

Abstract
Although sex is now accepted as a ubiquitous and ancestral feature of eukaryotes, direct observation of sex is still lacking in most unicellular eukaryotic lineages. Evidence of sex is frequently indirect and inferred from the identification of genes involved in meiosis from whole genome data and/or the detection of recombination signatures from genetic diversity in natural populations. In haploid unicellular eukaryotes, sex-related chromosomes are named mating-type (MTs) chromosomes and generally carry large genomic regions where recombination is suppressed. These regions have been characterized in Fungi and Chlorophyta and determine gamete compatibility and fusion. Two candidate MT+ and MT− alleles, spanning 450–650 kb, have recently been described in Ostreococcus tauri, a marine phytoplanktonic alga from the Mamiellophyceae class, an early diverging branch in the green lineage. Here, we investigate the architecture and evolution of these candidate MT+ and MT− alleles. We analyzed the phylogenetic profile and GC content of MT gene families in eight different genomes whose divergence has been previously estimated at up to 640 Myr, and found evidence that the divergence of the two MT alleles predates speciation in the Ostreococcus genus. Phylogenetic profiles of MT trans-specific polymorphisms in gametologs disclosed candidate MTs in two additional species, and possibly a third. These Mamiellales MT candidates are likely to be the oldest mating-type loci described to date, which makes them fascinating models to investigate the evolutionary mechanisms of haploid sex determination in eukaryotes.
Keywords: sex-determining chromosome; recombination suppression; mating types; Chlorophyta; picoeucaryote, Mamiellophyceae
Significance
Direct evidence of sexual reproduction is difficult to observe in many unicellular eukaryotes, whereas indirect evidence relies on gene content or recombination signatures. Here, we report the gene content of two candidate mating-type loci in a unicellular phytoplanktonic eukaryote. Identification and phylogenetic analyses of the gametologs shared between the two mating types suggest signatures of trans-specific evolution, that is, an ancient divergence, prior to the speciation events within the Ostreococcus lineage. The divergence between gametologs can be leveraged to assign strains from distantly related species to each of the two mating types. Thus, they are likely to be the oldest mating-type loci described to date, which makes them fascinating models to investigate the evolutionary mechanisms of haploid sex determination in eukaryotes.
Introduction
Meiotic sex and its associated intra- and interchromosomal recombination events are considered ubiquitous, ancestral features of eukaryotes (Speijer et al. 2015). Across the eukaryotic tree of life, meiotic sex has been reported in many algal lineages (reviewed in Umen and Coelho 2019), such as chlorophytes (Sager and Granick 1954; Suda et al. 1989; Fučíková et al. 2015), bacillariophytes (Chepurnov et al. 2004), chlorarachniophytes (Beutlich and Schnetter 1993), cryptophytes (Hill and Wetherbee 1986; Kugrens and Lee 1988), cyanidiophytes (Malik et al. 2007), dinoflagellates (Pfiester 1989), and euglenoids (Ebenezer et al. 2019).
There have been intense efforts to study sex-determining mechanisms and underlying genetic make-up in multicellular animals and plants (Bachtrog et al. [2014] for a review). However, less is known about sex-determining mechanisms in microbial eukaryotes. Ancestral sex-determining mechanisms have evolved in unicellular eukaryotes, so that “it is clear that the evolution of different sexes in its most basic form is represented by the evolution of mating types” (Hoekstra 1987). Obviously, it is less straightforward to identify morphological differences between sexes in microorganisms than in macro-organisms. The term “mating type” describes different “sexual types” in unicellular eukaryotes, and was first coined by Tracy Sonneborn. He used this term to indicate that only certain lines (or “stocks”) of the ciliate Paramecium aurelia mated with each other, but never with themselves (Sonneborn 1937). He noted that the Paramecium mating system was “strikingly similar to the sexual differences between gametes in some of the unicellular green alga.” He referred to earlier work by Strehlow (1929) on “plus” and “minus” “sexes” reported in unicellular soil and freshwater green algae from the order Chlamydomonadales. In the Fungal kingdom, there has been a rapidly growing experimental evidence of mating types for many species (reviewed in Billiard et al. [2012]; Wolfe and Butler [2017]), initially in the yeasts Saccharomyces cerevisiae(Astell et al. 1981) and Neurospora crassa(Staben and Yanofsky 1990). Mating types were identified later in the green algal lineage, as in Chlamydomonas reinhardtii(Ferris et al. 2002), and across the eukaryotic tree of life (reviewed in Umen and Coelho [2019]). Interestingly, the evolutionary link between mating types and male and female sexes has been unambiguously demonstrated in the volvocine green lineage (Nozaki et al. 2006; Ferris et al. 2010; Hamaji et al. 2018). However, the origin of mating types remains unresolved. Three main hypotheses have been formulated for the origin and maintenance of this genetic setup, which requires outcrossing. First, it may mediate the prevention of genetic conflicts (Hurst and Hamilton 1992); second, the prevention of haploid selfing, that is mating among clonal cells (Billiard et al. 2011, 2012). A third proximate hypothesis is that this genetic system has evolved from a cell signaling system for partner recognition and pairing by producing recognition/attraction molecules and their receptors, as initially suggested by Hoekstra (1987) and expanded by Hadjivasiliou and Pomiankowski (2016). Common themes of mating-type loci were quickly noticed: they often come in two types (with notable exceptions in fungi, e.g., Billiard et al. [2011] for a review) with hardly any sequence conservation. Although orthologous genes may be identified between the two mating-type regions, gametologs, mating-type regions share little synteny as a consequence of rearrangements and insertion of repetitive DNA (Ferris and Goodenough 1997; Lengeler et al. 2002; Ferris et al. 2010; Badouin et al. 2015; Fontanillas et al. 2015; Hamaji et al. 2016; Geng et al. 2018). Moreover, mating-type loci may also experience recombination suppression both in diploid sexual system, as well as in haploid sexual systems and the UV sex chromosomes (Bachtrog et al. 2011; Coelho et al. 2018). Recombination suppression may be stepwise and thus generate “evolutionary strata” of differentiation between the two mating types (Hartmann et al. [2021] for a review in Fungi). The consequence of recombination suppression are manifold (Charlesworth and Charlesworth 2000; Charlesworth 2016) and may include a higher probability of fixation of deleterious mutations, massive rearrangements, which may be associated to lower gene density (Yamamoto et al. 2021), GC composition changes, as well as differential gene expression (Ma et al. 2020). GC composition results from the balance between mutation biases, selection, and GC-biased gene conversion (Galtier et al. 2001), a molecular process linked to recombination. Therefore, regions with suppressed recombination are expected to display a significant lower GC content as compared with recombining regions, and a 4–10% lower GC content over the mating-type locus has been reported in the mating-type region of four species of volvocine algae (Hamaji et al. 2018).
The genomic features associated to mating-type regions may thus guide the identification of candidate mating-type loci in lineages in which genomic data are available, whereas the experimental conditions eliciting syngamy and meiosis have not yet been found, precluding experimental validation. Although there is no direct evidence of sexual reproduction in the cosmopolitan marine picoeukaryote Ostreococcus tauri (Mamiellophyceae, Chlorophyta) there are three lines of indirect evidence for sexual reproduction (Grimsley et al. 2010). The first line of evidence comes from screening the whole genome sequence for genes encoding proteins involved in meiosis. These proteins have been described in all Mamiellophyceae species for which full genomes sequences are available, including O. tauri (Derelle et al. 2006), O. lucimarinus (Palenik et al. 2007), Micromonas pusilla, Micromonas commoda (Worden et al. 2009), and in Bathycoccus spp. metagenomes from the Arctic (Joli et al. 2017). The second line of evidence comes from population polymorphism data that indicate inter- and intrachromosomal recombination (Grimsley et al. 2010). Indeed, when sequencing can be performed in several strains from the same population, analyses of the polymorphism spectrum allow the estimation of the frequency of sex in natural populations (Tsai et al. 2008; Grimsley et al. 2010; Drott et al. 2020; Hasan and Ness 2020; Koufopanou et al. 2020). Finally, the third line of evidence comes from a population genomic analysis that demonstrated the existence of a candidate mating-type loci (450 and 650 kb) in O. tauri (Grimsley et al. 2010). Ostreococcus tauri RCC4221 was suggested to represent the candidate “minus” mating type (hereafter MT−) together with O. lucimarinus CCE9901, because of the presence of a gene encoding for a plant-specific transcription factor from the RWP-RK gene family (GF) (Worden et al. 2009). This GF includes the “sex-determining gene” (minus dominance MID) of minus mating-type loci in Volvocales algae (Ferris and Goodenough 1997; Umen 2011). The candidate opposite mating type (hereafter MT+) was identified from the genome analysis of 12 O. tauri strains lacking sequence homology with O. tauri RCC4221 over the 650-kb region. These strains also lacked a gene containing an RWP-RK domain (Blanc-Mathieu et al. 2017). Phylogenetic analysis of five gametologs revealed that O. tauri MT− and MT+ genes clustered with different Ostreococcus species of the same mating type, respectively. This suggests that mating-type differentiation predates speciation within Ostreococcus, suggesting that Ostreococcus MT+ and MT− are remarkably ancient. However, the total number of gametologs, their synteny, and sequence conservation among Mamiellales and Mamiellophyceae remains unknown.
