

ABSTRACT
Noncoding small RNAs (sRNAs) are crucial for the posttranscriptional regulation of gene expression in all organisms and are known to be involved in the regulation of bacterial virulence. In the human pathogen Bordetella pertussis, which causes whooping cough, virulence is controlled primarily by the master two-component system BvgA (response regulator)/BvgS (sensor kinase). In this system, BvgA is phosphorylated (Bvg+ mode) or nonphosphorylated (Bvg− mode), with global transcriptional differences between the two. B. pertussis also carries the bacterial sRNA chaperone Hfq, which has previously been shown to be required for virulence. Here, we conducted transcriptomic analyses to identify possible B. pertussis sRNAs and to determine their BvgAS dependence using transcriptome sequencing (RNA-seq) and the prokaryotic sRNA prediction program ANNOgesic. We identified 143 possible candidates (25 Bvg+ mode specific and 53 Bvg− mode specific), of which 90 were previously unreported. Northern blot analyses confirmed all of the 10 ANNOgesic candidates that we tested. Homology searches demonstrated that 9 of the confirmed sRNAs are highly conserved among B. pertussis, Bordetella parapertussis, and Bordetella bronchiseptica, with one that also has homologues in other species of the Alcaligenaceae family. Using coimmunoprecipitation with a B. pertussis FLAG-tagged Hfq, we demonstrated that 3 of the sRNAs interact directly with Hfq, which is the first identification of sRNA binding to B. pertussis Hfq. Our study demonstrates that ANNOgesic is a highly useful tool for the identification of sRNAs in this system and that its combination with molecular techniques is a successful way to identify various BvgAS-dependent and Hfq-binding sRNAs.
IMPORTANCE Noncoding small RNAs (sRNAs) are crucial for posttranscriptional regulation of gene expression in all organisms and are known to be involved in the regulation of bacterial virulence. We have investigated the presence of sRNAs in the obligate human pathogen B. pertussis, using transcriptome sequencing (RNA-seq) and the recently developed prokaryotic sRNA search program ANNOgesic. This analysis has identified 143 sRNA candidates (90 previously unreported). We have classified their dependence on the B. pertussis two-component system required for virulence, namely, BvgAS, based on their expression in the presence/absence of the phosphorylated response regulator BvgA, confirmed several by Northern analyses, and demonstrated that 3 bind directly to B. pertussis Hfq, the RNA chaperone involved in mediating sRNA effects. Our study demonstrates the utility of combining RNA-seq, ANNOgesic, and molecular techniques to identify various BvgAS-dependent and Hfq-binding sRNAs, which may unveil the roles of sRNAs in pertussis pathogenesis.
KEYWORDS: small RNA, Hfq, BvgAS regulon, pertussis, RNA-seq, ANNOgesic
INTRODUCTION
Small noncoding RNA (sRNA) regulators function as major players in posttranscriptional regulation, affecting a broad spectrum of cellular processes in bacterial physiology (reviewed in references 1–6). In many systems, sRNAs are specifically expressed as cells adapt to environmental conditions, including oxidative stress, DNA damage, iron and nutrition starvation, and lower temperatures. A major class of these sRNAs exert their actions via base pairing with multiple target genes and modulate translation and/or mRNA stability (5, 7). This function can depend upon the RNA chaperone Hfq, a member of the Lsm/Sm family of proteins that bind sRNAs and mRNAs and facilitate their interaction (8–10). Disruption of the hfq gene shows pleotropic physiological effects in Escherichia coli, such as growth defects and increased sensitivity to stresses (11). In addition, hfq mutants exhibit significant virulence attenuation in a wide variety of pathogens, affecting invasion of epithelial cells, secretion of virulence factors, and survival in macrophages, suggesting that Hfq is essential for both physiological fitness and pathogenesis (9, 12). Hfq-dependent sRNAs have also been reported to be involved in bacteria/host interactions through modulating the expression of various virulence determinants, such as those involved in quorum sensing, type III secretion systems (T3SSs), iron transport, and biofilm formation (9, 12–14).
Bordetella pertussis is a Gram-negative bacterial pathogen that causes the highly contagious, acute respiratory illness whooping cough (pertussis). Despite a high coverage rate of vaccination (∼85% worldwide according to WHO [15]), infection by this pathogen has resurged globally, in particular among vaccinated populations in developed countries. This resurgence has been attributed in part to the shorter-lived immunity achieved by the current acellular vaccine relative to the previously used whole-cell vaccine (16). Therefore, there is an urgent need for a comprehensive understanding of the molecular mechanisms of B. pertussis pathogenesis in order to develop better vaccines that engender longer-lasting and more effective immunity.
In B. pertussis, the majority of virulence genes are regulated by BvgAS, a master two-component system composed of BvgS, the histidine sensor kinase, and BvgA, the response regulator (17). Under standard growth conditions at 37°C, BvgS is autophosphorylated and transfers phosphate to BvgA via a phosphorelay. Phosphorylated BvgA (BvgA∼P) dimers then activate virulence-activated genes (vags), such as pertussis toxin (ptx), filamentous hemagglutinin/adhesion (fha), and fimbriae (fim). This is known as the Bvg+ mode (18–20). BvgA∼P dimers also activate the promoter for bvgR, which encodes a cyclic di-GMP (c-di-GMP) phosphodiesterase, which converts c-di-GMP to GMP (21, 22). The secondary messenger c-di-GMP, together with another response regulator RisA, is needed to activate a set of virulence-repressed genes (vrgs) (21). In the presence of BvgR, the level of c-di-GMP dramatically decreases, which in turn leads to a loss of RisA activation. As a consequence, vrgs are repressed in the Bvg+ mode. On the other hand, switching cells to a lower temperature (25°C) or growth in a high concentration (20 to 50 mM) of either nicotinic acid or magnesium sulfate (MgSO4) modulates gene expression, by inducing the Bvg− mode (18–20, 23). Under these modulated conditions, BvgA is not phosphorylated. Without BvgA∼P, BvgR remains inactive, and subsequently, the expression of vrgs increases. These observations suggest sophisticated networks among various regulators during the B. pertussis life cycle.
Our previous transcriptomic study identified >550 genes (∼15% of the B. pertussis genome), which are affected by BvgAS (24). However, this number does not include any RNAs that were omitted from the annotation of the Tohama I reference genome, such as sRNAs and transcripts for small open reading frames (ORFs). Given that Hfq and sRNA regulators play a major role in the virulence of various pathogens (12, 25, 26), and that the hfq deletion mutation in the clinical strain B. pertussis 18323 affects the expression of various virulence genes, including those within the T3SS (14), the involvement of sRNAs in gene regulation during B. pertussis pathogenesis seems likely.
Previous efforts to investigate sRNAs in B. pertussis have been limited. An in silico search for B. pertussis sRNAs using sRNA identification protocols and high-throughput technologies (SIPHT) (27) identified 14 sRNAs, which were then confirmed by Northern blotting (28). More recently, transcriptome sequencing (RNA-seq) analyses combined with terminator 5′-phosphate-dependent exonuclease (TEX)-treated and -untreated RNA samples were used to define the transcriptome architecture of B. pertussis Tohama I, revealing >500 “orphan” RNAs, which presumably include sRNAs; however, in this case, the candidates, which were collected using one condition, were not confirmed experimentally (29). Other work identified one of these candidates, RgtA, as a Bvg− sRNA that is involved in glutamate metabolism (30). Finally, 9 additional sRNAs have been identified during the colonization of mouse trachea by the clinical strain B. pertussis 18323 (31).
Here, we conducted a genome-wide transcriptomic search for sRNAs using RNA-seq data that we previously used to define the BvgAS regulon in B. pertussis Tohama I (24). In this case, wild-type (WT) and ΔbvgAS strains were grown both without and with MgSO4, yielding the nonmodulating Bvg+ mode and the modulating Bvg− mode, respectively. To process the data, we performed a computational analysis using the prokaryotic sRNA search program ANNOgesic, a comprehensive tool for generating bacterial genome annotations based on RNA-seq data (32). ANNOgesic was recently developed to surpass the limitations of current bacterial sRNA search programs. It can detect more than 20 genomics features, including sRNAs. The overall detection rate is ∼80%; specifically, for sRNAs in Helicobacter pylori 26695, Campylobacter jejuni 81116, and E. coli K-12, the sensitivity has reached 90%, 84%, and 90%, respectively.
