

Abstract
Long non-coding RNA has emerged as a key regulator of myriad gene functions. One such lncRNA mrhl, reported by our group, was found to have important role in spermatogenesis and embryonic development in mouse. Recently, its human homolog, Hmrhl was shown to have differential expression in several type of cancers. In the present study, we further characterize molecular features of Hmrhl and gain insight into its functional role in leukemia by gene silencing and transcriptome-based studies. Results indicate its high expression in CML patient samples as well as in K562 cell line. Silencing experiments suggest role of Hmrhl in cell proliferation, migration & invasion. RNA-seq and ChiRP-seq data analysis further revealed its association with important biological processes, including perturbed expression of crucial TFs and cancer-related genes. Among them ZIC1, PDGRFβ and TP53 were identified as regulatory targets, with high possibility of triplex formation by Hmrhl at their promoter site. Further, overexpression of PDGRFβ in Hmrhl silenced cells resulted in rescue effect of cancer associated cellular phenotypes. In addition, we also found TAL-1 to be a potential regulator of Hmrhl expression in K562 cells. Thus, we hypothesize that Hmrhl lncRNA may play a significant role in the pathobiology of CML.
Graphical Abstract
Graphical Abstract.
Regulation of cancer phenotype genes by lncRNA Hmrhl in K562 cells.
INTRODUCTION
Long non-coding RNA (lncRNA) has emerged as the most studied gene regulatory element of the last decade. As the name implies, long non-coding RNAs are transcripts with a length longer than 200 nucleotides, which have no functional protein coding ability whatsoever (1,2). Reported to be expressed from unicellular eukaryotic organism to humans, lncRNAs show tissue/cell/stage specific differential expression, are not well conserved, exhibit different sub-cellular localization and shows enormous diversity in its role and mechanism to regulate expression of protein coding genes (3–7). With the recent developments in deep sequencing technologies, their repertoire is exponentially increasing however, biological significance and characterization of most of lncRNA remains elusive (8,9). Some well-studied lncRNAs are reported to be the key players in distinct processes like dosage compensation, genomic imprinting, epigenetic regulator, pluripotency, post transcription regulator of mRNA, modulator of stability/translation of mRNA (10–16). LncRNAs have thus become the focal point of genomic research as they keep signifying their role as crucial regulator in vital biological processes like development and differentiation, cell cycle progression, and most importantly in pathology and progression of many human diseases including cancer (7,16–21). There is now enough evidence and knowledge that cellular stage/type specificity of these lncRNAs can provide us better understanding, identification, prognostic value and even therapeutic options for many incurable diseases especially cancer (22–25).
Transcriptome analysis of normal and cancerous tissues over the years has revealed differential expression of at least 2000 lncRNAs, with some been specific to certain type of cancers (26). For example, expression of differential display 3, also known as PCA3 and SCHLAP1 were found to be specific for human prostate cancer with PCA3 been later approved by the Food and Drug Administration (FDA) to be used as a biomarker for prostate cancer (27,28). SAMMSON which has melanoma‐specific expression provides a potential target for therapeutics with fewer side effects (29). Contrary to this, many well-known lncRNAs are reported to be involved in various cancers, such as deregulation of HOTAIR is associated with 26 different human tumors (30). LncRNA, MALAT1 was found to be overexpressed in several human cancers, including lung, breast, prostate, hepatocellular and ovarian cancer while H19 in human cancers, including liver, breast, gastric, colorectal, esophageal and lung cancer (31–33). In addition, differential expression of many lncRNAs like ANRIL, NEAT1, LUNAR1 and PCGEM1 are reported to be associated with several different cancer types (34–37).
LncRNA Mrhl (meiotic recombination hot spot locus RNA) was first reported in mouse by our group and has been extensively studied since then to reveal its biological significance (38–43). This 2.4 kb, intronic, nuclear restricted lncRNA was found to be a negative regulator of WNT signaling via its interaction with p68/Ddx5 RNA helicase in mouse spermatogonial cells (39,40). In addition, the role of Mrhl in meiotic commitment of spermatogonial cells through regulation of SOX8 at the chromatin level has been documented (41). More recently, transcriptomics and genome-wide occupancy studies of Mrhl in mouse embryonic stem cells (mESCS) revealed its role as a chromatin regulator of cellular differentiation and development genes along with its probable importance in maintenance of stemness in mESCs (43).
In our latest study, we have identified the human homolog of mouse Mrhl (Hmrhl) which shares 65% homology with that of mouse Mrhl and is encoded in an identical syntenic locus within the phkb gene (44). This study has revealed a functional role of Hmrhl as an enhancer RNA for its host gene, phkb, in chronic myelogenous leukemia (CML) cell line. Additionally, expression profile of Hmrhl among various tumor and normal samples confirmed its deregulation in several cancers (44). Among these, significant upregulation of Hmrhl in lymphoma tumor samples was evidently notable. This observation prompted us to gain insights into the possible functional molecular role of Hmrhl in leukemia. We started with the expression and characterization of Hmrhl in CML cell line, K562. Our sub-cellular localization study specified its enrichment within the nucleus and its association with chromatin which was consistent with our earlier observation with mouse Mrhl RNA. In-vitro studies demonstrated that depletion of Hmrhl influences cancer related phenotypes like cell migration, proliferation, and cell invasion in K562 cells. We used RNA-seq based approach to address the effect of silencing Hmrhl on the global transcriptome of K562 cells which revealed deregulation of 831 genes with significant perturbation of developmental processes and enrichment in chemokines and cytokine-mediated signaling pathway. Additionally, genome-wide occupancy study of Hmrhl RNA indicated its association with several loci throughout the genome, particularly at the intergenic and repetitive elements sites along with the other regions. Interestingly, by intersecting data from RNA seq and ChIRP seq, we found possible involvement of Hmrhl in the regulation of PDGFRβ and ZIC1 genes. Further overexpression of one of the target genes PDGFRβ in Hmrhl silenced K562 cells, shows a significant rescue effect of cancer associated cellular phenotypes (cellular proliferation, migration and invasion) evaluated earlier after Hmrhl knockdown. Observations from our data also suggested that regulation of Hmrhl RNA itself is mediated through TAL1, a key transcription factor involved in hematopoiesis. Deductions from this study suggest lncRNA Hmrhl as a chromatin regulator of genes associated with the phenotype of leukemia, mechanism of which needs to be explored further.
MATERIALS AND METHODS
Biological resources
K562 (chronic myelogenous leukemia/erythro leukemia) cell line was obtained from NCCS, Pune (India), whereas human lymphocyte cell line (ID no. GM12878) was from Coriell Institute, USA. Both cell lines were cultured in RPMI medium (Gibco) supplemented with 10% FBS (Gibco) for K562 and 15% non-heat inactivated FBS (Gibco) for GM12878 along with 100 units/ml penicillin-streptomycin solution (Sigma) at 37°C in a humidified chamber with 5% CO2.
Reagents
All chemicals were obtained from Sigma-Aldrich and Life Technologies unless otherwise specified.
Transfection, RNA isolation, cDNA synthesis and real-time PCR
Total RNA was isolated from cultured K562 and GM12878 cells with the help of RNAiso Plus (Takara Bio), following manufacturer's instructions. Isolated RNA was subjected to DNase treatment with RNase free DNase I (New England Biolabs, Catalog No# M0303S) to further remove DNA impurities. Concentration and purity of RNA was assessed by Nanodrop. 2–3 μg of total RNA was then reverse transcribed with the help of oligo(dT)17 primer and Super Script III/ Revertaid Reverse Transcriptase or with the help of cDNA synthesis kit from Biorad (iScript™ cDNA Synthesis Kit, Catalog #1708891).
For real-time quantitative PCR (qPCR), the cDNA (≤100 ng) was added to Syber green mix (TAKARA) with gene specific primers and the reaction (with total volume of 20 μl) was carried out in a Biorad CFX96 Real-Time System. The qRT-PCR reaction included an initial denaturation step at 95°C for 10 min, which was then followed by 35 cycles of 92°C for 15 s and 60°C for 1 min. Three technical replicates were set up for each sample. ΔΔCt method was used to determine expression fold changes by subsequent calculations.
Knockdown and co-transfection protocol
A pool of four Hmrhl specific siRNAs targeting the conserved region of Hmrhl and scrambled negative control siRNA were obtained from Dharmacon. K562 cells were transfected with 100 nM siRNA diluted in a serum-free media with lipofectamine 2000 reagent (Thermo Fischer) in a six-well plate as per the manufacturer's protocol. The transfected medium was replaced with complete medium (10% FBS and 1% penicillin/streptomycin) after 9 h of transfection. Cells were harvested for RNA isolation after 36 h of post-transfection, and the knockdown efficiency for Hmrhl was verified by real-time PCR.
For co-transfection experiment, lipofectamine 2000 reagent (Thermo Fischer) was used and instructions from manufacture was followed. Briefly, K562 cells were co-transfected with 1μg of PDGRF-β expression vector (Origene, Cat. #RC206377) and 30 pmol of siRNA-Hmrhl, diluted in serum free media with lipofectamine in a six-well plate. The transfected medium was replaced with complete medium (10% FBS and 1% penicillin/streptomycin) after 9 h of transfection. Cells were harvested for RNA isolation after 48 h of post-transfection, and real-time PCR was done to check the expression level of PDGRF-β and Hmrhl. Knockdown and transfected cells were used further for various phenotypic assays.
The list of all RT-PCR primers, si-RNA and probes used for the experiments are listed in Supplementary Table S1.
Hmrhl stability assay
To test the Hmrhl stability, 3 × 105 cells were cultured with complete media (10% FBS and 1% penicillin–streptomycin solution) in a six-well plate for 24 h. Subsequently, the media was replaced by fresh medium containing 10 ug/ml of Actinomycin D (Invitrogen) and was incubated for different time intervals at 37°C. RNA was isolated for each time point and real-time PCR was performed to measure the expression of Hmrhl along with 18s rRNA as control. Half-lives were calculated by fitting a nonlinear regression model of one-phase exponential decay (constraint = plateu > 0) using GraphPad Prism software version 5.04.
Subcellular fractionation
Subcellular fractionation was performed by following the previously described protocol by Gagnon et al. (45). Briefly, 5–10 million K562 cells were lysed in 380 μl of ice-cold HLB (Hypotonic lysis buffer: 10 mM Tris, pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.3% NP-40 and 10% glycerol, supplemented with 100 U of Ribolock RNase inhibitor and incubated on ice for 10min followed by centrifugation at 1000g at 4°C for 3 min. The supernatant containing cytoplasmic fraction was kept separately, and the nuclear pellet was washed with 1 ml of ice-cold HLB buffer three times and centrifuged at 300 g at 4°C for 2 min. To isolate total nuclear and cytoplasmic RNA, 1 ml of TRIzol reagent (Thermo Fisher Scientific) was added directly to the fractions and proceeded for RNA extraction. To separate nucleoplasmic and chromatin fractions, the nuclear pellet was resuspended in 380 μl of MWS buffer (Modified Wuarin-Schibler buffer: 10 mM Tris–HCl, pH 7.0, 4 mM EDTA, 0.3 M NaCl, 1 M urea, and 1% NP-40), supplemented with 100 U of Ribolock RNase inhibitor and incubated on ice for 10 min followed by centrifugation at 1000g for 3 min at 4°C. The supernatant was kept as the nucleoplasmic fraction and the chromatin pellet was washed with ice-cold MWS buffer, centrifuged at 500g for 2 min at 4°C. TRIzol was then added to both nucleoplasmic fraction and chromatin pellet and proceeded for RNA extraction, as described earlier.
Stellaris RNA fluorescent in situ hybridization (FISH)
RNA-FISH was carried out by following the protocol from Biosearch Technologies (https://www.biosearchtech.com/products/rna-fish). A set of Stellaris RNA FISH Probes labeled with Cy5 dye specific to Hmrhl targeted transcript was purchased from Biosearch Technologies. Briefly, 5 × 106 cells were washed with PBS, followed by fixation in 1 ml fixation buffer (37% formaldehyde in PBS) for 10 min at room temperature. Cells were then centrifuged and washed thrice with PBS before permeabilizing with 70% ethanol for at least 1 h at 2–8°C. Cell pellet was washed once with washed buffer A (Biosearch Technologies, Cat #SMF-WA1-60) and then proceeded for hybridization with 100 ul hybridization buffer (Biosearch Technologies, Cat #SMFHB1-10) containing Hmrhl probe (working concentration 125 nM) for overnight at 37°C. Next day, cells were washed once with wash buffer A and resuspended in the same to be incubated for 30 min at 37°C in the dark. After incubation, cells were centrifuged and resuspended in wash buffer A containing DAPI stain and incubated further for 30 min at 37°C in the dark. Next, cells were washed with wash buffer B (Biosearch Technologies, Cat #SMF-WB1-20) and resuspend in a small drop (∼30 μl) of Vectashield Mounting Medium. Subsequently, 10 ul of cell suspension was mounted on a slide for microscopic observation.
Cell proliferation assay
Cell proliferation was evaluated using the Cell Counting Kit-8 (CCK8) assay (Dojindo). Cells (5000 per 100 μl) with or without Hmrhl silencing were used, and cell proliferation was detected at every 24 h up to 96 h according to the manufacturer's protocol. Briefly, 10 μl of CCK 8 solution was added to each sample (5000 cells/100 μl) and was incubated for 2 h at 37°C. The colour developed was then measured spectrophotometrically (VERSAmax microplate reader) at 450 nm. Each sample was taken in triplicates, and the experiment was repeated with three biological replicates. After subtraction of background, the cell proliferation was represented as net absorbance.
Transwell assays
Migration assay
The migration of K562 cells was assessed using 8μm inserts (Corning Incorporated, Corning, NY, USA) according to the manufacturer's instructions. Cells (1.5 × 105) with/without silenced Hmrhl were suspended in the 500 μl of serum-free RPMI 1640 media and added onto the upper chambers of the inserts. Then, the inserts were placed in a 24-well plate, and the lower chambers were filled with 750 μl of RPMI 1640 media containing 20% FBS, which served as the chemoattractant. Cells were then allowed to migrate for 24–48 h. Thereafter, the migrated cells in the bottom chamber were stained with 0.4% Trypan Blue (Sigma) for 1 min, counted in a hemocytometer under an inverted microscope (at 10× in Olympus CKX41), and expressed as a percentage of cells migrated as compared with the control. Also, migrated cells were quantified using CCK-8 assay as described above, and the results were represented as percentage cell migration for each group compared to control.
