

Abstract
Epilepsy is a neurological disorder afflicting ~65 million people worldwide. It is caused by aberrant synchronized firing of populations of neurons primarily due to imbalance between excitatory and inhibitory neurotransmission. Hence, the historical focus of epilepsy research has been neurocentric. However, the past two decades have enjoyed an explosion of research into the role of glia in supporting and modulating neuronal activity, providing compelling evidence of glial involvement in the pathophysiology of epilepsy. The mechanisms by which glia, particularly astrocytes and microglia, may contribute to epilepsy and consequently could be harnessed therapeutically are discussed in this Review.
Since the time of the Greek philosopher Hippocrates, epilepsy, previously called the ‘shaking palsy’, has been a fascinating, yet often horrifying, syndrome. Once believed to be the result of demonic possession, the invention of electroencephalography in 1929 transformed epilepsy into a well-defined clinical syndrome that became synonymous with the presence of electrographic seizures (that is, abnormal brain waves resulting from hypersynchronous discharge of large populations of neurons in the brain)1. As per the current standard of the International League Against Epilepsy, an epilepsy diagnosis requires at least two unprovoked (or reflex) seizures occurring >24 h apart, one unprovoked (or reflex) seizure and a probability of further seizures similar to the general recurrence risk after two unprovoked seizures occurring over the next 10 years, or a diagnosis of an epilepsy syndrome2. Seizures can range from subtle, absent staring into space (absence) to profound convulsions of the entire body (tonic–clonic). Seizures can be focal, affecting only a discrete part of the brain, or generalized, encompassing larger brain regions in both hemispheres. Epilepsy affects >1% of the worldwide population, making it the most common neurological illness.
The invention of phenobarbital in 1912 was a turning point in the management of epilepsy, providing at least two-thirds of all affected individuals with seizure freedom and an excellent quality of life. Active research now focuses on providing the same for treatment-resistant patients. Much of this research has focused on how to dampen hypersynchronous discharge and to explain how changes in intrinsic neuronal activity cause such discharge. This research focus led to the widely accepted hypothesis that an imbalance of excitatory and inhibitory currents is largely responsible for epileptic seizures. Possible causes for these imbalances are manifold and include changes in neurotransmitters and mutations in their receptors, ion channels and ion transporters as well as altered network connectivity. Meaningful insight has also come from the study of rare genetic causes of epilepsy, which showed that even subtle changes in a single protein can drive the disease3; however, in most instances epilepsy appears to be a polygenic disorder.
Phenobarbital.
An anticonvulsant that acts by activating inhibitory postsynaptic neuronal GABA type A (GABAA) receptors. Phenobarbital can activate GABAA receptors independent of GABA, but it also potentiates the effects of GABA.
On the basis of the presumed underlying causes, epilepsy is classified into two broad categories: genetic, or idiopathic, epilepsy is due to a genetic predisposition of the brain to generate seizures, whereas acquired epilepsy results from a known lesion or acute insult that establishes a series of alterations in cellular, molecular and physiological properties that give rise to seizures. The gradual transformation from a formerly healthy brain into one plagued by spontaneous seizures is called epileptogenesis and can be divided into three phases4. First, a brief event or insult, for example, stroke or head trauma, initiates cellular and molecular changes in the affected brain regions. Second, a latent period starts with (tumour or encephalitis) or immediately after (stroke or trauma) the insult and lasts until chronic, unprovoked seizures occur. This can be months or even years later, and behavioural seizures are not apparent during this period. This is the time window during which non-neuronal changes, discussed extensively below, may profoundly contribute to the eventual disease presentation. Third, after the manifestation of recurring seizures, molecular and cellular changes often continue to exacerbate the severity of the disease.
Idiopathic.
Any condition or disease that occurs spontaneously or with an unknown aetiology.
Various biological changes have been described to occur during epileptogenesis and include gliosis5; uncontrolled inflammation6; an impaired blood–brain barrier (BBB)7; neurodegeneration8; aberrant neurogenesis9; axonal and dendritic plasticity; changes in neural circuits10; structural and functional changes in receptors, ion channels, transporters and enzymes implicated in excitatory and inhibitory neurotransmission11; reorganization of the extracellular matrix (ECM)12; and epigenetic reprogramming13. These changes may differ qualitatively and quantitatively depending on the precipitating insult or event. Although a majority of epilepsy research has focused on intrinsic neuronal properties and neuronal network function, over the past two decades, evidence for non-neuronal contributions to epilepsy has surfaced with increasing frequency. The evidence that the neuronal environment may be critically altered in epilepsy and even involved in disease aetiology now warrants closer examination, and this is the principal objective of this Review. Here, we highlight evidence that supports the viewpoint that non-neuronal cells may contribute to disease development, disease progression or both. We hope to stir broader conversation to consider astrocytes, microglia, vascular and immune cells and even the ECM produced by neurons and non-neuronal cells as important players in epilepsy and possibly even as potential therapeutic targets in the future.
Gliosis
The presence of a prominent gliotic scar has become a hallmark of chronic focal epilepsy5; indeed, gliosis is probably omnipresent in all forms of epilepsy. Gliosis refers to a spectrum of physicochemical and physiological changes in glial cells, particularly astrocytes and microglia, that occur universally in response to diverse forms of CNS insults and diseases5. The extent and nature of these changes vary with the type and severity of the injury. Defining features of gliosis include hypertrophy of the cell bodies and processes; upregulated expression of several proteins, including intermediate filament proteins (glial fibrillary acidic protein (GFAP) and vimentin) in astrocytes and ionized calcium-binding adaptor molecule 1 (IBA1) and CD68 in microglia; cellular proliferation; and the formation of a tenacious scar rich in ECM molecules such as chondroitin sulfate proteoglycans (CSPGs)14. Gliosis may impart either beneficial or detrimental effects on nearby cells in a highly context-dependent manner5. Reactive glia release cytokines, chemokines, growth factors and other molecules to mitigate or prevent the brain damage due to insult by controlling inflammation, removing debris and inducing synaptic plasticity and tissue remodelling. However, unrestrained reactive gliosis may cause excessive inflammation, neuronal death and tissue damage. Recent findings suggest that reactive astrocytes can assume distinct neuroprotective or neurotoxic phenotypes explaining the diametric role of reactive glia in CNS pathophysiology15.
Gliotic scar.
A dense fibrous mass of reactive glia formed in response to CNS injury. It is a part of the tissue-remodelling process that occurs following injury and provides both beneficial and detrimental effects in a context-dependent manner.
The question of whether gliosis is a cause or consequence of seizures is debatable. In the case of trauma-associated epilepsy, gliosis precedes the development of epilepsy and hence could be causative. The best evidence for gliosis being causative of epilepsy comes from studies in which gliosis was induced by the conditional astrocyte-specific deletion of the β1 integrin gene Itgb1. These animals showed widespread gliosis without other pathologies16 and developed spontaneous, recurrent seizures, suggesting that astrogliosis alone was sufficient to cause epilepsy. An unrelated study similarly found that virally induced astrogliosis in the hippocampus was sufficient to cause neuronal hyperexcitability17. Furthermore, astrocyte-specific deletion of the Tsc1 gene, which causes tuberous sclerosis complex, induces abnormal neuronal organization and astrogliosis, and this disease also presents with epileptic seizures18. Lastly, seizures are one of the most common symptoms of Alexander disease, caused by heterozygous mutations in the astrocyte-specific GFAP gene. Astrocytes in affected individuals express insufficient glutamate transporters, causing excitotoxic neuronal loss and seizures, also supporting a causal link between astrocytic dysfunctions and epileptic seizures19,20. Note that reactive astrogliosis appears to be present in most animal models of acquired epilepsies as well as in tissues from people with epilepsy and, as the above studies suggest, is typically associated with epilepsy. However, there are examples in the literature in which the interdependence between seizures and astrogliosis appears absent or questionable. For instance, one study that examined tissue resected from people with focal cortical dysplasia found an absence of gliosis in the nonlesional epileptogenic cortex21. In addition, in pilocarpine-treated epileptic mice, seizure frequency did not correlate with reactive gliosis but instead with a loss of GABAergic interneurons in the dentate gyrus22. Taken together, these findings show that gliosis has been unequivocally acknowledged as an integral part of epilepsy histopathology and as a likely contributor to epileptogenesis in many, but not all, forms of epilepsy. How gliosis contributes to seizures is an area of active investigation, and the possible mechanisms are discussed extensively below and summarized in FIG. 1 and TABLE 1.
Fig. 1 |. Interplay between CNS insult, reactive gliosis and seizures.
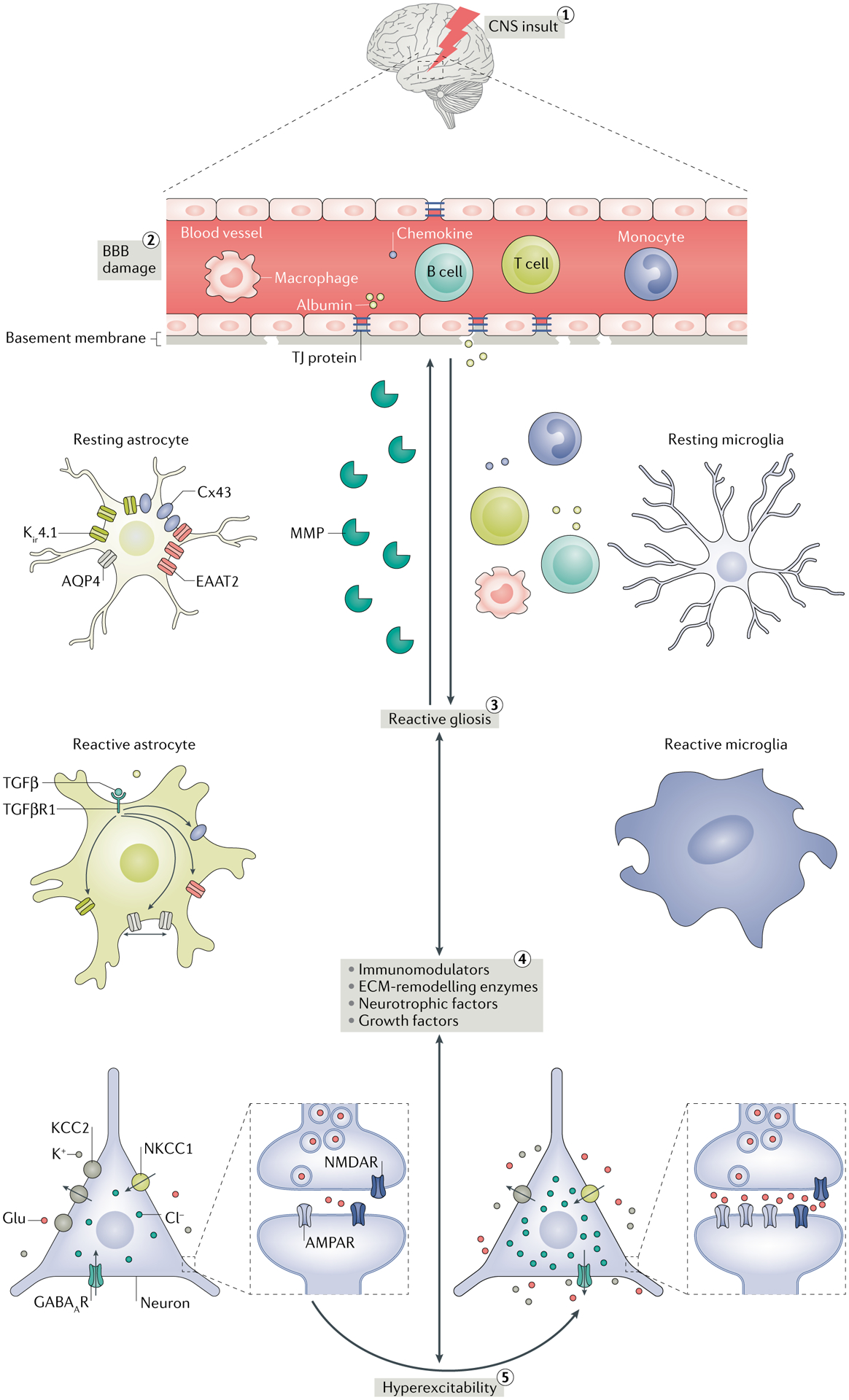
(1) Various CNS insults such as stroke, infection, trauma, tumours and hypoxia can cause blood–brain barrier (BBB) damage. (2) The resulting increased permeability of the BBB promotes extravasation of serum albumin, peripheral immune cells (macrophages, monocytes, B cells and T cells) and chemokines into the brain parenchyma, where, along with direct CNS insult, they induce reactive gliosis. (3) Reactive gliosis refers to morphological (hypertrophy of the cell bodies and processes) and physiological changes (changes in the expression and functions of many glial proteins) in glial cells in response to various CNS injuries. (4) Reactive glia release extracellular matrix (ECM)-remodelling enzymes such as matrix metalloproteinases (MMPs), neurotrophic factors such as brain-derived neurotrophic factor (BDNF), growth factors such as transforming growth factor-β (TGFβ) and immunomodulators such as cytokines that modulate the expression and function of receptors, transporters, enzymes and other molecules implicated in the functions of excitatory and inhibitory neurotransmission by a variety of mechanisms. MMPs can aggravate BBB impairment by damaging the tight junction (TJ) connections and promote the infiltration of serum proteins in the brain. In addition to TGFβ, serum albumin is known to activate TGFβ receptor 1 (TGFβR1) signalling in astrocytes by directly interacting with TGFβR1. Accordingly, the membrane expression of inwardly rectifying potassium channel 4.1 (Kir4.1), excitatory amino acid transporter 2 (EAAT2) and the gap junction protein connexin 43 (Cx43) are decreased. Activation of TGFβR1 also changes the trafficking and surface expression of aquaporin 4 (AQP4). As a result, extracellular concentrations of K+ and glutamate (Glu) increase, which promotes network hyperexcitability. Control of Cl− concentration is also crucial for regulating hyperexcitability. The K+–Cl− cotransporter (KCC2; a Cl− exporter) and the Na+–K+–Cl− cotransporter 1 (NKCC1; a Cl− importer) play a crucial role in maintaining a low concentration of intracellular Cl−, which is essential for maintaining the inhibitory activity of GABAergic neurotransmission. BDNF decreases the membrane expression of KCC2, causing net accumulation of Cl− intracellularly and reversal of Cl− flux through GABA type A receptor (GABAAR). Therefore, the activation of GABAAR paradoxically causes hyperexcitation. Cytokines (for example, tumour necrosis factor (TNF)) released from reactivated glia can increase the postsynaptic density of AMPA receptors (AMPARs) and excitatory neurotransmission. (5) As a consequence, network hyperexcitability and seizures occur owing to impairment in K+, Cl− and glutamate buffering and imbalance in excitatory and inhibitory neurotransmission. Importantly, seizures can aggravate the gliotic condition by further increasing the release of neuromodulator molecules, setting the vicious cycle of gliosis and seizures. NMDAR, NMDA receptor.