We picked several candidates to analyze by Northern blots and Hfq-binding studies. Our study demonstrates that combining RNA-seq, ANNOgesic, and molecular techniques is a successful approach for identifying various BvgAS-dependent and Hfq-binding sRNAs, which may reveal the roles of sRNAs in pertussis pathogenesis.
RESULTS
RNA-seq analysis using ANNOgesic reveals sRNA candidates.
To search for B. pertussis sRNAs that were absent from the genomic annotation, we employed ANNOgesic, a recently developed RNA-seq-based genome annotation suite for bacteria and archaea, which contains a module for prokaryotic sRNA detection (32). This toolkit has not been used previously with B. pertussis RNA. The input fastq files were obtained from our previous RNA-seq study, which had identified the BvgAS-dependent regulon (GEO no. GSE98155) (24). Duplicates, collected under each condition, consisted of the following: (i) WT strain grown without MgSO4 (BvgA∼P is present), (ii) WT strain grown with MgSO4 (BvgA∼P is absent), (iii) the ΔbvgAS strain grown without MgSO4, and (iv) the ΔbvgAS strain grown with MgSO4.
To analyze the sequencing data, reads were first trimmed by Trimmomatic and aligned to the B. pertussis Tohama I reference genome with an RNA-seq pipeline, READemption (33), to generate the coverage wiggle (WIG) format files. These files were then processed by ANNOgesic to predict sRNAs from each condition.
In an ANNOgesic analysis “average coverage” is the number of mapped reads for an sRNA divided by the length of the designated gene (sRNA), a value that is similar to reads per kilobase per million (RPKM) mapped reads. As the average coverage value increases, the probability of a valid sRNA increases. Consequently, we used the average coverage value to evaluate the expression of a particular sRNA (see Table S1 in the supplemental material). After eliminating RNAs corresponding to the abundant SsrA and rRNAs (which had ANNOgesic rankings of 1 to 3), predicted sRNAs, whose best average coverage values were more than 30 in any of the 8 samples, were considered significant. This yielded the 143 candidates shown in Table S1.
Previously, various B. pertussis sRNAs present in the Tohama I strain have been reported. Early in silico analyses identified 14 sRNAs, which were confirmed by Northern analyses (28). In a later global transcriptomics analysis of the strain grown under standard laboratory conditions, >500 orphan and antisense RNAs were identified; it was speculated that some of these RNAs would represent sRNA candidates (29). One of these candidates, RgtA, was shown to be dependent on the RNA chaperone Hfq, to be more abundant in the Bvg− mode, and to be involved in glutamate transport (30).
To compare our ANNOgesic candidates to those in these previous reports, we classified any of our sRNA candidates as previously identified if they were transcribed in the same intergenic region with the same directionality. We found that 49 of our 143 candidates were previously identified in the study by Amman et al. (Table S1) (29), while 4 other candidates were found in the study by Hot et al. (28). Consequently, 90 possible sRNAs were newly identified by our ANNOgesic analysis.
BvgAS dependence of predicted sRNAs.
To classify the BvgAS dependence of the 143 sRNAs predicted by ANNOgesic, we compared the average coverage of the following: set (i), WT without or with MgSO4; and set (ii), WT versus the ΔbvgAS strain without MgSO4. We considered a difference of 1.5-fold or greater in the coverage score in the average reads in either set as significant. There are 25 candidates that are more abundant in the presence of BvgA∼P (see Table S2 in the supplemental material) and 53 candidates that are more abundant in the absence of BvgA∼P (see Table S3 in the supplemental material).
As a comparison, we also used an independent method to visually identify sRNAs within our transcriptomic data set using Signal Map (Roche). Twenty sRNAs found by this visual analysis are shown in Fig. S1 in the supplemental material. Within this list, 17 sRNAs, S1 to S17, were among those predicted by ANNOgesic. About one-half of them (S2, S4, S6, S8, S10, S14, and S17) had been predicted, but not confirmed, in previous work. In addition, S9 has been recognized previously as a riboswitch and involved in flavin mononucleotide (FMN) biosynthesis (29, 34, 35), and we confirmed this analysis using the Rfam database (36).
The following three additional candidates shown in Fig. S1E were not predicted by ANNOgesic: S18, which had been previously identified as RgtA, a sRNA whose expression was reported to be activated by the B. pertussis response regulator RisA in the Bvg− mode (30); and S19 and S20, which were previously unrecognized.
Northern analyses confirm ANNOgesic sRNA predictions with high sensitivity.
Twelve of the sRNA candidates in Fig. 1 were chosen for validation by Northern blotting analysis, namely, S1, S2, S3, S4, S8, S9, S10, S11, S12, and S17, which were identified by ANNOgesic; and S18 and S20, which were identified by the Signal Map analysis (Table 1). These candidates were selected because they were carried wholly within intergenic regions (except S4), they represented very highly ranked ANNOgesic sRNAs (S1 to S4 and S17), and/or they represented a mixture of predicted Bvg+, Bvg−, or Bvg-independent transcripts. In addition, the 3′ end of S4 (Fig. S1) was antisense to the 3′ end of gene BP3392, the transposase present within a particular copy of an IS481 element. As hundreds of copies of this element are present within the B. pertussis genome (37), the presence of an sRNA that might regulate transcription from this element could provide an important regulatory feature. Finally, S18 and S20 represented two possible sRNAs that were found by the Signal Map analysis, but they were not predicted by ANNOgesic.
FIG 1.

Northern analyses confirm sRNA candidates. Representative gels show the presence of the indicated sRNAs isolated from cells growing with (+) or without (−) MgSO4, as indicated. In each case, the first lane contains RNA size markers with the indicated sizes in nucleotides. The results of rehybridization of the blot with a primer for 5S RNA as a control is shown underneath. Bands in S1 and S12 that are seen only in the absence or presence of MgSO4 are indicated with a short line and asterisk.
TABLE 1.
sRNA candidates in B. pertussis
| Namea | Rankb | ANNOgesic IDb | Neighboring genesc |
Directiond | Expression modee |
|||
|---|---|---|---|---|---|---|---|---|
| ANNOgesicb | Northern blotf | |||||||
| 5’ ORF | 3’ ORF | |||||||
| S1 | 4 | 212 | <BP2496 | BP2497> | < | Bvg(−) | Bvg(−)/Bvg(+) | |
| S2 | 5 | 128 | BP1548> | dnaX> | > | Bvg(−) | Independent | |
| S3 | 6 | 188 | <ndk | valS> | < | Independent | Independent | |
| S4 | 7 | 296 | BP3391> | <BP3392 | > | Independent | Independent | |
| S5 | 8 | 16 | <phoB | ubiE> | < | Bvg(+) | ||
| S6 | 104 | 294 | <BP3385 | <BP3386 | < | Bvg(−) | ||
| S7 | 11 | 168 | <BP1907 | cysS > | > | Independent | ||
| S8 | 16 | 123 | <BP1496 | thrS> | > | Independent | Bvg(+) | |
| S9 | 17 | 45 | BP0470> | ribB> | > | Bvg(+) | Bvg(+) | |
| S10 | 25 | 298 | <BP3409 | <BP3410 | < | Bvg(+) | Bvg(+) | |
| S11 | 26 | 242 | <BP2886 | BP2887> | > | Bvg(+) | Bvg(+) | |
| S12 | 28 | 17 | ubiE> | BP0162> | > | Independent | Bvg(−)/Bvg(+) | |
| S13 | 30 | 239 | panB> | BP2851> | < | Bvg(−) | ||
| S14 | 34 | 95 | BP1164> | BP1165> | > | Independent | ||
| S15 | 86 | 288 | BP3352> | BP3353> | < | Independent | ||
| S16 | 52 | 42 | <BP0405 | <tRNA Val | < | Bvg(+) | ||
| S17 | 9 | 276 | <BP3151 | <BP3152 | < | Bvg(+) | Bvg(+) | |
| S18 | <BP2735 | BP2736> | < | Bvg(−) | ||||
| S19 | <dnaA | rpmH> | > | |||||
| S20 | <BP0920 | <citB | > | Independent | ||||
Names in bold were confirmed by Northern blot analyses.
ANNOgesic rank, ID, and expression mode (see Table S1). S18, S19, and S20 were not identified by ANNOgesic so this information is not available for them.
Neighboring genes of the sRNA; > corresponds to top strand gene; < corresponds to bottom strand gene.
Directionality of the sRNA transcription; >, top strand; <, bottom strand.
Expression mode of sRNA in RNA-seq or Northern blot analyses. Bvg(+), observed in the presence of BvgA∼P; Bvg(−), observed in the absence of BvgA∼P; (−), not detected; CONST, similar levels in BvgA(+) and BvgA(−).