Here, we investigated the architecture and phylogenetic profiles of the MT+ and MT− alleles to unfold their evolutionary history. We analysed the gene set of the two candidate mating-type loci, and identified the complete set of gametologs between them. This allowed us to define the set of orthologous genes located inside each of the available candidate MT loci in Mamiellales. This data set was then leveraged: 1) to investigate the presence of evolutionary strata, 2) construct gene genealogies to search for trans-specific evolution signatures, and 3) identify the opposite mating types from additional Mamiellophyceae sequence data. This allowed to trace back the age of the divergence of the MT+ and MT− alleles in this early diverging branch of the green lineage.
Results
Sorting Out GFs in O. tauri MT according to Their Prevalence across Species
The GC content can be used as a predictor of recombination rates in genomes undergoing GC-biased gene conversion (Meunier and Duret 2004; Charlesworth et al. 2020), and it was suggested that there is an inverse relationship between chromosome length and GC content, which is consistent with GC-biased GC conversion in Ostreococcus(Jancek et al. 2008). The genome-wide spontaneous mutation rate is GC->AT biased, which is consistent with a mechanism like GC-biased gene conversion that could explain the difference between the observed 0.60 GC frequency in the genome and the expected equilibrium 0.36 GC frequency under mutation bias (Krasovec et al. 2017). The detection of the sharp (∼9–17%) decrease in GC content on the big outlier chromosome was used to define MT boundaries in O. tauri RCC4221 (MT−), O. tauri RCC1115 (MT+), and six Mamiellales genomes (fig. 1 and supplementary table S1, Supplementary Material online). Using OrthoFinder, we assigned genes from the Ostreococcus spp., Bathycoccus prasinos, M. commoda, and M. pusilla to GFs. Mating-type GFs were defined as GFs with members located within the MT region of either O. tauri RCC4221 (MT−) or O. tauri RCC1115 (MT+). The presence/absence of the genes of these GFs in the lineage provides important information about MT+ and MT− specific GFs, as well as four additional distinct nonoverlapping GF categories (table 1).
Fig. 1.

Size and GC content in the candidate mating-type chromosomes (candidate mating-type locus positions as in supplementary table S1, Supplementary Material online) in the eight Mamiellales genomes. Sequences of CH02 of Ostreococcus lucimarinus and Micromonas pusilla have been reversed complemented to take colinearity of flanking regions as described in Palenik et al. (2007) and Worden et al. (2009) into account. Node divergence estimations are from Šlapeta et al. (2006) for Micromonas and Parfrey et al. (2011) for the basal node.
Table 1.
Classification, Description, and Number of Genes and GFs in Ostreococcus tauri RCC4221 (MT−) and RCC1115 (MT+) Strains
| GF Class | Features of Included Genes | RCC4221 (MT−) | RCC1115 (MT+) |
|---|---|---|---|
| MT-specific GFs | Present in either all Ostreococcus MT− or all Ostreococcus MT+ | 6 genes in 6 GFs | 2 genes in 2 GFs |
| Core MT GFs | Present in all Mamiellales genomes and located only in MT region | 23 genes in 23 GFs | 23 genes in 23 GFs |
| Shared MT GFs (noncore) | Present in both Ostreococcus MT loci, but not in all Mamiellales MT regions | 75 genes in 69 GFs | 79 genes in 69 GFs |
| GFs extending outside MT | Present in one Ostreococcus MT locus but with homologous genes in other regions in the opposite strain | 28 genes in 27 GFs | 8 genes in 4 GFs |
| GFs not retained for analysis | Present in only one Ostreococcus MT locus and Mamiellales genomes but absent from the genomes of the opposite strains/MT; divergent GFs or singletons | 112 genes | 128 genes |
| Total number of genes | 244 | 240 |
The “MT-specific” GF class contains genes that are shared only by Ostreococcus genomes from the same MT. The MT-specific GFs contain the smallest number of genes: six and two genes for MT− and MT+, respectively. These GFs are expected to contain genes involved in sex determination and functional control associated with each MT, as well as dispensable genes trapped into this locus (Wilson et al. [2019] for a review in Ascomycetes). Functional annotation revealed that most of these genes encode for hypothetical proteins or do not have any predicted function. The MT− specific GFs contain a gene with an RWP-RK domain (ostta02g01710), as previously reported (Worden et al. 2009), and a gene (ostta02g00990) that encodes for an SRP-dependent cotranslational protein involved in targeting proteins to the membrane. Within the MT+-specific GFs, there are only two genes, which encode for hypothetical proteins annotated with Gene Ontology terms linked to mismatch repair, protein binding, and transport (supplementary table S2, Supplementary Material online).
The “core MT” GF class contains GFs exclusively composed of gametologs that are located inside the boundaries of all candidate MT regions in all eight Mamiellales genomes (supplementary table S1, Supplementary Material online). There are 23 “core MT” GF, which make up less than 10% of genes of the MT (table 1) and these likely belonged to the ancestral locus which evolved into a MT in the lineage. Functional annotation indicates that these genes have housekeeping functions, such as ATP and DNA binding, transcription, glycolipid biosynthesis, protein transport, and RNA methylation, but no obvious link to mating (supplementary table S3, Supplementary Material online).
The largest GF class (69 GFs) regroups gametologs that are shared by both Ostreococcus MT loci, and that can be absent from the MT regions in some Mamiellales species (Shared MT GFs, noncore). A fourth class of GFs contains genes located within the O. tauri MT locus or on standard chromosomes (GF extending outside MT), and provides evidence of translocations between standard chromosomes and the MT loci. The remaining GFs are present in only one O. tauri MT locus and other Mamiellales genomes, or contain genes that are too divergent to generate phylogenies, as the alignments are too short. Therefore, they were excluded from further analyses, together with singleton genes (except the MT-specific GFs).
Although the core and specific GFs categories should contain the most ancient genes on the MT, the other GF categories likely reflect gain, loss, and translocation of genes in and out of the MT. This prompted us to undertake synteny and phylogenetic profiling of each GF to understand its evolutionary dynamics.
Genomic Architecture of O. tauri Mating-Type Regions
Syntenic regions outside the MT loci have been reported between species of the same genus: O. tauri and O. lucimarinus (Palenik et al. 2007), M. pusilla and M. commoda (Worden et al. 2009). Within O. tauri, regions outside the MT locus have been shown to be perfectly syntenic and share >99% nucleotide identity, in sharp contrast with the MT region (O. tauri Chromosome 2, fig. 1), which cannot be aligned at the nucleotide level between MT− (RCC4221) and MT+ (RCC1115) (Blanc-Mathieu et al. 2017). We further investigated the relative position of orthologous genes in the MT+ and MT− regions, but found no evidence for synteny in genes from shared and core GFs between both regions (fig. 2A): MT-specific genes do not cluster but are interspersed throughout the MT+ and MT− loci.
