

Abstract
Juvenile polyposis syndrome (JPS) is a clinically diagnosed hamartomatous polyposis syndrome that increases the risk of gastrointestinal cancer. Approximately 40–50% of JPS is caused by a germline disease-causing variant (DCV) in the SMAD4 or BMPR1A genes. The aim of this study is to characterize the phenotype of DCV-negative JPS and compare it to DCV-positive JPS. Herein we analyze a cohort of 145 individuals with JPS from nine institutions, including both pediatric and adult centers. Data analyzed included age at diagnosis, family history, cancer history, need for colectomy/gastrectomy, and polyp number and location. Compared to DCV-positive JPS, DCV-negative JPS was associated with younger age at diagnosis (p<0.001), lower likelihood of having a family history of JPS (p<0.001), and a lower risk of colectomy (p=0.032). None of the DCV-negative individuals had gastric or duodenal polyps, and polyp burden decreased after the first decade compared to DCV-positive JPS. Subgroup analysis between SMAD4 and BMPR1A carriers showed that SMAD4 carriers were more likely to have a family history of JPS and require gastrectomy. Taken together, these data provide the largest phenotypic characterization of individuals with DCV-negative JPS to date, showing that this group has distinct differences compared to JPS due to a SMAD4 or BMPR1A variant. Better understanding of phenotype and cancer risk associated with JPS both with and without a DCV may ultimately allow for individualized management of polyposis and cancer risk.
Keywords: Juvenile polyposis syndrome, SMAD4, BMPR1A, hereditary gastrointestinal cancer
Introduction
Juvenile polyposis syndrome (JPS) is a rare hereditary gastrointestinal polyposis syndrome with an increased risk of gastrointestinal cancer. Clinical diagnostic criteria for JPS include having 5 or more cumulative, pathologically defined juvenile polyps in the colon, at least one pathologically defined juvenile polyp from both the upper and lower intestinal tracts, or any number of juvenile polyps with a family history of JPS.(1) In some cases, JPS is caused by a pathogenic or likely pathogenic (P/LP) germline variant in either SMAD4 or BMPR1A, whose protein products are important components of the TGFβ-BMP signaling pathway.(2) The frequency of a SMAD4 or BMPR1A P/LP variant – henceforward collectively referred to as a disease-causing variant (DCV) - in individuals who meet clinical criteria for JPS is reported to be between 40–50%.(3)
Although several cohorts of JPS patients have been described (Table 1), these studies have either focused largely on patients with a known SMAD4 or BMPR1A DCV(4,5) or have not differentiated between those with or without an identifiable DCV.(6) These prior JPS cohorts have demonstrated variable cancer risk associated with JPS, ranging from 11% and 86%, in part due to changes in screening recommendations over time, the heterogeneous populations included in each study, as well as likely selection bias.(4–10) Although some studies support that SMAD4 DCV carriers have a more severe upper gastrointestinal polyposis and malignancy phenotype,(4,5,7,11) it remains difficult to determine the precise risk of gastrointestinal cancer and optimal risk-reducing strategies for JPS patients with and without a DCV given small sample sizes and cohort heterogeneity. For this reason, patients are recommended to undergo lifelong surveillance with endoscopy and colonoscopy, starting at time of diagnosis.(12–15)
Table 1:
Prior reported JPS cohorts
| Total (n) | DCV positive | Age at diagnosis (average, range) | Family history of JPS | Required colectomy or gastrectomy | Cancer prevalence | Age of cancer diagnosis | |
|---|---|---|---|---|---|---|---|
| Coburn 1995(9) | 218 | NR | 19 y (9 mo – 67 y) | 50% (n = 109) | 45% (n = 99) | 17% (n = 36) | 36 y (4 y – 60 y) |
| Howe 1998(10) | 29 | NR | 32 y (6 y – 68 y) | 100% | NR | 55% (n = 16) | 42 y colorectal (17 y – 68 y) 58 y upper (20 y – 72 y) |
| Brosens 2007(8) | 84 | NR | NR | NR | NR | 18% (n = 8) | 44 y |
| Latchford 2012(7) | 44 | 64% (n = 28) | 27 y (4 y – 57 y) | NR | 30% (n = 13) | 14% (n = 6) | 47 y |
| Aytac 2015(5) | 35 | 100% | 17 y (3 y – 65 y) | 83% (n = 29) | 69%(n = 24) | 11% (n = 4) | 38 y (29 y – 70 y) |
| Ishida 2017(6) | 171 | NR | 28 y (1 – 80 y) | NR | NR | 86% (n = 147) | NR |
NR = not recorded; y = years; mo = months
Quantifying cancer risk in individuals with JPS without a DCV remains particularly challenging given the paucity of data on clinical characteristics and cancer risks in this population. This multi-institutional study is the largest study to date comparing DCV-positive and DCV-negative JPS patients. The aim of this study is to characterize the differences between these two groups in order to understand the associated clinical phenotypes and cancer risks, and to help better inform risk management strategies for JPS.
