

 This work is licensed under a
This work is licensed under a Abstract
Context
Pubertal delay can be the clinical presentation of both idiopathic hypogonadotropic hypogonadism (IHH) and self-limited delayed puberty (SLDP). Distinction between these conditions is a common but important diagnostic challenge in adolescents.
Objective
To assess whether gene panel testing can assist with clinical differential diagnosis and to allow accurate and timely management of delayed puberty patients.
Design
Retrospective study.
Methods
Patients presenting with delayed puberty to UK Paediatric services, followed up to final diagnosis, were included. Whole-exome sequencing was analysed using a virtual panel of genes previously reported to cause either IHH or SLDP to identify rarely predicted deleterious variants. Deleterious variants were verified by in silico prediction tools. The correlation between clinical and genotype diagnosis was analysed.
Results
Forty-six patients were included, 54% with a final clinical diagnosis of SLDP and 46% with IHH. Red flags signs of IHH were present in only three patients. Fifteen predicted deleterious variants in 12 genes were identified in 33% of the cohort, with most inherited in a heterozygous manner. A fair correlation between final clinical diagnosis and genotypic diagnosis was found. Panel testing was able to confirm a diagnosis of IHH in patients with pubertal delay. Genetic analysis identified three patients with IHH that had been previously diagnosed as SLDP.
Conclusion
This study supports the use of targeted exome sequencing in the clinical setting to aid the differential diagnosis between IHH and SLDP in adolescents presenting with pubertal delay. Genetic evaluation thus facilitates earlier and more precise diagnosis, allowing clinicians to direct treatment appropriately.
Introduction
Delayed puberty is a common problem in the paediatric endocrinology clinic, affecting over 2% of adolescents. This condition is diagnosed when children enter puberty 2–2.5 s.d. later than the population average (traditionally, after the age of 14 years in boys and 13 years in girls) (1). Several underlying aetiologies cause pubertal delay, including idiopathic hypogonadotropic hypogonadism (IHH) and hypergonadotropic hypogonadism. However, the most common cause of pubertal delay is self-limited, or constitutional, delayed puberty (SLDP), a functional hypogonadotropic state where individuals enter puberty late but are post-pubertal by the time they reach adulthood. Hypergonadotropic hypogonadism can be easily excluded by hormonal profiles. The differential diagnosis between SLDP and IHH, however, is often difficult, as both conditions may present with essentially the same clinical and hormonal features (2). Whilst a variety of clinical and biochemical investigations are available to make the diagnosis, none of these can reliably distinguish between those patients who will spontaneously enter and progress in a normal manner through puberty (i.e. SLDP), and those who will require medical induction of puberty and reproductive therapies (i.e. IHH) (3, 4).
This is a vital clinical distinction to make, as if IHH is diagnosed, treatment modalities to allow optimisation of future fertility (particularly for boys) can be used – in the form of gonadotropins rather than sex steroids for induction of puberty (5) and commenced earlier than the puberty induction regimen used for SLDP patients (6).
Observational studies have shown that SLDP is a familial condition, with the majority of pedigrees displaying autosomal dominant inheritance (with or without complete penetrance) (7, 8). Additionally, 79% of patients have a positive family history of SLDP without any family members with IHH, providing clinicians with further evidence of the diagnosis (7, 9, 10, 11). In contrast, IHH can be inherited via several modes of inheritance including autosomal dominant, autosomal recessive, X-linked or de novo mutation (12, 13). Moreover, in IHH families variable penetrance is commonly seen, probably due in part to the oligogenic inheritance of the disease (14, 15). To date, more than 40 genes have been identified that carry mutations which lead or contribute to conditions of IHH (13). Similarly, over the last 5 years, a smaller but increasing number of genes have been discovered that underlie SLDP by our and other groups through next-generation sequencing (16, 17). Crucially, whilst there is some overlap in the genetic background of these conditions, the majority of mutations are distinct between the two diseases (15). Therefore, genetic analysis using exome sequencing of a panel of known genes could be used to assist a clinician in distinguishing those adolescents with severe gonadotropin deficiency from those with isolated delayed puberty, allowing delivery of accurate and timely treatment to patients. However, this potential utility has yet to be assessed in a clinical cohort of patients presenting in adolescence with delayed puberty of undiagnosed aetiology.
Therefore, in this study, we investigated the burden of genetic variants in a real world, mixed ethnicity cohort of UK adolescent patients presenting with pubertal delay, in order to validate the use of genetic analysis of known causal genes to confirm the diagnosis of IHH or SLDP. We also evaluated the utility of genetic criteria to assist clinicians in confirming the diagnosis of IHH.
Subjects and methods
Patients
This study investigated a cohort of patients who were referred for genetic evaluation for delayed puberty from Paediatric Endocrinology and Paediatric services around the UK from 2015 to 2020 under a NIHR clinical research network portfolio study (Genetic Factors Affecting the Timing of Puberty, CPMS ID 30730).
Delayed puberty was defined as the onset of Tanner stage G2 (testicular volume > 3 mL) at >14 years in boys or Tanner stage B2 at > 13.0 year in girls (i.e. two s.d. later than average pubertal development). Chronic illness was excluded by detailed medical history, physical examination, and routine laboratory investigations, and hormonal investigations (basal and stimulated serum LH and FSH, serum testosterone, serum oestradiol, inhibin B) were evaluated to determine the clinical diagnosis. Pubertal progression was also assessed to establish the clinical diagnosis: those who were near to completion or had completed pubertal development (Tanner staging of at least G4 or B4) by 18 years of age and not requiring further sex steroid treatment were diagnosed as SLDP, whereas patients who had not completed puberty by the age of 18 years or had arrested puberty during prior to the age of 18 years were defined as IHH. Patients who did not have a definite diagnosis of SLDP or IHH, as they were still undergoing a period of clinical follow-up, were excluded from this analysis (n = 1).
DNA sequencing and bioinformatics
This study utilised whole-exome sequencing (WES) data of 46 patients with central pubertal delay (i.e. SLDP or IHH), and WES data of 35 healthy control individuals with normal pubertal timing. WES was performed on DNA extracted from peripheral blood leukocytes, using an Agilent V5 platform and Illumina HiSeq 2000 sequencing. Fraction of target regions with coverage > 4× was 99.4–99.8%. The exome sequences were aligned to the UCSC hg19 reference genome using the Burrows–Wheeler Aligner software (BWA-MEM (bwa-0.7.12)). Picard tools (picard-tools-1.119) were used to sort alignments and mark PCR duplicates. The genome analysis toolkit (GATK-3.4-46) was used to realign around indels and recalibrate quality scores using dbSNP, Mills and 1000 genomes as reference resources. Variant calling and joint genotyping using pedigree information were performed using HaplotypeCaller in GVCF mode from the genome analysis toolkit. The resulting variants were filtered using the variant quality score recalibration (VQSR) function from GATK.
