

 This work is licensed under a
This work is licensed under a Abstract
Summary
In July 2017, a 35-year-old woman was referred to our care for treatment of a large pituitary mass with an unusually high growth rate. She presented with right-sided ptosis and diplopia (n. III palsy), increasing retrobulbar pain and vertigo. Although laboratory investigations were consistent with acromegaly, she exhibited no clear phenotypic traits. During transsphenoidal surgery aimed at biopsy, typical adenomatous tissue was encountered, upon which it was decided to proceed to debulking. Histopathological analysis demonstrated a poorly differentiated plurihormonal Pit-1-positive adenoma with focal growth hormone (GH) and prolactin positivity, positive SSTR2 staining and a Ki-67 of 20–30%. Postoperative magnetic resonance imaging (MRI) examination revealed a large tumour remnant within the sella invading the right cavernous sinus with total encasement of the internal carotid artery and displacement of the right temporal lobe. As a consequence, she was treated additionally with radiotherapy, and a long-acting first-generation somatostatin analogue was prescribed. Subsequently, the patient developed secondary hypocortisolism and diabetes mellitus despite adequate suppression of GH levels. In September 2019, her symptoms recurred. Laboratory evaluations indicated a notable loss of biochemical control, and MRI revealed tumour progression. Lanreotide was switched to pasireotide, and successful removal of the tumour remnant and decompression of the right optic nerve was performed. She received adjuvant treatment with temozolomide resulting in excellent biochemical and radiological response after three and six courses. Symptoms of right-sided ptosis and diplopia remained. Evidence for systemic therapy in case of tumour progression after temozolomide is currently limited, although various potential targets can be identified in tumour tissue.
Learning points
Poorly differentiated plurihormonal Pit-1-positive adenoma is a potentially aggressive subtype of pituitary tumours.
This subtype can express somatostatin receptors, allowing treatment with somatostatin analogues.
A multidisciplinary approach involving an endocrinologist, neurosurgeon, pituitary pathologist, neuroradiologist, radiation oncologist and medical oncologist is key for the management of patients with aggressive pituitary tumours, allowing the successful application of multimodality treatment.
Temozolomide is first-line chemotherapy for aggressive pituitary tumours and carcinomas.
Further development of novel targeted therapies, such as peptide receptor radionuclide therapy (PRRT), vascular endothelial growth factor (VEGF) receptor-targeted therapy, tyrosine kinase inhibitors, mammalian target of rapamycin (mTOR) inhibitors and immune checkpoint inhibitors, is needed.
Patient Demographics: Adult, Female, White, Netherlands
Clinical Overview: Pituitary, Pituitary
Related Disciplines: Oncology
Publication Details: Unique/unexpected symptoms or presentations of a disease, October, 2021
Background
Pituitary tumours are heterogeneous in their origin, growth pattern and biological behaviour. They account for 10–15% of all intracranial neoplasms (1) and generally present either due to hypo- or hypersecretion of pituitary hormones and/or due to local space-occupying effects. Histopathologically, they can be divided into six main categories (somatotroph, lactotroph, thyrotroph, corticotroph, gonadotroph and plurihormonal tumours) (2). Adenomas are considered benign with pituitary carcinomas being exceptionally rare. However, many pituitary tumours are large and invasive neoplasms that cause significant morbidity, and the 2016 European Society of Endocrinology (ESE) survey described that aggressive pituitary tumours are clinically and pathologically similar to carcinomas (3, 4).
This case is of interest to other clinicians in the field, as it provides an excellent example of the challenges involved in care for patients with aggressive pituitary tumours, characterized by resistance to medical treatment and multiple recurrences despite standard therapies. In addition, it emphasizes the importance of consultation of a multidisciplinary team as recommended by the ESE (5). Finally, it underscores the need for further investigation of novel targeted therapies for patients with aggressive pituitary tumours and carcinomas.