Northerns were performed for sRNAs in bold.
Northern analyses (probes listed in Table S4 in the supplemental material) confirmed the presence of all the candidates (Fig. 1, Table 1), indicating that ANNOgesic correctly predicted all 10 of its candidates that we tested. This finding indicates that the sensitivity, or the true positive rate, of ANNOgesic for detecting sRNA was quite high, namely, in this case 10 out of 10 or 100%. Although the presence of many different-sized species for S4 was unusual, heterogenous species were also observed in the S4 Signal Map (Fig. S1A).
The Bvg mode assigned by the average coverage of WT ± MgSO4 or WT versus ΔbvgAS (Table 1; Table S2, S3) matched that observed with the Northern blot analyses except for 4 sRNAs, as follows: S1, S2, S8, and S12. However, in the cases of S1 and S12, the situation was complicated by the presence of multiple species, of which some were more abundant in one mode than another (Fig. 1, Table 1). Discrepancies between the visual Signal Map results and ANNOgesic can arise because the Signal Map result is based on a visual examination that does not provide any statistical information, whereas ANNOgesic provides the statistical information of the expression level for each potential sRNA signal.
Gene conservation among B. pertussis sRNAs.
Gene conservation among closely related organisms is an important consideration for the validation of potential sRNAs and has been used as a method to identify sRNAs in various bacterial genomes (38). Consequently, we examined the sequence conservation of the sRNAs that were predicted by ANNOgesic using NCBI BLAST together with the multiple ortholog alignment tool of BioCyc. This strategy revealed that except for S4, all of these sRNAs are highly conserved in sequence and genomic location among B. pertussis, B. bronchiseptica, and B. parapertussis (Fig. 2, Fig. S2A to H). For S2, extensive conservation extends to other Bordetella spp. and even to Achromobacter xylosoxidans within the Alcaligenaceae family (Fig. S2B). In addition, for several of the sRNAs, including the previously unidentified sRNA S3, there is also limited homology outside the Bordetella genus (Fig. 2; Fig. S2A, D, and E). These observations suggest that these sRNAs may have similar functions in various related species. For S4, which is antisense to the 3′ end of a particular IS481 element transposase, conservation was seen only among various B. pertussis genomes.
FIG 2.
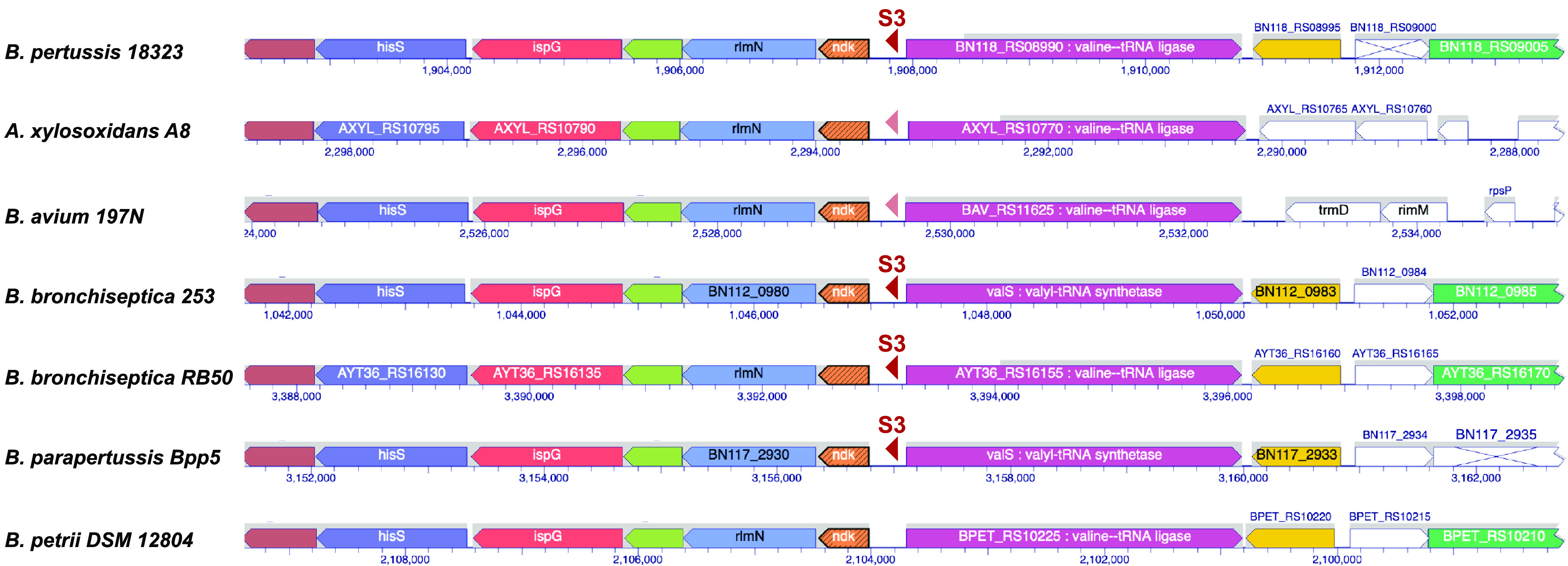
Conservation of the sRNA genomic neighborhoods surrounding S3. The diagram was created by the multigene alignment tool in BioCyc and shows individual genes, their directions, and their chromosomal positions in Bordetella pertussis 18323, Achromobacter xylosoxidans A8, Bordetella avium 197N, Bordetella bronchiseptica 253, Bordetella bronchiseptica RB50, Bordetella parapertussis Bpp5, and Bordetella petrii DSM 12804. Homologous genes are shown in the same color; nonhomologous genes are in white (the position of S3 within the Bordetella pertussis Tohama I genome is given in Fig. S1A). S3 is depicted by the red triangles. Dark red reflects a very high level of homology (B. pertussis 18323 at 100%, B. parapertussis Bpp5 and B. bronchiseptica RB50 at 98%, B. bronchiseptica 253 at 98% [99% coverage]) while light red reflects some sequence conservation (Achromobacter xylosoxidans A8 at 93% [71% coverage] and B. avium 197N at 82% [58% coverage]).
Identification of 3 Hfq-associated sRNAs.
The bacterial RNA chaperone Hfq is a major player in posttranscriptional gene regulation, modulating the stability of mRNAs via base pairing with trans-encoded sRNAs (8, 9). As Hfq is intimately involved in sRNA regulation, we would expect that at least some pertussis sRNAs would be dependent on B. pertussis Hfq (HfqBp). In fact, previous work has demonstrated that an hfq deletion mutation in the clinical strain B. pertussis 18323 affected the expression of various virulence genes, including those associated with the T3SS (14).
Based on this assumption, we investigated whether any of our identified sRNAs were associated with Hfq using a pulldown assay in a strain in which the chromosomal hfq of B. pertussis was tagged with a 3× FLAG epitope, generating HfqBp-FLAG. Hfq of the Bordetella genus lacks a C-terminal extension found in many other proteobacteria, including E. coli, Salmonella enterica, and Yersinia pestis (Fig. 3A). As it was important to confirm that neither the natural truncation nor a C-terminal tag eliminated Hfq function, we investigated the ability of a tagged HfqBp to complement an E. coli hfq deletion strain, which had previously been used to screen the effect of E. coli hfq mutants on sRNAs (39).
FIG 3.

C-terminally tagged HfqBp is active. (A) Amino acid sequences of bacterial Hfq proteins among various species from both beta- and gammaproteobacteria phyla. Identical residues throughout all the species are colored in yellow; identical residues found in a subset of species are colored similarly in either cyan, pink, green, white, or orange. (B) Schematic showing the presence of the translational fusion PBAD-rpoS-lacZ and the tagged hfqBp within the chromosome of the E. coli Δhfq strain SG30200 (57). (C) Plate showing the indicated strains after growth on a MacConkey plate containing 0.0001% arabinose for 24 h.
The E. coli strain used to assay the tagged HfqBp carries a translational fusion of rpoS-lacZ in the chromosome under the control of the arabinose inducible promoter PBAD (Fig. 3B). Complementation by a plasmid-borne hfq is assayed by an increase in the rpoS-lacZ fusion protein, as manifested by an increase in β-galactosidase. This arises because translation of the rpoS mRNA is positively regulated by Hfq together with the sRNAs DsrA, RprA, and ArcZ (40–42).