Methods
Individuals were identified at nine different institutions including the Children’s Hospital of Philadelphia, Hospital of the University of Pennsylvania, Ann & Robert H. Lurie Children’s Hospital of Chicago, Texas Children’s Hospital, Dana-Farber Cancer Institute, University of Wisconsin, University of Pittsburgh Medical Center, Memorial Sloan Kettering Cancer Center, and Yale University. Each institution collected data for this study in accordance with an institution-specific IRB protocol and the US common rule, and patients were included per the individual institution recruitment period and exclusion/inclusion criteria. Where required by the institutional IRB protocol and US common rule regulations, written consent was obtained from participants. Deidentified data were transferred to the Children’s Hospital of Philadelphia and University of Pennsylvania group within established data use agreements.
Individuals with a diagnosis of JPS were identified from each institution; for consistency, a uniform data collection template and data dictionary was used by all centers in order to standardize measures and ensure consistency of data, and the resulting quality of the data was checked by the Children’s Hospital of Philadelphia investigators. Patients were included if they met clinical diagnostic criteria for JPS, including one of the following: 1) having 5 or more cumulative pathologically defined juvenile polyps in the colon, 2) at least one pathologically defined juvenile polyp from both the upper and lower intestinal tract, or 3) any number of juvenile polyps with a family history of JPS.(1) Complete genetic testing was defined as sequencing, deletion, and duplication analysis for the BMPR1A and SMAD4 genes; those without complete genetic testing were excluded from the analysis.
Data collected included gender, age at last follow-up, age at time of JPS diagnosis, genetic testing results, family history of a JPS diagnosis (defined as having a family member meeting clinical criteria for a JPS diagnosis), personal history of cancer, personal history of colectomy, and personal history of gastrectomy. All cancer types were included in the data set, but only gastrointestinal cancers were included in the analysis. The pediatric age group was defined as age of last follow up under 20 years. Information on gastrointestinal polyp burden was also obtained, and was defined per decade as none, low (1 to 10 polyps), and high (>10 polyps); these numbers included both upper and lower gastrointestinal (GI) polyps. Polyps were classified as upper GI if they were gastric or duodenal, and classified as lower GI if they were located in the colon or rectum; other small intestinal polyps were not tracked. The cutoff of 10 polyps was used for data consistency, as each institution recorded and obtained data from different clinical reporting materials (e.g., colonoscopy reports, pathology reports, clinical notes) and therefore in some circumstances the exact polyp number could not be determined. In addition, data on location of polyps and the presence of adenomas were collected; a distinction was not made between adenomas arising independently versus those arising from juvenile polyps.
Patients were only included in the final analysis if a pathogenic or likely pathogenic (P/LP) variant was identified in either SMAD4 or BMPR1A, or if there was documented negative genetic testing for both SMAD4 and BMPR1A, with no pathogenic, likely pathogenic, or variant of uncertain significance (VUS) identified in either gene. Patients with a VUS in SMAD4 or BMPR1A were excluded from analysis. No patients were known to have a pathogenic or likely pathogenic variant in any other polyposis gene, however comprehensive polyposis genetic testing outside of SMAD4 and BMPR1A was not required for inclusion. Data were analyzed in Stata statistical analysis software (v.16) using Wilcoxon Rank-Sum and Chi-squared analyses, where appropriate. For variables whose outcome might be biased by age (personal history of cancer, gastrectomy, and colectomy) analysis was completed through logistic regression controlling for age at last follow up.
Results
A total of 145 individuals with JPS from 137 families were collected from 9 institutions (Figure 1). Twenty-five patients were excluded given incomplete genetic testing, and two individuals were excluded for variants of uncertain significance in BMPR1A (c.1433G>A and c.1559_1560insTT). After exclusions, 118 individuals remained for analysis. Of the included individuals, 64 (54%) had no P/LP variant identified in SMAD4 or BMPR1A, hereafter referred to as disease-causing variant (DCV) negative JPS, and 54 (46%) had a P/LP variant identified in either SMAD4 (n= 27) or BMPR1A (n= 27), hereafter referred to as DCV-positive JPS. Of the BMPR1A P/LP variants identified, three individuals had a 10q23 deletion involving both PTEN and BMPR1A. When the pediatric JPS group (n=71) was analyzed separately, 22% had a DCV identified, whereas in the adults (n=47) 83% had a DCV identified (p<0.001).