Table 1 |.
Glial mechanisms that contribute to ictogenesis and epileptogenesis
| Glial factor and/or pathway | Function | Biochemical changes in epilepsy | Mechanism of hyperexcitability | Refs |
|---|---|---|---|---|
| Kir4.1 |
|
|
|
28,29,35 |
| AQP4 |
|
|
|
25,46 |
| Connexins (GJ proteins) | Ionic and biochemical coupling between CNS cells |
|
Impaired spatial redistribution of K+ and glutamate | 29,50,51 |
| NKCC1 | Moves Cl− into the cell | Increased expression | Impaired GABAergic inhibition due to increased intracellular Cl− and depolarizing shift in the reversal potential of GABAAR | NA |
| KCC2 | Moves Cl− into the extracellular space | Decreased expression | Impaired GABAergic inhibition due to increased intracellular Cl− and depolarizing shift in the reversal potential of GABAAR | NA |
| GLT1 or EAAT2 | Clearance of extracellular glutamate | Results of expression studies in human epileptic tissues inconsistent | Increased extracellular glutamate | 25,57 |
| GS |
|
|
|
17,66,67 |
| Adenosine kinase | Phosphorylates adenosine (endogenous anticonvulsant) and terminates its action | Increased expression | Decreased level of adenosine | 95 |
| Gliotransmitters | Regulation of neurotransmission and synaptic plasticity | Increased release of glutamate and ATP | Increased activation of glutamatergic and purinergic signalling | 91,134 |
| BBB | Astrocyte end feet support the maintenance of BBB integrity |
|
|
123,125,135 |
AQP4, aquaporin 4; BBB, blood–brain barrier; EAAT2, excitatory amino acid transporter 2; GABAAR, GABA type A receptor; GJ, gap junction; GLT1, glutamate transporter 1; GS, glutamine synthetase; KCC2, K+–Cl− cotransporter; Kir 4.1, inwardly rectifying potassium channel 4.1; NA, not available; NKCC1, Na+–K+–Cl− cotransporter 1; MTLE, mesial temporal lobe epilepsy; TGFβ, transforming growth factor-β.
Sclerosis.
A pathological stiffening of tissue at the injury site due to overgrowth of fibrous connective tissue that replaces original tissue.
Ion and water homeostasis
Neuronal activity hinges on Na+ and K+ ions moving across the cell membrane into or out of the extracellular space (ECS). This space is very small in volume and therefore even modest ion fluxes can change ion concentrations considerably. Indeed, a single action potential raises extracellular K+ concentration ([K+]o) by almost 1 mM, and during seizures repeated neuronal firing increases [K+]o from a resting level of ~3 mM to ~12 mM (REFS23,24). In normal brain, [K+]o is quickly readjusted to ~3 mM by the neuronal Na+/K+-ATPase, and if necessary excessive extracellular K+ can also be cleared via astrocytic inwardly rectifying potassium (Kir) channels. Once inside the astrocyte, K+ is spatially buffered via redistribution through gap junctions (GJs) into the syncytial network of GJ-coupled astrocytes25.
A failure to adequately buffer K+ has been extensively discussed as one way in which gliotic astrocytes may contribute to epilepsy, particularly because simply increasing [K+]o is sufficient to induce robust epileptiform activity in hippocampal slices from animals or humans26,27. Astrocytic K+ buffering is mainly attributed to Kir4.1, the most abundant Kir channel in the CNS that is predominantly expressed in astrocytes28. Loss of Kir4.1 expression and function has been reported in multiple animal models of acquired epilepsy and in tissues from people with epilepsy (reviewed in REF.29). In individuals with mesial temporal lobe epilepsy (MTLE) and hippocampal sclerosis, downregulation of Kir4.1 has been found most prominently in the perivascular domains of astrocytic end feet30, and patch clamp studies show a reduction in inwardly rectifying K+ currents in the astrocytes of sclerotic hippocampus samples from patients with pharmacoresistant TLE31. Furthermore, astrocyte-specific deletion of Kir4.1 in mice impairs K+ uptake and causes ataxia, seizures and premature death32. However, even stronger evidence for the role of Kir4.1 in epilepsy comes from quantitative trait loci studies that identified an association between variations in the KCNJ10 gene that encodes Kir4.1 and seizure susceptibility in humans and mice33,34. Note that, in addition to activity-dependent K+ clearance, Kir4.1 also contributes substantially to setting the resting membrane potential of astrocytes28, which provides thermodynamic energy for the clearance of glutamate via Na+-dependent electrogenic glutamate transporters. Indeed, RNAi-mediated knockdown of Kir4.1 decreased both K+ and glutamate uptake in cultured cortical astrocytes35. Taken together, these findings indicate that reactive astrocytes associated with seizure foci show decreased expression and function of Kir4.1, thereby causing dysregulated clearance of extracellular K+ and glutamate, which both directly contribute to neuronal hyperexcitability (FIG. 1).
Similar to that of K+, the transmembrane distribution of Cl− ions is crucial in the maintenance of normal neuronal activity, as the inhibitory action of GABA type A receptors (GABAARs) is governed by the transmembrane Cl− gradient. Two transporters, the Na+–K+–Cl− cotransporter (NKCC1) and the K+–Cl− cotransporter 2 (KCC2), are responsible for shuttling Cl− in and out of the cell, respectively. In mature neurons, intracellular Cl− concentration ([Cl−]i) is low as it is being shuttled into the ECS via KCC2 (REF.36). Loss-of-function mutations in KCC2 are known to cause an increased risk of epilepsy in humans37, and human and animal studies show reduced expression of KCC2 in the brain during epileptogenesis37. Glutamate is one of among several factors that can affect the expression and function of KCC2. Excessive levels of glutamate, present during seizures, activate NMDA receptors (NMDARs) and can cause dephosphorylation of the KCC2 protein via protein phosphatase 1 and, in turn, decrease membrane expression of KCC2 (REF.38). As a result, the efflux of Cl− into the ECS decreases, [Cl−]i increases and results in a depolarizing shift in the reversal potential of GABAAR, reducing its inhibitory activity and hence contributing to hyperexcitability36. As astrocytes are crucially important in maintaining the extracellular levels of glutamate, it is likely that astrocyte dysfunction in the epileptic condition may contribute to hyperexcitability via elevated levels of glutamate and impaired GABAAR-mediated inhibition (FIG. 1). In addition to glutamate, reactive glia, especially microglia, are known to increase the synthesis and release of brain-derived neurotrophic factor (BDNF), which has been shown likewise to reduce the membrane expression of KCC2 via TrkB-mediated signalling39.
Ion movement across the cell membrane is accompanied by obligate transmembrane water flux to maintain osmotic balance. As a consequence, neuronal activity is directly associated with water flux, which changes the composition and size of the ECS. Water transport across the membrane occurs through channels known as aquaporins (AQPs). AQP4 is highly expressed by astrocytes, particularly in the neuropil and their end-foot processes where they connect with the blood vessels40. Changes in AQP4 expression have been observed in cases of animal and human epilepsy (reviewed in REF.41), and elimination of AQP4 by genetic deletion or by RNAi impairs water transport in astrocytes42,43. AQP4 is anchored to the astrocytic membrane through a dystrophin-associated protein complex composed of α-syntrophin or dystrophin. Mice with genetic deletions of α-syntrophin or dystrophin show reduced expression of AQP4 in the astrocyte end feet around the blood vessels44,45 and present with severe seizures when challenged by hyperthermia or pentylenetetrazole. Given that AQP4 and Kir4.1 colocalize in astrocyte end feet46 and that the flux of ions and water through them changes the size of the ECS, it is commonly suggested that AQP4 functions in concert with Kir4.1 for both K+ and water homeostasis. This is important, as changing the volume of the ECS alone is sufficient to alter seizure susceptibility and epileptogenesis47. Indeed, loss or mislocalization of AQP4 expression in the perivascular end feet of astrocytes in Aqp4−/− or Snta1−/− mice is associated with decreased clearance of synaptic K+, impaired K+ buffering and severe and prolonged seizures48,49. Taken together, increasing evidence suggests that the colocalization of Kir4.1 and AQP4 at astrocytic end feet on blood vessels is important to mediate effective K+ and water clearance into blood vessels and that functional loss of either Kir4.1 or AQP4 enhances seizure susceptibility.
Pentylenetetrazole.
A convulsant that acts by directly antagonizing GABA type A receptor-mediated inhibitory neurotransmission and therefore is often used experimentally to induce seizures in animals.
