effects of SNAP on whole-cell TRPM4 currents (n = 5 cells from three animals).
As an alternative approach, we patch-clamped SMCs using the conventional whole-cell configuration, which, unlike the perforated-patch configuration, allows free diffusion between the pipette solution and the intracellular compartment. In these experiments, 200 µM free Ca2+ was included in the pipette solution to activate TRPM4 currents, and tetraethylammonium (TEA; 10 mM) was included in the bath solution to block large-conductance Ca2+-activated K+ (BK) channel currents. The cation gradient established by intracellular and extracellular solutions was essentially symmetrical. Under these conditions, voltage ramps (−100 to +100 mV from a holding potential of −60 mV) activated outwardly rectifying cation currents that reversed at ∼0 mV. Currents developed approximately 20–30 s after breaking into the cell and reached stable maximal amplitude after 100 s (Figure 2E). These currents were completely inhibited by the TRPM4 blocker 9-phenanthrol (30 μM), identifying them as TRPM4 currents (Figure 2F and G). Whole-cell TRPM4 currents were unaffected by SNAP (Figure 2H–J), indicating that NO does not act directly on TRPM4 channels to diminish TICC activity.
NO/cGMP/PKG Signaling Blocks TRPM4 Activity By Impairing IP3R-Mediated Release of SR Ca2+
TRPM4 channels in SMCs are activated by Ca2+ released from the SR through IP3Rs 4,5. The contribution of NO/cGMP/PKG signaling to IP3R-mediated Ca2+ release was investigated by imaging IP3R-mediated Ca2+ signals using spinning-disk confocal microscopy. Native SMCs were loaded with the Ca2+-sensitive fluorophore Fluo-4-AM, and Ca2+ imaging was performed during addition of the Gq-protein–coupled thromboxane A2 agonist, U46619 in the absence of extracellular Ca2+. U46619 (100 nM) stimulated an increase in the global intracellular [Ca2+] in vehicle-treated SMCs (Figure 3A and C, Movie S1). In control experiments, this increase in Ca2+ was blocked by the IP3R-specific blocker, Xestospongin C (3 µM; Figure S3). Notably, the U46619-induced Ca2+ response was nearly abolished by pretreatment with dibutyryl-cGMP (Figure 3B and C, Movie S2). In control experiments designed to assess SR Ca2+ load, we applied a bolus of caffeine (10 mM) to SMCs treated with vehicle or dibutyryl-cGMP to induce the release of Ca2+ from the SR through ryanodine receptors (Figure 3A and B). The peak amplitude of caffeine-evoked global increases in cytosolic [Ca2+] did not differ between vehicle- and dibutyryl-cGMP–treated groups (Figure 3D), indicating that inhibition of U46619-induced Ca2+ release by dibutyryl-cGMP treatment was not due to a reduction in total SR [Ca2+]. These data indicate that cGMP diminishes IP3R activity in SMCs.
Figure 3.

cGMP impairs IP3R-mediated release of Ca2+ from the SR and activation of TRPM4. (A) Representative confocal images of a time course of Ca2+ signals in a Fluo-4-AM–loaded SMC treated with vehicle, and a trace showing changes in fractional fluorescence (F/F0) in response to U46619 (100 nM) and caffeine (10 mM) Scale bar = 10 µm. (B) Representative confocal images of a time course of Ca2+ signals in an SMC treated with dibutyryl-cGMP (1 µM), and a trace showing changes in fractional fluorescence (F/F0) in response to U46619 (100 nM) and caffeine (10 mM) Scale bar = 10 µm. (C) Summary data showing the change in fractional fluorescence (ΔF/F0) in response to U46619 in SMCs treated with vehicle or dibutyryl-cGMP (*P < .05; vehicle, n = 11 cells from five animals; dibutyryl-cGMP, n = 14 cells from five animals). (D) Summary data showing no significant difference in the change in fractional fluorescence (ΔF/F0) in response to caffeine (10 mM) between SMCs treated with vehicle and those treated with dibutyryl-cGMP (vehicle, n = 7 cells from three animals; dibutyryl-cGMP, n = 9 cells from three animals). (E) Representative trace from a perforated patch-clamp experiment demonstrating that TICC activity is significantly increased by Bt3IP3-AM (10 µM) and inhibited by SNAP (30 µM). (F) Summary data showing that the increase in TICC activity in response to Bt3IP3 is diminished by SNAP (*P < .05; n = 9 cells from five animals). (G) Representative trace from a perforated patch-clamp experiment demonstrating that TICC activity induced by negative pressure (−20 mmHg), applied through the patch pipette, is inhibited by dibutyryl-cGMP (0.5 µM) and cannot be rescued by Bt3IP3-AM (10 µM). (H) Summary data showing that inhibition of TICC activity in response to dibutyryl-cGMP is not restored by Bt3IP3-AM (*P < .05; n = 7 cells from four animals).
We also investigated the effects of NO on IP3R-mediated activation of TRPM4 channels using patch-clamp electrophysiology. In agreement with a previous study 4, we found that directly stimulating IP3Rs by applying Bt3IP3-AM, a stable, membrane-permeable analog of IP3, increased TICC activity (Figure 3E and F). Subsequent administration of SNAP reversed the stimulatory effects of Bt3IP3 (Figure 3E and F), indicating that NO blocks TRPM4 currents by inhibiting the activity of IP3Rs. To further explore this mechanism, we evoked TICC activity by applying negative pressure (−20 mmHg) to the patch pipette and then tested whether adding dibutyryl-cGMP blunted this response (Figure 3G and H). In the presence of dibutyryl-cGMP, Bt3IP3 did not increase TICC activity (Figure 3G and H). Collectively, the findings of these Ca2+ imaging and patch-clamp electrophysiology experiments indicate that increased cGMP levels inhibit the activity of IP3Rs, thereby diminishing TRPM4 channel activity.
