

SUMMARY
Pancreatic ductal adenocarcinoma (PDAC) is characterized by notorious resistance to current therapies attributed to inherent tumor heterogeneity and highly desmoplastic and immunosuppressive tumor microenvironment (TME). Unique proline isomerase Pin1 regulates multiple cancer pathways, but its role in the TME and cancer immunotherapy is unknown. Here we find that Pin1 is overexpressed both in cancer cells and cancer-associated fibroblasts (CAFs), and correlates with poor survival in PDAC patients. Targeting Pin1 using clinically available drugs induces complete elimination or sustained remissions of aggressive PDAC by synergizing with anti-PD-1 and gemcitabine in diverse model systems. Mechanistically, Pin1 drives the desmoplastic and immunosuppressive TME by acting on CAFs, and induces lysosomal degradation of the PD-1 ligand PD-L1 and the gemcitabine transporter ENT1 in cancer cells, besides activating multiple cancer pathways. Thus, Pin1 inhibition simultaneously blocks multiple cancer pathways, disrupts the desmoplastic and immunosuppressive TME, and upregulates PD-L1 and ENT1, rendering PDAC eradicable by immunochemotherapy.
Graphical Abstract
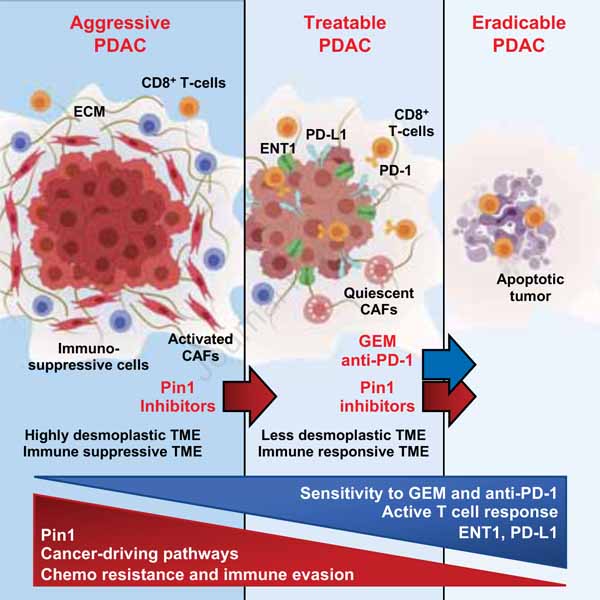
In brief:
Pin1 can drive the desmoplastic and immunosuppressive tumor microenvironment in addition to its known oncogenic functions.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive solid malignancies, with near uniform mortality (Siegel et al., 2021), and is projected to be the 2nd leading cause of cancer deaths by 2030 (Rahib et al., 2014). PDAC is notoriously resistant to chemotherapy, targeted therapies, and even immunotherapy (Brahmer et al., 2012; Kleeff et al., 2016), attributed to inherent intratumor heterogeneity and a highly desmoplastic and immunosuppressive tumor microenvironment (TME) (Binnewies et al., 2018; Ho et al., 2020; McGranahan and Swanton, 2017). Tumor heterogeneity renders tumors resistant to targeted therapies aiming at blocking individual pathways because multiple pathways are often activated simultaneously and/or rapidly upregulated as a compensatory mechanism (Gerlinger et al., 2012; Hanahan and Weinberg, 2011; Luo et al., 2009), especially in PDAC where they are highly heterogeneous and continuously evolving (Biankin et al., 2012; Samuel and Hudson, 2011).
The PDAC TME is dominated by dense desmoplasia and immunosuppressive cell populations, which limit cytotoxic T-cell response (Balachandran et al., 2019; Laklai et al., 2016). Cancer-associated fibroblasts (CAFs) play a central role in promoting the desmoplastic and immunosuppressive TME by producing extracellular matrix proteins and cytokines, as well as interacting with cancer cells to promote tumor growth and malignancy (Sahai et al., 2020; Whittle and Hingorani, 2019). Recent strategies targeting the stroma reduce tumor growth and increase tumor response to chemo- and/or immunotherapy; however, they rarely lead to obvious tumor regression (Jiang et al., 2016; Sherman et al., 2014). Moreover, some of those approaches even lead to disease acceleration and more aggressive tumors (Ozdemir et al., 2014; Rhim et al., 2014) and clinical trials have not yet produced promising results (Ho et al., 2020; Hosein et al., 2020; Sahai et al., 2020), suggesting that targeting the TME might not be sufficient.
Targeting immune checkpoints such as the one mediated by programmed cell death protein 1 (PD-1) and its ligand PD-L1 has improved patient survival in various cancers. However, the response rate is very low in PDAC patients (Brahmer et al., 2012), due to diminished tumor immunogenicity, including low PD-L1 expression and immunosuppressive TME, but the underlying mechanisms are not well understood (Gotwals et al., 2017; Mahoney et al., 2015; Sharma and Allison, 2015; Zou et al., 2016). PD-L1 expression is tightly controlled at the transcriptional and post-translational levels, but is aberrantly altered in human cancers (Burr et al., 2017; Cha et al., 2019; Dorand et al., 2016; Lim et al., 2016; Zhang et al., 2018). Importantly, although PD-L1 has been well studied for its engagement with PD-1 on T-cells to evade antitumor immunity, recent studies have shown that the presence of PD-L1-expressing cancer cells within tumors is known to be an important predictor of response to immune checkpoint blockade (ICB) therapy in patients (Herbst et al., 2014). Moreover, upregulating PD-L1 expression in cancer cells using different approaches improves ICB efficacy in experimental models (Herter-Sprie et al., 2016; Jiao et al., 2017; Zhang et al., 2018). Since most PDAC tumors are negative for PD-L1 (Tessier-Cloutier et al., 2017), it is critical to understand the mechanisms and signaling pathways behind the regulation of PD-L1 levels to improve ICB response and efficacy.
A central common signaling mechanism in cancer is proline-directed phosphorylation regulating numerous oncoproteins and tumor suppressors (Blume-Jensen and Hunter, 2001; Ubersax and Ferrell, 2007), many of which are further regulated by a unique proline isomerase, Pin1 (Lu and Hunter, 2014; Zhou and Lu, 2016). Aberrant Pin1 overactivation promotes tumorigenesis by activating over 60 oncoproteins and inactivating over 30 tumor suppressors in various cancers, including numerous substrates in oncogenic Kras signaling (Lu and Hunter, 2014; Zhou and Lu, 2016), which is dominant in PDAC (Waters and Der, 2018). Furthermore, PIN1−/− mice develop normally, but are highly resistant to tumorigenesis (Girardini et al., 2011; Liou et al., 2002; Wulf et al., 2004; Zhou and Lu, 2016). Moreover, genetic polymorphisms that reduce Pin1 expression are also associated with reduced risk for multiple cancers in humans (Li et al., 2013). These data suggest that targeting Pin1 in PDAC might simultaneously block multiple oncogenic signaling pathways without major toxicity (Lu and Hunter, 2014; Zhou and Lu, 2016). This notion has been corroborated by the recent unexpected identification of all-trans retinoic acid and arsenic trioxide (ATRA + ATO) as synergistic Pin1 inhibitors that block multiple cancer-driving pathways, eliminate cancer stem cells, and increase response to chemotherapy, targeted therapy, and radiation in various cancers (Kozono et al., 2018; Liu et al., 2019; Luo et al., 2020; Mugoni et al., 2019; Wei et al., 2015; Zhou and Lu, 2016). These findings identify Pin1 as a major cellular target for ATRA + ATO to safely cure deadly acute promyelocytic leukemia (APL) (de The and Chen, 2010 ; Kozono et al., 2018; Wei et al., 2015; Zhou and Lu, 2016). However, it is unknown whether Pin1 has any role in the TME and cancer immunotherapy and whether Pin1 inhibitors could render a solid malignancy curable.
Here we report that in PDAC, Pin1 drives the desmoplastic and immunosuppressive TME by acting on CAFs, and induces PD-L1 and ENT1 endocytosis and lysosomal degradation in cancer cells by acting on HIP1R, in addition to activating multiple oncogenic pathways. Consequently, targeting Pin1 using including ATRA + ATO simultaneously blocks multiple cancer pathways, disrupts the desmoplastic and immunosuppressive TME, and upregulates PD-L1 and ENT1, thereby rendering aggressive PDAC eradicable by synergizing with immunochemotherapy. These findings may have immediate therapeutic impact on PDAC patients as some Pin1 inhibitors are approved drugs.
RESULTS
Targeting Pin1 Disrupts the Desmoplastic and Immunosuppressive TME and Renders PDAC Tumors Curable by Immunochemotherapy in KPC Mouse-Derived Allografts
To evaluate the effects of targeting Pin1 on the TME and tumor growth in PDAC, we used two different Pin1 inhibitors (Pin1i-1 and −2) to treat three different PDAC mouse models. Pin1i-1 is a combination of ATRA + ATO (Kozono et al., 2018), and Pin1i-2 is Sulfopin, a highly Pin1-specific covalent inhibitor that targets the ATO-binding pocket and has no detectable side effect (Dubiella et al., 2021) (Tables S1 and S2). The three mouse models were patient-derived tumor orthotopic xenograft (PDTX) model (Day et al., 2015; Rubio-Viqueira et al., 2006), patient-derived PDAC organoid and CAFs orthotopic xenograft (PDOX) model (Koikawa et al., 2018a), and the LSL-K-RasG12D/+; LSL-p53R172H/+;Pdx1-Cre (KPC) genetically engineered mouse model (GEMM) derived orthotopic allograft (GDA) model in syngeneic immunocompetent mice (Day et al., 2015; Hingorani et al., 2005; Li et al., 2019). Treatment of overt tumor (>0.5 cm)-bearing PDTX or GDA mice with Pin1i-1 or −2 significantly reduced tumor growth (Figures S1A-S1C, and S2B), collagen deposition (Figures 1A and S1D), and CAF activation and proliferation(Figures 1B, S1F, S1G, and S1J). Compared with vehicle controls, Pin1 inhibitors reduced the proliferation of cancer cells (Figure S1E) and resulted in a more differentiated histology (Figures S1D and S1I), as evidenced by reduced vimentin and increased E-cadherin, markers for epithelial mesenchymal transition (EMT) (Figure S1H). Both Pin1 inhibitors also increased tumor-infiltrating CD8α+ T-cells, specifically CD8α+Granzyme B+ cytotoxic T-cells (CTLs) and decreased immunosuppressive CD4+FOXP3+ regulatory T-cells (Tregs), Ly6G+CD11b+ myeloid cells, and F4/80+CD206+ tumor associated macrophages (TAMs) in GDA mice (Figures 1C and S2D-S2G). Thus, Pin1 inhibitors suppress tumor growth and progression, and block CAF activation, as well as disrupt the desmoplastic and immunosuppressive TME in PDAC mouse models.
Figure 1. Targeting Pin1 Disrupts the Desmoplastic and Immunosuppressive TME and Renders PDAC Tumors Curable by Immunochemotherapy in GDA Mice.
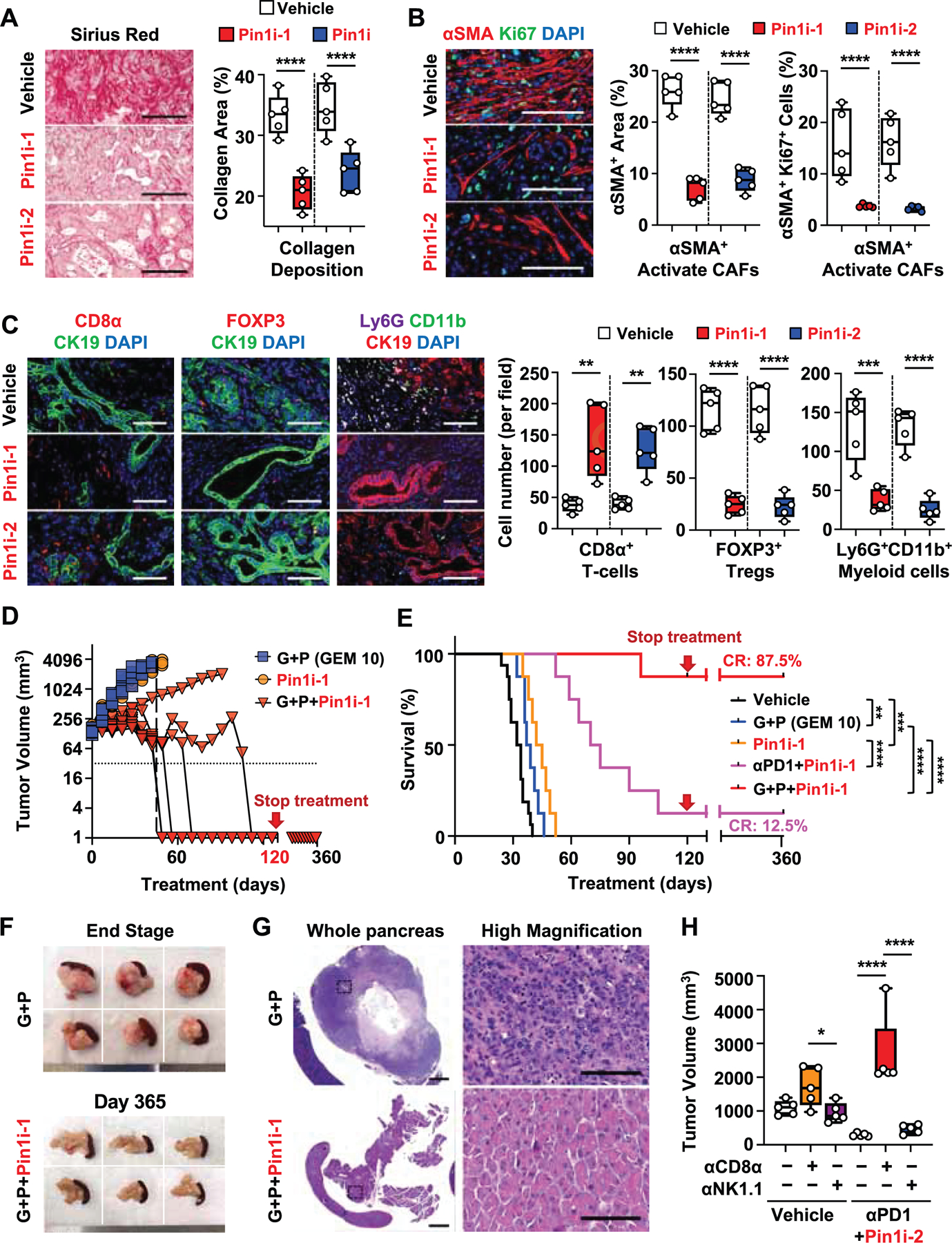
(A-C) Overt tumor-bearing (>0.5 cm) GDA mice were treated with Pin1i-1 or −2 for 4 weeks, followed by examining collagen deposition using Sirius Red staining (A), CAF proliferation using double immunofluorescence (IF) for αSMA and Ki67 (B), and tumor-infiltrating immune cell populations (CD8α+ T-cells, FOXP3+ Tregs, and Ly6G+ CD11b+ Myeloid cells) using IF (C). (D-G) Tumor-bearing GDA mice were treated with Pin1i, low dose (10 mg/kg, weekly) GEM + αPD1 (G+P), Pin1i + αPD1, or Pin1i + G+P for up to 120 days and monitored for tumor growth (D), and overall survival using Kaplan-Meier survival analysis (E) for up to 365 days, as well as examining macroscopic tumors (F) or microscopic tumors (G) after autopsy (n = 8–16). Median survival; vehicle 33 days, G+P 38 days, Pin1i-1 43.5 days, and Pin1i-1 + αPD1 72.4 days (E). CR, complete remission.
(H) Tumor-bearing GDA mice were treated with vehicle, Pin1i-2 + αPD1, anti-CD8α (αCD8α), anti-NK1.1 (αNK1.1) or their combination for 3 weeks, followed by assaying tumor volume. Scale bars, 200 μm (A), 50 μm (B), 100 μm (C), and 2000 and 100 μm (high magnification) (G). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by student’s t test (A-C), long-rank test (E), or one-way ANOVA (H).
See also Figures S1 and S2.
The above results suggest that Pin1 inhibitors might increase PDAC responses to chemo- and/or immunotherapy. Thus, we first treated overt tumor-bearing PDTX, PDOX or GDA mice with Pin1i and/or low dose (20 mg/kg, weekly) GEM, with or without starting Pin1i treatment 3 days before starting GEM (Figure S1A). Pin1i-1 and −2 were only slightly superior to GEM treatment in inhibiting tumor growth and improving survival, but the effects were significantly enhanced by their combination in each of mouse models assessed (Figures S1K-M, and S2A). The combination effects were even more profound in a group of tumor-bearing mice with Pin1i treatment started at 3 days before GEM (Figure S1K). This prompted us to routinely start a Pin1i treatment at 3 days before others in combination therapies. In addition, unlike GEM treatment alone, Pin1i also fully prevented liver metastasis of PDAC in PDTX mice (Figure S1N).
Next, we treated tumor-bearing GDA mice with Pin1i, and/or anti-PD-1 (αPD1), or low dose (10 mg/kg, weekly) GEM + αPD1 (G+P) (Figure S1A). Treatment with αPD1 or G+P partially inhibited tumor growth and marginally increased overall survival (Jiang et al., 2016), and both Pin1i-1 and −2 as single agents were slightly more potent than above treatments (Figures 1D-1E, S2B, and S2I). Notably, Pin1i-1 + αPD1 combination dramatically reduced tumor growth and more than doubled median survival, with 12.5% complete regression (Figure 1E). The most pronounced effect was seen in a group treated with the triple combination. Pin1i-1 and G+P combination markedly disrupted the desmoplastic and immunosuppressive TME (Figures S2C, S2F, and S2G), and fundamentally changed overall survival, leading to 87.5% complete regression (Figures 1D and 1E). These mice survived without macroscopic (Figure 1F) or microscopic (Figure 1G) evidence for residual PDAC for at least for 1 year in good health, even though the treatments were stopped at 120 days (Figures 1E and S2H). Similar results were obtained with Pin1i-2 in combination with αPD1 or G+P, although less impressive during long-term treatment (Figures S2B, S2C, S2E, S2F, S2H, and S2I).
To examine the relevance of cytotoxic and natural killer cells for the synergy between Pin1 inhibition and immunotherapy, we examined the efficacy of Pin1i-2 and αPD1 combination in the absence or presence of CD8α+ T-cells or NK1.1+ cells in GDA mice. When CD8α+ T-cells were depleted, PDAC tumor growth accelerated considerably and the synergistic efficacy of Pin1i-2 and αPD1 treatment was completely offset (Figure 1H). αPD1 treatment alone also accelerated tumor growth in CD8α+ T-cell depleted GDA mice (Figure S2J), reminiscent of hyper-progression observed in clinical trials of cancer patients with αPD1 therapy (Wang et al., 2020). Immune cell profiling showed that while Pin1i increased the number of tumor infiltrating CD8α+ T-cells and their Granzyme B expression, Pin1i alone also increased their expression of the activation/exhaustion markers, PD-1, lymphocyte activation gene-3 (LAG-3), and cytotoxic T-lymphocyte antigen 4 (CTLA-4), but the later effects tended to be reduced by combination with αPD1 (Figure S2E and S2G), indicative of reversion of lymphocyte exhaustion (Ruscetti et al., 2020). In contrast, PD-L1 expression on myeloid and dendritic cells (DCs), which contribute to anti-tumor immune response (Oh et al., 2021), was no changed after Pin1 inhibition (Figure S2E). Thus, CD8+ CTLs are pivotal for the synergy between Pin1 inhibition and immunotherapy.
To investigate whether the synergy of immunochemotherapy with Pin1 inhibition in PDAC is specific to GEM or αPD1, we tested the microtubule stabilizing paclitaxel (PTX), non-nucleoside-based chemotherapy, and anti-CTLA-4 (αCTLA4), an alternative ICB. Pin1 inhibition did not potentiate PTX anti-tumor activity and tended to increase αCTLA4 anti-tumor effect, but the effect was small and without statistical significance (Figure S2K). Thus, although Pin1 inhibitors have moderate single agent anticancer efficacy, they potently disrupt the desmoplastic and immunosuppressive TME, and remarkably render most aggressive PDAC tumors curable when combined with GEM and αPD1 in GDA mice.
Pin1 is Overexpressed both in Cancer Cells and CAFs in Human PDAC, and Correlates with the Desmoplastic and Immunosuppressive TME, and Poor Patient Survival
Given such unexpected and striking potency of Pin1i in rendering PDAC curable, we evaluated Pin1 expression in human tumor tissues surgically resected from 167 PDAC patients and its relationships with TME changes and patient survival using IHC with a validated Pin1 monoclonal antibody (Bao et al., 2004). Pin1 was overexpressed in cancer cells and correlated positively with cancer progression (Figures 2A and S3A). Furthermore, Pin1 was overexpressed in tumor stromal CAFs, which expressed various CAF markers, α-smooth muscle actin (αSMA), fibroblast activation protein (FAP), human leukocyte antigen (HLA) class II histocompatibility antigen DP beta 1 (HLA-DPB1), or CD44 (Figures 2B-2D, and S3B) (Hosein et al., 2020). By classifying our cohorts into Pin1-High and Pin1-Low groups based on their IHC intensity and area, we not only found that Pin1 was overexpressed in cancer cells in 71.5% of the patients and correlated with poor survival, as reported (Chen et al., 2019), but also uncovered that Pin1 was overexpressed in CAFs in 51.9% of the patients and correlated with poor survival (Figure 2E). More impressively, high Pin1 both in cancer cells and CAFs, as compared with low Pin1 group, strongly correlated with reduced median overall survival from 60 to 16 months in this cohort (Figure 2F). To ask whether Pin1 overexpression is relevant to the desmoplastic and immunosuppressive TME, we evaluated collagen deposition and tumor-infiltrating immune cell populations in human PDAC tissues. Pin1 overexpression in CAFs, but not in cancer cells, significantly correlated with collagen deposition (Figure 2G). Pin1 overexpression in cancer cells and CAFs also correlated with fewer infiltrated CD8+ T-cells and more CD163+ TAMs (Figures 2H and 2I). Thus, Pin1 is overexpressed in cancer cells and CAFs in human PDAC, and correlates well with the desmoplastic and immunosuppressive TME, and poor survival.
Figure 2. Pin1 is Overexpressed in Cancer Cells and CAFs in Human PDAC, and Strongly Correlates with the Desmoplastic and Immunosuppressive TME, and Poor Patient Survival.
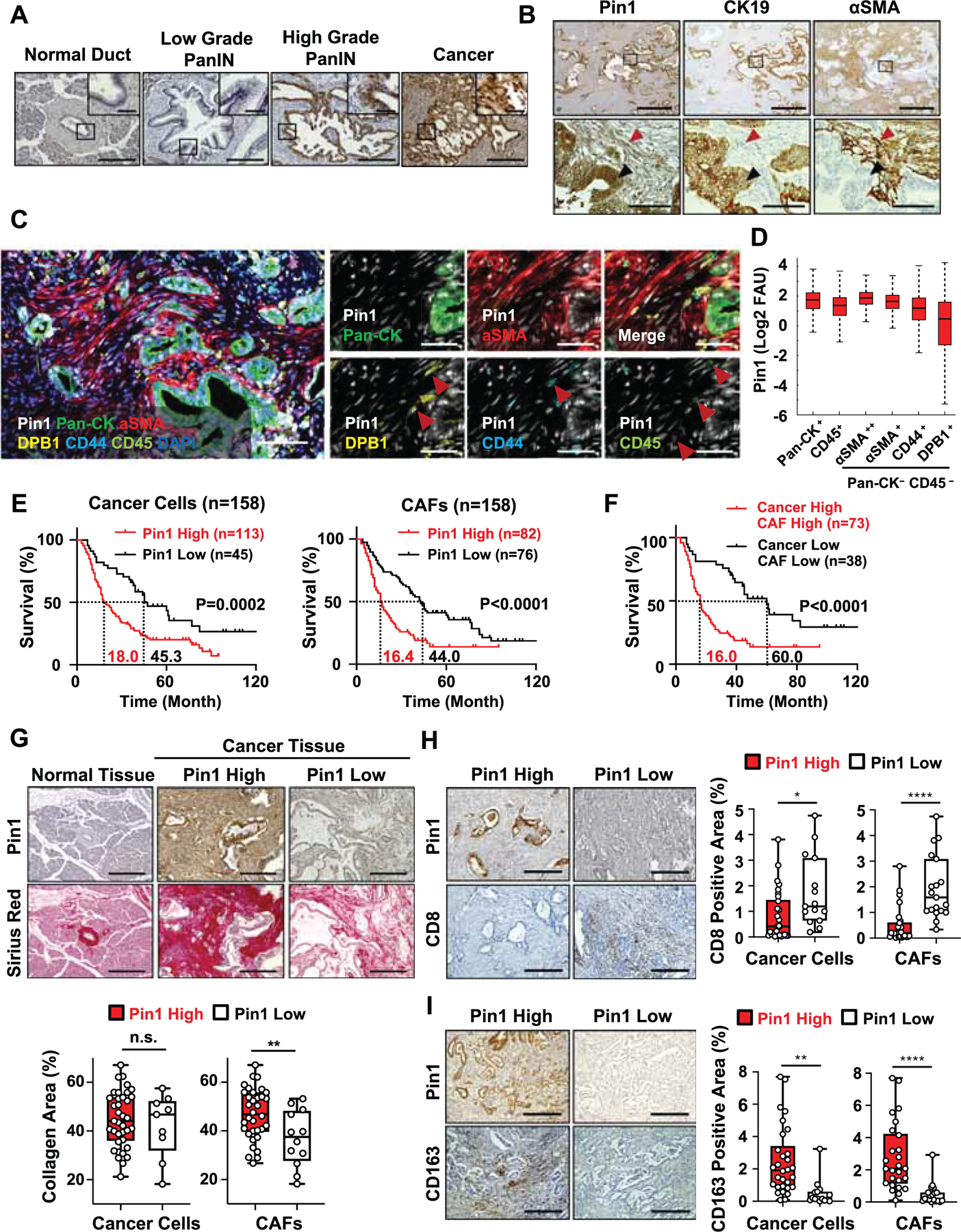
(A) Representative images of Pin1 IHC staining in human PDAC tissues at different stages of cancer progression. Quantification of Pin1 expression is shown in Figure S3A.