Figure 1: JPS inclusion cohort.
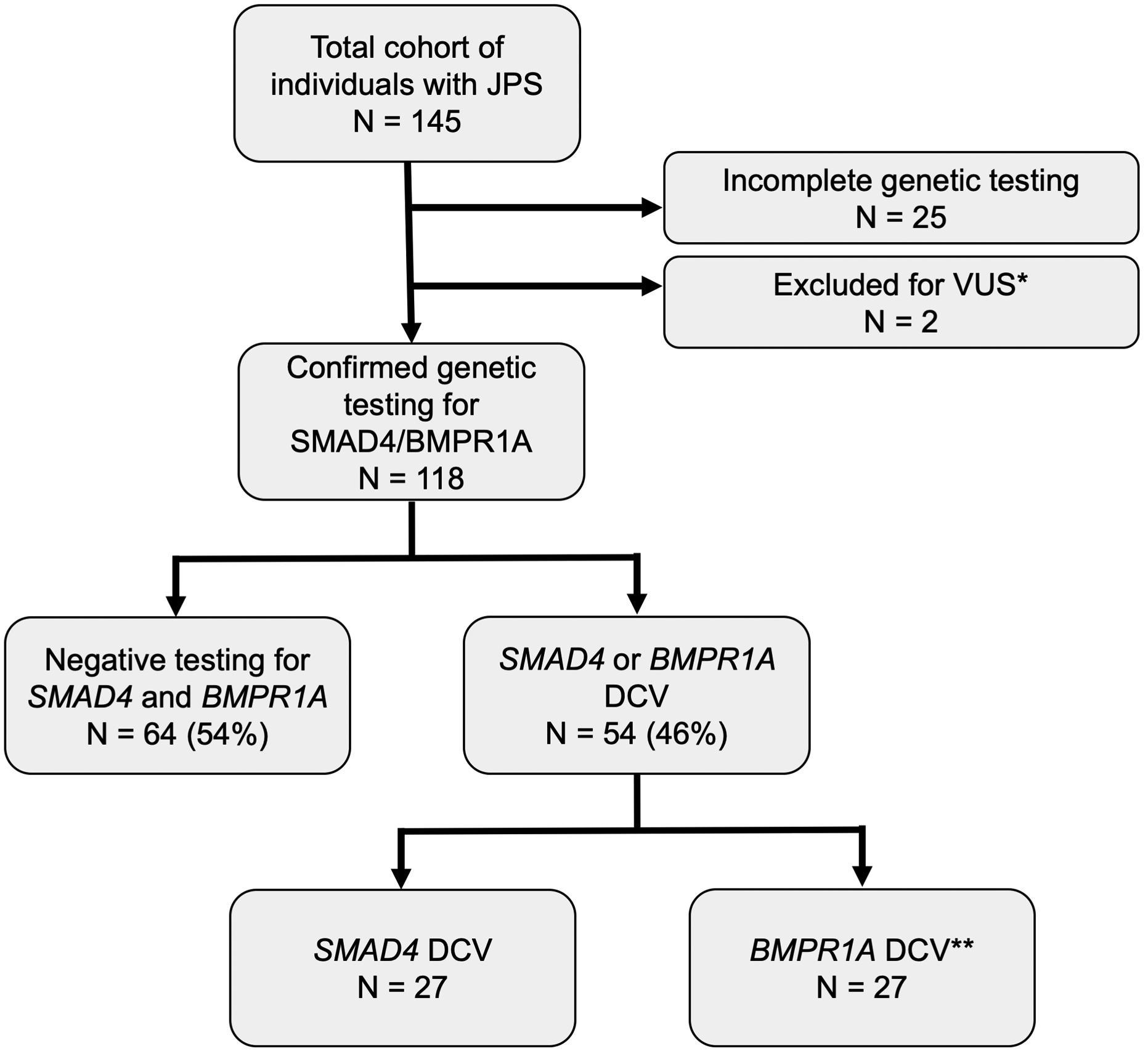
Schematic representation of individuals included in the JPS cohort for final analysis; DCV = disease-causing (pathogenic/likely pathogenic) variant
*BMPR1A VUS in two patients, excluded from analysis
**Includes three individuals with BMPR1A/PTEN inclusive 10q23 deletion
There were no statistically significant differences in gender between DCV-positive and DCV-negative individuals (Table 2). DCV-negative JPS patients were on average younger at diagnosis (median 5 years vs 18 years; p<0.001) and were less likely to have a family history of JPS compared to DCV-positive individuals (p<0.001) (Table 2). Individuals with DCV-negative JPS were also on average younger at last follow up (median 11 years, range 3–57 years) than those with a DCV (median 33.5 years, range 2–73 years, p<0.001), however the overall years of follow up were similar (median 4.5 years vs 5 years, p=0.350). In assessing polyp location, none of the DCV-negative group had upper GI (gastric/duodenal) polyps, whereas all had polyps in the lower GI tract (Table 2, p<0.001 for lower GI polyps in DCV-negative versus DCV-positive individuals). Additionally, no individuals in the DCV-negative JPS group developed adenomas, as compared to almost half (45%) of those in the DCV-positive group (p<0.001). For individuals with a DCV, DCV-specific subgroup analysis was also performed (Table 2). When SMAD4 carriers were compared to BMPR1A carriers, there were no statistically significant differences between gender, age at diagnosis, age at last follow up, location of polyps, and presence of adenomas. However, SMAD4 carriers were more likely to have a family history of JPS (p=0.016).
Table 2:
JPS demographic data and clinical history
| Disease-causing Variant Negative JPS (n=64) | Disease-causing Variant Positive JPS (n=54) | p-value | ||
|---|---|---|---|---|
| SMAD4 (n=27) | BMPR1A (n=27) | |||
| Female (%) | 42 | 56 | 0.123 | |
| 61 | 52 | 0.511 | ||
| Age at Diagnosis: Median (range) (years) | 5 (2 – 55) | 18 (1 – 72) | <0.001 | |
| 19 (2 – 54) | 18 (1 – 72) | 0.996 | ||
| Age at Last Follow Up: Median (range) (years) | 11 (3 – 57) | 33.5 (2 – 73) | <0.001 | |