Analysis of the called variants was performed using Ingenuity Variant Analysis (QIAGEN Redwood City, www.qiagen.com/ingenuity). Filtering for potential causal variants was carried out using a classic bioinformatic pipeline (Fig. 1). A virtual panel of 47 genes, previously reported in the literature to have a causal role in the pathogenesis of delayed puberty (Table 1), including IHH (13, 18) and SLDP (9, 10, 14, 19, 20, 21), was applied as a filter across the whole-exome sequencing dataset. Quality control filters included thresholds for call quality, read depth and Phred strand bias, and only variants with minor allele frequency (MAF) < 0.5% in the Genome Aggregation Database (gnomAD) database (accessed March 2021) were retained. Predicted functional annotation prioritised nonsense, exonic missense, splice site variants, structural or promoter changes. Case-control analysis identified variants present in affected individuals and not present in controls, using a control group of 35 individuals previously whole-exome sequenced using the same bioinformatics pathway, who have been accurately phenotyped as having timing of puberty within the normal range. To identify likely disease-causing variants in these 47 genes, only variants that met the ACMG criteria (22) for pathogenicity, likely pathogenicity, or variants of uncertain significance (VUS) were selected in the analysis.
Figure 1.

The analytic pipeline used for identifying genetic defects in patients with pubertal delay, using known genes reported in IHH or SLDP. MAF, minor allele frequency; DP, delayed puberty.
Table 1.
List of the genes previously reported causing IHH and SLDP used as a virtual panel during WES analysis (9, 10, 12, 13, 14, 18, 19, 20, 21, 41).
| Phenotype | n | Genes |
|---|---|---|
| IHH | 36 | ANOS1, CCDC141, DCC/NTN1, DMXL2, FEZF1, FGF17, FGF8, FGFR1, FSHB, GNRH1, KISS1, KISS1R, KLB, LEP, LEPR, LHB, NSMF, NR0B1, NTN1, OTUD4, PCSK1, PLXNA1, PNPLA6, POLR3A/B, PROK2, PROKR2, RNF216, SEMA3E, SMCHD1, SOX10, SOX2, STUB1, TUBB3, WDR11 |
| IHH and SLDP | 7 | HS6ST1, GNRHR, IL17RD, TAC3, TACR3, SEMA3A, CHD7 |
| SLDP | 4 | EAP1, IGSF10, LGR4, FTO |
VUS identified were further selected for being potentially deleterious using in silico analysis software tools: SIFT (23), PolyPhen-2 (24), Mutation Taster (25), REVEL (26), and MetaLR (27). Variants predicted to be deleterious by ≥3 from five tools were retained (Fig. 1). Promoter variants were annotated using RegulomeDB (https://regulomedb.org/regulome-search) and Haploreg (https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php) resources.
Genotypic criteria for the diagnosis of IHH
Using the filtered WES data, the genotype of each patient was determined to support a genotypic diagnosis of either SLDP or IHH. This categorisation was done without consideration of clinical data to minimise the risk of bias in genotype interpretation. The genotype interpretation was assisted by bioinformatic tools and published literature. Information pertaining to previously reported mutations, including variant location in the protein structure and inheritance characteristics of variants in each gene, was reviewed. The pattern of inheritance for each variant in both the proband with pubertal delay and family relatives was used to determine whether the variant might cause IHH or SLDP (Supplementary Table 1, see section on supplementary materials given at the end of this article).
A genotypic diagnosis of IHH was made if the patient carried (i) a known deleterious variant in a known IHH gene with the same zygosity as previously demonstrated to cause IHH, (ii) a new predicted deleterious variant in a known IHH gene with the same zygosity as previously demonstrated to cause IHH, (iii) homozygosity for a new predicted deleterious variant where heterozygosity or homozygosity of this gene is reported in IHH.
A genotypic diagnosis of SLDP was made if the patient carried a known or new predicted deleterious variant in a known SLDP gene with the same zygosity as previously demonstrated to cause SLDP.
An inconclusive genotype was called if (i) no variants were found, or (ii) a new predicted deleterious variant in a known IHH gene with discordant zygosity to that previously demonstrated for example, heterozygous where homozygous carriage has been shown to cause IHH, or (iii) the oligogenic carriage of two separately predicted deleterious variants in two IHH genes or a mix of IHH and SLDP genes were identified.
Ethical approval and consent to participate
Ethical approval was granted by the London–Chelsea NRES committee (13/LO/0257). All participants provided written informed consent prior to study participation. The study was conducted in accordance with the guidelines of The Declaration of Helsinki.
Statistical analysis
Continuous data were expressed as mean and s.d. when normally distributed or median with interquartile range (IQR) otherwise. Fisher’s exact test was used to compare categorical variables in epidemiological data. Unpaired t-test (two-tail) or, for multiple comparisons, the Mann–Whitney U-test, was used to compare continuous variables as appropriate. Cohen’s kappa coefficient (κ) was used to demonstrate the correlation between genotypic and clinical diagnoses. Statistical differences were deemed significant at a P-value < 0.05. Statistical analysis was performed using GraphPad Prism 8 (GraphPad Software).
Results
Clinical characteristics of SLDP and IHH patient groups are similar at presentation
From 46 patients presenting at initial assessment with delayed puberty, 54.3% (n = 25) had SLDP, and 45.7% (n = 21) had IHH as their final clinical diagnosis. The majority were male accounting for 87% (n = 40/46) and gender distributions were not different between SLDP and IHH groups. All clinical details are shown in Table 2. The median age at presentation was 16.0 (IQR 15.0, 17.1) years, with the majority of patients in early puberty, demonstrated by 40% of male patients with ≤3 mL and 47.5% with 4–9 mL testicular volumes, 34.8% of male and female patients with prepubertal pubic hair (PH1) Tanner stage and 50% with PH2, and 50% of female patients with prepubertal breast (B1) stage and 16.7% with B2 at first clinical assessment. Age at the first clinical sign of puberty (achievement of G2 or B2) was 15.6 vs 16.4 years in males and 11.8 vs 16.3 years in females for SLDP and IHH groups, respectively. A family history of pubertal delay was identified in 72% of those in the SLDP final clinical diagnostic group and 47.6% of the IHH group (P-value = 0.2). Micropenis with bilateral cryptorchidism was found in two patients with a final clinical diagnosis of IHH, and of these two patients, one also demonstrated synkinesis, whilst a third patient had a cleft palate. Anosmia was found in five patients, all in the IHH group (0% vs 25%, P-value = 0.03); anosmia was, therefore, present in 25% of IHH patients. All patients had normal basal pituitary function (thyroid function tests, cortisol, IGF1, and prolactin). Thus, most patients in this cohort presented with isolated delayed puberty without clear clinical signs of IHH (e.g. micropenis, cryptorchidism or synkinesis), representative of the DP patients in whom the distinction between SLDP and IHH is considered most difficult.