(B) Pin1, Cytokeratin (CK) 19, and αSMA IHC staining in human PDAC tissues. Black arrows indicate Pin1+CK19+αSMA− cancer cell and red arrows indicate Pin1+CK19-αSMA+ CAF.
(C and D) Tissue-based cyclic immunofluorescence (t-CyCIF) for Pin1 (white), pan-cytokeratin (Pan-CK) (green), αSMA (red), HLA-DBP1 (yellow), CD44 (cyan) and CD45 (green) in human PDAC tissues (C) and quantification of Pin1 expression (D). Red arrows indicate Pin1+ and DPB1+ or CD44+ CAFs or Pin1+ and CD45+ cells (C).
(E and F) Human PDAC tissues were classified into cancer cell Pin1-High or Pin1-Low groups, CAFs Pin1-High or Pin1-Low (E), or cancer Pin1-High and CAF Pin1-High or cancer Pin1-Low and CAF Pin1-Low groups (F) based on Pin1 IHC, followed by examining patient overall survival using Kaplan–Meier survival analysis.
(G) Pin1 IHC and Sirius red staining in human normal pancreas and PDAC tissues, and quantification of collagen deposition using Sirius red staining (n = 46).
(H and I) Pin1 and CD8 (H) or CD163 (I) IHC staining in human PDAC tissues, and quantification of CD8 or CD163 positive cells area per field. (n=45) (H, I).
Scale bars, 300 and 50 μm (inset) (A), 1000 and 100 μm (high magnification) (B), 100 (left panel) and 50 μm (C), and 300 μm (G-I). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by student’s t test (G-I) or log-rank test (E, F).
Pin1 Promotes Oncogenic Signaling Pathways, CAF Activation and Crosstalk with Cancer Cells to Enhance Tumor Growth and Malignancy in Human Organoids and PDOXs
To determine the functional significance of Pin1 overexpression in CAFs in human PDAC tumors, we derived primary CAFs from two different PDAC patients (Koikawa et al., 2018a; Koikawa et al., 2018b) and inhibited their Pin1 function using Pin1i, genetic knockdown (KD) or CRISPR knockout (KO). Both Pin1 inhibitors dose-dependently reduced Pin1 and its many substrate oncoproteins in CAFs (Figures 3A and S3C), with ATRA and ATO synergistically targeting and degrading Pin1, as shown (Kozono et al., 2018; Wei et al., 2015). We demonstrated that Pin1 inhibition suppressed CAF proliferation (Figures 3B, S3D and S3E), induced CAF quiescent phenotype and inhibited their ability to secrete a wide range of cytokines (Figures 3C-E, and S3F). Notably, we found suppression of IL-6 and TGF-β release by cancer cells and IL-6, TGF-β, LIF and CXCL12 secretion by CAFs in response to Pin1 inhibition (Figures S3G and S3H). Those factors promote cancer cell progression (Erkan et al., 2012; Shi et al., 2019), prevent T cell recruitment into the TME (Garg et al., 2018), induce the desmoplastic and immunosuppressive TME (Mace et al., 2018), and suppress response to anti-PD-L1 (αPDL1) (Feig et al., 2013; Mariathasan et al., 2018). Thus, Pin1 activates CAFs and may contribute to the desmoplastic and immunosuppressive TME.
Figure 3. Pin1 Promotes Oncogenic Signaling Pathways, CAF Activation and Crosstalk with Cancer Cells to Enhance Tumor Growth and Malignancy in Organoids and PDOXs.
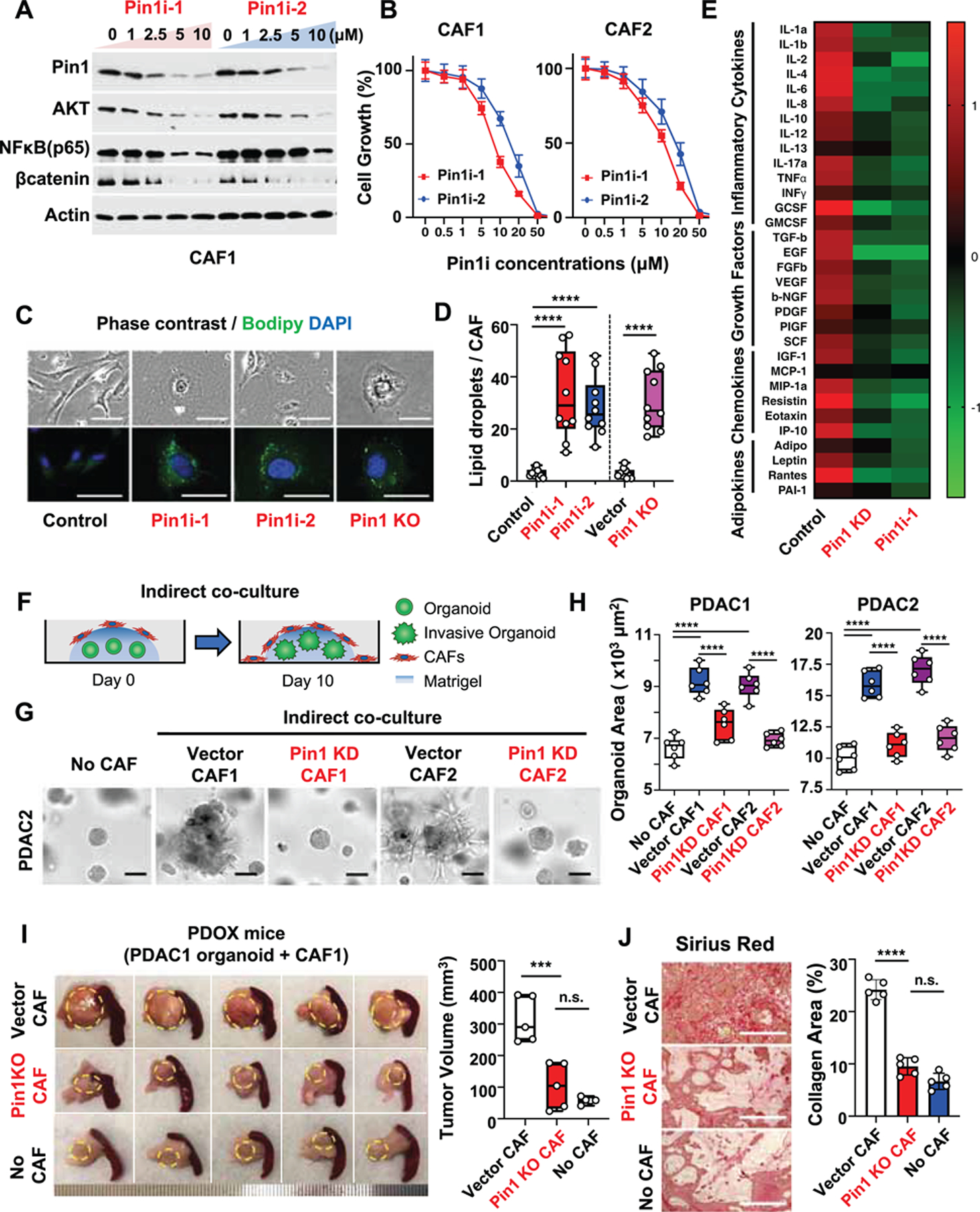
(A-E) Primary human CAFs derived from two different PDAC patients (CAF1 or CAF2) were treated with Pin1i-1 or −2 for 72 hrs, or subjected to stable Pin1 KD or KO, followed by examining Pin1 and its substrate oncoproteins using IB (A), cell growth using proliferation assay (B), quiescent phenotype as measured by lipid droplets/CAF cell using BODIPY (C, D), and cytokine production using cytokine array (E). ATRA and ATO were used in 10:1 ratio as Pin1i-1 and only ATRA concentrations were shown (A, B). Pin1i-1 (10 μM) or Pin1i-2 (5 μM) (C-E).
(F-H) Pin1 KD or vector control CAFs were seeded to the top of Matrigel that contained pre-formed PDAC organoids and co-cultured for 10 days (F), followed by examining organoid growth and invasion into Matrigel using microscopic imaging analysis (G, H). Human PDAC organoids were derived from two different PDAC patients (PDAC1 and PDAC2 organoids). (I and J) Human PDAC organoids were orthotopically transplanted alone or with Pin1 KO CAFs or CRISPR control CAFs into the pancreas of NSG mice for 5 weeks, followed by examining tumor growth (l), and fibrosis using Sirius red staining (J) (n = 5).
Scale bars, 100 (upper panels) and 50 μm (lower panels) (C), 100 μm (G), and 200 μm (J). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by one-way ANOVA.
See also Figure S3.
A continuous crosstalk between cancer cells and CAFs enhances tumor growth and invasion (Erkan et al., 2012). To address whether Pin1 inhibition in CAFs affects their ability to act on cancer cells, we first indirectly co-cultured pre-formed human PDAC organoids with CAFs to analyze the effects of CAF-derived humoral factors on PDAC organoid growth and invasion (Figure 3F). Unlike control CAFs, which promoted PDAC organoid growth and invasion, Pin1 KD CAFs failed to promote PDAC organoid growth and invasion similar to the level of those without added CAFs (Figures 3G and 3H). To analyze the effects of cell-cell interactions, we then directly co-cultured PDAC organoids with Pin1i-treated or KO CAFs, followed by assaying organoid growth and invasion using time-lapse imaging (Koikawa et al., 2018a) (Figure S3I). The results further confirmed that control CAFs, but neither Pin1i-treated nor Pin1 KO CAFs, promoted PDAC organoid growth and invasion (Figures S3J and S3K, and Video S1). Finally, to examine whether Pin1 is important for CAFs to promote the TME and tumor growth of PDAC in vivo, we orthotopically co-transplanted human PDAC organoids with Pin1 KO or control CAFs into the mouse pancreas (PDOX model). In contrast to control CAFs, Pin1 KO CAFs completely failed to promote tumor fibrosis, cancer cell proliferation, and tumor growth and progression, similar to those without co-transplanted CAFs (Figures 3I, 3J, S3L, and S3M). Thus, Pin1 in CAFs is necessary to promote the pro-tumorigenic TME and enhance the growth and malignant phenotype of PDAC in human organoids and mouse tumors.
Pin1 Promotes Oncogenic Signaling Pathways and Reduces the Expression of PD-L1 and ENT1 at the Cell Surface of PDAC Cells
The above results show that targeting Pin1 disrupts the TME and renders PDAC tumors curable by immunochemotherapy, but the previous attempts to target stroma cells in PDAC were only modestly effective (Ho et al., 2020; Sahai et al., 2020; Whittle and Hingorani, 2019). We reasoned that Pin1 inhibitors might also act on cancer cells, given its overexpression in cancer cells (Figures 2A-2D). Thus, we examined the effects of Pin1i, Pin1 KD or KO in primary PDAC cells and organoids derived from two different PDAC patients (Koikawa et al., 2018a). Indeed, like Pin1 KD or KO, both Pin1 inhibitors dose-dependently reduced Pin1 and its substrate oncoproteins including those in Kras signaling (Figures S4A-S4C, and S4E), as well as PDAC cell proliferation (Figures S4D, S4F, and S4G), but the effects was largely abolished by Pin1 KO (Figure S4H), confirming their specificity. Furthermore, like Pin1 KD, both Pin1 inhibitors also reduced human PDAC organoid growth and enhanced the ability of GEM to inhibit organoid growth and proliferation (Figures S4I-S4L). Moreover, when human PDAC organoids were pre-treated with Pin1i-1 or −2, followed by treatment with GEM, both Pin1 inhibitors greatly potentiated GEM-mediated organoid apoptosis, whereas neither Pin1i nor GEM alone had obvious effect (Figures 4A and 4B).
Figure 4. Pin1 Promotes Oncogenic Signaling Pathways and Reduces the Expression of PD-L1 and ENT1 at the Cell Surface of PDAC Cells.
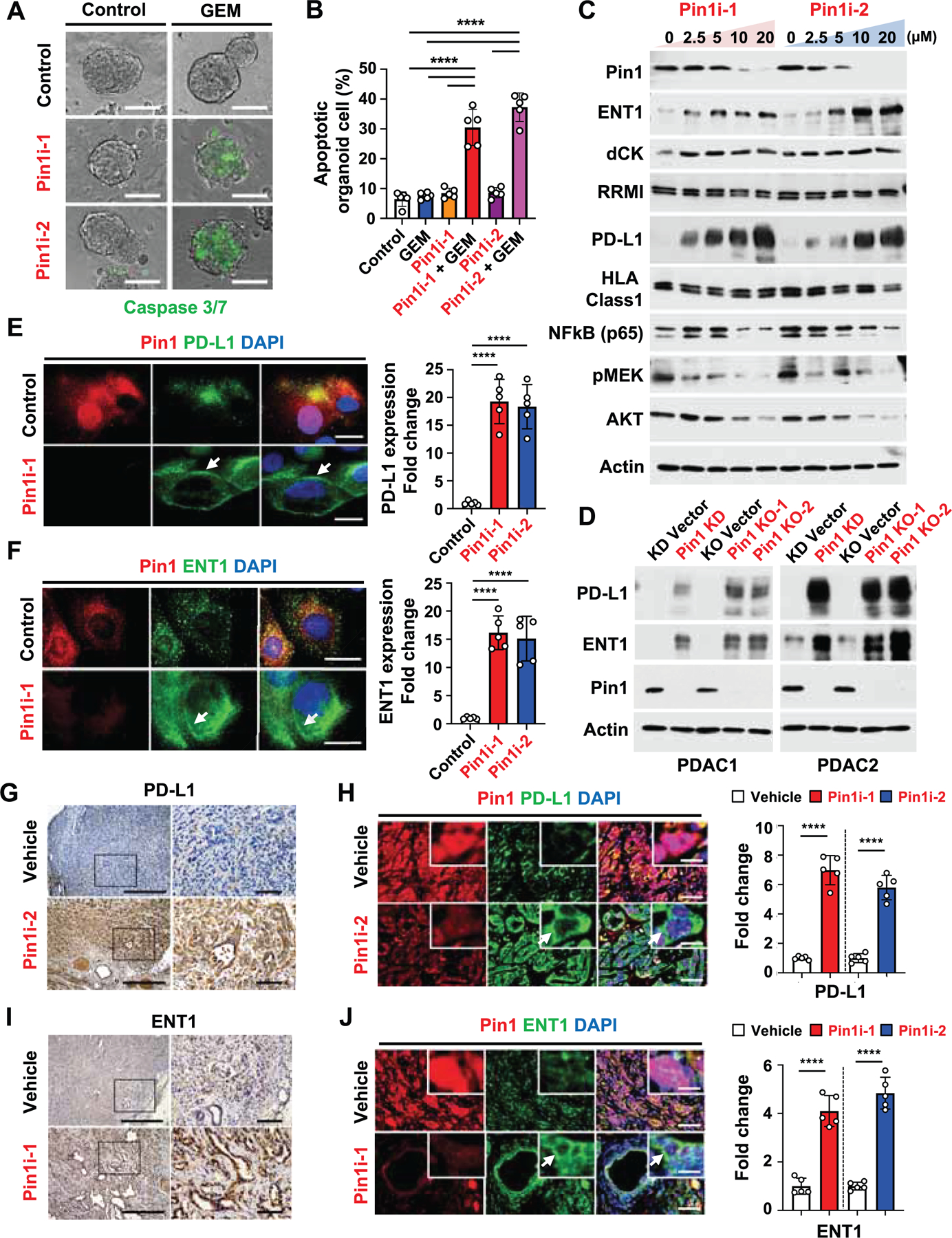
(A and B) Human PDAC2 organoids were pre-treated with control (DMSO), Pin1i-1 (10 μM) or Pin1i-2 (5 µM) for 72 hrs, and then treated with control (PBS) or GEM (25 nM) for 24 hrs, followed by examining organoid apoptosis using caspase 3/7 green fluorescence reagent.
(C) Human PDAC2 cells were treated with Pin1i-1 or −2 at various concentrations for 72 hrs, followed by IB for different proteins indicated. ATRA and ATO were used in 10:1 ratio as Pin1i-1 and only ATRA concentrations are shown.
(D) PDAC1 or 2 cells were subjected to Pin1 KD or KO, followed by IB for different proteins indicated.
(E and F) PDAC2 cells were treated with control (DMSO), Pin1i-1 (10 μM), or Pin1i-2 (5μM), followed by IF for Pin1 (red), DAPI (blue), and PD-L1 (green) (E) or ENT1 (green) (F). White arrows point to PD-L1 (E) and ENT1 (F) at the cell surface.
(G-J) Tumor-bearing GDA mice were treated with vehicle, Pin1i-1 or −2 for 4 weeks, followed by IHC for PD-L1 (G) or ENT1 (I), or IF for Pin1 (red), DAPI (blue), and PD-L1 (green) (H) or ENT1 (green) (J) (n = 5). White arrows point to PD-L1 (H) and ENT1 (J) at the cell surface.
Scale bars, 100 μm (A), 50 μm (E, F), 500 and 100 μm (right panels) (G, I), and 50 and 12.5 μm (inset) (H, J). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by one-way ANOVA (B, E, F) or student’s t test (H, J).
See also Figures S4 and S5.
Given the striking ability of Pin1 inhibitor pretreatment to potentiate the GEM sensitivity of PDAC cells, we reasoned that Pin1 inhibitors might affect the expression of some determinants of the GEM sensitivity. Notably, equilibrative nucleoside transporter 1 (ENT1) is required for GEM uptake and is a therapeutic response marker for GEM, deoxycytidine kinase (dCK) is involved in the first phosphorylation cascade in GEM activation, and ribonucleotide reductase subunit 1 (RRM1) is a main nuclear target for GEM (Amrutkar and Gladhaug, 2017; Farrell et al., 2009). Both Pin1 inhibitors dose-dependently increased the levels of ENT1, but neither dCK nor RRM1 was affected (Figure 4C). Since Pin1 inhibition potently enhanced αPD1 efficacy against PDAC (Figure 1E), we hypothesized that Pin1 inhibitors might also affect the expression of ICB response biomarkers, whose upregulation increases ICB efficacy such as PD-L1 (Jiao et al., 2017; Zhang et al., 2018) and HLA class 1 (McGranahan et al., 2017; Yamamoto et al., 2020). Indeed, both Pin1 inhibitors dose-dependently upregulated PD-L1, but not HLA class 1 level (Figure 4C). The upregulation of PD-L1 and ENT1 was also confirmed by Pin1 KD or KO (Figure 4D), and was notable on the cell surface of Pin1i-treated PDAC cells in vitro (Figures 4E and 4F) and Pin1i-treated PDTXs and GDAs in mice (Figures 4G-4J, S5A, and S5B). Moreover, Pin1 expression inversely correlated with that of PD-L1 and ENT1 in human PDAC tissues (Figures S5C-S5E). Thus, Pin1 inhibition not only blocks multiple cancer pathways but also induces PD-L1 and ENT1 expression on the surface of PDAC cells in vitro and in vivo.
Pin1 Promotes Endocytosis and Lysosomal Degradation of PD-L1 and ENT1 by Acting on the pS929-Pro Motif in HIP1R
Our findings that Pin1 inhibition in PDAC cells increased PD-L1 and ENT1 protein levels (Figure 4C-4F), but decreased their mRNA levels (Figure S5F) suggest that Pin1 inhibition might stabilize ENT1 and PD-L1 proteins. The proteolysis of both PD-L1 (Burr et al., 2017; Cha et al., 2019; Dorand et al., 2016; Lim et al., 2016; Zhang et al., 2018) and ENT1 (Hu et al., 2017) is highly regulated, and to modulate proteolysis is a dominant mechanism for Pin1 to regulate dozens of oncogenic proteins (Zhou and Lu, 2016). To assess the mechanisms behind Pin1-mediated effects, we treated PDAC cells with chemical inhibitors for the lysosomal degradation pathway (3-MA, Bafilomycin and Chloroquine) or proteasomal degradation pathway (MG132 and MLN4924). Inhibition of the proteasomal pathway increased PD-L1, but not ENT1, as shown (Hu et al., 2017; Zhang et al., 2018), while inhibition of the lysosomal pathway increased both PD-L1 and ENT1 (Figure S5G), suggesting that lysosomal degradation may be a common mechanism for Pin1-mediated regulation of PD-L1 and ENT1.
To support this possibility, we investigated Huntingtin interacting protein 1-related (HIP1R) (Wang et al., 2019) and CKLF Like MARVEL Transmembrane Domain Containing 6 (CMTM6) (Burr et al., 2017), which promote and inhibit the endocytosis and lysosomal degradation of PD-L1, respectively, and also contain putative Pin1 substrate recognition sites (pSer/Thr-Pro motifs), whereas such Pin1 recognition sites are not present in PD-L1. Both Pin1 inhibitors dose-dependently increased HIP1R with a slower mobility and presumably in phosphorylated form, which was also induced by Pin1 KD or KO (Figures 5A-5D), but was undetectable after treating cell lysates with calf intestinal phosphatase (CIP) (Figure 5E). Co-immunoprecipitation (Co-IP) demonstrated that Pin1 bound to phosphorylated HIP1R under endogenous conditions, and that this interaction was phosphatase-sensitive (Figure 5E). By contrast, Pin1 inhibitors neither affected the PD-L1 stabilizing CMTM6, nor did Pin1 interact with CMTM6 (Figures 5A, 5C, and 5E). To determine the importance of HIP1R in regulating PD-L1 and ENT1 levels, we stably knocked down HIP1R in PDAC cells and found that HIP1R KD increased both PD-L1 and ENT1 levels (Figure 5F). These results together suggest that Pin1 promotes PD-L1 and ENT1 degradation by acting on phosphorylated HIP1R.
Figure 5. Pin1 Promotes the Endocytosis and Lysosomal Degradation of PD-L1 and ENT1 by Acting on the pS929-Pro Motif in HIP1R.
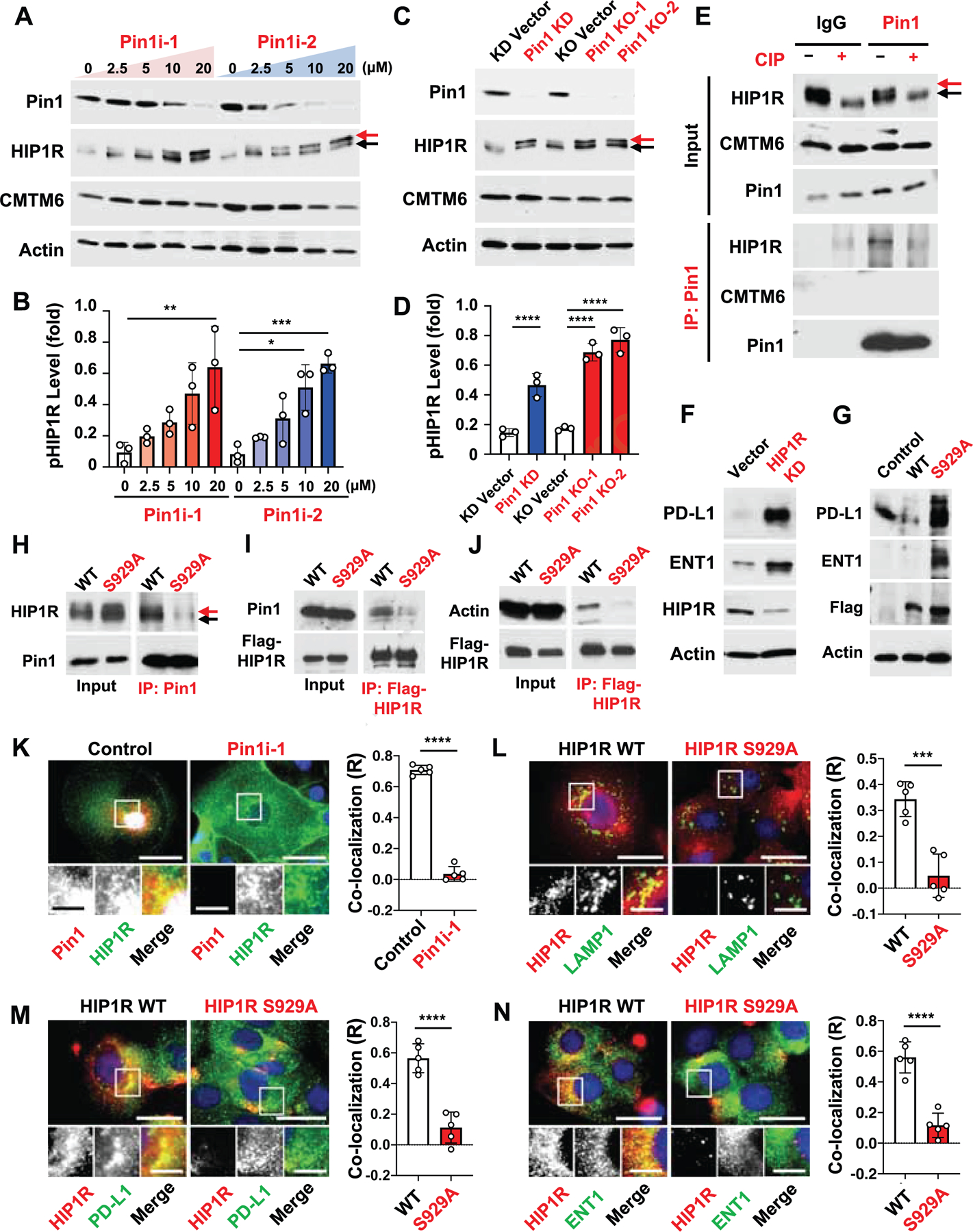
(A-D) Human PDAC2 cells were treated with Pin1i-1 or −2 at various concentrations for 72 hrs (A, B) or subjected to Pin1 KD or KO (C, D), followed by IB for different proteins indicated (A, C) and quantifying phosphorylated HIP1R (B, D). A red arrow points to phosphorylated HIP1R and a black arrow to non-phosphorylated HIP1R. ATRA and ATO were used in 10:1 ratio as Pin1i-1 and only ATRA concentrations are shown (A, B).