We assayed the complementation ability of two C-terminally tagged HfqBp proteins, namely, HfqBp-FLAG and His-tagged HfqBp-HIS; the isogenic E. coli WT and hfq deletion strains served as controls. We tested HfqBp containing the FLAG tag as well as one with a His tag to ask whether the nature of the tag would affect function. As expected, the β-galactosidase level was very low in the E. coli hfq deletion mutant (Fig. 3C). In contrast, when either HfqBp-FLAG or HfqBp-HIS was present in the E. coli hfq deletion strain, the level of β-galactosidase was comparable to that obtained in the E. coli WT strain. These results indicated that the C-terminally tagged HfqBp proteins are active in this construct.
Based on this result, we used B. pertussis containing the hfqBp-flag within its chromosome to coimmunoprecipitate RNA with Hfq. Both the WT and the hfqBp-flag strains were grown with and without MgSO4. After coimmunoprecipitation of the HfqBp-FLAG complexes with an anti-FLAG antibody, RNA was extracted for Northern blot analyses to monitor the levels of sRNAs. Using this system, we confirmed the HfqBp dependence of particular species within three sRNAs. The Bvg-independent S4, which contains heterogenous transcripts, had species of ∼350 nucleotide (nt), 200 nt, 150 nt, and 75 nt long that precipitated with tagged Hfq, while the Bvg+ S17 species of ∼50 nt long and the Bvg- S18 (RgtA) species of ∼200 nt long were enriched in the Hfq pulldown (designated by short lines in Fig. 4).
FIG 4.

Northern analyses indicate a direct B. pertussis Hfq interaction with S4, S17, and S18 (RgtA). Gels show the results of a Northern analysis after a pulldown of RNA using an antibody to the FLAG-tag epitope of HfqBp-FLAG. As indicated, RNA was isolated from B. pertussis WT cells (−) or cells containing HfqBp-FLAG (F) grown in the presence or absence of MgSO4. Lane 1, RNA size markers with the indicated sizes in nucleotides; lanes 2 to 5, RNA before pulldown; lanes 6 to 9, RNA after pulldown. Far right gel indicates the control using 5S RNA. Bands that are only seen in the Hfq pulldown are indicated by a short line.
Identification of various genes in Tohama I that are affected by Hfq.
We next investigated whether the expression of particular B. pertussis genes was dependent on Hfq. Total RNA samples, collected from WT or the isogenic hfq deletion strain after growing in media without MgSO4, were analyzed using real-time quantitative PCR (RT-qPCR) (Fig. 5). When comparing Δhfq versus WT, genes with a fold change (FC) of ≥2 or ≤0.5 and a P value of <0.05 were considered to be significant (see Table S5 in the supplemental material). We selected a variety of genes that previous analyses had assigned as Bvg+ mode, Bvg− mode, or Bvg independent (24, 43). We included 5 Bvg+ mode genes that encode known or putative transcriptional regulators (BP2142 [GntR family member], BP3239 and BP2878 [putative LysR family members], brpL [an ECF sigma factor], and BP0319 [a putative regulator]) based on our previous observation that the BvgAS regulon controls dozens of proteins involved in transcription (24). Our list also included 3 classic vrgs (vrg18, vrg24, and vrg73) and 4 proteins involved in transport, as follows BP3831 (amino acid ABC transporter substrate-binding protein), BP3862 (peptide ABC transporter substrate-binding protein), vag8 (autotransporter), and sphB1 (autotransporter subtilisin-like protease). Finally, we included 4 flagellar genes, namely, fliA (sigma factor), fliF (flagellar M-ring protein), fliD (flagellar hook-associated protein 2), and fliO (flagellar protein), based on the recent discovery that B. pertussis can be motile (44) and our previous finding that all of these genes are BvgA independent in B. pertussis (24) despite their being Bvg− in B. bronchiseptica (17).
FIG 5.

RT-qPCR analyses of selected B. pertussis genes from RNA isolated from Δhfq versus WT B. pertussis cells. The mean FC comparing Δhfq/WT (with error bars reflecting ± SE) is shown for each gene. Analyses were performed using biological triplicates except for BP3841, which is the average of 2 biological replicates. Genes in red indicate upregulation in the absence of hfq with a mean FC of >2 and P value of <0.05; genes in green indicate downregulation in the absence of hfq with a mean FC of <0.5 and P value of <0.05. Red and green horizontal lines show the FC values of 2 and 0.5, respectively. Table S5 lists all the determined FC values, the mean FC, the SE, the t statistic, and the determined P value for each gene. Genes marked with an asterisk (*) had P values of <0.05. The mode of each gene (Bvg+, Bvg−, or Bvg independent [ind]) is indicated.
Of the 23 B. pertussis Tohama I genes we tested, the absence of HfqBp upregulated mRNA levels of 8 genes with a mean FC of ≥2.0 and downregulated 2 genes with a mean FC of ≤0.5 (Fig. 5, Table S5). For BP3862, encoding a peptide ABC transporter substrate-binding protein, the P value was >0.05 and for BP3841 (not annotated), there was 1 analysis using 2 biological replicates, so statistics could not be performed. Consequently, we could not conclude that these two genes were significantly affected.
The two identified downregulated genes were the Bvg+ genes vag8 (autotransporter) and BP1005 (conserved hypothetical protein). The identified upregulated genes were two Bvg+ mode genes (BP2171 [ccoQ, encoding a Cytochrome c oxidase subunit] and BP2878 [encoding a putative LysR-family transcriptional regulator]) and the Bvg− mode genes vrg24, vrg73, BP3871 (cold shock like protein), and BP3831 (encoding an amino acid ABC transporter substrate binding protein) (Fig. 5). BP3831 is the Bvg− mode gene that has been shown to be a likely target of S18 (RgtA) (29). The finding that BP3831 is affected by an hfq deletion together with our demonstration that S18 (RgtA) interacts directly with Hfq (Fig. 4) suggests the interaction between RgtA and BP3831 mRNA is mediated by Hfq.
We compared these findings to a previous comprehensive study of genes affected by a hfq deletion in the B. pertussis clinical strain 18323 (14). In this case, FCs of Δhfq/WT were determined by microarray analyses in exponentially growing and stationary-phase cells as well as cells at 0 and 12 days after mouse lung infection. Among our significantly affected genes, BP1005 and vag8 were also downregulated under all growth conditions of B. pertussis 18323, and BP3871 and BP3831 were also upregulated during exponential growth of B. pertussis 18323. However, two upregulated genes (BP2171 and BP2878) were downregulated in the clinical strain during exponential growth and at 12 days postinfection, respectively, and two others (vrg24 and vrg73) were not dysregulated under any condition. It appears then that the B. pertussis hfq regulon is sensitive to the particular strain and growth conditions, suggesting myriad ways that sRNAs can influence B. pertussis gene expression.
DISCUSSION
Transcriptomic analysis by ANNOgesic reveals new B. pertussis sRNAs.
While sRNA regulators have been investigated intensively in various bacterial pathogens, B. pertussis has remained an incomplete research area in this regard. A previous study using SIPHT identified 14 sRNAs, which were then confirmed by Northern blotting analyses (28). A later RNA-seq analysis, using samples collected under one condition, defined the transcriptome architecture of B. pertussis Tohama I and revealed >500 orphan and antisense RNAs, which could include sRNAs (29). One of the candidates, RgtA, was confirmed as a Bvg− mode sRNA that is involved in glutamate metabolism (30). Finally, 9 additional sRNAs have been identified during the colonization of mouse trachea by the clinical strain B. pertussis 18323 (31).
In this study, we tested the ability of the recently developed prokaryotic genome annotation toolkit ANNOgesic to correctly identify sRNAs within a B. pertussis RNA-seq data set. This analysis integrates high-throughput RNA sequencing data to predict unannotated features that have not been readily identified previously. In addition to predicting sRNA and sRNA targets from RNA-seq data, ANNOgesic offers a variety of tools for detecting the transcription start site (TSS), processing site, transcript, promoter, terminator, untranscribed region (UTR), small open reading frame (sORF), operon, riboswitch, CRISPR, and circular RNA in bacteria and archaea (32), making it a pioneer in the field. It is modular, has comprehensive documentation, and is easy to use. In recent work, ANNOgesic has been used to help define the transcriptome map of Bacteroides thetaiotaomicron (45).