Invasion assay
The invasiveness of K562 cells was monitored using invasion chambers (24well) coated with BD Matrigel™ matrix (8μm pore size, Corning). The invasion was measured by determining the ability of K562 cells to migrate through the Matrigel (a reconstituted basement membrane). Briefly, the matrigel membrane was first rehydrated by adding 500 μl of warm (37°C) media (RMPI-1640 with 10% FBS) in both the chambers of the insert and incubating it for 2 h at 37°C. After rehydration 750 μl RPMI-1640 with 20% FBS, was added to the lower compartment of the invasion chamber while K562 cells (2.5 × 105, with/without silenced Hmrhl) were suspended in 500 μl serum-free RPMI-1640 and were seeded in the upper compartment of the invasion chambers and incubated for 48 h. After 48 h, cells that invaded to the bottom chamber were analyzed by manual cell counting using 0.4% trypan blue as well as by CCK8 assay. Results are represented as the percentage of invaded cells in different groups as compared to the control sample.
Cell apoptosis assay
For analysis of cellular apoptosis, instructions provided by the kit (Annexin V-FITC Apoptosis Detection Kit, Sigma, Catalog No. APOAF-50TST) was followed. Cells (with/without Hmrhl silencing) were collected, rinsed twice with cold PBS, and then resuspended in 1× Binding Buffer at a concentration of 1 × 106 cells/ml. Cells were then stained with annexin-FITC and PI and incubation at RT (25°C) for 15 min in the dark. FACS analysis was performed using flow cytometer (BD FACS Calibur). 10 000 reads were taken in the flow cytometer. For positive control of apoptosis, cells were treated with Imatinib (1 μM) for 48 h. Cells that stained positive for annexin-FITC were either in the end stage of apoptosis or undergoing necrosis.
Cell cycle analysis
K562 cells (1 × 106 cells/ml) from different experimental groups were collected and washed twice with PBS. Subsequently, the cells were fixed in 70% ethanol for 30 min. After washing again twice with PBS, cells were finally resuspended in PBS and treated with 2 μl RNase A (10 mg/ml, boiled for 5 min, aliquoted and stored frozen at –20°C) for 2 h at 37°C. 10 μl of Propidium Iodide (50 μg/ml, BD Biosciences) was added to the mixture. Flow cytometry was performed to detect and analyze changes in the cell cycle distribution using a flow cytometer (BD FACS Calibur).
Chromatin immunoprecipitation (ChIP)
For the assay, cells (1 × 106) were fixed with 1% formaldehyde for 10 min at RT and the crosslinking was subsequently quenched with 125 mM glycine for 5 min at RT. After washing with PBS containing EDTA-free Complete Protease Inhibitor Cocktail (Roche) twice, cell pellets were resuspended in 300 μl of lysis buffer (50 mM Tris, pH 8.0; 10 mM EDTA; 1% SDS containing Protease Inhibitor Cocktail) and were allowed to sit on ice for 10 min. Cell extracts were then sonicated at high intensity for 30 min with 30 s on/off cycles in a Bioruptor sonicator (Diagenode). Sonicated samples were diluted (1:10) in ChIP dilution buffer (1.1% Triton X-100; 1.2 mM EDTA; 16.7 mM Tris, pH 8.0; 167 mM NaCl, Protease Inhibitor Cocktail) and centrifuged at 20 000g for 10 min at 4°C. An aliquot (50 μl) of the soluble chromatin was kept as input. The chromatin was precleared with beads and immunoprecipitated with TAL1 antibody (Diagenode, Cat #C15200012) overnight at 4°C. Next day 40 μl of protein A dynabeads (Invitrogen) were added to the immunocomplex and incubated at 4°C for 2 h. Beads were washed with Low salt buffer (150 mM NaCl; 0.1% SDS; 1% Triton X-100; 2 mM EDTA; 20 mM Tris, pH 8.0), high salt buffer (500 mM NaCl; 0.1% SDS; 1% Triton X-100; 2 mM EDTA; 20 mM Tris, pH 8.0), lithium chloride buffer (0.25 M LiCl; 1% NP-40; 1% sodium deoxycholate; 1 mM EDTA; 10 mM Tris, pH 8.0) and TE buffer (10 mM Tris, pH 8.0; 1 mM EDTA). The immunocomplex was eluted in elution buffer (1% SDS and 0.1 M NaHCO3) and reverse crosslinked overnight at 65°C followed by proteinase K digestion at 45°C for 1 h. Subsequently, the DNA was extracted and subjected to real-time PCR.
Chromatin isolation by RNA purification (ChIRP)
ChIRP was carried out according to the previously described protocol by Chu et al. (46). Hmrhl specific anti-sense probes with BiotinTEG at 3′ end was designed using the Biosearch Technologies ChIRP Probe Designer tool (https://www.biosearchtech.com/chirpdesigner/). As a negative control for ChIRP, the LacZ probe with BiotinTEG at its 3′ end was used. About 60 million cells were harvested for a single ChIRP reaction. In brief, cells were cross-linked with 1% glutaraldehyde for 10 min at room temperature. Crosslinking cells were then quenched with 0.125 M glycine for 5 min, followed by centrifugation at 2000g to collect the cell pellet. Cells were then lysed using freshly prepared lysis buffer (50 mM Tris–Cl, pH 7.0, 10 mM EDTA, 1% SDS) supplemented with 1 mM PMSF, 1× protease inhibitor cocktail (PI) and Superase. The suspension was then sonicated in a Biorupture sonicator with 30 s ON and 30 s OFF cycle for 60 min. Sonicated chromatin was then centrifuged at 16 000g for 10 min at 4°C. For a typical ChIRP reaction, 1 ml of sonicated lysate was used, in which 5% was kept aside as RNA and DNA input. Sonicated chromatin was hybridized in buffer (750 mM NaCl, 1% SDS, 50 mM Tris–Cl pH 7.0, 1 mM EDTA, 15% formamide) supplemented with 1 mM PMSF, 1× PI and Superase. Hmrhl probe (100 pmol), as well as LacZ probe was added per 1 ml of chromatin lysate in two separate tubes and was incubated for 4 h at 37°C with constant mixing. Approximately, 100 μl C-1 magnetic beads were then added to the 1ml chromatin lysate and incubated further for 30 min at 37°C with constant mixing. The beads were then washed five times with wash buffer (2× SSC, 0.5% SDS, 1 mM PMSF). From the last wash, 100 μl was set aside for RNA isolation and 900 μl for DNA isolation. Hmrhl pull-down efficiency was verified by qRT-PCR analysis. For ChIRP sequencing, DNA was isolated according to the said protocol and sent for high throughput sequencing in duplicates.
RNA-Seq analysis
GRCh38 (hg38) Genome was downloaded from GENCODE and indexed using Bowtie2-build with default parameters. Adapter ligation was done using Trim Galore (v0.4.4) and each of the raw Fastq files were passed through a quality check using the Fast QC. PCR duplicates were removed using the Samtools 1.3.1 with the help of rmdup option. Each of the raw files was then aligned to GRCh38 (hg38) genome assembly using TopHat with default parameters for paired end sequencing as described in Trapnell et al. (47). After aligning, quantification of transcripts was performed using Cufflinks and then Cuffmerge was used to create merged transcriptome annotation. Finally, differentially expressed (DE) genes were identified using Cuffdiff. The threshold for DE genes was log2(fold change) >1.5 for up-regulated genes and log2(fold change) <1.5 for down-regulated genes.
GO enrichment analysis
Gene Ontology (GO) analysis was performed in PANTHER (48). Significant enrichment test was performed with the set of differentially expressed genes in PANTHER.
Cluster analysis
The Hierarchical clustering method was performed using Cluster 3.0 (49). Gene expression data (FPKM of all samples, that is, scrambled and siRNA treated) was taken and log2 transformed. Low expressed (FPKM < 0.05) and invariant genes were removed. Then genes were centred, and clustering was performed based on the differential expression pattern of genes and fold change. Genes were grouped in 11 clusters and visualized as a network in Cytoscape (50). The Functional enrichment of each cluster was performed using the Gene Mania Tool (51). Heatmaps were generated in Java TreeView 3.0 for both up and down-regulated genes.
TF network analysis
Among all DE protein-coding genes, the transcription factors were identified using PANTHER protein class module. Motifs were downloaded for all transcription factors from JASPAR (52) and sequence of interest for each TF (1.5 kb upstream and 500 bp downstream of TSS) was extracted using BedtoFasta of the Bedtools suite (53). Then each motif was scanned across the sequence of all TFs to create the table matrix that reflects the number of binding sites for each TF across the other TFs using MEME suite (54) with an e-value of 1E–04. Finally, the heatmap was generated from the table matrix using R 3.3.2.
ChIRP-Seq analysis
GRCh38 (hg38) Genome was downloaded from GENCODE and indexed using Bowtie2-build with default parameters. Adapter ligation was done using Trim Galore (v 0.4.4) and each of the raw Fastq files were passed through a quality check using the FastQC. PCR duplicates were removed using the Samtools 1.3.1 with the help of ‘rmdup’ option. Each of the raw files was then aligned to GRCh38 (hg38) genome assembly using Bowtie 2 with default parameters for paired-end sequencing as described in Trapnell et al. (47). Replicates of both control and treated were merged respectively using the Samtools 1.3.1. Peaks were called using MACS2. Final peaks were selected, giving the criteria of above 5-fold change and P value <0.05.
Hmrhl motif prediction motifs were identified using MEME, based on the criteria of one occurrence per sequence (OOPS) and significance of 1E–04 for all ChIRP-Seq genomic loci. Sequence for each locus was extracted from GRCh38 (hg38) genome using bedtofasta of bedtools suite. After feeding sequences from the genomic loci obtained from MACS2, two significant motifs were identified in MEME motif discovery suite.
RNA-DNA triplex prediction
Sequences from the Hmrhl occupied regions (in addition extended upto ±25 bp) of the selected genes (TP53, PDGFRβ, ZIC1) were used to assess the compatibility of potential TFO/TTS pairs according to the canonical triplex formation rules between the entire stretch of Hmhrl sequence (Triplex Forming Oligo Nucleotide) and the aforementioned Hmrhl occupied regions of a respective genes (Triplex Target Sites) using the software Triplexator with default parameters (55).
RESULTS
Expression of LncRNA Hmrhl is co-related with chronic myelogenous leukemia (CML)
In our recently published study on Hmrhl, we had observed that most significant upregulation in the expression of Hmrhl was in lymphocyte tumor samples among all the other cancers (44). We went ahead and looked for the expression pattern of Hmrhl in patient samples of the two types of leukemia namely chronic myelogenous leukaemia (CML, slow developing) and acute myeloid leukaemia (AML, fast developing and more severe). For this purpose, we took advantage of the publicly available transcriptome data of both CML and AML patients (from https://www.ebi.ac.uk/ega/dacs/EGAC00001000481) and calculated the corresponding transcript FPKM values. Based on our data analysis, Hmrhl expression was indeed found to be upregulated in CML patient samples in contrast to the samples from AML patients (Figure 1A). We had previously shown that Hmrhl expression is elevated in K562 cells (derived from myelogenous leukemia cell line) in comparison with the control cell line GM12878. We have reconfirmed this observation as shown in Figure 1B. Therefore, we have selected K562 cell line to delineate potential molecular function of Hmrhl in CML in the present study.
Figure 1.
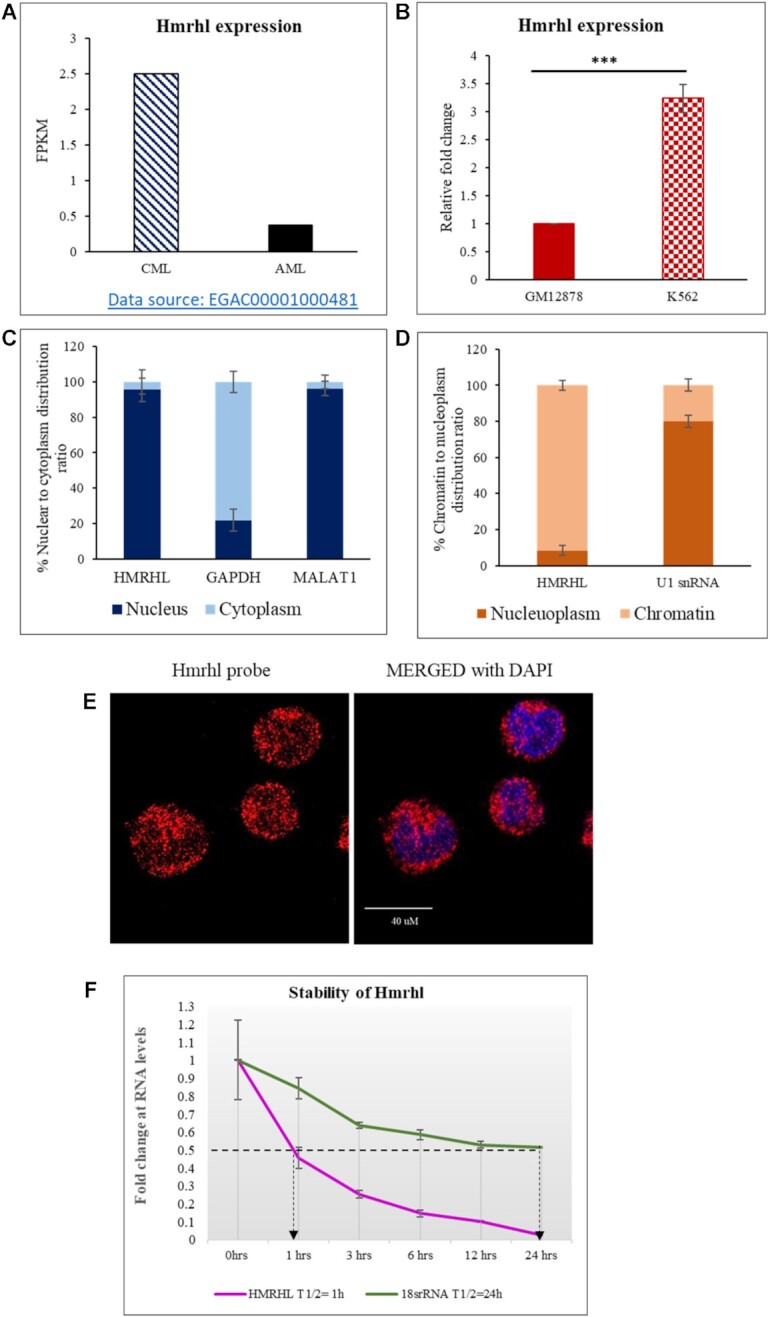
General characterization of LncRNA Hmrhl. (A) FPKM values depicting level of lncRNA Hmrhl in AML and CML patient samples as obtained from public data source ‘https://www.ebi.ac.uk/ega/dacs/EGAC00001000481’. (B) RT-qPCR analysis of Hmrhl RNA levels with respect to internal control GAPDH in CML cell line K562 and control cell line GM12878. (C) Subcellular fractionation showing nuclear restricted expression of Hmrhl RNA with levels of GAPDH and MALAT1 serving as authentication of purity for each fraction. (D) Chromatin association of Hmrhl RNA as represented by sub nuclear distribution of Hmrhl compared to nucleoplasm associated U1snRNA. (E) RNA-FISH using lncRNA Hmrhl specific probe reconfirms nuclear restriction of Hmrhl in K562 cells. (F) RNA stability assay of Hmrhl RNA and 18S rRNA in the presence of actinomycin D (10 ug/ml). The data are presented as mean ± SD from three independent experiments, *P< 0.05, **P< 0.01, ***P< 0.001 by Student's t-test. See also Supplementary Figure S1: Cells with different magnification after RNA-FISH using lncRNA Hmrhl specific probe which reconfirms Hmrhl nuclear restriction in K562 cells.