Case presentation
A 35-year-old woman was referred to our care with a large pituitary mass with an unusually high growth rate. Her medical history included hypothyroidism due to Hashimoto’s disease, for which she was receiving levothyroxine replacement 100 µg once daily, and familial hypercholesterolemia, for which she was prescribed atorvastatin 40 mg and ezetimibe 10 mg once daily. In recent years, she was diagnosed with a mild perceptive hearing loss. She presented to a neurologist in March 2017 with symptoms of headache, dizziness and tinnitus, upon which a CT was made, reported as not containing any explanatory abnormalities.
In July 2017, she developed right-sided ptosis and diplopia (n. III palsy), upon which the CT from March was reassessed, revealing a right parasellar mass, centred in the cavernous sinus but partly extending into the sella, measuring ca. 22 × 14 × 9 mm. There was remodelling of adjoining ossal structures (hypophyseal fossa and dorsum sellae) and mild left deviation of the pituitary stalk.
An MRI was made, revealing fast growth. The T2 iso-intense mass was now measuring 34 × 26 × 20 mm and extended further laterally into the middle cranial fossa, medially into the sella and superiorly into the superior orbital fossa, not into the orbita. There was full encasement (Knosp grade 4) and anterior–medial displacement of the internal carotid artery. Initial laboratory investigations carried out in the peripheral hospital had revealed an increased insulin-like growth factor 1 (IGF-1) of 52.3 nmol/L (SD 4.51), consistent with a biochemical diagnosis of acromegaly, although our patient did not exhibit any clear phenotypic traits. There was no galactorrhea, and prolactin (PRL) was 389 mU/L. Our patient was referred to a gynaecologist because of secondary amenorrhoea in 2014 and diagnosed with polycystic ovary syndrome in 2015. She was prescribed medroxyprogesterone (Provera) 10 mg daily from December 2015 to April 2017 but was not on this drug or on the combined oral contraceptive at referral to our centre in July 2017 and had clear central hypogonadism (follicle-stimulating hormone: 4.3 U/L, luteinizing hormone: 1.9 U/L, estradiol: <40 nmol/L). There was no clear hypothyroidism or hypocortisolism (thyroid-stimulating hormone: 1.05 mU/L (reference range (RR): 0.5–4 mU/L), free T4 (fT4): 15.4 pmol/L (RR: 11–19.4 pmol/L), morning cortisol: 345 nmol/L – sufficient at >330 nmol/L)).
The patient presented to our neurosurgical referral center with right-sided ptosis and diplopia (n. III palsy) present since 3–4 weeks, increasing retrobulbar pain and vertigo. She was referred for transsphenoidal surgery aimed at biopsy. During the procedure, typical adenomatous tissue was encountered, upon which it was decided to proceed to full sellar debulking.
Investigation
Histopathological analysis was consistent with a poorly differentiated plurihormonal Pit-1-positive adenoma with focal GH and PRL positivity, positive SSTR2 staining, a high mitotic index and a Ki-67 of 20–30% (Fig. 1). Other transcription factors and pituitary hormones including TSH beta (β-TSH) and estrogen receptor alpha (ERα/ESR1), GFAP, neurofilament, CK8/18, CK-AE1/3 and CD45 were negative.
Figure 1.

Immunohistochemistry (stains as labelled).
Postoperatively, IGF-1 decreased to 42 nmol/L (s.d. 3.37), random growth hormone (GH) was 5.80 mU/L (RR: 0–30 mU/L), PRL was 212 mU/L (RR: <500 mU/L). Morning cortisol was 357 nmol/L, TSH 1.42 mU/L (RR: 1.5–4 mU/L), fT4 17.1 pmol/L (RR: 11–19.4 pmol/L). Results of a subsequent oral glucose tolerance test are shown in Table 1. It was concluded there was remaining autonomous production of GH, insufficiently suppressed in response to a 75 g bolus of glucose and therefore persisting acromegaly.
Table 1.