IRAG, PKG, and IP3Rs Form a Nanoscale Signaling Complex on the SR
Upon stimulation by increases in intracellular cGMP, PKGs phosphorylate specific substrate proteins, regulating diverse functions. One of these targets is IRAG, which suppresses the activity of IP3Rs following phosphorylation by PKG1β. Previous studies using coimmunoprecipitation assays have reported that IRAG forms a macrocomplex with IP3Rs and PKG1β 10. To visualize this structure on the SR of isolated SMCs in situ, we utilized GSDIM (ground state depletion followed by individual molecule return) superresolution microscopy. We previously used DNA-origami–based nanorulers to show that our GSDIM system has a lateral resolution of 20–40 nm 21–23. Freshly isolated SMCs from cerebral pial arteries were co-immunolabeled for IP3R and the SR marker protein calnexin, IP3R and IRAG, or IP3R and PKG1. The resulting superresolution colocalization maps showed that all of these proteins were present as defined clusters (Figure 4A–C). Using an object-based analysis (OBA) approach 24,25, as described in our previous publications 21,23,26,27, we generated new maps of IP3R clusters that overlapped at the resolution limit of our microscope system with the centroid of each calnexin cluster, and calnexin clusters that overlapped with the centroid of each IP3R cluster. These two maps were then merged to reveal calnexin-IP3R protein clusters that were colocalized at or below the resolution of our GSDIM (Figure 4A). The same analysis was used to reveal colocalized IP3R-IRAG protein clusters (Figure 4B) and colocalized IP3R-PKG1 protein clusters (Figure 4C). Particle analysis revealed that the sizes of individual IP3R (1401 ± 5 nm2), IRAG (1679 ± 8), and PKG1 (1255 ± 5 nm2; Figure 4D–F) protein clusters were exponentially distributed, as were IP3R-IRAG and IP3R-PKG1 protein complexes (Figure 4G and H). An analysis comparing the original superresolution localization maps of IP3R-IRAG and IP3R-PKG1 protein clusters with a simulated random distribution of these elements showed that the amount of colocalization between proteins was significantly higher in the original distribution than in the random simulation (Figure 4G and H). These data suggest that IP3R, IRAG, and PKG1 colocalize on the SR of SMCs to form a nanoscale signaling complex.
Figure 4.

IRAG, PKG, and IP3Rs form a nanoscale signaling complex on the SR. (A–C) Superresolution localization maps of freshly isolated cerebral artery SMCs immunolabeled for IP3R (red) and calnexin (CANX; green) (A), IP3R and IRAG (cyan) (B), and IP3R and PKG1 (white) (C). Colocalized clusters were identified by object-based analysis (OBA) and mapped (yellow). Scale bar = 3 μm. Panels to the right show enlarged areas of the original superresolution maps indicated by the white boxes. Scale bar = 500 nm. (D–F) Frequency distribution of sizes of individual IP3R (D), IRAG (E), and PKG1 (F) protein clusters (n = 74 774 IP3R clusters; n = 34 782 IRAG clusters; and n = 46 570 PKG1 clusters). (G and H) Summary data showing the % colocalization of actual versus randomized superresolution colocalization maps and the frequency distribution of sizes of colocalized IP3R-IRAG clusters (G) and colocalized IP3R-PKG1 clusters (H). *P < .05; n = 21 cells from three animals for IP3R-IRAG colocalization (n = 2763 IP3R-IRAG colocalized clusters); n = 17 cells from thre animals for IP3R-PKG1 colocalization (n = 2106 IP3R-PKG1 colocalized clusters).
NO/cGMP/PKG Signals Through IRAG to Reduce TRPM4 Activity
Next, we used a knockdown approach to determine if the inhibitory effect of NO/cGMP/PKG signaling on TRPM4 activity is dependent on IRAG (Figure 5A). To this end, we knocked down IRAG expression in SMCs by treating isolated cerebral arteries with silencing morpholinos targeting Mrvi1. IRAG protein was readily immunodetected by Wes capillary electrophoresis in cerebral arteries transfected with non-silencing control morpholinos (Figure 5B), but its expression levels, normalized to total protein load, were significantly lower in arteries treated with IRAG-targeting morpholinos (Figure 5C), demonstrating the efficacy of the knockdown procedure.
Figure 5.

NO/cGMP/PKG signals through IRAG to reduce TRPM4 activity. (A) Diagram showing Ca2+ release via IP3Rs mediated by PKG1β-activated IRAG, which in turn regulates TRPM4 channels. (B) Blot representation of a Wes protein capillary electrophoresis experiment showing IRAG protein expression in cerebral arteries treated with standard control or IRAG-targeted morpholinos. (C) Summary data showing densitometry analyses of IRAG protein expression in cerebral arteries (*P < .05; n = 5 arteries from five animals). (D) Representative traces from perforated patch-clamp experiments demonstrating that SNAP (30 µM) significantly reduced TICC activity induced by negative pressure (−20 mmHg), applied via a patch pipette, in an SMC treated with standard control morpholinos (top), but had no effect following IRAG knockdown (bottom). (E) Summary data showing a reduction in TICC activity in response to SNAP (*P < .05; n = 7 cells from five animals). (F) Summary data showing no reduction in TICC activity in response to SNAP following knockdown of IRAG (n = 7 cells from five animals).
The effects of IRAG knockdown on NO-mediated suppression of TRPM4 activity were evaluated in patch-clamp electrophysiology experiments using SMCs isolated from cerebral arteries treated with control or IRAG-targeting morpholinos. TICC activity stimulated by application of negative pressure was inhibited by SNAP in cells from arteries treated with control morpholinos (Figure 5D and E). However, the inhibitory effects of SNAP were absent in cells treated with IRAG-targeting morpholinos (Figure 5F and G). These data demonstrate that IRAG is vital for inhibition of TRPM activity by the NO/cGMP/PKG signaling cascade.