Table 2.
Demographic data and clinical details of pubertal delay patients both from the total cohort and compared between final clinical diagnosis of IHH and SLDP.
| Total (n =46) | Final clinical diagnosis | P-value | ||
|---|---|---|---|---|
| SLDP (n =25) | IHH (n =21) | |||
| Patients with identified variants, n (%) | 15 (32.6) | 6 (24.0) | 9 (42.9) | 0.2 |
| Gender, n (%) | 0.1 | |||
| Male | 40 (87.0) | 24 (96.0) | 16 (76.2) | |
| Female | 6 (13.0) | 1 (4.0) | 5 (23.8) | |
| Ethnicity | 0.5 | |||
| European | 29 | 16 | 13 | |
| South Asian | 6 | 2 | 4 | |
| Ashkenazi Jewish | 5 | 5 | 0 | |
| Middle Eastern | 2 | 2 | 0 | |
| Turkish | 2 | 0 | 2 | |
| African | 2 | 0 | 2 | |
| Age at Tanner stage II (years) | ||||
| Male | 15.9 (1.5) | 15.6 (1.4) | 16.4 (1.6) | 0.1 |
| Female | 15.4 (2.3) | 11.8 (0.0) | 16.3 (1.2) | 0.03* |
| Hormonal treatment, n (%) | 40 (87.0) | 21 (84.0) | 19 (90.5) | 0.8 |
| Anosmia, n (%) | 5 (10.9) | 0 (0.0) | 5 (23.8) | 0.04* |
| MPH | 173.4 (7.9) | 173.5 (7.77) | 173.3 (8.44) | 0.9 |
| MPH SDS | −0.41 (0.93) | −0.48 (1.06) | −0.30 (0.73) | 0.6 |
| Family history of DP, n (%) | 28 (60.9) | 18 (72.0) | 10 (47.6) | 0.2 |
| Consanguinity, n (%) | 1 (2.2) | 0 (0.0) | 1 (4.8) | 0.9 |
| IHH red flag signs, n (%) | 2 (6.5) | 0 (0.0) | 3 (14.3) | 0.9 |
| First visit* | ||||
| Age (years)ǂ | 16.0 (15.0, 17.1) | 15.6 (14.9, 16.3) | 16.5 (15.7, 17.2) | 0.05 |
| Weight SDS | −1.19 (1.78) | −1.58 (1.79) | −0.69 (1.69) | 0.1 |
| Height SDS | −1.67 (1.15) | −1.80 (1.10) | −1.51 (1.22) | 0.4 |
| BMI SDS | −0.29 (1.87) | −0.67 (1.79) | 0.18 (1.90) | 0.139 |
| Tanner staging | ||||
| TV (mL), n (%) | 0.2 | |||
| ≤ 3 | 16 (40.0) | 7 (29.2) | 9 (56.2) | |
| 4–9 | 19 (47.5) | 14 (58.3) | 5 (31.2) | |
| 10–15 | 5 (12.5) | 3 (12.5) | 2 (12.6) | |
| Genital staging, n (%) | 0.1 | |||
| I | 16 (40.0) | 11 (45.8) | 5 (31.2) | |
| II | 16 (40.0) | 6 (25.0) | 10 (62.5) | |
| III | 5 (12.5) | 5 (20.8) | 0 (0.0) | |
| IV | 2 (5.0) | 1 (4.2) | 1 (6.2) | |
| V | 1 (2.5) | 1 (4.2) | 0 (0.0) | |
| Pubic hair staging, n (%) | 0.3 | |||
| I | 16 (34.8) | 9 (36.0) | 7 (35.0) | |
| II | 23 (50.0) | 14 (56.0) | 9 (45.0) | |
| III | 3 (6.5) | 0 (0.0) | 3 (15.0) | |
| IV | 1 (2.2) | 1 (4.0) | 0 (0.0) | |
| V | 3 (6.5) | 1 (4.0) | 1 (5.0) | |
| Breast staging, n (%) | 0.8 | |||
| I | 3 (60.0) | 1 (100.0) | 2 (50.0) | |
| II | 1 (20.0) | 0 (0.0) | 1 (25.0) | |
| III | 1 (20.0) | 0 (0.0) | 1 (25.0) | |
| S-LH (IU/L)ǂ | 0.8 (0.2, 1.8) | 0.8 (0.2, 2.1) | 1.0 (0.1, 1.6) | 0.6 |
| S-FSH (IU/L)ǂ | 1.8 (1.1, 2.8) | 2.0 (1.6, 3.4) | 1.5 (0.7, 2.7) | 0.07 |
| S-Test (nmol/L)ǂ | 0.7 (0.4, 1.5) | 1.0 (0.6, 4.9) | 0.4 (0.4, 0.7) | 0.007* |
| Peak S-LH (IU/L)ǂ | 9.1 (1.7, 13.3) | 11.70 (8.12, 14.25) | 6.00 (1.17, 10.72) | 0.09 |
| Inhibin B (pg/mL)ǂ | 51.0 (18.5, 115.0) | 149.0 (104.0, 183.0) | 36.0 (14.5, 69.8) | 0.009* |
*First visit data were missing from one female with IHH; ǂData presented as median (IQR), otherwise presented as mean (s.d.).
DP, delayed puberty; MPH, mid parental height; SDS, standard deviation score; S-FSH, serum follicle-stimulating hormone; S-LH, serum luteinizing hormone; S-Test, serum testosterone; TV, testicular volume; BII/GII, Tanner stage II.