(E) PDAC2 cell lysates were incubated with CIP (+ CIP) or CIP plus phosphatase inhibitors (- CIP), followed by IB directly (input) or after IP with Pin1 antibodies.
(F) HIP1R in PDAC2 cells was stably knocked down using shRNA or vector control, followed by IB.
(G) Flag-HIP1R WT or its S922A mutant was stably expressed in PDAC2 cells, followed by IB.
(H-J) PDAC2 cells stably expressing Flag-HIP1R WT or S929A mutant were subjected to IB directly (input) or after IP with Pin1 antibody (H) or Flag antibody (I, J). A red arrow points to phosphorylated HIP1R and a black arrow to non-phosphorylated HIP1R.
(K) PDAC2 cells were treated with control (DMSO) or Pin1i-1 (10 μM), and then subjected to IF for Pin1 (red), HIP1R (green), and DAPI (blue).
(L-N) HIP1R WT or S929A stably transfected PDAC2 cells were subjected to IF for HIP1R (red), DAPI (blue), and LAMP1 (green) (L), PD-L1 (green) (M), or ENT1 (green) (N), followed by assaying co-localization of Pin1-HIP1R (K), HIP1R-LAMP1 (L), HIP1R-PDL1 (M), and HIP1R-ENT1 (N) using Image J program, with R value being Pearson’s coefficient for co-localization.
Scale bars, 50 and 5 μm (inset) (K), and 20 and 5 μm (inset) (L-N). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by one-way ANOVA (B, D) or student’s t test (K-N).
See also Figure S5.
To identify the Pin1-binding site in HIP1R, we generated an Ala substitution (S929A) at the only putative and conserved S/P site, pSer929-Pro motif in HIP1R (Figure S5H). Unlike wild-type (WT) HIP1R, when introduced into PDAC cells, the S929A mutant not only failed to interact with Pin1, but also increased levels of phosphorylated HIP1R, as well as PD-L1 and ENT1 (Figures 5G-5I), similar to Pin1 inhibitors, Pin1 KD or KO (Figures 5A-5D). Notably, the pSer929-Pro in HIP1R is located within its actin-binding domain (Figure S5H), which is critical for HIP1R-mediated endocytosis and lysosomal degradation (Gottfried et al., 2010; Messa et al., 2014). These results suggest that the S929A mutation might impair the ability of HIP1R to bind actin and promote the endocytosis of PD-L1 and ENT1 to lysosomes. Indeed, WT HIP1R, but not its S929A mutant, interacted with actin (Figure 5J) and colocalized with Pin1, PD-L1 and ENT1 to peri-nuclear lysosomes, as marked by the lysosomal marker LAMP1 (Figures 5K-5N). Cycloheximide (CHX) chase assay demonstrated that the HIP1R, but not its S929A mutant, was unstable (Figure S5I). Thus, Pin1 promotes the endocytosis and lysosomal degradation of PD-L1 and ENT1 presumably by acting on the pSer929-Pro motif in HIP1R to facilitate actin binding.
Targeting Pin1 Synergizes with Immunochemotherapy to Induce PDAC Organoid Apoptosis
Given that targeting Pin1 robustly induces the cell surface expression of ENT1 and PD-L1 in PDAC cells, a critical question is whether targeting Pin1 would affect the therapeutic response to GEM and αPD1. Thus, we targeted Pin1 in established human PDAC organoids without any stromal cells to avoid indirect effects (Koikawa et al., 2018a), followed by the organoid apoptosis assay using time-lapse imaging to visualize and quantify the dynamic changes in organoid apoptosis (Figure 6A). As expected, both Pin1 inhibitors time-dependently induced ENT1 and PD-L1 levels (Figure 6B), and time- and dose-dependently sensitized PDAC organoids to GEM-induce apoptosis, with highly synergistic effects (Figures 6C-6E, and S6A). The similar effect was observed in Pin1i with ENT1 dependent 5-FU (Figures S6G and S6H). Pin1 KO or introduction of HIP1R929A mutant into the PDAC organoids resulted in similar synergy with GEM treatment (Figure S5J), further supporting our model. To examine the importance of ENT expression for modulating GEM responsiveness by Pin1i, we treated Pin1i pre-treated WT or ENT1-KD PDAC organoids with GEM. Pin1i-2 induced apoptosis in WT PDAC organoids, but not in ENT1-KD PDAC organoids (Figures S6B and S6C), supporting the role of ENT1 in GEM response..
Figure 6. Targeting Pin1 Synergizes with Immunochemotherapy to Induce PDAC Organoid Apoptosis.
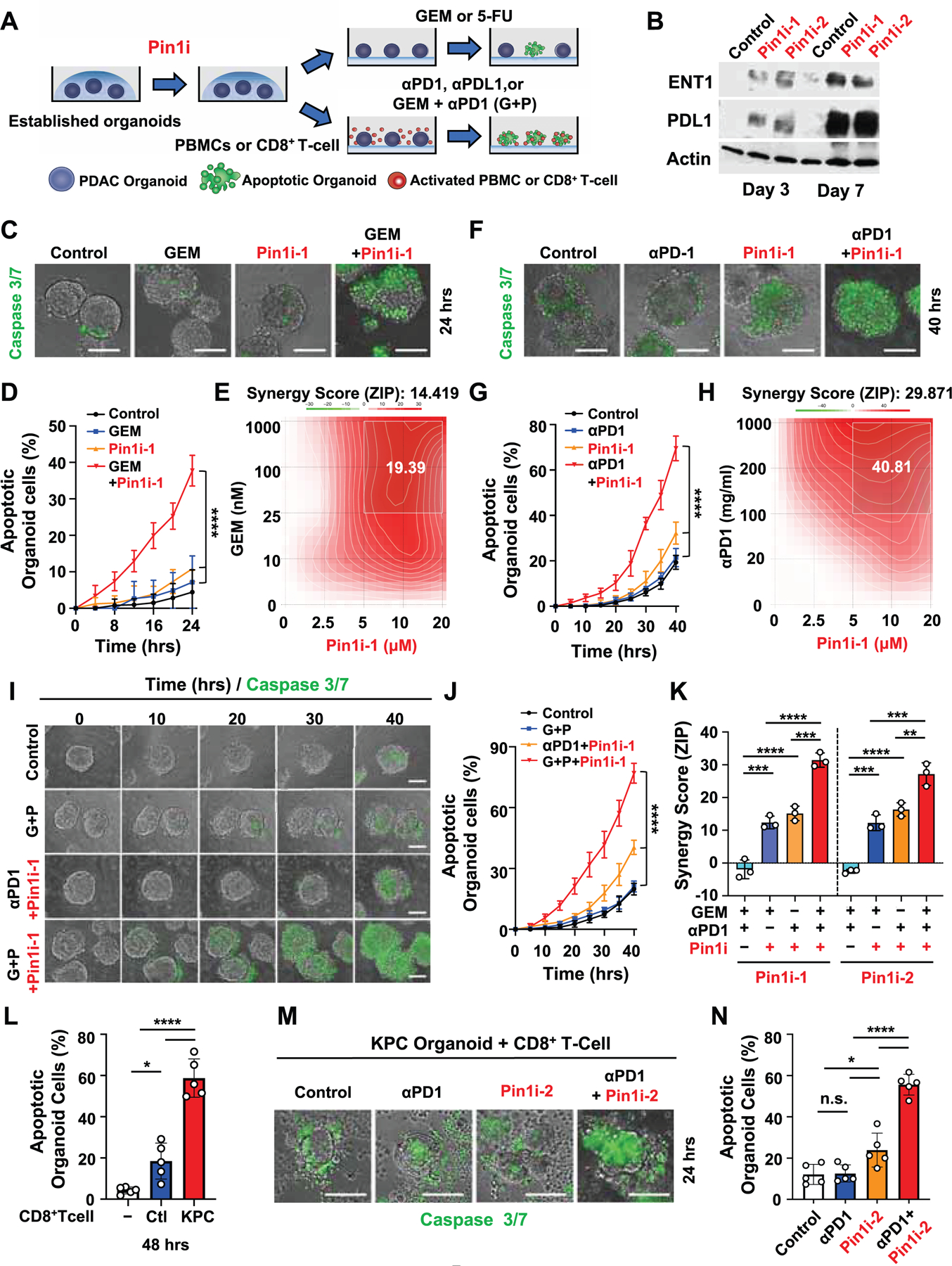
(A) Experimental setup of PDAC organoid apoptosis assay. Established primary human or KPC mouse PDAC organoids were pre-treated with control or Pin1i for 3 days, followed by i) treating with control, GEM or 5-FU, or ii) co-culturing with activated human PBMCs or mouse derived CD8+ T-cells, and treating with control, αPD1, αPDL1, or GEM + αPD1 (G+P). Apoptosis of PDAC organoid was measured using caspase 3/7 green fluorescent reagent and time-lapse imaging.
(B) Human PDAC2 organoids were treated with control (DMSO), Pin1i-1 (10 μM) or Pin1i-2 (5 μM) for 3 or 7 days, followed by IB.
(C-E) Pin1i-1 pre-treated PDAC2 organoids were treated with GEM, followed by examining PDAC organoid apoptosis at constant concentrations (Pin1i-1 10 μM and GEM 25 nM) for different times (C, D) or at different concentrations for 24 hrs and analyzing the synergy score of Pin1i and GEM using Synergy finder (E).
(F-H) Pin1i-1 pre-treated PDAC2 organoids were co-cultured with activated human PBMCs and then treated with αPD1, followed by examining PDAC organoid apoptosis at constant concentrations (Pin1i-1 10 μM and αPD1 200 μg/ml) for different times (F, G) or at different concentrations indicated for 40 hrs and analyzing synergy score of Pin1i and αPD1 using Synergy finder (H).
(I-K) Pin1i pre-treated PDAC2 organoids were co-cultured with activated human PBMCs, and then treated with GEM and/or αPD1, followed by assaying PDAC organoid apoptosis at constant concentrations (Pin1i-1 10 μM, GEM 10 nM and αPD1 100 μg/ml) for different times (I, J) or at different concentrations indicated for 40 hrs and analyzing synergy score of Pin1i, GEM and/or αPD1 using Synergy finder (K).
(L-N) KPC organoids derived from KPC mouse PDAC tumors were co-cultured with/without activated CD8+ T-cells derived from the same KPC tumor-bearing mice or their tumor-free littermates that did not have all the three transgene for 48 hrs (L), or the KPC organoids were pre-treated with control (DMSO) or Pin1i-2 (5 μM) for 3 days and co-cultured with the same KPC tumor-bearing mouse derived activated CD8+ T-cells, and then treated with αPD1 (200 μg/ml) for 24 hrs (M, N), followed by examining PDAC organoid apoptosis.
Scale bars, 100 μm. Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by one-way ANOVA.
See also Figure S6.
To examine the effects of Pin1 inhibition on the ability of αPD1 to induce PDAC organoid apoptosis, Pin1i pre-treated human PDAC organoids and activated human primary peripheral blood mononuclear cells (PBMCs) were subjected to the organoid apoptosis assay (Figure 6A) (Dijkstra et al., 2018; Jiao et al., 2017). Both Pin1 inhibitors increased the ability of αPD1 or αPDL1 to induce organoid apoptosis, and the effects were again dose-dependent and highly synergistic (Figures 6F-6H, S6D, S6I, and S6J). To exclude possible off-target effects of Pin1 inhibitors, we conducted the same experiment with Pin1 KO and HIP1R929A PDAC organoids with activated human PBMCs and found a similar enhancement of αPD1 anti-tumor effects (Figure S5K). Pin1i-2 also promoted the apoptosis of WT PDAC organoids, but not of PD-L1-KD PDAC organoids (Figures S6E and S6F), supporting that the effects of Pin1 inhibition and αPD1 are dependent on PD-L1 expression. Moreover, both Pin1 inhibitors time- and dose-dependently enhanced the ability of G+P to induce apoptosis of PDAC organoids, and the effects were also highly synergistic (Figures 6I-6K, S6K, S6L, and Video S2).
To use an autologous organoid model to confirm the above findings that CD8+ cytotoxic T-cells are important factor for the response of Pin1i + αPD1 therapy in GDA mice (Figures 1H and S2E-G), we co-cultured KPC mouse PDAC organoids with activated CD8+ T-cells derived from the same KPC tumor-bearing mice or their tumor-free littermates. As expected, the CD8+ T-cells derived from tumor-bearing mice were significantly more effective in inducing organoid apoptosis than those from non-tumor-bearing controls (Figures 6L and S6M). More importantly, Pin1i synergized with αPD1 to induce PDAC organoid apoptosis (Figures 6M and 6N), and the synergistic effects were also upheld when GEM was used (Figures S6N and S6O), as in GDA mice (Figure 1E), confirming the critical role of CD8+ CTLs for Pin1 inhibitors to potentiate immunotherapy response. Thus, targeting Pin1 acts on PDAC cells not only to inhibit multiple cancer pathways and induce the cell surface expression of PD-L1 and ENT1, but also to synergize with immunochemotherapy to induce apoptosis of human and mouse PDAC organoids.
Targeting Pin1 Renders Primary PDAC Tumors Eradicable by Immunochemotherapy in KPC GEMM
To determine whether Pin1 inhibitors are able to disrupt the highly desmoplastic and immunosuppressive TME and render primary PDAC tumors eradicable by immunochemotherapy in a GEMM of PDAC, we used KPC mice expressing commonly occurring KrasG12D and p53R172H mutations in the pancreas. These mice developed the spectrum of aggressive PDAC with 100% penetrance, which recapitulate tumor heterogeneity, desmoplastic and immunosuppressive TME, poor immunogenicity and rapid progression typical of human disease (Bayne et al., 2012; Hingorani et al., 2005). We treated overt tumor-bearing KPC mice with Pin1i, G+P or their combination. As shown (Jiang et al., 2016), G+P neither affected the desmoplastic and immunosuppressive TME, nor increased survival, with most mice succumbing to the disease within 3 months (Figures 7B and 7D-7F). However, Pin1i-1 or −2 and G+P combination suppressed cancer cell proliferation, and CAF activation and proliferation (Figures 7A, 7C, S7A, and S7B), disrupted the desmoplastic and immunosuppressive TME, increased tumor infiltrating CD8α+Granzyme B+ CTLs (Figures 7B, 7D and 7E) and induced PD-L1 and ENT1 expression in cancer cells (Figures S7C and S7D). Importantly, Pin1i-1 or −2 and G+P combination drastically increased survival, with 60–70% of treated mice surviving for at least 6 months after treatment (Figure 7F). Upon necropsy, these surviving mice had no obvious macroscopic tumors, and only residual microscopic tumors were notable (Figures 7G and S7E), without any liver metastases (Figure S7F), indicating that tumors are disappearing despite continuous expression of oncogenic mutant Kras and p53 in the pancreas. In conclusion, Pin1 inhibitors not only block multiple cancer pathways, disrupt the desmoplastic and immunosuppressive TME, and induce PD-L1 and ENT1 expression, but also render PDAC tumors eradicable by immunochemotherapy in GDA and KPC mice (Figure S7G).
Figure 7. Targeting Pin1 Renders Primary PDAC Tumors Eradicable by Immunochemotherapy in KPC GEMM.
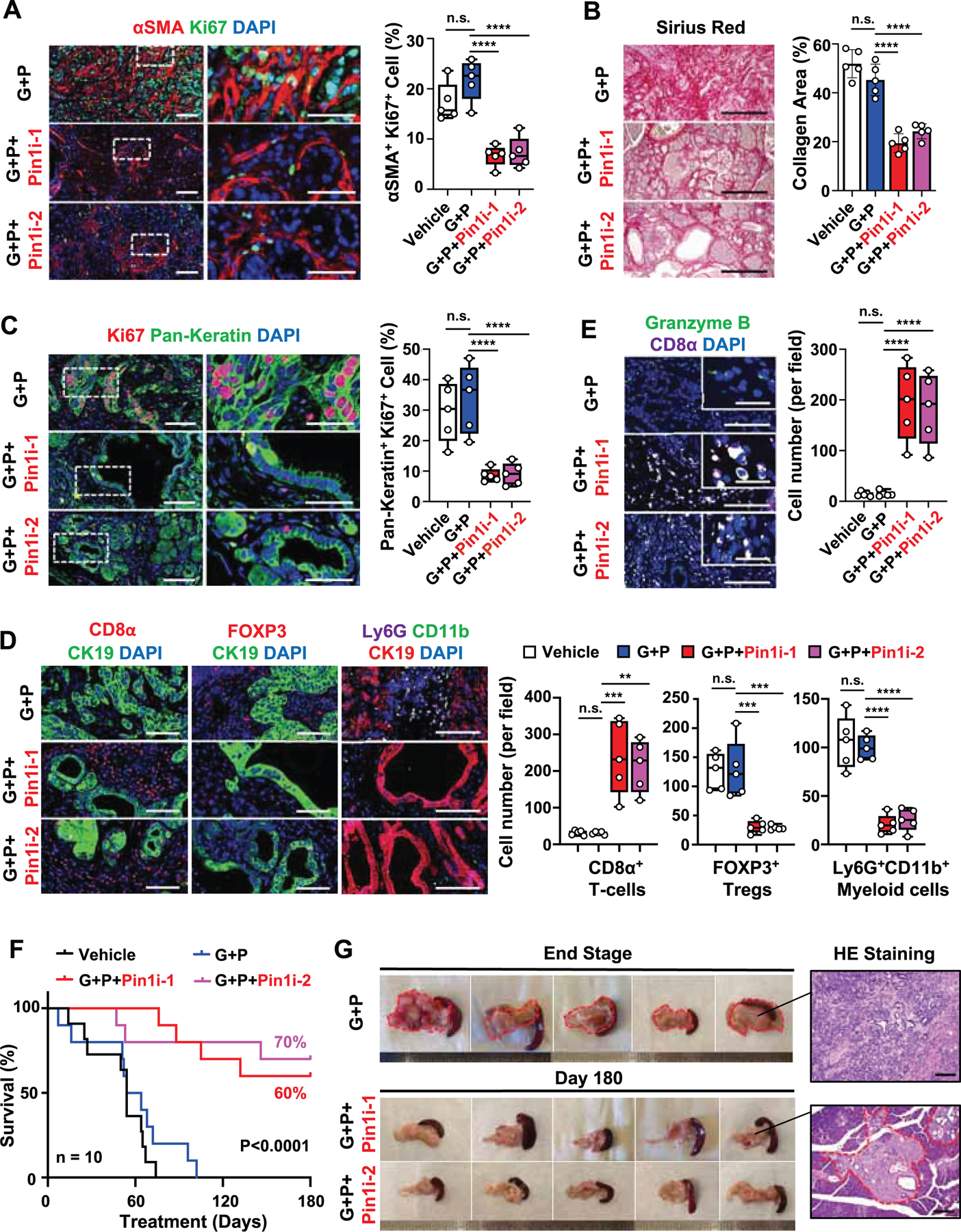
(A-E) Overt tumor-bearing KPC mice were treated with vehicle or low dose (10 mg/kg, weekly) GEM + αPD1 (G+P) with/without Pin1i-1 or −2 for a month, followed by assaying CAF activation using IF for αSMA and CAF proliferation using double IF for αSMA and Ki67 (A), cancer cell proliferation using double IF for Pan-Keratin and Ki67 (C), collagen deposition using Sirius red staining (B) and tumor-infiltrating immune cell populations, CD8α+ T-cells, FOXP3+ Tregs, and Ly6G+CD11b+ Myeloid cells (D) and CD8α+Granzyme B+ CTLs (E) using IF (n = 5).
(F and G) Tumor-bearing KPC mice were treated with vehicle or G+P with/without Pin1i-1 or −2 for up to 180 days, followed by monitoring overall survival (n = 10) (F), as well as detecting macroscopic and microscopic tumors with H&E staining (G) after autopsy.
Scale bars, 100 and 25 μm (inset) (A, C, E), 500 μm (B), 100 μm (D), and 200 μm (G). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by one-way ANOVA (A-E) or log-rank test (F).
See also Figure S7.
DISCUSSION
PDAC is notoriously resistant to current therapies due to inherent tumor heterogeneity and highly desmoplastic and immunosuppressive TME. Here we find that Pin1 is overexpressed both in cancer cells and CAFs in PDAC patients, and highly correlates with the desmoplastic and immunosuppressive TME and poor patient survival. Functionally, besides activating multiple cancer pathways, Pin1 drives the desmoplastic and immunosuppressive TME and promotes tumor malignancy and drug resistance by acting on stromal cells such as CAFs, and inducing endocytosis and degradation PD-L1 and ENT1 in cancer cells by acting on pS929-HIP1R. Consequently, targeting Pin1 offers a unique and promising approach to render this deadly cancer eradicable. These findings support a possible design of future clinical trials using Pin1 inhibitors in combination with immunochemotherapy for PDAC patients given that the ATRA+ATO therapy is a safe modality to cure most APL patients.
In PDAC, stromal CAFs play a vital role in promoting the desmoplastic and immunosuppressive TME, as well as tumor growth and malignancy, and have emerged as cancer targets (Sahai et al., 2020; Whittle and Hingorani, 2019). However, the mechanisms controlling CAF activation and function are still not fully understood. We show that Pin1 is overexpressed in CAFs and correlates with the desmoplastic and immunosuppressive TME and poor survival. Targeting Pin1 not only inhibits multiple oncogenic pathways in CAFs, but also suppresses their growth, activation, and production of cytokines implicated in immunosuppression (Erkan et al., 2012; Mace et al., 2018; Mariathasan et al., 2018). Furthermore, targeting Pin1 eliminates the ability of CAFs to promote the desmoplastic TME, tumor growth and malignancy in human PDAC organoids and/or PDOX mice. Moreover, Pin1 inhibitors also potently increase tumor-infiltrating CD8+ CTLs and decrease immunosuppressive cells in GDA and KPC mice. These results are consistent with the reports that ATRA reduces the desmoplastic and immunosuppressive TME, and tumor growth and malignancy via multiple cancer and CAF-related pathways (Chen et al., 2019; Ene-Obong et al., 2013; Froeling et al., 2011; Guan et al., 2014; Kocher et al., 2020), because many ATRA-mediated effects might be at least partially due to Pin1 inhibition (Wei et al., 2015; Zhou and Lu, 2016). Of note, CAFs are heterogenous and their functions are complex in PDAC. Our results suggest that Pin1 inhibition might target αSMA+ CAFs (myCAFs) and PDGFRα+ CAFs (iCAFs), with the latter contributing to desmoplastic immune suppressive TME by secreting collagens and cytokines (Garg et al., 2018; Hosein et al., 2020), but further work needs to define the role of Pin1 in various types of CAFs.
Protein degradation is a key mechanism to regulate not only numerous oncogenic proteins (Lu and Hunter, 2014; Zhou and Lu, 2016), but also many cancer therapeutic targets/receptors biomarkers, including PD-L1 (Burr et al., 2017; Wang et al., 2019; Zhang et al., 2018), and ENT1 (Hu et al., 2017). Notably, HIP1R is a key protein in lysosomal proteolysis by binding with a membrane protein such as PD-L1 and cytoplasmic actin for endocytosis (Gottfried et al., 2010; Messa et al., 2014; Wang et al., 2019). However, whether this PD-L1 degradation is further regulated is not known, which is especially important for PDAC because most PDAC patients have very low PD-L1 (Tessier-Cloutier et al., 2017). We demonstrated that Pin1 binds to the pSer929-Pro motif in HIP1R, and also promotes the HIP1R-actin interaction and HIP1R-mediated endocytosis and lysosomal degradation of PD-L1 and ENT1 in PDAC cells. Moreover, Pin1 inhibition highly synergizes with αPD1 to promote the ability of activated lymphocytes to induce human and KPC PDAC organoid apoptosis and to dramatically reduce tumor growth and increase overall survival of GDA mice. These results are consistent with the recent findings that human cancer patients with elevated PD-L1 levels tend to respond better to PD-1 blockade (Herbst et al., 2014). Moreover, induction of PD-L1 expression in cancer cells in response to radiation (Herter-Sprie et al., 2016 ), PARP inhibitors (Jiao et al., 2017), or inhibition of PD-L1 proteasomal proteolysis using CDK4/6 inhibitors (Zhang et al., 2018) also potentiates ICB efficacy.