Fig. 2.

Gene organization in the mating-type region of Ostreococcus tauri RCC4221 (MT−) and RCC1115 (MT+). (A) Location of gene pairs from the 23 core GFs and 57 shared GFs in the mating-type region of O. tauri RCC4221 (MT−, blue rectangle) and RCC1115 (MT+, red rectangle). Genes from core and shared GFs are represented by bright and dark ticks, respectively, and homologous gene pairs are connected by gray lines. Genes from MT+ or MT− specific GFs are also shown, represented by black ticks. Shared GFs having multiple copies in either O. tauri RCC4221 (MT−) and RCC1115 (MT+) are not depicted. (B, C) Location of homologous MT genes from GFs with copies outside of either O. tauri MT region in Mamiellales genomes, for O. tauri RCC4221 (MT−, 39 GFs, B) and RCC1115 (MT+, 16 GFs, C). Each peripheral segment represents a chromosome or scaffold of one of eight Mamiellales genomes. The MT genes from O. tauri RCC4221 (MT−, B) or RCC1115 (MT+, C) are connected to their homologs by gray lines. If a homolog is located within a MT locus, the link is colored in orange. CH, chromosome; CG, contig; UG, unitig.
Ancient inversion events are a well-known trigger for suppression of recombination in genome evolution, but the relative position of orthologous genes in MT− and MT+ regions provide no evidence of a past inversion event. Instead, visual examination of the global pattern suggested a large translocation of the [b, c] segment in 5′ followed by the [a, b] segment in 3′ (fig. 2A). To investigate this hypothesis, we defined a simple statistic, Sdist, based on the relative distance between orthologous genes on the MT+ and MT−: Sdist is equal to 0 for perfect colinearity (see Materials and Methods). Random permutations of the gene orders enabled the estimation of the null distribution. The observed Sdist was not significantly different from the average Sdist for orthologous genes placed randomly on the two MTs (10,000 permutations, P > 0.10). However, the translocation of the 5′ extremity of MT− (segment [b, c]) to the start of MT− (arrow on fig. 2A) was associated with a significantly smaller Sdist than the random Sdist (100,000 permutations P = 0.0054). This demonstrates that this translocation significantly improves the overall colinearity between MT+ and MT−, supporting the idea of a past large-scale translocation in one of the MT loci.
To track gene translocation events between the MTs and the autosomal regions, we located the positions of 46 (MT−) and 30 (MT+) genes from GFs sharing genes inside and outside the MT regions. Genes of the same GFs as MT− genes were located on diverse autosomes (fig. 2B). We also observed a similar patchy distribution for GFs of gene members extending outside the MT+ (fig. 2C). This provides evidence for past gene translocations between many autosomes and the MT regions.
To search for evidence of evolutionary strata, defined as discrete regions containing orthologous genes with similar substitution rates (Lahn and Page 1999), we computed the rate of synonymous substitutions (Ks) (Tzeng et al. 2004) of the genes belonging to the 69 shared MT GFs on MT− and MT+ in O. tauri (shared GFs). We were able to compute the number of nonsynonymous substitutions (Ka) for only 22 gene pairs, given that for other gene pairs Ks values were close to saturation. From these 22, 19 had a Ks<1, and only two were adjacent on both the MT+ and the MT− (supplementary table S4, Supplementary Material online). This is consistent with a scenario of independent gene conversion events between the two MTs, except for one event spanning two genes. Interestingly, within these recently diverged genes, only two pairs were adjacent in only one of the mating types (MT+). This suggests that the source or the destination of the conversion events between MTs tends to span several kb. These observations indicate an absence of evidence for strata throughout the large MT regions of O. tauri. However, this absence of evidence may be reconsidered in the future if additional genome data in novel species can be informative to infer the ancestral gene order on the mating type (Branco et al. 2017).
Phylogenetic Insights into Evolutionary Dynamics of Mating Types
The topology of each GF phylogenetic tree is informative about the relative chronology of the speciation and the divergence events between the MT+ and MT− alleles. We assessed whether the topology supported either of the two scenarios: 1) in the “mating-type allele diverged postspeciation” scenario: mating-type alleles diverged after speciation events within Ostreococcus (no mating type alleles=Post); or 2) in the “mating-type allele diverged ante speciation” scenario: mating-type alleles diverged prior to the speciation event (mating-type allele separation=Ante). This latter scenario has previously been coined as trans-specific evolution resulting from long term balancing selection (Richman 2000). Consequently, the variation within the genes following the “Ante” scenario may be named trans-specific polymorphisms (Devier et al. 2009). The number of GFs for each topology is displayed in figure 3. Interestingly, this dual phylogenetic signal (mating-type allele divergence ante vs. postspeciation) is mirrored by a GC3 content signature of the genes. Indeed, genes belonging to GFs that support ancient mating-type origin have a significantly lower GC3 content than genes whose evolutionary history is concordant with the speciation history of the genus. For the 23 core MT GFs (listed in supplementary table S3, Supplementary Material online), the majority of phylogenies (21 trees, supplementary fig. S1, Supplementary Material online) support the “ancient mating-type” evolutionary scenario that mating-type region diverged before the speciation events within Ostreococcus, whereas only two phylogenies support the scenario of a mating-type differentiation after the speciation events.
Fig. 3.
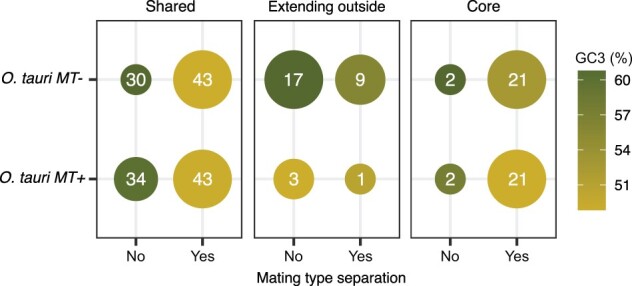
Phylogenetic signal and GC3 content of GF members in Ostreococcus tauri RCC4221 (MT−) and RCC1115 (MT+). “Post” for GF genes with mating-type separation after speciation and “Ante” for GF genes with mating-type separation prior to speciation. Circle size is proportional to the number of GF genes (numerical value within each circle), and circle color depicts the average GC3 content from low (yellow-golden) to high (green).
Thus, most core and shared MT GFs support an ancient mating-type origin (fig. 4A with mating-type separation and 4B without mating-type separation). In contrast, the phylogenies of most GFs containing paralogous genes outside the MT region are consistent with the speciation tree, suggesting their translocation inside the MT locus occurred recently.
Fig. 4.

Unrooted maximum-likelihood phylogenetic trees of representative core MT GFs 000581 (A) and 000945 (B). Genes from MT− strains are colored in blue, genes from MT+ strains are colored in red. Ultrafast bootstrap support values are denoted on branches. OT4221, Ostreococcus tauri RCC4221; OT1115, O. tauri RCC1115; OL, O. lucimarinus; O809, O. sp. RCC809; OMED, O. mediterraneus RCC2590; B1105, Bathycoccus prasinos RCC1105; MC299, Micromonas commoda RCC299; MPU, M. pusilla.
Expanding the Number of Mamiellales Species with Two Mating-Type Alleles
Since the core MT GFs allow MT+ and MT− delineation in the Mamiellales, we used the sequence data to screen 33 transcriptomes (MMETSP and 1KP data sets) from several Mamiellophyceae species for homologous sequences (listed in supplementary table S5, Supplementary Material online). The taxonomic affiliation of each transcriptome was inferred from 18S rDNA sequences (supplementary table S6 and fig. S2, Supplementary Material online). The phylogenetic range of the transcriptomes spanned from the early divergent freshwater species, such as Monomastix opisthostigma (Monomastigales), Crustomastix, and Dolichomastix (Dolichomastigales), to early Mamiellales, such as Mantoniella. It also included several Micromonas strains from novel species, such as Micromonas bravo and Micromonas polaris. In total, at least one homologous gene was recovered for each GF (with an average of 11 GFs per transcriptome) in 28 of 33 transcriptomes (fig. 5).