Oral glucose tolerance test 12-9-2017.
| 9:40 (t = −15) | 09:55 (t = 0) | 10:25 (t = 30) | 10:55 (t = 60) | 11:25 (t = 90) | 11:55 (t = 120) | |
|---|---|---|---|---|---|---|
| Glucose (mmol/L) | 5.2 | 5.3 | 8.6 | 10.6 | 11.1 | 11.1 |
| Growth hormone (mU/L) | 3.45 | 3.82 | 3.13 | 3.45 | 3.63 | 3.32 |
Early postoperative MRI (Fig. 2A) revealed that transsphenoidal surgery had resulted in incomplete debulking with a large tumour remnant remaining in the sella and right cavernous sinus, fully encasing the internal carotid artery and displacing the right temporal lobe to superiorly and laterally. The optic chiasm had been decompressed. The tumour remnant was metabolically active on Ga68-DOTATOC.
Figure 2.

Changes in tumour size over time on MRI (T1 post-contrast coronal section).
DNA analysis by next-generation sequencing revealed no pathogenic mutations in the AIP, CDKN1A, CDKN1B, CDKN2B, CDKN2C, MEN1, NF1 and PRKAR1A genes.
Treatment
Postoperatively, there was a substantial decrease of diplopia and ptosis. The patient was discussed in our multidisciplinary pituitary tumour working group, and it was decided to refer her for additional fractionated stereotactic radiotherapy (30 × 1.8 Gy in 6 weeks), as there was a clinically relevant invasive tumour remnant with pathological markers indicating aggressive behaviour. Given the persisting presence of acromegaly, lanreotide 120 mg subcutaneously every 4 weeks was prescribed.
During the following months, the patient developed secondary hypocortisolism requiring substitution with hydrocortisone 10 mg twice daily. Her weight increased substantially, and she developed diabetes mellitus despite adequate suppression of GH levels, requiring metformin 500 mg once daily from July 2018, three times daily by May 2019. It seems unlikely that the weight gain was secondary to the glucocorticoid, as it occurred largely before the hydrocortisone was initiated. The development of hyperglycemia/diabetes mellitus was interpreted as secondary to the weight gain, with the use of both lanreotide and hydrocortisone as possible contributing factors.
Outcome and follow-up
MRIs performed in February and July 2018 (Fig. 2B and C) and January 2019 indicated a clear decrease in volume of the intrasellar and right parasellar tumour remnant. The patient was clinically stable, and her IGF-1 decreased to 12.1 nmol/L (s.d. −1.15) by May 2019 (Fig. 3). However, by September 2019, she noted a recurrence of right-sided ptosis, diplopia and retrobulbar pain. Laboratory evaluations indicated a notable loss of biochemical control with IGF-1 increasing to 42.1 nmol/L (s.d. 3.41).
Figure 3.
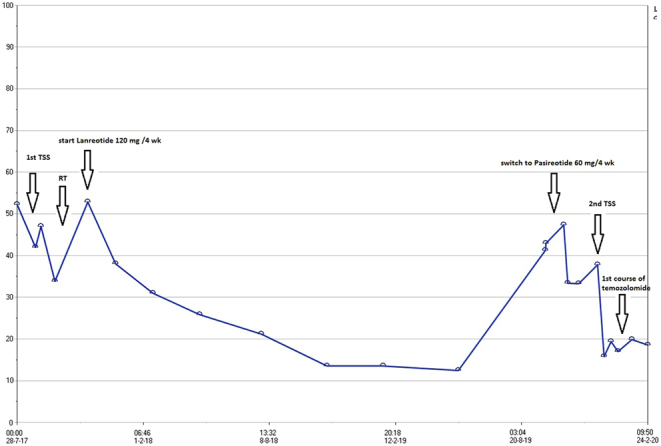
Course of IGF-1 (nmol/L) over time including applied treatments.