It is likely that without or before ICB therapy, tumors with high PD-L1 will gain an advantage by avoiding immune surveillance resulting in poor outcomes. However, in response to αPD1 therapy, immune cells might be activated as well as protected from exhaustion to recognize and kill cancer cells expressing high PD-L1 at the cell surface. This might be especially true if cancer cells sensitize to the therapy by Pin1 inhibitors, which block multiple cancer pathways and disrupt the desmoplastic and immunosuppressive TME, are combined with GEM, which is cytotoxic to cancer cells, as suggested by our results in human organoids, GDA mice and KPC mice. However, more experiments are needed to define the mechanisms by which high PD-L1 levels in cancer cells improve αPD1 response, and whether Pin1 in immune cells including CD8+ CTLs contributes to their function and tumor killing response. Notably, our results also indicate that combination treatment with Pin1 inhibitor and αPD1 induces only a small minority of tumors (12.5%) to completely regression, but leads to the complete regressions in the vast majority (87.5%) of tumors when given in combination with low-dose GEM in GDA mice, underscoring the importance of cytotoxic damage to cancer cells.
Our results together suggest a potential new treatment strategy using Pin1 inhibitors in combination with αPD1 and GEM to render aggressive PDAC eradicable. Our experimental design with a Pin1 inhibitor treatment for 3 days before addition of the combination of GEM and αPD1 treatment is consistent with the ability of Pin1 inhibitors to reduce multiple cancer pathways in cancer cells and CAFs, to induce the cell surface expression of PD-L1 and ENT in cancer cells, to synergize with GEM and αPD1 to induce organoid apoptosis, and to have better efficacy in various PDAC mice models. We believe prior to GEM and αPD1 treatments, it may be important to ‘prime’ the tumor with Pin1 inhibition to reduce multiple cancer pathways in cancer cells and CAFs and to induce the cell surface expression of PD-L1 and ENT in cancer cells, to prepare the tumor and its microenvironment for immunochemotherapy, but further work needs to define the exact impact and timing for such additional Pin1 inhibitor treatment.
We speculate that the Pin1 inhibitor-immunochemotherapy combination would add little and/or non-overlapping toxicity to immunochemotherapy. ATRA+ATO are well tolerated and render APL curable with an acceptable safety profile even in children (de The and Chen, 2010), and PIN1−/− mice develop normally (Liou et al., 2002; Zhou and Lu, 2016). A recent phase I study found that adding ATRA to GEM and nab-paclitaxel in the treatment of advanced PDAC is safe and tolerable, and results in an encouraging progression-free survival for over 11 months, as well as reprograms the TME (Kocher et al., 2020), consistent with our findings. The efficacy of ATRA is limited by a short half-life of 45 min in humans (Lefebvre et al., 1991) and requires addition of ATO to cure APL (de The and Chen, 2010). However, liposomal ATRA has a longer half-life with significant efficacy in APL patients as monotherapy (Jain et al., 2014). In this context, our data show that the covalent Pin1 inhibitor Sulfopin, which targets the ATO-binding site (Dubiella et al., 2021), matches ATRA+ATO with regard to efficacy in PDAC.
In summary, we uncover a unique therapeutic strategy and elucidate the underlying mechanisms by which targeting Pin1 renders aggressive PDAC eradicable by synergizing with immunochemotherapy, paving the way for clinical trials and further development of new Pin1 inhibitors to evaluate their clinical impact on patients with PDAC.
Limitations of the Study
Our studies present pre-clinical data that justify further development of Pin1 inhibitors in preparation for first-in-human trials, but they have some limitations. A potential concern is that PD-L1 expression induced by Pin1 inhibition might aid immune evasion of cancer cells, and hence combination of a Pin1 inhibitor with a ICB might be preferable to capitalize on increased PD-L1 expression to increase the synergy. Although we found complete resolution of a majority of PDAC tumors in the GDA model, and long sustained reemission in the majority of the spontaneous PDAC tumors in the genetic KPC mouse model, the KPC mice have a minimal residual disease and some KPC mice showed a resistance to Pin1i after long treatment.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled Lead Contact, Kazuhiro Koikawa (kkoikawa@bidmc.harvard.edu).
Materials availability
Any other unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
This study did not generate/analyze unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human PDAC tissue samples
Human PDAC tissue samples used in this study came from 167 patients who underwent surgical resections for pancreatic cancer at Kyushu University Hospital (Fukuoka, Japan). The study was approved by the Ethics Committee of Kyushu University and conducted by the Ethical Guidelines for Human Genome/Gene Research enacted by the Japanese Government and Helsinki Declaration.
Mouse models of PDAC
We used the following 4 different commonly used mouse models of human PDAC (Bleijs et al., 2019; Day et al., 2015). 1) The KPC (LSL-K-RasLSLG12D/+; LSL-p53R172H/+;Pdx1-Cre) genetically engineered mouse model (Hingorani et al., 2005). KPC mice were generated by crossbreeding LSL-KrasG12D (B6.129S4-Krastm4Tyj/J Stock No: 008179, a congenic C57BL/6J genetic background, Jackson Laboratories), LSL-p53 (129S-Trp53tm2Tyj/J, Stock No: 008652, a 129S4/SvJae background, Jackson Laboratories, also known As:p53LSL.R172H 129svj), and Pdx1-Cre (B6.FVB-Tg (Pdx1-cre) 6Tuv/J Stock No: 014647, a C57BL/6 genetic background. Allelic profiling of resulting KPC mice by the B6 panel (Jackson Laboratories) confirmed 129S1/SvImJ with average match of 84.2 %, 129S4SvJae with average match of 83.8%, B6N with average match of 65.7% and B6J, with average match of 65.1%. 2) the KPC Genetically engineered mouse-derived orthotopic allografts (GDA) mouse model (Hingorani et al., 2005; Li et al., 2019). Mouse PDAC cells were established from KPC mouse pancreatic tumor tissues, followed by orthotopically injecting 1 ×106 mouse PDAC cells in 50 μl Matrigel (356231, Coaning) into the pancreas of female 8 week-old syngeneic immunocompetent C57BL/6 mice (Jackson Laboratories). 3) PDAC Patient-derived Tumor orthotopic xenografted (PDTX) mouse model (Gilles et al., 2018; Rubio-Viqueira et al., 2006). PDAC PDX tumors were obtained from Dr. Muthuswamy and Dr. Hidalgo and divided into 4 × 2 mm pieces, followed by xenografting orthotopically into the pancreas of female 8-week-old immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NOD scid gamma; NSG) mice (Jackson Laboratories). 4) PDAC Patient-derived Organoid and CAFs orthotopic xenograft (PDOX) mouse model (Dantes et al., 2020; Koikawa et al., 2018a). 5 × 104 patient derived PDAC organoids and 5 ×104 patient derived CAFs in 50 μl of Matrigel were injected orthotopically into the pancreas of female 8 week-old immunodeficient NSG mice (Jackson Laboratories).
Human and mouse PDAC organoids
Primary human PDAC organoids (PDAC1 and PDAC2 organoid) were established form fresh surgically resected human PDAC tissues from two different patients in Kyushu University, and mouse PDAC organoids were established form KPC mice pancreatic tumor as previously described (Koikawa et al., 2018b; Okumura et al., 2019).These organoids were 3D cultured in Growth Factor reduced (GFR) Matrigel (356231, Corning) with complete organoid media, containing AdDMEM/F12 (12634–010, Invitrogen, CA, USA), medium supplemented with 1M HEPES (Invitrogen), GlutaMax (35050–061, Invitrogen), penicillin/streptomycin (15140122, Invitrogen), B27 (17504044, Invitrogen), N-acetyl-L-cysteine (9165, Sigma-Aldrich Co.), Wnt-3a (5036-WN-010, R&D Systems), R-Spondin 1 (120–38, Peprotech), Noggin (120–10C, Invitrogen), epidermal growth factor (EGF, AF-100–15, Peprotech), fibroblast growth factor (FGF, C100–26, Peprotech), Nicotinamide (N0636, Sigma-Aldrich Co.), Y- 27263 (Y0503, Sigma-Aldrich Co.) and A83–01 (2939/10, R&D Systems). To distinguish and visualize PDAC organoids and CAFs in live-cell imaging fluorescence microscopy, PDAC organoids were labeled with green fluorescent protein (GFP) or Cell Tracker™ Green (C7025, Life Technologies).
Human CAFs and human/mouse PDAC cells.
The human CAFs (CAF1 and CAF2) were established in Kyushu University from fresh surgically resected PDAC tissues from two different patients using the outgrowth method, as described previously (Bachem et al., 2005). The isolated cells were confirmed to be CAFs by their spindle-shaped morphology, and immunofluorescence staining for αSMA-, vimentin-, CD90-, glial fibrillary acidic protein-, and nestin-positive, and CK19-negative (Endo et al., 2017), and used within 6 passages. Human PDAC cells (PDAC1 and PDAC2) were isolated form PDAC organoids, which were established surgically resected human PDAC tissues from two different patients in Kyushu University, and Mouse PDAC cells were established from pancreatic tumors of KPC mice using the outgrowth method described previously (Okumura et al., 2019), and the cells were tested for CK19, SMA, Vimentin and CD45 to verify their identity and purity, and used within 8 passages for all experiments. Cell lines were maintained in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich Co.) supplemented with 5–10% fetal bovine serum, streptomycin (100 mg/ml), and penicillin (100 mg/ml) at 37℃ in a humidified atmosphere containing 10% CO2. All cell lines were tested negative for mycoplasma contamination. To distinguish and visualize PDAC cells and CAFs in live-cell imaging fluorescence microscopy, PDAC cells were labeled with green fluorescent protein (GFP) or Cell Tracker™ Green (C7025, Life Technologies), whereas CAFs were labeled with Cell Tracker™ Red (C34552, Life Technologies).
METHOD DETAILS
Mouse experiments.
Pin1i-1 (ATRA+ATO) treatment was described as previously (Kozono et al., 2018; Wei et al., 2015), so did Pin1i-2 (Sulphopin) treatment (Dubiella et al., 2021). Briefly, all mice were randomly selected to receive treatments groups. Tumor sizes were measured by weekly palpation with electronic caliper or ultrasound, and when tumors were reached >0.5cm, the treatment was started. Briefly, animals were placed under general anesthesia with 2–3% isoflurane once a week. Under general anesthesia, the abdominal wall became very flaccid, and the muscle tone of the mouse was very low. The mouse was scuffed with the left hand to allow for palpation and measurement with the right hand. This allowed to detect the firm pancreatic mass by palpation, and to determine its size by electronic caliper measurements. Mice in our hands did not develop ascites, and the firm upper abdominal mass was easily identified below the rib cage in the left abdomen. In the GDA model and PDX models, palpable tumors predictably were detectable within 4–7 days post implantation. Hence electronic caliper measurements under general anesthesia were our routine method, and confirmatory ultrasound was conducted in some cases (Figure S1B). In the transgenic KPC mice, where we expected spontaneous tumor development, we started weekly palpation from age 8 weeks. When we could not clearly detect the tumor by palpation after age 12 weeks, we monitored mice by ultrasound. Once we detected the maximum length >0.5 cm tumor, we randomized the mice to treatment, and then followed tumor growth by electronic caliper measurement. To validate the external caliper measurements, we measured the tumor size prior to euthanization of the mice and then measured the actual tumor size upon necropsy to ascertain concordance. Mice were treated with ATO (2 mg/kg, i.p., 3 times/week, Sigma) and subcutaneous implantation of 5 mg 21-day slow-releasing ATRA pellets (Innovative Research of America) (Pin1i-1) or placebo pellets (Innovative Research of America), or Sulfopin (40 mg/kg, i.p., every day) (Pin1i-2) or vehicle (Sulfopin diluted solution; 5% NMP, 5% Solutol, 20% DMSO), and/or Gemcitabine (10mg/kg or 20mg/kg, i.p., weekly) or vehicle (PBS), and/or anti-PD1 (200μg, i.p., every 3–4 days, BE0146, BioXcell) or vehicle (IgG isotype control, BE0090, BioXcell). To evaluate the effects of CD8+ T-cell or NK1.1+ cell depletion in GDA mice, mice were treated with anti-CD8 (200 μg, i.p., twice a week, BE0117, BioXcell) (Pantelidou et al., 2019) or anti-NK1.1 (25 μ g, i.p., twice a week, BE0036, BioXcell) (Waggoner et al., 2011), or vehicle (IgG isotype control), and/or Sulfopin (40 mg/kg, i.p., every day) (Pin1i-2) or vehicle (Sulfopin diluted solution; 5% NMP, 5% Solutol, 20% DMSO), and/or anti-PD1 (200 μg, i.p., every 3–4 days, BE0146, BioXcell) or vehicle (IgG isotype control). To test Sulfopin and/or PTX and/or anti-CTLA4 combination treatment in GDA mice, mice were treated with Sulfopin (40 mg/kg, i.p., every day) (Pin1i-2) or vehicle (Sulfopin diluted solution; 5% NMP, 5% Solutol, 20% DMSO), and/or PTX (10mg/kg i.p., weekly) or vehicle (PTX diluted solution; 10% DMSO, 40% PEG300, 5% Tween 80, 45% Saline), and/or anti-CTLA-4 (250 μg, i.p., every 3–4 days, BE0032, BioXcell) or vehicle (IgG isotype control). Tumor volumes were calculated using the formula L x W2 × 0.52, where L and W represent length and width, respectively (Kozono et al., 2018). Survival events were scored when mice lost over 10% body weight, tumor burden reached 2.0 cm in diameter or per absolute survival events. All animal experiments were approved by the IACUC of the Beth Israel Deaconess Medical Center, Boston, MA, USA.
PDAC organoid and CAF co-culture model
For indirect co-culture model, 5 × 103 primary human PDAC organoids were cultured in 50 μl GFR Matrigel (356231, Corning) with Organoid Media and 5 × 104 primary human CAFs, which were stably transfected with Pin1 shRNA or control vector, were then seeded on the top of Matrigel for 10 days, followed by monitoring organoid growth using Cyntellect Celigo (Cyntellect) and analyzing the organoid size using Cyntellect Celigo software (version 1.3, Cyntellect). Direct co-culture model of PDAC organoids and CAFs was as described (Koikawa et al., 2018b). Briefly, primary human PDAC organoid cells were transfected with GFP and subjected to organoid culture. Human primary CAFs were pre-treated with Pin1 inhibitor (Pin1-i1 or Pin1i-2), or stably transfected with Pin1 CRISPR KO or vector control. Before the start of co-culture, CAFs were stained in red with cell tracker red (C34452, Invitrogen) for visualization. 1 × 104 Organoids were co-cultured with 1 × 105 human primary CAFs on the Matrigel and collagen (354249, BD Biosciences) mixed gel coated 6 well plate, followed by observing time lapse images using BZ-X800 fluorescence microscope (KEYENCE) and examining the organoid area using BZ-X 800 analyzer software (KEYENCE).
Organoid apoptosis assay
For assaying the effects of Pin1 inhibition on GEM sensitivity, 5 × 103 primary human PDAC organoids were cultured in 50 μl GFR Matrigel (356231, Corning) for 7 days and then the organoids were treated with control (DMSO) or Pin1 inhibitors (Pin1i-1, or Pin1i-2) for 3 days. The organoids were isolated from Matrigel using collagenase, and seed Matrigel coated 96 well plate, and then treated with control (PBS) or GEM, or control (DMSO) or 5-FU, followed by assaying organoid apoptosis using fluorescent caspase 3/7 and live-cell time lapse imaging for 24 hr. At the start of coculture, a green-fluorescent caspase 3/7 probe reagent (R37111, Invitrogen) and Hoechst (135102, Invitrogen) was added to visualize cells undergoing apoptosis. Apoptotic organoids were monitored by time lapse imaging using BZ-X800 fluorescence microscope (KEYENCE) and quantified using BZ-X800 analyzer (ver. 1.1.1.8, KEYENCE).
For assaying the effects of Pin1 inhibition on anti-PD1 or PDL1 sensitivity, organoids and PBMCs were co-culture as described previously (Dijkstra et al., 2018; Jiao et al., 2017). Human PDAC organoids were treated with control (DMSO) or Pin1 inhibitors (Pin1i-1, or Pin1i-2) for 3 days, and then co-cultured with human PBMCs. Human PBMCs (Precision for Medicine) were stimulated by PDAC organoid culture media, and 8.0 × 104 PBMCs were incubated with 2 µl CD3/28 beads (11161D, Gibco) and 30 U recombinant IL-2 (200–02, Peprotech) per well in 96-well plates for 24 hours before starting co-culture. PDAC organoids and activated PBMCs were directly co-cultured at 5:1 ratio on Matrigel (356231, Coaning) coated 96 well plate and treated with control (IgG), anti-PD1, or anti-PDL1, or control (PBS + IgG) or GEM + anti-PD1. At the start of co-culture, a green-fluorescent caspase 3/7 probe reagent (R37111, Invitrogen) and Hoechst (135102, Invitrogen) were added to visualize cells undergoing apoptosis. Apoptotic organoids were monitored by live-cell time lapse imaging was started 2 hours after the start of co-culture using BZ-X800 fluorescence microscope (KEYENCE) and quantified using BZ-X800 analyzer (ver. 1.1.1.8, KEYENCE).
For assaying the effects of Pin1 inhibition on GEM and anti-PD1 sensitivity in mouse PDAC organoids, mouse PDAC organoids (KPC organoids) were established from KPC mouse PDAC tumors, and 1× 106 KPC organoid cells were orthotopically transplanted into their tumor-free littermates that did not have all the three transgene or female C57BL/6 WT (8 weeks of age, Jackson) mouse pancreas. 4 weeks after transplantation, mouse CD8+ T-cells were isolated from the KPC tumor-bearing mouse, or tumor-free littermate mouse or C57BL/6 WT mouse spleens using CD8+ T-cell Isolation Kit (19853, STEM CELL) according to the manufacturer’s instructions, and then 8.0 × 104 CD8+ T-cells were activated with 2 µl CD3/28 beads (11453D, Gibco) and 30 U recombinant IL-2 (212–12, Peprotech) per well in 96-well plate for 24 hours before the start of co-culture. At the start of co-culture, a green-fluorescent caspase 3/7 probe reagent (R37111, Invitrogen) and Hoechst (135102, Invitrogen) were added to visualize cells undergoing apoptosis. KPC organoids were treated with control (DMSO) or Pin1 inhibitors (Pin1i-1 or Pin1i-2) for 3 days, and KPC organoids and activated CD8+ T-cells were directly co-cultured 5:1 ratio on Matrigel (356231, Corning) coated 96 well plate, and then treated with control (IgG) or anti-PD1, or control (PBS + IgG) or GEM + anti-PD1. Apoptotic organoids were again monitored by time lapse imaging using BZ-X800 fluorescence microscope (KEYENCE) and quantified using BZ-X800 analyzer (ver. 1.1.1.8, KEYENCE).
Synergy score analysis
To analyze the synergistic effects of the combination therapy between Pin1i and chemotherapies (GEM or 5-FU) or checkpoint immunotherapies (anti-PD-1 or anti-PD-L1), using Organoid apoptosis assay (see Organoid apoptosis assay). Synergy scores were calculated by Synergyfinder ver 2 (https://synergyfinder.fimm.fi/) (Ianevski et al., 2020).
Production of stable Pin1 KD or KO cell lines.
Establishment of Pin1 KD and CRISPR KO cells were as descried previously (Kozono et al., 2018; Wei et al., 2015). Pin1 guide RNAs (gRNAs) were designed using the online CRISPR design tool (http://CRISPR.mit.edu/). The gRNA sequences were gRNA-1 AGTCACGGCGGCCCTCGTCC TGG, gRNA-2 CAGTGGTGGCAAAAACGGGC AGG. The pLentiCRISPR construction was performed according to the protocol provided by the Zhang Lab (http://genome-engineering.org/gecko/). Oligos, (F)— 5′-CACCC-gRNA and (R) AAAC-gRNA-C, were cloned into the gRNA Cloning Vector (Addgene, plasmid #49536). To obtain single clones of Pin1 KO cells, cells were transfected with the pLenti CRISPR plasmid containing each target gRNA sequence or empty vector, selected with puromycin for 3 days and isolated by colony formation assay or single cell culture. The single clones were validated by immunoblotting analysis and DNA sequencing.
Construct of HIP1R point mutant
Human HIP1R (Huntingtin Interacting Protein 1 Related) (Accession No. NM_003959.3; 3,204 bp ORF sequence) in pcDNA3.1+/C-(K)-DYK was purchased from GenScript (NJ, USA). For constructing the point mutants of the putative Pin1 binding serine929-proline site, Ser929 was substituted by alanine (S929A) using inverse PCR method with primer sets, as described before (Suizu et al., 2016). In brief, whole plasmid DNA was amplified by the polymerase chain reaction of 16 cycles with primer sets described below in Universe Hot Start High-Fidelity 2x PCR Master mix (Biotool). After the reaction, template plasmid DNAs (wild type) were digested by DpnI enzyme (NEB). The amplified mutated linear DNA fragments were self-ligated and circulized in the presence of T4 Polynucleotide kinase (NEB) and T4 DNA ligase (NEB). The mutation site of plasmid DNA was confirmed by sanger DNA sequencing analysis in DF/HCC DNA resource core facility. Protein expression was analyzed by immunoblotting with anti-FLAG (M2) antibody (Sigma). Primer sets for S929A, sense primer; 5’-CCCCCCACCTGAGCCGC-3’, anti-sense primer5’-; CGTGCTTGTTGGCCTTCACCTTGG-3’, Primer sets for S1017A, sense primer; 5’-cCCCTGGAGAGGAGGTGGCC −3’, anti-sense primer; 5’-cGCCTGATGCCCCAGCCAG-3’
Production of stable HIP1R expressing cell lines
To subclone human HIP1R into a lentivirus vector, wild type or point mutated HIP1R ORF including DYK tag sequence was amplified by the polymerase chain reaction of 20 cycles with primer sets described below in Q5 High-Fidelity DNA Polymerase reaction mix (NEB). The digested PCR fragments with restriction enzymes XhoI and NotI were ligated into lentivirus backbone plasmid vector pCSII-EF1α-MCS-IRES2-Blasticidin (Suizu et al., 2016). For lentivirus production, 293FT packaging cells were transfected with lentiviral plasmid (pCSII), packaging plasmid (pcDNA-ΔR8.91), and envelope plasmid (VSV-G/pMD2.G) by PEI (Polyscience) transfection method. After transfection for 24hrs, the transfection reagent was replaced by fresh medium. After incubation at 35℃, 5% CO2 for 48 hrs, the resulting lentivirus supernatant was collected and filtrated with 0.45 µm disc filter. Patient-derived PDAC cells were infected with the lentivirus supernatant and fresh media at 1:1 ratio with 8µg/mL polybrene. After infection for 48 hrs, the virus particles are replaced by fresh media and the stably HIP1R-expressing cells were selected in the presence of 2µg/ml of Blasticidin for at least 4 days. Protein expression was analyzed by immunoblotting with anti-FLAG (M2) antibody (Sigma). Primer sets for subcloning Hs HIP1R into pCSII lentivirus vector, sense primer with XhoI enzyme site; 5’-ATCATCCTCGAGCCACCATGAACAGCATCAAG-3’, anti-sense primer including DYK with NotI enzyme site; 5’-ATCGCGGCCGCTCACTTATCGTCGTCATCCTTGTAATCG-3’
Production of stable HIP1R KD, ENT KD and PD-L1 KD cell lines
For silencing endogenous HIP1, ENT1 or PD-L1 expression, lentivirus producing shRNA targeti ng human HIP1R, ENT1 or PD-L1 mRNA was utilized. Five individual clones from MISSION® shRNA target set HIP1R (NM_003959.1, Sigma), ENT1 (NM_004955.1, Sigame), PD-L1 (NM_014143.2, Sigma) (Table S3) or pLKO.1 empty vector was co-transfected with a lentivirus pack aging and envelope plasmid into 293FT cells as described above. The resulting lentiviral particle s were used to infect PDAC by the mix of lentivirus supernatant and fresh media at 1:1 ratio with 8 g/mL polybrene. After infection for 48 hrs., the virus particles are replaced by fresh media and the stably shRNA-expressing cells were selected in the presence of 2 μg/ml of Puromycin for at least 4 days. Protein expression was analyzed by immunoblotting with anti-HIP1R (16814–1-AP, Proteintech), ENT1 (11337–1-AP, Proteintech) or PD-L1 (13684, Cell Signaling Technology) an tibody.