| 37 (13 – 65) | 27 (2 – 73) | 0.394 | ||
| Years of Follow Up: Median (range) (years) | 4.5 (0–30) | 5 (0 – 48) | 0.350 | |
| 8 (0 – 48) | 2 (0 – 44) | <0.001 | ||
| Positive Family History for JPS (%) | 6 | 52 | <0.001 | |
| 56 | 48 | 0.016 | ||
| Presence of Adenomas (%) * | 0 | 45 | <0.001 | |
| 52 | 38 | 0.635 | ||
| Upper GI Polyps (%) * | 0 | 57 | <0.001 | |
| 60 | 55 | 0.839 | ||
| Lower GI Polyps (%) * | 100 | 87 | 0.003 | |
| 88 | 86 | 0.548 | ||
Demographic data, clinical history, polyp location, and adenoma frequency in individuals with JPS, subdivided by no DCV and SMAD4/BMPR1A DCV
Polyp and adenoma burden as measured by lifetime presence of upper (gastric/duodenal) polyps, lower (colonic/rectal) polyps, and adenomas (upper or lower)
Only two individuals (3%) in the DCV-negative group developed cancer, as compared with 14 individuals (26%) in the DCV-positive group; of the latter group, 10 developed 11 incidences of gastrointestinal cancer (19%) (Table 3, Table 4). In logistic regression controlling for age at last follow up, the difference in incidence of gastrointestinal cancer was not statistically significant (Table 3, 95% CI 0.4 – 45.5, p-value 0.208); non-gastrointestinal cancers were not included in this analysis. Two individuals (3%) in the DCV-negative group required a colectomy – one for polyp burden, one for cancer (ages 44 and 55) - and none required gastrectomy. In the DCV-positive group 18 individuals (33%, median age 24 years, range 5–72 years) required colectomy – one for cancer, 16 for polyp burden (including 3 with severe anemia), and one for diverticulitis - and 8 (15%, median age 38.5 years, range 15–58 years) required gastrectomy - five for polyp burden, 3 for cancer (Table 3). The difference in colectomy requirement was statistically significant (95% CI 1.2–34.2, p-value 0.03), whereas the need for gastrectomy was not statistically significant when analysis was controlled for age at last follow up. In subgroup analysis, SMAD4 carriers were more likely to require gastrectomy (p=0.02), but the incidence of gastrointestinal cancer and need for colectomy were not significantly different.