Fig. 5.

Presence–absence matrix of best BLAST hits (BBH) of core MT GFs in each Mamiellophyceae transcriptome. Species’ names of sequenced strains (left column) as inferred from 18S rDNA sequence analysis extracted from the transcriptome (supplementary fig. S2, Supplementary Material online). The color of each rectangle indicates the taxonomic affiliation of the BBH (color key at the bottom). Transcriptomes containing genes with a BBH affiliated to a different species are highlighted in gray.
The most striking pattern came from O. mediterraneus MMETSP0929 (strain RCC2572) and O. lucimarinus MMETSP0939 (strain BCC118000) transcriptomes. Although both data sets displayed hits for almost all core genes (17 out of 23), the taxonomic affiliation inferred for these genes by best BLAST hit (BBH) was not consistent with the 18S taxonomic affiliation. Instead, it suggested affiliation to a different species of the opposite mating type (supplementary fig. S3, Supplementary Material online). In O. mediterraneus MMETSP0929, 14 of 17 genes were affiliated to species from the opposite MT groups (MT−), such as O. tauri and O. lucimarinus, not to the reference genome O. mediterraneus RCC2590 MT+. Likewise, 15 of 17 BBHs of O. lucimarinus MMETSP0939 came from MT+ genomes, and not from the MT−O. lucimarinus reference genome. To confirm the taxonomic affiliation of these genes, we built maximum likelihood phylogenies, including homologs extracted from the transcriptomes (supplementary fig. S3, Supplementary Material online). From the 17 GFs with a BBH, 12 passed the alignment length and identity thresholds (see Materials and Methods). Of these, ten phylogenies included both O. mediterraneus MMETSP0929 and O. lucimarinus MMETSP0939, and two phylogenies included only O. lucimarinus MMETSP0939. From these, 11 phylogenies were consistent with ancient MT+ and MT− divergence (example in fig. 6A), whereas one phylogeny regrouped genes according to species (fig. 6B).
Fig. 6.

Unrooted maximum-likelihood phylogenetic trees of representative core MT GFs 001374 (A) and 003390 (B) including homologous sequences from Ostreococcus lucimarinus MMETSP0939 (strain BCC118000) and O. mediterraneus MMETSP0929 (strain RCC2572). Candidate mating-type genes MT+ are in red, MT− in blue. Topology (A) clusters genes according to mating type, whereas topology (B) corresponds to the species phylogeny. Ultrafast bootstrap support values are indicated on branches. OT4221, O. tauri RCC4221; OT1115, O. tauri RCC1115; OL, O. lucimarinus; OLMMETSP0939, O. lucimarinus BCC118000; O809, O. sp. RCC809; OMED, O. mediterraneus RCC2590; OMMMETSP0929, O. mediterraneus RCC2572; B1105, Bathycoccus prasinos RCC1105; MC299, Micromonas commoda RCC299; MPU, M. pusilla.
These phylogenetic analyses confirmed the taxonomic affiliation inferred from amino acid sequence conservation and support an ancient divergence of genes from two MT regions. This led us to conclude that O. lucimarinus strain RCC2572 and O. mediterraneus strain BCC118000 (MMETSP0929 and MMETSP0939, respectively) are of the opposite mating type to the strains for which the reference genome is available. This extends the evidence of the existence of two mating types in O. tauri to two additional Ostreococcus species.
Identification of Candidate Mating Types Based on Gene Genealogies in Micromonas commoda
Micromonas is the most represented Mamiellophyceae genus in the available transcriptomic data sets, with 14 transcriptomes. Therefore, we further examined the individual GF phylogenetic topologies and sequence similarities by using the core MT GF set (23 GFs) to search for clustering that might suggest an ancient divergence of MTs in Micromonas. To this end, we selected Micromonas transcriptomes with more than one positive hit with the GFs, and the highest number of hits in the majority of transcriptomes (nine transcriptomes), together with one outgroup from the genus (Mantoniella sp. MMETSP1468). Finally, we built individual GF phylogenies from these sequences and the core genes GF data set (supplementary fig. S4, Supplementary Material online).
A consistent subclustering of strains within the M. commoda group was observed. MMETSP 1084, 1387, 1403, and 1400 clustered together in 11 of 13 phylogenies, whereas MMETSP1404 and 1393 clustered with genes from the reference genome of M. commoda RCC299 (fig. 7A and supplementary fig. S4, Supplementary Material online). In only two phylogenies, there was no apparent subclustering (fig. 7C). Additionally, the branch lengths of the 11 phylogenies displaying subclustering were longer and similar to the branch lengths separating M. polaris from M. bravo, or M. commoda from M. pusilla. Consistent with this, the average pairwise amino acid identities between M. commoda genes from the two different subclusters ranged from 65% to 89% (supplementary table S7, Supplementary Material online). For comparison, we built phylogenies of the actin and β-tubulin genes (fig. 7B and D), which are highly conserved, and their phylogenetic topology showed a species topology signature, where these strains did not support two subclusters. Pairwise amino acid identity for the latter GFs between strains ranged from 98% to 99.4% (for actin and β-tubulin, respectively), as expected for strains from the same species. This phylogenetic signal was similar to the Ostreococcus core GF phylogenies, consistent with an ancient mating-type separation (fig. 4A). Despite the low number of genes (13 genes from 23 GFs), this subclustering suggests that there are two MTs in M. commoda: strains MMETSP1404, 1393, and M. commoda RCC299 (the reference genome); and strains MMETSP 1084, 1387, 1400, and 1403, representing the opposite MT. As Worden et al. (2009) suggested, M. commoda RCC299 would represent the MT−, given the presence of an RWP-RK motif gene in its candidate MT region. Thus, the strains MMETSP 1084, 1387, 1403, and 1400 would represent the MT+ type. Taken together, phylogenetic analyses of GFs are consistent with an ancient gene divergence of MT gametologs in the M. commoda lineage, as expected under recombination suppression.
Fig. 7.

Phylogenetic trees of representative core MT GFs 001102 (A) and 003908 (C), and actin (B) and β-tubulin (D) genes, for Micromonas commoda and M. pusilla reference genomes and homologous genes retrieved from diverse Micromonas spp. transcriptomes. In the phylogeny “A,” two M. commoda subclusters are highlighted in dark green (MMETSP1403, 1400, 1084, 1387) and light green (MMETSP1393, 1404, and the reference M. commoda RCC299). In phylogeny “C,” there is no “A type” subclustering. In the actin and β-tubulin trees, subclusters are absent and branch lengths are shorter than “A,” but parallel with phylogeny “C.”
Clues about Earlier Origin of Mating-Type Loci in Mamiellophyceae
As the phylogenetic signal may be lost over time as a consequence of the decay of similarity between orthologs (Jain et al. 2019), we investigated indirect signatures of MTs. MTs evolve without recombination, and this has been shown to decrease GC content. We therefore investigated whether a GC signature could be detected in homologous genes to the core GFs outside the Mamiellales (comprising Ostreococcus, Bathycoccus, and Micromonas). Thus, we analyzed the GC content of the synonymous third codon position (GC3) of core GF hits in several Mamiellophyceae species, and compared this with the GC3 content of genes from the background genome or transcriptome. Core MT GFs have significantly lower GC3 (∼20%) than genes of the background genome (or transcriptome) in Bathycoccus, Ostreococcus, and Micromonas (fig. 8 and supplementary table S8, Supplementary Material online). Interestingly, we found evidence of a similar difference in GC3 content between gene hits against the core MT GFs and the background transcriptome in Mantoniella squamata CCAP 1965/1 and the uncultured Mamiellophyceae (uncultured eukaryote RCC2288), with ∼10% and 20% differences between genes from the GFs and genes from the background transcriptome, respectively. This suggested that genes that are homologous to the core GFs are also located in a low GC chromosome region in these Mamiellophyceae species (fig. 8 and supplementary table S8, Supplementary Material online). However, there is no evidence for a GC3 content difference between homologous genes to the core GFs and the genes from the background transcriptome in Crustomastix or Monomastix (fig. 8).