Cell proliferation assay
Cells were seeded at a density of 5000 cells (PDAC1 and PDAC2), or 3000 cells (CAF1 and CAF2) per well in 96-well flat-bottomed plates and incubated for 24 h in culture medium. At 24 h, cells were treated with ATO, ATRA, their combination (Pin1i-1), or Sulfopin (Pin1i-2). Control cells received dimethyl sulfoxide (DMSO) at a concentration equal to that of drug-treated cells for 72 hours. The cell viability was determined by CellTiter-Glo® 2.0 Assay (Promega) following the manufacturer’s instructions.
ELISA assay and cytokine array
The concentrations of cytokines in culture media were evaluated using human IL-6 uncoated ELISA (88–7066), human TGF-β uncoated ELISA (88–50390), human SDF-1 (CXCL12) ELISA (EHCXCL12A), and human LIF ELISA (BMS242) from Invitrogen, human IFNα ELISA (41100–1, R&D Systems) and using human cytokine ELISA plate array I (EA-4001, Signosis), according to the manufacturer’s instructions.
Flow cytometric analysis
PDAC tumor tissues were finely sliced into 0.5–1.0 mm fragments and dissolved by collagenase/dispase (11097113001, Sigma) for 30 min at 37 ℃ or by Tumor dissociation Kit (130–960-730, Miltenyi Biotec) according to the manufacturer’s instructions. After filtered, cell lysate was collected. To assess cell surface expressions, cells were harvested by non-enzymatic cell dissociation solution, and resuspend in blocking solution (Cell Staining Buffer, BioLegend). Cells were incubated with following antibodies; CD45-Cy7 (25–0451-82, Invitrogen), CD3-Pacific blue (100214, BioLegend), CD4-APC (100516, BioLegend), CD8 α -PE (553033, BioLegend), CD11b-PE (101208, BioLegend), CD11c-PE (117308, BioLegend), CD206-APC (141708, BioLegend), F4/80-FITC (123108, BioLegend), CD279-APC (PD-1,135210, BioLegend), CD274-APC (PD-L1, 124312, BioLegend), H-2Kb-FITC (MHC-ClassI, 116506, BioLegend), 1-A/1-E-FITC (MHC-ClassII, 107606, BioLegend), NK1.1-PE (10870, BioLegend), anti-CD223-FITC (Lag-3, 11–2231-82, Invitrogen) for 60 min on ice. For FOXP3 staining, cells were fixed and permeabilized using Foxp3 / Transcription Factor Staining Buffer Set (00–5523, eBioscience), and incubated with FOXP3-FITC (11–5773-82, Invitrogen) for 30 mins at RT. For Granzyme B staining, cells were fixed and permeabilized using Intracellular Fixation & Permeabilization Kit (88–8824-00, eBioscience), and incubated with Granzyme B-FITC (515403, BioLegend) for 30 mins at RT. All antibodies were diluted according to manufacture instruction. Cells were analyzed using CytoFLEX flow cytometer and CytExpert software (Beckman Coulter, IN, USA).
Immunoblotting analysis (IB)
Cultured cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 1% NP40, 0.1% sodium dodecyl sulfate (SDS), 0.25% Na-deoxycholate) with freshly added phosphatase inhibitors containing 5 mM NaF and 0.2 mM sodium orthovanadate (NA3VO4), and proteinase inhibitors containing 2 μg/ml Aprotinin, 2 μg/ml Leupeptin, 2 μg/ml Pepstatin A, 1 mM DTT (dithiothreitol), 0.5 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF), and 20 μM Chymotrypsin, and then mixed with the SDS sample buffer and loaded onto a gel after boiling for 10 minutes at 95 °C. The proteins were resolved by polyacrylamide gel electrophoresis and transferred to PVDF membrane. The transferred membrane was washed three times with Tris-buffered saline containing 0.1% Tween 20 (TBST). After blocking with TBST containing 5% milk or 3% bovine serum albumin (BSA) for 1 h room temperature, the membrane was incubated with the appropriate primary antibody (diluted 1:500 – 1:10000) in 5% milk or 3% BSA-containing TBST at 4 °C overnight. After incubation with the primary antibody, the membrane was washed three times with TBST for a total of 30 min followed by incubation with horseradish peroxidase conjugated goat anti-rabbit or anti-mouse IgG (diluted 1:5000) for 1 h at room temperature. After three extensive washes with TBST for a total of 30 min, the immunoblots were visualized by enhanced chemi-luminescence. Immunoblotting results were quantified using ImageJ (NIH).
Immunoprecipitation analysis (IP)
Cells were lysed in IP lysis buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 1% Triton X-100 and 10% glycerol) or NP-40 IP lysis buffer (10 mM Tris HCl, pH7.5, 100mM NaCl, 0.5% NP-40) with freshly added phosphatase and protease inhibitors. After centrifugation at 13,000 g for 15 min, the supernatant was pre-cleaned with Protein A Sepahrose beads (L00210, GeneScript) for 60 min at 4℃, one-tenth of the supernatant was stored as input, and the remainder was incubated for 12 h with 2 µg anti-Pin1 or M2 Flag agarose (Sigma) at 4℃. The supernatants were incubated with Protein A Sepahrose beads for 60 min at 4℃, and then washed three times with the aforementioned lysis buffer. After brief centrifugation, immunoprecipitates were collected, suspended in 2 × SDS sample buffer (100 mM Tris-HCl, pH 6.8, 4% SDS, 5% β-mercaptoethanol, 20% glycerol, and 0.1% bromophenol blue), boiled for 10 min at 95℃, and subjected to immunoblotting analysis.
HIP1R protein stability assay
HIP1R WT or HIP1R S929A transfected PDAC cells were incubated with 300 μg/mL cycloheximide (C104450, Sigma) under existing culture conditions. Cells were harvested at the indicated time points with RIPA buffer (50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 1% NP40, 0.1% sodium dodecyl sulfate (SDS), 0.25% Na-deoxycholate) and a proteinase and phosphatase inhibitors (5 mM NaF and 0.2 mM sodium orthovanadate (NA3VO4), 2 μg/ml Aprotinin, 2 μg/ml Leupeptin, 2 μg/ml Pepstatin A, 1 mM DTT (dithiothreitol), 0.5 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF), and 20 μM Chymotrypsin). The supernatant was collected and subjected to western blot analysis using antibody for HIP1R.
Quantitative real-time PCR analysis.
The purification of high-quality RNA from cells was performed using The RNeasy Mini Kit (74104, QIAGEN) in accordance with the manufacturer’s protocols. Samples were performed in triplicates. SYBR Green PCR Master Mix (4309155, Applied Biosystems) was used for two-step real-time RT-PCR analysis on an Applied Biosystems StepOnePlus Real Time PCR instrument. Expression value of the targeted gene in a given sample was normalized to the corresponding ex pression of GAPDH. The 2-ΔΔCt method was used to calculate relative expression of the targeted genes. The primers were: GAPDH-F, 5’- AGCCTCAAGATCATCAGCAATG’, GAPDH-R 5’- TGATGGCATGGACTGTGGTCAT −3’, hPin1-F, 5’-GCCTCACAGTTCAGCGACT-3’, hPin1-R, 5’-ACTCAGTGCGGAGGATGATGT-3’, hENT1-F, 5’-CAGAAAGTGCCTTCGGCTAC-3’ , hENT1-R, 5’-TGGGCTGAGAGAGTTGGAGACT-3’, hPD-L1-F, 5’-TGGCATTTGCTGAAC GCATTT-3, hPD-L1-R, 5’-TGCAGCCAGGTCTAATTGTTTT-3’ (Zhang et al., 2018), hPD-L1 −2F, 5’-GGTGCCGACTACAAGCGAAT-3’, hPD-L1–2R, 5’-AGCCCTCAGCCTGACATGTC-3 ‘ (Burr et al., 2017), hPD-L1–3F, 5’-ATTTGGAGGATGTGCCAGAG-3’, hPD-L1–3R, 5’-CCAG CACACTGAGAATCAACA-3’ (Mezzadra et al., 2017), hPD-L1–4 F, 5’- CCTACTGGCATTTG CTGAACGCAT-3’, hPD-L1–4 R, 5’- ACCATAGCTGATCATGCAGCGGTA-3’
Lipid droplet accumulation assay
Lipid droplet accumulation assay was performed as described previously (Endo et al., 2017). Cells were stained with 1 mg/mL 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene (bodipy 493/503; #D-3922; Life Technologies, Carlsbad, CA), and the number of bodipy-positive punctures per cell in 20 cells was counted.
Immunohistochemistry analysis (IHC)
In vitro PDAC organoids or in vivo mouse pancreas tissues were cut into 4-μm-thick sections from paraffin-embedded samples, deparaffinized in Histoclear, and rehydrated through an ethanol gradient. Antigen retrieval was achieved by microwaving or autoclaving the sections in sodium citrate buffer (pH 6.0) or Tris-EDTA (pH 8.0). Endogenous peroxidase activity was blocked by incubation with 3% hydrogen for 15 min. After blocking with 5% Goat serum for 30 min at room temperature (RT), the sections were incubated with antibodies in 1% Goat serum buffer (1:1000) overnight at 4°C. Then the sections were incubated with HRP-conjugate secondary antibodies (1:1000, Mouse IgG; PI-2000 or Rabbit IgG; PI-1000, VECTOR) for 60 min at RT. Counterstaining was performed with hematoxylin. Sirius red staining was conducted using a Picrosirius Red Staining Kit (24901, Polysciences, Inc.), according to the manufacturer’s instructions. Whole-tissue slide scans at 4× magnification was performed on BZ-X800 fluorescence microscope (KEYENCE), and scanned at least three different representative areas at 10x magnification. Image analysis was performed by thresholding for positive staining and normalizing to total tissue area, using ImageJ (NIH) and BZ-X800 analyzer (KEYENCE). IHC intensity was semi-quantified manually in a double-blind manner as a 3-tier scale (0; negative to weak, 1; moderate, 2; strong) based on previous reports (Kozono et al., 2018; Marechal et al., 2012; Wei et al., 2015; Yamaki et al., 2017). For Pin1 analysis, Low staining was defined as a staining intensity of “0” or “1 in under 50% of cancer or CAF cells”, and high staining was defined as a staining intensity of “2” and “1 in >50% cancer or CAF cells”.
Immunofluorescence analysis (IF)
Cells were seeded on cover glass (#12–542A Fisher Scientific) at approximately 60% confluence and subsequently treated with Pin1i-1 or Pin1i-2 for 72 hours. At 72 hours, cells were fixed by 4% PFA for 20 min, and washed three times with PBS, each time for 5 min. Cells were then permeabilized with 0–0.1% Triton X-100 and blocked in 3% BSA in PBS for 1 hour at RT. Human PDAC or mouse tumor tissue sections were boiled in 10 mM sodium citrate (pH 6.0), for antigen retrieval after deparaffinization. The sections were permeabilized with PBS containing 0.1–0.5% Triton X-100 and blocked with PBS containing 5% Goat serum for 30 min RT. The primary antibodies were diluted in PBS containing 1% Goat serum (1:100) and incubated in slides for overnight at 4 °C. The cells were rinsed by PBS three times, each time for 5 min. Secondary antibodies were diluted in PBS (1:200) and incubated for 20 min at room temperature. 20 μg/ml DAPI was used to label nuclear of cells. Whole-tissue slide scans at 4× magnification was performed on BZ-X800 fluorescence microscope (KEYENCE), and scanned at least three different representative areas at 10x magnification (for tissue analysis) or 20x magnification (for cell analysis). Image analysis was performed by thresholding for positive staining and normalizing to total tissue area, using ImageJ (NIH) and BZ-X800 analyzer (KEYENCE).
t-CyCIF experimental protocol
t-CyCIF imaging consisted of multiple cycles of antibody incubation, imaging, and fluorophore inactivation. The t-CyCIF experimental protocol was conducted as previously described (Du et al., 2019; Lin et al., 2018). In brief, 5 micron sections from 9 FFPE pancreatic adenocarcinoma specimens were baked at 60°C for 30 min, dewaxed using Bond Dewax Solution (Leica Biosystems) at 72°C, and antigen retrieval was performed with Epitope Retrieval 1 Solution (Leica Biosystems) at 100°C for 20 minutes using the BOND RX Automated IHC/ISH Stainer (Leica Biosystems). All antibodies were diluted in Odyssey Intercept Buffer (plus Hoechst 33342 0.25 μg/mL; LI-COR Biosciences) and incubated overnight at 4°C in the dark. See the Key Resources Table for the complete list of antibodies (note that PDGFRα staining was non-specific and inadequate to detect iCAFs). Slides were coverslipped using 20–50% glycerol solution (Sigma-Aldrich) in PBS. Images were taken using DAPI, FITC, Cy3, and Cy5 channels on the RareCyte CyteFinder Instrument (20x/0.75NA objective lens, RareCyte Inc. Seattle WA). After imaging, the fluorophores were inactivated by incubating with photobleaching solution (4.5% H2O2 and 20 mM NaOH in PBS) for 30 minutes under LED lights (this step was repeated twice).
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-Pin1 | Bao et al., 2004 | N/A |
| Rabbit polyclonal anti-Pin1 | Ayala et al., 2003; Wulf et al., 2001 | N/A |
| Mouse monoclonal anti-Pin1 (clone G-8) | Santa Cruz | Cat# sc-46660; RRID: AB_628132 |
| Mouse monoclonal Alexa Fluor 647 anti-Pin1 (clone G-8) | Santa Cruz | Cat# sc-46660 AF647; RRID: AB_628132 |
| Rabbit polyclonal anti-Pin1 | Cell Signaling Technology | Cat# 3722; RRID: AB_10692654 |
| Mouse monoclonal anti-CD8 alpha | Abcam | Cat# ab17147; RRID: AB_443686 |
| Rabbit monoclonal anti-CD3 (clone SP7) | Abcam | Cat# ab16669; RRID: AB_443425 |
| Rabbit monoclonal anti-CD45 (clone EP322Y) | Abcam | Cat# ab40763; RRID: AB_726545 |
| Rabbit monoclonal anti-CD163 | Abcam | Cat# ab182422; RRID: AB_2753196 |
| Rabbit monoclonal anti-Cytokeratin 19 | Abcam | Cat# ab52625; RRID: AB_2281020 |
| Rabbit monoclonal anti-Fibroblast activation protein | Abcam | Cat# ab207178; RRID: AB_2864720 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Rabbit polyclonal anti-Granzyme B | Abcam | Cat# ab4059; RRID: AB_304251 |
| Recombinant Alexa Fluor 488 Anti-HLA-DPB1 antibody (clone EPR11226) | Abcam | Cat#ab201527; RRID: AB_2890211 |
| Recombinant Alexa Fluor 555 Anti-alpha smooth muscle Actin antibody (clone EPR5368) | Abcam | Cat# ab202509; RRID: AB_2868435 |
| Goat F(ab) Anti-Mouse IgG H&L | Abcam | Cat# ab6668; RRID: AB_955960 |
| Rabbit polyclonal anti-FBW7 | Bethyl | Cat# A301–720A; RRID: AB_1210897 |
| Rat monoclonal anti-Mouse FOXP3, Alexa Fluor 647 | BioLegend | Cat# 126408; RRID: AB_1089115 |
| Rat monoclonal anti-Mouse Ly-6G, Alexa Fluor 647 | BioLegend | Cat# 127610; RRID: AB_1134159 |
| Rat monoclonal anti-Mouse CD11c, Alexa Fluor 647 | BioLegend | Cat# 117312; RRID: AB_389328 |
| Rat monoclonal anti-Mouse CD8α, Alexa Fluor 647 | BioLegend | Cat# 100724; RRID: AB_389326 |
| Rabbit polyclonal anti-Akt | Cell Signaling Technology | Cat# 9272; RRID: AB_329827 |
| Rabbit monoclonal anti-c-Jun (clone 60A8) | Cell Signaling Technology | Cat# 9165; RRID: AB_2130165 |
| Mouse monoclonal anti-E-Cadherin (clone 4A2) | Cell Signaling Technology | Cat# 14472; RRID: AB_2728770 |
| Mouse monoclonal anti-Pan Keratin (clone C11) | Cell Signaling Technology | Cat# 4523S; RRID: AB_836889 |
| Rabbit monoclonal anti-NF-kappaB p65 (clone D14E12) | Cell Signaling Technology | Cat# 8242; RRID: AB_10859369 |
| Rabbit monoclonal anti-LAMP1 (clone D2D11) | Cell Signaling Technology | Cat# 9091; RRID: AB_2687579 |
| Rabbit monoclonal anti-PD-L1 (clone E1L3N) | Cell Signaling Technology | Cat# 13684; RRID: AB_2687655 |
| Rabbit monoclonal anti-SQSTM1/p62 (clone D1Q5S) | Cell Signaling Technology | Cat# 39749; RRID: AB_2799160 |
| Rabbit monoclonal anti-RRM1 (clone D12F12) | Cell Signaling Technology | Cat# 8637; RRID: AB_11217623 |
| Rabbit monoclonal anti-Vimentin (clone D21H3) | Cell Signaling Technology | Cat# 5741; RRID: AB_10695459 |
| Rabbit monoclonal anti-β-Catenin (clone D10A8) | Cell Signaling Technology | Cat# 8480; RRID: AB_11127855 |
| Rabbit monoclonal anti-Cleaved Caspase3 (clone 5A1E) | Cell Signaling Technology | Cat# 9664S; RRID: AB_2070042 |
| Rabbit monoclonal anti-STING | Cell Signaling Technology | Cat# 13647S; RRID: AB_2732796 |
| Mouse monoclonal PE anti-CD44 (clone 156–3C11) | Cell Signaling Technology | Cat# 8724; RRID: AB_10829611 |
| Rabbit monoclonal Alexa Fluor 555 anti-PDGF Receptor α (D13C6) XP | Cell Signaling Technology | Cat# 8893; RRID: AB_2797679 |
| Mouse monoclonal anti-Human Smooth Muscle Actin (clone 1A4) | Dako | Cat# M0851; RRID: AB_2223500 |
| Rat monoclonal Super Bright 600 anti-CD8α (clone 53–6.7) | eBioscience | Cat# 63–0081-82; RRID: AB_2637163 |
| Mouse monoclonal eFluor 660 anti-CD8a (clone AMC908) | eBioscience | Cat# 50–0008-80; RRID: AB_2574148 |
| Goat Polyclonal Alexa Fluor 488 anti-CD4 | R&D Systems | Cat# FAB8165G; RRID: AB_2728839 |
| Rabbit polyclonal anti-PD-L1 | Novus | Cat# NBP1–76769; RRID: AB_11024101 |
| Mouse monoclonal anti-AQP9 (clone G-3) | Santa Cruz | Cat# sc-74409; RRID: AB_1119055 |
| Mouse monoclonal anti-Cytokeratin 19 (clone A-3) | Santa Cruz | Cat# sc-376126; RRID: AB_10988034 |
| Mouse monoclonal anti-cyclin D1 (clone DCS-6) | Santa Cruz | Cat# sc-20044; RRID: AB_627346 |
| Mouse monoclonal anti-dCK (clone H-3) | Santa Cruz | Cat# sc-393099; RRID: AB_2864729 |
| Mouse monoclonal FITC anti-ENT1 (clone F12) | Santa Cruz | Cat#sc-377283FITC; RRID: N/A |
| Rabbit polyclonal anti-CMTM6 | Sigma-Aldrich | Cat# HPA026980; RRID: AB_10602801 |
| Mouse monoclonal anti-FLAG (clone M2) | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Mouse monoclonal anti-β-Actin | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| Rabbit polyclonal anti-ENT1 | Proteintech | Cat# 11337–1-AP; RRID: AB_2190784 |
| Rabbit polyclonal anti-HLA class I ABC | Proteintech | Cat# 15240–1-AP; RRID: AB_1557426 |
| Rabbit polyclonal anti-HIP1R | Proteintech | Cat# 16814–1-AP; RRID: AB_2117572 |
| Rat monoclonal PE-Cyanine7 antimouse CD45 | Invitrogen | Cat# 25–0451-82; RRID: AB_2734986 |
| Mouse monoclonal Pan Cytokeratin Alexa Fluor 488 (clone AE1/AE3) | Invitrogen | Cat# 53–9003-82; RRID: AB_1834350 |
| Rat monoclonal Pacific blue antimouse CD3 | BioLegend | Cat# 100214; RRID:AB_493645 |
| Rat monoclonal APC anti-mouse CD4 | BioLegend | Cat# 100516; RRID: AB_312719 |
| Rat monoclonal PE anti-CD8α | BioLegend | Cat# 553033; RRID: AB_394571 |
| Rat monoclonal PE anti-mouse/human CD11b | BioLegend | Cat# 101208; RRID: AB_312791 |
| Armenian Hamster monoclonal PE antimouse CD11c | BioLegend | Cat# ;117308; RRID: AB_313777 |
| Rat monoclonal PE anti-mouse NK1.1 | BioLegend | Cat# 108708; RRID: AB_313395 |
| Rat monoclonal APC anti-mouse CD206 | BioLegend | Cat# 141708; RRID: AB_10900231 |
| Rat monoclonal FITC anti-mouse F4/80 | BioLegend | Cat# 123108; RRID:AB_893502 |
| Rat monoclonal APC anti-mouse CD279 (PD-1) | BioLegend | Cat# 135210; RRID: AB_2159183 |
| Rat monoclonal APC anti-mouse CD274 (PD-L1) | BioLegend | Cat# 124312; RRID: AB_10612741 |
| Rat monoclonal FITC anti-mouse H-2Kb (MHC-ClassI) | BioLegend | Cat#116506; RRID: AB_313733 |
| Rat monoclonal FITC anti-mouse 1-A/1-E (MHC-ClassII) | BioLegend | Cat# 107606; RRID: AB_313321 |
| Rat monoclonal FITC anti-human/mouse Granzyme B | BioLegend | Cat# 515403; RRID: AB_2114575 |
| Rat monoclonal APC anti-mouse CD152 (CTLA4) | BioLegend | Cat# 106310; RRID: AB_2087653 |
| Mouse monoclonal Alexa Fluor 647 antihuman CD45 | BioLegend | Cat# 304056; RRID: AB_2564155 |
| Rat monoclonal FITC anti-FOXP3 | Invitrogen | Cat# 11–5773-82; RRID:AB_465243 |
| Rat monoclonal FITC anti-CD223 (Lag-3) | Invitrogen | Cat# 11–2231-82; RRID: AB_2572484 |
| Goat polyclonal anti-Mouse IgG, IgM (H+L), Alexa Fluor 488 | Invitrogen | Cat# A-10680; RRID: AB_2534062 |
| Goat polyclonal anti-Rabbit IgG (H+L), Alexa Fluor 488 | Invitrogen | Cat# A-11008; RRID: AB_143165 |
| Goat polyclonal anti-Mouse IgG (H+L), Alexa Fluor 594 | Invitrogen | Cat# A-11005; RRID: AB_2534073 |
| Goat polyclonal anti-Rabbit IgG (H+L), Alexa Fluor 594 | Invitrogen | Cat# A-11012; RRID: AB_2534079 |
| Goat polyclonal anti-Rat IgG (H+L), Alexa Fluor 594 | Invitrogen | Cat# A-11007; RRID: AB_10561522 |
| Goat polyclonal anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat# A-21235; RRID: AB_2535804 |
| Goat polyclonal anti-Rabbit IgG IRDye 800CW | LI-COR | Cat# 926–32211; RRID: AB_621843 |
| Horse anti-Mouse IgG (H+L) | Vector Laboratories | Cat# PI-2000; RRID: AB_2336177 |
| Goat anti-Rabbit IgG (H+L) | Vector Laboratories | Cat# PI-1000; RRID: AB_2336198 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Pin1 shRNA lentiviral particles | Kondo et al., 2015 | N/A |
| CRISPR Pin1 lentiviral particles | Kozono et al., 2018 | N/A |
| GFP (CMV Bsd) lentiviral particles | GeneTarge | Cat# LVP001 |
|
| ||
| Biological samples | ||
|
| ||
| Tissue specimens of patients with PDAC | Kyushu University Hospital, Fukuoka, Japan | Ethics board approval #28–189 |
| Archival tissue specimens of patients with PDAC | Brigham and Women’s Hospital, Boston, MA | IRB approval #1627 |
| PDAC patient derived xenograft (PDX) tumor | Gilles et al., 2018 | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| All-trans retinoic acid (ATRA) | Sigma | Cat. #R2625 |
| Arsenic trioxide (ATO) | Sigma-Aldrich | Cat# A1010 |
| ATRA-releasing pellet | Innovative Research of America | Cat# V-111 |
| Placebo pellet | Innovative Research of America | Cat# C-111 |
| Sulfopin | Dubiella et al., 2021 | N/A |
| NMP | Sigma-Aldrich | Cat# 328634 |
| Kolliphor HS 15 (Solutol) | Sigma-Aldrich | Cat# 42966 |
| DMSO | Corning | Cat# 25–950-CQC |
| Gemcitabine | FRESENIUS KABI | NDC 63323–102-13 |
| 5-FU | Sigma | Cat# F6627 |
| InVivoMAb anti-PD-1 | BioXcell | Cat# BE0146 |
| InVivoMAb anti-PD-L1 | BioXcell | Cat# BE0285 |
| InVivoMAb anti-CD8a | BioXcell | Cat# BE0117 |
| InVivoMAb anti-Nk1.1 | BioXcell | Cat# BE0036 |
| InVivoMAb anti-CTLA4 | BioXcell | Cat# BE0032 |
| InVivoMAb rat IgG2a isotype control | BioXcell | Cat# BE0089 |
| InVivo ph7.0 Dilution Buffer | BioXcell | Cat# IP0070 |
| Paclitaxel | MedChemExpress | Cat# HY-B0015 |
| PEG 300 | MedChemExpress | Cat# HY-Y0873 |
| Tween 80 | MedChemExpress | Cat# HY-Y1891 |
| Growth Factor reduced (GFR) Matrigel | Corning | Cat# 356231 |
| RPMI 1640 without L-glutamine and phenol red | Corning | Cat# 17–105-CV |
| AdDMEM/F12 | Invitrogen | Cat# 12634–010 |
| HEPES | Invitrogen | Cat# H4034 |
| GlutaMax | Invitrogen | Cat# 35050–061 |
| Penicillin/Streptomycin | Invitrogen | Cat# 15140122 |
| B27 | Invitrogen | Cat# 17504044 |
| N-acetyl-L-cysteine | Sigma-Aldrich | Cat# 9165 |
| Wnt-3a | R&D Systems | Cat# 5036-WN-010 |
| R-Spondin 1 | Peprotech | Cat# 120–38 |
| Noggin | Invitrogen | Cat# 120–10C |
| EGF | Peprotech | Cat# AF-100–15 |
| FGF | Peprotech | Cat# C100–26 |
| Nicotinamide | Sigma-Aldrich | Cat# N0636 |
| Y- 27263 | Sigma-Aldrich | Cat# Y0503 |
| A83–01 | R&D Systems | Cat# 2939/10 |
| Cell Tracker Green | Life Technologies | Cat# C7025 |
| Cell Tracker Red | Life Technologies | Cat# C34552 |
| Matrigel | Corning | Cat# 356231 |
| Collagen | BD Biosciences | Cat# 354249 |
| Collagenase/dispase | Sigma-Aldrich | Cat# 11097113001 |
| TrypLE Express | GIBCO | Cat# 12604–021 |
| Defined K-SFM | GIBCO | Cat# 10744019 |
| Dynabeads Human T-Activator CD3/CD28 for T Cell Expansion and Activation | GIBCO | Cat# 11161D |
| Dynabeads Mouse T-Activator CD3/CD28 for T Cell Expansion and Activation | GIBCO | Cat# 11456D |