Multiple deleterious variants in known pubertal delay genes are identified from whole-exome sequencing of patients with delayed puberty
Of the 46 patients analysed by WES, 15 patients (32.6%) were identified with potentially deleterious variants in a known gene reported in either IHH or SLDP (Fig. 2). In all, 15 potentially deleterious variants in 12 genes were identified, with two siblings (patient 13 and 14) carrying the same variant, (GNRHR c.317A>G; p.Q106R). All variants were rare with a MAF of less than 0.5%, and the majority, 73.3% (n = 11/15), were inherited in a heterozygous manner. The exception to this was variants in GNRHR (c.317A>G; p.Q106R), PROKR2 (c.809G>A; p.R270H) and TAC3 (c.209-1G>C), which displayed homozygous inheritance. One variant was identified in a gene confined to SLDP, IGSF10 (1 of 4 genes in this category, 25%). Five of the genes reported in both SLDP and IHH were identified with eight variants in total (5 of 7 genes in this category, 71%), and six variants were identified in six genes previously reported only in IHH (9 of 35 genes in this category, 17.1%) (Fig. 2A). The most common type of allelic variation was missense (63.2%) followed by variants affecting a splice site (15.8%), nonsense (10.6%), promoter (5.3%) and in-frame deletion (5.3%) variants (Fig. 2B). The KISS1R promoter variant (c.-249G>A) occurred within a region with a highly significant puberty-specific differential promoter methylation pattern (28), predicted to be a site of EZH2 transcription factor binding, a member of the polycomb group of transcriptional regulators (29). The details of each deleterious variant, including zygosity, MAF and predicted functional impact are described in Table 3.
Figure 2.
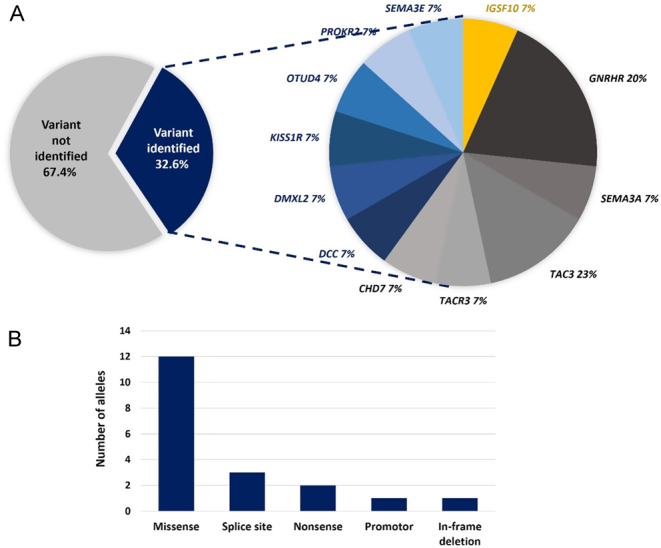
Variants identified in this cohort. (A) Proportion of variants identified in each gene group; yellow, genes confined to SLDP; blue, variants in genes reported in IHH only; grey, variants in genes reported in both SLDP and IHH. (B) Number of alleles in each variant category.
Table 3.
Identified variants in pubertal delay patients.
| Gene lists/genes | Variants | Protein variants | Reference | Translational impact | Zygosity | ACMG crtieria | SIFT | PolyPhen-2 | Mutation Taster | REVEL | MetaLR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SLDP only | |||||||||||
| IGSF10 | c.7124A>G | p.N2375S | Missense | Het | VUS | X | X | X | X | X | |
| Both SLDP and IHH | |||||||||||
| GNRHR | |||||||||||
| c.317A>G | p.Q106R | (42, 43) | Missense | Hom | P | X | X | X | X | O | |
| c.436C>T | p.P146S | (44) | Missense | Het | LP | X | X | X | X | X | |
| SEMA3A | c.1849C>T | p.R617Ter | Nonsense | Het | P | – | – | X | – | – | |
| TAC3 | |||||||||||
| c.209-1G>C | – | (31) | Splice site | Hom | P | – | – | X | – | – | |
| c.*2-1G>T | – | Splice site | Het | LP | – | – | – | – | – | ||
| TACR3 | c.1090C>T | p.R364Ter | Nonsense | Het | P | – | – | X | – | – | |
| CHD7 | c.3738G>A | p.M1246I | Missense | Het | VUS | X | X | X | X | X | |
| IHH only | |||||||||||
| DCC | c.1933C>T | p.P645S | (45) | Missense | Het | VUS | X | X | X | O | O |
| DMXL2 | c.2540C>T | p.T847I | Missense | Het | VUS | X | X | X | O | O | |
| KISS1R | c.-249G>A | – | Promoter | Het | VUS | – | – | – | – | – | |
| OTUD4 | c.458_460delCTG | p.A153del | In-frame deletion | Het | VUS | – | – | – | – | – | |
| PROKR2 | c.809G>A | p.R270H | (46) | Missense | Hom | VUS | X | X | X | X | X |
| SEMA3E | c.398G>T | p.C133F | Missense | Het | VUS | X | X | X | X | X | |
X, a potentially deleterious variant by prediction tools (deleterious by SIFT, probably or possibly damaging by PolyPhen2, disease causing by Mutation Taster, likely disease causing by REVEL, and damaging by MetaLR); O, non-pathogenic predicted by prediction tools (tolerated by SIFT, benign or unknown by PolyPhen2, polymorphism by Mutation Taster, likely benign by REVEL, and tolerated by MetaLR); Het, heterozygous; Hom, homozygous; P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance.
Genotypic characteristics are distinct between IHH and SLDP patient groups
We compared the genetic characteristics between SLDP and IHH patients. In patients with IHH, an underlying genetic variant was identified more frequently (9/21, 42.9%) than in those with SLDP (6/25, 24.0%), but the difference was not significant (P = 0.2). Deleterious variants previously reported only in SLDP were not identified in IHH patients. Homozygous, promoter and nonsense variants were identified only in patients with a final clinical diagnosis of IHH. Patients with a clinical diagnosis of SLDP were found to carry potentially deleterious variants in genes previously reported in IHH only, including DMXL2, OTUD4, and SEMA3E (Table 4).
Table 4.