Fig. 8.

GC3 content comparison between genes from core MT GFs (dark green) and overall genome or transcriptome (light green) in Mamiellophyceae species. Phylogenetic relationships are inferred from 18S rDNA phylogeny. An asterisk (*) indicates a significant GC3 content difference (Student’s t-test P value <0.05). Ostreococcus, O. tauri RCC4221; Bathycoccus, B. prasinos RCC1105; Micromonas, M. pusilla CCMP1545; Mantoniella squamata, Mantoniella squamata strain CCMP1436—MMETSP1468; Mantoniella squamata, Mantoniella squamata strain CCCAP 1965/1—QXSZ; Mantoniella antarctica, M. antarctica strain SL-175—MMETSP1106; Uncult. Mamiellophyceae, Uncultured eukaryote RCC2288—MMETSP1326; Crustomastix, C. stigmatica CCMP3273—MMETSP0803; Dolichomastix, D. tenuilepis CCMP3274—MMETSP0033; D. tenuilepis, D. tenuilepis strain M1680—XOAL; Monomastix, Monomastix opisthostigma CCAC 0206—BTFM.
Discussion
Direct evidence of meiosis is not available for most marine planktonic microbial eukaryotes. This is either due to the difficulty in culturing certain species, or because experimental studies are hampered by a lack of knowledge about sex determination and the conditions required to induce a sexual cycle. In the case of haploid green picoalgae (cell diameter <2 µm) of the Mamiellales lineage, population genomics data in one species allowed the identification of two candidate mating-type alleles with suppressed recombination (Blanc-Mathieu et al. 2017). Here, comparative genomics of seven related species within the Mamiellales lineage unraveled different facets in the mode and tempo of evolution in this enigmatic locus.
First, although no MT+ and MT− specific genes could be identified for all seven species, MT+ and MT− specific genes could be identified within the Ostreococcus genus. MT− specific genes may be implicated in mating-type differentiation, such as the previously identified gene encoding an RWP-RK domain (Worden et al. 2009). The two MT+ specific genes that have been identified in Ostreococcus encode for unknown proteins. One of these proteins (gm1.767_g) harbors WD40 repeats and is predicted to bind to other proteins. The second protein has a DNA binding domain, which is also found in DNA mismatch repair proteins (gm1.689_g, PF00488). A WD40 protein has been shown to regulate mating in the fungus Ustilago maydis(Wang et al. 2011). Nevertheless, the functional range of WD40 proteins is too wide to confidently infer a role of the Ostreococcus protein to act as a MT+ signal protein.
Second, comparative phylogenetics of core gametologs allowed the identification the opposite mating types in two additional species for which transcriptomes were available: the MT− in O. mediterraneus and the MT+ in O. lucimarinus. This mating-type profiling is made possible by the high divergence between the MT+ and MT− regions, as gametologs cluster by MT and not by species. By screening available environmental data from the TARA Oceans project for the presence of these gametologs, we previously found that, in fact, both mating types of O. lucimarinus were present at the stations where this species had been detected (Leconte et al. 2020). Mating-type profiling was also suggested between strains from M. commoda: phylogenies of the gametologs suggest two clusters of strains, in contrast with phylogenies of highly conserved housekeeping genes (actin, β-tubulin, and 18 s rDNA) (fig. 7 and supplementary table S7 and fig. S2, Supplementary Material online).
Third, analyzing additional transcriptome data from early diverging branches of the Mamiellophyceae class, we could detect orthologous genes to the Mamiellales gametologs in eight additional transcriptomes. However, we could not detect any significant difference in GC3 signatures in the earliest Mamiellophyceae, as would be expected under suppressed recombination; on the contrary, GC3 values appear to be higher in homologous genes in Dolichomastigales. This suggests the Mamiellales gametologs are not part of a lower GC region in earlier branching Mamiellophyceae. The conservation of 23 gametologs within the Mamiellales lineage prompted us to investigate the dynamic of these genes. The additional gametologs within the Ostreococcus lineage support an ancient large translocation event. Inversions have been previously suggested to trigger recombination suppression and have been recently reported in the origin of a young sex-determining chromosome (Natri et al. 2019). However, translocations are also expected to disrupt recombination (McKim et al. 1988).
One intriguing feature of sex-determining chromosomes is their organization as multiple discrete regions, where genes can be clustered by genetic divergence (measured by the rate of nonsynonyms substitutions), defined as “evolutionary strata.” In humans, strata were first described by Lahn and Page (1999), who suggested that suppression of recombination was initiated in one region (stratum) and later expanded in discrete steps, by strata. This could happen through additional chromosomal inversions, which are known to suppress recombination in mammalian chromosomes. Only a few X–Y sequence similarities persist, and these alleles are orderly stratified by age in the X chromosome and scrambled in the Y. Although strata have been observed in several vertebrates, plants, and fungi (Bachtrog et al. 2014; Badouin et al. 2015; Coelho et al. 2018), they do not appear to be a common feature of algal mating types and sex chromosomes. Indeed, we found no evidence of evolutionary strata in Ostreococcus MTs, as neither ancient nor recent genes cluster in any of the MTs This may be due to their ancient divergence, associated with a limited more recent expansion dynamic, as suggested in the UV chromosomes of the brown algae Ectocarpus(Ahmed et al. 2014). Alternatively, it could also be due to the lack of information about the ancestral gene order on the mating type (Branco et al. 2017).
To counteract the effects of reduced recombination inside MTs, gene conversion between mating types has been suggested to act as a homogenizing force in Chlamydomonas(De Hoff et al. 2013). In fungal mating types, the suppression of recombination maintains linkage of mating-type genes within each locus, which is required for correct mating-type determination (Kües 2000; Branco et al. 2017). However, gene flow between mating-type loci and gene conversion events has recently been reported in several species (Sun et al. 2012; Hartmann et al. 2020). This suggests an important difference in the evolutionary processes of haploid sex-determining systems versus diploid sex-determining systems, where gene flow between sex-determining regions is rare (Hartmann et al. 2020).
The diversification within Mamiellales is estimated to have occurred between 330 and 640 Ma (Lang et al. 2010; Parfrey et al. 2011; Blank 2013), much earlier than the diversification within Volvocales where deep homology of mating-type loci has been reported (Ferris et al. 2010), and with a higher upper limit to the estimated 370 Myr divergence of the STE3-like pheromone receptors from basidiomycete fungi (Devier et al. 2009). Therefore, our data suggest the Mamiellales mating-type sex-determining region to be among the oldest mating type locus reported.
In conclusion, we analyzed the phylogenetic profiles of the GFs within the Ostreococcus mating types, and gained insights into the evolutionary history of this sex-determining region in one of the earliest diverging orders of Chlorophytes. The identification of strains from the two opposite mating types in three species will guide future experimental approaches for mating and strain crossing, since a highly efficient transformation protocol is now available in Ostreococcus(Sanchez et al. 2019). Complete genome sequences in additional Mamiellophyceae are now essential to investigate the early dynamics of the sex-determining regions in the green lineage.
Materials and Methods
Mating-Type GF Definition
The full set of predicted genes from eight Mamelliales genomes (supplementary table S1, Supplementary Material online) was loaded into a custom version of the pico-PLAZA framework (Proost et al. 2009; Vandepoele et al. 2013) to define and analyze GFs. Following an “all-against-all” protein sequence similarity search, performed with BLASTP (version 2.6.0+, maximum E-value threshold 1e-4, keeping up to 2,500 hits), we delineated GFs using OrthoFinder version 2.1.2 (Emms and Kelly 2015).
The boundaries of the mating-type (MT) region of Ostreococcus tauri RCC4221 (Blanc-Mathieu et al. 2014) and RCC1115 (Blanc-Mathieu et al. 2017) served as a starting point for defining candidate MT GFs (supplementary table S1, Supplementary Material online). All genes located within either MT region were extracted, based on the coordinates of their coding sequence (CDS). For each gene included in these two gene sets, the GF they were assigned to was subsequently retrieved, consisting of a validated homologous group of ortholog and paralog genes in eight available genomes. Based on the location of the GF members (chromosome or scaffold and coordinates), a “MT signal” value was then computed for every genome in which the GF was represented. This value corresponds to the fraction of members located within the MT region (for the given genome-GF combination), and was used to filter and classify the list of candidate GFs. The complete list of MT GFs is reported in supplementary table S9, Supplementary Material online.