Table 3:
Gastrointestinal cancer and colectomy/gastrectomy incidence
| Disease-causing Variant Negative JPS (%) (n=64) | Disease-causing Variant Positive JPS (%) (n=54) | Odds Ratio | 95% Confidence Interval | p-value | |
|---|---|---|---|---|---|
| History of GI cancer (%) | 1.6 (n = 1) | 18.5 (n = 10) | 4.45 | 0.4 – 45.5 | 0.208 |
| Colectomy (%) | 3.1 (n = 2) | 33.3 (n = 18) | 6.40 | 1.2 – 34.3 | 0.030 |
| Gastrectomy (%) | 0 (n = 0) | 14.8 (n = 8) | 3.80 | 0.4 – 41.2 | 0.272 |
History of gastrointestinal cancer, need for colectomy, and need for gastrectomy in those with and without a disease-causing variant
Table 4:
Cancers reported in the JPS cohort
| Gene | P/LP Variant | Age of cancer diagnosis (years) | Type of cancer |
|---|---|---|---|
| BMPR1A | c.1433G>A | 26 | Colon |
| c.44_47delTGTT | 68 | Pancreas | |
| c.182G>A | 44 | Rectosigmoid | |
| SMAD4 | c.372_373dupTA | 45;64 | Gastric; Colon |
| c.692dupG | 29 | Gastric | |
| c.692dupG | 39 | Cervical | |
| c.692dupG | 46 | Esophageal | |
| c.1507_1508insATCC | 44 | Breast cancer | |
| c.1206dupT | 24 | Colon | |
| c.1447+2T>C | 48 | Papillary thyroid cancer | |
| c.403C>T | 39 | Colon | |
| c.1308+2T>G | 47 | Colon | |
| c.1231_1232delAG | 28 | Colon | |
| c.1245_1248delGACA | 15 | Gastric | |
| No disease-causing variant | NA | 55 | Colon |
| NA | 34 | Bladder |
Bolded cancers were considered within the JPS spectrum
To quantify differences in polyp burden in the JPS cohort, cumulative upper and lower gastrointestinal polyp burden was defined as absent, low (1 to 10 polyps), or high (>10 polyps) over each decade of life. The data on polyp burden is included in Figure 2, which delineates polyp burden over each decade of life, as available. Similar polyp burden was noted in the first decade of life. However, DCV-negative individuals had a lower polyp burden in the second and third decade of life. After the age of 40 the DCV-negative group was not large enough to track polyp burden.
Figure 2: JPS polyp burden by age.
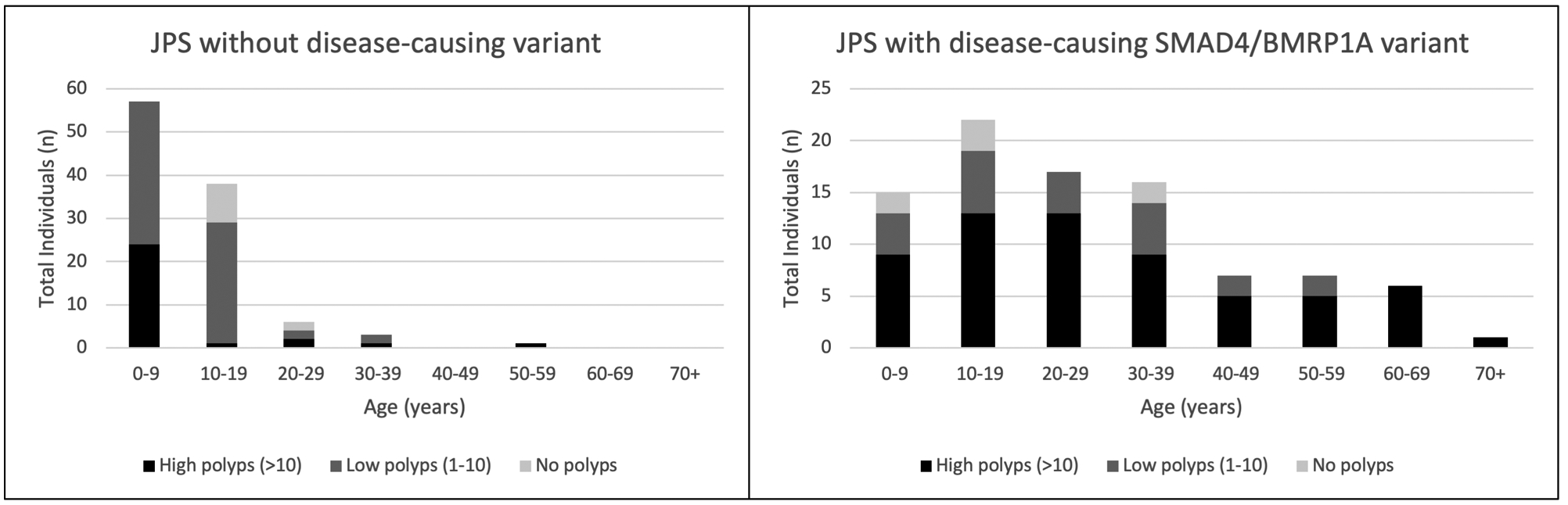
Representation of polyps present at high (>10), low (1–10), and none (0) polyps present (both upper and lower) in the 10-year age range
Discussion
JPS is a gastrointestinal cancer risk syndrome whose diagnosis and risk management are important for both pediatric and adult practitioners. Although prior studies have focused on the cancer risks associated with JPS,(6,7) there still remain limited data on the cancer risks of individuals with DCV-negative JPS, a subpopulation that accounts for at least half the JPS population. In this study, we present the findings from a large multi-institutional evaluation of individuals with JPS, which is the largest cohort to date to compare affected individuals with or without a DCV in SMAD4 or BMPR1A. As such, this cohort provides important insight into the under-characterized group of individuals with JPS who lack an identifiable BMPR1A or SMAD4 DCV.