| Recombinant IL-2 | Peprotech | Cat# 200–02 |
| Recombinant IL-2 | Peprotech | Cat# 212–12 |
| Green-fluorescent caspase 3/7 probe reagent | Invitrogen | Cat# R37111 |
| Image-iT LIVE Red Caspase-3 and 7 Detection Kit, for microscopy | Invitrogen | Cat# I35101 |
| Cell Staining Buffer | BioLegend | Cat. #420201 |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | Cat# 00–5523-00 |
| Intracellular Fixation & Permeabilization Kit | eBioscience | Cat# 88–8824-00 |
| RIPA Lysis and Extraction Buffer | Thermo Scientific | Cat# 89901 |
| NaF | Sigma-Aldrich | Cat# S1504 |
| NA3VO4 | Sigma-Aldrich | Cat# S6508 |
| Aprotinin | Sigma-Aldrich | Cat# A1153 |
| Leupeptin | Sigma-Aldrich | Cat# 62070 |
| Pepstatin | Sigma-Aldrich | Cat# P5318 |
| DTT | Fisher Scientific | Cat# BP172–25 |
| AEBSF | Sigma-Aldrich | Cat# A8456 |
| Chymotrypsin | Sigma-Aldrich | Cat# C7268 |
| Tween 20 | VWR LIFE SCIENCE | Cat# M147 |
| HEPES | Sigma-Aldrich | Cat# H3375 |
| Triton X-100 | Sigma-Aldrich | Cat# T9284 |
| Cycloheximide | Sigma-Aldrich | Cat# C104450 |
| Glycerol | Fisher Chemical | Cat# G33–4 |
| Protein A Sepahrose beads | GeneScript | Cat# L00210 |
| β-mercaptoethanol | Millipore | Cat# ES-007-E |
| 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene | Life Technologies | Cat# D-3922 |
| Hematoxylin | Fisher Chemical | Cat# CS401–1D |
| Picrosirius Red Staining Kit | Polysciences | Cat# 24901 |
| Blasticidin | InvivoGen | Cat# ant-bl-05 |
| Puromycin | Sigma | Cat#P8833 |
| MG132 | Sigma | Cat#1211877–36-9 |
| NAE inhibitor, MLN4929 | Calbiochem | Cat# 5.05477.000 |
| 3-MA | Sigma | Cat# M9281 |
| Bafilomycin A1 | Sigma | Cat# 19–148 |
| Chloroquine | CAYMAN CHEMICAL COMPANY | Cat# 14194 |
| EasySep Mouse CD8+ T Cell Isolation Kit | STEM CELL | Cat# 19853 |
| EasySep Buffer | STEM CELL | Cat# 20144 |
| Tumor dissociation Kit Mouse | Miltenyi Biotec | Cat# 130–960-730 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4309155 |
|
| ||
| Critical commercial assays | ||
|
| ||
| CellTiter-Glo 2.0 Assay | Promega | Cat# G9242 |
| Human IL-6 uncoated ELISA Kit | Invitrogen | Cat# 88–7066-88 |
| Human TGF-β uncoated ELISA Kit | Invitrogen | Cat# 88–50390-88 |
| Human SDF1α (CXCL12) ELISA Kit | Invitrogen | Cat# EHCXCL12A |
| Human LIF ELISA Kit | Invitrogen | Cat# BMS242 |
| Human IFNα ELISA kit | R&D Systems | Cat# 41100–1 |
| Human cytokine ELISA plate array I | Signosis | Cat# EA-4001 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human: Patient derived PDAC organoid | Koikawa et al., 2018a | N/A |
| Human: Patient derived PDAC cell | Koikawa et al., 2018a | N/A |
| Human: Patient derived PSC cell | Endo et al., 2017; Koikawa et al., 2018b | N/A |
| Human: PBMC | Precision for Medicine | Lot# 2010113876 |
| Mouse: KPC derived PDAC organoid | Koikawa et al., 2018a | N/A |
| Mouse: KPC derived PDAC cell | Okumura et al., 2019 | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6 | Jackson Laboratories | Stock # 000664 IMSR_JAX:000664 |
| Mouse: NOD.Cg- prkdcscidll2rgtm1Wjl/Szj | Jackson Laboratories | Stock # 005557 IMSR_JAX:005557 |
| Mouse: LSL-KRasLSLG12D/+ | Jackson Laboratories | Stock # 008179 IMSR_JAX:008179 |
| Mouse: LSL-p53R172H/+ | Jackson Laboratories | Stock # 008652 IMSR_JAX:008652 |
| Mouse: Pdx1-Cre | Jackson Laboratories | Stock # 014647 IMSR_JAX:014647 |
|
| ||
| Oligonucleotides | ||
|
| ||
| MISSION shRNA clone for human HIP1R | Sigma-Aldrich | Table S3 |
| MISSION shRNA clone for human CD274 (PD-L1) | Sigma-Aldrich | Table S3 |
| MISSION shRNA clone for human SLC29A1 (ENT1) | Sigma-Aldrich | Table S3 |
|
| ||
| Software and algorithms | ||
|
| ||
| BZ-X 800 analyzer software ver. 1.1.1.8 | KEYENCE | https://www.keyence.com/ss/products/microscope/bz-x800_long/ |
| Synergyfinder ver. 2 | Ianevski et al., 2020 | https://synergyfinder.fimm.fi |
| CRISPR design tool | ZHANG LAB | http://CRISPR.mit.edu/ |
| Guide Design Resources | ZHANG LAB | https://zlab.bio/guide-design-resources |
| CytExpert software | Beckman Coulter | Model #B90883 |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| BioRender | BioRender.com. | https://app.biorender.com |
t-CyCIF image processing
The image processing of tissue cyclic immunofluorescence is organized in the following steps, each of which is described in detail below: i) the software ASHLAR is used to stitch, register, and correct for image acquisition artifacts (using the BaSiC algorithm). The output of ASHLAR is a single pyramid ome.tiff file for each region imaged; ii) the ome.tiff file is re-cut into tiles (typically 5000×5000 pixels) containing only the highest resolution image for all channels. One random cropped image (250×250 pixels) per tile is outputted for segmentation training (using ImageJ/Fiji); iii) using the ilastik software the labelling of nuclear, cytoplasmic and background areas are trained on the cropped images. Based on the user training the Ilastik software outputs a 3-color RGB image with label probabilities; iv) the RBG probability images are thresholded and watershed in MATLAB to segment the nuclear area. The cytoplasmic measurements are derived by dilating the nuclear mask; v) single-cell measurements are extracted for each channel (cell pixel median and mean for both nuclear and cytoplasmic area) as well as morphological measurements of area, solidity, and cell coordinates location.
BaSiC; The BaSiC ImageJ plugin tool was used to perform background and shading correction of the original images (Peng et al., 2017). The BaSiC algorithm calculates the flatfield, the change in effective illumination across an image, and the darkfield, which captures the camera offset and thermal noise. The dark field correction image is subtracted from the original image, and the result is divided by the flatfield image correction to obtain the final image.
ASHLAR; Alignment by Simultaneous Harmonization of Layer/Adjacency Registration (ASHLAR) is used to stitch together image tiles and register image tiles in subsequent layers to those in the first layer (Lin et al., 2018; Rashid et al., 2019). For the first image layer, neighboring image tiles are aligned to one another via a phase correlation algorithm that corrects for local state positioning error. A similar method is applied for subsequent layers to align tiles to their corresponding tile in the first layer. ASHLAR outputs an OME-TIFF file containing a multi-channel mosaic of the full image across all imaging cycles. Full codes available at: https://github.com/labsyspharm/ashlar.
Ilastik; ilastik is a machine learning based bioimage analysis tool that is used to obtain nuclear and cytoplasmic segmentation masks from OME-TIFF files (Berg et al., 2019). For increased processing speed, randomly selected 250 × 250 pixel regions from the original OME-TIFF are used as training data. ilastik’s interactive user interface allows the user to provide training annotations on the cropped regions. Users are presented with a subset of the channels stacked images and label pixels as either nuclear area, cytoplasmic area, or background area. The annotations are used to train non-linear classifiers that are applied to the entire image to obtain probability masks describing the probabilities of each pixel belonging to the nuclear, cytoplasmic, or background area. A MATLAB (version 2018a) script uses these masks to construct binary masks for nuclear and cytoplasmic area.
t-CyCIF data analysis workflow
The data analysis is divided in a set of pre-processing steps in which data from different tissues is i) log2-transformed and aggregated together, ii) filtered for image analysis errors, and iii) normalized on a channel-by-channel basis across the entire data from a single experiment. All the steps are performed in MATLAB.
Data aggregation.
The image processing workflow outputs one ome.tiff image and one data file (.mat) for each tissue area imaged. The data matrices from each .mat file are concatenated into a single matrix for each metric measured (median/mean, nuclear/cytoplasmic) into a single structure (“AggrResults”). The morphological data (i.e., area, solidity, and centroid coordinates) is concatenated into a single structure (“MorpResults”), which also contains the indexing vector to keep track of the tissue of origin within the dataset.
Data filtering.
Single cells are filtered to identify and potentially exclude from subsequent analysis errors in segmentation and cells lost through the rounds of imaging. Two types of criteria are used to filter cells: morphological criteria based on cell object segmented area, which are applied to all the rounds for the cell object, and DAPI-based criteria which are applied to the DAPI measurement for each imaging round. The latter corrects for cell loss during cycling and computational misalignment, which are both round specific.
Morphological filtering criteria are: 1) nuclear area within a user-input range; 2) cytoplasmic area within a user-input range; 3) nuclear object solidity above a user-input threshold. DAPI-based criteria are: 1) nuclear DAPI measurement above a user-input threshold; 2) ratio between nuclear and cytoplasmic DAPI measurement above a user-input threshold. The filter information for the criteria is allocated to a logical (0–1) structure ‘Filter’, which is used to select the cells to analyze in the further analysis by indexing. The threshold selection is dataset dependent and is performed by data inspection. The values used in each dataset are available with the codes used for data analysis in the github repository https://github.com/santagatalab/2021_Koikawa_et_al_CyCIF_codes.
Data normalization.
Each channel distribution is normalized by probability density function (pdf) centering and rescaling. The aim is to center the distribution of the log2 fluorescent signal at 0 and rescale the width of the distribution to be able to compare across channels. The data is first log-transformed (base 2). The standard normalization is performed using a 2-component Gaussian mixture model, each model capturing the negative and the positive cell population. If the 2-component model fails to approximate the channel distribution, two other strategies are attempted: i) a 3-component model is used assuming the components with the two highest means are the negative and positive distribution (i.e., discarding the lowest component) or ii) the user selects a percentage ‘x’ of assumed positive cells and a single Gaussian distribution fit is performed on the remainder of the data to capture the negative distribution. The single Gaussian fit is then used as the lower component in a 2-component model to estimate the distribution of the positive population. The strategy chosen for each channel in each dataset is available in the github repository https://github.com/santagatalab/2021_Koikawa_et_al_CyCIF_codes. The “add_coeff” is defined as the intersection of the negative and positive distributions. The “mult_coeff” is defined as the difference between the mean of the negative and positive distributions. The full distribution is normalized by subtracting the add_coeff and dividing by the mult_coeff. The normalization is performed on the nuclear and cytoplasmic single-cell, single-channel distributions individually. The data preprocessing workflow is performed on all datasets. The individual analyses used in the paper are performed only in select datasets as follows.
Isolation of CAF subsets
Cells from tissue-based experiments are classified into lineage compartments by cell type markers, by gating on the sign of the normalized values of cell type markers. Stromal cells were defined as double negatives for pan-cytokeratin and CD45. Stromal cells were subtyped by k-means clustering based on normalized values of alpha smooth muscle actin, CD44 and DPB1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical analysis
Biochemical experiments in vitro were routinely repeated at least three times, and the repeat number was increased according to effect size or sample variation. We estimated the sample size considering the variation and mean of the samples. No statistical method was used to predetermine sample size. No animals or samples were excluded from any analysis. Animals were randomly assigned groups for in vivo studies; no formal randomization method was applied when assigning animals for treatment. Group allocation and outcome assessment was not done in a blinded manner, including for animal studies. A computer program Prism 8 (GraphPad Software, CA, USA) was used for statistical analysis. All data are presented as the means ± s.d., followed by determining significant differences using the unpaired Student’s t test or one-way analysis of variance (ANOVA) test or Pearson’s chi-square test. Kaplan-Meier survival analysis was used for all survival studies, and the groups were compared using the log-rank test. Differences of *P < 0.05, **P < 0.01, ***P = 0.001, and ****P < 0.001 were considered statistically significant.
Supplementary Material
Video S1. Pin1-inhibited or CRISPR KO CAFs fail to promote PDAC growth and invasion in human 3D PDAC organoid direct co-cultures, related to Figure S3
Pin1i-treated CAF1 (Pin1i1 10 μM or Pin1i-2 5 μM for 72 hrs), or Pin KO CAF1 or control CAF1, labeled in red using cell tracker red, were cocultured with established PDAC1 organoids, labeled by GFP (green), followed by live-cell time-lapse movies to visualize their interactions and analyze organoid growth and invasion.
Video S2. Pin1 inhibitors synergize with immunochemotherapy (GEM + αPD1) to induce human PDAC organoid apoptosis, related Figures 6 and S6
Pin1i pre-treated human PDAC2 organoids were co-cultured with activated human PBMCs, and then then treated with GEM and/or αPD1, followed by assaying PDAC organoid apoptosis using green fluorescent Caspase 3/7 reagent and live-cell time-lapse imaging for 40 hrs (Pin1i-1 10 μM, Pin1i-2 5 μM, GEM 10 nM, and αPD1 100 μg/ml).
Table S1. Blood test results and body weight change of Pin1i-2 treated non-tumor-bearing B6 mice, related to Figure 1, S1 and S2
WT B6 mice (non-tumor-bearing mice) were treated with vehicle or Pin1i-2 (20, 40, or 60 mg/kg, i.p., daily) for 5 weeks, followed by examining the hematopoietic (RBC, Hb, Plate, and kidney and live function) using blood test, and body weight at 3 weeks and 5 weeks (n=5).
Table S2. Immune profiling of splenocytes from Pin1i-2 treated non-tumor-bearing B6 mice, related to Figure 1, S1 and S2
WT B6 mice (non-tumor-bearing mice) were treated with vehicle or Pin1i-2 (40 mg/kg, i.p., daily) for 5 weeks, followed by examining the immune profile of splenocytes using flow cytometry (n=5).
Table S3. shRNA sequence information, related to STAR Methods
Figure S7. Targeting Pin1 Renders Primary PDAC Tumors Eradicable by Immuno- chemotherapy in KPC GEMM, Related to Figure 7
(A-B) Pin1 inhibitors disrupt the desmoplastic and immunosuppressive TME in KPC mice. Overt tumor-bearing KPC mice were treated with vehicle or G+P with/without Pin1i-1 or −2, followed by assaying CAF activation using αSMA IF (A) or PDGFRα IF (B) and CAF proliferation using double IF for PDGFRα and Ki67 (B) (n = 5).
(C-D) Pin1 inhibitors increased PD-L1 and ENT1 expression in KPC mice. Overt tumor-bearing KPC mice were treated with vehicle or G+P with/without Pin1i-1 or −2, followed by assaying PDL1 (C) and ENT1 (D) expression using IF after autopsy (n = 5).
(E-F) Pin1 inhibitors render PDAC tumors eradicable by immunochemotherapy in KPC mice. Overt tumor-bearing KPC mice were treated with vehicle or G+P with/without Pin1i-1 or −2, for up to 180 days, followed by detecting macroscopic and microscopic tumors (E) and liver metastasis (F) after autopsy (n = 10).
(G) Left, in PDAC, Pin1 acts on cancer cells to induce HIP1R-mediated endocytosis and lysosomal degradation of PD-L1 and ENT1, and on stromal cells such as CAFs to drive the desmoplastic and immunosuppressive TME, as well as activating multiple oncogenic pathways, including many in oncogenic Kras signaling in both cells, thereby inducing drug resistance to immunochemotherapy. Right, Pin1 inhibitors suppress multiple oncogenic pathways, increase the cell surface expression of PD-L1 and ENT1 in cancer and disrupt the desmoplastic and immunosuppressive TME, together rendering aggressive PDAC tumors eradicable by immunochemotherapy (GEM + αPD1) in a synergistic manner.
Scale bars, 100 and 50 μm (B), 100 μm (C, D), 5000 and 200 μm (E), and 200 μm (F). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by oneway ANOVA (A-D).
{kind=link}
Figure S6. Targeting Pin1 Synergizes with Immunochemotherapy to Induce Human PDAC Organoid Apoptosis, Related to Figure 6
(A) Pin1 inhibitors synergize with GEM to induce human PDAC organoid apoptosis. Pin1i-2 pre-treated PDAC2 organoids were treated with GEM, followed by examining organoid apoptosis at different concentrations for 24 hrs and analyzing synergy score of Pin1i-2 and GEM using Synergy finder. We have tested 2 different patient-derived PDAC organoids (PDAC1 and PDAC2 organoid) and obtained consistent results. Subsequent experiments were shown with one organoid (PDAC2 organoid) with high expression of PD-L1 and ENT.
(B-C) ENT1 KD impairs the synergic effect of Pin1i and GEM. Control (DMSO) or Pin1i-2 (5 μM) pre-treated ENT1 WT or KD PDAC2 organoids were treated with control (PBS) or GEM (25 nM), followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 24 hrs.
(D) Pin1 inhibitors synergize with αPD1 to induce human PDAC organoid apoptosis. Pin1i-2 pre-treated PDAC2 organoids were co-cultured with activated human PBMCs, and then treated with αPD1, followed by examining organoid apoptosis at different concentrations for 40 hrs and analyzing synergy score of Pin1i-2 and αPD1 using Synergy finder.
(E-F) PD-L1 KD impaired the synergic effect of Pin1i and αPD1. Control (DMSO) or Pin1i-2 (5 μM) pre-treated PD-L1 WT or KD PDAC2 organoids were co-cultured with activated human PBMCs, and then treated with control (IgG) or αPD1 (200 μg/ml), followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 40 hrs.
(G-J) Pin1 inhibitors synergize with 5-FU or αPD1 to induce human PDAC organoid apoptosis. Control (DMSO), Pin1i-1 (10 μM) or Pin1i-2 (5 μM) pre-treated PDAC2 organoids were treated with control (DMSO) or 5-FU (25 nM), followed by examining PDAC organoid apoptosis using time lapse imaging for 24 hrs (G, H), or were co-cultured with activated human PBMCs, and then treated with control (IgG) or αPD-L1 (200 μg/ml), followed by examining PDAC organoid apoptosis using time lapse imaging for 40 hrs (I, J).
(K and L) Pin1 inhibitors synergize with immunochemotherapy (GEM + αPD1) to induce human PDAC organoid apoptosis. Control (DMSO) or Pin1i pre-treated PDAC2 organoids were co-cultured with activated human PBMCs, and then treated with control (PBS and/or IgG), GEM and/or αPD1, followed by assaying PDAC organoid apoptosis for different times at constant concentrations (Pin1i-1 10 μM, Pin1i-2 5 μM, GEM 10 nM, and αPD1 100 μg/ml).
(M) KPC tumor-bearing mouse derived CD8+ T-cells induce KPC organoid apoptosis. KPC mouse PDAC tumor derived organoids were co-cultured with/without activated CD8+ T-cells derived from KPC tumor-bearing mice or their tumor-free controls, followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 40 hrs.
(N-O) Pin1 inhibitors synergize with GEM and αPD1 to induce KPC PDAC organoid apoptosis. Control (DMSO) or Pin1i-2 pre-treated KPC tumor derived organoids were co-cultured with KPC tumor-bearing mouse derived activated CD8+ T-cells, and then treated with control (PBS + IgG), or GEM and αPD1, followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 24 hrs at constant concentrations (Pin1i-2 5 μM, GEM 10 nM, and αPD1 200 μg/ml).
Scale bars, 100μm (B, E, G, I, K, M, N). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by one-way ANOVA (C, F, H, J, O) or student’s t test (L).
{kind=link}
Figure S5. Pin1 promotes GEM Resistance and Reduces the Expression of PD-L1 and ENT1 at the Cell Surface of PDAC Cells, Related to Figures 4, 5 and 6
(A and B) Pin1 inhibition increases the expression of PD-L1 or ENT1 notably at the cell surface in PDTX mice. PDTX mice were treated with vehicle, Pin1i-1, or Pin1i-2 for 4 weeks, followed by IF for Pin1 (red), DAPI (blue), and PD-L1 (green) (A), or ENT1 (green) (B) (n = 5). White arrows point to PD-L1 (A) and ENT1 (B) at the cell surface.
(C-E) Pin1 overexpression is correlated with reduced PD-L1 or ENT1 levels in human PDAC tissues. Human PDAC tissues were subjected to IHC for Pin1 and PD-L1 (n = 53) (C), or for Pin1 and ENT1 (n=46) (D), followed by semi-quantifying their expression as high or low and examining their correlations by Pearson’s chi-square test (PD-L1: p=0.013, ENT1: p=0.008) (E).
(F) Pin1 inhibition suppresses mRNA expression of Pin1, ENT1 and PD-L1 in human PDAC cells. PDAC2 cells were treated with vehicle (DMSO), Pin1i-1 (10 μM) or Pin1i-2 (5 μM) for 72 hrs, followed by RT-PCR.
(G) Both PD-L1 and ENT levels are regulated by the lysosome-dependent proteolysis. PDAC2 cells were treated with 3-MA (15 mM), Bafilomycin A1 (30 nM), Chloroquine (20 μM), MLN4929 (1 μM), or MG132 (5 μM) for 12 hrs, followed by analyzing PD-L1 and ENT1 levels using IB.
(H) A schematic for the structural domains of HIP1R and the location of only one putative Pin1 recognition Ser929-Pro motif that is identical in humans and mice.
(I) Cycloheximide (CHX) chase assay shows the degradation of HIP1R in HIP1R WT or S929A PDAC2 cells.
(J-K) Pin1 KO or HIP1RS929A synergizes with GEM or αPD1 to induce human PDAC organoid apoptosis as well as Pin1i. Pin1i-2 (5 μM) pre-treated PDAC2 organoids or Pin1 KO or HIP1RS929A PDAC2 organoids were treated with GEM (25 nM), followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 48 hrs (J). Pin1i-2 (5 μM) pre-treated PDAC2 organoids or Pin1 KO or HIP1RS929A PDAC2 organoids were co-cultured with activated human PBMCs, and then treated with αPD1 (200 μg/ml), followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 40 hrs (K).
Scale bars, 50 μm and 12.5 μm (inset) (A, B), 500 μm (C, D) and 100 μm (J, K). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by student’s t test (A, B) or one-way ANOVA (F, J, K).