IRAG Knockdown Impairs NO-Mediated Vasodilation
The effects of IRAG knockdown on NO-induced vasodilation were investigated using ex vivo pressure myography. Constriction of intact cerebral pial arteries in response to a depolarizing concentration (60 mM) of extracellular KCl did not differ between arteries treated with control or IRAG-targeting morpholinos (Figure 6), suggesting that IRAG knockdown did not grossly alter voltage-dependent Ca2+ influx or underlying contractile processes. In addition, myogenic tone did not differ between groups (Figure 6B). Application of SNAP (100 nM–100 µM) to arteries with predeveloped myogenic tone (60 mmHg) caused a concentration-dependent vasodilation in both groups (Figure 6C). However, SNAP-induced dilation was significantly blunted in arteries treated with IRAG-targeting morpholinos compared with controls. The EC50 for SNAP-induced dilation was 0.2 µM for control arteries and 1.3 µM for arteries treated with IRAG-targeting morpholinos (Figure 6D). These data indicate that IRAG is vital for NO-induced dilation of cerebral arteries. We conclude that signaling via the NO/cGMP/PKG pathway decreases TRPM4 activity through IRAG-mediated inhibition of SR Ca2+ release through IP3Rs. The subsequent loss of the depolarizing effects of TRPM4 channels dilates cerebral arteries (Figure 6E).
Figure 6.

IRAG knockdown impairs NO-mediated vasodilation. (A) Summary data showing no significant difference in the constriction response to elevated levels of extracellular KCl (60 mM) between cerebral arteries transfected with control morpholinos and those transfected with IRAG-targeted morpholinos (n = 9 arteries from three animals). (B) Summary data showing no difference in the myogenic tone of cerebral arteries between those transfected with control morpholinos and those transfected with IRAG-targeted morpholinos (n = 3 arteries from three animals). (C) Representative traces showing concentration-dependent dilation of cerebral arteries transfected with control or IRAG-targeted morpholinos in response to SNAP. (D) SNAP concentration-response curve (100 nM–100 µM) for dilation of cerebral arteries transfected with control or IRAG-targeted morpholinos. The EC50 for control arteries was 0.2 µM and that for IRAG KD was 1.3 µM (n = 6 arteries from four animals in each group). (E) Proposed mechanism of NO-induced inhibition of TRPM4. NO, nitric oxide; sGC, soluble guanylyl cyclase; GTP, guanosine triphosphate; cGMP, cyclic guanosine monophosphate; PKG, cGMP-dependent protein kinase; IP3R, inositol 1,4,5-triphosphate receptor; IRAG, IP3R-associated cGMP-kinase substrate; SR, sarcoplasmic reticulum; and TRPM4, transient receptor potential melastatin 4.
Discussion
Despite decades of investigation, the signaling pathways activated by the endogenous vasodilator NO that relax SMCs remain incompletely understood. Here, we demonstrate that the NO/cGMP/PKG cascade decreases the activity of TRPM4 channels in SMCs. Our findings show that NO does not act directly on the channel but instead inhibits Ca2+-dependent activation of TRPM4 by blocking the IP3R-mediated release of Ca2+ from the SR. Using superresolution imaging to investigate potential mechanisms, we found that IP3Rs on the SR form a trinary nanoscale complex with PKG and IRAG in SMCs. Our findings also showed that knockdown of IRAG diminished the ability of NO to inhibit TRPM4 activity and dilate cerebral arteries. We, thus, conclude that the NO/cGMP/PKG cascade signals through IRAG to eliminate the depolarizing influence of TRPM4 channels and cause vasodilation.
TRPM4 channels are vital for regulating SMC membrane potential and contractility, the development of myogenic tone by cerebral arteries and arterioles, and the autoregulation of blood flow in the brain 1–3,7. TRPM4 is Ca2+-activated, selective for monovalent cations, and impermeant to Ca2+ and other divalent cations 28–30. The membrane potential of vascular SMCs in situ (approximately −40 mV) is hyperpolarized compared with the Na+ reversal potential (ENa ≈ +52 mV), and the primary TRPM4 current under these conditions reflects an influx of Na+ ions that depolarizes the membrane. TRPM4 channels in vascular SMCs are stimulated by stretch- or agonist-induced activation of GqPCRs, which act through stimulation of phospholipase Cγ and subsequent hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to generate diacylglycerol (DAG) and inositol triphosphate (IP3) 5,6,15. Diacylglycerol activates protein kinase Cδ, which increases trafficking of TRPM4 protein to the plasma membrane 2,31–33, and IP3 stimulates IP3R-mediated release of SR Ca2+, which activates resident TRPM4 channels 4,5. Our data show that the NO/cGMP/PKG pathway counteracts stretch-induced TRPM4 activity, thereby attenuating the depolarizing effects of the channel and relaxing SMCs. Thus, our findings indicate that the opposing effects of two omnipresent stimuli–intraluminal pressure and endothelium-derived NO—maintain appropriate vascular tone by regulating TRPM4 channel activity in SMCs.