The percentage of our study population with a DCV is similar to the prevalence of DCVs previously reported in JPS studies.(7,16) However, when divided into the pediatric and adult JPS based on age of last follow up, the pediatric group has a strikingly lower rate of DCVs identified (22%), compared to the adult group in which a large majority (83%) had a DCV. This difference was borne out in the polyposis data in adults, as few of the individuals followed at adult institutions were DCV-negative, which was accounted for in the statistical analysis of adult-onset outcomes such as cancer and need for gastrectomy or colectomy. This age difference could be explained by increasing diagnosis of DCV-negative JPS in pediatric populations, or decreased follow-up of these individuals with adult providers. It is also possible that in DCV-negative individuals the long-term cancer risk is lower or their polyp burden decreases after adolescence. Conversely, this could be a population that requires, but does not always receive, more intensive surveillance.
Our data also demonstrate that individuals with DCV-negative JPS were less likely to have a family history of JPS and were diagnosed at a younger age; a smaller study from 1998 also supported these findings.(10) However, given the small sample size, other differences between the groups in this prior study did not reach statistical significance.(10) We further demonstrate that individuals with DCV-negative JPS were significantly less likely to undergo colectomy compared to individuals with a DCV-positive JPS. However, given the younger age at last follow up of the DCV-negative group, this study was under-powered to show a difference in cancer incidence or need for gastrectomy. More extensive study of disease burden and cancer incidence of the DCV-negative group into adulthood will be necessary to better elucidate this population’s risk.
Differences in polyp burden over time between DCV-positive and DCV-negative groups was another notable phenotypic difference in our data. Although both the DCV-positive and DCV-negative JPS groups had significant polyp burden in the first decade of life, the DCV-negative JPS group had decreased polyp burden in the second and third decades, whereas the DCV-positive group maintained a persistently elevated polyp burden. Furthermore, DCV-negative individuals did not have upper gastrointestinal polyposis, a difference in polyp presentation that has not previously been noted. To further understand the lifetime polyposis risk, individuals with DCV-negative JPS need longer-term follow up in research registries with close documentation and tracking of polyp histology, location and number. While upper and lower GI polyps can often be managed endoscopically, some individuals in this JPS cohort also underwent gastrectomy or colectomy for polyp control. A higher percentage of those undergoing colectomy did so for polyp control compared to those undergoing gastrectomy, where a higher percentage did so for cancer. Although the severity of disease at time of gastrectomy/colectomy was not captured in this study, these differing rates may be attributed to the different morbidities, or perceived-morbidities, of these procedures, or may be due to differing effectiveness of endoscopic surveillance in the upper versus lower GI tract as well as differing thresholds for recommending surgery amongst different centers.