Hmrhl is a chromatin associated LncRNA with a short half-life
Since, we had shown earlier that the mouse Mrhl RNA is nuclear restricted and has regulatory function at chromatin level (43), we were curious to examine the sub cellular localization of Hmrhl RNA in K562 cells. Results from biochemical fractionation studies indicate the presence of Hmrhl predominantly in the nuclear fraction which was in parallel with MALAT1, a well-known highly abundant nuclear enriched lncRNA in human (Figure 1C) (31). GAPDH was used as a negative control to authenticate the purity of the nuclear fraction (Figure 1C). Hmrhl was further found to be predominately associated with the chromatin fraction with very little in the nucleoplasm fraction (Figure 1D). U1snRNA served as a positive control for nucleoplasm in the subnuclear fractionation study (Figure 1D). RNA FISH experiment further validated Hmrhl nuclear localization and it appears as bright red punctuate foci on blue DAPI background of chromatin mass (Figure 1E, Supplementary Figure S1). We also observed some spots to be of brighter intensity as compared to other foci suggesting that Hmrhl might be highly concentrated at some of the genomic loci (Figure 1E, Supplementary Figure S1). This corroborated with our earlier study where Hmrhl was also found to be nuclear restricted and chromatin associated in HEK-293T cells (44).
The stability of noncoding RNAs (ncRNAs) like protein-coding mRNAs, is found to be closely associated with their physiological function, in general regulatory RNAs been less stable than with housekeeping function (56). In addition, global measurement of the half-lives of lncRNAs in mouse has revealed that spliced lncRNAs are more stable than unspliced lncRNAs (single exonic), cytoplasmic lncRNAs are more stable than nuclear and antisense-overlapping lncRNAs are more stable than those transcribed from the intron (57). Since Hmrhl is an unspliced, nuclear, intronic lncRNA, we presumed Hmrhl to be a short lived lncRNA. To find out the exact half-life of Hmrhl, we used actinomycin D method and evaluated the half-life against highly stable 18S rRNA as positive control (Figure 1F). Results shown in Figure 1F indicate that Hmrhl certainly is a short lived lncRNA with a half-life of approximately 1 hr.
Knockdown of Hmrhl RNA effects cell proliferation, migration and invasion properties of K562 cells
We next investigated the cellular role of Hmrhl in cancer associated cellular phenotype of K562 cells. Cell proliferation, migration and invasion are processes readily associated with cancer phenotype and studied widely to establish the role of a particular gene in cancer (58). Therefore, we started with CCK-8 assay which demonstrates significant suppression of K562 cell proliferation 48 hrs after Hmrhl silencing which continued till the time of observation. that is, 96 h (Figure 2A). We performed flow cytometric analysis to further answer the question whether down regulated Hmrhl condition affected proliferation of K562 cells is caused due to alteration in cellular apoptosis or cell cycle progression. We observed that silencing Hmrhl RNA results in significant accumulation of cells at the G0/G1 phase (∼70%, P value < 0.01) as compared to control (∼50%) with reduction at S phase, from ∼30% in si-Hmrhl treated sample to ∼17% in control (P value < 0.001) and si-scramble (Figure 2B and C). However, no effect of Hmrhl silencing could be seen in apoptosis assay as all samples showed similar percentage of cells in each quadrant with maximum number of live cells and very few apoptotic cells (Supplementary Figure S2A).
Figure 2.

LncRNA Hmrhl contributes to cancer phenotype in K562. (A) CCK-8 assay shows decline in cell proliferation up on Hmrhl silencing measured over a time period of 96 h. (B, C) Bar graph and Flow cytometry histogram showing G0/G1 arrest represented as % of cells in each cell cycle phase after knockdown of Hmrhl compared to scramble RNA treatment and control samples. (D, E) Transwell assay followed by CCK-8 assay to evaluate the endpoint reveals inhibition of cellular migration and invasion after si-Hmrhl treatment in K562 cells respectively. (F, G) Reconfirmation of effect of Hmrhl knockdown on cell migration and invasion respectively using manual cell counting as the end point assay. The data are presented as mean ± SD from three independent experiments, *P< 0.05, **P< 0.01, ***P< 0.001 by Student's t-test. See also Supplementary Figure S2: (A) evaluation of cell death by apoptosis as an effect of Hmrhl silencing using annexin V-FITC/PI staining. Dot plot profile of (i) control cells (ii) si-Hmrhl treated K562 cells (iii) scramble-RNA treated cells. (B, C) Cell migration evaluated by CCK-8 assay and manual counting respectively shows no difference over the time period of 24 and 48 h.
The results of the transwell assay showed that siRNA treatment significantly impaired the migration capability of treated cells as compared to control (Figure 2D). With a 70% inhibition in cell migration observed after 24 h of Hmrhl silencing, no further decline was noted till 48 h (Supplementary Figure S2B). Hmrhl downregulated K562 cells also inhibited their invasion ability by ∼80% when compared to control cells (Figure 2E). Both the assays were further verified using manual counting approach which showed similar results as obtained by CCK-8 after transwell assay (Figure 2F, G, Supplementary Figure S2C).
Perturbation of Hmrhl affects genes involved in key signaling pathway in leukemia as well as in the developmental process
To elucidate the functional role of Hmrhl, we took the RNA-seq approach to examine whether it regulates expression of protein coding genes in these cells. Hmrhl was silenced using a pool of four targeted siRNAs in K562 cells which gave us an average down regulation efficiency of ∼60% as compared to the scrambled control siRNA (Figure 3A and B). Both Hmrhl siRNA and scramble siRNA treated cells were then subjected to RNA-seq analysis. Our analysis with a fold cut-off of 1.5 ± revealed a total of 1584 dysregulated genes (DE) up on Hmrhl silencing, out of which 831 genes were found to be protein coding (Figure 3C, Supplementary file 1). Further evaluation of protein coding DE genes indicated that majority of them are down regulated (592) compared to upregulated genes (239) (Figure 3C).
Figure 3.

Transcriptome analysis reveals role of lncRNA Hmrhl in biological processes related to development and signaling. (A) Diagrammatic representation of targeted siRNA binding sites used for silencing Hmrhl. (B) Knockdown efficiency of Hmrhl shown as relative expression of Hmrhl after si-Hmrhl and scramble-RNA treated K562 cells with GAPDH as internal control. (C) Diagrammatic representation of percentage of DE genes in each category obtained after RNA-seq analysis of transcriptome after perturbation of Hmrhl RNA. (D–F) Gene ontology (GO) enrichment analysis of DE genes showing number of genes associated with significant pathways, molecular functions and key biological processes respectively. Error bars indicate standard deviation from three independent experiments. *P< 0.05, **P< 0.01, ***P< 0.001 by Student's t-test.
Following the identification of differentially expressed genes, Gene Ontology (GO) enrichment analysis of protein coding genes with a P-value <0.05 was carried out to identify pathways that are perturbed upon Hmrhl silencing. Notably, several pathways that are associated with key signaling processes in cancer were found to be affected such as chemokine and cytokine signaling pathway (P00031, 22 genes) followed by the WNT signaling pathway (P00057, 21 genes) and integrin signaling pathway (P00034, 14 genes) which were found be most enriched along with heterotrimetric G-protein signaling pathway (P00027, 11 genes) and 5HT2 type receptor mediated signaling pathway (P04374, 11 genes) in the top five list (Figure 3D, Table 1). Moreover, GO analysis also revealed diverse molecular functions and a number of biological processes to be altered upon Hmrhl silencing. Figure 3E shows top 10 of such enriched molecular functions which includes enzyme inhibitor activity (19 genes, P-value = 0.0154), transcription factor binding (12 genes, P-value = 0.0136), Rho GTPase binding (9 genes, P-value = 0.0329), nuclease activity (5 genes, P-value = 0.00225), voltage-gated calcium channel activity (5 genes, P-value = 0.0214) and cell adhesion mediator activity (5 genes, P-value = 0.0374). We also observed from the analysis that most of the genes with altered expression pattern belongs to biological processes of cellular development and differentiation such as system development (GO:0048731), developmental regulation process (GO:0050793), nervous system developmental process (GO:0007399), regulation of cell differentiation (GO:0045595) (Figure 3F). We could conclude, from our observations therefore, that Hmrhl is involved in key signaling pathways and processes of cell development and differentiation in K562 cells.
Table 1.
Detail description of DE genes in biological pathways as obtained after GO analysis
| Gene | Fold change | Biological function |
|---|---|---|
| Inflammation mediated by cytokines and chemokines signaling pathway | ||
| RGS1 | 1.57636 | Regulates G protein-coupled receptor signalling cascades |
| CCL3 | −1.71239 | Monokine with inflammatory and chemokinetic properties. |
| ITGB2 | −1.22457 | Integral cell-surface proteins that participate in cell adhesion as well as cell-surface mediated signalling |
| IL6 | 1.57968 | Cytokine with a wide variety of biological functions. |
| GNA15 | −5.38507 | Involved as modulators or transducers in various transmembrane signalling systems |
| CCR4 | 3.77073 | Function as a chemoattractant homing receptor |
| FPR2 | −1.78241 | Powerful neutrophil chemotactic factors |
| MYH7B | −2.16246 | Involved in muscle contraction. |
| PRKCE | −2.62241 | Known to be involved in diverse cellular signalling pathways |
| MYH11 | −2.30486 | Involved in muscle contraction. |
| CXCL10 | −2.27738 | Involved in a wide variety of processes such as chemotaxis, differentiation, and activation of peripheral immune cells, regulation of cell growth, apoptosis |
| IL1B | −3.97255 | An important mediator of the inflammatory response, and is involved in a variety of cellular activities, including cell proliferation, differentiation, and apoptosis. |
| ITGA2 | −1.27507 | It is responsible for adhesion of platelets and other cells to collagens |
| GNA14 | −1.21527 | Involved as modulators or transducers in various transmembrane signalling systems |
| GNG7 | −3.67311 | |
| GNB3 | −1.60354 | |
| FPR1 | −1.25003 | Powerful neutrophil chemotactic factors |
| RELB | −1.27448 | Involved in many biological processed such as inflammation, immunity, differentiation, cell growth, tumorigenesis and apoptosis. |
| PLCB4 | 2.48726 | Important role in the intracellular transduction of many extracellular signals in the retina |
| PTAFR | −2.05699 | A chemotactic phospholipid mediator that possesses potent inflammatory, smooth-muscle contractile and hypotensive activity. |
| MYLK2 | −2.0379 | Implicated in the level of global muscle contraction and cardiac function |
| MYH3 | 2.22508 | Involved in cell movement and transport of materials within and between cells. |
| WNT signaling pathway | ||
| DCHS1 | −1.27603 | Calcium-dependent cell-adhesion protein. Mediates functions in neuroprogenitor cell proliferation and differentiation. |
| WNT8B | −1.68662 | May play an important role in the development and differentiation of certain forebrain structures, notably the hippocampus |
| PCDH12 | 2.29283 | Acts as a regulator of cell migration, probably via increasing cell-cell adhesion |
| GNA15 | −5.38507 | Involved as modulators or transducers in various transmembrane signalling systems. |
| MYH7B | −2.16246 | Involved in muscle contraction. |
| PRKCE | −2.62241 | Plays essential roles in cancer cell invasion and regulation of apoptosis, regulation of multiple cellular processes such as cell adhesion, motility, migration and cell cycle |
| ANKRD6 | 3.17182 | Allows efficient phosphorylation of beta-catenin, thereby inhibiting beta-catenin/Tcf signals. |
| CTNNA2 | 1.67243 | Regulate cell-cell adhesion and differentiation in the nervous system |
| CTNNA3 | −2.54244 | May be involved in formation of stretch-resistant cell-cell adhesion complexes. |
| GNA14 | −1.21527 | Involved as modulators or transducers in various transmembrane signalling systems. |
| CDH12 | −2.78544 | Calcium-dependent cell adhesion proteins with a role during a critical period of neuronal development |
| GNG7 | −3.67311 | Involved as a modulator or transducer in various transmembrane signalling systems. |
| GNB3 | −1.60354 | Involved as a modulator or transducer in various transmembrane signalling systems |
| FZD9 | −1.53141 | Involved in beta-catenin canonical signalling pathway |
| TP53 | −1.89693 | Acts as a tumour suppressor in many tumour types; induces growth arrest or apoptosis depending on the physiological circumstances and cell type. |
| FAT2 | −1.23901 | Involved in the regulation of cell migration and proliferation |
| PLCB4 | 2.48726 | Plays an important role in the intracellular transduction of many extracellular signals in the retina |
| PCDH11Y | 1.70719 | Play a role in cell-cell recognition during development of the central nervous system |
| FRZB | −1.71826 | Function as modulators of Wnt signalling through direct interaction with Wnts. |
| MYH3 | 2.22508 | Involved in cell movement and transport of materials within and between cells. |
| SFRP5 | −2.18751 | Have a role in regulating cell growth and differentiation in specific cell types. |
| Interigin signaling pathway | ||
| ITGB2 | −1.22457 | Involved in leukocyte adhesion and transmigration of leukocytes including T-cells and neutrophils |