Similar to neuronal networks, astrocytes are connected into a syncytial network formed by GJ channels composed of connexin proteins (Cxs)50. GJ coupling enables intercellular movement of ions (ionic coupling) and small molecules (biochemical coupling) between astrocytes as well as between astrocytes, neurons and the vasculature50. Several important functions have been attributed to astrocytic GJ coupling, including spatial buffering of K+ and glutamate, trafficking and delivery of energetic metabolites from blood vessels to neurons, intercellular propagation of Ca2+ waves and regulation of cell volume (reviewed in REF.29). Naturally, any change in the GJ coupling could impact any one of these astrocytic functions and may contribute, in turn, to epilepsy. At the expression level, both increases and decreases in transcripts and Cxs have been reported in animal models and in humans with epilepsy (reviewed in REFS50,51). However, whether these changes translate into changes in function is not clear. Post-translational modifications of Cxs can alter trafficking of Cxs, assembly and subcellular localization of the GJs, as well as gating of the GJ channels52. Functional studies of coupling between astrocytes of people with MTLE with hippocampus sclerosis found a complete absence of functional GJ coupling in the sclerotic hippocampus in tissue from 75 individuals, yet abundant coupling was present in the nonsclerotic hippocampal specimens53. A reduction in astrocytic coupling may cause hyperexcitability owing to impaired spatial buffering of K+ and glutamate, whereas reduced coupling may also restrict the supply of energy metabolites and hinder the propagation of Ca2+ waves, thereby dampening the initiation and synchronization of neuronal activity. Pharmacological approaches have also been used to investigate the role of astrocytic GJ coupling in the pathophysiology of epilepsy and generally report that the GJ inhibitors reduce seizures51; however, one study found a proconvulsive effect54. Note however that the GJ inhibitors cannot differentiate between astrocytic and neuronal Cx isoforms, making it difficult to ascribe an unequivocal role to astrocytic GJs from such studies51. In conclusion, an abundance of evidence suggests changes in astrocytic GJ expression and function, but ascribing a clear mechanistic role to these changes as drivers of epileptogenesis remains elusive.
Neurotransmitters and energy metabolites
Increased extracellular glutamate is a biochemical hallmark of animal and human epileptic tissues and is believed to cause neurotoxicity and seizures55,56. One of the most widely accepted roles of astrocytes in normal brain physiology is the clearance of neuronally released glutamate57. This is accomplished via one of two Na+-coupled electrogenic excitatory amino acid transporters, EAAT1 and EAAT2, that couple the import of one glutamate with three Na+ ions and the release of one K+ ion (REF.58). EAAT1 is primarily expressed early during development, whereas EAAT2 is the principal astrocyte-specific glutamate transporter in the adult brain. The rodent versions of EAAT1 and EAAT2 are known as glutamate aspartate transporter (GLAST) and glutamate transporter 1 (GLT1), respectively. Rapid astrocytic clearance of excess synaptic glutamate following synaptic transmission terminates its action, controls excitatory neurotransmission and prevents accidental activation of extrasynaptic NMDARs that are coupled to proapoptotic pathways. Once taken up by astrocytes, glutamate must be sequestered and degraded because the glutamate transporters are bidirectional and increased glutamate concentration within astrocytes will eventually cause its release into synapses. Some of the intracellular glutamate is degraded by conversion via glutamate dehydrogenase into α-ketoglutarate, which is then fuelled into the tricarboxylic acid (TCA) cycle, and thereby glutamate serves as an energy source57. A large portion of the remaining intracellular glutamate is converted to glutamine via the glutamine synthetase (GS) pathway and released into the interstitial space where adjacent neurons import it for glutamate synthesis via a phosphate-activated glutaminase (PAG). This is commonly dubbed the glutamate–glutamine cycle57 (FIG. 2). In excitatory neurons, glutamate is packaged into secretory vesicles to be released into the synapse, whereas in GABAergic inhibitory neurons, glutamate is converted into GABA by glutamate decarboxylase (GAD). Hence, astrocytes are in the supply chain for both vesicular glutamate and GABA, and interfering with the glutamate–glutamine cycle at any stage will crucially impact extracellular glutamate levels, GABA and glutamate supply and synaptic function and thus may be causally associated with epileptogenesis.
Fig. 2 |. Regulation of synaptic transmitter homeostasis and energy metabolites by astrocytes.
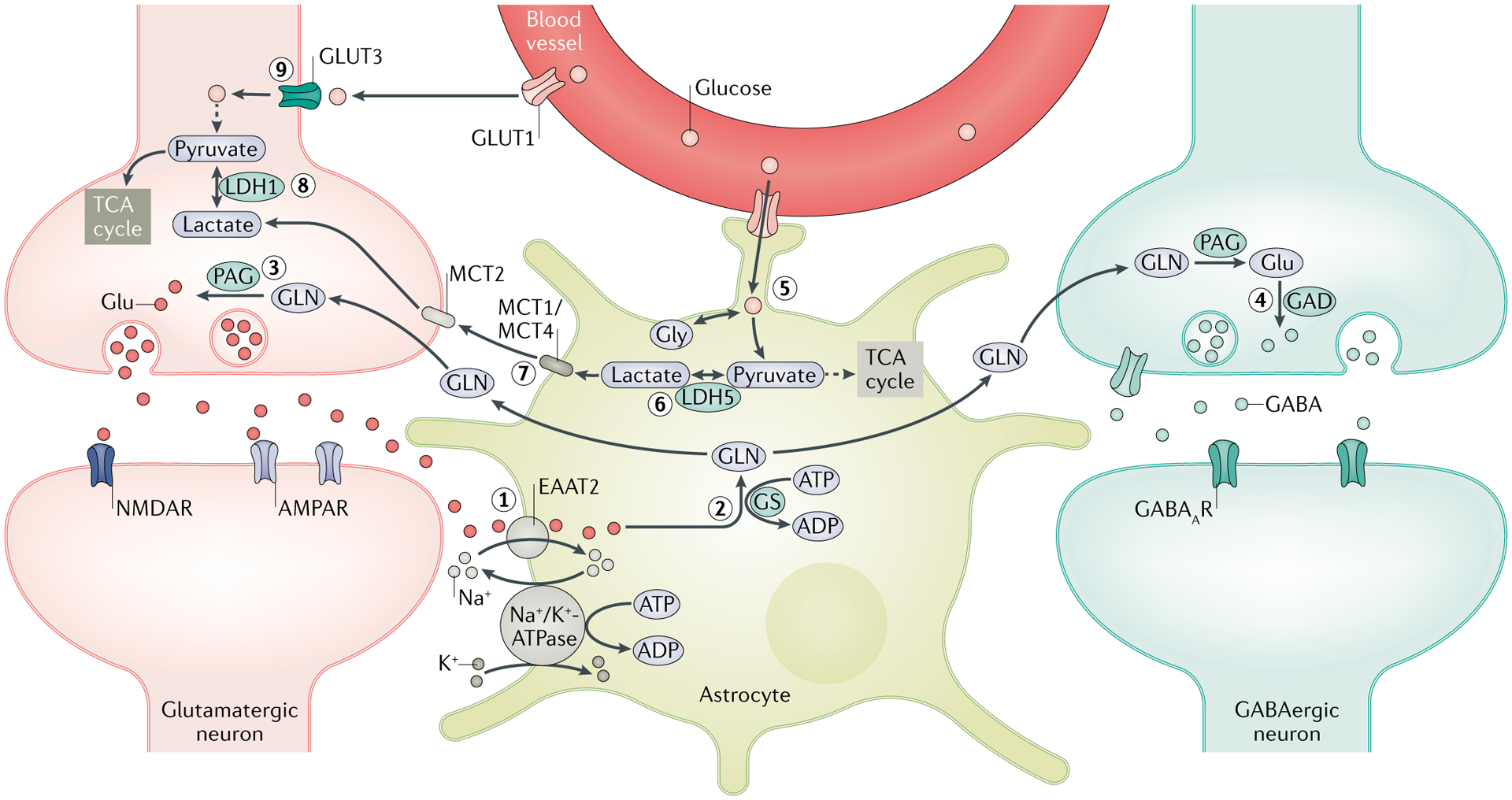
A crucial function of astrocytes is to maintain the optimal level of extracellular glutamate (Glu) through the Glu–glutamine (GLN) cycle. (1) Following synaptic transmission, excess extracellular Glu is absorbed by astrocytes through excitatory amino acid transporter 2 (EAAT2). (2) A large amount of Glu is converted by GLN synthetase (GS) into GLN, which is shuttled into the surrounding neurons. (3) GLN is then metabolized by phosphate-activated glutaminase (PAG) into Glu, which is packaged into synaptic vesicles in the glutamatergic neurons. (4) In GABAergic neurons, Glu is further converted by Glu decarboxylase (GAD) into GABA, which is released in the synapse during inhibitory neurotransmission. Rapid clearance of extracellular Glu following increased activity of synaptic transmission exerts an energetic burden on astrocytes owing to the energy-intensive nature of the Glu–GLN cycle, as both EAAT2 and GS require ATP. (5) The energy deficit that results stimulates the glycolytic oxidation of glucose imported from the blood through glucose transporter 1 (GLUT1) and is enzymatically derived from glycogen stores in astrocytes to generate energy metabolites — lactate and pyruvate. (6) Owing to the limited oxidative capacity of astrocytes, pyruvate is primarily converted into lactate via lactate dehydrogenase 5 (LDH5) and (7) shuttled into the surrounding neurons via monocarboxylate transporters (MCTs). (8) In neurons, LDH1 catalyses the conversion of lactate into pyruvate, which is subsequently used as an energy source through the tricarboxylic acid (TCA) cycle. This pathway is termed the astrocyte neuron lactate shuttle (ANLS) and is crucial in meeting the energy demands of neurons during high synaptic activity, such as during seizures. (9) Glucose is also taken up by neurons from the blood vessels through GLUT3; however, owing to the limited glycolytic capability of neurons and the lack of glycogen stores, neurons derive pyruvate primarily from astrocytes via the ANLS pathway. Disruptions in the expressions and functions of enzymes and transporters implicated in both the Glu–GLN cycle and the ANLS are associated with the development of neuronal hyperexcitability and seizures. AMPAR, AMPA receptor; GABAAR, GABA type A receptor; Gly, glycine; NMDAR, NMDA receptor.
Indeed, the findings that GLT1-knockout mice exhibit severe spontaneous seizures59 whereas mice overexpressing glial GLT1 are resistant to pilocarpine-induced epileptogenesis60 illustrate the importance of GLT1 in maintaining synaptic glutamate homeostasis and seizure threshold. Interestingly, GLT1 expression is modulated by the antibiotic ceftriaxone, which acts as a transcriptional stimulator61. Early treatment with ceftriaxone before the onset of epilepsy increased astrocytic expression of GLT1, decreased the extracellular glutamate level and reduced seizure frequency in a mouse model of tuberous sclerosis complex62. Another finding yielding a compelling new treatment strategy originated from the finding that GLT1 is downregulated in reactive astrocytes by heat shock protein 90β (HSP90β)-mediated proteasomal degradation63. Chronic treatment with an HSP90β inhibitor suppressed kainic-acid-induced seizures63. Changes in the functions of GLT1 have also been found prominently in lesional epilepsies64,65.
Efforts to measure the expression of EAATs in human epileptic tissues have yielded inconsistent results, albeit pathophysiological increases in glutamate can be measured in patients before and during seizures55, suggesting that astrocytes are unable to clear neuronally released glutamate. Abnormally high levels of extracellular glutamate during ictal and interictal periods and the changes in the expression and function of EAATs in astrocytes are often accompanied by impairment in the function of GS. A significant reduction in the expression and enzymatic activity of GS has been observed in the hippocampus of individuals with MTLE, and this loss of GS activity was particularly pronounced in areas with astrogliosis66,67. Continuous infusion of the GS antagonist, methionine sulfoximine (MSO), into the hippocampus of rats caused drastic reduction in GS activity, neuropathological changes similar to MTLE and epilepsy in a majority of the MSO-treated rats, suggesting that a deficiency in astroglial GS activity is a potential molecular mechanism for seizure generation in MTLE68.
The role of astrogliosis in disrupting the glutamate–glutamine cycle and consequently causing hyperexcitability has been elegantly demonstrated in a mouse model of virally induced reactive gliosis17. Here local infection of astrocytes with adeno-associated virus (AAV) led to reactive gliosis in AAV-infected astrocytes, evident by cellular hypertrophy and increased GFAP and vimentin expression. Reactive astrocytes also had decreased expression of GS. Tissue slices isolated from infected animals showed epileptiform hippocampal activity. Importantly, all of these consequences of astrogliosis could be mimicked by inhibiting the glutamate–glutamine cycle and were reversed by providing exogenous glutamine. Similar findings have since been reported in rat spinal dorsal horn neurons69 and in an astrocyte neuron co-culture system70, suggesting that a disruption of neuronal GABA synthesis solely by disrupting the astrocytic supply of glutamine can induce the neural hyperexcitability underlying epilepsy.