Comparison of genetic characteristics between patients clinically diagnosed as SLDP and IHH.
| Final clinical diagnosis | ||
|---|---|---|
| SLDP (%) | IHH (%) | |
| Patient with variant identified | ||
| Yes | 6 (24.0) | 9 (42.9) |
| No | 19 (76.0) | 12 (57.1) |
| Variants from gene list | ||
| SLDP only | 1 (14.2) | 0 (0.0) |
| SLDP and IHH | 2 (42.9) | 6 (66.7) |
| IHH only | 3 (42.9) | 3 (33.3) |
| Zygosity of the variants | ||
| Heterozygous | 6 (100.0) | 5 (55.6) |
| Homozygous | 0 (0.0) | 4 (44.4) |
| Variant category (n = 20 alleles) | ||
| Predicted to affect protein structure/expression (nonsense, splice site, promoter) | 1 (16.7) | 5 (38.5) |
| Predicted not to affect protein structure/expression (missense, in-frame variant) | 5 (83.3) | 8 (61.5) |
Genetic criteria can be utilised for the diagnosis of IHH and SLDP in patients presenting with delayed puberty
After a list of qualified variants was filtered, each patient’s genotypic diagnosis was determined based on the criteria outlined in methods. One patient was found to have a genotypic diagnosis of SLDP, seven patients had a genotypic diagnosis of IHH, and the remaining seven had an inconclusive genotype (Table 5). Notably, three patients who were initially given a clinical diagnosis of SLDP (patients 10, 13, and 14) were shown to have a diagnosis of IHH by genetic analysis. At final diagnosis, these three patients had a confirmed clinical diagnosis of IHH after completion of follow-up. These three cases are in keeping with the published literature of these known pathogenic mutations, where cases with IHH can present initially with simple delayed puberty (30, 31). Parents of these cases, as in the literature, were either unaffected carriers or manifested self-limited delayed puberty, Supplementary Table 3 (30, 32). Autosomal recessive inheritance is a known inheritance pattern in IHH, but phenotypically, these cases are at the milder end of the spectrum, not associated classically with red flag signs for IHH, and thus are more likely to be misdiagnosed clinically. Homozygosity in the absence of consanguinity for these families is due, for the GNRHR_Q106R variant, to its MAF of 0.4% in the non-Finnish European population, and, for the TAC3 splice variant to it being a founder mutation in the Congolese population, from where the patient’s parents both originated, Supplementary Table 3.
Table 5.
The association between final clinical and genotypic diagnosis. Het, heterozygous; Hom, homozygous. Genotypic diagnosis is shown in bold, where discordant with the initial clinical diagnosis but concordant with final clinical diagnosis.
| No. | Initial clinical diagnosis | Final clinical diagnosis | Sex | Age at 1st visit (years) | DP in family | Gene | Ethnicity | Nucleotide change | Protein change | MAF (%) by ethnicity | Zygosity | Genotypic diagnosis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Uncertain | SLDP | M | 18.3 | No | IGSF10 | Non-Finnish European | c.7124A>G | p.N2375S | 0.0256 | Het | SLDP |
| 2 | Uncertain | SLDP | M | 13.3 | No | DMXL2 | Askenazi Jewish | c.2540C>T | p.T847I | 0 | Het | Inconclusive |
| 3 | Uncertain | SLDP | M | 17.2 | No | OTUD4 | Non-Finnish European | c.458_460delCTG | p.A153del | 0.00558 | Het | Inconclusive |
| 4 | Uncertain | SLDP | M | 17.4 | No | CHD7 | Non-Finnish European | c.3738G>A | p.M1246I | 0 | Het | Inconclusive |
| 5 | Uncertain | SLDP | M | 17.2 | Father, uncle | SEMA3E | Non-Finnish European | c.398G>T | p.C133F | 0 | Het | Inconclusive |
| 6 | Uncertain | SLDP | M | 15.0 | Father, uncle | TAC3 | African | c.*2-1G>T | Splice site | 0.0259 | Het | Inconclusive |
| 7 | Uncertain | IHH | M | 15.6 | Father | PROKR2 | South Asian | c.809G>A | p.R270H | 0.0588 | Hom | IHH |
| 8 | Uncertain | IHH | M | 14.8 | Mother | KISS1R | Non-Finnish European | c.-249G>A | Promoter | 0 | Het | Inconclusive |
| 9 | Uncertain | IHH | M | 15.9 | Parents, brother | SEMA3A | Non-Finnish European | c.1849C>T | p.R617Ter | 0.00088 | Het | IHH |
| 10 | SLDP | IHH | F | 14.9 | Mother, sister | TAC3 | African | c.209-1G>C | Splice site | 0.112 | Hom | IHH |
| 11 | Uncertain | IHH | M | 18.1 | Father | GNRHR | Non-Finnish European | c.436C>T | p.P146S | 0.119 | Het | Inconclusive |
| 12 | Uncertain | IHH | M | 17.1 | No | DCC | South Asian | c.1933C>T | p.P645S | 0.173 | Het | IHH |
| 13 | SLDP | IHH | M | 16.5 | Mother, brother | GNRHR | Non-Finnish European | c.317A>G | p.Q106R | 0.418 | Hom | IHH |
| 14 | SLDP | IHH | M | 13.5 | Mother, brother | GNRHR | Non-Finnish European | c.317A>G | p.Q106R | 0.418 | Hom | IHH |
| 15 | Uncertain | IHH | M | 16 | No | TACR3 | South Asian | c.1090C>T | p.R364Ter | 0.00327 | Het | IHH |
A correlation between genotypic and clinical diagnosis was identified for patients presenting with delayed puberty
The correlation between final clinical diagnosis and genotype was assessed in 15 patients who were definitively diagnosed with either SLDP or IHH following the diagnostic criteria outlined above. Notably, the patients who were diagnosed with SLDP and IHH by genotyping had SLDP and IHH phenotypes, respectively, with a Cohen’s kappa (k) = 0.327 (95% IC, 0.137–0.517), demonstrating that genotype diagnosis has a fair agreement with the final clinical diagnosis (Fig. 3). Patients diagnosed as IHH by genotypic criteria are thus very likely to have IHH as a final clinical diagnosis. Likewise, patients with a genotype of SLDP are likely to have a clinical diagnosis of SLDP. For those patients who were diagnosed with an inconclusive genotype by WES, they may belong to the clinical phenotypic group of either SLDP or IHH.
Figure 3.

Clinical diagnosis of the patients who underwent WES with variants identified and grouped by genotypic criteria (n = 19). X-axis shows three groups of genotypic diagnosis in patients who underwent WES. Y-axis shows the percentage of patients in each genotype diagnosis. Clinical diagnosis is shown by bars as indicated.
Using this genotypic diagnostic framework, 7 of 21 patients with a final clinical diagnosis of IHH can be diagnosed with IHH, and all patients with a final clinical diagnosis of SLDP had a genetic result that was not compatible with IHH. Applying these criteria resulted in a sensitivity and specificity of 33.3% and 100%, respectively, of using genotypic criteria for diagnosis of IHH, with a positive predictive value (PPV) of 100%, and negative predictive value (NPV) of 64.10% (Supplementary Table 2).