For every retained GF, protein sequences were aligned using MAFFT version 7.187 (Katoh and Standley 2013) with the L-INS-i alignment method and a maximum of 1,000 iterative refinements. We edited the multiple sequence alignments (MSAs) using several filters on both sequences and positions, implemented in the PLAZA framework and described by Proost (Proost et al. 2009). Briefly, highly divergent and partial sequences were filtered out, and positions containing gaps in minimum 10% of the sequences or containing potentially misaligned amino acids removed. We also applied a minimum length cut-off to the edited MSA: the edited MSA had to be 50 amino acid long at least, otherwise we ignored it. In case the original unedited MSA was shorter, we used this length as a cut-off value instead. Finally, we retained only MSAs that showed at least 50% alignment of amino acid identity in half of the sequences of the MSA. The circular plots depicting the location of homologous genes from GFs having copies outside of the O. tauri MT loci (fig. 2B and C) were generated with the circlize package in R (Gu et al. 2014; https://r-project.org/).
To test different gene order rearrangement scenarios between the MT+ and MT− regions, we defined Sdist, which is the absolute value of the difference of the position of orthologous genes on the MT+ and MT− regions. If there are n orthologous genes between the two loci with pi− the position (in rank) of gene i on MT− and pi+ the position of its ortholog on MT+, . Sdist=0 if all orthologs are perfectly collinear. The expected Sdist under random position of orthologous genes in the two mating types was assessed by simulations. If there has been an inversion of gene order between the two regions, Sdist is maximal, Sdist=z(2n−2z), with z = n/2 if n is even, and z = (n−1)/2 if z is odd.
GF Clustering and Phylogeny
For each GF MSA that passed our filtering criteria, we built an ML phylogenetic tree using IQ-TREE version 1.6.5 (Nguyen et al. 2015). Trees were built under the best-fitting substitution model selected by ModelFinder (Kalyaanamoorthy et al. 2017), chosen among commonly used models (JTT, LG, WAG, Blosum62, VT, and Dayhoff). Empirical amino acid frequencies were calculated from the data, the FreeRate model (Yang 1995; Soubrier et al. 2012) was used to account for rate heterogeneity across sites, and branch supports were assessed using ultrafast bootstrap approximation (UFBoot) (Soubrier et al. 2012) with 1,000 bootstrap replicates.
We used similar alignment, MSA editing, and phylogenetic tree building procedures when considering sequences from external sources (e.g., transcripts from MMETSP samples). The divergent gene removal criterion was based on the results of the all-against-all protein sequence similarity search performed using data from the eight reference genomes only (supplementary table S1, Supplementary Material online). Therefore, it was not used to filter out these sequences from the MSAs. Phylogenetic trees were built for full alignments in case the editing was deemed too stringent, for instance discarding transcripts flagged as partial sequences. Finally, when investigating the molecular phylogeny of the 18S rDNA genes, we used IQ-TREE’s ModelFinder Plus parameter to select the best DNA substitution model.
GF Phylogenetic Tree Classification
We visualized and inspected the MT GF trees using FigTree version 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/). We examined ultrafast bootstrap support values and topology type, and counted the number of times genes clustered by mating type or according to their taxonomic classification (by species).
Searching for Homologs in Publicly Available Transcriptomes
We used sequences of core MT GF members as queries to search for homologs in Mamiellophyceae transcriptomes (33 transcriptomes in total, listed in supplementary table S5, Supplementary Material online). Transcriptomes were retrieved from the MMETSP (Keeling et al. 2014; Johnson et al. 2019) and 1KP data sets (Matasci et al. 2014). Reassembled MMETSP transcriptomes were downloaded from https://doi.org/10.5281/zenodo.251828 (version 1; January 2017) and 1KP transcriptomes via 1KP’s R interface (https://github.com/ropensci/onekp). CDS from each Mamiellophyceae MMETSP transcriptome were predicted using TransDecoder (Haas et al. 2013) with default parameters. Sequence similarity searches were performed using TBLASTX (maximum E-value threshold 1e-4) and results were filtered to retain hits with alignment length >50 and amino acid identity >60%. In-depth phylogenetic analyses of individual hits from O. mediterraneus strain RCC2572 (MMETSP0929), O. lucimarinus strain BCC118000 (MMETSP0939), Micromonas MMETSP transcriptomes (1084, 1327, 1387, 1393, 1400, 1401, 1402, 1403, 1404), and Mantoniella MMETSP transcriptomes (1106, 1468) were performed as previously described for the reference genomes. The presence/absence matrix of each informative orthologous group against the transcriptomes was generated using the ggplot2 package in R (Wickham 2011).
To validate and elucidate each MMETSP transcriptome’s taxonomic affiliation, we downloaded Mamiellophyceae 18S rDNA sequences from reference genomes in GenBank, the SILVA database (Quast et al. 2013), and Micromonas spp. sequences provided in Simon et al. (2017) (supplementary table S6, Supplementary Material online). Transcripts matching selected 18S sequences were extracted with BLASTN (maximum E-value 1e-5) and 18S rDNA sequences were subsequently predicted using RNammer (Lagesen et al. 2007). A ML phylogenetic tree was built using IQ-TREE and following each clustering of this Mamiellophyceae reference tree (rooted in Monomastix spp.), transcriptomes were tentatively classified according to a species clustering (supplementary fig. S2, Supplementary Material online). Phylogeny indicated that MMETSP transcriptomes matched their species classification, and transcriptomes from novel Micromonas species as M. polaris and M. bravo were designated using the data and new classification of Simon et al. (2017).
Compositional Analysis (GC3) of GFs in Mamiellophyceae
To evaluate compositional differences between third codon positions (GC3) of GF members and CDS from the overall genome or transcriptome (supplementary table S8, Supplementary Material online), we used a custom python script to perform GC3 calculations. We subsequently evaluated the results using Student’s t-test as implemented in R.
Synonymous and Nonsynonymous Divergence of Shared MT GFs
We used homologous pairs of the 69 shared MT GFs to calculate sequence genetic divergence with the seqinr package v3.4-5 (kaks function) using (Li 1993) method (LWL85) in R.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work was funded by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie ITN project SINGEK (H2020-MSCA-ITN-2015-675752 to L.F.B. and F.B.). We thank the Moore foundation for sequencing most of the Mamiellophycean transcriptomes analyzed in this study and the Genotoul Bioinformatic platform for providing computing and data storage resources.
Data Availability
All genomic and transcriptomic sequence data have been deposited at GenBank under the accession numbers CAID00000000.1 (Ostreococcus tauri), PRJNA337288 (O. lucimarinus), PRJNA15676 (Micromonas commoda), PRJNA15678 (M. pusilla), PRJNA394752 (Bathycoccus prasinos), and PRJNA248394 (MMESTP). The accession numbers of the 18S rDNA sequences are summarized in supplementary table S6, Supplementary Material online.