Astrocytes also serve the crucial function of regulating the energy homeostasis essential for optimal synaptic activity71. Clearance of extracellular glutamate and its conversion into glutamine are energy-intensive processes. As mentioned above, astrocytes sequester extracellular glutamate via EAATs utilizing the electrochemical gradient of Na+ as a driving force with a stoichiometry of one glutamate to three Na+ ions. The transmembrane distribution of Na+ ions is then re-established by the energy-consuming Na+/K+-ATPase. The conversion of glutamate into glutamine by GS also requires ATP. Thus, the glutamate–glutamine cycle imposes an energetic burden on astrocytes that triggers an uptake of glucose from the blood vessel through the glucose transporter GLUT1 expressed on the membranes of both blood capillary endothelial cells and astrocytes. Glucose is also taken up by neurons from the blood vessels through GLUT3. However, the glucose-handling pathways differ between astrocytes and neurons owing to the cell-specific expression of glucose-metabolizing machinery. Glycolytic oxidation of glucose into pyruvate is predominant in astrocytes and can be upregulated, but the utilization of pyruvate through the TCA cycle is limited. By contrast, neurons have a limited capacity for glycolysis but an active TCA cycle and oxidative phosphorylation71. In fact, neurons predominantly process glucose through the pentose phosphate pathway to maintain the redox status necessary to counter the oxidative stress that results from their high mitochondrial activity. Glycolytically produced pyruvate in astrocytes is converted into lactate by lactate dehydrogenase 5 (LDH5). Lactate is then shuttled via monocarboxylate transporters (MCTs) to neurons, where it is utilized as an energy substrate via the TCA cycle after being converted into pyruvate by LDH1. This pathway is often called the astrocyte neuron lactate shuttle (ANLS) (FIG. 2), and its activity has been shown to positively correlate with neuronal activity (reviewed in REF.72). Astrocytes (but not neurons) store glycogen as an energy source to meet the high energy demand of neurons during prolonged neuronal activity by augmenting glycolysis and exporting lactate to neurons via the ANLS pathway. Any perturbations in this metabolic pathway may modulate neuronal excitability. Strategies such as a ketogenic diet that switch the energy source of the brain from glucose to ketone bodies by using high-fat and low-carbohydrate diets are often employed successfully in controlling refractory seizures73. The ketogenic diet tampers with energy metabolism and is likely modulating the ANLS pathway. A study by Sada et al. demonstrated that substituting glucose with ketone bodies (β-hydroxybutyrate or acetoacetate) hyperpolarized excitatory neurons in mouse brain slices74. The stabilizing effects of glucose restriction on neuronal excitability were abrogated by an exogenous provision of lactate, pyruvate and its downstream energy metabolites. It was further shown that pharmacological inhibition of LDH, specifically in astrocytes, decreased the excitability of surrounding excitatory neurons in the hippocampus, suggesting that lactate from astrocytes is a source of ictogenic pyruvate in neurons. Although the downstream mechanisms are not clear, the authors suggested that the activation of KATP channels could contribute to neuronal hyperpolarization because inhibitors of KATP channels abolished the effects of LDH inhibition. Importantly, an analogue of stiripentol, a clinically used antiepileptic drug that also inhibits LDH, potently suppressed kainic acid-induced seizures in mice.
Taken together, these findings show that astrocytes serve a pivotal function in supplying neurons with high-energy substrates to meet their energy demand and amino acid precursors for the synthesis of neurotransmitters. Thus, astrocytes are crucial in maintaining normal excitation–inhibition balance. A detailed mechanistic understanding of the role of astrocytes in providing metabolic support to neurons may enable the development of novel metabolic interventions for the treatment of epilepsy.
Synaptic structure and function
Overwhelming evidence suggests that epilepsy is a disease in which neural circuits are chronically altered. As these circuits are formed by synapses between neurons, it is reasonable to consider epilepsy as a disease resulting from impairment in synaptic structure and/or function. Glial cells influence synaptic structure and function by regulating long-term processes such as synaptogenesis, synaptic pruning and synaptic maturation as well as by releasing gliotransmitters.
Synaptic structure.
Although a role for glia in the development of the brain has been well recognized for decades, more recent studies have identified astrocytes and microglia as essential players in normal synaptogenesis, and many of the molecules involved in synapse development and activity-dependent pruning have been identified75. These include BDNF, cholesterol, thrombospondins (TSPs), oestradiol, protocadherins, integrins, ephrins, glypicans, transforming growth factor-β (TGFβ) and hevin, among others75. Some of these factors, for example, TSPs, are primarily expressed in the developing brain, and subsequently their expressions are downregulated. However, after CNS injury, reactive astrocytes secrete excessive TSPs, which have been shown to interact with α2δ1, an auxiliary calcium channel subunit, to promote aberrant synaptogenesis, which can alter the neuronal circuitry to generate epileptiform activity76. Interestingly, the anticonvulsant drug gabapentin disrupts the interaction between TSP and α2δ1, attenuating epileptiform activity following epileptogenic freeze lesion77, suggesting that reactive astrocyte-triggered aberrant synaptogenesis might be a potential mechanism of network hyperexcitability in neurodevelopment-disorder-associated epilepsy.
Glial cells also regulate critical signalling pathways associated with synaptic pruning. For example, during the development of retinothalamic circuitry, axons from the retinal ganglion cells undergo eye-specific segregation through elimination of overlapping inputs by microglia and astrocytes in an activity-dependent manner78. Astrocytes induce the expression of complement component C1q at synapses that, in turn, activates the complement pathway to stimulate microglia for phagocytosing the synapses79. Mice deficient in C1q exhibit impaired synaptic pruning, enhanced synaptic connectivity and eventually epileptic activity80. Recent studies suggest that astrocytes not only trigger phagocytosis via microglia but also exhibit phagocytic activity themselves under physiological and pathological conditions81. The complement components are upregulated in rodent models of epilepsy and may revitalize the synaptic-pruning functions of microglia during adolescence or adulthood82.
Astrocyte-released molecules are also important for the regulation of synaptic strength. For example, glial-derived tumour necrosis factor (TNF) regulates trafficking of glutamate and GABAARs onto postsynaptic membranes, cholesterol enhances presynaptic neurotransmitter release and glypican 4 and glypican 6 increase the number of AMPA receptors (AMPARs) at postsynaptic sites75. Presynaptic and postsynaptic differentiation and regulation of GABAAR expression at developing inhibitory synapses can also be modulated by astrocyte-released molecules83; however, their molecular characterization is yet to be done.
Although there is little direct evidence of seizures resulting from impaired glia-mediated regulation of synaptogenesis and pruning, a few studies have reported aberrant synaptogenesis and synaptic pruning in neurodevelopmental disorders that are associated with epilepsy78. For example, astrocytes contribute to abnormal dendritic morphology and the reduced synapse density commonly found in fragile X syndrome, which is tightly associated with epileptic seizures81,84. Similarly, microglia-induced disruptions in neuronal circuits due to impaired synaptic pruning can contribute to seizures in the mouse model of Rett syndrome85. Finally, impaired synaptic pruning is a major driving force for the development of TLE in individuals with a mutation in the gene LGI1, encoding leucine-rich glioma-inactivated protein 1 (REF.78).
Gliotransmission and epilepsy.
A number of astrocyte-released molecules including glutamate, ATP and adenosine act as neurotransmitters and neuromodulators of neurons in a process called gliotransmission. As each of these neuromodulators can potently induce neuronal excitability86, gliotransmission may contribute to epileptogenesis. Astrocyte-released glutamate exerts a multitude of effects on neurons including increased neuronal excitability and synchronization of activity, synaptic plasticity, regulation of GABAergic neurotransmission and generation of slow inward currents in neurons87. However, glutamate release from astrocytes is typically induced experimentally by photolysis of caged calcium or inositol 1,4,5-trisphosphate (IP3) infusion. Although these stimulations induce slow inward currents that cause transient depolarizations reminiscent of paroxysmal depolarizations86,88, the physiological relevance of such astrocyte stimulation has been questioned89. Nevertheless, as these neuronal paroxysmal depolarizations occur entirely in response to astrocytic calcium signalling and precede neuronal hyperactivity, it illustrates that, at least in principle, astrocytic glutamate release is capable of inducing neuronal hyperexcitability.
More compelling evidence for glutamate being the driver of epilepsy exists for malignant glia. Astrocyte-derived tumours (that is, gliomas) are notorious for causing seizures, with >70% of people presenting with seizures before diagnosis. Approximately one-third of all these individuals develop tumour-associated epilepsy, which is often pharmacoresistant. Both in tumour-bearing mice and humans, glutamate is released from the tumour as a result of the elevated expression of the system Xc, a cystine–glutamate antiporter encoded by the SLC7A11 gene, which is upregulated in a majority of individuals with glioma90. This transporter is primarily responsible for the synthesis of the cellular antioxidant glutathione (GSH) from the uptake of cystine. However, the obligatory release of glutamate leads to chronic elevation of extracellular glutamate, which induces excitotoxicity, in turn, on the surrounding principal neurons91. In addition to glutamate-mediated excitotoxicity, matrix metalloproteinases (MMPs) released by the glioma cause preferential death of GABAergic interneurons by disintegrating the protective coat of the perineuronal nets (PNNs) around them92. The resulting increase in excitatory drive and decrease in inhibitory drive eventually lead to spontaneous electrographic and behavioural seizures91,93,94.
Astrocytes release ATP and express a wide range of ion channels and G protein-coupled receptors activated by extracellular nucleotides and their cleavage products by ectonucleotidases95. The pro-hyperexcitability consequences of the purinergic signalling are mainly effectuated by astrogliosis, release of glutamate and pro-inflammatory factors, remodelling of the ECM and synaptogenesis. By contrast, ATP-mediated suppression of synaptic activity is largely mediated by its cleavage product adenosine and a subset of ATP receptors, eventually reducing excitatory drive either by enhancing inhibitory neurotransmission96 or by suppressing excitatory neurotransmission97. Adenosine activates presynaptic and postsynaptic Gαi/o protein-coupled adenosine A1 receptors (A1Rs), and studies in humans with TLE and rodent models of epilepsy have shown downregulation of A1Rs95. At the presynaptic terminal, activation of A1Rs reduces calcium influx by inhibiting N-type calcium channels, thereby curtailing excitatory synaptic transmission. Moreover, miniature events at excitatory synapses are also decreased by presynaptic A1R-mediated reduced sensitivity of the neurotransmitter release machinery98. At postsynaptic terminals, A1R activation decreases neuronal excitability by enhancing the outflow of potassium through G protein-coupled Kir channels95.
Adenosine has been widely accepted as an endogenous anticonvulsant owing to its A1R-mediated synaptic inhibition and neuroprotective effects. Moreover, the physiological relevance of adenosine has been evident by an increase in the level of adenosine triggered by seizure activity99. Additional studies on the anticonvulsive effects of adenosine have elucidated an interesting role of the astrocyte-based adenosine cycle in acquired epilepsies. Astrocyte-released ATP is rapidly degraded into adenosine in the ECS and subsequently cleared by astrocytic nucleoside transporters and phosphorylated by adenosine kinase (ADK) predominantly expressed in astrocytes95. As ADK determines extracellular adenosine levels, any alteration in ADK expression can limit the adenosine levels and accordingly curtail its anticonvulsive and neuroprotective functions. A possible ADK dysfunction in epilepsy has been seen in epilepsies of animals and humans, which show robust overexpression of ADK in reactive astrocytes100, suggesting adenosine deficiency as a contributing factor to seizures. Furthermore, ADK overexpression is sufficient to trigger seizures even in the absence of astrogliosis or any other epileptogenic event95. Importantly, these findings support a therapeutic approach through adenosine augmentation using either A1R agonists or ADK inhibitors, which are reported to reduce seizures in a wide range of epilepsy models, including those resistant to conventional antiepileptic drugs95.