| CAV1 | 2.14717 | scaffolding protein promotes cell cycle progression |
| COL9A2 | 2.03169 | extracellular matrix structural constituent conferring tensile strength |
| SH3D21 | −1.33154 | structural component of nucleus and plasma membrane |
| COL11A2 | 3.24925 | May play an important role in fibrillogenesis by controlling lateral growth of collagen II fibrils. |
| ITGA2 | −1.27507 | responsible for adhesion of platelets and other cells to collagens |
| COL17A1 | 2.04848 | May play a role in the integrity of hemidesmosome |
| COL4A3 | 1.66658 | major structural component of glomerular basement membranes |
| COL9A3 | −1.43053 | Structural component of hyaline cartilage and vitreous of the eye |
| ACTN2 | −1.3298 | anchor actin to a variety of intracellular structures. |
| ITGA10 | 2.5105 | Acts as a receptor for collagen. |
| LAMA2 | 2.37888 | mediate the attachment, migration and organization of cells into tissues during embryonic development by interacting with other extracellular matrix components |
| LAMC1 | −2.12482 | mediate the attachment, migration and organization of cells into tissues during embryonic development by interacting with other extracellular matrix components |
| ELMO1 | 1.7877 | Involved in cytoskeletal rearrangements required for phagocytosis of apoptotic cells and cell motility. |
| Heterotrimetric G-protein signalling pathway | ||
| CHRM4 | −1.26805 | Mediates various cellular responses, including inhibition of adenylate cyclase, breakdown of phosphoinositides and modulation of potassium channels through the action of G proteins |
| RGS1 | 1.57636 | Regulates G protein-coupled receptor signalling cascades |
| PLCB4 | 2.48726 | Plays an important role in the intracellular transduction of many extracellular signals in the retina. |
| PRKCE | −2.62241 | Plays essential roles in the regulation of multiple cellular processes linked to cytoskeletal proteins, such as cell adhesion, motility, migration and cell cycle |
| CACNA1A | 3.46911 | Involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell motility, cell division and cell death. |
| CACNA1E | −1.17684 | Involved in a variety of calcium-dependent processes, including muscle contraction, hormone or neurotransmitter release, gene expression, cell motility, cell division and cell death. |
| GNA15 | −5.38507 | Involved as modulators or transducers in various transmembrane signalling systems. |
| GNGT1 | 2.02369 | |
| GNA14 | −1.21527 | |
| GNG7 | −3.67311 | |
| GNB3 | −1.60354 | |
Gene co-expression and TF interaction analysis reveals distinct gene networks associated with Hmrhl RNA
We observed subsets of important transcription factors (TF) and common cancer related genes to be dysregulated up on of Hmrhl silencing (Figure 4A, B), implying probable role of Hmrhl in regulation of these key TFs and cancer associated genes. A thorough literature search revealed many of these TFs and cancer related DE genes do have significant biological role in leukemia, development and signaling pathways. In TFs group (Figure 4B) for example, KLF12, an important regulator of gene expression during vertebrate development has been reported to be upregulated in K562 cells (59), while hypermethylation of TFAP2E, which is considered as a tumor suppressor gene was found to be associated with K562 cell line and shortened survival of CML patients (60,61). Others, like KLF2, MAFA, STAT4, ASCL2, KLF4, BATF, TP63 were all linked to leukemia and lymphomas in various studies (62–70). We would like to mention here the fact that although p53 has crucial role in cancer, it is reported to be mutated in K562 cells. An insertion in exon 5 leads to a nonfunctional truncated protein of p53 in K562 cells (71). While this mutation allowed researchers to detect the p53 transcript by RT-PCR, they have failed to detect the truncated protein using western blotting or immunohistochemical staining. Many TFs, like TBX8, FEV, SP8 with their crucial role in development were also identified (72–74). Expressed only in fetal hematopoietic cells and essential for hematopoietic stem cell renewal, FEV, was reported to be expressed in infant leukemia samples with involvement in maintenance and propagation of leukemic stem cells (74). Beside these, others TFs were found to be associated with various signaling pathways which directly or indirectly modulates the cell growth, proliferation and apoptosis. For example, ASCL2, which is hypermethylated in lymphocytic leukemias and SOX15, a tumor suppressor, both act via WNT signaling pathway to modulate cell proliferation and invasion (65,75,76). Transcription factors, STAT4 and IRF5 both were reported to be expressed abnormally low in leukemia (64,77). STAT4, which has association with neoplasia, is part of STAT signaling pathway critical for mediating the response of hematopoietic cells to a diverse spectrum of cytokines (78) while IRF5, which modulates expression of genes controlling cell growth and apoptosis, was recently demonstrated to be a target of BCR-ABL kinase activity which reduces CML cell proliferation (79). Another transcription factor KLF4 was reported to be critically regulating human myeloid leukemia cell proliferation and its dysregulated expression alters cell division, differentiation and apoptosis in myeloid leukemias (66,67,80). TP63, which regulates cellular activities like division, adhesion and apoptosis, was established to have a pro-survival effect with a role in blast crisis in CML (69,70). This detailed analysis of DE TFs further supports our GO outcomes of RNA seq analysis where most effected pathways and biological processes belongs to development and signaling (Figure 3D & F). This was found true for the DE genes associated with cancer as well (Figure 4A). For example, both GLI1 and PTCH1 are associated with hedgehog signaling, important for embryonic development and tumorigenesis (81,82). Dysregulated expression and mutation in these genes has been corelated with phenotype and cancer stemness in CML (81,82). CD274, also known as PDL1, is a receptor found on CML specific T-cells and are stipulated to increase immune suppressive signals leading to reduce antitumor activity within tumor microenvironment (83). It was also found to be associated with the phenotype and maintenance of leukemic stem cells in CML and AML (84,85). Other DE genes, like CCR4, ITK, LRP1B, GPC5, PDGRFβ, PRDM16, PTPRK, ZIC1 were all found to be involved in signaling pathways with fundamental role in development, immune response, cell growth, proliferation, migration etc. (86–93). Fusion transcripts, mutation and dysregulated expression of PDGRFB and PRDM16 were reported in many leukemias (91). Another study has reported the abnormal promoter methylation of PTPRK in 47% of ALL and B-cell lymphoma patients and was associated with decreased survival in the cohort (92).
Figure 4.
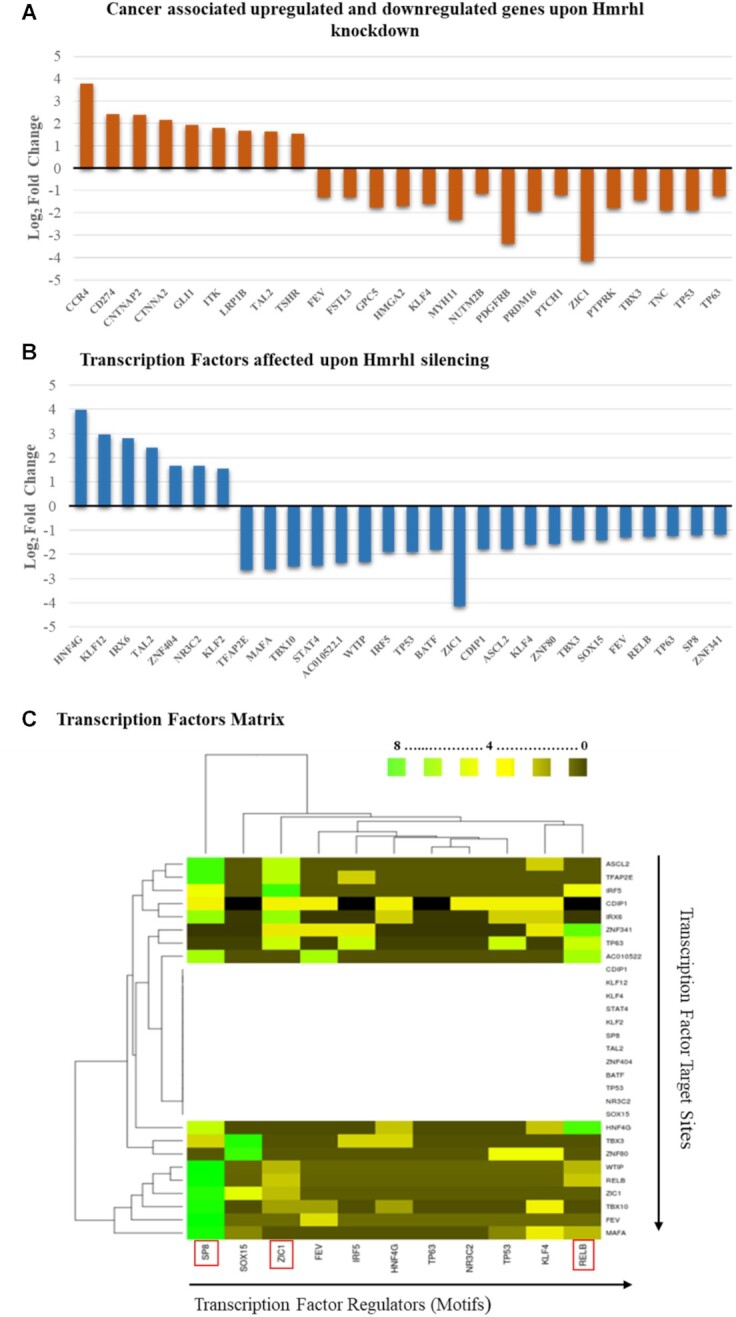
Further analysis of DE genes obtained after Hmrhl knockdown revealed some important (A) cancer associated genes and (B) transcription factors. (C) Heat map visualization of DE Transcription factors matrix. Heat map was generated from the table matrix which was obtained using MEME suit. The empty region observed in the middle of the transcription factor matrix is due to the unavailability of known motifs of transcription factor (TF) regulators in the motif database, i.e., JASPAR. This shall not be confused with the absence (#0) or zero occurrence of a motif on a query site of the target TF.
We further carried out analysis of important TF regulators among all DE transcription factors obtained after Hmrhl silencing (Figure 4C, Supplementary file 2). Transcription factors (TFs) in large part, determine the connectivity of gene regulatory networks as well as the quantitative level of gene expression. And it is evident from literature survey that dysregulation of TFs is associated with cancers (94). This allowed us to develop a TF matrix which shows SP8, ZIC1 and RELB as the top three dominating TFs (marked in red boxes, Figure 4C) as they have maximum number of motifs for binding across the promoters of all DE TFs (Supplementary Table S2). Both, SP8 and ZIC1 genes have been shown to be essential for proper embryonic development, with SP8 playing a key role in limb development whereas ZIC1 is involved in neurogenesis (95–97). Dysregulation of both of these genes has been found to be associated with various types of cancer where they promote metastasis by regulating cell cycle and growth (93,98). RELB on the other hand codes for a member of the nuclear factor kappa-B (NF-κB) family of TFs and is involved in many biological processes such as inflammation, immunity, differentiation, cell growth, tumorigenesis and apoptosis (99,100).
To understand the functional relevance of perturbed genes upon Hmrhl silencing in K562, we further analyzed DE genes for gene-gene interactions to be identified as co-expression modules. We carried out the hierarchical clustering and obtained eleven clusters using the Cytoscape platform (Table 2, Supplementary Figure S3A&B, Supplementary file 3). We subjected each of these gene cluster modules for functional enrichment with the Gene Mania tool, to obtain different cellular pathways to which these genes belong. Results from this analysis showed signal transduction and developmental activities for clusters 1; cluster 4 and 5 demonstrate ion channel and adhesion molecular activity. Nervous system functions for cluster 6 and kinase signaling processes for cluster 7 were obtained, whereas cluster 2, 9 and 10 were associated with activities for muscle contraction and potassium channel. Cluster 11 was found to be involved in the extracellular signaling process (Table 2, Supplementary Figure S3A, B), whereas clusters 3 and 8 did not indicate any functional relevance to the cellular pathway (Supplementary Figure S4).
Table 2.
Gene co-expression modules (clusters 1, 2, 4, 5, 6, 7, 9, 10 and 11) with their distinct functional profiles, obtained after hierarchical clustering using Cytoscape platform. Also see, Supplementary Figure S3A&B for detail co-expression modules and Supplementary Figure S1: showing clusters 3 and 8 with no specific functional enrichment after gene co-expression analysis
| S.No. | Gene cluster | Related function |
|---|---|---|
| 1 | Cluster 1 | Signal transduction activity |
| • voltage-gated calcium channel activity | ||
| • synaptic transmission, glutamatergic | ||
| • regulation of neuron projection development | ||
| • adult behavior | ||
| • ion channel complex | ||
| • voltage-gated calcium channel | ||
| 2 | Cluster 2 | Muscle contraction activity |
| • muscle system process | ||
| • muscle contraction | ||
| • structural constituent of cytoskeleton | ||
| • contractile fiber | ||
| 3 | Cluster 4 | Ion channel activity |
| • actin binding | ||
| • striated muscle thin filament | ||
| • myofilament | ||
| • zinc ion binding | ||
| • oxidoreductase activity, acting on a sulfur group of donors | ||
| • contractile fiber | ||
| • transition metal ion binding | ||
| 4 | Cluster 5 | Cell adhesion activity |
| • cardiac right ventricle morphogenesis | ||
| • regulation of heterotypic cell-cell adhesion | ||
| • skin development | ||
| • primary cilium | ||
| • heterotypic cell-cell adhesion | ||
| 5 | Cluster 6 | Nervous system functions |
| • suckling behaviour | ||
| • axon guidance | ||
| • neuron projection guidance | ||
| • multi-organism behaviour | ||
| • locomotory behaviour | ||
| • copper ion homeostasis | ||
| 6 | Cluster 7 | Kinase signaling processes |
| • synaptic transmission, dopaminergic | ||
| • protein kinase C signalling | ||
| • regulation of behaviour | ||
| 7 | Cluster 9 | Muscle contraction functions |
| • myofilament | ||
| • muscle system process | ||
| • striated muscle thin filament | ||
| • protein ADP-ribosylation | ||
| • transferase activity, transferring pentosyl groups | ||
| • muscle contraction | ||
| • NAD+ ADP-ribosyltransferase activity | ||
| 8 | Cluster 10 | Potassium channel activity |
| • potassium channel activity | ||
| • potassium ion transmembrane transporter activity | ||
| • potassium ion transmembrane transport | ||
| • cellular potassium ion transport | ||
| • cation channel activity | ||
| • ion channel activity | ||
| • potassium ion transport | ||
| 9 | Cluster 11 | Extracellular signaling process |
| • extracellular negative regulation of signal transduction | ||
| • determination of dorsal identity | ||
| • extracellular regulation of signal transduction | ||
| • flavin adenine dinucleotide binding | ||
| • negative regulation of BMP signaling pathway | ||
| • regulation of BMP signaling pathway | ||
| • alpha-amino acid metabolic process |
Hmrhl binding to chromatin regulates differential expression of target gene via triplex formation
Since Hmrhl is associated with chromatin, we were curious to identify the genomic binding sites by the ChIRP-seq technique. Chromatin isolation by RNA purification (ChIRP) coupled with high-throughput sequencing (ChIRP-seq) is a robust technique that allows one to identify genome-wide chromatin binding sites for lncRNAs (46). We used biotinylated antisense oligonucleotides probes to pull down Hmrhl, specificity of which was confirmed by qRT-PCR against non-specific control probes against lacZ (Figure 5A). Sequencing of the ChIRP DNA indicated association of Hmrhl with thousands of genomic loci on chromatin. Highly significant sequence reads selected on the bases of 5-fold enrichment over input DNA and a P-value <0.01, gave us a total of 68 177 peaks (Supplementary file 4). Further annotation of these peaks revealed enrichment of Hmrhl to be significantly higher at some chromatin regions as compared to other loci, which includes intergenic, introns, repeat elements as well as promoter regions (Figure 5B).
Figure 5.