Literature Cited
- Ahmed S, et al. 2014. A haploid system of sex determination in the brown alga Ectocarpus sp. Curr Biol. 24(17):1945–1957. [DOI] [PubMed] [Google Scholar]
- Astell CR, et al. 1981. The sequence of the DNAs coding for the mating-type loci of Saccharomyces cerevisiae. Cell 27(1 Pt 2):15–23. [DOI] [PubMed] [Google Scholar]
- Bachtrog D, et al. 2011. Are all sex chromosomes created equal? Trends Genet. 27(9):350–357. [DOI] [PubMed] [Google Scholar]
- Bachtrog D, et al. 2014. Sex determination: why so many ways of doing it? PLoS Biol. 12(7):e1001899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badouin H, et al. 2015. Chaos of rearrangements in the mating-type chromosomes of the Anther-Smut fungus Microbotryum lychnidis-dioicae. Genetics 200:1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutlich A, Schnetter R.. 1993. The life cycle of Cryptochlora perforans (Chlorarachniophyta). Bot Acta. 106(5):441–447. [Google Scholar]
- Billiard S, et al. 2011. Having sex, yes, but with whom? Inferences from fungi on the evolution of anisogamy and mating types. Biol Rev Camb Philos Soc. 86(2):421–442. [DOI] [PubMed] [Google Scholar]
- Billiard S, López-Villavicencio M, Hood ME, Giraud T.. 2012. Sex, outcrossing and mating types: unsolved questions in fungi and beyond. J Evol Biol. 25(6):1020–1038. [DOI] [PubMed] [Google Scholar]
- Blanc-Mathieu R, et al. 2014. An improved genome of the model marine alga Ostreococcus tauri unfolds by assessing Illumina de novo assemblies. BMC Genomics 15:1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc-Mathieu R, et al. 2017. Population genomics of picophytoplankton unveils novel chromosome hypervariability. Sci Adv. 3(7):e1700239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank CE. 2013. Origin and early evolution of photosynthetic eukaryotes in freshwater environments: reinterpreting proterozoic paleobiology and biogeochemical processes in light of trait evolution. J Phycol. 49(6):1040–1055. [DOI] [PubMed] [Google Scholar]
- Branco S, et al. 2017. Evolutionary strata on young mating-type chromosomes despite the lack of sexual antagonism. Proc Natl Acad Sci U S A. 114(27):7067–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D.. 2000. The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci. 355(1403):1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D. 2016. Plant sex chromosomes. Annu Rev Plant Biol. 67:397–420. [DOI] [PubMed] [Google Scholar]
- Charlesworth D, et al. 2020. Using GC content to compare recombination patterns on the sex chromosomes and autosomes of the guppy, Poecilia reticulata, and its close outgroup species. Mol Biol Evol. 37(12):3550–3562. [DOI] [PubMed] [Google Scholar]
- Chepurnov VA, Mann DG, Sabbe K, Vyverman W.. 2004. Experimental studies on sexual reproduction in diatoms. Int Rev Cytol. 237:91–154. [DOI] [PubMed] [Google Scholar]
- Coelho SM, Gueno J, Lipinska AP, Cock JM, Umen JG.. 2018. UV chromosomes and haploid sexual systems. Trends Plant Sci. 23(9):794–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Hoff PL, et al. 2013. Species and population level molecular profiling reveals cryptic recombination and emergent asymmetry in the dimorphic mating locus of C. reinhardtii. PLoS Genet. 9(8):e1003724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derelle E, et al. 2006. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci U S A. 103(31):11647–11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devier B, Aguileta G, Hood ME, Giraud T.. 2009. Ancient trans-specific polymorphism at pheromone receptor genes in Basidiomycetes. Genetics 181(1):209–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drott MT, et al. . 2020. The Frequency of Sex: Population Genomics Reveals Differences in Recombination and Population Structure of the Aflatoxin-Producing Fungus Aspergillus flavus. mBio. 11(4):e00963-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebenezer TE, et al. 2019. Transcriptome, proteome and draft genome of Euglena gracilis. BMC Biol. 17(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S.. 2015. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris P, et al. 2010. Evolution of an expanded sex-determining locus in Volvox. Science 328(5976):351–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris PJ, Armbrust EV, Goodenough UW.. 2002. Genetic structure of the mating-type locus of Chlamydomonas reinhardtii. Genetics 160(1):181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris PJ, Goodenough UW.. 1997. Mating type in Chlamydomonas is specified by mid, the minus-dominance gene. Genetics 146(3):859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanillas E, et al. 2015. Degeneration of the nonrecombining regions in the mating-type chromosomes of the anther-smut fungi. Mol Biol Evol. 32(4):928–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fučíková K, Pažoutová M, Rindi F.. 2015. Meiotic genes and sexual reproduction in the green algal class Trebouxiophyceae (Chlorophyta). J Phycol. 51(3):419–430. [DOI] [PubMed] [Google Scholar]
- Galtier N, Piganeau G, Mouchiroud D, Duret L.. 2001. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics 159(2):907–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng S, Miyagi A, Umen JG.. 2018. Evolutionary divergence of the sex-determining gene MID uncoupled from the transition to anisogamy in volvocine algae. Dev Camb Engl. 145(7):dev162537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsley N, Péquin B, Bachy C, Moreau H, Piganeau G.. 2010. Cryptic sex in the smallest eukaryotic marine green alga. Mol Biol Evol. 27(1):47–54. [DOI] [PubMed] [Google Scholar]
- Gu Z, Gu L, Eils R, Schlesner M, Brors B.. 2014. circlize Implements and enhances circular visualization in R. Bioinformatics 30(19):2811–2812. [DOI] [PubMed] [Google Scholar]
- Haas BJ, et al. 2013. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nat Protoc. [Internet]. 8. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3875132/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjivasiliou Z, Pomiankowski A.. 2016. Gamete signalling underlies the evolution of mating types and their number. Philos Trans R Soc Lond B Biol Sci. 371(1706):20150531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaji T, et al. 2016. Sequence of the Gonium pectorale mating locus reveals a complex and dynamic history of changes in volvocine algal mating haplotypes. G3 (Bethesda) 6(5):1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaji T, et al. 2018. Anisogamy evolved with a reduced sex-determining region in volvocine green algae. Commun Biol. 1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann FE, et al. 2020. Higher gene flow in sex-related chromosomes than in autosomes during fungal divergence. Mol Biol Evol. 37(3):668–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann FE, et al. 2021. Recombination suppression and evolutionary strata around mating-type loci in fungi: documenting patterns and understanding evolutionary and mechanistic causes. New Phytol. 229(5):2470–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan AR, , Ness RW.. 2020. Recombination Rate Variation and Infrequent Sex Influence Genetic Diversity in Chlamydomonas reinhardtii. Genome Biol Evol. 12(4):370–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DRA, Wetherbee R.. 1986. Proteomonas sulcata gen. et sp. nov. (Cryptophyceae), a cryptomonad with two morphologically distinct and alternating forms. Phycologia 25(4):521–543. [Google Scholar]
- Hoekstra RF. 1987. The evolution of sexes. Exp Suppl. 55:59–91. [DOI] [PubMed] [Google Scholar]
- Hurst LD, Hamilton WD.. 1992. Cytoplasmic fusion and the nature of sexes. Proc R Soc Lond B Biol Sci. 247:189–194. [Google Scholar]
- Jain A, Perisa D, Fliedner F, von Haeseler A, Ebersberger I.. 2019. The evolutionary traceability of a protein. Genome Biol Evol. 11(2):531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jancek S, Gourbière S, Moreau H, Piganeau G.. 2008. Clues about the genetic basis of adaptation emerge from comparing the proteomes of two Ostreococcus ecotypes (Chlorophyta, Prasinophyceae). Mol Biol Evol. 25(11):2293–2300. [DOI] [PubMed] [Google Scholar]