MRI was repeated in October 2019 (Fig. 2D), demonstrating a large increase in tumour size, with the sagittal diameter increasing from 29 to 37 mm and the coronal diameter from 10 to 27 mm. There was compression of the right optic nerve and the temporal lobe. The patient was again discussed in our multidisciplinary pituitary tumour working group, where it was decided to repeat transsphenoidal surgery aimed at debulking to be followed by temozolomide. In addition, it was decided to switch lanreotide to pasireotide 60 mg subcutaneously every 4 weeks. Less than 3 weeks after the MRI was performed, the patient was admitted to our endocrinology ward because of loss of food intake due to jaw pain and loss of right-sided facial sensation as a result of trigeminal nerve involvement. She had again developed complete right-sided ptosis and hyperglycemia as a side effect of pasireotide, for which insulin (glargin) was added to her diabetes treatment regimen. Her pain was managed with amitryptilin 25 mg once daily. She was discharged after 10 days receiving part-time home nursing.
Fluorodeoxyglucose-PET performed in December 2019 revealed no metastases. Repeated transsphenoidal surgery was performed that same month, resulting in successful debulking of tumour to the medial and lateral side of the right internal carotid artery and decompression of the right pre-chiasmatic optic nerve. Postoperatively, the patient noted a substantial decrease in right-sided retrobulbar pain, but the functioning of her third and fifth cranial nerves did not significantly improve. Histopathological analysis was again consistent with a poorly differentiated plurihormonal Pit-1-positive adenoma with partly overlapping and patchy GH and PRL positivity. Ki-67 was found to be 10–20% in hotspots. Molecular analysis performed at the Erasmus Medmethylguanine-DNA methyltransferase (MGMT) promotor methylation. Early postoperative MRI revealed clear decrease of suprasellar and right parasellar tumour volume, but remaining tumour tissue at the dorsal and right side of the sella (Fig. 2F).
At the end of January 2020, the patient started three courses of palliative temozolomide (5 days orally every 4 weeks, 150 mg/m2 for the first course, 200 mg/m2 from the second course).
In April 2020, follow-up MRI indicated an excellent response with clear decrease in tumour size (Fig. 2G). She received her fourth course of temozolomide with a short delay due to thrombocytopenia. In May 2020, she developed symptomatic hypoglycaemia as a result of decreasing GH levels in response to treatment, upon which her insulin doses were reduced. Temozolomide dose reduction to 150 mg/m2 was applied from the fifth course because of bone marrow toxicity. MRI performed after six courses in July 2020 indicated further radiological response with a reduction in size of the remaining tumour tissue (Fig. 2H). The patient received her ninth course of palliative temozolomide in September 2020, and it is intended to complete the standard 12 courses. Symptoms of ptosis and diplopia of the right eye remain, and the patient was suggested tarsorrhaphy by our ophthalmologist, a procedure consisting of surgical fusion of the upper and lower eyelid margins.
Discussion
Plurihormonal Pit-1-positive adenomas, previously known as silent subtype III, have been recognized as a biologically aggressive subtype (3, 6). Unlike gonadotroph adenomas, the most common subtype of clinically non-functioning tumours, they are clustered in a much younger age group and also show an association with MEN1 syndrome (7). Published series showed that these neoplasms are not always silent and may present with acromegaly, amenorrhea and galactorrhea, and hyperthyroidism (7, 8, 9, 10). Their hallmark is the expression of Pit-1 and presence of nuclear spheridia (nuclear inclusions) that can be identified on hematoxylin and eosin-stained slides (7). In contrast to differentiated Pit-1 lineage adenomas, these tumours often show a distinct immunohistochemical staining characterized by very focal or scattered positivity for one or more Pit-1 family hormones including GH, PRL, and β-TSH (7). They can express somatostatin receptors, allowing treatment with somatostatin analogues. Variable expression for ERα and alpha subunit can also be identified. The distinction of these aggressive tumours from other pituitary tumours is important given their aggressive biology along with their sensitivity to radiation (9) and temozolomide (11).
Development of resistance to first-generation somatostatin analogues is recognized in the treatment of neuroendocrine tumours but is considered to be very rare in acromegaly. Possible mechanisms are formation of antibodies against somatostatin analogues and desensitisation at a receptor or post-receptor level, that is, alterations in somatostatin receptor number and/or receptor–effector coupling (12). No data are available on how much more common this finding is in plurihormonal Pit-1-positive adenomas compared to regular somatotroph adenomas due to the low prevalence of the former subtype.