A recent comparison of different available sRNA prediction tools (46) indicated that three methods (APERO, ANNOgesic, and TLA from RNA-eXpress) outperformed others. Within these three tools, APERO worked for paired-end sequencing data sets only, while TLA was not specific for bacteria. Therefore, ANNOgesic was an excellent choice for sRNA detection in this study. Our results validated this choice. Not only did ANNOgesic provide a high true positive rate on sRNA prediction but also its results inferred correctly the expression mode of those sRNAs in most cases.
Using ANNOgesic, we found 143 putative sRNAs, including 90 candidates that had not been reported previously. Northern blot analyses confirmed all 10 of the sRNA candidates that we tested. Furthermore, our homology searches indicated that 9 of the confirmed candidates are highly conserved within various Bordetella spp. (Fig. 2, Fig. S2), a feature that is consistent with their providing an important function within the Bordetella genus. Overall, we conclude that ANNOgesic is a highly useful tool for the identification and classification of sRNAs in this system. We expect that the vast majority of the 90 previously unrecognized B. pertussis sRNAs that were predicted by ANNOgesic will be valid.
Despite this success, the program did miss RgtA (S18), which was identified in a previous study (30), and S19 and S20, which we found by a visual analysis of our data set. It is possible that ANNOgesic missed these RNAs because we set a stringent cutoff of a best average coverage value greater than 30 in at least one sample. In addition, in the case of S19, ANNOgesic may have assigned this RNA as a part of the leader region of rpmH rather than a unique sRNA. For example, various sRNAs, which are transcribed from the leader region (5′ UTR) of genes, such as graL in E. coli and EutX, a part of the riboswitch of eut genes in Enterococcus faecalis, were also not initially recognized as sRNAs (47, 48).
Dependence of B. pertussis sRNAs on Hfq.
The RNA chaperone Hfq is found in multiple bacterial species, from nitrogen-fixing symbionts to Enterobacteriaceae pathogens, and is a major player in posttranscriptional gene regulation, modulating the stability of mRNAs mostly via base-pairing trans-encoded sRNAs (5, 8–10). HfqBp has been shown to play an essential role in the expression of various virulence genes (14, 49), as an isogenic hfq deletion mutation of the B. pertussis clinical strain 18323 produces smaller amounts of toxins (49), and the transcriptional levels of the T3SS and other virulence factors (vag8, bsp22, and tcfA) are downregulated by an hfq deletion (14).
HfqBp (8.8 kDa) is smaller than E. coli Hfq (11.2 kDa) because the C-terminal domain (CTD) of E. coli Hfq is missing in HfqBp and in other members of the betaproteobacteria phyla (Fig. 3A) Nonetheless, previous work has shown that a truncated E. coli Hfq, harboring residues 1 to 65, maintains an active RNA chaperon activity, binding single-stranded RNA similarly to WT (50). In addition, we find that HfqBp is functionally active in E. coli (Fig. 3C). Other work has found that the CTD of E. coli Hfq is essential for the rapid release of the double-stranded RNA products, although its presence does not change the rate of RNA binding, suggesting that the CTD may be important for rapid recycling of Hfq (10, 50). The evolutionary significance for why Hfq proteins within the betaproteobacteria phyla lack the CTD has yet to be revealed.
Using an in vivo pulldown assay, we found 3 sRNAs that interact with HfqBp: S4, which is expressed independently of the Bvg mode; S17, which is enhanced in the Bvg+ mode; and RgtA (S18), which is enhanced in the Bvg− mode. To our knowledge, this is the first demonstration of the direct interaction of any B. pertussis sRNAs with HfqBp. Previous reports have indicated that the expression of RgtA is Hfq dependent (30), and we have confirmed that this sRNA binds directly to HfqBp.
S4 is located just upstream of a particular IS481 in an orientation in which its 3′ end is adjacent to or overlaps the 3′ end of the RNA for this particular element’s transposase (BP3392 in B. pertussis Tohama I, BN118_RS00705 in B. pertussis 18323). B. pertussis contains hundreds of copies of IS481, and it is thought that its transposition has been and continues to be a major factor for B. pertussis gene rearrangement and evolution. S4 RNA is quite heterogenous (Fig. 1), and the 3′ end(s) of S4 have not been precisely mapped, but the RNA-seq data indicate some S4 RNAs may contain 3′ ends that are antisense to the BP3392 transcript (Fig. S1A), suggesting that S4 might be able to regulate expression of the IS481 transposase RNA. Previous work has detected various antisense sRNA signals that appeared to arise from the activities of inward and outward promoters of the IS481 element (29). However, in this case, the S4 transcript is derived from a promoter that is independent of the IS element. These observations, therefore, raise the possibility that S4 might affect the level of the transposase by a different manner.
In the past few years, it has become increasingly apparent that both the Bvg+ and Bvg− modes contribute to the lifestyle of B. pertussis; Bvg+ contributes through the expression of adhesin and virulence factors and the Bvg− through the expression of genes involved in survival, transmission, and/or persistence (24, 51, 52). The known roles of bacterial sRNAs in regulating lifestyle and pathogenesis suggest that these factors will be important determinants for B. pertussis growth under various conditions and during infection. Our work here establishes the use of ANNOgesic as a valuable tool for detecting valid sRNAs as well as the presence of Hfq-binding sRNAs in both the Bvg+ and Bvg− modes. We expect that future work will identify the targets of these sRNAs and how they affect the B. pertussis life cycle.
MATERIALS AND METHODS
Bacterial strains and cell culture.
B. pertussis Tohama I BP536 (53) and its ΔbvgAS derivative have been described (24). To create B. pertussis hfq::3xflag, allelic exchange was performed utilizing an 898-bp fragment containing, in order, 161-bp of the upstream flanking sequence, the entire 234-bp hfq gene, a 99-bp sequence (GTCGACAAGCTTGCGGCCGCACTCGAGGACTACAAAGACGATGACGACAAGGACTACAAAGACGATGACGACAAGGACTACAAAGACGATGACGACAAG) (3× FLAG coding sequence underlined) inserted between the last codon and the stop codon of the hfq gene, and the 404-bp downstream flanking sequence. This fragment was synthesized and cloned between the KpnI and BamHI sites of the allelic exchange plasmid pSS4894 (21) by GenScript USA, Inc. The resulting plasmid pQC2314 was then transformed into E. coli strain RHO3 (54) to generate a donor strain for conjugation. Using wild-type B. pertussis BP536 as a recipient, allelic exchange was carried out as described previously (21) to obtain the B. pertussis strain QC4594 (B. pertussis BP536, hfq::3xflag) which was further verified by PCR/sequencing using primers outside the allelic exchange construct.
To delete hfq in B. pertussis, we used the allelic exchange plasmid pSS4661, which contains a vegetative origin of replication (oriV); an origin of transfer (oriT); the genes encoding antibiotic resistance to ampicillin (bla), kanamycin (kan), bleomycin (ble), and streptomycin (str); a gene for the endonuclease I-SceI driven by Pptx of B. pertussis,; an I-SceI cleavage site; and a multiple cloning site. Using genomic DNA of B. pertussis BP536 as the template, the upstream flanking region (amplified by primers 3533-hfqUSOE-BsaNotI [TATAGGTCTCCGGCCGCGGTGGACGAGGCCTATGTCGACTTC] and 3534-hfqUSIE [TATAGGTCTCCTGATTGCTCATTGGCCAGGCTCCATTTG]) and the downstream flanking region (amplified by primers 3535-hfqDSIE [TATAGGTCTCAATCAGGTGGAAGTTCCCGCTGAATAATC] and 3536-hfqDSOE-BsaBamHI [TATAGGTCTCGGATCCCCCGCTGGCGCTCGACCCGATCCAG]) were digested with BsaI and ligated together into a vector cleaved with NotI and BamHI (BsaI recognition sites are in bold and cohesive ends created by digestion are underlined). The net result was an insertion of a NotI-BamHI fragment, containing flanking sequences of the hfq gene and an in-frame deletion of codons 4 to 71 of the hfq gene itself, inserted between the NotI and BamHI sites of pSS4661, yielding plasmid pQC1798. To perform allelic exchange, plasmid pQC1798 was transformed into the E. coli donor strain S17.1 (55) with selection on LB agar plus 20 μg/ml kanamycin. In a subsequent conjugation, the recipient was B. pertussis BP536 that had been grown for 3 days at 37°C on Bordet Gengou (BG) agar supplemented with 50 μg/ml streptomycin and 50 mM MgSO4 to induce the Bvg− mode, thus preventing Pptx-driven I-SceI expression upon transfer of pQC1798. Plasmid cross-in conjugation was performed by swabbing the E. coli donor strain together with the Bordetella recipient strain onto BG agar supplied with 50 mM MgSO4. After incubation at 37°C for 3 h, bacteria were recovered by swabbing and were reswabbed onto BG agar plus 20 μg/ml kanamycin, 50 μg/ml streptomycin, and 50 mM MgSO4. Exconjugants, obtained after 3 to 4 days of incubation at 37°C, were resteaked onto BG agar plus 50 μg/ml streptomycin but without MgSO4, to allow Pptx-driven I-SceI expression, the cleavage of the I-SceI restriction site, and plasmid cross-out, resulting in B. pertussis strain QC2910 (B. pertussis BP536, Δhfq) which was further verified by PCR using primers outside the allelic exchange construct and sequencing of the PCR product.