ChIRP-Seq analysis of Hmrhl in K562 cells. (A) Plot showing efficiency for Hmrhl pulldown over LacZ in ChIRP experiment. (B) Annotation of peaks (C) Venn diagram representing overlapped genes obtained from intersection of RNA-seq and ChIRP-seq dataset. (D) Gene Ontology of common genes thus obtained. (E) FPKRM values depicting levels of ZIC1, PDGRFβ and TP53 after Hmrhl knockdown as obtained from RNA-seq data analysis. (F) expression pattern of ZIC1, PDGRFβ and TP53 determined by RT-qPCR after Hmrhl silencing in K562 cells. The data are presented as mean ± SD from three independent experiments, *P< 0.05, **P< 0.01, ***P< 0.001 by Student's t-test.
To get a better understanding of the genes which are perturbed upon Hmrhl RNA silencing because of physical association of Hmrhl RNA at the chromatin level, we overlapped the high throughput sequencing data from both RNA-seq and ChIRP-seq study. For this analysis we used –10 kb upstream of transcription start site (TSS) and +5 kb downstream of TSS of genes as our domain of target genes to explore the occupancy of Hmrhl RNA. With a stringent cut-off of >10-fold enrichment for ChIRP-seq, we obtained 62 overlapped genes between two sets of data (Figure 5C, Supplementary file 5). Gene ontology of these common genes revealed that most of them belong to the biological processes like signal transduction (P-value = 0.00506), cell development and differentiation (P-value = 0.0425) along with others like cellular adhesion (P-value = 0.04478) and migration (P-value = 0.0866) (Figure 5D, Table 3). These results very well corelate with our GO analysis of RNA-seq genes where signaling and developmental pathways were prominently enriched (Figure 3D–F). Interestingly, the biological processes and pathways obtained after GO analysis of common genes like signaling, cell adhesion, migration etc. are well associated with the cancer development and malignances. We further analysed the subset of common genes based on their role in signaling pathways in cancer as well as role in the developmental process and subsequently identified PDGFRβ, TP53 and ZIC1 as our top hits among the genes that were physically occupied with Hmrhl and also significantly dysregulated upon Hmrhl silencing (Figure 5C). We also calculated the FPKM value of each corresponding genes from RNA seq data, and this analysis shows that the expression of TP53, PDGFRβ and ZIC1 are significantly altered upon Hmrhl silencing in comparison to control (Figure 5E). We further confirmed the expression of PDGFRβ, ZIC1 and TP53 genes at RNA expression level upon Hmrhl silencing, by real-time PCR (Figure 5F). Our results are consistent with what we observed from RNA-seq data analysis.
Table 3.
Biological pathways obtained after GO enrichment of common genes between RNA-seq and ChIRP-seq datasets. Detail description of genes under each pathway is given below
| Gene | Fold change in expression (from RNAseq dataset) | Distance to TSS (from ChIRP-seq dataset) | Biological function |
|---|---|---|---|
| Signal Transduction (GO:0007165, p.val: 0.005062523) | |||
| TG | 2.56054 | -734 | hormone activity and carboxylic ester hydrolase activity. |
| FGD1 | 1.63956 | -319 | Activates CDC42. Plays a role in regulating the actin cytoskeleton and cell shape. |
| ANK2 | 1.69588 | -3310 | protein kinase binding and structural constituent of cytoskeleton |
| PDE1B | −1.77852 | -1499 | calmodulin binding and 3',5'-cyclic-AMP phosphodiesterase activity |
| NRXN2 | 1.51534 | -2035 | transmembrane signaling receptor activity and calcium channel regulator activity |
| NRXN3 | −2.08903 | -661 | cell adhesion molecule binding |
| PDGFRB | −3.39665 | -162 | regulation of many biological processes including embryonic development, angiogenesis, cell proliferation and differentiation. |
| CHN1 | −2.19144 | -194 | GTPase activator activity and ephrin receptor binding |
| PTCH2 | 4.70794 | -7374 | hedgehog receptor activity and hedgehog family protein binding. |
| HPGDS | −1.58043 | -1303 | calcium ion binding and magnesium ion binding |
| Neurotransmitter secretion (GO:0007269, p. val:0.009331) | |||
| NRXN2 | 1.51534 | -2035 | transmembrane signaling receptor activity and calcium channel regulator activity |
| NRXN3 | −2.08903 | -661 | cell adhesion molecule binding |
| DGKI | −5.02254 | 8365 | NAD+ kinase activity and diacylglycerol kinase activity |
| Cytoskeleton organization (GO:0007010, p.val: 0.010913659) | |||
| FGD1 | 1.63956 | -319 | Activates CDC42. Plays a role in regulating the actin cytoskeleton and cell shape. |
| ANK2 | 1.69588 | -3310 | protein kinase binding and structural constituent of cytoskeleton |
| WTIP | −2.31319 | -7255 | involved in several cellular processes such as cell fate determination, cytoskeletal organization, repression of gene transcription, cell-cell adhesion, cell differentiation, proliferation and migration |
| CTNNA2 | 1.67243 | -1990 | structural molecule activity and structural constituent of cytoskeleton |
| Positive regulation of phospholipase C activity (GO:0010863, p. val: 0.025440324) | |||
| PDGFRB | −3.39665 | -162 | regulation of many biological processes including embryonic development, angiogenesis, cell proliferation and differentiation. |
| PTAFR | −2.05699 | -3443 | G protein-coupled receptor activity and lipopolysaccharide binding |
| Vocalization behavior (GO:0071625, p.val:0.039299) | |||
| NRXN2 | 1.51534 | -2035 | transmembrane signaling receptor activity and calcium channel regulator activity |
| NRXN3 | −2.08903 | -661 | cell adhesion molecule binding |
| Cell differentiation (GO:0030154, p.val: 0.042536642) | |||
| IGSF10 | 1.70255 | -5900 | control of early migration of neurons expressing gonadotropin-releasing hormone |
| CPLX2 | −1.76176 | -9666 | Negatively regulates the formation of synaptic vesicle. Positively regulates a late step in exocytosis of various cytoplasmic vesicles |
| TDRD6 | −2.02059 | -4622 | involved in germ cell development |
| TP53 | −1.89693 | -148 | Acts as a tumour suppressor in many tumour types; induces growth arrest or apoptosis depending on the physiological circumstances and cell type. |
| ZIC1 | −4.16215 | 1079 | transcriptional activator. Involved in neurogenesis |
| Neuron cell-cell adhesion (GO:0007158, p. val:0.044788079) | |||
| NRXN2 | 1.51534 | -2035 | transmembrane signaling receptor activity and calcium channel regulator activity |
| NRXN3 | −2.08903 | -661 | cell adhesion molecule binding |
| Negative regulation of smoothened signaling pathway (GO:0045879, p.val:0.05296437) | |||
| SERPINE2 | −2.36671 | -3485 | signaling receptor binding and serine-type endopeptidase inhibitor activity. |
| PTCH2 | 4.70794 | -7374 | hedgehog receptor activity and hedgehog family protein binding. |
| Regulation of small GTPase mediated signal transduction (GO:0051056, p.val:0.056180682) | |||
| FGD1 | 1.63956 | -319 | Activates CDC42. Plays a role in regulating the actin cytoskeleton and cell shape. |
| CHN1 | −2.19144 | -194 | GTPase activator activity and ephrin receptor binding |
| ARHGAP29 | −2.32394 | -1824 | GTPase activator activity and PDZ domain binding |
These results therefore suggested that Hmrhl might function as a potential regulator of PDGFRB, TP53 and ZIC1. We were further interested to investigate the mechanism that facilitates lncRNA Hmrhl to select the chromatin region throughout the genome for its association. For this, we first looked for common sequence motifs enriched in the Hmrhl-bound genomic regions in our ChIRP-seq dataset and identified a strong GA-rich sequence, motif 1 (motif e-value: 1.00E–04) that accounts for 80.36% of these sequence reads and a second motif 2 (motif e-value: 1.00E–04), a CT-rich sequence which covers the rest of 24.54% (Figure 6A, B). This indicates that GA-rich sequence might play a dominating role in the targeting of Hmrhl lncRNAs across the genome. Next, we analysed the promoter region of our genes of interest (i.e. PDGFRβ, TP53 and ZIC1) and observed the enrichment of the ChIRP-seq peak for the presence of motifs region by FIMO in the MEMO suite (Figure 6C). Then we generated the potential triplex formation structures at these sites using the Triplexator program (55). Interestingly, triplex forming oligonucleotides (TFOs) thus obtained for all the three genes were also GA-enriched with high TFO score (Figure 6D, Table 4). Recently, using different techniques, several studies had validated the formation of RNA–DNA triplex structure by GA-rich homopurine sequences (101–103). These reports also established gene regulatory mechanism by lncRNA via RNA–DNA triplex formation, where the triplex acts as an anchor for the recruitment of chromatin modifiers in proximity to the gene promoters (104,105). In our study, triplex forming oligonucleotides (TFOs) as scanned by the Triplexator software within the Hmrhl RNA detected TFOs with high score (Table 4) with overrepresentation of GA-rich sequences (Figure 6D). This raises the possibility of triplex formation by the GA-rich sequences between target genes and Hmrhl RNA. Therefore, we hypothesize that this might be the probable regulatory mechanism employed by Hmrhl RNA.
Figure 6.

Regulation of target genes expression via Hmrhl by possible triplex formation at chromatin level. (A) Predicted motifs enriched in all Hmrhl peaks in ChIRP-seq. (B) Distribution of Motifs for genome occupancy for Hmrhl on target genes. (C) Enrichment of Hmrhl at the promoter region of ZIC1, PDGRFβ and TP53 in ChiRP- seq analysis. (D) Predicted DN-RNA triplex formation at the target sites using Triplextor.
Table 4.
Triplex forming oligonucleotides (TFOs) predicted by Triplextor program
| OligoID | TFOs 5′-3′ | Score |
|---|---|---|
| TP53 | GAAAGtAGAGcAG | 11 |
| PDGFRβ Triplex 1 | GGGGGagGAgGGGGG | 12 |
| PDGRFβ Triplex 2 | GGGGGaGgAGGGGGgG | 13 |
| ZIC1 Triplex 1 | AAGAAAGGAAGgGAGGGAGGgaGGgAgGGA | 25 |
| ZIC1 Triplex 2 | AAGgAAGGAAGgaAGGaAGGAGGGAAgG | 25 |
| ZIC1 Triplex 3 | GaAggAAGAAAGGAAGgGAGGGAGG | 21 |
Overexpression of one of the Hmrhl targets PDGRF-β to analyse the rescue effect upon Hmrhl knockdown
In order to gain a further insight into the possible role of Hmrhl RNA in cancer associated cellular phenotypes, we selected PDGRF-β, which is known to have essential role in activation of the intracellular signaling pathway that leads to cell migration, invasion, and proliferation, for our further experiments. To see whether overexpression of PDGRF-β in trans can reverse the effects of Hmrhl RNA downregulation, we co-transfect PDGRF-β plasmid and si-Hmrhl in K562 cells (Figure 7A) and evaluated the rescue effect it had on cancer associated phenotypes. In case of cellular proliferation, we observed that overexpression of PDGRF-β was able to restore the decline in cell-proliferation after Hmrhl downregulation at time points 72 h (P value = 0.0629) and 96 h (P value = 0.0681). However, migration assay showed a statistically significant increase in cellular migration in case of co-transfection as compared to both control (∼50% increase) and si-Hmrhl only samples (∼120% increase) (Figure 7C). While in case of invasion assay, the rescue effect was only partial (Figure 7D). Although % cell invasion was significantly higher in case of co-transfected samples when compared to si-Hmrhl only, a significant difference between % cell invasion in case of control and co-transfected samples still remains (Figure 7D). These results further confirm PDGRF-β to be an important target of Hmrhl in leukemia.
Figure 7.

Overexpression of PDGRFβ in Hmrhl downregulated cells. (A) expression levels of PDGRFβ and Hmrhl after co-transfection of PDGRFβ plasmid and si-Hmrhl RNA in K562 cells. (B) CCK-8 assay showing rescued cell proliferation after PDGRFβ overexpression as compared to Hmrhl silencing cells measured over a time period of 96 h. (C, D) Transwell assays followed by CCK-8 assay after 48 h to evaluate the endpoint, reveals increased cellular migration (C) and partial recovery of cellular invasion (D) in co-transfected cells as compared to control. The data are presented as mean ± SD from three independent experiments, **P< 0.01, ***P< 0.001 by Student's t-test.
TAL1 regulates Hmrhl expression in K562 cells
Since Hmrhl RNA plays a pivotal role in regulating key genes and pathways related to cancer pathobiology, we wanted to examine how Hmrhl RNA itself is regulated in K562 cells. We first tried to identify all possible TFs that has potential binding site within the promoter region (–3 kb to +0.5 kb) of Hmrhl with the help of GP-Miner program (Figure 8A). We found out that most of them have potential role in leukemia, hematopoiesis as well as in other type of cancers (Figure 8B). In order to determine the actual physical occupancy of various TFs, particularly in K562 cell lines, we took a look at the promoter region of Hmrhl from ENCODE project consortium, and interestingly, we came up with certain transcription factors like TAL1 and GATA2 that are highly enriched within the upstream of Hmrhl (Figure 8C). Among them, TAL1, is a primary regulator of erythroid differentiation with an established role in genesis of hemopoietic malignancies (106). We also took advantage of the TAL1 ChIP-seq dataset for K562 available from ENCODE. We used the Integrative Genomic Viewer (IGV) software tool to visualized TAL1 enrichment which clearly showed that, TAL1 is indeed enriched at the promoter region of Hmrhl (Figure 8C). To further experimentally validate this, we performed TAL1 Chromatin Immunoprecipitation study (ChIP) to pull-down TAL1 associated chromatin and scored for the promoter region of Hmrhl. Our real-time PCR data clearly indicates higher enrichment of TAL1 in comparison to mock control (IgG) (Figure 8D), thus confirming our observation from publicly available ChIP-seq dataset. Enrichment of TAL1 at the promoter of Hmrhl prompted us to ask the next obvious question that whether TAL1 regulates the expression of Hmrhl. To address this, we silenced Tal1 using siRNA in K562 cell line (Figure 8E). Interestingly, we did observe an ∼2-fold down regulation of Hmrhl after silencing Tal1 (Figure 8F). Coherently, we parallelly evaluated Tal1 expression upon Hmrhl down regulation and found no significant change in the expression level of Tal1 between si-scramble and si-Hmrhl (Figure 8G, H). Thus, these observations suggest that TAL1 could be a potential regulator of Hmrhl RNA expression in K562 cells and therefore in chronic myelogenous leukemia.
Figure 8.