How does NO inhibit TRPM4 channel activity? The canonical NO/cGMP/PKG signaling pathway relaxes SMCs by lowering cytosolic Ca2+ levels. However, the mechanisms that promote the movement of Ca2+ out of the cytosol remain uncertain. Some studies suggest that PKG directly phosphorylates BK channels to increase their open probability, leading to membrane hyperpolarization and diminished Ca2+ influx 34,35. Other reports indicate that PKG phosphorylates phospholamban to increase the activity of the SR Ca2+-ATPase and enhance sequestration of Ca2+ by the SR 36. However, data showing that the NO donor SNAP does not affect Ca2+ spark-activated BK channel activity suggests that neither of these mechanisms fully accounts for the rapid and potent vasodilator properties of NO. Our findings indicate that the NO/cGMP/PKG pathway, acting through IRAG, inhibits TRPM4 channel activity by blocking the IP3R-mediated release of Ca2+ from the SR. IRAG is abundant in SMCs and is a primary target of the PKG1β splice variant 10. Our data show that IRAG is located on the SR membrane in a nanoscale signaling complex with IP3Rs and PKG in cerebral artery SMCs, a finding in agreement previously reported results obtained using immunoprecipitation 10. These proteins are less than 40 nm apart, suggesting that PKG can rapidly phosphorylate IRAG in response to increases in cGMP. Similarly, IRAG is in a preformed complex with IP3Rs and can swiftly inhibit SR Ca2+ release upon phosphorylation. Decreasing IP3R-mediated Ca2+ release decreases Ca2+-dependent TRPM4 channel activity and associated depolarizing effects. Thus, the loss of TRPM4 currents reduces voltage-dependent Ca2+ influx through CaV1.2 channels, thereby relaxing SMCs and dilating arteries. Presumably, IRAG is subsequently dephosphorylated by an as yet unidentified protein phosphatase when cGMP levels drop, resulting in disinhibition of IP3Rs and reengagement of the TRPM4 pathway.
In conclusion, this study showed that the NO/cGMP/PKG pathway, acting through IRAG and IP3Rs, inhibits TRPM4 channels in SMCs to dilate cerebral arteries. These findings provide further evidence that modulation of TRPM4 activity is vital for regulating SMC membrane potential and contractility. Moreover, impaired endothelial cell function and loss of NO synthesis is a hallmark of hypertension and many other cardiovascular diseases. Our findings show that TRPM4 is an indirect target of the NO signaling cascade, suggesting that pharmacological manipulation of the channel may be a viable therapeutic strategy for treatment of vascular dysfunction
Materials and Methods
Chemical and Reagents
All chemicals and other reagents were obtained from Sigma-Aldrich, Inc. (St. Louis, MO) unless otherwise specified.
Animals
Adult (8–10-weeks-old) male and female C57BL/6J mice (Jackson Labs, Bar Harbor, ME) were used for all experiments. All animal procedures used in this study were approved by the Institutional Animal Care and Use Committee of the University of Nevada, Reno School of Medicine. Animals were anesthetized with isoflurane and euthanized by decapitation and exsanguination.
Isolation of Cerebral Arteries and SMCs
Cerebral arteries were gently removed from the brain and washed in Ca2+-free physiological saline solution (PSS) consisting of 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, 10 mM HEPES, and 10 mM glucose (pH 7.4, adjusted with NaOH), with 0.5% bovine serum albumin (BSA). Native SMCs for patch-clamp and superresolution imaging experiments were obtained by initially digesting isolated arteries in 1 mg/mL papain (Worthington Biochemical Corporation, Lakewood, NJ), 1 mg/mL dithiothreitol (DTT), and 1 mg/mL BSA in Ca2+-free PSS at 37°C for 12 min, followed by a 14-min incubation with 1 mg/mL type II collagenase (Worthington Biochemical Corporation). A single-cell suspension was prepared by washing digested arteries three times with Ca2+-free PSS and triturating with a fire-polished glass pipette. All cells used for this study were freshly dissociated on the day of experimentation.
Electrophysiological Recordings
For all patch-clamp electrophysiology experiments, currents were recorded at room temperature using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA) equipped with an Axon CV 203BU headstage and Digidata 144A digitizer (Molecular Devices).
TICCs produced by the influx of Na+ through TRPM4 channels were recorded in the perforated-patch configuration, in which amphotericin B (200 µg/mL) is included in the patch pipette to create pores in the plasma membrane. Cells were voltage-clamped at −70 mV, and membrane stretch was delivered by applying negative pressure through the recording electrode using a Clampex-controlled pressure clamp system (HSPC-1; ALA Scientific Instruments Inc., Farmingdale, NY). TICCs were recorded in a bath solution consisting of 134 mM NaCl, 6 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, and 10 mM glucose (pH 7.4, adjusted with NaOH). The patch pipette solution contained 110 mM K-aspartate, 1 mM MgCl2, 30 mM KCl, 10 mM NaCl, 10 mM HEPES, and 0.005 mM EGTA (pH 7.2, adjusted with NaOH). TICC activity was calculated as the sum of the open channel probability (NPo) of multiple open states of 1.75 pA 4.
Whole-cell TRPM4 currents were recorded in a bath solution consisting of 156 mM NaCl, 1.5 mM CaCl2, 10 mM glucose, 10 mM HEPES, and 10 mM TEACl (pH 7.4, adjusted with NaOH). The patch pipette solution contained 156 mM CsCl, 8 mM NaCl, 1 mM MgCl2, 10 mM HEPES (pH 7.4, adjusted with NaOH), and a free Ca2+ concentration of 200 µM, adjusted using the appropriate amount of CaCl2 and EGTA, as determined with Max-Chelator software (WEBMAXC standard, available at https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/webmaxc/webmaxcS.htm). Whole-cell cation currents were evoked by applying 400 ms voltage ramps from −100 to +100 mV from a holding potential of −60 mV, ending with a 300 ms step at +100 mV. Voltage ramps were repeated every 2 s for 300 s. The selective TRPM4 inhibitor 9-phenanthrol (30 μM) was applied after 200 s of recording. Whole-cell TRPM4 current amplitude was expressed as the 9-phenanthrol-sensitive current at +100 mV.
Ca2+ Imaging
A suspension of freshly isolated SMCs was placed in a glass-bottom 35 mm dish, and cells were allowed to adhere to the glass coverslip for 20 min at room temperature. Smooth muscle cells were then loaded with the Ca2+-sensitive fluorophore, Fluo-4-AM (1 µM; Invitrogen, Waltham, MA), in the dark for 20 min at room temperature in Ca2+-free PSS. Cells were subsequently washed three times with Ca2+-containing PSS and incubated at room temperature for 20 min in the dark to allow sufficient time for Fluo-4 de-esterification and loading of Ca2+ stores. Immediately before imaging, cells were washed three times and imaged in Ca2+-free PSS to eliminate signals resulting from the influx of Ca2+. U46619 (Enzo Biochem, Farmingdale, NY) and caffeine (Sigma) were bath applied. Images were acquired at an acquisition rate of 33 frames per second using an iXon 897 EMCCD camera (Andor, Belfast, UK) coupled to a spinning-disk confocal head (CSU-X1; Yokogawa, Tokyo, Japan) with a 100x oil-immersion objective (Olympus, Tokyo, Japan).