- Johnson LK, Alexander H, Brown CT.. 2019. Re-assembly, quality evaluation, and annotation of 678 microbial eukaryotic reference transcriptomes. GigaScience [Internet]. 8(4):giy158. Available from: https://academic.oup.com/gigascience/article/8/4/giy158/5241890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joli N, Monier A, Logares R, Lovejoy C.. 2017. Seasonal patterns in Arctic prasinophytes and inferred ecology of Bathycoccus unveiled in an Arctic winter metagenome. ISME J. 11(6):1372–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS.. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 30(4):772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, et al. 2014. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 12(6):e1001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koufopanou V, et al. . 2020. Population Size, Sex and Purifying Selection: Comparative Genomics of Two Sister Taxa of the Wild Yeast Saccharomyces paradoxus. Genome Biol Evol. 12(9):1636–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovec M, Eyre-Walker A, Sanchez-Ferandin S, Piganeau G.. 2017. Spontaneous mutation rate in the smallest photosynthetic eukaryotes. Mol Biol Evol. 34(7):1770–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kües U. 2000. Life history and developmental processes in the basidiomycete Coprinus cinereus. Microbiol Mol Biol Rev. 64(2):316–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugrens P, Lee RE.. 1988. Ultrastructure of fertilization in a Cryptomonad1. J Phycol. 24(3):385–393. [Google Scholar]
- Kumar S, Stecher G, Suleski M, Hedges SB.. 2017. TimeTree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol. 34(7):1812–1819. [DOI] [PubMed] [Google Scholar]
- Lagesen K, et al. . 2007. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35(9):3100–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahn BT, Page DC.. 1999. Four evolutionary strata on the human X chromosome. Science 286(5441):964–967. [DOI] [PubMed] [Google Scholar]
- Lang D, et al. 2010. Genome-wide phylogenetic comparative analysis of plant transcriptional regulation: a timeline of loss, gain, expansion, and correlation with complexity. Genome Biol Evol. 2:488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leconte J, et al. 2020. Genome resolved biogeography of Mamiellales. Genes 11(1):66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengeler KB, et al. 2002. Mating-type locus of Cryptococcus neoformans: a step in the evolution of sex chromosomes. Eukaryot Cell. 1(5):704–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W-H. 1993. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 36(1):96–99. [DOI] [PubMed] [Google Scholar]
- Ma W-J, Carpentier F, Giraud T, Hood ME.. 2020. Differential gene expression between fungal mating types is associated with sequence degeneration. Genome Biol Evol. 12(4):243–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S-B, Ramesh MA, Hulstrand AM, Logsdon JM.. 2007. Protist homologs of the meiotic Spo11 gene and topoisomerase VI reveal an evolutionary history of gene duplication and lineage-specific loss. Mol Biol Evol. 24(12):2827–2841. [DOI] [PubMed] [Google Scholar]
- Matasci N, et al. 2014. Data access for the 1,000 Plants (1KP) project. Gigascience 3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim KS, Howell AM, Rose AM.. 1988. The effects of translocations on recombination frequency in Caenorhabditis elegans. Genetics 120(4):987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier J, Duret L.. 2004. Recombination drives the evolution of GC-content in the human genome. Mol Biol Evol. 21(6):984–990. [DOI] [PubMed] [Google Scholar]
- Natri HM, Merilä J, Shikano T.. 2019. The evolution of sex determination associated with a chromosomal inversion. Nat Commun. 10(1):145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki H, Mori T, Misumi O, Matsunaga S, Kuroiwa T.. 2006. Males evolved from the dominant isogametic mating type. Curr Biol. 16(24):R1018–R1020. [DOI] [PubMed] [Google Scholar]
- Palenik B, et al. 2007. The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc Natl Acad Sci U S A. 104(18):7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, Lahr DJG, Knoll AH, Katz LA.. 2011. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc Natl Acad Sci U S A. 108(33):13624–13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfiester LA. 1989. Dinoflagellate sexuality. In: Bourne GH, Jeon KW, Friedlander M, editors. International review of cytology. Vol. 114. New York: Academic Press. p. 249–272. [Google Scholar]
- Proost S, et al. 2009. PLAZA: a comparative genomics resource to study gene and genome evolution in plants. Plant Cell 21(12):3718–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, et al. . 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41(Database issue):D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman A. 2000. Evolution of balanced genetic polymorphism. Mol Ecol. 9(12):1953–1963. [DOI] [PubMed] [Google Scholar]
- Sager R, Granick S.. 1954. Nutritional control of sexuality in Chlamydomonas reinhardi. J Gen Physiol. 37(6):729–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez F, et al. 2019. Simplified transformation of Ostreococcus tauri using polyethylene glycol. Genes 10(5):399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon N, et al. . 2017. Revision of the Genus Micromonas Manton et Parke (Chlorophyta, Mamiellophyceae), of the Type Species M. pusilla (Butcher) Manton & Parke and of the Species M. commoda van Baren, Bachy and Worden and Description of Two New Species Based on the Genetic and Phenotypic Characterization of Cultured Isolates. Protist. 168(5):612–635. [DOI] [PubMed] [Google Scholar]
- Šlapeta J, López-García P, Moreira D.. 2006. Global dispersal and ancient cryptic species in the smallest marine eukaryotes. Mol Biol Evol. 23(1):23–29. [DOI] [PubMed] [Google Scholar]
- Sonneborn TM. 1937. Sex, sex inheritance and sex determination in Paramecium aurelia. Proc Natl Acad Sci U S A. 23(7):378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubrier J, et al. 2012. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol Biol Evol. 29(11):3345–3358. [DOI] [PubMed] [Google Scholar]
- Speijer D, Lukes J, Elias M.. 2015. Sex is a ubiquitous, ancient, and inherent attribute of eukaryotic life. Proc Natl Acad Sci U S A. 112(29):8827–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staben C, Yanofsky C.. 1990. Neurospora crassa a mating-type region. Proc Natl Acad Sci U S A. 87(13):4917–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehlow K. 1929. Über die Sexualität einiger Volvocales. Z Bot. 21(625):692. [Google Scholar]
- Suda S, Watanabe MM, Inouye I.. 1989. Evidence for sexual reproduction in the primitive green alga Nephroselmis olivacea (Prasinophyceae). J Phycol. 25(3):596–600. [Google Scholar]
- Sun Y, et al. 2012. Large-scale introgression shapes the evolution of the mating-type chromosomes of the filamentous ascomycete Neurospora tetrasperma. PLoS Genet. 8(7):e1002820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai IJ, , BensassonD, , BurtA, , Koufopanou V.. 2008. Population genomics of the wild yeast Saccharomyces paradoxus: Quantifying the life cycle. Proc Natl Acad Sci U S A. 105(12):4957–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzeng Y-H, Pan R, Li W-H.. 2004. Comparison of three methods for estimating rates of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 21(12):2290–2298. [DOI] [PubMed] [Google Scholar]
- Umen J, Coelho S.. 2019. Algal sex determination and the evolution of anisogamy. Annu Rev Microbiol. 73:267–291. [DOI] [PubMed] [Google Scholar]
- Umen JG. 2011. Evolution of sex and mating loci: an expanded view from Volvocine algae. Curr Opin Microbiol. 14(6):634–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandepoele K, et al. 2013. pico-PLAZA, a genome database of microbial photosynthetic eukaryotes. Environ Microbiol. 15(8):2147–2153. [DOI] [PubMed] [Google Scholar]
- Wang L, Berndt P, Xia X, Kahnt J, Kahmann R.. 2011. A seven-WD40 protein related to human RACK1 regulates mating and virulence in Ustilago maydis. Mol Microbiol. 81(6):1484–1498. [DOI] [PubMed] [Google Scholar]
- Wickham H. 2011. ggplot2. WIREs Comp Stat. 3(2):180–185. [Google Scholar]
- Wilson AM, Wilken PM, van der Nest MA., Wingfield MJ, Wingfield BD.. 2019. It’s all in the genes: the regulatory pathways of sexual reproduction in filamentous ascomycetes. Genes 10(5):330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Butler G.. 2017. Evolution of mating in the Saccharomycotina. Annu Rev Microbiol. 71:197–214. [DOI] [PubMed] [Google Scholar]
- Worden AZ, et al. 2009. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science 324(5924):268–272. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, et al. 2021. Three genomes in the algal genus Volvox reveal the fate of a haploid sex-determining region after a transition to homothallism. Proc Natl Acad Sci U S A. 118(21):e2100712118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 1995. A space-time process model for the evolution of DNA sequences. Genetics 139(2):993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic and transcriptomic sequence data have been deposited at GenBank under the accession numbers CAID00000000.1 (Ostreococcus tauri), PRJNA337288 (O. lucimarinus), PRJNA15676 (Micromonas commoda), PRJNA15678 (M. pusilla), PRJNA394752 (Bathycoccus prasinos), and PRJNA248394 (MMESTP). The accession numbers of the 18S rDNA sequences are summarized in supplementary table S6, Supplementary Material online.