To obtain cells for RNA isolation, cells were grown on BG agar (56) containing streptomycin (50 μg/ml) at 37°C in the absence (Bvg+ mode) or in the presence (Bvg− mode) of 50 mM MgSO4. After 3 days, cells were scraped from plates and resuspended in PLB medium (56) with or without MgSO4 to obtain an initial optical density at 600 nm (OD600) between 0.1 and 0.2. Cells (in duplicates) were incubated at 37°C with shaking at 250 rpm until the OD600 reached a value between 0.5 and 0.6. Aliquots were then collected by centrifugation.
The gene encoding hfqBp-flag or hfqBp-his was inserted into the E. coli Δhfq strain SG30200 (57) as follows. Gene fragments harboring hfqBp with either a 3× FLAG or His-tag fused at the C terminus before the hfq stop codon were generated by PCR using primers that were designed to give PCR products with 40 nt upstream and 40 nt downstream, which were then introduced into DJS2814 (MG1655 lacX74 malP::lacIq, Δhfq::trpAterminator-kan-PBAD-ccdB cassette mini λ-tet; gift of Susan Gottesman, National Cancer Institute, Bethesda, MD) to replace the Δhfq::trpAterminator-kan-PBAD-ccdB cassette. Recombinants were selected by growth on arabinose at 37°C (to select against the mini-λ). The resulting mutants, which were verified by PCR and sequencing, yielded either KM3032 (harboring Bp hfq-his) or KM3033 (harboring Bp hfq-flag). After P1 lysates of both KM3032 and KM3033 were prepared, the fragments harboring either Bp hfq-his or Bp hfq-flag were replaced with the Δhfq::cat-sacB cassette in SG30200 (MG1655, Δhfq::cat-sacB, ΔpurA::kan) by P1 transduction, generating the final strains as either KM3040 (MG1655, Bp hfq-his) or KM3041 (MG1655, Bp hfq-flag). The strains were verified by PCR and checked by the loss of the kanamycin cassette. PM1409 (58), which contains E. coli hfq and is otherwise isogenic with SG30200, served as the positive control. It is referred to as WT E. coli in Fig. 3.
Pipeline for sRNA prediction by ANNOgesic.
Illumina reads in fastq format from our previously published RNA-seq analyses (24) were first trimmed by Trimmomatic (59) version 0.36 and then processed by the RNA-seq pipeline READemption (33) version 0.4.5. Reads longer than 12 nt were aligned to the reference genome sequences for B. pertussis Tohama I (GenBank accession no. NC_002929.2) using segemehl (60) version 0.2.0. The subcommands “gene_quanti” and “coverage” in READemption were used to calculate the gene expression.
sRNA predictions were made by ANNOgesic (32) version 0.6.27 using the subcommand “srna.” The input files included the coverage wiggle files generated by READemption, the GFF annotation format, and the reference genome file (NC_002929.2). Default parameters were used, and the predicted sRNAs were ranked by the average coverage among the replicates. The average coverage score equal or greater than 30 in at least one condition was used as the cutoff.
The expression profiles of the sRNAs were designated Bvg+, Bvg−, or Bvg independent based on the average coverage of the WT ± MgSO4 or WT-MgSO4/ΔbvgAS-MgSO4 data sets. An FC of ≥1.5 or ≤0.67 in either data set was designated Bvg+ or Bvg−, respectively.
Visualization of sRNAs by Signal Map.
The transcriptomic data for the previously reported RNA-seq analyses used for this work are available in the NCBI database (GEO no. GSE98155] (24). A transcriptomic analysis of the fastq files generated from these data were first performed using the open source software Rockhopper with the default parameter. This analysis generated a strand-specific and normalized wiggle file from each fastq file. The wiggle files were then converted into the GFF format, and SignalMap software (Roche) was used to visualize with the reference genome annotation (NC_002929.2).
RNA isolation and Northern blot analysis.
Total RNA was isolated by the hot-phenol method as described (61, 62). Briefly, each frozen cell pellet was suspended with 600 μl ice-cold phosphate-buffered saline (PBS) and 100 μl 8× lysis solution (320 mM Na-acetate, 8% SDS, and 16 mM EDTA [pH 8]), and 800 μl UltraPure 25:24:1 phenol/chloroform/isoamyl alcohol (Invitrogen) were added. The mixture was incubated at 65°C for 5 min with maximum shaking, and the aqueous phase was separated by centrifugation. The aqueous phase was then reextracted using 25:24:1 phenol/chloroform/isoamyl alcohol twice. The RNA was precipitated by adding 3 volumes of ethanol to the aqueous phase and chilled at –70°C overnight. The RNA precipitate was collected by centrifugation at 4°C and washed with 70% ethanol. Each RNA pellet was dissolved in diethyl pyrocarbonate (DEPC)-treated water.
For Northern blot analyses, RNA (7 μg) was electrophoresed on 10% polyacrylamide, 7 M urea gels in 1× Tris-borate-EDTA (TBE) buffer at 180 V for ∼1 h. The RNA was then electroblotted (200 V for 1.5 h at 4°C) onto a Zeta probe GT blotting membrane (Bio-Rad). Membranes were cross-linked by UV light, and the RNA was hybridized with biotinylated oligomers (Table S4) (Integrated DNA Technologies [IDT]) overnight at 42°C. Chemiluminescent signals were visualized with the Amersham Imager 600 (GE Healthcare) (61). Size marker lanes contained the Biotinylated sRNA ladder (Kerafast). Northern analyses were performed at least in duplicate.
RT-qPCR analysis.
Real-time PCR analyses were performed as previously described (24). Oligonucleotides used to amplify target genes and the internal control are available upon request. In each RT-qPCR analysis, which contained biological duplicates or triplicates as indicated in Table S5, the cycle in which fluorescence was detected (the quantitation cycle [Cqgene]) was observed for the gene of interest as well as the Cq of the reference gene rpoD (CqrpoD); the ΔCq was determined as Cqgene − CqrpoD; the ΔΔCq was determined as ΔCq for the RNA from the Δhfq strain − ΔCq for the RNA from the WT strain; the fold change (FC) was determined as 2-ΔΔCq. For the statistical analyses, the mean FC and standard error (SE) from the values for a particular gene were first determined using the website https://ncalculators.com/statistics/standard-error-calculator.htm. The t statistic was calculated as the mean FC/SE. The P value was then determined using the one-tailed hypothesis and a “degrees of freedom” value of n − 1 for the number of replicates (n) performed for a gene analyzed in a single RT-qPCR analysis and (n1 − 1) + (n2 − 1) + … for genes in which multiple analyses were performed using the website https://www.socscistatistics.com/pvalues/tdistribution.aspx. These values are listed in Table S5.
Hfq coimmunoprecipitation.
Wild-type B. pertussis Tohama I BP536 and the 3× FLAG-tagged hfq strain (QC4594) were collected by centrifugation at 4°C to an equivalence of an OD600 of 40 to 50. Pellets were stored at −80°C. Lysates were obtained as previously described (39, 63). Briefly, cells were resuspended with 0.8 ml of lysis buffer (20 mM Tris-Cl [pH 8], 150 mM NaCl, 1 mM MgCl2, 1 mM DTT, 1× cOmplete protease inhibitor cocktail [Roche], and 0.05 U SUPERase.In [ThermoFisher]) and then disrupted with 1.2 ml glass beads (0.3 mm diameter; Sigma) by sonication. Cleared lysates were collected by centrifugation in an Eppendorf microcentrifuge at 14,000 rpm at 4°C for 10 min. The supernatant was mixed with 50 μl EZview red anti-FLAG M2 affinity gel (Sigma) by rotation for 1 h at 4°C. The beads were collected by centrifugation and then washed sequentially with low salt buffer (20 mM Tris-Cl [pH 8], 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, and 0.1% SDS), high salt buffer (20 mM Tris-Cl [pH 8], 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, and 0.1% SDS), LiCl buffer (10 mM Tris-Cl [pH 8], 250 mM LiCl, 1 mM EDTA, 1% deoxycholate, and 1% NP-40), and twice with Tris-EDTA (TE) buffer (10 mM Tris-Cl [pH 8] and 1 mM EDTA). RNA was eluted from the beads by the addition of 0.3 ml of elution buffer (100 mM NaHCO3 and 1% SDS, containing 0.05 U SUPERase.In). The solution was extracted with 25:24:1 phenol/chloroform/isoamyl alcohol and precipitated with ethanol, and the RNA was dissolved in diethyl pyrocarbonate (DEPC)-treated water. For the Northern analysis, 5.5 μg and 0.16 μg of the supernatant and eluant were used, respectively, and samples were treated as described above. Northern analyses were performed at least in duplicate.