Discussion
Pubertal delay can be the presentation of a broad spectrum of clinical phenotypes ranging from IHH, which is a pathological condition and needs intensive medical therapy, to SLDP, a more benign condition usually compatible with normal reproductive capacity post-puberty. Many clinical and biochemical parameters have been applied to try to distinguish these two conditions; however, these all have limitations in terms of specificity and sensitivity (3). Diagnostic uncertainty is associated with increased psychological stress for both adolescents and their parents (33). In this study, we investigated for the first time whether the identification of a genetic defect in patients with pubertal delay, through WES combined with a virtual panel, can distinguish these two conditions.
Prompt diagnosis will aid clinical management, by ameliorating the need for patients to undergo unnecessary investigations or inappropriate treatment. Furthermore, early diagnosis of IHH can facilitate the use of optimal therapeutic modalities for pubertal induction, such as the use of gonadotropins in males with IHH and commencement of therapy at an earlier age than the standard sex steroid therapy indicated for SLDP patients (6). Our clinical data highlight this issue, as the mean age of development of secondary sexual characteristics, following hormonal induction, in the patients with a final diagnosis of IHH was over 16 years.
The findings of this study point to a fair correlation between genotypic diagnosis and final clinical diagnosis, with a 100% specificity and PPV of genetic testing for the diagnosis of patients with IHH. This study also found that patients who carry homozygous or loss-of-function variants in genes reported in IHH will be very likely to have a final clinical diagnosis of IHH. On the other hand, patients carrying variants reported only in SLDP with heterozygous inheritance are likely to have a clinical diagnosis of SLDP. From this cohort review, we found that testing through WES with a virtual panel can make a definitive diagnosis for 17.4% (n = 8/46) of patients presenting with pubertal delay, and that of these patients, seven out of eight had IHH. This supports the use of genetic investigation in the clinical setting, combined with clinical and biochemical criteria, to confirm the diagnosis of IHH in adolescents presenting with pubertal delay.
Moreover, genetic testing also has a benefit for initial diagnosis in patients who do not manifest obvious clinical signs of IHH as we identified a genotypic diagnosis of IHH in three patients in whom there had been an initial clinical diagnosis of SLDP, who went on to have a final clinical diagnosis of IHH. In these patients, who presented with isolated DP without red flags for IHH, where there was also a family history of DP, a default diagnosis of SLDP was made. Such individuals may also have reasonable prepubertal testes volumes (e.g. 3 mL) or have entered puberty and then arrested, and without genetic analysis, it is only after several years of careful follow-up that they can be diagnosed as IHH.
This study also identified deleterious variants in three other genes known to contribute to IHH, namely DMXL2, OTUD4 and SEMA3E, in three patients with a final clinical diagnosis of SLDP. Although these patients had clinical characteristics of SLDP, they may have a more significant defect in their GnRH neuroendocrine system, including impact on fertility or timing of menopause/andropause, which may need monitoring into adulthood. Moreover, it suggests that in a subset of patients with pubertal delay, there may be some overlap of genetic and pathophysiological mechanisms between SLDP and IHH, or lie along a spectrum of GnRH deficiency. This sub-category may be reflected by the ‘inconclusive genotypic’ group identified in this study, in whom a moderate burden of mutations in GnRH deficiency genes may lead to a SLDP phenotypic pattern, whilst a severe mutational burden, such as homozygous or loss-of-function mutation, may lead to a more severe reproductive phenotype, that is, IHH. It is likely that with further genetic discovery and a better understanding of the pathophysiology underlying these conditions, the genotype–phenotype correlation in this inconclusive group will become clearer. We believe that this group merits careful observation of their clinical progression.
Our study has some limitations. The number of genes in the virtual panel associated with SLDP (n = 4) was far smaller than those associated with IHH (n = 42). Only a few causal genes have been identified in SLDP to date, leading to a lower pick-up rate for SLDP mutations in this study (deleterious variants were identified in 25% (1 in 4) SLDP genes and 33% (14 in 43) of IHH genes in this study). Therefore, our group is working to better characterise the genetic basis of SLDP. Identification of a larger number of SLDP genes and their use in the virtual panel will improve the ability to make a genetic diagnosis in SLDP patients at presentation. Moreover, genotypic interpretation in patients with oligogenic inheritance is complex because of our lack of knowledge of variant–variant interaction. We have described such patients’ genotypes as inconclusive to minimise potential bias. Furthermore, although some patients had a family history of DP, DNA from the majority of these family members was not available to enable an analysis of genotype–phenotype correlation in the wider pedigree. Finally, this genetic analysis shows lower sensitivity and negative predictive value than some biochemical modalities (3, 34, 35, 36, 37, 38). Given that a combination of investigations can increase the sensitivity and specificity to diagnosis IHH (39, 40), this type of genetic analysis is at present likely to be best combined with biochemical profiling (e.g. basal LH, FSH, inhibin B, AMH) in order to maximise the diagnostic accuracy.
To our knowledge, this is the first study to demonstrate the correlation between genotypic diagnosis and final clinical diagnosis in a cohort of adolescent patients with severe pubertal delay, validating the use of genetic analysis to support the distinction between the clinical diagnosis of SLDP and IHH. We have also described a set of genotypic criteria for interpreting WES results from a virtual panel using curated information from previous reports. The use of early genetic diagnosis in this condition has the potential for significant cost savings as it can prevent unnecessary investigations and lead to improved health and fertility outcomes for patients. In summary, our analysis shows that WES analysis using a virtual panel in patients with delayed puberty is a useful tool to give a definite diagnosis in an uncertain clinical presentation.
Supplementary Material
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this study.
Funding
T S is funded by Faculty of Medicine, Prince of Songkla University. M T D receives funding from the Great Ormond Street Hospital (GOSH) Children’s Charity and the Medical Research Foundation, UK (grant# 535963). Research at GOSH benefits from funding received from the NIHR Biomedical Research Centre (MTD). S R H is funded by the National Institute for Health Research [CL-2017-19-002] and the Rosetrees Trust [M222-F1].
Acknowledgements
The authors are very grateful to the patients and families who contributed their time, medical information, and DNA samples to this study.