{kind=link}
Figure S4. Pin1 Promotes Oncogenic Signaling Pathways and Growth of PDAC Cells, Related to Figure 4
(A) ATRA and ATO cooperatively reduce Pin1 and its substrate oncoproteins in human PDAC cells. Primary human PDAC cells (PDAC1 and PDAC2) derived from two different PDAC patients were treated with ATRA, ATO, or ATRA + ATO (Pin1i-1) for 72 hrs, followed by examining Pin1 and its substrate oncoproteins using IB. ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations were shown.
(B and C) Pin1i-1, Pin1 KD or Pin1 KO reduces many Pin1 substrates in oncogenic Kras signaling networks in PDAC cells. PDAC2 cells were treated with Pin1i-1 for 72 hrs (B) or subjected to Pin1 KD or KO (C) followed by examining Pin1 and its substrate oncoproteins in Kras signaling using IB. ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations are shown (B).
(D-F) ATRA, ATO, Pin1i-1 and Pin1i-2 dose-dependently suppress cell growth and ablate Pin1. PDAC cells were treated with ATRA, ATO, Pin1i-1 or Pin1i-2 for 72 hrs, followed by examining cell growth using proliferation assay (D, F) and Pin1 using IB (E). ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations are shown (E).
(G) Pin1 KD inhibits human PDAC organoid growth. Pin1 KD or vector control human PDAC cells were subjected to 3D organoid cultures, followed by assaying organoid growth curve.
(H) Pin1i-2 fails to suppress Pin1 KO cell growth. Pin1-KO or CRISPR control PDAC2 cells were treated with Pin1i-2 for 72 hrs, followed by examining cell growth using proliferation assay (H).
(I-L) Pin1 inhibitors enhance the ability of GEM to inhibit human PDAC organoid growth. PDAC organoids were treated with Pin1i-1 or −2 and/or GEM at various concentrations for 7 days, followed by examining organoid growth using microscope (I, J; PDAC2 organoid) and H&E staining, and organoid proliferation using double IF for Pan-Keratin and Ki67 IF (K, L; PDAC1 organoid, Pin1i-1 10 μM and GEM 10 nM).
Scale bars, 100 µm (G, I) and 100 and 20 μm (inset) (K). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by student’s t test (G) or one-way ANOVA (H, J, L).
{kind=link}
Figure S3. Pin1 is Overexpressed in CAFs, and Pin1 Promotes Oncogenic Signaling Pathways, CAF Activation and Crosstalk with Cancer Cells to Enhance Tumor Growth and Malignancy in Human Organoids and PDOXs, Related to Figure 2 and 3
(A-B) Pin1 is overexpressed in cancer cells and CAFs, and correlated with PDAC progression. Quantification of Pin1 expression at different stages of human PDAC progression by deterring Pin1 IHC score. Normal-Adjacent normal (Normal, n=12), Low grade PanIN (Low, n=20), High grade PanIN (High, n=35), PDAC (n=167) (A). Representative images of Co-IF for Pin1 (red) and FAP (green) in human PDAC tissue (B).
(C-F) ATRA and ATO synergistically reduce Pin1 and its substrate oncoproteins, suppress cell growth and induce quiescent phenotype in CAFs, like Pin1 KD. CAF1 and CAF2 cells derived from two different human PDAC patients were treated with ATRA, ATO, or their combination (Pin1i-1) for 72 hrs (C, D, F) or genetically knocked down of Pin1 (E), followed by examining Pin1 and its substrate oncoproteins using IB (C), cell growth using cell proliferation assay (D, E), quiescent phenotype as measured by lipid droplets/CAF cell using BODIPY staining (F). ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations were shown to simplify labeling (C). CAFs were treated with control (DMSO), ATRA (10 μM) or Pin1i-1 (10 μM) (F).
(G and H) Pin1 inhibitors reduce cytokine production in primary human PDAC cells (G) and CAFs (H). Primary PDAC2 or CAF1 cells were treated with Pin1i-1 or −2 for 72 hrs, before assaying IL-6, TGFβ, LIF and CXCL12 using ELISA. Pin1i-1 10 μM (G). ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations were shown (H).
(I-K) Pin1-inhibited or CRISPR KO CAFs fail to promote PDAC growth and invasion in human 3D PDAC organoid direct co-cultures. Control (DMSO) or Pin1i-treated CAF1 (Pin1i-1 10 μM or Pin1i-2 5 μM for 72 hrs), or CRISPER control or Pin KO CAF1, labeled in red using cell tracker red, were cocultured with established human PDAC1 organoids, labeled by GFP (green) (I), followed by time-lapse imaging to visualize their interactions and analyze organoid growth and invasion (J, K).
(L and M) Pin1-KO CAFs fail to promote PDAC tumor progression and proliferation in PDOX mice. Human PDAC1 organoids were orthotopically co-transplanted with or without CRISPER control or Pin1 KO CAF1 into NSG mouse pancreas for 5 weeks, followed by examining histology using H&E staining (L) and cell proliferation using Ki67 IHC (M) derived from PDAC1 organoids (n = 5).
Scale bars, 100 and 20 μm (inset) (B), 100 μm (J), 100 and 50 μm (right panels) (L), and 50 μm (M). Error bars, mean ± s.d.; **p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; multiple comparisons: by student’s t test (E, G) or one-way ANOVA (F, H, K, M).
{kind=link}
Figure S2. Targeting Pin1 Disrupts the desmoplastic and/or immunosuppressive TME and Render PDAC Sensitive to GEM in PDTX, PDOX and GDA mice, Related to Figure 1
(A) Pin1 inhibitors render PDAC sensitive to GEM in GDA mice. Overt tumor-bearing GDA mice were treated with vehicle, Pin1i and/or GEM (20 mg/kg) (n = 6), followed by monitoring overall survival by Kaplan–Meier survival analysis. Median survival; vehicle 34 days, GEM 42 days, Pin1i-2 56 days, GEM + Pin1i-2 83.5 days.
(B-H) Pin1 inhibitors render PDAC sensitive to GEM and αPD1 by disrupting the desmoplastic and immunosuppressive TME in GDA mice. Tumor-bearing GDA mice were treated with vehicle, Pin1i and/or αPD1 or GEM (10mg/kg) +αPD1 (G+P), followed by measuring tumor volumes (B), assaying collagen deposition using Sirius Red staining (C), tumor-infiltrated immune cell profiling using flow cytometry (D, E), and tumor-infiltrating immune cell populations, CD8α+ T-cells, FOXP3+ Tregs, Ly6G+CD11b+ Myeloid cells (F), and CD8α+Granzyme B+ CTLs (G) using IF after 4 weeks treatment (n = 5), or measuring body weight after 120 days treatment (Control/No transplantation: n = 3, G+P + Pin1i-1: n = 7, G+P + Pin1i-2: n = 6) (H).
(I) Pin1 inhibitors render PDAC curable by immunochemotherapy in GDA mice. Overt tumor-bearing GDA mice were treated with vehicle, Pin1i-2, GEM (10 mg/kg) + αPD1 (G+P), Pin1i-2 + αPD1, or Pin1i-2 + G+P for up to 150 days and followed by monitoring overall survival by Kaplan–Meier survival analysis up to 250 days (n=8). Median survival; vehicle 36.5 days, G+P 41 days, Pin1i-2 54 days, αPD1 + Pin1i-2 83.5 days, G+P + Pin1i-2 133.5 days (I).
(J) αPD1 renders GDA tumors hyperprogressive in the absence of CD8+ T-cells. Tumor-bearing GDA mice were treated with αCD8α and vehicle, αPD1, Pin1i-2, or αPD1 + Pin1i-2 for 9 days, followed by examining tumor volume.
(K) Pin1 inhibition does not significantly potentiate PTX or αCTLA4 therapy in GDA mice. Tumor-bearing GDA mice were treated with vehicle, Pin1i-2, PTX, αCTLA4, or their combination for 4 weeks, followed by assaying tumor volume.
Scale bars, 200 μm (C), 100 μm (F), and 100 and 25 μm (inset) (G). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by log-rank test (A, I), student’s t test (D), or one-way ANOVA (B, C, E-H, J, K).
{kind=link}
Figure S1. Targeting Pin1 Disrupts the desmoplastic and/or immunosuppressive TME and Inhibits Tumor Growth and Progression in PDTX and GDA mice, Related to Figure 1
(A-B) Experimental setup and treatment schedule. Tumor-bearing (>0.5 cm) PDTX, PDOX or GDA mice were treated with Pin1i, GEM and/or αPD1, starting Pin1i 3 days before others unless stated otherwise. Tumor sizes were detected by ultrasound or palpation with electronic caliper (A). Representative ultrasound images of the initial tumor on Day −3 (B).
(C-H) Pin1 inhibitors suppress tumor growth and progression, and desmoplastic TME in PDTX mice. Overt tumor-bearing PDTX mice were treated with vehicle or Pin1i-1 for 4 weeks, followed by assaying tumor growth and volume (C), collagen deposition and cancer cell differentiation using Sirius Red and H&E staining (D), cancer cell proliferation using Ki67 IHC (E), tumor-associated CAF activation and proliferation using double IF for αSMA and Ki67 (F) or platelet-derived growth factor receptor α (PDGFRα) and Ki67 (G), and EMT using double IF for E-cadherin and Vimentin (H) (n = 5). White arrows indicate αSMA and Ki67 double positive CAFs (F), Ki67 and PDGFRα double positive CAFs (G), or Vimentin positive cancer cell (H).
(I and J) Pin1 inhibitors suppress cancer cell proliferation and tumor progression, and desmoplastic and immunosuppressive TME in GDA mice. Tumor-bearing GDA mice were treated with vehicle, Pin1i-1 or −2 for 4 weeks, followed by examining cancer cell differentiation using H&E staining (I), and CAF proliferation using double IF for PDGFRα and Ki67 (J) using IF (n = 5).
(K-N) Pin1 inhibitors render PDAC sensitive to GEM in PDOX and PDTX mice. Patient-derived PDAC1 organoids and CAFs (K; PDOX mice, n = 5) or patient-derived PDAC tumors (L-N; PDTX mice, n = 6) were orthotopically transplanted into NSG mouse pancreas and when tumors reached 0.5 cm, mice were treated with vehicle, Pin1i and/or GEM (20 mg/kg), followed by measuring tumor volumes (K and L) and liver metastasis (N) after autopsy at 4 weeks after treatment, or monitoring overall survival by Kaplan–Meier survival analysis (M). Median survival; vehicle 36 days, GEM 42.5 days, Pin1i-1 43 days, and Pin1i-1 + GEM 71 days (M).
Scale bars, 500 and 100 μm (right panel) (D), 100 and 25 μm (inset) (F, G), 50 μm (H, J), and 100 μm (I), and 200 μm (N). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by student’s t test (C-H, J), one-way ANOVA (K, L, N), or log-rank test (M).
{kind=link}
HIGHLIGHTS.
Pin1 is overexpressed both in PDAC cells and CAFs, and correlates with poor patient survival.
Targeting Pin1 disrupts the desmoplastic and immunosuppressive TME by acting on CAFs.
Targeting Pin1 increases PD-L1 and ENT1 expression in cancer cells by acting on HIP1R.
Targeting Pin1 renders aggressive PDAC eradicable by synergizing with immunochemotherapy.
ACKNOWLEDGMENTS
We are grateful to Stephanie K Dougan for her expert advice and Terri Woo for her assistance with t-CyCIF and animal facility staffs at Beth Israel Deaconess Medical Center (BIDMC) for mouse experiments. The graphically abstract and schemas were created with BioRender.com. F.S was supported by JSPS KAKENHI JP18KK044, AMED grant JP20gm5910024. C.Q. is Alzheimer’s Association Research Fellow. The work is supported by the funding from the Ludwig Center at Harvard, NIH grants U54-CA225088 to S.S., T32-HL007627 to G.G., the Breast Cancer Research Foundation (BCRF), SPORE grant 1P50CA168504, and 5R01CA226776 to G.M.W., R01CA205153 to K.P.L., N.S.G. and X.Z.Z., R01AG055559 to K.P.L., E.H.L. and X.Z.Z., and U01NS096835 to K.P.L., as well as gift donations from the Owens Family Foundation to X.Z.Z. and K.P.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
K.K., G.M.W., T.D.M, B.J.P., N.S.G., X.Z.Z. and/or K.P.L. are inventors of a number of issued patents and/or pending patent applications on Pin1, Pin1 biomarkers, Pin1 inhibitors and Pin1 inhibitor combination to treat human diseases; X.Z.Z and K.P.L. are inventors of cis P-tau antibody technology, which was licensed by BIDMC to the startup Pinteon Therapeutics. X.Z.Z. and K.P.L. are the scientific founders and former scientific advisors of and own equity in Pinteon. Their interests were reviewed and are managed by BIDMC in accordance with its conflict of interest policy. G.M.W. reports research funding from Glaxo Smith Kline (institutional funding). N.S.G. is a founder, science advisory board member (SAB) and equity holder in Gatekeeper, Syros, C4, Allorion, Jengu, Inception, B2S, EoCys, Larkspur and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Deerfield and Sanofi. All other authors do not have any competing interest.
REFERENCES
- Amrutkar M, and Gladhaug IP (2017). Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers (Basel) 9, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachem MG, Schunemann M, Ramadani M, Siech M, Beger H, Buck A, Zhou S, Schmid-Kotsas A, and Adler G (2005). Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 128, 907–921. [DOI] [PubMed] [Google Scholar]
- Balachandran VP, Beatty GL, and Dougan SK (2019). Broadening the Impact of Immunotherapy to Pancreatic Cancer: Challenges and Opportunities. Gastroenterology 156, 2056–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Sauter G, Sowadski J, Lu KP, and Wang D (2004). Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am. J. Pathol 164, 1727–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, and Vonderheide RH (2012). Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 21, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, et al. (2019). ilastik: interactive machine learning for (bio)image analysis. Nat Methods 16, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. (2012). Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. (2018). Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleijs M, van de Wetering M, Clevers H, and Drost J (2019). Xenograft and organoid model systems in cancer research. EMBO J 38, e101654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, and Hunter T (2001). Oncogenic kinase signalling. Nature 411, 355–365. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366, 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, Lam EYN, Henderson MA, Bell CC, Stolzenburg S, et al. (2017). CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, Chan LC, Li CW, Hsu JL, and Hung MC (2019). Mechanisms Controlling PD-L1 Expression in Cancer. Mol Cell 76, 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xu X, Wen X, Xu S, Wang L, Lu W, Jiang M, Huang J, Yang D, Wang J, et al. (2019). Targeting PIN1 exerts potent antitumor activity in pancreatic ductal carcinoma via inhibiting tumor metastasis. Cancer Sci 110, 2442–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantes Z, Yen HY, Pfarr N, Winter C, Steiger K, Muckenhuber A, Hennig A, Lange S, Engleitner T, Ollinger R, et al. (2020). Implementing cell-free DNA of pancreatic cancer patient-derived organoids for personalized oncology. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP, Merlino G, and Van Dyke T (2015). Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 163, 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de The H, and Chen Z (2010). Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer 10, 775–783. [DOI] [PubMed] [Google Scholar]
- Dijkstra KK, Cattaneo CM, Weeber F, Chalabi M, van de Haar J, Fanchi LF, Slagter M, van der Velden DL, Kaing S, Kelderman S, et al. (2018). Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 174, 1586–1598 e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, Chirieleison SM, Pareek TK, Abbott DW, Stearns DS, Letterio JJ, et al. (2016). Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 353, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z, Lin JR, Rashid R, Maliga Z, Wang S, Aster JC, Izar B, Sorger PK, and Santagata S (2019). Qualifying antibodies for image-based immune profiling and multiplexed tissue imaging. Nat Protoc 14, 2900–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiella C, Pinch BJ, Koikawa K, Zaidman D, Poon E, Manz TD, Nabet B, He S, Resnick E, Rogel A, et al. (2021). Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat Chem Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, Koikawa K, Okumura T, Sada M, Horioka K, et al. (2017). Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 152, 1492–1506 e1424. [DOI] [PubMed] [Google Scholar]
- Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, Marshall JF, Chin-Aleong J, Chelala C, Gribben JG, et al. (2013). Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 145, 1121–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkan M, Adler G, Apte MV, Bachem MG, Buchholz M, Detlefsen S, Esposito I, Friess H, Gress TM, Habisch HJ, et al. (2012). StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut 61, 172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, Abrams R, Benson AB, Macdonald J, Cass CE, et al. (2009). Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology 136, 187–195. [DOI] [PubMed] [Google Scholar]
- Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, Connell CM, Roberts EW, Zhao Q, Caballero OL, et al. (2013). Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 110, 20212–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, Clevers H, Hart IR, and Kocher HM (2011). Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology 141, 1486–1497, 1497 e1481–1414. [DOI] [PubMed] [Google Scholar]
- Garg B, Giri B, Modi S, Sethi V, Castro I, Umland O, Ban Y, Lavania S, Dawra R, Banerjee S, et al. (2018). NFkappaB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology 155, 880–891 e888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, et al. (2012). Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 366, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilles ME, Hao L, Huang L, Rupaimoole R, Lopez-Casas PP, Pulver E, Jeong JC, Muthuswamy SK, Hidalgo M, Bhatia SN, and Slack FJ (2018). Personalized RNA Medicine for Pancreatic Cancer. Clin Cancer Res 24, 1734–1747. [DOI] [PubMed] [Google Scholar]
- Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A, et al. (2011). A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 20, 79–91. S1535–6108(11)00226–1 [pii] [DOI] [PubMed] [Google Scholar]
- Gottfried I, Ehrlich M, and Ashery U (2010). The Sla2p/HIP1/HIP1R family: similar structure, similar function in endocytosis? Biochem Soc Trans 38, 187–191. [DOI] [PubMed] [Google Scholar]
- Gotwals P, Cameron S, Cipolletta D, Cremasco V, Crystal A, Hewes B, Mueller B, Quaratino S, Sabatos-Peyton C, Petruzzelli L, et al. (2017). Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat Rev Cancer 17, 286–301. [DOI] [PubMed] [Google Scholar]
- Guan J, Zhang H, Wen Z, Gu Y, Cheng Y, Sun Y, Zhang T, Jia C, Lu Z, and Chen J (2014). Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer letters 345, 132–139. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. (2014). Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herter-Sprie GS, Koyama S, Korideck H, Hai J, Deng J, Li YY, Buczkowski KA, Grant AK, Ullas S, Rhee K, et al. (2016). Synergy of radiotherapy and PD-1 blockade in Kras-mutant lung cancer. JCI Insight 1, e87415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, and Tuveson DA (2005). Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483. [DOI] [PubMed] [Google Scholar]
- Ho WJ, Jaffee EM, and Zheng L (2020). The tumour microenvironment in pancreatic cancer - clinical challenges and opportunities. Nat Rev Clin Oncol 17, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosein AN, Brekken RA, and Maitra A (2020). Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol 17, 487–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Qin Y, Zhang B, Liang C, Ji S, Shi S, Xu W, Xiang J, Liang D, Ni Q, et al. (2017). FBW7 increases the chemosensitivity of pancreatic cancer cells to gemcitabine through upregulation of ENT1. Oncol Rep 38, 2069–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianevski A, Giri AK, and Aittokallio T (2020). SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res 48, W488–W493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain P, Kantarjian H, Estey E, Pierce S, Cortes J, Lopez-Berestein G, and Ravandi F (2014). Single-agent liposomal all-trans-retinoic Acid as initial therapy for acute promyelocytic leukemia: 13-year follow-up data. Clinical lymphoma, myeloma & leukemia 14, e47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. (2016). Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med 22, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, et al. (2017). PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res 23, 3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, Neale RE, Tempero M, Tuveson DA, Hruban RH, and Neoptolemos JP (2016). Pancreatic cancer. Nat Rev Dis Primers 2, 16022. [DOI] [PubMed] [Google Scholar]
- Kocher HM, Basu B, Froeling FEM, Sarker D, Slater S, Carlin D, deSouza NM, De Paepe KN, Goulart MR, Hughes C, et al. (2020). Phase I clinical trial repurposing all-trans retinoic acid as a stromal targeting agent for pancreatic cancer. Nature communications 11, 4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koikawa K, Ohuchida K, Ando Y, Kibe S, Nakayama H, Takesue S, Endo S, Abe T, Okumura T, Iwamoto C, et al. (2018a). Basement membrane destruction by pancreatic stellate cells leads to local invasion in pancreatic ductal adenocarcinoma. Cancer letters 425, 65–77. [DOI] [PubMed] [Google Scholar]
- Koikawa K, Ohuchida K, Takesue S, Ando Y, Kibe S, Nakayama H, Endo S, Abe T, Okumura T, Horioka K, et al. (2018b). Pancreatic stellate cells reorganize matrix components and lead pancreatic cancer invasion via the function of Endo180. Cancer letters 412, 143–154. 10.1016/j.canlet.2017.10.010. [DOI] [PubMed] [Google Scholar]
- Kozono S, Lin YM, Seo HS, Pinch B, Lian X, Qiu C, Herbert MK, Chen CH, Tan L, Gao ZJ, et al. (2018). Arsenic targets Pin1 and cooperates with retinoic acid to inhibit cancer-driving pathways and tumor-initiating cells. Nature communications 9, 3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, et al. (2016). Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med 22, 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre P, Thomas G, Gourmel B, Agadir A, Castaigne S, Dreux C, Degos L, and Chomienne C (1991). Pharmacokinetics of oral all-trans retinoic acid in patients with acute promyelocytic leukemia. Leukemia 5, 1054–1058. [PubMed] [Google Scholar]
- Li J, Qian W, Qin T, Xiao Y, Cheng L, Cao J, Chen X, Ma Q, and Wu Z (2019). Mouse-Derived Allografts: A Complementary Model to the KPC Mice on Researching Pancreatic Cancer In Vivo. Comput Struct Biotechnol J 17, 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Dong Z, Lin Y, Jia X, Li Q, Jiang H, Wang L, and Gao Y (2013). The rs2233678 polymorphism in PIN1 promoter region reduced cancer risk: a meta-analysis. PLoS One 8, e68148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, et al. (2016). Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JR, Izar B, Wang S, Yapp C, Mei S, Shah PM, Santagata S, and Sorger PK (2018). Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou YC, Ryo R, Huang HK, Lu PJ, Bronson R, Fujimori F, Uchidafl U, Hunter T, and Lu KP (2002). Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc. Natl. Acad. Sci. USA 99, 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PH, Shah RB, Li Y, Arora A, Ung PM, Raman R, Gorbatenko A, Kozono S, Zhou XZ, Brechin V, et al. (2019). An IRAK1-PIN1 signalling axis drives intrinsic tumour resistance to radiation therapy. Nat Cell Biol 21, 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, and Hunter T (2014). Pin1 and cancer. Cell Res 24, 1033–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, and Elledge SJ (2009). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ML, Zheng F, Chen W, Liang ZM, Chandramouly G, Tan J, Willis NA, Chen CH, de Oliveira Taveira M, Zhou XZ, et al. (2020). Inactivation of the prolyl isomerase Pin1 sensitizes BRCA1-proficient breast cancer to PARP inhibition. Cancer Res 80, 3033–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, Nordquist E, Cruz-Monserrate Z, Yu L, Young G, et al. (2018). IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut 67, 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney KM, Rennert PD, and Freeman GJ (2015). Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 14, 561–584. [DOI] [PubMed] [Google Scholar]
- Marechal R, Bachet JB, Mackey JR, Dalban C, Demetter P, Graham K, Couvelard A, Svrcek M, Bardier-Dupas A, Hammel P, et al. (2012). Levels of gemcitabine transport and metabolism proteins predict survival times of patients treated with gemcitabine for pancreatic adenocarcinoma. Gastroenterology 143, 664–674 e666. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. (2017). Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 171, 1259–1271 e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, and Swanton C (2017). Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 168, 613–628. [DOI] [PubMed] [Google Scholar]
- Messa M, Fernandez-Busnadiego R, Sun EW, Chen H, Czapla H, Wrasman K, Wu Y, Ko G, Ross T, Wendland B, and De Camilli P (2014). Epsin deficiency impairs endocytosis by stalling the actin-dependent invagination of endocytic clathrin-coated pits. eLife 3, e03311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, Logtenberg MEW, Slagter M, Rozeman EA, Hofland I, et al. (2017). Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugoni V, Panella R, Cheloni G, Chen M, Pozdnyakova O, Stroopinsky D, Guarnerio J, Monteleone E, Lee JD, Mendez L, et al. (2019). Vulnerabilities in mIDH2 AML confer sensitivity to APL-like targeted combination therapy. Cell Res 29, 446–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SA, Wu DC, Cheung J, Navarro A, Xiong H, Cubas R, Totpal K, Chiu H, Wu Y, Comps-Agrar L, et al. (2021). PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nature Cancer 1, 681–691. [DOI] [PubMed] [Google Scholar]
- Okumura T, Ohuchida K, Kibe S, Iwamoto C, Ando Y, Takesue S, Nakayama H, Abe T, Endo S, Koikawa K, et al. (2019). Adipose tissue-derived stromal cells are sources of cancer-associated fibroblasts and enhance tumor progression by dense collagen matrix. Int J Cancer 144, 1401–1413. [DOI] [PubMed] [Google Scholar]
- Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, et al. (2014). Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 25, 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelidou C, Sonzogni O, De Oliveria Taveira M, Mehta AK, Kothari A, Wang D, Visal T, Li MK, Pinto J, Castrillon JA, et al. (2019). PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov 9, 722–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T, Thorn K, Schroeder T, Wang L, Theis FJ, Marr C, and Navab N (2017). A BaSiC tool for background and shading correction of optical microscopy images. Nature communications 8, 14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, and Matrisian LM (2014). Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74, 2913–2921. [DOI] [PubMed] [Google Scholar]
- Rashid R, Gaglia G, Chen YA, Lin JR, Du Z, Maliga Z, Schapiro D, Yapp C, Muhlich J, Sokolov A, et al. (2019). Highly multiplexed immunofluorescence images and single-cell data of immune markers in tonsil and lung cancer. Sci Data 6, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, Dekleva EN, Saunders T, Becerra CP, Tattersall IW, et al. (2014). Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 25, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C, Shi C, Danenberg K, Danenberg PV, Kuramochi H, et al. (2006). An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 12, 4652–4661. [DOI] [PubMed] [Google Scholar]
- Ruscetti M, Morris J.P.t., Mezzadra R, Russell J, Leibold J, Romesser PB, Simon J, Kulick A, Ho YJ, Fennell M, et al. (2020). Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 181, 424–441 e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T, et al. (2020). A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel N, and Hudson TJ (2011). The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol 9, 77–87. [DOI] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, et al. (2014). Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 159, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Gao W, Lytle NK, Huang P, Yuan X, Dann AM, Ridinger-Saison M, DelGiorno KE, Antal CE, Liang G, et al. (2019). Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 569, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Fuchs HE, and Jemal A (2021). Cancer Statistics, 2021. CA Cancer J Clin 71, 7–33. [DOI] [PubMed] [Google Scholar]
- Suizu F, Hirata N, Kimura K, Edamura T, Tanaka T, Ishigaki S, Donia T, Noguchi H, Iwanaga T, and Noguchi M (2016). Phosphorylation-dependent Akt-Inversin interaction at the basal body of primary cilia. EMBO J 35, 1346–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier-Cloutier B, Kalloger SE, Al-Kandari M, Milne K, Gao D, Nelson BH, Renouf DJ, Sheffield BS, and Schaeffer DF (2017). Programmed cell death ligand 1 cut-point is associated with reduced disease specific survival in resected pancreatic ductal adenocarcinoma. BMC Cancer 17, 618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubersax JA, and Ferrell JE Jr. (2007). Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol 8, 530–541. [DOI] [PubMed] [Google Scholar]
- Waggoner SN, Cornberg M, Selin LK, and Welsh RM (2011). Natural killer cells act as rheostats modulating antiviral T cells. Nature 481, 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yao H, Li C, Shi H, Lan J, Li Z, Zhang Y, Liang L, Fang JY, and Xu J (2019). HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat Chem Biol 15, 42–50. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang F, Zhong M, Yarden Y, and Fu L (2020). The biomarkers of hyperprogressive disease in PD-1/PD-L1 blockage therapy. Mol Cancer 19, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AM, and Der CJ (2018). KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harbor perspectives in medicine 8, a031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Kats L, Li W, Nechama M, Kondo A, Luo M, Yao Y, Moerke NJ, Cao S, Reschke M, et al. (2015). Active Pin1 as a target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nature Med 21, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle MC, and Hingorani SR (2019). Fibroblasts in Pancreatic Ductal Adenocarcinoma: Biological Mechanisms and Therapeutic Targets. Gastroenterology 156, 2085–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulf G, Garg P, Liou YC, Iglehart D, and Lu KP (2004). Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis. EMBO J. 23, 3397–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaki S, Yanagimoto H, Tsuta K, Ryota H, and Kon M (2017). PD-L1 expression in pancreatic ductal adenocarcinoma is a poor prognostic factor in patients with high CD8(+) tumor-infiltrating lymphocytes: highly sensitive detection using phosphor-integrated dot staining. International journal of clinical oncology 22, 726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ, et al. (2020). Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, et al. (2018). Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XZ, and Lu KP (2016). The isomerase Pin1 controls numerous cancer-driving pathways and is a unique drug target. Nat Rev Cancer 16, 463–478. [DOI] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, and Chen L (2016). PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Science translational medicine 8, 328rv324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Pin1-inhibited or CRISPR KO CAFs fail to promote PDAC growth and invasion in human 3D PDAC organoid direct co-cultures, related to Figure S3
Pin1i-treated CAF1 (Pin1i1 10 μM or Pin1i-2 5 μM for 72 hrs), or Pin KO CAF1 or control CAF1, labeled in red using cell tracker red, were cocultured with established PDAC1 organoids, labeled by GFP (green), followed by live-cell time-lapse movies to visualize their interactions and analyze organoid growth and invasion.