ACKNOWLEDGMENTS
We thank Susan Gottesman for the strains SG30200, PM1409, and DJS2814; Rawan Elaksher and Nadim Majdalani for technical assistance; and Bokyung Son, Melissa Arroyo-Mendoza, Abraham Correa-Medina, Rawan Elaksher, and Nadim Majdalani for helpful discussions. We also thank the reviewers and Maxwell Lee (Center for Cancer Research, National Cancer Institute) for help with the statistical analyses needed for the RT-qPCR data.
This work was funded by the intramural program of the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (K.M., M.S., K.Y., C.M., D.D.K., J.L., L.G.K., and D.M.H.); by the National Cancer Institute (C.-H.T.); by the Food and Drug Administration, Center for Biologics Evaluation and Research (Q.C. and S.S.); and SFB/Transregio 34 (CRC-TRR34), grant number 16524344 (S.-H.Y. and K.U.F.)
The work utilized the computational resources of the NIH HPC Biowulf cluster.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Deborah M. Hinton, Email: dhinton@helix.nih.gov.
Kathryn T. Elliott, College of New Jersey
REFERENCES
- 1.Dutta T, Srivastava S. 2018. Small RNA-mediated regulation in bacteria: a growing palette of diverse mechanisms. Gene 656:60–72. doi: 10.1016/j.gene.2018.02.068. [DOI] [PubMed] [Google Scholar]
- 2.Papenfort K, Vanderpool CK. 2015. Target activation by regulatory RNAs in bacteria. FEMS Microbiol Rev 39:362–378. doi: 10.1093/femsre/fuv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bossi L, Figueroa-Bossi N. 2016. Competing endogenous RNAs: a target-centric view of small RNA regulation in bacteria. Nat Rev Microbiol 14:775–784. doi: 10.1038/nrmicro.2016.129. [DOI] [PubMed] [Google Scholar]
- 4.Nitzan M, Rehani R, Margalit H. 2017. Integration of bacterial small RNAs in regulatory networks. Annu Rev Biophys 46:131–148. doi: 10.1146/annurev-biophys-070816-034058. [DOI] [PubMed] [Google Scholar]
- 5.Hor J, Matera G, Vogel J, Gottesman S, Storz G. 2020. Trans-acting small RNAs and their effects on gene expression in Escherichia coli and Salmonella enterica. EcoSal Plus 9:1–24. doi: 10.1128/ecosalplus.ESP-0030-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holmqvist E, Wagner EGH. 2017. Impact of bacterial sRNAs in stress responses. Biochem Soc Trans 45:1203–1212. doi: 10.1042/BST20160363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogel J, Luisi BF. 2011. Hfq and its constellation of RNA. Nat Rev Microbiol 9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chao Y, Vogel J. 2010. The role of Hfq in bacterial pathogens. Curr Opin Microbiol 13:24–33. doi: 10.1016/j.mib.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Santiago-Frangos A, Woodson SA. 2018. Hfq chaperone brings speed dating to bacterial sRNA. Wiley Interdiscip Rev RNA 9:e1475. doi: 10.1002/wrna.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsui HC, Feng G, Winkler ME. 1996. Transcription of the mutL repair, miaA tRNA modification, hfq pleiotropic regulator, and hflA region protease genes of Escherichia coli K-12 from clustered Esigma32-specific promoters during heat shock. J Bacteriol 178:5719–5731. doi: 10.1128/jb.178.19.5719-5731.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papenfort K, Vogel J. 2014. Small RNA functions in carbon metabolism and virulence of enteric pathogens. Front Cell Infect Microbiol 4:91. doi: 10.3389/fcimb.2014.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pusic P, Tata M, Wolfinger MT, Sonnleitner E, Haussler S, Blasi U. 2016. Cross-regulation by CrcZ RNA controls anoxic biofilm formation in Pseudomonas aeruginosa. Sci Rep 6:39621. doi: 10.1038/srep39621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bibova I, Hot D, Keidel K, Amman F, Slupek S, Cerny O, Gross R, Vecerek B. 2015. Transcriptional profiling of Bordetella pertussis reveals requirement of RNA chaperone Hfq for Type III secretion system functionality. RNA Biol 12:175–185. doi: 10.1080/15476286.2015.1017237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.World Health Organization. 2015. Weekly epidemiological record. World Health Organization, Geneva, Switzerland. https://www.who.int/wer/2015/wer9035.pdf?ua=1. [Google Scholar]
- 16.Tan T, Dalby T, Forsyth K, Halperin SA, Heininger U, Hozbor D, Plotkin S, Ulloa-Gutierrez R, Wirsing von Konig CH. 2015. Pertussis across the globe: recent epidemiologic trends from 2000 to 2013. Pediatr Infect Dis J 34:e222–e232. doi: 10.1097/INF.0000000000000795. [DOI] [PubMed] [Google Scholar]
- 17.Cummings CA, Bootsma HJ, Relman DA, Miller JF. 2006. Species- and strain-specific control of a complex, flexible regulon by Bordetella BvgAS. J Bacteriol 188:1775–1785. doi: 10.1128/JB.188.5.1775-1785.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lacey BW. 1960. Antigenic modulation of Bordetella pertussis. J Hyg (Lond) 58:57–93. doi: 10.1017/s0022172400038134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Decker KB, James TD, Stibitz S, Hinton DM. 2012. The Bordetella pertussis model of exquisite gene control by the global transcription factor BvgA. Microbiology (Reading) 158:1665–1676. doi: 10.1099/mic.0.058941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulanger A, Chen Q, Hinton DM, Stibitz S. 2013. In vivo phosphorylation dynamics of the Bordetella pertussis virulence-controlling response regulator BvgA. Mol Microbiol 88:156–172. doi: 10.1111/mmi.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Q, Ng V, Warfel JM, Merkel TJ, Stibitz S. 2017. Activation of Bvg-repressed genes in Bordetella pertussis by RisA requires cross talk from noncooperonic histidine kinase RisK. J Bacteriol 199:e00475-17. doi: 10.1128/JB.00475-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Romling U, Galperin MY, Gomelsky M. 2013. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Merkel TJ, Barros C, Stibitz S. 1998. Characterization of the bvgR locus of Bordetella pertussis. J Bacteriol 180:1682–1690. doi: 10.1128/JB.180.7.1682-1690.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon K, Bonocora RP, Kim DD, Chen Q, Wade JT, Stibitz S, Hinton DM. 2017. The BvgAS Regulon of Bordetella pertussis. mBio 8:e01526-17. doi: 10.1128/mBio.01526-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Chavarria LC, Vadyvaloo V. 2015. Yersinia pestis and Yersinia pseudotuberculosis infection: a regulatory RNA perspective. Front Microbiol 6:956. doi: 10.3389/fmicb.2015.00956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhatt S, Egan M, Jenkins V, Muche S, El-Fenej J. 2016. The tip of the iceberg: on the roles of regulatory small RNAs in the virulence of enterohemorrhagic and enteropathogenic Escherichia coli. Front Cell Infect Microbiol 6:105. doi: 10.3389/fcimb.2016.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Livny J, Teonadi H, Livny M, Waldor MK. 2008. High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS One 3:e3197. doi: 10.1371/journal.pone.0003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hot D, Slupek S, Wulbrecht B, D'Hondt A, Hubans C, Antoine R, Locht C, Lemoine Y. 2011. Detection of small RNAs in Bordetella pertussis and identification of a novel repeated genetic element. BMC Genomics 12:207. doi: 10.1186/1471-2164-12-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amman F, D'Halluin A, Antoine R, Huot L, Bibova I, Keidel K, Slupek S, Bouquet P, Coutte L, Caboche S, Locht C, Vecerek B, Hot D. 2018. Primary transcriptome analysis reveals importance of IS elements for the shaping of the transcriptional landscape of Bordetella pertussis. RNA Biol 15:967–975. doi: 10.1080/15476286.2018.1462655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keidel K, Amman F, Bibova I, Drzmisek J, Benes V, Hot D, Vecerek B. 2018. Signal transduction-dependent small regulatory RNA is involved in glutamate metabolism of the human pathogen Bordetella pertussis. RNA 24:1530–1541. doi: 10.1261/rna.067306.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hiramatsu Y, Suzuki K, Motooka D, Nakamura S, Horiguchi Y. 2020. Expression of small RNAs of Bordetella pertussis colonizing murine tracheas. Microbiol Immunol 64:469–475. doi: 10.1111/1348-0421.12791. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32.Yu SH, Vogel J, Forstner KU. 2018. ANNOgesic: a Swiss army knife for the RNA-seq based annotation of bacterial/archaeal genomes. Gigascience 7:giy096. doi: 10.1093/gigascience/giy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forstner KU, Vogel J, Sharma CM. 2014. READemption—a tool for the computational analysis of deep-sequencing-based transcriptome data. Bioinformatics 30:3421–3423. doi: 10.1093/bioinformatics/btu533. [DOI] [PubMed] [Google Scholar]