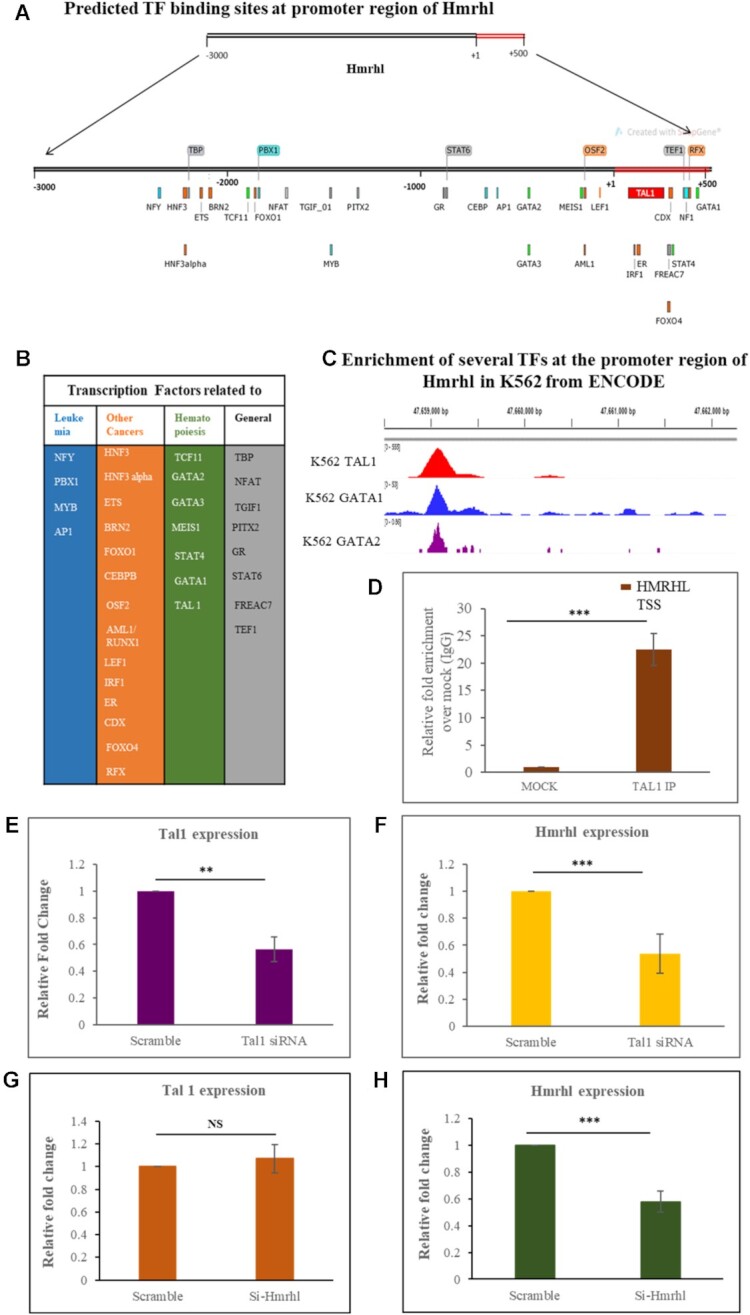
Regulation of expression of lncRNA Hmrhl by TAL1. (A) Binding location of all the possible TFs at the promoter site of Hmrhl (–3 kb to +500 bp) as predicted by GP Miner program. (B) Categorization of predicted TFs according to their role and involvement in leukemias, other cancers, hematopoiesis and others biological processes. (C) ChIP seq data in K562 from ENCODE establishing enrichment of few of the predicted TFs within the upstream of Hmrhl as viewed in Integrative Genomic Viewer (IGV) tool. (D) TAL1 ChIP shows high enrichment of Hmrhl promoter site when compared to mock control (IgG). (E, F) Expression levels of TAL1 and Hmrhl respectively as determine by RT-qPCR after TAL1 silencing in K562 cells. (G, H) Evaluation of relative expression pattern of TAL1 and Hmrhl respectively up on Hmrhl knockdown. The data are presented as mean ± SD from three independent experiments, *P < 0.05, **P < 0.01, ***P < 0.001 by Student's t-test.
DISCUSSION
Exploring lncRNAs as novel drivers of tumorigenesis holds a strong platform and reports establishing its correlation with cancer has increased exponentially in the last decade (22,107). Although there are experimentally supported association of 1614 human lncRNAs with various types of cancer (ln2cancer, http://www.bio-bigdata.com/lnc2cancer/statistics.html) only few of them are well documented (like H19, Xist, HOTAIR, ANRIL, NEAT1, MALAT1 etc). Most of them are not fully characterized and their functional relevance and regulatory mechanisms are still elusive (26). Following the lead from our previous study where human lncRNA Hmrhl was first reported, we tried to explore the potential of Hmrhl RNA in context of cancer phenotype in leukemia and to fully understand its regulatory targets and mechanism involved (44). Findings from the current study that Hmrhl is nuclear restricted and chromatin associated, strengthen our idea of Hmrhl RNA as a gene regulator (Figure 1C, D, E). Most of the nuclear lncRNAs associated with chromatin influence gene expression directly or indirectly by interacting with chromatin and associated proteins. For example, PINCR (p53-induced lncRNA) promotes the upregulation of a subset of p53 target genes involved in G1 arrest and apoptosis. It does so by association with the enhancer region of the candidate genes along with Matrin 3 and p53, modulating their expression upon DNA damage (108). In another example, low-irradiation-induced lncRNA, PARTICLE (promoter of MAT2A-antisense radiation-induced circulating lncRNA) represses MAT2A expression by forming a DNA–RNA triplex at the MAT2A locus, and by recruiting transcription repressive complex proteins G9a and SUZ12 (a subunit of PRC2) to the MAT2A promoter for methylation (109).
We have shown the functional relevance of Hmrhl with knockdown and transcriptome analysis using comprehensive bioinformatics approaches. This exercise revealed many interesting candidate pathways to be affected upon down regulation in K562 cell line. Inflammation mediated by chemokine and cytokines signaling and WNT pathway been the top two hits with most number of genes to be effected (Figure 3D, Table 1). There is now overwhelming evidence to support the relationship between cancer and inflammation involving chemokines and cytokines. Predominant involvement of chemokines and cytokines in progression of cancer can be linked to their many functional capacities in metastasis, providing tumor micro-environment, survival and proliferation (110,111). In our study also most of the dysregulated genes in this group like CCL3, CCR4, CXCL10, IL-6, ITGB, ITGA, MYLK2, ReLB are found to be associated with various cancers with their substantiated role in cell growth and proliferation, migration and metastasis (Table 1). Observations that Hmrhl promotes cell proliferation, migration and invasion in CML cell line can also be attributed to the preceded fact (Figure 2).
One interesting observation to be noticed was aberrant expression of 21 genes from WNT signaling pathway up on Hmrhl silencing, given the fact that in our previous study no link between the two was found, as translocation of β-catenin to nucleus was not observed under the same conditions in HeK293T cells (44). It will also be fascinating to mention here that mouse counter-part of Hmrhl, Mrhl was found to be inversely co-related with WNT-signaling in mouse spermatogonial cell line, GC1-Spg, while in the same species no regulation was observed between the two in mouse embryonic stem cells (42,43). This adds to the already evident fact that functional aspect of lncRNAs are highly cellular context driven and are rarely conserved. In chronic myelogenous leukemia, Hmrhl seems to influence a completely different set of genes belonging to WNT signaling group majority of which belongs to cadherin superfamily (e.g. DCHS1, PCDH12, CDH12, FZD9, FAT2, PCDH11Y, FRZB, SFRP5), catenin family (e.g. CTNNA2, CTNNA3) and signaling molecules having role in development and differentiation (Table 1). WNT signaling pathway is crucial for the development and homeostasis of blood and immune cells. Involvement of WNT signaling in the malignant transformation of cells has been reported earlier and its deregulation becomes apparent in the malignancies of the hematopoietic system for example aberrant activation of the canonical WNT signaling is crucial for CLL pathogenesis (112). Beside affecting many important genes and TFs related to cancer, evaluation from GO analysis also shows its possible role in development and differentiation especially in neuronal development. Further exploration in this direction will be interesting as role of Mrhl (mouse homolog of Hmrhl) in development of neuronal cell lineage is recently established by our group (manuscript under revision in Journal of Cell Science, bioRxiv 2021.06.23.449546; doi: https://doi.org/10.1101/2021.06.23.449546).
Using chromatin isolation by RNA purification sequencing (ChIRP-seq) technique, it was revealed that Hmrhl RNA has efficiency to bind to a significant number of functional chromatin regions such as promoters. Although a large number of genome loci were found to be associated with Hmrhl RNA in our ChIRP-seq analysis, only a small number of intersected genes were obtained after overlapping the RNA-seq and ChIRP-seq data sets. It could be attributed to the fact that binding of lncRNA to chromatin is a physical phenomenon while regulation of genes by lncRNA, a mechanical phenomenon, is highly cell and context dependent. Therefore, a lncRNA might not regulate all the physically associated genomic region in every cells. Furthermore, examples from our own studies on lncRNA Mrhl, has shown that the gene targets of Mrhl lncRNA are quite different in the mouse spermatogonial cells and mouse embryonic stem (ES) cells (41,43) highlighting that the gene targets and consequent perturbation of gene expression will be cell type specific and context dependent even in the same species. Integrating whole transcriptome analysis and genome-wide chromatin association of Hmrhl, we systematically identified three genes (ZIC1, PDGRFβ and TP53) as potential regulatory targets of Hmrhl RNA. Promoters of all of the three genes show high affinity for Hmrhl binding and a significant downregulated expression after Hmrhl depletion (Figures 5E, F & 6C, D, Table 4). ZIC1, which was one of the dominating TFs in our TF matrix analysis (Figure 4C, Supplementary Table S2), has been reported for its oncogenic as well as tumor suppressive behavior in various cancers (113–115). ZIC1 is also proclaimed as a one of the members of transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line (116,117). Dysregulation of ZIC1 expression may be thus associated with the outcome of our cancer phenotype study indicating Hmrhl as probable promoter of leukemogenesis with a role in cell proliferation, migration and invasion (Figure 2). As one of the most downregulated gene in si-Hmrhl RNAseq dataset, PDGRFβ was our another gene of interest to be regulated by Hmrhl. PDGFs signaling is crucial during normal development as well as has a significant role in human cancers. Stimulation of the PDGRFβ leads to the activation of the intracellular signaling pathway that further promotes cell migration, invasion, and proliferation (118,119). This fact is evident from our PDGRF-β overexpression study in Hmrhl downregulated cells which shows complete to partial rescue effects for different cancer associated cellular phenotype assays (Figure 7). Previous observation demonstrates that autocrine PDGF signaling involved in various types of malignancies, such as gliomas and leukemia (120,121). A recent study has demonstrated metastatic and proliferative effect in murine model of pancreatic cancer induced via p53 mutation through PDGRFβ (122). This is particularly fascinating as p53 is our third gene of interest and is also mutated in K562 cell line to be translated into a non-functional protein. It will be intriguing to investigate further whether Hmrhl directly regulate PDGRFβ or via regulating p53 or both. As a well-known tumor suppressor, importance of p53 regulation by Hmrhl further extends its role in other cancers like breast cancer where contrary to leukemia, Hmrhl expression is highly downregulated (44). It would be interesting to see whether Hmrhl regulates p53 expression in wild type p53 tumor cells also.
Towards regulatory mode of Hmrhl, we postulate DNA–RNA triplex formation at the regulatory sites of target gene as a major mechanism. It is now known that DNA–RNA triplexes, where lncRNAs acts as a third strand is one of the mechanisms initiates/repress the expression of target gene via recruitment or stabilization of various chromatin modulators or RNA-binding proteins to the regulatory sequences (103,123). Numerous evidences show that triplex formations at chromatin level are by motifs that are GA rich symmetrical sequences and is often considered for trans-acting lncRNA that target distant genes. For example, lncRNA MEG3 is guided to the target site via GA enriched motifs, where it facilitates the recruitment of PRC2 to the target sites (103). Other recent examples include HOTAIR lncRNA, which was found to preferentially occupy AG-rich DNA motifs across the genome. These authors predicted that triplex formation by HOTAIR recruit PRC2 and LSD1 which leads to target gene silencing (104). As predicted bioinformatically, GA rich triplex formation motifs of Hmrhl RNA at target site certainly strengthen the fact of trans regulatory mechanism of lncRNA Hmrhl however, further experimental verification remains to be documented (Figure 6, Table 4). The versatility of Hmrhl function in CML needs to be explored with a thorough screening of Hmrhl-bound protein partners particularly since many of the lncRNA are known to interact with chromatin through protein partners in addition to RNA-DNA triplex formation (109,124).
We also found that expression of Hmrhl is regulated by transcription factor TAL1. TAL1 is the master regulator of haematopoiesis. It is essential for HSC renewal and commitment of haematopoietic lineages and hence its fundamental relevance in leukemogenesis evident (106). A very recent study shows that disrupting -31CBS (CTCF binding sites) relative to TSS of TAL1 cause reduce cell survival, cell cycle arrest leading to apoptosis in erythroleukemia cell line K562 (125). Also, a study by Ngoc et. al. has shown TAL1 to regulate 57 putative lncRNA in eight different T-ALL cell lines by directly binding near or at the lncRNA loci, either alone or in coordination with its regulatory partners (126). Enrichment of TAL1 at Hmrhl promoter and its downregulation on TAL1 depletion supports our notion of functional significance of Hmrhl in CML. Binding sites of many TFs related to haematopoiesis at the promoter region of Hmrhl certainly sparks the thought of relevant biological significance of Hmrhl in the context of haematopoiesis and erythroid differentiation.
In summation, our study provides abundant evidence to elucidate significance of lncRNA Hmrhl as an oncogene in erythroleukemia and its regulation by TAL1. Mechanistically, our data suggests its role in elevation of cell growth and migration in vitro possibly by involvement of signalling pathway genes and target gene regulation via triplex formation by binding at chromatin level. Of course, more in‐depth research is required to fully comprehend the underlying mechanisms of Hmrhl RNA not only in leukemia but in other cancers which might reveal some potential therapeutic targets. Also, as indicated from the current study, involvement of Hmrhl in development and differentiation especially in neurogenesis and haematopoiesis needs further exploration to understand the complexity behind these processes.
DATA AVAILABILITY
Sequence reads have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under accession number GSE167440.
Supplementary Material
ACKNOWLEDGEMENTS
We thank central facility, JNCASR. We thank Dr Narendra Nala for his assistance in flow cytometry. We acknowledge Suma B.S. for her assistance with confocal imaging facility.
Notes
Present address: Subhendu Roy Choudhury, University of Maryland, School of Medicine, Institute for Genome Science, Baltimore, MD, USA.
Present address: Utsa Bhaduri, Department of Life Sciences, University of Trieste, Italy, and EU Horizon 2020 TRIM-NET Innovative Training Network (ITN) of Marie Sklodowska-Curie Actions (MSCA) and Division of Medical Genetics, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo (Foggia), Italy.
Contributor Information
Subhendu Roy Choudhury, Molecular Biology and Genetics Unit, Jawaharlal Nehru Centre for Advance Scientific Research, Bangalore, India.
Sangeeta Dutta, Molecular Biology and Genetics Unit, Jawaharlal Nehru Centre for Advance Scientific Research, Bangalore, India.