Video S2. Pin1 inhibitors synergize with immunochemotherapy (GEM + αPD1) to induce human PDAC organoid apoptosis, related Figures 6 and S6
Pin1i pre-treated human PDAC2 organoids were co-cultured with activated human PBMCs, and then then treated with GEM and/or αPD1, followed by assaying PDAC organoid apoptosis using green fluorescent Caspase 3/7 reagent and live-cell time-lapse imaging for 40 hrs (Pin1i-1 10 μM, Pin1i-2 5 μM, GEM 10 nM, and αPD1 100 μg/ml).
Table S1. Blood test results and body weight change of Pin1i-2 treated non-tumor-bearing B6 mice, related to Figure 1, S1 and S2
WT B6 mice (non-tumor-bearing mice) were treated with vehicle or Pin1i-2 (20, 40, or 60 mg/kg, i.p., daily) for 5 weeks, followed by examining the hematopoietic (RBC, Hb, Plate, and kidney and live function) using blood test, and body weight at 3 weeks and 5 weeks (n=5).
Table S2. Immune profiling of splenocytes from Pin1i-2 treated non-tumor-bearing B6 mice, related to Figure 1, S1 and S2
WT B6 mice (non-tumor-bearing mice) were treated with vehicle or Pin1i-2 (40 mg/kg, i.p., daily) for 5 weeks, followed by examining the immune profile of splenocytes using flow cytometry (n=5).
Table S3. shRNA sequence information, related to STAR Methods
Figure S7. Targeting Pin1 Renders Primary PDAC Tumors Eradicable by Immuno- chemotherapy in KPC GEMM, Related to Figure 7
(A-B) Pin1 inhibitors disrupt the desmoplastic and immunosuppressive TME in KPC mice. Overt tumor-bearing KPC mice were treated with vehicle or G+P with/without Pin1i-1 or −2, followed by assaying CAF activation using αSMA IF (A) or PDGFRα IF (B) and CAF proliferation using double IF for PDGFRα and Ki67 (B) (n = 5).
(C-D) Pin1 inhibitors increased PD-L1 and ENT1 expression in KPC mice. Overt tumor-bearing KPC mice were treated with vehicle or G+P with/without Pin1i-1 or −2, followed by assaying PDL1 (C) and ENT1 (D) expression using IF after autopsy (n = 5).
(E-F) Pin1 inhibitors render PDAC tumors eradicable by immunochemotherapy in KPC mice. Overt tumor-bearing KPC mice were treated with vehicle or G+P with/without Pin1i-1 or −2, for up to 180 days, followed by detecting macroscopic and microscopic tumors (E) and liver metastasis (F) after autopsy (n = 10).
(G) Left, in PDAC, Pin1 acts on cancer cells to induce HIP1R-mediated endocytosis and lysosomal degradation of PD-L1 and ENT1, and on stromal cells such as CAFs to drive the desmoplastic and immunosuppressive TME, as well as activating multiple oncogenic pathways, including many in oncogenic Kras signaling in both cells, thereby inducing drug resistance to immunochemotherapy. Right, Pin1 inhibitors suppress multiple oncogenic pathways, increase the cell surface expression of PD-L1 and ENT1 in cancer and disrupt the desmoplastic and immunosuppressive TME, together rendering aggressive PDAC tumors eradicable by immunochemotherapy (GEM + αPD1) in a synergistic manner.
Scale bars, 100 and 50 μm (B), 100 μm (C, D), 5000 and 200 μm (E), and 200 μm (F). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by oneway ANOVA (A-D).
Figure S6. Targeting Pin1 Synergizes with Immunochemotherapy to Induce Human PDAC Organoid Apoptosis, Related to Figure 6
(A) Pin1 inhibitors synergize with GEM to induce human PDAC organoid apoptosis. Pin1i-2 pre-treated PDAC2 organoids were treated with GEM, followed by examining organoid apoptosis at different concentrations for 24 hrs and analyzing synergy score of Pin1i-2 and GEM using Synergy finder. We have tested 2 different patient-derived PDAC organoids (PDAC1 and PDAC2 organoid) and obtained consistent results. Subsequent experiments were shown with one organoid (PDAC2 organoid) with high expression of PD-L1 and ENT.
(B-C) ENT1 KD impairs the synergic effect of Pin1i and GEM. Control (DMSO) or Pin1i-2 (5 μM) pre-treated ENT1 WT or KD PDAC2 organoids were treated with control (PBS) or GEM (25 nM), followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 24 hrs.
(D) Pin1 inhibitors synergize with αPD1 to induce human PDAC organoid apoptosis. Pin1i-2 pre-treated PDAC2 organoids were co-cultured with activated human PBMCs, and then treated with αPD1, followed by examining organoid apoptosis at different concentrations for 40 hrs and analyzing synergy score of Pin1i-2 and αPD1 using Synergy finder.
(E-F) PD-L1 KD impaired the synergic effect of Pin1i and αPD1. Control (DMSO) or Pin1i-2 (5 μM) pre-treated PD-L1 WT or KD PDAC2 organoids were co-cultured with activated human PBMCs, and then treated with control (IgG) or αPD1 (200 μg/ml), followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 40 hrs.
(G-J) Pin1 inhibitors synergize with 5-FU or αPD1 to induce human PDAC organoid apoptosis. Control (DMSO), Pin1i-1 (10 μM) or Pin1i-2 (5 μM) pre-treated PDAC2 organoids were treated with control (DMSO) or 5-FU (25 nM), followed by examining PDAC organoid apoptosis using time lapse imaging for 24 hrs (G, H), or were co-cultured with activated human PBMCs, and then treated with control (IgG) or αPD-L1 (200 μg/ml), followed by examining PDAC organoid apoptosis using time lapse imaging for 40 hrs (I, J).
(K and L) Pin1 inhibitors synergize with immunochemotherapy (GEM + αPD1) to induce human PDAC organoid apoptosis. Control (DMSO) or Pin1i pre-treated PDAC2 organoids were co-cultured with activated human PBMCs, and then treated with control (PBS and/or IgG), GEM and/or αPD1, followed by assaying PDAC organoid apoptosis for different times at constant concentrations (Pin1i-1 10 μM, Pin1i-2 5 μM, GEM 10 nM, and αPD1 100 μg/ml).
(M) KPC tumor-bearing mouse derived CD8+ T-cells induce KPC organoid apoptosis. KPC mouse PDAC tumor derived organoids were co-cultured with/without activated CD8+ T-cells derived from KPC tumor-bearing mice or their tumor-free controls, followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 40 hrs.
(N-O) Pin1 inhibitors synergize with GEM and αPD1 to induce KPC PDAC organoid apoptosis. Control (DMSO) or Pin1i-2 pre-treated KPC tumor derived organoids were co-cultured with KPC tumor-bearing mouse derived activated CD8+ T-cells, and then treated with control (PBS + IgG), or GEM and αPD1, followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 24 hrs at constant concentrations (Pin1i-2 5 μM, GEM 10 nM, and αPD1 200 μg/ml).
Scale bars, 100μm (B, E, G, I, K, M, N). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by one-way ANOVA (C, F, H, J, O) or student’s t test (L).
Figure S5. Pin1 promotes GEM Resistance and Reduces the Expression of PD-L1 and ENT1 at the Cell Surface of PDAC Cells, Related to Figures 4, 5 and 6
(A and B) Pin1 inhibition increases the expression of PD-L1 or ENT1 notably at the cell surface in PDTX mice. PDTX mice were treated with vehicle, Pin1i-1, or Pin1i-2 for 4 weeks, followed by IF for Pin1 (red), DAPI (blue), and PD-L1 (green) (A), or ENT1 (green) (B) (n = 5). White arrows point to PD-L1 (A) and ENT1 (B) at the cell surface.
(C-E) Pin1 overexpression is correlated with reduced PD-L1 or ENT1 levels in human PDAC tissues. Human PDAC tissues were subjected to IHC for Pin1 and PD-L1 (n = 53) (C), or for Pin1 and ENT1 (n=46) (D), followed by semi-quantifying their expression as high or low and examining their correlations by Pearson’s chi-square test (PD-L1: p=0.013, ENT1: p=0.008) (E).
(F) Pin1 inhibition suppresses mRNA expression of Pin1, ENT1 and PD-L1 in human PDAC cells. PDAC2 cells were treated with vehicle (DMSO), Pin1i-1 (10 μM) or Pin1i-2 (5 μM) for 72 hrs, followed by RT-PCR.
(G) Both PD-L1 and ENT levels are regulated by the lysosome-dependent proteolysis. PDAC2 cells were treated with 3-MA (15 mM), Bafilomycin A1 (30 nM), Chloroquine (20 μM), MLN4929 (1 μM), or MG132 (5 μM) for 12 hrs, followed by analyzing PD-L1 and ENT1 levels using IB.
(H) A schematic for the structural domains of HIP1R and the location of only one putative Pin1 recognition Ser929-Pro motif that is identical in humans and mice.
(I) Cycloheximide (CHX) chase assay shows the degradation of HIP1R in HIP1R WT or S929A PDAC2 cells.
(J-K) Pin1 KO or HIP1RS929A synergizes with GEM or αPD1 to induce human PDAC organoid apoptosis as well as Pin1i. Pin1i-2 (5 μM) pre-treated PDAC2 organoids or Pin1 KO or HIP1RS929A PDAC2 organoids were treated with GEM (25 nM), followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 48 hrs (J). Pin1i-2 (5 μM) pre-treated PDAC2 organoids or Pin1 KO or HIP1RS929A PDAC2 organoids were co-cultured with activated human PBMCs, and then treated with αPD1 (200 μg/ml), followed by examining apoptotic organoids using time lapse imaging and green fluorescent caspase 3/7 reagent for 40 hrs (K).
Scale bars, 50 μm and 12.5 μm (inset) (A, B), 500 μm (C, D) and 100 μm (J, K). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by student’s t test (A, B) or one-way ANOVA (F, J, K).
Figure S4. Pin1 Promotes Oncogenic Signaling Pathways and Growth of PDAC Cells, Related to Figure 4
(A) ATRA and ATO cooperatively reduce Pin1 and its substrate oncoproteins in human PDAC cells. Primary human PDAC cells (PDAC1 and PDAC2) derived from two different PDAC patients were treated with ATRA, ATO, or ATRA + ATO (Pin1i-1) for 72 hrs, followed by examining Pin1 and its substrate oncoproteins using IB. ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations were shown.
(B and C) Pin1i-1, Pin1 KD or Pin1 KO reduces many Pin1 substrates in oncogenic Kras signaling networks in PDAC cells. PDAC2 cells were treated with Pin1i-1 for 72 hrs (B) or subjected to Pin1 KD or KO (C) followed by examining Pin1 and its substrate oncoproteins in Kras signaling using IB. ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations are shown (B).
(D-F) ATRA, ATO, Pin1i-1 and Pin1i-2 dose-dependently suppress cell growth and ablate Pin1. PDAC cells were treated with ATRA, ATO, Pin1i-1 or Pin1i-2 for 72 hrs, followed by examining cell growth using proliferation assay (D, F) and Pin1 using IB (E). ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations are shown (E).
(G) Pin1 KD inhibits human PDAC organoid growth. Pin1 KD or vector control human PDAC cells were subjected to 3D organoid cultures, followed by assaying organoid growth curve.
(H) Pin1i-2 fails to suppress Pin1 KO cell growth. Pin1-KO or CRISPR control PDAC2 cells were treated with Pin1i-2 for 72 hrs, followed by examining cell growth using proliferation assay (H).
(I-L) Pin1 inhibitors enhance the ability of GEM to inhibit human PDAC organoid growth. PDAC organoids were treated with Pin1i-1 or −2 and/or GEM at various concentrations for 7 days, followed by examining organoid growth using microscope (I, J; PDAC2 organoid) and H&E staining, and organoid proliferation using double IF for Pan-Keratin and Ki67 IF (K, L; PDAC1 organoid, Pin1i-1 10 μM and GEM 10 nM).
Scale bars, 100 µm (G, I) and 100 and 20 μm (inset) (K). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by student’s t test (G) or one-way ANOVA (H, J, L).
Figure S3. Pin1 is Overexpressed in CAFs, and Pin1 Promotes Oncogenic Signaling Pathways, CAF Activation and Crosstalk with Cancer Cells to Enhance Tumor Growth and Malignancy in Human Organoids and PDOXs, Related to Figure 2 and 3
(A-B) Pin1 is overexpressed in cancer cells and CAFs, and correlated with PDAC progression. Quantification of Pin1 expression at different stages of human PDAC progression by deterring Pin1 IHC score. Normal-Adjacent normal (Normal, n=12), Low grade PanIN (Low, n=20), High grade PanIN (High, n=35), PDAC (n=167) (A). Representative images of Co-IF for Pin1 (red) and FAP (green) in human PDAC tissue (B).
(C-F) ATRA and ATO synergistically reduce Pin1 and its substrate oncoproteins, suppress cell growth and induce quiescent phenotype in CAFs, like Pin1 KD. CAF1 and CAF2 cells derived from two different human PDAC patients were treated with ATRA, ATO, or their combination (Pin1i-1) for 72 hrs (C, D, F) or genetically knocked down of Pin1 (E), followed by examining Pin1 and its substrate oncoproteins using IB (C), cell growth using cell proliferation assay (D, E), quiescent phenotype as measured by lipid droplets/CAF cell using BODIPY staining (F). ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations were shown to simplify labeling (C). CAFs were treated with control (DMSO), ATRA (10 μM) or Pin1i-1 (10 μM) (F).
(G and H) Pin1 inhibitors reduce cytokine production in primary human PDAC cells (G) and CAFs (H). Primary PDAC2 or CAF1 cells were treated with Pin1i-1 or −2 for 72 hrs, before assaying IL-6, TGFβ, LIF and CXCL12 using ELISA. Pin1i-1 10 μM (G). ATRA and ATO were used in 10: 1 ratio as Pin1i-1 and only ATRA concentrations were shown (H).
(I-K) Pin1-inhibited or CRISPR KO CAFs fail to promote PDAC growth and invasion in human 3D PDAC organoid direct co-cultures. Control (DMSO) or Pin1i-treated CAF1 (Pin1i-1 10 μM or Pin1i-2 5 μM for 72 hrs), or CRISPER control or Pin KO CAF1, labeled in red using cell tracker red, were cocultured with established human PDAC1 organoids, labeled by GFP (green) (I), followed by time-lapse imaging to visualize their interactions and analyze organoid growth and invasion (J, K).
(L and M) Pin1-KO CAFs fail to promote PDAC tumor progression and proliferation in PDOX mice. Human PDAC1 organoids were orthotopically co-transplanted with or without CRISPER control or Pin1 KO CAF1 into NSG mouse pancreas for 5 weeks, followed by examining histology using H&E staining (L) and cell proliferation using Ki67 IHC (M) derived from PDAC1 organoids (n = 5).
Scale bars, 100 and 20 μm (inset) (B), 100 μm (J), 100 and 50 μm (right panels) (L), and 50 μm (M). Error bars, mean ± s.d.; **p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; multiple comparisons: by student’s t test (E, G) or one-way ANOVA (F, H, K, M).
Figure S2. Targeting Pin1 Disrupts the desmoplastic and/or immunosuppressive TME and Render PDAC Sensitive to GEM in PDTX, PDOX and GDA mice, Related to Figure 1
(A) Pin1 inhibitors render PDAC sensitive to GEM in GDA mice. Overt tumor-bearing GDA mice were treated with vehicle, Pin1i and/or GEM (20 mg/kg) (n = 6), followed by monitoring overall survival by Kaplan–Meier survival analysis. Median survival; vehicle 34 days, GEM 42 days, Pin1i-2 56 days, GEM + Pin1i-2 83.5 days.
(B-H) Pin1 inhibitors render PDAC sensitive to GEM and αPD1 by disrupting the desmoplastic and immunosuppressive TME in GDA mice. Tumor-bearing GDA mice were treated with vehicle, Pin1i and/or αPD1 or GEM (10mg/kg) +αPD1 (G+P), followed by measuring tumor volumes (B), assaying collagen deposition using Sirius Red staining (C), tumor-infiltrated immune cell profiling using flow cytometry (D, E), and tumor-infiltrating immune cell populations, CD8α+ T-cells, FOXP3+ Tregs, Ly6G+CD11b+ Myeloid cells (F), and CD8α+Granzyme B+ CTLs (G) using IF after 4 weeks treatment (n = 5), or measuring body weight after 120 days treatment (Control/No transplantation: n = 3, G+P + Pin1i-1: n = 7, G+P + Pin1i-2: n = 6) (H).
(I) Pin1 inhibitors render PDAC curable by immunochemotherapy in GDA mice. Overt tumor-bearing GDA mice were treated with vehicle, Pin1i-2, GEM (10 mg/kg) + αPD1 (G+P), Pin1i-2 + αPD1, or Pin1i-2 + G+P for up to 150 days and followed by monitoring overall survival by Kaplan–Meier survival analysis up to 250 days (n=8). Median survival; vehicle 36.5 days, G+P 41 days, Pin1i-2 54 days, αPD1 + Pin1i-2 83.5 days, G+P + Pin1i-2 133.5 days (I).
(J) αPD1 renders GDA tumors hyperprogressive in the absence of CD8+ T-cells. Tumor-bearing GDA mice were treated with αCD8α and vehicle, αPD1, Pin1i-2, or αPD1 + Pin1i-2 for 9 days, followed by examining tumor volume.
(K) Pin1 inhibition does not significantly potentiate PTX or αCTLA4 therapy in GDA mice. Tumor-bearing GDA mice were treated with vehicle, Pin1i-2, PTX, αCTLA4, or their combination for 4 weeks, followed by assaying tumor volume.
Scale bars, 200 μm (C), 100 μm (F), and 100 and 25 μm (inset) (G). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by log-rank test (A, I), student’s t test (D), or one-way ANOVA (B, C, E-H, J, K).
Figure S1. Targeting Pin1 Disrupts the desmoplastic and/or immunosuppressive TME and Inhibits Tumor Growth and Progression in PDTX and GDA mice, Related to Figure 1
(A-B) Experimental setup and treatment schedule. Tumor-bearing (>0.5 cm) PDTX, PDOX or GDA mice were treated with Pin1i, GEM and/or αPD1, starting Pin1i 3 days before others unless stated otherwise. Tumor sizes were detected by ultrasound or palpation with electronic caliper (A). Representative ultrasound images of the initial tumor on Day −3 (B).
(C-H) Pin1 inhibitors suppress tumor growth and progression, and desmoplastic TME in PDTX mice. Overt tumor-bearing PDTX mice were treated with vehicle or Pin1i-1 for 4 weeks, followed by assaying tumor growth and volume (C), collagen deposition and cancer cell differentiation using Sirius Red and H&E staining (D), cancer cell proliferation using Ki67 IHC (E), tumor-associated CAF activation and proliferation using double IF for αSMA and Ki67 (F) or platelet-derived growth factor receptor α (PDGFRα) and Ki67 (G), and EMT using double IF for E-cadherin and Vimentin (H) (n = 5). White arrows indicate αSMA and Ki67 double positive CAFs (F), Ki67 and PDGFRα double positive CAFs (G), or Vimentin positive cancer cell (H).
(I and J) Pin1 inhibitors suppress cancer cell proliferation and tumor progression, and desmoplastic and immunosuppressive TME in GDA mice. Tumor-bearing GDA mice were treated with vehicle, Pin1i-1 or −2 for 4 weeks, followed by examining cancer cell differentiation using H&E staining (I), and CAF proliferation using double IF for PDGFRα and Ki67 (J) using IF (n = 5).
(K-N) Pin1 inhibitors render PDAC sensitive to GEM in PDOX and PDTX mice. Patient-derived PDAC1 organoids and CAFs (K; PDOX mice, n = 5) or patient-derived PDAC tumors (L-N; PDTX mice, n = 6) were orthotopically transplanted into NSG mouse pancreas and when tumors reached 0.5 cm, mice were treated with vehicle, Pin1i and/or GEM (20 mg/kg), followed by measuring tumor volumes (K and L) and liver metastasis (N) after autopsy at 4 weeks after treatment, or monitoring overall survival by Kaplan–Meier survival analysis (M). Median survival; vehicle 36 days, GEM 42.5 days, Pin1i-1 43 days, and Pin1i-1 + GEM 71 days (M).
Scale bars, 500 and 100 μm (right panel) (D), 100 and 25 μm (inset) (F, G), 50 μm (H, J), and 100 μm (I), and 200 μm (N). Error bars, mean ± s.d.; *p < 0.05, **p <0.001, ***p = 0.0001, ****p< 0.0001; n.s., not significant; by student’s t test (C-H, J), one-way ANOVA (K, L, N), or log-rank test (M).
Data Availability Statement
This study did not generate/analyze unique datasets or code.