- 34.Winkler WC, Cohen-Chalamish S, Breaker RR. 2002. An mRNA structure that controls gene expression by binding FMN. Proc Natl Acad Sci USA 99:15908–15913. doi: 10.1073/pnas.212628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pedrolli D, Langer S, Hobl B, Schwarz J, Hashimoto M, Mack M. 2015. The ribB FMN riboswitch from Escherichia coli operates at the transcriptional and translational level and regulates riboflavin biosynthesis. FEBS J 282:3230–3242. doi: 10.1111/febs.13226. [DOI] [PubMed] [Google Scholar]
- 36.Kalvari I, Nawrocki EP, Argasinska J, Quinones-Olvera N, Finn RD, Bateman A, Petrov AI. 2018. Non-coding RNA analysis using the Rfam database. Curr Protoc Bioinformatics 62:e51. doi: 10.1002/cpbi.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MT, Churcher CM, Bentley SD, Mungall KL, Cerdeno-Tarraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O'Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, et al. 2003. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet 35:32–40. doi: 10.1038/ng1227. [DOI] [PubMed] [Google Scholar]
- 38.Wassarman KM, Repoila F, Rosenow C, Storz G, Gottesman S. 2001. Identification of novel small RNAs using comparative genomics and microarrays. Genes Dev 15:1637–1651. doi: 10.1101/gad.901001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moon K, Gottesman S. 2011. Competition among Hfq-binding small RNAs in Escherichia coli. Mol Microbiol 82:1545–1562. doi: 10.1111/j.1365-2958.2011.07907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Majdalani N, Cunning C, Sledjeski D, Elliott T, Gottesman S. 1998. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc Natl Acad Sci USA 95:12462–12467. doi: 10.1073/pnas.95.21.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Majdalani N, Chen S, Murrow J, St John K, Gottesman S. 2001. Regulation of RpoS by a novel small RNA: the characterization of RprA. Mol Microbiol 39:1382–1394. doi: 10.1111/j.1365-2958.2001.02329.x. [DOI] [PubMed] [Google Scholar]
- 42.Mandin P, Gottesman S. 2010. Integrating anaerobic/aerobic sensing and the general stress response through the ArcZ small RNA. EMBO J 29:3094–3107. doi: 10.1038/emboj.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Q, Stibitz S. 2019. The BvgASR virulence regulon of Bordetella pertussis. Curr Opin Microbiol 47:74–81. doi: 10.1016/j.mib.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 44.Hoffman CL, Gonyar LA, Zacca F, Sisti F, Fernandez J, Wong T, Damron FH, Hewlett EL. 2019. Bordetella pertussis can be motile and express flagellum-like structures. mBio 10:e00787-19. doi: 10.1128/mBio.00787-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryan D, Jenniches L, Reichardt S, Barquist L, Westermann AJ. 2020. A high-resolution transcriptome map identifies small RNA regulation of metabolism in the gut microbe Bacteroides thetaiotaomicron. Nat Commun 11:3557. doi: 10.1038/s41467-020-17348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leonard S, Meyer S, Lacour S, Nasser W, Hommais F, Reverchon S. 2019. APERO: a genome-wide approach for identifying bacterial small RNAs from RNA-Seq data. Nucleic Acids Res 47:e88. doi: 10.1093/nar/gkz485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dylewski M, Ćwiklińska M, Potrykus K. 2018. A search for the in trans role of GraL, an Escherichia coli small RNA. Acta Biochim Pol 65:141–149. doi: 10.18388/abp.2018_2562. [DOI] [PubMed] [Google Scholar]
- 48.DebRoy S, Gebbie M, Ramesh A, Goodson JR, Cruz MR, van Hoof A, Winkler WC, Garsin DA. 2014. A riboswitch-containing sRNA controls gene expression by sequestration of a response regulator. Science 345:937–940. doi: 10.1126/science.1255091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bibova I, Skopova K, Masin J, Cerny O, Hot D, Sebo P, Vecerek B. 2013. The RNA chaperone Hfq is required for virulence of Bordetella pertussis. Infect Immun 81:4081–4090. doi: 10.1128/IAI.00345-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santiago-Frangos A, Kavita K, Schu DJ, Gottesman S, Woodson SA. 2016. C-terminal domain of the RNA chaperone Hfq drives sRNA competition and release of target RNA. Proc Natl Acad Sci USA 113:E6089–E6096. doi: 10.1073/pnas.1613053113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trainor EA, Nicholson TL, Merkel TJ. 2015. Bordetella pertussis transmission. Pathog Dis 73:ftv068. doi: 10.1093/femspd/ftv068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Karataev GI, Sinyashina LN, Medkova AY, Semin EG, Shevtsova ZV, Matua AZ, Kondzariya IG, Amichba AA, Kubrava DT, Mikvabia ZY. 2016. [Insertional Inactivation of Virulence Operon in Population of Persistent Bordetella pertussis Bacteria]. Genetika 52:422–430. [PubMed] [Google Scholar]
- 53.Stibitz S, Yang MS. 1991. Subcellular localization and immunological detection of proteins encoded by the vir locus of Bordetella pertussis. J Bacteriol 173:4288–4296. doi: 10.1128/jb.173.14.4288-4296.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopez CM, Rholl DA, Trunck LA, Schweizer HP. 2009. Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomallei. Appl Environ Microbiol 75:6496–6503. doi: 10.1128/AEM.01669-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
- 56.Chen Q, Decker KB, Boucher PE, Hinton D, Stibitz S. 2010. Novel architectural features of Bordetella pertussis fimbrial subunit promoters and their activation by the global virulence regulator BvgA. Mol Microbiol 77:1326–1340. doi: 10.1111/j.1365-2958.2010.07293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang A, Schu DJ, Tjaden BC, Storz G, Gottesman S. 2013. Mutations in interaction surfaces differentially impact E. coli Hfq association with small RNAs and their mRNA targets. J Mol Biol 425:3678–3697. doi: 10.1016/j.jmb.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soper T, Mandin P, Majdalani N, Gottesman S, Woodson SA. 2010. Positive regulation by small RNAs and the role of Hfq. Proc Natl Acad Sci USA 107:9602–9607. doi: 10.1073/pnas.1004435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoffmann S, Otto C, Doose G, Tanzer A, Langenberger D, Christ S, Kunz M, Holdt LM, Teupser D, Hackermuller J, Stadler PF. 2014. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol 15:R34. doi: 10.1186/gb-2014-15-2-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moon K, Gottesman S. 2009. A PhoQ/P-regulated small RNA regulates sensitivity of Escherichia coli to antimicrobial peptides. Mol Microbiol 74:1314–1330. doi: 10.1111/j.1365-2958.2009.06944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Masse E, Escorcia FE, Gottesman S. 2003. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev 17:2374–2383. doi: 10.1101/gad.1127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. 2012. An atlas of Hfq-bound transcripts reveals 3' UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 31:4005–4019. doi: 10.1038/emboj.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download SPECTRUM00044-21_Supp_1_seq9.xlsx, XLSX file, 0.1 MB (59.7KB, xlsx)
Supplemental material. Download SPECTRUM00044-21_Supp_2_seq10.pdf, PDF file, 2.1 MB (2.2MB, pdf)