It has long been suspected that there may be additional disease-causing germline variants that have yet to be identified in patients with a clinical diagnosis of DCV-negative JPS. However, the lack of family history and younger age of diagnosis also suggest that there may be a low penetrance autosomal dominant variant, an autosomal recessive variant, mosaicism, or epigenetic change that is responsible for driving the phenotype. Additional genomic studies of this DCV-negative population, in particular, are required to better understand the underlying risk for JPS. Furthermore, this suggests that pediatric providers should consider underlying JPS in the setting of juvenile polyps, regardless of whether a family history of JPS exists. Should an individual have DCV-negative JPS, testing for other pathogenic germline variants, in genes such as ENG and PTEN should also be considered, especially in the setting of physical features such as macrocephaly. It is also possible that DCV-negative JPS is a distinct clinical phenotype and should be treated as such; these individuals could have an acquired phenotype rather than a syndromic phenotype, as isolated juvenile polyps in children are common. Should this be the case, it would imply that there is a population of patients with juvenile polyps that are not at high risk of cancer and do not require close longitudinal follow up throughout adulthood. This underscores the importance of designation and close longitudinal follow-up of DCV-negative JPS individuals in research registries to help better clarify their follow-up patterns in adulthood, as well as their long-term outcomes and cancer risks.
Although the main focus of our study was comparison of JPS with and without a DCV in SMAD4 or BMPR1A, subset analysis of individuals with a SMAD4 P/LP variant compared to those with a BMPR1A variant suggested that patients with a SMAD4 P/LP variant have a higher rate of malignancy, although this difference did not quite reach statistical significance (p=0.051). The overall cancer risk in the study population (13.4% of total patients, 8.4% when restricted to gastrointestinal cancers) is similar to risk estimates previously reported in other studies.(7) Of note, patients with 10q23 deletions involving both PTEN and BMPR1A are known to have a more severe JPS phenotype, with onset often in infancy and requiring early colectomy.(17,18) None of the three individuals with 10q23 deletions included in this study developed cancer, and their removal from the analysis led to no change in statistical significance of other measures, and thus they were included in the final analysis presented.
Limitations to this study include that data was collected from multiple different centers with differing levels of data granularity, which limits analysis of certain potential endpoints including: exact polyp number, origin of adenomas (isolated or arising from juvenile polyps), the severity of polyposis that led to decision to pursue gastrectomy or colectomy, other clinical features including history of hereditary hemorrhagic telangiectasia, and the detailed family history of relatives with reported JPS. Additionally, given the younger age of last follow-up of the DCV-negative JPS cohort, we had less data available on this cohort in older decades compared to the DCV-positive JPS cohort. Long term follow up of DCV-negative patients will be crucial to improved understanding of this cohort.
This study provides the largest characterization of a cohort of DCV-negative JPS to date. Our data provides evidence to suggest that given their different phenotype, DCV-negative JPS may be able to be surveyed and/or managed differently than those with JPS with an identifiable DCV, as has also been suggested by most recent update to recommendation by the National Comprehensive Cancer Network.(15) Additional research is needed to clinically distinguish those DCV-negative patients who may remain at risk for cancer throughout their lifetimes, and likely other germline changes remain to be identified in some DCV-negative individuals. At this time, we would still recommend following clinical guidelines for JPS as developed by expert consortia.(12,14) However, these data do suggest that there is a subset of individuals who meet clinical criteria for JPS, but have no identifiable DCV in SMAD4 or BMPR1A, who may not have the same polyp distribution and long-term risks as individuals with DCV-positive JPS.
Grant Support:
This work is supported by the Precious Jules Foundation (SPM), Alex’s Lemonade Stand Foundation (SPM), The Donaldson Fund (MBY, SSy, CU), the National Institutes of Health (K08DK106489 and R03DK120946, BWK), and the Lustgarten Family Colon Cancer Research Fund (BWK).
Abbreviations:
- DCV
Disease-causing Variant
- JPS
Juvenile Polyposis Syndrome
- GI
Gastrointestinal
- P/LP
Pathogenic/Likely Pathogenic
Footnotes
Conflicts of interest: BWK for Janssen Pharmaceuticals (travel) and Exact Sciences (consulting); SSy for Myriad Genetics and DC Health (consulting); no other potential conflicts of interest to declare.