References
- 1.Palmert MR, Dunkel L. Clinical practice: delayed puberty. New England Journal of Medicine 2012366443–453. ( 10.1056/NEJMcp1109290) [DOI] [PubMed] [Google Scholar]
- 2.Howard SR.Genes underlying delayed puberty. Molecular and Cellular Endocrinology 2018476119–128. ( 10.1016/j.mce.2018.05.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrington J, Palmert MR. Clinical Review: Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. Journal of Clinical Endocrinology and Metabolism 2012973056–3067. ( 10.1210/jc.2012-1598) [DOI] [PubMed] [Google Scholar]
- 4.Varimo T, Miettinen PJ, Kansakoski J, Raivio T, Hero M. Congenital hypogonadotropic hypogonadism, functional hypogonadotropism or constitutional delay of growth and puberty? An analysis of a large patient series from a single tertiary center. Human Reproduction 201732147–153. ( 10.1093/humrep/dew294) [DOI] [PubMed] [Google Scholar]
- 5.Rohayem J, Hauffa BP, Zacharin M, Kliesch S, Zitzmann M. & German Adolescent Hypogonadotropic Hypogonadism Study Group. Testicular growth and spermatogenesis: new goals for pubertal hormone replacement in boys with hypogonadotropic hypogonadism? A multicentre prospective study of hCG/rFSH treatment outcomes during adolescence. Clinical Endocrinology 20178675–87. ( 10.1111/cen.13164) [DOI] [PubMed] [Google Scholar]
- 6.Howard S, Dunkel L. Sex steroid and gonadotropin treatment in male delayed puberty. Endocrine Development 201629185–197. ( 10.1159/000438891) [DOI] [PubMed] [Google Scholar]
- 7.Wehkalampi K, Widen E, Laine T, Palotie A, Dunkel L. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care. Journal of Clinical Endocrinology and Metabolism 200893723–728. ( 10.1210/jc.2007-1786) [DOI] [PubMed] [Google Scholar]
- 8.Sedlmeyer IL, Hirschhorn JN, Palmert MR. Pedigree analysis of constitutional delay of growth and maturation: determination of familial aggregation and inheritance patterns. Journal of Clinical Endocrinology and Metabolism 2002875581–5586. ( 10.1210/jc.2002-020862) [DOI] [PubMed] [Google Scholar]
- 9.Howard SR, Guasti L, Ruiz-Babot G, Mancini A, David A, Storr HL, Metherell LA, Sternberg MJ, Cabrera CP, Warren HRet al. IGSF10 mutations dysregulate gonadotropin-releasing hormone neuronal migration resulting in delayed puberty. EMBO Molecular Medicine 20168626–642. ( 10.15252/emmm.201606250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mancini A, Howard SR, Cabrera CP, Barnes MR, David A, Wehkalampi K, Heger S, Lomniczi A, Guasti L, Ojeda SRet al. EAP1 regulation of GnRH promoter activity is important for human pubertal timing. Human Molecular Genetics 2019281357–1368. ( 10.1093/hmg/ddy451) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mancini A, Howard SR, Marelli F, Cabrera CP, Barnes MR, Sternberg MJE, Leprovots M, Hadjidemetriou I, Monti E, David Aet al. LGR4 deficiency results in delayed puberty through impaired Wnt/β-catenin signaling. JCI Insight 20205e133434. ( 10.1172/jci.insight.133434) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howard SR, Dunkel L. Delayed puberty: phenotypic diversity, molecular genetic mechanisms and recent discoveries. Endocrine Reviews 2019401285–1317. ( 10.1210/er.2018-00248) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Young J, Xu C, Papadakis GE, Acierno JS, Maione L, Hietamaki J, Raivio T, Pitteloud N. Clinical management of congenital hypogonadotropic hypogonadism. Endocrine Reviews 201940669–710. ( 10.1210/er.2018-00116) [DOI] [PubMed] [Google Scholar]
- 14.Zhu J, Choa RE, Guo MH, Plummer L, Buck C, Palmert MR, Hirschhorn JN, Seminara SB, Chan YM. A shared genetic basis for self-limited delayed puberty and idiopathic hypogonadotropic hypogonadism. Journal of Clinical Endocrinology and Metabolism 2015100E646–E654. ( 10.1210/jc.2015-1080) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cassatella D, Howard SR, Acierno JS, Xu C, Papadakis GE, Santoni FA, Dwyer AA, Santini S, Sykiotis GP, Chambion Cet al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. European Journal of Endocrinology 2018178377–388. ( 10.1530/EJE-17-0568) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howard SR.The genetic basis of delayed puberty. Frontiers in Endocrinology 201910 423. ( 10.3389/fendo.2019.00423) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barroso PS, Jorge AAL, Lerario AM, Montenegro LR, Vasques GA, Lima Amato LG, Gontijo Silveira LF, Mendonca BB, Latronico AC. Clinical and genetic characterization of a constitutional delay of growth and puberty cohort. Neuroendocrinology 2020110959–966. ( 10.1159/000504783) [DOI] [PubMed] [Google Scholar]
- 18.Amato LGL, Montenegro LR, Lerario AM, Jorge AAL, Guerra Junior G, Schnoll C, Renck AC, Trarbach EB, Costa EMF, Mendonca BBet al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. European Journal of Endocrinology 2019181103–119. ( 10.1530/EJE-18-0764) [DOI] [PubMed] [Google Scholar]
- 19.Gianetti E, Tusset C, Noel SD, Au MG, Dwyer AA, Hughes VA, Abreu AP, Carroll J, Trarbach E, Silveira LFet al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. Journal of Clinical Endocrinology and Metabolism 2010952857–2867. ( 10.1210/jc.2009-2320) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howard SR, Guasti L, Poliandri A, David A, Cabrera CP, Barnes MR, Wehkalampi K, O'Rahilly S, Aiken CE, Coll APet al. Contributions of function-altering variants in genes implicated in pubertal timing and body mass for self-limited delayed puberty. Journal of Clinical Endocrinology and Metabolism 2018103649–659. ( 10.1210/jc.2017-02147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howard SR, Oleari R, Poliandri A, Chantzara V, Fantin A, Ruiz-Babot G, Metherell LA, Cabrera CP, Barnes MR, Wehkalampi Ket al. HS6ST1 insufficiency causes self-limited delayed puberty in contrast with other GnRH deficiency genes. Journal of Clinical Endocrinology and Metabolism 20181033420–3429. ( 10.1210/jc.2018-00646) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector Eet al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine 201517405–424. ( 10.1038/gim.2015.30) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature Protocols 200941073–1081. ( 10.1038/nprot.2009.86) [DOI] [PubMed] [Google Scholar]
- 24.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods 20107248–249. ( 10.1038/nmeth0410-248) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nature Methods 201411361–362. ( 10.1038/nmeth.2890) [DOI] [PubMed] [Google Scholar]