Utsa Bhaduri, Molecular Biology and Genetics Unit, Jawaharlal Nehru Centre for Advance Scientific Research, Bangalore, India.
Manchanahalli R Satyanarayana Rao, Molecular Biology and Genetics Unit, Jawaharlal Nehru Centre for Advance Scientific Research, Bangalore, India.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
Department of Biotechnology, India [BT/01/COE/07/09]; M.R.S.R. acknowledges Department of Science and Technology for J.C. Bose and S.E.R.B. distinguished fellowships and the Year of Science Chair professorship; S.R.C. acknowledge S.E.R.B. for National postdoctoral fellowship; S.D. acknowledges D.B.T. for postdoctoral fellowship.
Conflict of interest statement. None declared.
REFERENCES
- 1. Wilusz J.E., Sunwoo H., Spector D.L. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009; 23:1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jarroux J., Morillon A., Pinskaya M.. Rao M.R.S. History, discovery, and classification of lncRNAs. Long Non Coding RNA Biology. 2017; 1008:Springer; 1–46. [DOI] [PubMed] [Google Scholar]
- 3. Niederer R.O., Hass E.P., Zappulla D.C. Long noncoding RNAs in the yeast S. cerevisiae. Long Non Coding RNA Biology. 2017; Springer; 119–132. [DOI] [PubMed] [Google Scholar]
- 4. Iyer M.K., Niknafs Y.S., Malik R., Singhal U., Sahu A., Hosono Y., Barrette T.R., Prensner J.R., Evans J.R., Zhao S. et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015; 47:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kopp F., Mendell J.T. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018; 172:393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marchese F.P., Raimondi I., Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017; 18:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wapinski O., Chang H.Y. Long noncoding RNAs and human disease. Trends Cell Biol. 2011; 21:354–361. [DOI] [PubMed] [Google Scholar]
- 8. Frese K.S., Katus H.A., Meder B. Next-generation sequencing: from understanding biology to personalized medicine. Biology. 2013; 2:378–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun M., Kraus W.L. From discovery to function: the expanding roles of long noncoding RNAs in physiology and disease. Endocr. Rev. 2015; 36:25–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Penny G.D., Kay G.F., Sheardown S.A., Rastan S., Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature. 1996; 379:131–137. [DOI] [PubMed] [Google Scholar]
- 11. Ilik I.A., Quinn J.J., Georgiev P., Tavares-Cadete F., Maticzka D., Toscano S., Wan Y., Spitale R.C., Luscombe N., Backofen R. et al. Tandem stem-loops in roX RNAs act together to mediate X chromosome dosage compensation in Drosophila. Mol. Cell. 2013; 51:156–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sleutels F., Zwart R., Barlow D.P. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature. 2002; 415:810–813. [DOI] [PubMed] [Google Scholar]
- 13. Mercer T.R., Mattick J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol. 2013; 20:300–307. [DOI] [PubMed] [Google Scholar]
- 14. Rosa A., Ballarino M. Long noncoding RNA regulation of pluripotency. Stem Cells International. 2016; 2016:1797692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yoon J.H., Abdelmohsen K., Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J. Mol. Biol. 2013; 425:3723–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang F., Xue X., Zheng L., Bi J., Zhou Y., Zhi K., Gu Y., Fang G. Long non-coding RNA GHET1 promotes gastric carcinoma cell proliferation by increasing c-Myc mRNA stability. FEBS J. 2014; 281:802–813. [DOI] [PubMed] [Google Scholar]
- 17. Lü M.H., Tang B., Zeng S., Hu C.J., Xie R., Wu Y.Y., Wang S.M., He F.T., Yang S.M. Long noncoding RNA BC032469, a novel competing endogenous RNA, upregulates hTERT expression by sponging miR-1207-5p and promotes proliferation in gastric cancer. Oncogene. 2016; 35:3524–3534. [DOI] [PubMed] [Google Scholar]
- 18. Schmitz S.U., Grote P., Herrmann B.G. Mechanisms of long noncoding RNA function in development and disease. Cell. Mol. Life Sci. 2016; 73:2491–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ramos A.D., Andersen R.E., Liu S.J., Nowakowski T.J., Hong S.J., Gertz C.C., Salinas R.D., Zarabi H., Kriegstein A.R., Lim D.A. The long noncoding RNA Pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell. 2015; 16:439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo G., Wang M., Wu X., Tao D., Xiao X., Wang L., Min F., Zeng F., Jiang G. Long non-coding RNA MEG3 inhibits cell proliferation and induces apoptosis in prostate cancer. Cell. Physiol. Biochem. 2015; 37:2209–2220. [DOI] [PubMed] [Google Scholar]
- 21. Mitra S.A., Mitra A.P., Triche T.J. A central role for long non-coding RNA in cancer. Front. Genet. 2012; 3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lorenzi L., Avila Cobos F., Decock A., Everaert C., Helsmoortel H., Lefever S., Verboom K., Volders P.J., Speleman F., Vandesompele J. et al. Long noncoding RNA expression profiling in cancer: challenges and opportunities. Genes Chromosomes Cancer. 2019; 58:191–199. [DOI] [PubMed] [Google Scholar]
- 23. Sahu A., Singhal U., Chinnaiyan A.M. Long noncoding RNAs in cancer: from function to translation. Trends Cancer. 2015; 1:93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmitt A.M., Chang H.Y. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016; 29:452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bartonicek N., Maag J.L., Dinger M.E. Long noncoding RNAs in cancer: mechanisms of action and technological advancements. Mol. Cancer. 2016; 15:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao Y., Wang P., Wang Y., Ma X., Zhi H., Zhou D., Li X., Fang Y., Shen W., Xu Y. et al. Lnc2Cancer v2. 0: updated database of experimentally supported long non-coding RNAs in human cancers. Nucleic Acids Res. 2019; 47:D1028–D1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bussemakers M.J., Van Bokhoven A., Verhaegh G.W., Smit F.P., Karthaus H.F., Schalken J.A., Debruyne F.M., Ru N., Isaacs W.B. Dd3: a new prostate-specific gene, highly overexpressed in prostate cancer. Cancer Res. 1999; 59:5975–5979. [PubMed] [Google Scholar]
- 28. Rittenhouse H., Blase A., Shamel B., Schalken J., Groskopf J. The long and winding road to FDA approval of a novel prostate cancer test: our story. Clin. Chem. 2013; 59:32–34. [DOI] [PubMed] [Google Scholar]
- 29. Leucci E., Vendramin R., Spinazzi M., Laurette P., Fiers M., Wouters J., Radaelli E., Eyckerman S., Leonelli C., Vanderheyden K. et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016; 531:518–522. [DOI] [PubMed] [Google Scholar]
- 30. Hajjari M., Salavaty A. HOTAIR: an oncogenic long non-coding RNA in different cancers. Cancer Biol. Med. 2015; 12:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yoshimoto R., Mayeda A., Yoshida M., Nakagawa S. MALAT1 long non-coding RNA in cancer. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2016; 1859:192–199. [DOI] [PubMed] [Google Scholar]
- 32. Zhang K., Luo Z., Zhang Y., Zhang L., Wu L., Liu L., Yang J., Song X., Liu J. Circulating lncRNA H19 in plasma as a novel biomarker for breast cancer. Cancer Biomarkers. 2016; 17:187–194. [DOI] [PubMed] [Google Scholar]
- 33. Si X., Zang R., Zhang E., Liu Y., Shi X., Zhang E., Shao L., Li A., Yang N., Han X. et al. LncRNA H19 confers chemoresistance in ERα-positive breast cancer through epigenetic silencing of the pro-apoptotic gene BIK. Oncotarget. 2016; 7:81452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aguilo F., Di Cecilia S., Walsh M.J. Long non-coding RNA ANRIL and polycomb in human cancers and cardiovascular disease. Long Non-coding RNAs in human disease. 2015; Springer; 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu X., Li Z., Zheng H., Chan M.T., Wu W.K.K. NEAT 1: a novel cancer-related long non-coding RNA. Cell Prolif. 2017; 50:12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peng W., Feng J. Long noncoding RNA LUNAR1 associates with cell proliferation and predicts a poor prognosis in diffuse large B-cell lymphoma. Biomed. Pharmacother. 2016; 77:65–71. [DOI] [PubMed] [Google Scholar]
- 37. Xue Y., Wang M., Kang M., Wang Q., Wu B., Chu H., Zhong D., Qin C., Yin C., Zhang Z. et al. Association between lncrna PCGEM1 polymorphisms and prostate cancer risk. Prostate Cancer Prostatic Dis. 2013; 16:139–144. [DOI] [PubMed] [Google Scholar]
- 38. Nishant K.T., Ravishankar H., Rao M.R.S. Characterization of a mouse recombination hot spot locus encoding a novel non-protein-coding RNA. Mol. Cell. Biol. 2004; 24:5620–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ganesan G., Rao M.R.S. A novel noncoding RNA processed by Drosha is restricted to nucleus in mouse. RNA. 2008; 14:1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arun G., Akhade V.S., Donakonda S., Rao M.R.S. mrhl RNA, a long noncoding RNA, negatively regulates Wnt signaling through its protein partner Ddx5/p68 in mouse spermatogonial cells. Mol. Cell. Biol. 2012; 32:3140–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Akhade V.S., Arun G., Donakonda S., Rao M.R.S. Genome wide chromatin occupancy of mrhl RNA and its role in gene regulation in mouse spermatogonial cells. RNA Biology. 2014; 11:1262–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Akhade V.S., Dighe S.N., Kataruka S., Rao M.R.S. Mechanism of Wnt signaling induced down regulation of mrhl long non-coding RNA in mouse spermatogonial cells. Nucleic Acids Res. 2016; 44:387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pal D., Neha C.V., Bhaduri U., Zenia Z., Dutta S., Chidambaram S., Rao M.R.S. LncRNA Mrhl orchestrates differentiation programs in mouse embryonic stem cells through chromatin mediated regulation. Stem Cell Res. 2021; 53:102250. [DOI] [PubMed] [Google Scholar]
- 44. Fatima R., Choudhury S.R., Divya T.R., Bhaduri U., Rao M.R.S. A novel enhancer RNA, Hmrhl, positively regulates its host gene, phkb, in chronic myelogenous leukemia. Non-coding RNA Res. 2019; 4:96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gagnon K.T., Li L., Janowski B.A., Corey D.R. Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat. Protoc. 2014; 9:2045–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chu C., Quinn J., Chang H.Y. Chromatin isolation by RNA purification (ChIRP). JoVE (J. Visual. Exp.). 2012; 3912:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012; 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thomas P.D., Campbell M.J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003; 13:2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De Hoon M.J., Imoto S., Nolan J., Miyano S. Open source clustering software. Bioinformatics. 2004; 20:1453–1454. [DOI] [PubMed] [Google Scholar]
- 50. Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Warde-Farley D., Donaldson S.L., Comes O., Zuberi K., Badrawi R., Chao P., Franz M., Grouios C., Kazi F., Lopes C.T. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010; 38:W214–W220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.Y., Chou A., Ienasescu H. et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014; 42:D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Buske F.A., Bauer D.C., Mattick J.S., Bailey T.L. Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 2012; 22:1372–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ayupe A.C., Reis E.M. Evaluating the stability of mRNAs and noncoding RNAs. Enhancer RNAs. 2017; Humana Press; 139–153. [DOI] [PubMed] [Google Scholar]
- 57. Clark M.B., Johnston R.L., Inostroza-Ponta M., Fox A.H., Fortini E., Moscato P., Dinger M.E., Mattick J.S. Genome-wide analysis of long noncoding RNA stability. Genome Res. 2012; 22:885–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eccles S.A., Box C., Court W. Cell migration/invasion assays and their application in cancer drug discovery. Biotechnol. Annu. Rev. 2005; 11:391–421. [DOI] [PubMed] [Google Scholar]
- 59. Wang H., Hu H., Zhang Q., Yang Y., Li Y., Hu Y., Ruan X., Yang Y., Zhang Z., Shu C. et al. Dynamic transcriptomes of human myeloid leukemia cells. Genomics. 2013; 102:250–256. [DOI] [PubMed] [Google Scholar]
- 60. Jelinek J., Gharibyan V., Estecio M.R., Kondo K., He R., Chung W., Lu Y., Zhang N., Liang S., Kantarjian H.M. et al. Aberrant DNA methylation is associated with disease progression, resistance to imatinib and shortened survival in chronic myelogenous leukemia. PLoS One. 2011; 6:e22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Polakova K.M., Koblihova J., Stopka T. Role of epigenetics in chronic myeloid leukemia. Curr. Hematol. Malign. Rep. 2013; 8:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wang C., He H., Liu G., Ma H., Li L., Jiang M., Lu Q., Li P., Qi H. DT-13 induced apoptosis and promoted differentiation of acute myeloid leukemia cells by activating AMPK-KLF2 pathway. Pharmacol. Res. 2020; 158:104864. [DOI] [PubMed] [Google Scholar]
- 63. Eychène A., Rocques N., Pouponnot C. A new MAFia in cancer. Nat. Rev. Cancer. 2008; 8:683–693. [DOI] [PubMed] [Google Scholar]
- 64. Lin T.S., Mahajan S., Frank D.A. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene. 2000; 19:2496–2504. [DOI] [PubMed] [Google Scholar]
- 65. Potter C., Moorman A.V., Relton C.L., Ford D., Mathers J.C., Strathdee G., McKay J.A. Maternal red blood cell folate and infant vitamin B12 status influence methylation of genes associated with childhood acute lymphoblastic leukemia. Mol. Nutr. Food Res. 2018; 62:1800411. [DOI] [PubMed] [Google Scholar]
- 66. Huang Y., Chen J., Lu C., Han J., Wang G., Song C., Zhu S., Wang C., Li G., Kang J. et al. HDAC1 and Klf4 interplay critically regulates human myeloid leukemia cell proliferation. Cell Death. Dis. 2014; 5:e1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li Z., Zhao J., Li Q., Yang W., Song Q., Li W., Liu J. KLF4 promotes hydrogen-peroxide-induced apoptosis of chronic myeloid leukemia cells involving the bcl-2/bax pathway. Cell Stress Chaperones. 2010; 15:905–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Liao J., Humphrey S.E., Poston S., Taparowsky E.J. Batf promotes growth arrest and terminal differentiation of mouse myeloid leukemia cells. Mol. Cancer Res. 2011; 9:350–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yamaguchi H., Inokuchi K., Sakuma Y., Dan K. Mutation of the p51/p63 gene is associated with blastic crisis in chronic myelogenous leukemia. Leukemia. 2001; 15:1729–1734. [DOI] [PubMed] [Google Scholar]