In essence, glial cells release a wide range of substances and gliotransmitters to shape the synaptic structure and function with high fidelity, and the disorders with defective synaptic structure and function, including epilepsy, are highly associated with the dysfunctional glial cells.
ECM remodelling and BBB integrity
Although most epilepsy research studies have focused on physiological changes in neurons and glia, emerging literature now suggests that the ECM into which neurons and glia are embedded may significantly influence neuronal excitability and indeed contribute to epilepsy (TABLE 2). Both glia and neurons release a heterogeneous mixture of proteins, glycoproteins and proteoglycans that form an amorphous interstitial matrix filling the ECS and highly specialized lattice-like assemblies known as PNNs (BOX 1) ensheathing mainly fast-spiking GABAergic interneurons101. These molecules collectively constitute the ECM, which also includes a highly organized basement membrane surrounding the blood vessels and the entire pial surface. The ECM primarily provides support for the cellular arrangement; however, interactions with membrane receptors including integrins, osteopontin and CD44 suggest that their physiological role may be much more complex101.
Table 2 |.
Extracellular matrix remodelling in epileptogenesis
| ECM component and/or remodelling molecules | Source | Functions | Pathology and/or epileptogenic effects | Refs |
|---|---|---|---|---|
| CSPG and/or CS-GAGs | Neurons, astrocytes and radial glia |
|
|
110,118,133,136 |
| HSPGs and/or glypicans | Astrocytes, neurons and endothelial cells | Synaptogenesis and synaptic maturation |
|
136–138 |
| Tenascin C | Astrocytes, neurons, radial glial and epithelial cells |
|
|
12,103,139,140 |
| Tenascin R | Neurons and oligodendrocytes |
|
|
12,103,140–142 |
| HA | Neurons and astrocytes |
|
|
103,108,109,136 |
| MMPs | Astrocytes, endothelial cells, neurons, microglia and oligodendrocytes |
|
|
12,105,106,143,144 |
| ADAMTs | Neurons, astrocytes, microglia, monocytes and macrophages |
|
|
103,145–147 |
| tPA | Neurons and microglia |
|
|
103,146,148 |
| TSPs | Astrocytes and macrophages |
|
|
12,140 |
| Reelin | Cajal-Retzius cells and interneurons |
|
|
103,149 |
| Hevin | Vasculature cells, astrocytes, radial glia and neurons |
|
|
15,103,140 |
| SPARC | Radial glia, vasculature cells, astrocytes and microglia |
|
|
140,150 |
ADAMTs, a disintegrin and metalloproteinase with thrombospondin motifs; CS–GAG, chondroitin sulfate–glycosaminoglycan; CSPG, chondroitin sulfate proteoglycan; DGC, dentate granule cell; ECM, extracellular matrix; HA, hyaluronic acid; HSPG, heparin sulfate proteoglycan; LTP, long-term potentiation; MMP, matrix metalloproteinase; PNN, perineuronal net; PV+, parvalbumin positive; SPARC, secreted protein acidic and rich in cysteine; TLE, temporal lobe epilepsy; tPA, tissue plasminogen activator; TSP, thrombospondin.
Box 1 |. Perineuronal nets.
A specialized extracellular matrix (eCM) structure, termed the perineuronal net (PNN), has fascinated histologists since its discovery in the 1900s. PNNs are formed by lattice-like assemblies of chondroitin sulfate proteoglycans (CSPGs), hyaluronic acid (HA), tenascin R and link proteins and are tightly applied to the cell somas, apical dendrites and axon initial segments101. The chemical constituents of PNNs interact with receptors on the cell membrane and harbour various growth factors and guidance molecules, thereby controlling several cellular processes. For instance, semaphorin 3A, an axon guidance molecule, is anchored to the PNN via chondroitin sulfate e, and fibroblast growth factor 2 binds to the phosphacan core protein of PNN–CSPG132. PNNs are believed to serve a wide range of functions, including synaptic stabilization, neuroprotection and ionic buffering101. Their importance in CNS function is most well documented during the development of the visual system, for which the establishment of ocular dominance columns occurs through the stabilization of synapses in the visual cortex by PNNs133. The enzymatic destruction of PNNs dissolves the ocular dominance, enabling renewed synaptic plasticity, suggesting that the PNNs do not permit synaptic plasticity.
ECM remodelling in epilepsy.
The ECM is subject to remodelling by a number of matrix-remodelling enzymes such as MMPs and tissue inhibitors of metalloproteinases (TIMPs) that are released by neurons and glial cells. During the course of development, substantial ECM remodelling transforms the immature brain ECM that supports structural and functional plasticity into mature brain ECM, which restricts the structural remodelling of neuronal network, axonal outgrowth and synaptogenesis and hence is nonpermissive to synaptic plasticity101. Pathological changes in the remodelling of the ECM and PNNs that can potentially alter synaptic circuits in a maladaptive way have been observed in animal models of epilepsy and in human epileptic tissues102–104 (FIGS 3,4). As the biochemical changes in the ECM and PNN molecules correlate positively with upregulated levels of MMPs and tissue plasminogen activator (tPA)104–106, it is likely that the abnormal enzymatic activity causes the degradation of these structures. Indeed, at least in the rodent models, PNN breakdown and seizure activity can be inhibited by treatment with doxycycline, which is a broad-spectrum inhibitor of MMPs107.
Fig. 3 |. Disintegration of perineuronal nets in glioma-associated epilepsy.
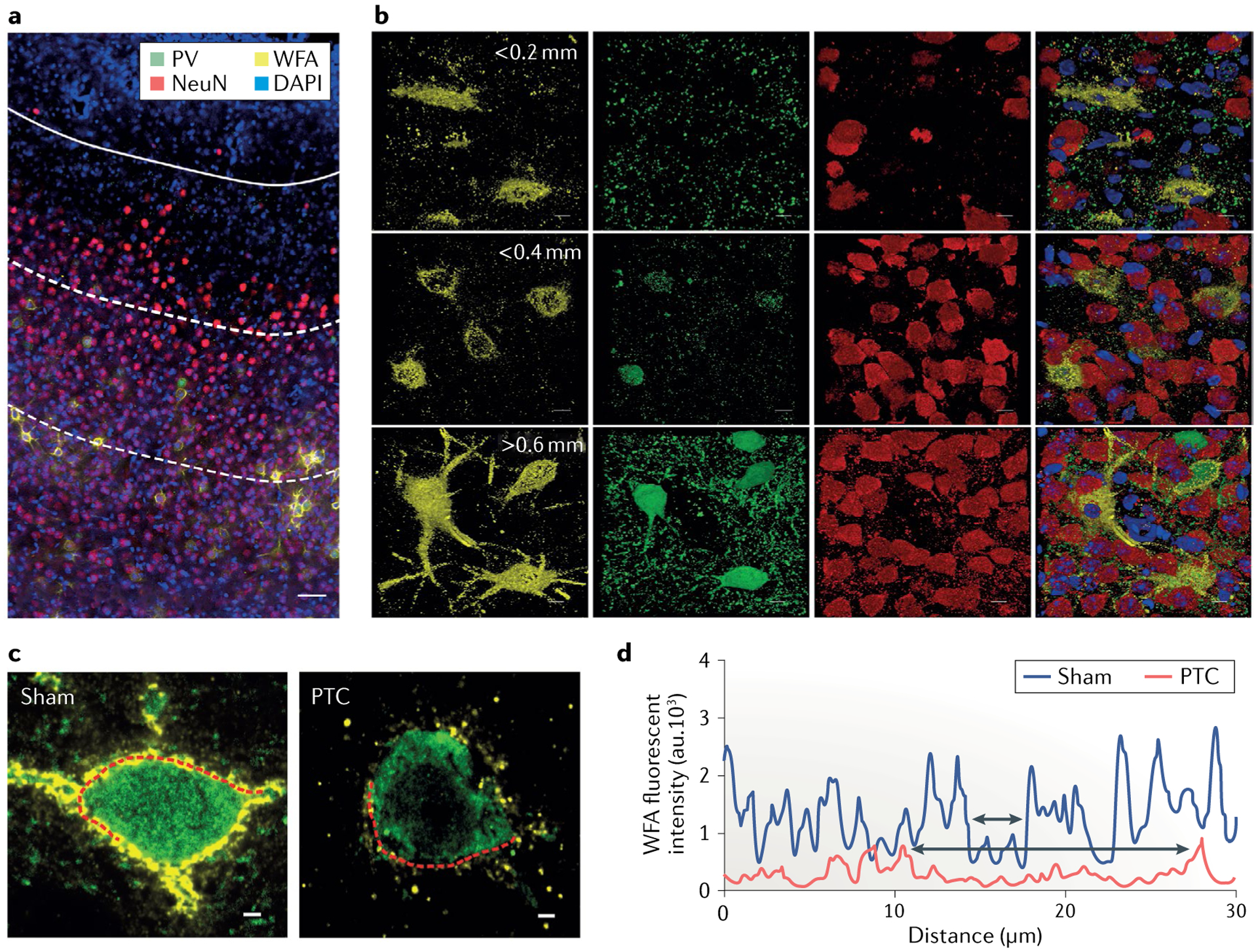
a,b | Immunohistochemical staining of the cerebral cortex from a glioma-implanted mouse (part a) shows 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei (blue), Wisteria floribunda agglutinin (WFA)-stained perineuronal nets (PNNs; yellow), parvalbumin-positive (PV+) interneurons (green) and NeuN-stained neurons (red) (scale = 50 μm). The presence of glioma is identified by densely packed DAPI-stained cells indicated in the upper part of the image demarcated by a white solid line (tumour border). The distance between the tumour border and each of the white dotted lines below is ~0.2 mm. Magnified individual images of PNNs, PV+ interneurons and NeuN+ neurons and their merged images corresponding to their distance from the tumour border are shown on the right (upper panels <0.2 mm, middle panels 0.2–0.4 mm and lower panels 0.4–0.6 mm; scale = 5 μm). c | Confocal images of PV+ interneurons (green) show disintegrated architecture of PNNs (yellow) in the peritumoural cortex (PTC) and are compared to sham-treated mouse cortex (scale = 2 μm). d | The WFA fluorescence intensity of a line drawn along the periphery of a PV+ interneuron in the peritumoural cortex (left panel of part c) and sham (right panel of part c) shows many high-intensity WFA peaks in sham compared with those in the peritumoural cortex. Upper and lower two-headed arrows within the two nearest WFA peaks indicate the size of a hole in the PNN from the sham and peritumoural cortex, respectively. au, arbitrary units. Adapted from REF.92, Springer Nature Limited.
Fig. 4 |. Changes in the extracellular matrix as contributors to epilepsy.