- 26.Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi Det al. Revel: an ensemble method for predicting the pathogenicity of rare missense variants. American Journal of Human Genetics 201699877–885. ( 10.1016/j.ajhg.2016.08.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Human Molecular Genetics 2015242125–2137. ( 10.1093/hmg/ddu733) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wyatt AK, Zavodna M, Viljoen JL, Stanton JA, Gemmell NJ, Jasoni CL. Changes in methylation patterns of Kiss1 and Kiss1r gene promoters across puberty. Genetics and Epigenetics 2013. 5 51–62. ( 10.4137/GEG.S12897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright H, Aylwin CF, Toro CA, Ojeda SR, Lomniczi A. Polycomb represses a gene network controlling puberty via modulation of histone demethylase Kdm6b expression. Scientific Reports 2021111996. ( 10.1038/s41598-021-81689-4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin L, Conway GS, Hill NR, Dattani MT, Hindmarsh PC, Achermann JC. A homozygous R262Q mutation in the gonadotropin-releasing hormone receptor presenting as constitutional delay of growth and puberty with subsequent borderline oligospermia. Journal of Clinical Endocrinology and Metabolism 2006915117–5121. ( 10.1210/jc.2006-0807) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young J, Bouligand J, Francou B, Raffin-Sanson ML, Gaillez S, Jeanpierre M, Grynberg M, Kamenicky P, Chanson P, Brailly-Tabard Set al. TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. Journal of Clinical Endocrinology and Metabolism 2010952287–2295. ( 10.1210/jc.2009-2600) [DOI] [PubMed] [Google Scholar]
- 32.Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, Dwyer A, Plummer L, Hughes V, Seminara S, Cheng YZet al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. Journal of Clinical Investigation 2007117457–463. ( 10.1172/JCI29884) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stewart JL, Mishel MH. Uncertainty in childhood illness: a synthesis of the parent and child literature. Scholarly Inquiry for Nursing Practice 200014299–319, dicussion 32, 1–32. [PubMed] [Google Scholar]
- 34.Gao Y, Du Q, Liu L, Liao Z. Serum inhibin B for differentiating between congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty: a systematic review and meta-analysis. Endocrine 202172633–643. ( 10.1007/s12020-020-02582-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Persani L, Bonomi M, Cools M, Dattani M, Dunkel L, Gravholt CH, Juul A. ENDO-ERN expert opinion on the differential diagnosis of pubertal delay. Endocrine 202171681–688. ( 10.1007/s12020-021-02626-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown DC, Stirling HF, Butler GE, Kelnar CJ, Wu FC. Differentiation of normal male prepuberty and hypogonadotrophic hypogonadism using an ultrasensitive luteinizing hormone assay. Hormone Research 19964683–87. ( 10.1159/000185001) [DOI] [PubMed] [Google Scholar]
- 37.Wu FC, Brown DC, Butler GE, Stirling HF, Kelnar CJ. Early morning plasma testosterone is an accurate predictor of imminent pubertal development in prepubertal boys. Journal of Clinical Endocrinology and Metabolism 19937626–31. ( 10.1210/jcem.76.1.8421096) [DOI] [PubMed] [Google Scholar]
- 38.Wu FC, Butler GE, Kelnar CJ, Stirling HF, Huhtaniemi I. Patterns of pulsatile luteinizing hormone and follicle-stimulating hormone secretion in prepubertal (midchildhood) boys and girls and patients with idiopathic hypogonadotropic hypogonadism (Kallmann’s syndrome): a study using an ultrasensitive time-resolved immunofluorometric assay. Journal of Clinical Endocrinology and Metabolism 1991721229–1237. ( 10.1210/jcem-72-6-1229) [DOI] [PubMed] [Google Scholar]
- 39.Dunkel L, Perheentupa J, Virtanen M, Mäenpää J. GnRH and HCG tests are both necessary in differential diagnosis of male delayed puberty. American Journal of Diseases of Children 1985139494–498. ( 10.1001/archpedi.1985.02140070068036) [DOI] [PubMed] [Google Scholar]
- 40.Segal TY, Mehta A, Anazodo A, Hindmarsh PC, Dattani MT. Role of gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests in differentiating patients with hypogonadotropic hypogonadism from those with constitutional delay of growth and puberty. Journal of Clinical Endocrinology and Metabolism 200994780–785. ( 10.1210/jc.2008-0302) [DOI] [PubMed] [Google Scholar]
- 41.Topaloglu AK, Kotan LD. Genetics of hypogonadotropic hypogonadism. Endocrine Development 20162936–49. ( 10.1159/000438841) [DOI] [PubMed] [Google Scholar]
- 42.Fathi AK, Hu S, Fu X, Huang S, Liang Y, Ning Q, Luo X. Molecular defects of the GnRH-receptor gene in Chinese patients with idiopathic hypogonadotropic hypogonadism and the severity of hypogonadism. Journal of Pediatric Endocrinology and Metabolism 201225659–668. ( 10.1515/jpem-2012-0087) [DOI] [PubMed] [Google Scholar]
- 43.Choi JH, Balasubramanian R, Lee PH, Shaw ND, Hall JE, Plummer L, Buck CL, Kottler ML, Jarzabek K, Wołczynski Set al. Expanding the spectrum of founder mutations causing isolated gonadotropin-releasing hormone deficiency. Journal of Clinical Endocrinology and Metabolism 2015100E1378–E1385. ( 10.1210/jc.2015-2262) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vagenakis GA, Sgourou A, Papachatzopoulou A, Kourounis G, Papavassiliou AG, Georgopoulos NA. The gonadotropin-releasing hormone (GnRH)-1 gene, the GnRH receptor gene, and their promoters in patients with idiopathic hypogonadotropic hypogonadism with or without resistance to GnRH action. Fertility and Sterility 2005841762–1765. ( 10.1016/j.fertnstert.2005.06.031) [DOI] [PubMed] [Google Scholar]
- 45.Bouilly J, Messina A, Papadakis G, Cassatella D, Xu C, Acierno JS, Tata B, Sykiotis G, Santini S, Sidis Yet al. DCC/NTN1 complex mutations in patients with congenital hypogonadotropic hypogonadism impair GnRH neuron development. Human Molecular Genetics 201827359–372. ( 10.1093/hmg/ddx408) [DOI] [PubMed] [Google Scholar]
- 46.Zhao Y, Wu J, Jia H, Wang X, Zheng R, Jiang F, Chen DN, Chen Z, Li JD. PROKR2 mutations in idiopathic hypogonadotropic hypogonadism: selective disruption of the binding to a Gα-protein leads to biased signaling. FASEB Journal 2019334538–4546. ( 10.1096/fj.201801575R) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.