Superresolution Microscopy
Ground state depletion followed by individual molecule return (GSDIM) superresolution microscopy was performed as described previously 21,23,26,27,37. Briefly, freshly isolated cerebral artery SMCs were allowed to adhere onto poly-L-lysine–coated glass coverslips for 30 min. The cells were then fixed for 20 min with 2% paraformaldehyde, quenched with 0.4 mg/mL NaBH4, and permeabilized with 0.1% Triton X-100. Thereafter, cells were blocked with 50% SEABLOCK blocking buffer (Thermo Fisher Scientific, Waltham, MA) for 2 h and incubated overnight at 4°C with primary antibody diluted in phosphate-buffered saline (PBS) containing 20% SEABLOCK, 1% BSA, and 0.05% Triton X-100. The following antibodies were obtained from the indicated vendors and used as described in the text: anticalnexin (ab22595; Abcam, Cambridge, UK), anti-IP3R (ab252536; Abcam), anti-IRAG (LS-C205780; LSBio, Seattle, WA), and anti-PKG1 (3248S; Cell Signaling Technology, Danvers, MA). Cells were washed three times with 1X PBS after each step. After overnight incubation, unbound primary antibody was removed by washing four times with 20% SEABLOCK, after which cells were incubated at room temperature for 2 h in the dark with Alexa-Fluor 647- or Alexa-Fluor 568-conjugated goat antirabbit or goat antimouse secondary antibodies, as appropriate. After washing with 1X PBS, coverslip-plated cells were mounted onto glass depression slides in a thiol-based photo-switching imaging buffer consisting of 50 mM Tris/10 mM NaCl (pH 8), 10% glucose, 10 mM mercaptoethylamine, 0.48 mg/mL glucose oxidase, and 58 μg/mL catalase. Coverslips were sealed to depression slides with Twinsil dental glue (Picodent, Wipperfurth, Germany) to exclude oxygen and prevent rapid oxidation of the imaging buffer. Superresolution images were acquired in epifluorescence mode using a GSDIM imaging system (Leica, Wetzlar, Germany) equipped with an oil-immersion 160x HCX Plan-Apochromat (NA 1.47) objective, an electron-multiplying charge-coupled device camera (EMCCD; iXon3 897; Andor Technology, Belfast, UK), and 500-mW, 532-, and 642-nm laser lines. Localization maps were constructed from images acquired at 100 Hz for 25 000 frames using Leica LAX software. Postacquisition image analyses of cluster size distribution were performed using binary masks of images in NIH Image J software.
Object-Based Colocalization Analysis
Object-based analysis was used to establish the co-localization of IP3R with Calnexin, IRAG or PKG1 proteins in superresolution localization maps. For this analysis, we used NIH ImageJ software with the JACoP co-localization analysis plug-in 24,25. The JACoP plug-in was used to analyze the two channels representing fluorophores detected by Alexa-Fluor 568 or Alexa-Fluor 647. Contiguous objects in both channels were identified by systematically inspecting the neighboring 8 pixels (in 2D) of a reference pixel. All adjacent pixels with intensities above a user defined threshold limit were considered to be part of the same structure as the reference pixel and were segmented as individual objects. The superresolution localization maps were previously thresholded by the detection algorithm incorporated into the LAX software used for image acquisition. Therefore, the threshold level in JACoP was set to 1 (nearly the minimum) for all images. After thresholding, centroids (defined as the single-pixel geometric centers of the defined objects) were determined for each object. Clusters in the other wavelength within 20 nm (resolution limit of our GSDIM system) of the centroid were considered as “colocalized.” The percentage of colocalizing clusters was calculated by determining the number of colocalizing clusters as a percentage of the total number of clusters analyzed. The percentage of colocalizing clusters in each superresolution map was compared with simulations of randomly distributed clusters. Particle analysis of the colocalization maps was performed to quantify cluster size and density.
IRAG Knockdown With Morpholino Oligonucleotides
Morpholino oligonucleotides, which provide specific knockdown of targeted proteins by inhibiting mRNA translation, were used to reduce IRAG expression in intact cerebral arteries, as described previously 6,38,39. A morpholino targeting the 5’-untranslated region through the first 25 bases of the coding sequence of Mrvi1 was designed and synthesized to block translation initiation of IRAG (Gene Tools, LLC; Philomath, OR). A non-silencing morpholino with no known binding targets in mice was used as a standard control morpholino for all experiments. The nucleotide sequence of IRAG-targeted morpholinos used in this study was 5’-AGC TCT TTG GTT TTG CAT CAA CAC C 3’, and that of standard control morpholinos was 5’-CCT CTT ACC TCA GTT ACA ATT TAT A 3’.
Morpholinos were prepared for delivery to intact cerebral arteries by first mixing a solution of morpholinos dissolved in Opti-MEM (50 µM) with an equal volume of a solution of Lipofectamine 2000 (0.017 mg/mL; Thermo Fisher Scientific) in Opti-MEM, followed by incubation for 2 h at room temperature. This solution was then combined with 1 mL of serum-free Dulbecco's modified Eagle's medium (Thermo Fisher Scientific) containing penicillin-streptomycin (1%), after which isolated cerebral arteries were immersed in the morpholino-containing solution and incubated for 36 h at 37°C in a humidified 5% CO2 environment. Successful morpholino-mediated knockdown of IRAG was verified by measuring protein levels using the Wes capillary electrophoresis immunoassay protein detection system. IRAG protein levels were quantified and compared between cerebral artery tissues transfected with standard control and IRAG-targeted morpholinos.