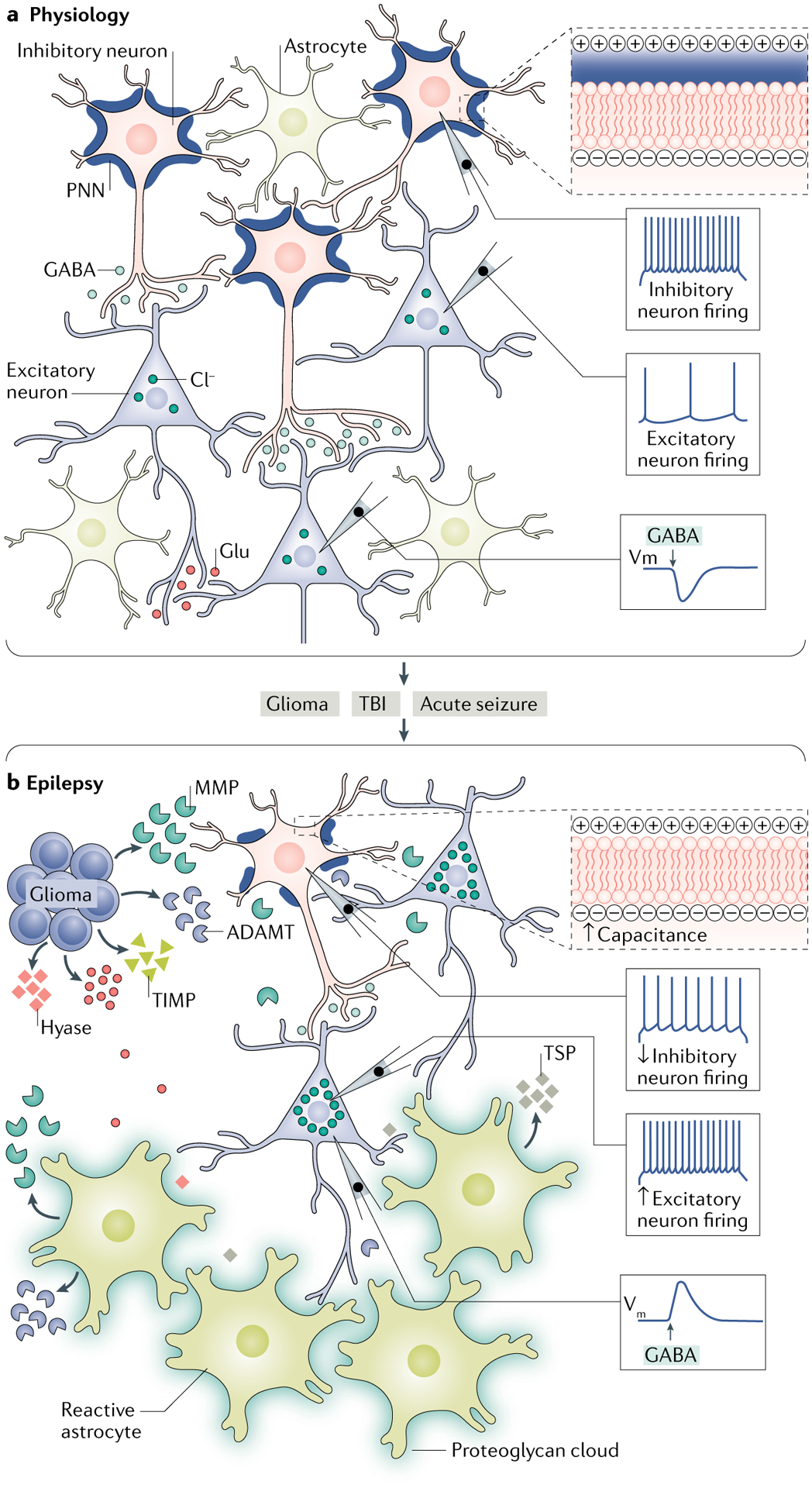
a | Under physiological conditions, the extracellular space is filled with a dense amorphous interstitial matrix and highly organized perineuronal nets (PNNs) surrounding parvalbumin-positive (PV+) inhibitory interneurons. The presence of intact PNNs around the cell reduces the effective membrane capacitance of the cell by increasing the charge separation between intracellular and extracellular compartments (upper right panel). This enables interneurons to sustain high firing frequencies to maintain a steady-state inhibitory drive by releasing GABA onto the excitatory cells, which helps to maintain optimal firing of the excitatory neurons. Intact structure of the extracellular matrix (ECM) also helps to maintain a low concentration of intracellular chloride; therefore, activation of the GABA type A receptor (GABAAR) causes hyperpolarization owing to the chloride influx. b | Alterations in the ECM that occur during several pathological conditions such as glioma, traumatic brain injury (TBI) and acute seizures can contribute to the development of epilepsy. Glioma cells release glutamate (Glu) and ECM-remodelling molecules, including matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs), hyaluronidase (hyase) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTs). These ECM-remodelling molecules cause degradation of the PNNs around inhibitory interneurons and thinning of the interstitial matrix. The loss of PNNs increases the membrane capacitance of inhibitory interneurons by decreasing the charge separation between intracellular and extracellular compartments (upper right panel), thereby slowing their firing frequency and, as a consequence, reducing their GABA release and lowering inhibition of excitatory neurons. Hence, the firing of excitatory neurons increases, contributing to network hyperexcitability. Glioma cells also cause selective loss of PV+ inhibitory interneurons owing to Glu excitotoxicity further lowering the inhibitory drive, thereby leading to epileptiform activity. Impairment in the ECM structure also alters chloride homeostasis, which results in an increased intracellular concentration of chloride and a depolarizing shift in the activation of the GABAAR. In a manner similar to that of glioma, following TBI and acute seizures, reactive astrocytes become the primary source of the ECM-remodelling molecules (MMPs and ADAMTs), synaptogenic molecules (thrombospondins (TSPs)) and proteoglycans, which can contribute to epileptogenesis by degrading PNNs and the interstitial matrix, thinning the interstitial matrix and causing aberrant synaptogenesis. Vm, membrane potential.
A major unanswered question is whether ECM remodelling is a cause or consequence of seizures. A partial answer is provided by a recent animal study in which the genetic deficiency of hyaluronic acid (HA), a component of PNNs, was sufficient to cause behavioural and electrographic seizures108. Deficiency of one of the major PNN components, tenascin C, increases the number of GFAP-expressing astrocytes and increases seizure susceptibility in mice, whereas depletion of another PNN component, tenascin R, induces aggravated astrogliosis, dysregulation of synaptic plasticity and perisomatic GABAergic inhibition103. In addition, in vivo enzymatic breakdown of the ECM using hyaluronidase causes electrographic-seizure-like events109, and the disruption of the PNNs using chondroitinase increases the frequency of myoclonic seizures110. These studies suggest that an intact ECM suppresses development of seizures and that disruptions in the ECM alone may be sufficient for at least some seizure activity. TABLE 2 presents a summary of the most common ECM components, enzymes involved in their remodelling and the effects of ECM remodelling on epileptogenesis.
How might the ECM composition and/or the structural integrity of PNNs affect neuronal excitability? There are several ways in which ECM molecules can specifically regulate the localization, mobility and function of ion channels and neurotransmitter receptors, namely, by regulating channels and receptors in the membrane, by acting as buffers for extracellular ions and ligands or by altering the intrinsic properties of neurons. For example, HA has been shown to regulate the activity of voltage-activated calcium channels111, whereas brevican112 and tenasin113 regulate the localization of voltage-gated potassium and sodium channels, respectively. The ECM molecules may also modulate synaptic plasticity by limiting the lateral mobility of receptors as shown for AMPAR114. Furthermore, experimental disruption of the ECM decreases the excitability of fast-spiking interneurons and thus inhibitory drive115.
One of the salient features of the ECM constituents is a very high density of negative charges contained in their CSPG side chains116. As these are attached to a protein backbone, they resemble stationary charges with little diffusional mobility in the ECS and complete impermeability to the neuronal membrane. When intact, stationary charges may act as a buffering system for extracellular cations, including Ca2+ (REF.117). Moreover, they may affect the transmembrane chloride gradient and thereby the excitatory or inhibitory nature of GABAergic neurotransmission118. Indeed, Cl− measurements in hippocampal slices using a genetically encoded Cl− sensor show an increase in [Cl−]i upon digestion of the ECM using chondroitinase. This, in turn, is sufficient to facilitate cellular epileptiform activity118.
We recently reported an additional mechanism whereby PNNs may regulate neuronal firing and contribute to epilepsy92. We propose that by virtue of the high-density negative charges, the PNNs increase the charge separation distance across the neuronal membrane, thereby decreasing the effective capacitance of the cell. The reduced capacitance decreases the time constant of the action potential and enhances the maximal achievable spike frequency. As PNNs are most prominent on fast-spiking parvalbumin-expressing GABAergic interneurons, we propose that the PNNs act akin to the myelin sheath on axons to facilitate rapid firing of action potentials at the axon hillock and more rapid transmission of electrical signals along the apical dendrites and somas. Indeed, in a study of tumour-associated epilepsy, we find that MMPs released from the tumour cells degrade the PNNs, which increases the membrane capacitance of GABAergic interneurons, thereby slowing their firing frequency. We show that peritumoural hyperexcitability can be induced as a result and that the PNN disruptions alone can phenocopy the presence of the tumour (FIG. 4).
As MMPs are commonly released after CNS infection, stroke or trauma, it is reasonable to hypothesize that similar degradation of PNNs may occur in association with other forms of acquired epilepsy, particularly those associated with reactive gliosis in which astrocytes are known to secrete the components and the remodelling enzymes of the ECM12,106,119. An excessive release of CSPGs from reactive astrocytes has been documented in spinal cord injury in which the newly formed ECM retards axonal sprouting120. In addition, the polarized expression of astrocyte membrane proteins including Kir4.1, AQP4 and GLT1 is controlled by the ECM molecules; hence, disruptions of the ECM can dramatically alter the functions of astrocytes and contribute to epileptogenesis120–122.
BBB integrity in epilepsy.
The basement membrane is another component of the ECM that is primarily known for the maintenance of BBB integrity101. However, it is also subject to degradation by MMPs, as is the case in cerebral ischaemia. The resulting leakiness of BBB may contribute directly to epileptogenesis. Acquired forms of epilepsies are invariably present with an impaired BBB, and the breakdown of the BBB contributes to the progression of epilepsy7. BBB disruption exposes the brain to invading immune system cells and serum components, most importantly albumin7,123. Albumin alone can trigger neuronal hyperexcitability by activating TGFβ receptors in astrocytes, which results in reduced expression of Kir channels, glutamate transporters and GJ proteins124 (FIG. 1). TGFβ also changes the trafficking and surface expression of AQP4 (REF.125). Together, this results in impaired K+ and glutamate buffering, which is likely to contribute to neuronal hyperexcitability. Activation of TGFβ signalling also induces aberrant neurogenesis, increased excitatory synaptogenesis and the release of pro-inflammatory molecules from astrocytes that cause excitotoxic damage to neurons and other surrounding cells126. Extravasation of albumin in the brain following BBB damage may also contribute to the development of pharmacoresistance to antiseizure drugs in refractory epilepsy by binding to antiseizure drugs in the brain and thereby decreasing their bioavailability and efficacy127. In addition, activation of TGFβ signalling in astrocytes following BBB disruption initiates transcriptional activation of various ECM-remodelling molecules, which potentially break the PNNs, thereby affecting the inhibitory activity of GABAergic interneurons and inducing the excitatory synaptogenesis contributing to epileptogenesis12,128. The critical contribution of TGFβ signalling in dysregulating astrocyte functions during BBB-disrupting injuries and epileptogenesis is supported by the findings that losartan, a US Food and Drug Administration-approved drug that inhibits TGFβ signalling, decreased the number of rats developing albumin-induced chronic seizures and inhibited epileptogenesis in CNS vascular injury-induced models of epilepsy129. The molecular mechanisms of BBB disruption during epileptogenesis are not clearly understood. In this regard, it has been proposed that MMPs critically contribute to the breakdown of the BBB by digesting tight junctions and basement membrane proteins130,131. Recently, it has been suggested that glutamate released during seizures increases the levels of MMP2 and MMP9 in the BBB capillaries via NMDARs and cytosolic phospholipase A2-mediated signalling, which promotes, in turn, reduction in the number of tight junction proteins and BBB leakage109.
Taken together, disruption of the physiochemical properties of the ECM, basement membrane and PNNs by ECM-remodelling molecules may significantly contribute to epileptogenesis by altering the membrane expression or function of ion channels and receptors; by disrupting glutamate, potassium and chloride homeostasis; or by changing the intrinsic firing properties of fast-spiking GABAergic interneurons.
Summary and conclusions
An important and rapidly growing body of scientific evidence, some of which is discussed above, leads us to suggest that the neuronal microenvironment contributes meaningfully to the aetiology of epilepsy. This microenvironment is dynamic and greatly influenced by glial cells. There remains little doubt that glia contribute to ictogenesis and epileptogenesis, and the many ways in which this occurs are summarized in TABLE 1. Glial cells are not just passive bystanders providing structural and tropic support to neurons but can actively participate in neuronal physiology. Although in some examples their contribution has been unequivocally demonstrated, many important questions regarding specific changes in glia and their contribution to the onset and progression of epilepsy remain, as summarized below.
Gliosis is a defining histological feature in epileptic tissue. Is it a cause or consequence of epilepsy? Although some studies have provided evidence that gliosis is a driving factor of epileptogenesis, additional studies are needed to test the causal relationship between gliosis and epileptogenesis under different experimental conditions.