- 70. Papakonstantinou N., Ntoufa S., Tsagiopoulou M., Moysiadis T., Bhoi S., Malousi A., Psomopoulos F., Mansouri L., Laidou S., Papazoglou D. et al. Integrated epigenomic and transcriptomic analysis reveals TP63 as a novel player in clinically aggressive chronic lymphocytic leukemia. Int. J. Cancer. 2019; 144:2695–2706. [DOI] [PubMed] [Google Scholar]
- 71. Law J. C., Ritke M. K., Yalowich J. C., Leder G. H., Ferrell R. E. Mutational inactivation of the p53 gene in the human erythroid leukemic K562 cell line. Leuk. Res. 1993; 17:1045–1050. [DOI] [PubMed] [Google Scholar]
- 72. Stigler B., Chamberlin H.M. A regulatory network modeled from wild-type gene expression data guides functional predictions in Caenorhabditis elegans development. BMC Syst. Biol. 2012; 6:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wei S., Du H., Li Z., Tao G., Xu Z., Song X., Shang Z., Su Z., Chen H., Wen Y. et al. Transcription factors Sp8 and Sp9 regulate the development of caudal ganglionic eminence-derived cortical interneurons. J. Comp. Neurol. 2019; 527:2860–2874. [DOI] [PubMed] [Google Scholar]
- 74. Liu T.H., Tang Y.J., Huang Y., Wang L., Guo X.L., Mi J.Q., Liu L.G., Zhu H., Zhang Y., Chen L. et al. Expression of the fetal hematopoiesis regulator FEV indicates leukemias of prenatal origin. Leukemia. 2017; 31:1079–1086. [DOI] [PubMed] [Google Scholar]
- 75. Jubb A.M., Chalasani S., Frantz G.D., Smits R., Grabsch H.I., Kavi V., Maughan N.J., Hillan K.J., Quirke P., Koeppen H. Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene. 2006; 25:3445–3457. [DOI] [PubMed] [Google Scholar]
- 76. Zhang D., Guo S., Wang H., Hu Y. SOX15 exerts antitumor function in glioma by inhibiting cell proliferation and invasion via downregulation of Wnt/β-catenin signaling. Life Sci. 2020; 255:117792. [DOI] [PubMed] [Google Scholar]
- 77. Manzella L., Tirrò E., Pennisi M.S., Massimino M., Stella S., Romano C., Vitale S.R., Vigneri P. Roles of interferon regulatory factors in chronic myeloid leukemia. Curr. Cancer Drug Targets. 2016; 16:594–605. [DOI] [PubMed] [Google Scholar]
- 78. Cattaneo F., Patrussi L., Capitani N., Frezzato F., D’Elios M.M., Trentin L., Semenzato G., Baldari C.T. Expression of the p66Shc protein adaptor is regulated by the activator of transcription STAT4 in normal and chronic lymphocytic leukemia B cells. Oncotarget. 2016; 7:57086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Massimino M., Consoli M.L., Mesuraca M., Stagno F., Tirrò E., Stella S., Pennisi M.S., Romano C., Buffa P., Bond H.M. et al. IRF5 is a target of BCR-ABL kinase activity and reduces CML cell proliferation. Carcinogenesis. 2014; 35:1132–1143. [DOI] [PubMed] [Google Scholar]
- 80. Morris V.A., Cummings C.L., Korb B., Boaglio S., Oehler V.G. Deregulated KLF4 expression in myeloid leukemias alters cell proliferation and differentiation through microRNA and gene targets. Mol. Cell. Biol. 2016; 36:559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Long B., Wang L.X., Zheng F.M., Lai S.P., Xu D.R., Hu Y., Lin D.J., Zhang X.Z., Dong L., Long Z.J. et al. Targeting GLI1 suppresses cell growth and enhances chemosensitivity in CD34+ enriched acute myeloid leukemia progenitor cells. Cell. Physiol. Biochem. 2016; 38:1288–1302. [DOI] [PubMed] [Google Scholar]
- 82. Abd Elrhman H.E., Ebian H.F. Patched homolog 1 (PTCHI) gene mutations can predict the outcome of chronic myeloid leukemia patients. Am. J. Blood Res. 2019; 9:15. [PMC free article] [PubMed] [Google Scholar]
- 83. Li Y., Lin C., Schmidt C.A. New insights into antigen specific immunotherapy for chronic myeloid leukemia. Cancer Cell Int. 2012; 12:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Valent P., Sadovnik I., Eisenwort G., Bauer K., Herrmann H., Gleixner K.V., Schulenburg A., Rabitsch W., Sperr W.R., Wolf D. Immunotherapy-based targeting and elimination of leukemic stem cells in AML and CML. Int. J. Mol. Sci. 2019; 20:4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ramsay A.G. Immune checkpoint blockade immunotherapy to activate anti-tumour T-cell immunity. Br. J. Haematol. 2013; 162:313–325. [DOI] [PubMed] [Google Scholar]
- 86. Ishida T., Ueda R. Immunopathogenesis of lymphoma: focus on CCR4. Cancer Sci. 2011; 102:44–50. [DOI] [PubMed] [Google Scholar]
- 87. Attygalle A.D., Feldman A.L., Dogan A. ITK/SYK translocation in angioimmunoblastic T-cell lymphoma. Am. J. Surg. Pathol. 2013; 37:1456–1457. [DOI] [PubMed] [Google Scholar]
- 88. Zhu H., Yang B., Liu J., Wu W., Ling Y. Case Report of acute myeloid leukemia with “WT1, ATRX, CEBPA, CSMD1, IKZF1, and LRP1B mutation and translocation between chromosome 1 and 19” developing from Philadelphia-negative chronic myeloid leukemia after TKI therapy. Medicine (Baltimore). 2020; 99:e18888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sun Y., Xu K., He M., Fan G., Lu H. Overexpression of Glypican 5 (GPC5) inhibits prostate cancer cell proliferation and invasion via suppressing Sp1-mediated EMT and activation of Wnt/β-catenin signaling. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018; 26:565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Montano-Almendras C.P., Essaghir A., Schoemans H., Varis I., Noel L.A., Velghe A.I., Latinne D., Knoops L., Demoulin J.B. ETV6-PDGFRB and FIP1L1-PDGFRA stimulate human hematopoietic progenitor cell proliferation and differentiation into eosinophils: the role of nuclear factor-κB. Haematologica. 2012; 97:1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Corrigan D.J., Luchsinger L.L., de Almeida M.J., Williams L.J., Strikoudis A., Snoeck H.W. PRDM16 isoforms differentially regulate normal and leukemic hematopoiesis and inflammatory gene signature. J. Clin. Invest. 2018; 128:3250–3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stevenson W.S., Best O.G., Przybylla A., Chen Q., Singh N., Koleth M., Pierce S., Kennedy T., Tong W., Kuang S.Q. et al. DNA methylation of membrane-bound tyrosine phosphatase genes in acute lymphoblastic leukaemia. Leukemia. 2014; 28:787–793. [DOI] [PubMed] [Google Scholar]
- 93. Zhong J., Chen S., Xue M., Du Q., Cai J., Jin H., Si J., Wang L. ZIC1 modulates cell-cycle distributions and cell migration through regulation of sonic hedgehog, PI 3 K and MAPK signaling pathways in gastric cancer. BMC Cancer. 2012; 12:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bushweller J.H. Targeting transcription factors in cancer—from undruggable to reality. Nat. Rev. Cancer. 2019; 19:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bell S.M., Schreiner C.M., Waclaw R.R., Campbell K., Potter S.S., Scott W.J. Sp8 is crucial for limb outgrowth and neuropore closure. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:12195–12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Kasberg A.D., Brunskill E.W., Potter S.S. SP8 regulates signaling centers during craniofacial development. Dev. Biol. 2013; 381:312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Aruga J., Inoue T., Hoshino J., Mikoshiba K. Zic2 controls cerebellar development in cooperation with Zic1. J. Neurosci. 2002; 22:218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Safe S., Abdelrahim M. Sp transcription factor family and its role in cancer. Eur. J. Cancer. 2005; 41:2438–2448. [DOI] [PubMed] [Google Scholar]
- 99. Burkly L., Hession C., Ogata L., Reilly C., Marconl L.A., Olson D., Tizard R., Gate R., Lo D. Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995; 373:531–536. [DOI] [PubMed] [Google Scholar]
- 100. Sharfe N., Merico D., Karanxha A., Macdonald C., Dadi H., Ngan B., Herbrick J.A., Roifman C.M. The effects of RelB deficiency on lymphocyte development and function. J. Autoimmun. 2015; 65:90–100. [DOI] [PubMed] [Google Scholar]
- 101. Khomyakova E.B., Gousset H., Liquier J., Huynh-Dinh T., Gouyette C., Takahashi M., Florentiev V.L., Taillandier E. Parallel intramolecular DNA triple helix with G and T bases in the third strand stabilized by Zn2+ ions. Nucleic Acids Res. 2000; 28:3511–3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Besch R., Giovannangeli C., Kammerbauer C., Degitz K. Specific inhibition of ICAM-1 expression mediated by gene targeting with Triplex-forming oligonucleotides. J. Biol. Chem. 2002; 277:32473–32479. [DOI] [PubMed] [Google Scholar]
- 103. Mondal T., Subhash S., Vaid R., Enroth S., Uday S., Reinius B., Mitra S., Mohammed A., James A.R., Hoberg E. et al. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA–DNA triplex structures. Nat. Commun. 2015; 6:7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Li Y., Syed J., Sugiyama H. RNA-DNA triplex formation by long noncoding RNAs. Cell Chem. Biol. 2016; 23:1325–1333. [DOI] [PubMed] [Google Scholar]
- 105. Kalwa M., Hänzelmann S., Otto S., Kuo C.C., Franzen J., Joussen S., Fernandez-Rebollo E., Rath B., Koch C., Hofmann A. et al. The lncRNA HOTAIR impacts on mesenchymal stem cells via triple helix formation. Nucleic Acids Res. 2016; 44:10631–10643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Porcher C., Chagraoui H., Kristiansen M.S. SCL/TAL1: a multifaceted regulator from blood development to disease. Blood. 2017; 129:2051–2060. [DOI] [PubMed] [Google Scholar]
- 107. Renganathan A., Felley-Bosco E.. Rao M.R.S. Long noncoding RNAs in cancer and therapeutic potential. Long Non Coding RNA Biology. 2017; 1008:Springer; 199–222. [DOI] [PubMed] [Google Scholar]
- 108. Chaudhary R., Gryder B., Woods W.S., Subramanian M., Jones M.F., Li X.L., Jenkins L.M., Shabalina S.A., Mo M., Dasso M. et al. Prosurvival long noncoding RNA PINCR regulates a subset of p53 targets in human colorectal cancer cells by binding to Matrin 3. Elife. 2017; 6:e23244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. O’Leary V.B., Ovsepian S.V., Carrascosa L.G., Buske F.A., Radulovic V., Niyazi M., Moertl S., Trau M., Atkinson M.J., Anastasov N. PARTICLE, a triplex-forming long ncRNA, regulates locus-specific methylation in response to low-dose irradiation. Cell Rep. 2015; 11:474–485. [DOI] [PubMed] [Google Scholar]
- 110. O’Hayre M., Salanga C.L., Handel T.M., Allen S.J. Chemokines and cancer: migration, intracellular signalling and intercellular communication in the microenvironment. Biochem. J. 2008; 409:635–649. [DOI] [PubMed] [Google Scholar]
- 111. Lazennec G., Richmond A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol. Med. 2010; 16:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Staal F.J., Sen M.J. The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur. J. Immunol. 2008; 38:1788–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Han W., Cao F., Gao X.J., Wang H.B., Chen F., Cai S.J., Zhang C., Hu Y.W., Ma J., Gu X. et al. ZIC1 acts a tumor suppressor in breast cancer by targeting survivin. Int. J. Oncol. 2018; 53:937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ge Q., Hu Y., He J., Chen F., Wu L., Tu X., Qi Y., Zhang Z., Xue M., Chen S. et al. Zic1 suppresses gastric cancer metastasis by regulating Wnt/β-catenin signaling and epithelial-mesenchymal transition. FASEB J. 2020; 34:2161–2172. [DOI] [PubMed] [Google Scholar]
- 115. Brill E., Gobble R., Angeles C., Lagos-Quintana M., Crago A., Laxa B., DeCarolis P., Zhang L., Antonescu C., Socci N.D. et al. ZIC1 overexpression is oncogenic in liposarcoma. Cancer Res. 2010; 70:6891–6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Suzuki H., Forrest A.R., Van Nimwegen E., Daub C.O., Balwierz P.J., Irvine K.M., Lassmann T., Ravasi T., Hasegawa Y., De Hoon M.J. et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat. Genet. 2009; 41:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zhong J., Chen S., Xue M., Du Q., Cai J., Jin H., Si J., Wang L. ZIC1 modulates cell-cycle distributions and cell migration through regulation of sonic hedgehog, PI 3 K and MAPK signaling pathways in gastric cancer. BMC Cancer. 2012; 12:290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Heldin C.H., Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999; 79:1283–1316. [DOI] [PubMed] [Google Scholar]
- 119. Steller E.J., Ritsma L., Raats D.A., Hoogwater F.J., Emmink B.L., Govaert K.M., Laoukili J., Borel Rinkes I.H., Van Rheenen J., Kranenburg O. The death receptor CD95 activates the cofilin pathway to stimulate tumour cell invasion. EMBO Rep. 2011; 12:931–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Wallmann T., Zhang X.M., Wallerius M., Bolin S., Joly A.L., Sobocki C., Leiss L., Jiang Y., Bergh J., Holland E.C. et al. Microglia induce PDGFRB expression in glioma cells to enhance their migratory capacity. IScience. 2018; 9:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Pardanani A., Tefferi A. Imatinib targets other than bcr/abl and their clinical relevance in myeloid disorders. Blood. 2004; 104:1931–1939. [DOI] [PubMed] [Google Scholar]
- 122. Weissmueller S., Manchado E., Saborowski M., Morris J.P. IV, Wagenblast E., Davis C.A., Moon S.H., Pfister N.T., Tschaharganeh D.F., Kitzing T. et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell. 2014; 157:382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Vance K.W., Ponting C.P. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014; 30:348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Blank-Giwojna A., Postepska-Igielska A., Grummt I. lncRNA KHPS1 activates a poised enhancer by triplex-dependent recruitment of epigenomic regulators. Cell Rep. 2019; 26:2904–2915. [DOI] [PubMed] [Google Scholar]
- 125. Li Y., Liao Z., Luo H., Benyoucef A., Kang Y., Lai Q., Dovat S., Miller B., Chepelev I., Li Y. et al. Alteration of CTCF-associated chromatin neighborhood inhibits TAL1-driven oncogenic transcription program and leukemogenesis. Nucleic Acids Res. 2020; 48:3119–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Ngoc P.C.T., Tan S.H., Tan T.K., Chan M.M., Li Z., Yeoh A.E., Tenen D.G., Sanda T. Identification of novel lncRNAs regulated by the TAL1 complex in T-cell acute lymphoblastic leukemia. Leukemia. 2018; 32:2138–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence reads have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (NCBI GEO) under accession number GSE167440.