Wes Capillary Electrophoresis
Cerebral arteries in ice-cold RIPA buffer (25 mM Tris pH 7.6, 150 mM NaCl, 1% Igepal CA-630, 1% sodium deoxycholate, and 0.1% SDS) containing a protease inhibitor cocktail (Cell Biolabs, Inc., San Diego, CA) were homogenized using a mechanical homogenizer followed by sonication. The resulting homogenate was then centrifuged at 14 000 rpm for 20 min, and the supernatant containing proteins was collected. Protein concentration was quantified with a BCA protein assay kit (Thermo Fisher Scientific) by measuring absorbance using a 96-well plate reader. Proteins were then resolved by capillary electrophoresis using the Wes system (ProteinSimple, San Jose, CA) and probed with an anti-IRAG primary antibody (LS-C205780; LSBio). Protein bands were analyzed using Compass for SW (ProteinSimple).
Pressure Myography
The effects of IRAG knockdown on SMC contractility and NO-induced dilation were assessed using cerebral pial arteries treated with IRAG-targeting or control morpholinos. Current best practices guidelines for pressure myography experiments were followed 40. Arteries were mounted between two glass cannulas (outer diameter, 40–50 μm) in a pressure myograph chamber (Living Systems Instrumentation, St Albans City, VT) and secured by a nylon thread. Intraluminal pressure was controlled using a servo-controlled peristaltic pump (Living Systems Instrumentation), and preparations were visualized with an inverted microscope (Accu-Scope Inc., Commack, NY) coupled to a USB camera (The Imaging Source LLC, Charlotte, NC). Changes in luminal diameter were assessed using IonWizard software (version 7.2.7.138; IonOptix LLC, Westwood, MA). Arteries were bathed in warmed (37°C), oxygenated (21% O2, 6% CO2, and 73% N2) and PSS (119 mM NaCl, 4.7 mM KCl, 21 mM NaHCO3, 1.17 mM MgSO4, 1.8 mM CaCl2, 1.18 mM KH2PO4, 5 mM glucose, and 0.03 mM EDTA) at an intraluminal pressure of 5 mmHg. Following equilibration for 15 min, intraluminal pressure was increased to 110 mmHg, and vessels were stretched to their approximate in vivo length, after which pressure was reduced back to 5 mmHg for an additional 15 min. Vessel viability was assessed for each preparation by evaluating vasoconstrictor responses to high extracellular [K+] PSS, made isotonic by adjusting the [NaCl] (60 mM KCl, 63.7 mM NaCl). Arteries that showed less than 10% constriction in response to elevated [K+] were excluded from further investigation.
Relaxation to the NO donor SNAP was assessed in arteries pressurized to 60 mmHg. Once spontaneous myogenic tone had developed and stabilized, SNAP concentration–response relationships were determined by cumulative addition of SNAP (100 nM–100 µM) to the superfusing bath solution; vasorelaxation was allowed to develop at each concentration step. At the end of this process, the passive diameter was obtained by bathing vessels in Ca2+-free PSS solution, supplemented with EGTA (2 mM) and the voltage-dependent Ca2+ channel blocker diltiazem (10 μM; Tocris Bioscience, Minneapolis, MN), included to inhibit SMC contraction. Data were calculated as vasodilation (%) = [(lumen diameter at dilation—lumen diameter at baseline]/developed myogenic tone] × 100.
Statistical Analysis
All summary data are presented as means ± SEM. Statistical analyses were performed and graphs were constructed using Prism version 8.3.0 (GraphPad Software, San Diego, CA). The value of n refers to the number of cells for patch-clamp electrophysiology, superresolution microscopy and Ca2+-imaging, and arteries for myography experiments. Statistical analyses were performed using unpaired Student's t-test or two-way analysis of variance (ANOVA). IC50 and EC50 values for SNAP were calculated using non-linear regression analysis of concentration–response curves. A P-value < .05 was considered statistically significant for all analyses.
Data and Materials Availability
All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
Supplementary Material
Contributor Information
Sher Ali, Department of Pharmacology, Center for Molecular and Cellular Signaling in the Cardiovascular System, Reno School of Medicine, University of Nevada, Reno, NV 89557-0318, USA.
Alfredo Sanchez Solano, Department of Pharmacology, Center for Molecular and Cellular Signaling in the Cardiovascular System, Reno School of Medicine, University of Nevada, Reno, NV 89557-0318, USA.
Albert L Gonzales, Department of Physiology and Cell Biology, Center for Molecular and Cellular Signaling in the Cardiovascular System, University of Nevada, Reno School of Medicine, University of Nevada, Reno, NV 89557-0318, USA.
Pratish Thakore, Department of Pharmacology, Center for Molecular and Cellular Signaling in the Cardiovascular System, Reno School of Medicine, University of Nevada, Reno, NV 89557-0318, USA.
Vivek Krishnan, Department of Pharmacology, Center for Molecular and Cellular Signaling in the Cardiovascular System, Reno School of Medicine, University of Nevada, Reno, NV 89557-0318, USA.
Evan Yamasaki, Department of Pharmacology, Center for Molecular and Cellular Signaling in the Cardiovascular System, Reno School of Medicine, University of Nevada, Reno, NV 89557-0318, USA.
Scott Earley, Department of Pharmacology, Center for Molecular and Cellular Signaling in the Cardiovascular System, Reno School of Medicine, University of Nevada, Reno, NV 89557-0318, USA.