Stereotactic radiotherapy was applied in this case according to the ESE clinical practice guidelines for aggressive pituitary tumours, as there was a clinically relevant invasive tumour remnant with pathological markers (Ki67 index, mitotic count) indicating aggressive behaviour. As mentioned in the guidelines, fractionated external beam radiation therapy is highly effective in pituitary tumours, but little data are available in the more aggressive phenotypes. Comparison of the reported success rates is hindered by varying techniques and doses used for radiation therapy, as well as by different imaging protocols to assess tumour volume (3).
The ESE guidelines suggest the Stupp protocol in patients with rapid tumour growth in whom maximal doses of radiotherapy have not been reached (very low level of evidence +ooo). Therefore, this treatment option could have been considered for our patient only after repeated transsphenoidal surgery in December 2019.
However, evidence for application of the Stupp protocol in pituitary tumours is very limited. As mentioned by the guidelines, only 17 cases are reported in the literature.
The response rate of 76% may be higher than that reported with temozolomide alone, but
- some of the patients had not received prior radiotherapy (RT) and some also received drugs other than temozolomide.
- there are no data on whether progression-free survival is longer than when the two treatments (RT and temozolomide) are given sequentially.
In addition, there were concerns regarding damage to the optic nerve of our patient when applying a second course of radiotherapy, and for these combined reasons, it was decided not to do so at this stage.
Temozolomide is recommended as first-line chemotherapy for aggressive pituitary tumours and carcinomas following documented tumour growth. It is associated with an overall response rate of 37–51% and increased survival rates (3, 13). However, 30% of patients show radiological progression despite temozolomide treatment and even fewer patients respond to retreatment with temozolomide after completion of the standard 12 courses (3). Several recent articles have highlighted the promise of novel targeted therapies to treat aggressive pituitary tumours and carcinomas, including PRRT, VEGF receptor-targeted therapy, tyrosine kinase inhibitors, mTOR inhibitors and immune checkpoint inhibitors (14, 15, 16). These patients should be able to get access to basket trials in oncology or medical need programs to advance their care.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written informed consent has been obtained from the patient for publication of the submitted article and accompanying images.
Author contribution statement
M R Postma is a PhD candidate in endocrinology and was the main author. J M A Kuijlen and A G W Korsten-Meijer were the neurosurgeon and ENT surgeon involved. H E Westerlaan was the neuroradiologist involved and selected the images for Figure 2. A C M van den Bergh was the radiation oncologist involved. J Nuver was the medical oncologist involved. W F A den Dunnen was the pathologist who interpreted the tumour pathology and provided the microscopic images for publication. G van den Berg was the main endocrinologist responsible for the patient and revised the draft. All authors contributed to the reviewing and editing process and approved the final version of the manuscript.
References
- 1.Dworakowska D, Grossman AB. Aggressive and malignant pituitary tumours: state-of-the-art. Endocrine-Related Cancer 201825R559–R575. ( 10.1530/ERC-18-0228) [DOI] [PubMed] [Google Scholar]
- 2.Osamura RY, Grossman A, Korbonits M, Kovacs K, Lopes MBS, Matsuno A, Trouillas J. Pituitary adenoma. In WHO Classification of Tumours of Endocrine Organs; Chapter 1: Tumors of the Pituitary Gland, pp. 14–18. Lyon, France: WHO, 2017. [Google Scholar]