Are the functional changes that occur in gliotic tissue restricted to the affected brain region, or does gliosis also alter distant brain regions?
Do glial cells preferentially contribute to lesional epilepsies, or is there a more generalizable glial dysfunction present across all forms of epilepsy?
Epilepsy is a chronic disorder, yet neuro–glial interactions are highly dynamic. Do these interactions evolve during epileptogenesis? Do they change with initiation and termination of seizures?
Current biochemical and pathophysiological analyses of human epileptic tissues examine the end point of disease and fail to address the dynamics of epileptogenesis. Are there any biomarkers released from glia that may predict disease progression?
Activated glia are important to resolve neuropathological conditions following CNS insult. However, uncontrolled, severe, reactive gliosis may contribute notably to the neuropathology associated with epilepsy. Are there factors that can turn off activated glia? Could such factors be harnessed to develop antiepileptic therapies?
Can strategies aimed at maintaining the integrity of the BBB control seizures? To what extent does the infiltration of blood serum proteins and peripheral immune cells influence neuroglial functions in the brain?
Glial cells secrete a wide range of ECM-modifying molecules that critically influence neuronal functions. To what extent do these molecules affect the neuronal environment such that they tip the excitation–inhibition balance? Might such changes be common among many forms of epilepsy?
Acknowledgements
This work was supported by grants from the US National Institutes of Health (RO1-NS036692, RO1-NS082851 and RO1-NS052634).
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Sontheimer H Diseases of the Nervous System 61–95 (Elsevier, 2015). [Google Scholar]
- 2.Fisher RS et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55, 475–482 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Kaplan DI, Isom LL & Petrou S Role of sodium channels in epilepsy. Cold Spring Harb. Perspect. Med 6, a022814 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loscher W, Hirsch LJ & Schmidt D The enigma of the latent period in the development of symptomatic acquired epilepsy - traditional view versus new concepts. Epilepsy Behav.52, 78–92 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Sofroniew MV Astrogliosis. Cold Spring Harb. Perspect. Biol 7, a020420 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vezzani A, French J, Bartfai T & Baram TZ The role of inflammation in epilepsy. Nat. Rev. Neurol 7, 31–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Vliet EA et al. Blood-brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130, 521–534 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Dingledine R, Varvel NH & Dudek FE When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv. Exp. Med. Biol 813, 109–122 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jessberger S & Parent JM Epilepsy and adult neurogenesis. Cold Spring Harb. Perspect. Biol 7, a020677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg EM & Coulter DA Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat. Rev. Neurosci 14, 337–349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devinsky O, Vezzani A, Najjar S, De Lanerolle NC & Rogawski MA Glia and epilepsy: excitability and inflammation. Trends Neurosci.36, 174–184 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Kim SY, Porter BE, Friedman A & Kaufer D A potential role for glia-derived extracellular matrix remodeling in postinjury epilepsy. J. Neurosci. Res 94, 794–803 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Hauser RM, Henshall DC & Lubin FD The epigenetics of epilepsy and its progression. Neuroscientist 24, 186–200 (2018). [DOI] [PubMed] [Google Scholar]
- 14.Silver J & Miller JH Regeneration beyond the glial scar. Nat. Rev. Neurosci 5, 146–156 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robel S et al. Reactive astrogliosis causes the development of spontaneous seizures. J. Neurosci 35, 3330–3345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that genetically induced astrogliosis is sufficient to cause epileptic seizures in mice without any other CNS pathologies.
- 17.Ortinski PI et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci 13, 584–591 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study demonstrates that selective induction of astrogliosis in mice using a virus is sufficient to cause network hyperexcitability by impairing neuronal inhibition.
- 18.Uhlmann EJ et al. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann. Neurol 52, 285–296 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Sosunov AA et al. Phenotypic conversions of “protoplasmic” to “reactive” astrocytes in Alexander disease. J. Neurosci 33, 7439–7450 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Messing A, Brenner M, Feany MB, Nedergaard M & Goldman JE Alexander disease. J. Neurosci 32, 5017–5023 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rossini L et al. Seizure activity per se does not induce tissue damage markers in human neocortical focal epilepsy. Ann. Neurol 82, 331–341 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Buckmaster PS, Abrams E & Wen X Seizure frequency correlates with loss of dentate gyrus GABAergic neurons in a mouse model of temporal lobe epilepsy. J. Comp. Neurol 525, 2592–2610 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moody WJ, Futamachi KJ & Prince DA Extracellular potassium activity during epileptogenesis. Exp. Neurol 42, 248–263 (1974). [DOI] [PubMed] [Google Scholar]
- 24.Heinemann U & Lux HD Ceiling of stimulus induced rises in extracellular potassium concentration in the cerebral cortex of cat. Brain Res.120, 231–249 (1977). [DOI] [PubMed] [Google Scholar]
- 25.Coulter DA & Steinhauser C Role of astrocytes in epilepsy. Cold Spring Harb. Perspect. Med 5, a022434 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Traynelis SF & Dingledine R Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J. Neurophysiol 59, 259–276 (1988). [DOI] [PubMed] [Google Scholar]
- 27.Gabriel S et al. Stimulus and potassium-induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J. Neurosci 24, 10416–10430 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olsen ML & Sontheimer H Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J. Neurochem 107, 589–601 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steinhauser C, Seifert G & Bedner P Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 60, 1192–1202 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Heuser K et al. Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J. Neuropathol. Exp. Neurol 71, 814–825 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hinterkeuser S et al. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur. J. Neurosci 12, 2087–2096 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Djukic B, Casper KB, Philpot BD, Chin LS & McCarthy KD Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci 27, 11354–11365 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buono RJ et al. Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res.58, 175–183 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Ferraro TN et al. Fine mapping of a seizure susceptibility locus on mouse chromosome 1: nomination of Kcnj10 as a causative gene. Mamm. Genome 15, 239–251 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Kucheryavykh YV et al. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia 55, 274–281 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Doyon N, Vinay L, Prescott SA & De Koninck Y Chloride regulation: a dynamic equilibrium crucial for synaptic inhibition. Neuron 89, 1157–1172 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Moore YE, Kelley MR, Brandon NJ, Deeb TZ & Moss SJ Seizing control of KCC2: a new therapeutic target for epilepsy. Trends Neurosci.40, 555–571 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Lee HH, Deeb TZ, Walker JA, Davies PA & Moss SJ NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci 14, 736–743 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rivera C et al. BDNF-induced TrkB activation downregulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol 159, 747–752 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Papadopoulos MC & Verkman AS Aquaporin water channels in the nervous system. Nat. Rev.: Neurosci 14, 265–277 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Binder DK, Nagelhus EA & Ottersen OP Aquaporin-4 and epilepsy. Glia 60, 1203–1214 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Solenov E, Watanabe H, Manley GT & Verkman AS Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am. J. Physiol 286, C426–C432 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Nicchia GP, Frigeri A, Liuzzi GM & Svelto M Inhibition of aquaporin-4 expression in astrocytes by RNAi determines alteration in cell morphology, growth, and water transport and induces changes in ischemia-related genes. FASEB J.17, 1508–1510 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Vajda Z et al. Delayed onset of brain edema and mislocalization of aquaporin-4 in dystrophin-null transgenic mice. Proc. Natl Acad. Sci. USA 99, 13131–13136 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neely JD et al. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc. Natl Acad. Sci. USA 98, 14108–14113 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagelhus EA, Mathiisen TM & Ottersen OP Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 129, 905–913 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Hochman DW The extracellular space and epileptic activity in the adult brain: explaining the antiepileptic effects of furosemide and bumetanide. Epilepsia 53 (Suppl. 1), 18–25 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Amiry-Moghaddam M et al. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc. Natl Acad. Sci. USA 100, 13615–13620 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Binder DK et al. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 53, 631–636 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Giaume C, Koulakoff A, Roux L, Holcman D & Rouach N Astroglial networks: a step further in neuroglial and gliovascular interactions. Nat. Rev. Neurosci 11, 87–99 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Mylvaganam S, Ramani M, Krawczyk M & Carlen PL Roles of gap junctions, connexins, and pannexins in epilepsy. Frontiers Physiol 5, 172 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deshpande T et al. Subcellular reorganization and altered phosphorylation of the astrocytic gap junction protein connexin43 in human and experimental temporal lobe epilepsy. Glia 65, 1809–1820 (2017). [DOI] [PubMed] [Google Scholar]
- 53.Bedner P et al. Astrocyte uncoupling as a cause of human temporal lobe epilepsy. Brain 138, 1208–1222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Voss LJ, Jacobson G, Sleigh JW, Steyn-Ross A & Steyn-Ross M Excitatory effects of gap junction blockers on cerebral cortex seizure-like activity in rats and mice. Epilepsia 50, 1971–1978 (2009). [DOI] [PubMed] [Google Scholar]
- 55.During MJ & Spencer DD Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341, 1607–1610 (1993). [DOI] [PubMed] [Google Scholar]
- 56.Eid T, Williamson A, Lee TS, Petroff OA & de Lanerolle NC Glutamate and astrocytes—key players in human mesial temporal lobe epilepsy? Epilepsia 49 (Suppl. 2), 42–52 (2008). [DOI] [PubMed] [Google Scholar]
- 57.Schousboe A, Scafidi S, Bak LK, Waagepetersen HS & McKenna MC Glutamate metabolism in the brain focusing on astrocytes. Adv. Neurobiol 11, 13–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vandenberg RJ & Ryan RM Mechanisms of glutamate transport. Physiol. Rev 93, 1621–1657 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Tanaka K et al. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702 (1997). [DOI] [PubMed] [Google Scholar]; This is the first study to show that the homozygous knockout of GLT1 in mice causes lethal spontaneous seizures.
- 60.Kong Q et al. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol. Dis 47, 145–154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rothstein JD et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433, 73–77 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Zeng LH, Bero AW, Zhang B, Holtzman DM & Wong M Modulation of astrocyte glutamate transporters decreases seizures in a mouse model of tuberous sclerosis complex. Neurobiol. Dis 37, 764–771 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sha L et al. Pharmacologic inhibition of Hsp90 to prevent GLT-1 degradation as an effective therapy for epilepsy. J. Exp. Med 214, 547–563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Campbell SL, Hablitz JJ & Olsen ML Functional changes in glutamate transporters and astrocyte biophysical properties in a rodent model of focal cortical dysplasia. Front. Cell. Neurosci 8, 425 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Campbell SL & Hablitz JJ Decreased glutamate transport enhances excitability in a rat model of cortical dysplasia. Neurobiol. Dis 32, 254–261 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eid T et al. Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363, 28–37 (2004). [DOI] [PubMed] [Google Scholar]; This study establishes a key role of GS in astrocytes for maintaining an optimal level of extracellular glutamate through the glutamate–glutamine cycle.
- 67.van der Hel WS et al. Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology 64, 326–333 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Eid T et al. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 131, 2061–2070 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang E, Yan X & Weng HR Glial glutamate transporter and glutamine synthetase regulate GABAergic synaptic strength in the spinal dorsal horn. J. Neurochem 121, 526–536 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kaczor P, Rakus D & Mozrzymas JW Neuron-astrocyte interaction enhance GABAergic synaptic transmission in a manner dependent on key metabolic enzymes. Front. Cell. Neurosci 9, 120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Magistretti PJ & Allaman I A cellular perspective on brain energy metabolism and functional imaging. Neuron 86, 883–901 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Pellerin L & Magistretti PJ Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab 32, 1152–1166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gano LB, Patel M & Rho JM Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res 55, 2211–2228 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sada N, Lee S, Katsu T, Otsuki T & Inoue T Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Verkhratsky A & Nedergaard M Physiology of astroglia. Physiol. Rev 98, 239–389 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tyzack GE et al. Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nat. Commun 5, 4294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andresen L et al. Gabapentin attenuates hyperexcitability in the freeze-lesion model of developmental cortical malformation. Neurobiol. Dis 71, 305–316 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Neniskyte U & Gross CT Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat. Rev. Neurosci 18, 658 (2017). [DOI] [PubMed] [Google Scholar]
- 79.Stevens B et al. The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007). [DOI] [PubMed] [Google Scholar]; This study demonstrates that astrocytes regulate complement-mediated synaptic pruning by phagocytic microglia.