Author Contributions
S.E. and A.L.G. conceived of and initiated the project. S.E. supervised the project and designed experiments. S.A. and A.S.S. performed patch-clamp electrophysiological recordings. P.T conducted pressure myography experiments. V.K. performed superresolution imaging and Wes protein-detection experiments. E.Y. conducted Ca2+ imaging experiments. S.A., A.S.S., A.L.G., P.T., V.K., E.Y., and S.E. analyzed the data. S.A. and S.E. wrote the manuscript and prepared the figures. S.A., A.S.S., A.L.G., and S.E., revised the manuscript. All authors have read and approved the final version of the manuscript and agree to be accountable for all aspects of the work and ensuring that questions related to the accuracy of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those eligible for authorship are listed.
Funding
This study was supported by grants from the National Institutes of Health (NHLBI R35HL155008, R01HL137852, R01HL091905, R01HL139585, R01HL122770, R01HL146054, NINDS RF1NS110044, R61NS115132, and NIGMS P20GM130459 to SE; NHLBI K01HL138215 to ALG). The High Spatial and Temporal Resolution Imaging Core at the COBRE Center for Molecular and Cellular Signaling in the Cardiovascular System, University of Nevada, Reno, is maintained by a grant from NIH/NIGMS (P20GM130459 Sub#5452).
Conflict of Interest
The authors have no conflicts of interest to report, financial or otherwise.
References
- 1. Gonzales AL, Garcia ZI, Amberg GC, Earley S. Pharmacological inhibition of TRPM4 hyperpolarizes vascular smooth muscle. Am J Physiol Cell Physiol. 2010;299(5):C1195–C1202. doi: 10.1152/ajpcell.00269.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res. 2004;95(9):922–929. doi: 10.1161/01.RES.0000147311.54833.03. [DOI] [PubMed] [Google Scholar]
- 3. Reading SA, Brayden JE. Central role of TRPM4 channels in cerebral blood flow regulation. Stroke. 2007;38(8):2322–2328. doi: 10.1161/STROKEAHA.107.483404. [DOI] [PubMed] [Google Scholar]
- 4. Gonzales AL, Amberg GC, Earley S. Ca2+ release from the sarcoplasmic reticulum is required for sustained TRPM4 activity in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. 2010;299(2):C279–C288. <Go to ISI>://MEDLINE:20427713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gonzales AL, Yang Y, Sullivan MNet al. . A PLCgamma1-dependent, force-sensitive signaling network in the myogenic constriction of cerebral arteries. Sci Signal. 2014;7(327):ra49. doi: 10.1126/scisignal.2004732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pires PW, Ko E-A, Pritchard HAT, Rudokas M, Yamasaki E, Earley S. The angiotensin II receptor type 1b is the primary sensor of intraluminal pressure in cerebral artery smooth muscle cells. J Physiol. 2017;595(14):4735–4753. <Go to ISI>://MEDLINE:28475214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Y, Baylie RL, Tavares MJ, Brayden JE. TRPM4 channels couple purinergic receptor mechanoactivation and myogenic tone development in cerebral parenchymal arterioles. J Cereb Blood Flow Metabol. 2014;34(10):1706–1714. doi: 10.1038/jcbfm.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288(5789):373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 9. Hofmann F, Bernhard D, Lukowski R, Weinmeister P. cGMP regulated protein kinases (cGK). Handb Exp Pharmacol. 2009;191(191):137–162. doi: 10.1007/978-3-540-68964-5_8. [DOI] [PubMed] [Google Scholar]
- 10. Schlossmann J, Ammendola A, Ashman Ket al. . Regulation of intracellular calcium by a signalling complex of IRAG, IP3 receptor and cGMP kinase Ibeta. Nature. 2000;404(6774):197–201. doi: 10.1038/35004606. [DOI] [PubMed] [Google Scholar]
- 11. Shaughnessy JD Jr, Largaespada DA, Tian Eet al. . Mrvil, a common MRV integration site in BXH2 myeloid leukemias, encodes a protein with homology to a lymphoid-restricted membrane protein Jaw1. Oncogene. 1999;18(12):2069–2084. doi: 10.1038/sj.onc.1202419. [DOI] [PubMed] [Google Scholar]
- 12. Geiselhoringer A, Werner M, Sigl Ket al. . IRAG is essential for relaxation of receptor-triggered smooth muscle contraction by cGMP kinase. EMBO J. 2004;23(21):4222–4231. <Go to ISI>://MEDLINE:15483626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ammendola A, Geiselhoringer A, Hofmann F, Schlossmann J. Molecular determinants of the interaction between the inositol 1,4,5-trisphosphate receptor-associated cGMP kinase substrate (IRAG) and cGMP kinase Ibeta. J Biol Chem. 2001;276(26):24153–24159. <Go to ISI>://MEDLINE:11309393. [DOI] [PubMed] [Google Scholar]
- 14. Desch M, Sigl K, Hieke Bet al. . IRAG determines nitric oxide- and atrial natriuretic peptide-mediated smooth muscle relaxation. Cardiovasc Res. 2010;86(3):496–505. <Go to ISI>://MEDLINE:20080989. [DOI] [PubMed] [Google Scholar]
- 15. Gonzales AL, Earley S. Endogenous cytosolic Ca(2+) buffering is necessary for TRPM4 activity in cerebral artery smooth muscle cells. Cell Calcium. 2012;51(1):82–93. doi: 10.1016/j.ceca.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brisbois EJ, Handa H, Major TC, Bartlett RH, Meyerhoff ME. Long-term nitric oxide release and elevated temperature stability with S-nitroso-N-acetylpenicillamine (SNAP)-doped Elast-eon E2As polymer. Biomaterials. 2013;34(28):6957–6966. doi: 10.1016/j.biomaterials.2013.05.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wo Y, Li Z, Brisbois EJet al. . Origin of long-term storage stability and nitric oxide release behavior of CarboSil polymer doped with S-nitroso-N-acetyl-d-penicillamine. ACS Appl Mater Interfaces. 2015;7(40):22218–22227. doi: 10.1021/acsami.5b07501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nelson MT, Cheng H, Rubart Met al. . Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270(5236):633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 19. Mironneau J, Arnaudeau S, Macrez-Lepretre N, Boittin FX. Ca2+ sparks and Ca2+ waves activate different Ca(2+)-dependent ion channels in single myocytes from rat portal vein. Cell Calcium. 1996;20(2):153–160. doi: 10.1016/s0143-4160(96)90104-9. [DOI] [PubMed] [Google Scholar]