References
- 1.Cichy W, Klincewicz B, Plawski A. Juvenile polyposis syndrome. Archives of medical science : AMS 2014;10(3):570–7 doi 10.5114/aoms.2014.43750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haug U, Kuntz KM, Knudsen AB, Hundt S, Brenner H. Sensitivity of immunochemical faecal occult blood testing for detecting left- vs right-sided colorectal neoplasia. Br J Cancer 2011;104(11):1779–85 doi 10.1038/bjc.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calva-Cerqueira D, Chinnathambi S, Pechman B, Bair J, Larsen-Haidle J, Howe JR. The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clin Genet 2009;75(1):79–85 doi 10.1111/j.1399-0004.2008.01091.x. [DOI] [PubMed] [Google Scholar]
- 4.Blatter R, Tschupp B, Aretz S, Bernstein I, Colas C, Evans DG, et al. Disease expression in juvenile polyposis syndrome: a retrospective survey on a cohort of 221 European patients and comparison with a literature-derived cohort of 473 SMAD4/BMPR1A pathogenic variant carriers. Genet Med 2020. doi 10.1038/s41436-020-0826-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aytac E, Sulu B, Heald B, O’Malley M, LaGuardia L, Remzi FH, et al. Genotype-defined cancer risk in juvenile polyposis syndrome. The British journal of surgery 2015;102(1):114–8 doi 10.1002/bjs.9693. [DOI] [PubMed] [Google Scholar]
- 6.Ishida H, Ishibashi K, Iwama T. Malignant tumors associated with juvenile polyposis syndrome in Japan. Surg Today 2017. doi 10.1007/s00595-017-1538-2. [DOI] [PubMed] [Google Scholar]
- 7.Latchford AR, Neale KF, Phillips RK, Clark SK. Juvenile polyposis syndrome: A study of genotype, phenotype, and long-term outcome. Diseases of the Colon and Rectum 2012;55(10). [DOI] [PubMed] [Google Scholar]
- 8.Brosens LA, van Hattem A, Hylind LM, Iacobuzio-Donahue C, Romans KE, Axilbund J, et al. Risk of colorectal cancer in juvenile polyposis. Gut 2007;56(7):965–7 doi 10.1136/gut.2006.116913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coburn MC, Pricolo VE, DeLuca FG, Bland KI. Malignant potential in intestinal juvenile polyposis syndromes. Annals of Surgical Oncology 1995;2(5):386–91. [DOI] [PubMed] [Google Scholar]
- 10.Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Annals of Surgical Oncology 1998;5(8):751–6. [DOI] [PubMed] [Google Scholar]
- 11.Sayed M, Ahmed A, Ringold J, Anderson M, Blair J, Mitros F, et al. Germline SMAD4 or BMPR1A mutations and phenotype of juvenile polyposis. Annals of Surgical Oncology 2002;9(9):901–6 doi 10.1245/ASO.2002.03.021. [DOI] [PubMed] [Google Scholar]
- 12.Cohen S, Hyer W, Mas E, Auth M, Attard TM, Spalinger J, et al. Management of Juvenile Polyposis Syndromes in Children and Adolescents: A Position Paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr 2018. doi 10.1097/MPG.0000000000002246. [DOI] [PubMed] [Google Scholar]
- 13.Achatz MI, Porter CC, Brugieres L, Druker H, Frebourg T, Foulkes WD, et al. Cancer Screening Recommendations and Clinical Management of Inherited Gastrointestinal Cancer Syndromes in Childhood. Clin Cancer Res 2017;23(13):e107–e14 doi 10.1158/1078-0432.CCR-17-0790. [DOI] [PubMed] [Google Scholar]
- 14.Genetic/Familial High-Risk Assessment: Colorectal. NCCN Clinical Practice Guidelines in Oncology 2018;V1.2018. [Google Scholar]
- 15.Genetic/Familial High-Rish Assessment: Colorectal. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology 2020;V1.2020. [Google Scholar]
- 16.Howe JR. The prevalence of MADH4 and BMPR1A mutations in juvenile polyposis and absence of BMPR2, BMPR1B, and ACVR1 mutations. Journal of Medical Genetics 2004;41(7):484–91 doi 10.1136/jmg.2004.018598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alimi A, Weeth-Feinstein LA, Stettner A, Caldera F, Weiss JM. Overlap of Juvenile polyposis syndrome and Cowden syndrome due to de novo chromosome 10 deletion involving BMPR1A and PTEN: implications for treatment and surveillance. American journal of medical genetics Part A 2015;167(6):1305–8 doi 10.1002/ajmg.a.36876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oliveira PH, Cunha C, Almeida S, Ferreira R, Maia S, Saraiva JM, et al. Juvenile polyposis of infancy in a child with deletion of BMPR1A and PTEN genes: surgical approach. Journal of pediatric surgery 2013;48(1):e33–7 doi 10.1016/j.jpedsurg.2012.09.067. [DOI] [PubMed] [Google Scholar]