- 3.McCormack A, Dekkers OM, Petersenn S, Popovic V, Trouillas J, Raverot G, Burman P. & ESE Survey Colla borators. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. European Journal of Endocrinology 2018178265–276. ( 10.1530/EJE-17-0933) [DOI] [PubMed] [Google Scholar]
- 4.Trouillas J, Burman P, McCormack A, Petersenn S, Popovic V, Dekkers O, Raverot G. Aggressive pituitary tumours and carcinomas: two sides of the same coin? European Journal of Endocrinology 2018178C7–C9. ( 10.1530/EJE-18-0250) [DOI] [PubMed] [Google Scholar]
- 5.Raverot G, Burman P, McCormack A, Heaney A, Petersenn S, Popovic V, Trouillas J, Dekkers OM. & European Society of Endocrinology. European Society of Endocrinology Clinical Practice Guidelines for the management of aggressive pituitary tumours and carcinomas. European Journal of Endocrinology 2018178G1–G24. ( 10.1530/EJE-17-0796) [DOI] [PubMed] [Google Scholar]
- 6.Mete O, Lopes MB. Overview of the 2017 WHO classification of pituitary tumors. Endocrine Pathology 201728228–243. ( 10.1007/s12022-017-9498-z) [DOI] [PubMed] [Google Scholar]
- 7.Mete O, Gomez-Hernandez K, Kucharczyk W, Ridout R, Zadeh G, Gentili F, Ezzat S, Asa SL. Silent subtype 3 pituitary adenomas are not always silent and represent poorly differentiated monomorphous plurihormonal Pit-1 lineage adenomas. Modern Pathology 201629131–142. ( 10.1038/modpathol.2015.151) [DOI] [PubMed] [Google Scholar]
- 8.Horvath E, Kovacs K, Smyth HS, Killinger DW, Scheithauer BW, Randall R, Laws ER, Jr, Singer W. A novel type of pituitary adenoma: morphological features and clinical correlations. Journal of Clinical Endocrinology and Metabolism 1988661111–1118. ( 10.1210/jcem-66-6-1111) [DOI] [PubMed] [Google Scholar]
- 9.Horvath E, Kovacs K, Smyth HS, Cusimano M, Singer W. Silent adenoma subtype 3 of the pituitary – immunohistochemical and ultrastructural classification: a review of 29 cases. Ultrastructural Pathology 200529511–524. ( 10.1080/01913120500323514) [DOI] [PubMed] [Google Scholar]
- 10.Erickson D, Scheithauer B, Atkinson J, Horvath E, Kovacs K, Lloyd RV, Young Jr WF. Silent subtype 3 pituitary adenoma: a clinicopathologic analysis of the Mayo Clinic experience. Clinical Endocrinology 20097192–99. ( 10.1111/j.1365-2265.2008.03514.x) [DOI] [PubMed] [Google Scholar]
- 11.Fealey ME, Scheithauer BW, Horvath E, Erickson D, Kovacs K, McLendon R, Lloyd RV. MGMT immunoexpression in silent subtype 3 pituitary adenomas: possible therapeutic implications. Endocrine Pathology 201021161–165. ( 10.1007/s12022-010-9120-0) [DOI] [PubMed] [Google Scholar]
- 12.Wahid ST, Marbach P, Stolz B, Miller M, James RA, Ball SG. Partial tachyphylaxis to somatostatin (SST) analogues in a patient with acromegaly: the role of SST receptor desensitisation and circulating antibodies to SST analogues. European Journal of Endocrinology 2002146295–302. ( 10.1530/eje.0.1460295) [DOI] [PubMed] [Google Scholar]
- 13.Lasolle H, Cortet C, Castinetti F, Cloix L, Caron P, Delemer B, Desailloud R, Jublanc C, Lebrun-Frenay C, Sadoul JLet al. Temozolomide treatment can improve overall survival in aggressive pituitary tumors and pituitary carcinomas. European Journal of Endocrinology 2017176769–777. ( 10.1530/EJE-16-0979) [DOI] [PubMed] [Google Scholar]
- 14.Ilie MD, Lasolle H, Raverot G. Emerging and novel treatments for pituitary tumors. Journal of Clinical Medicine 201981107. ( 10.3390/jcm8081107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamb LS, Sim HW, McCormack AI. Exploring the role of novel medical therapies for aggressive pituitary tumors: a review of the literature-‘are we there yet?’ Cancers 202012308. ( 10.3390/cancers12020308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petersenn S, Heaney AP. Targeted systemic and peptide radio-ligand therapy for aggressive pituitary tumors and carcinomas. Reviews in Endocrine and Metabolic Disorders 202021277–286. ( 10.1007/s11154-020-09554-9) [DOI] [PubMed] [Google Scholar]