- 20. ZhuGe R, Tuft RA, Fogarty KE, Bellve K, Fay FS, Walsh JV Jr. The influence of sarcoplasmic reticulum Ca2+ concentration on Ca2+ sparks and spontaneous transient outward currents in single smooth muscle cells. J Gen Physiol. 1999;113(2):215–228. doi: 10.1085/jgp.113.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pritchard HAT, Griffin CS, Yamasaki Eet al. . Nanoscale coupling of junctophilin-2 and ryanodine receptors regulates vascular smooth muscle cell contractility. Proc Natl Acad Sci. 2019;116(43):21874–21881. <Go to ISI>://MEDLINE:31591206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Griffin CS, Alvarado MG, Yamasaki Eet al. . The intracellular Ca(2+) release channel TRPML1 regulates lower urinary tract smooth muscle contractility. Proc Natl Acad Sci. 2020;117(48):30775–30786. doi: 10.1073/pnas.2016959117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thakore P, Pritchard HAT, Griffin CSet al. . TRPML1 channels initiate Ca2+ sparks in vascular smooth muscle cells. Sci Signal. 2020;13(637):eaba1015. <Go to ISI>://MEDLINE:32576680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224(3):213–232. <Go to ISI>://MEDLINE:17210054. [DOI] [PubMed] [Google Scholar]
- 25. Lachmanovich E, Shvartsman DE, Malka Y, Botvin C, Henis YI, Weiss AM. Co-localization analysis of complex formation among membrane proteins by computerized fluorescence microscopy: application to immunofluorescence co-patching studies. J Microsc. 2003;212(2):122–131. <Go to ISI>://MEDLINE:14629561. [DOI] [PubMed] [Google Scholar]
- 26. Pritchard HAT, Pires PW, Yamasaki E, Thakore P, Earley S. Nanoscale remodeling of ryanodine receptor cluster size underlies cerebral microvascular dysfunction in Duchenne muscular dystrophy. Proc Natl Acad Sci. 2018;115(41):E9745–E9752. <Go to ISI>://MEDLINE:30181262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pritchard HAT, Gonzales AL, Pires PWet al. . Microtubule structures underlying the sarcoplasmic reticulum support peripheral coupling sites to regulate smooth muscle contractility. Sci Signal. 2017;10(497):eaan2694. <Go to ISI>://MEDLINE:28928237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. 2002;109(3):397–407. doi: 10.1016/s0092-8674(02)00719-5. [DOI] [PubMed] [Google Scholar]
- 29. Murakami M, Xu F, Miyoshi I, Sato E, Ono K, Iijima T. Identification and characterization of the murine TRPM4 channel. Biochem Biophys Res Commun. 2003;307(3):522–528. doi: 10.1016/s0006-291x(03)01186-0. [DOI] [PubMed] [Google Scholar]
- 30. Nilius B, Prenen J, Droogmans Get al. . Voltage dependence of the Ca2+-activated cation channel TRPM4. J Biol Chem. 2003;278(33):30813–30820. doi: 10.1074/jbc.M305127200. [DOI] [PubMed] [Google Scholar]
- 31. Garcia ZI, Bruhl A, Gonzales AL, Earley S. Basal protein kinase Cdelta activity is required for membrane localization and activity of TRPM4 channels in cerebral artery smooth muscle cells. Channels. 2011;5(3):210–214. doi: 10.4161/chan.5.3.15111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Crnich R, Amberg GC, Leo MDet al. . Vasoconstriction resulting from dynamic membrane trafficking of TRPM4 in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2010;299(3):C682–C694. doi: 10.1152/ajpcell.00101.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Earley S, Straub SV, Brayden JE. Protein kinase C regulates vascular myogenic tone through activation of TRPM4. Am J Physiol Heart Circul Physiol. 2007;292(6):H2613–H2622. doi: 10.1152/ajpheart.01286.2006. [DOI] [PubMed] [Google Scholar]
- 34. Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. Am J Physiol Cell Physiol. 1993;265(1):C299–C303. <Go to ISI>://MEDLINE:8338137. [DOI] [PubMed] [Google Scholar]
- 35. Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci. 1994;91(16):7583–7587. <Go to ISI>://MEDLINE:7519783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Colyer J. Phosphorylation states of phospholamban. Ann N Y Acad Sci. 1998;853(1 CARDIAC SARCO):79–91. <Go to ISI>://MEDLINE:10603938. [DOI] [PubMed] [Google Scholar]
- 37. Thakore P, Alvarado MG, Ali Set al. . Brain endothelial cell TRPA1 channels initiate neurovascular coupling. Elife. 2021;10: 10:e63040. <Go to ISI>://MEDLINE:33635784. doi: 10.7554/eLife.63040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jepps TA, Bentzen BH, Stott JBet al. . Vasorelaxant effects of novel Kv 7.4 channel enhancers ML213 and NS15370. Br J Pharmacol. 2014;171(19):4413–4424. doi: 10.1111/bph.12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jepps TA, Carr G, Lundegaard PR, Olesen SP, Greenwood IA. Fundamental role for the KCNE4 ancillary subunit in Kv7.4 regulation of arterial tone. J Physiol. 2015;593(24):5325–5340. doi: 10.1113/JP271286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wenceslau CF, McCarthy CG, Earley Set al. . Guidelines for the measurement of vascular function and structure in isolated arteries and veins. Am J Physiol Heart Circul Physiol. 2021;321(1):H77–H111. doi: 10.1152/ajpheart.01021.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.