- 80.Chu Y et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc. Natl Acad. Sci. USA 107, 7975–7980 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Clarke LE & Barres BA Emerging roles of astrocytes in neural circuit development. Nat. Rev. Neurosci 14, 311–321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aronica E et al. Complement activation in experimental and human temporal lobe epilepsy. Neurobiol. Dis 26, 497–511 (2007). [DOI] [PubMed] [Google Scholar]
- 83.Hughes EG, Elmariah SB & Balice-Gordon RJ Astrocyte secreted proteins selectively increase hippocampal GABAergic axon length, branching, and synaptogenesis. Mol. Cell. Neurosci 43, 136–145 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eroglu C & Barres BA Regulation of synaptic connectivity by glia. Nature 468, 223–231 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schafer DP et al. Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. eLife 5, e15224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tian G-F et al. An astrocytic basis of epilepsy. Nat. Med 11, 973 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is the first study to show that glutamate released from astrocytes induces neuronal firing and seizure activity.
- 87.Clasadonte J & Haydon PG in Jasper’s Basic Mechanisms of the Epilepsies (eds Noebels JL, Avoli M, Rogawski MA, Olsen RW & Delgado-Escueta AV) (National Center for Biotechnology Information (US; ), 2012). [PubMed] [Google Scholar]
- 88.Kang N, Xu J, Xu Q, Nedergaard M & Kang J Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J. Neurophysiol 94, 4121–4130 (2005). [DOI] [PubMed] [Google Scholar]
- 89.Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G & Haydon PG Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. J. Neurosci 26, 9312–9322 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Robert SM et al. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci. Transl Med 7, 289ra86 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Buckingham SC et al. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med 17, 1269–1274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tewari BP et al. Perineuronal nets decrease membrane capacitance of peritumoral fast spiking interneurons in a model of epilepsy. Nat. Commun 9, 4724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Campbell SL et al. GABAergic disinhibition and impaired KCC2 cotransporter activity underlie tumor-associated epilepsy. Glia 63, 23–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pallud J et al. Cortical GABAergic excitation contributes to epileptic activities around human glioma. Sci. Transl Med 6, 244ra89 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boison D Adenosinergic signaling in epilepsy. Neuropharmacology 104, 131–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bowser DN & Khakh BS ATP excites interneurons and astrocytes to increase synaptic inhibition in neuronal networks. J. Neurosci 24, 8606 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang J-M et al. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron 40, 971–982 (2003). [DOI] [PubMed] [Google Scholar]
- 98.Gomes CV, Kaster MP, Tomé AR, Agostinho PM & Cunha RA Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim. Biophys. Acta 1808, 1380–1399 (2011). [DOI] [PubMed] [Google Scholar]
- 99.During MJ & Spencer DD Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann. Neurol 32, 618–624 (1992). [DOI] [PubMed] [Google Scholar]
- 100.Aronica E et al. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 52, 1645–1655 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lau LW, Cua R, Keough MB, Haylock-Jacobs S & Yong VW Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat. Rev. Neurosci 14, 722–729 (2013). [DOI] [PubMed] [Google Scholar]
- 102.McRae PA, Baranov E, Rogers SL & Porter BE Persistent decrease in multiple components of the perineuronal net following status epilepticus. Eur. J. Neurosci 36, 3471–3482 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dityatev A & Fellin T Extracellular matrix in plasticity and epileptogenesis. Neuron Glia Biol.4, 235–247 (2008). [DOI] [PubMed] [Google Scholar]
- 104.Dityatev A Remodeling of extracellular matrix and epileptogenesis. Epilepsia 51, 61–65 (2010). [DOI] [PubMed] [Google Scholar]
- 105.Dubey D et al. Increased metalloproteinase activity in the hippocampus following status epilepticus. Epilepsy Res.132, 50–58 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mizoguchi H & Yamada K Roles of matrix metalloproteinases and their targets in epileptogenesis and seizures. Clin. Psychopharmacol. Neurosci 11, 45 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pollock E, Everest M, Brown A & Poulter MO Metalloproteinase inhibition prevents inhibitory synapse reorganization and seizure genesis. Neurobiol. Dis 70, 21–31 (2014). [DOI] [PubMed] [Google Scholar]
- 108.Arranz AM et al. Hyaluronan deficiency due to Has3 knock-out causes altered neuronal activity and seizures via reduction in brain extracellular space. J. Neurosci 34, 6164–6176 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reports the first direct evidence for the development of seizures due to degradation of the ECM.
- 109.Rempe RG et al. Matrix metalloproteinase-mediated blood-brain barrier dysfunction in epilepsy. J. Neurosci 38, 4301–4315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rankin-Gee EK et al. Perineuronal net degradation in epilepsy. Epilepsia 56, 1124–1133 (2015). [DOI] [PubMed] [Google Scholar]
- 111.Kochlamazashvili G et al. The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca2+ channels. Neuron 67, 116–128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Favuzzi E et al. Activity-dependent gating of parvalbumin interneuron function by the perineuronal net protein brevican. Neuron 95, 639–655 (2017). [DOI] [PubMed] [Google Scholar]
- 113.Srinivasan J, Schachner M & Catterall WA Interaction of voltage-gated sodium channels with the extracellular matrix molecules tenascin-C and tenascin-R. Proc. Natl Acad. Sci. USA 95, 15753–15757 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Frischknecht R et al. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat. Neurosci 12, 897–904 (2009). [DOI] [PubMed] [Google Scholar]; This is one of the first studies to explain the molecular mechanism of ECM-regulated synaptic plasticity.
- 115.Balmer TS Perineuronal nets enhance the excitability of fast-spiking neurons. eNeuro 10.1523/ENEURO.0112-16.2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Morawski M et al. Ion exchanger in the brain: quantitative analysis of perineuronally fixed anionic binding sites suggests diffusion barriers with ion sorting properties. Sci. Rep 5, 16471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Härtig W et al. Cortical neurons immunoreactive for the potassium channel Kv3. 1b subunit are predominantly surrounded by perineuronal nets presumed as a buffering system for cations. Brain Res.842, 15–29 (1999). [DOI] [PubMed] [Google Scholar]
- 118.Glykys J et al. Local impermeant anions establish the neuronal chloride concentration. Science 343, 670–675 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This is one of the first studies to show the regulation of neuronal chloride homeostasis by the ECM.
- 119.Zamanian JL et al. Genomic analysis of reactive astrogliosis. J. Neurosci 32, 6391–6410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Struve J et al. Disruption of the hyaluronan-based extracellular matrix in spinal cord promotes astrocyte proliferation. Glia 52, 16–24 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Ye Z-C & Sontheimer H Modulation of glial glutamate transport through cell interactions with the extracellular matrix. Int. J. Dev. Neurosci 20, 209–217 (2002). [DOI] [PubMed] [Google Scholar]
- 122.Guadagno E & Moukhles H Laminin-induced aggregation of the inwardly rectifying potassium channel, Kir4. 1, and the water-permeable channel, AQP4, via a dystroglycan-containing complex in astrocytes. Glia 47, 138–149 (2004). [DOI] [PubMed] [Google Scholar]
- 123.Seiffert E et al. Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J. Neurosci 24, 7829–7836 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provides the first direct evidence that BBB disruption contributes to epileptogenesis.
- 124.Ivens S et al. TGF-β receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 130, 535–547 (2007). [DOI] [PubMed] [Google Scholar]; This study demonstrates the epileptogenic role of serum albumin and the TGFβ signalling in astrocytes following BBB disruption.
- 125.Kim SY, Buckwalter M, Soreq H, Vezzani A & Kaufer D Blood-brain barrier dysfunction-induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia 53 (Suppl. 6), 37–44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Weissberg I et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-β/ALK5 signaling in a model of acquired epilepsy following blood-brain barrier dysfunction. Neurobiol. Dis 78, 115–125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Salar S et al. Blood-brain barrier dysfunction can contribute to pharmacoresistance of seizures. Epilepsia55, 1255–1263 (2014). [DOI] [PubMed] [Google Scholar]
- 128.Kim SY et al. TGFβ signaling is associated with changes in inflammatory gene expression and perineuronal net degradation around inhibitory neurons following various neurological insults. Sci. Rep 7, 7711 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bar-Klein G et al. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann. Neurol 75, 864–875 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang Y, Estrada EY, Thompson JF, Liu W & Rosenberg GA Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab 27, 697–709 (2007). [DOI] [PubMed] [Google Scholar]; This study provides direct evidence that MMPs impair the integrity of the BBB by degrading tight junction proteins.
- 131.Feng S et al. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLOS ONE 6, e20599 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Djerbal L, Lortat-Jacob H & Kwok J Chondroitin sulfates and their binding molecules in the central nervous system. Glycoconj. J 34, 363–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pizzorusso T et al. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298, 1248–1251 (2002). [DOI] [PubMed] [Google Scholar]; This is the first study to demonstrate the functional role of PNNs in the visual cortex as a determinant of experience-dependent plasticity.
- 134.Engel T, Alves M, Sheedy C & Henshall DC ATPergic signalling during seizures and epilepsy. Neuropharmacology 104, 140–153 (2016). [DOI] [PubMed] [Google Scholar]
- 135.Cacheaux LP et al. Transcriptome profiling reveals TGF-β signaling involvement in epileptogenesis. J. Neurosci 29, 8927–8935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Dzyubenko E, Gottschling C & Faissner A Neuronglia interactions in neural plasticity: contributions of neural extracellular matrix and perineuronal nets. Neural Plast.2016, 5214961 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Naffah-Mazzacoratti M et al. Selective alterations of glycosaminoglycans synthesis and proteoglycan expression in rat cortex and hippocampus in pilocarpine-induced epilepsy. Brain Res. Bull 50, 229–239 (1999). [DOI] [PubMed] [Google Scholar]
- 138.Gottschall PE & Howell MD ADAMTS expression and function in central nervous system injury and disorders. Matrix Biol.44, 70–76 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mataga N, Mizuguchi Y & Hensch TK Experience-dependent pruning of dendritic spines in visual cortex by tissue plasminogen activator. Neuron 44, 1031–1041 (2004). [DOI] [PubMed] [Google Scholar]
- 140.Bast T, Ramantani G, Seitz A & Rating D Focal cortical dysplasia: prevalence, clinical presentation and epilepsy in children and adults. Acta Neurol. Scand 113, 72–81 (2006). [DOI] [PubMed] [Google Scholar]
- 141.Iseki K et al. Increased syndecan expression by pleiotrophin and FGF receptor-expressing astrocytes in injured brain tissue. Glia 39, 1–9 (2002). [DOI] [PubMed] [Google Scholar]
- 142.Hoffmann K et al. Retarded kindling progression in mice deficient in the extracellular matrix glycoprotein tenascin-R. Epilepsia 50, 859–869 (2009). [DOI] [PubMed] [Google Scholar]
- 143.Muir E et al. Matrix metalloproteases and their inhibitors are produced by overlapping populations of activated astrocytes. Mol. Brain Res 100, 103–117 (2002). [DOI] [PubMed] [Google Scholar]
- 144.Wilczynski GM et al. Important role of matrix metalloproteinase 9 in epileptogenesis. J. Cell Biol 180, 1021–1035 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kochlamazashvili G et al. The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca 2+ channels. Neuron 67, 116–128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Evers MR et al. Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. J. Neurosci 22, 7177–7194 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Konopka A et al. Cleavage of hyaluronan and CD44 adhesion molecule regulate astrocyte morphology via Rac1 signalling. PLOS ONE 11, e0155053 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Perosa S et al. Glycosaminoglycan levels and proteoglycan expression are altered in the hippocampus of patients with mesial temporal lobe epilepsy. Brain Res. Bull 58, 509–516 (2002). [DOI] [PubMed] [Google Scholar]
- 149.Jones EV & Bouvier DS Astrocyte-secreted matricellular proteins in CNS remodelling during development and disease. Neural Plast.2014, 321209 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu P, Takai K, Weaver VM & Werb Z Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol 3, a005058 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]