

Abstract
The increasing burden of cancer requires identifying and protecting individuals at highest risk. The epigenome provides an indispensable complement to genetic alterations for a risk stratification approach for the following reasons: gene transcription necessary for cancer onset is directed by epigenetic modifications and many risk factors studied so far have been associated with alterations related to the epigenome. The risk level depends on the plasticity of the epigenome during phases of life particularly sensitive to environmental and dietary impacts. Modifications in the activity of DNA regulatory regions and altered chromatin compaction may accumulate, hence leading to the increase of cancer risk. Moreover, tissue architecture directs the unique organization of the epigenome for each tissue and cell type, which allows the epigenome to control cancer risk in specific organs. Investigations of epigenetic signatures of risk should help identify a continuum of alterations leading to a threshold beyond which the epigenome cannot maintain homeostasis. We propose that this threshold may be similar in the population for a given tissue, but the pace to reach this threshold will depend on the combination of germline inheritance and the risk and protective factors encountered, particularly during windows of epigenetic susceptibility, by individuals.
Graphical Abstract
Graphical Abstract.
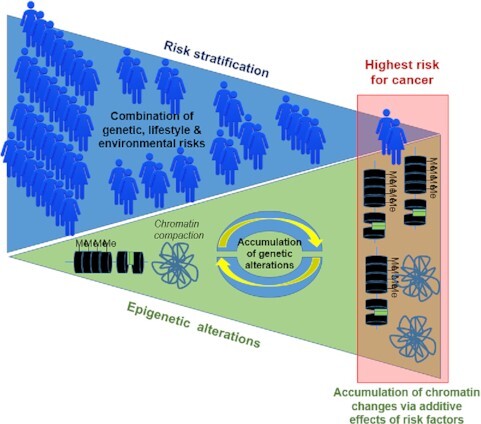
The accumulation of alterations in the epigenome and chromatin organization via additive effects of risk factors participates in risk stratification in populations.
INTRODUCTION
Our ability to prevent cancer development relies on the detection and mitigation of risk. An important challenge is to identify people at the highest risk for a particular cancer through a risk stratification process. Although epidemiologically the risk has been defined on multiple levels (e.g. family, lifestyle, genetic), biological investigations have shown that for a cancer to occur, changes in gene transcription prevail. But chemical modifications on the DNA and histones that direct gene transcription and define the epigenome are not yet part of a risk stratification scheme.
The need for risk stratification
Humans are not equal when dealing with cancer onset. It is of utmost importance to stratify the risk in order to develop targeted interventions. For most types of cancer there is not one causative factor but rather a multiplicity of factors, and their relative contribution to building 100% risk is not understood. A striking example is the increase of breast cancer incidence in young women (1). Although hormone levels might be useful to include for risk stratification in postmenopausal women, it does not seem to be the case for younger women (2), and the reasons for increased premenopausal cancer risk are still mostly unknown.
Genetics has been the driver for risk stratification, encompassing high risk variants (e.g. BRCA), intermediate risk variants (e.g. PALB2, ATM, CHEK2) and low risk single nucleotide polymorphisms (SNPs). The use of polygenic risk scores (PRS) linked to SNPs may stratify breast cancer risk regardless of the family history (3). However, even when integrating all the known genetic factors it only explains 45% of observed familial aggregation (4). It was proposed that associating genetic risk (including 313 SNPs from European ancestry (5) in addition to known mutations (6)) and other risk factors might improve risk stratification for a population with known breast cancer incidence (4). The other risk factors included lifestyle, reproductive information and mammographic density. This combination of factors revealed that for the population of women in the United Kingdom, 1.1% might have a lifetime risk of at least 30% (high risk) and 14.7% might have a risk below 30% but at least 17%. Women at high risk for breast cancer also included those with a 10-year risk at age 40 of at least 8%. Interestingly, the biggest contributors to this risk stratification model were PRS and mammographic density (7), both having relationships with the epigenome (8,9).
The participation of mammographic density in risk stratification reinforces the importance of tissue architecture in directing cancer onset. More than two decades ago, it was demonstrated that even in the presence of genetic alterations linked to cancer behavior, a tumor does not develop when the architecture of the normal tissue is maintained (10–12). Tissue architecture that encompasses the organization of cells and their components is controlling the epigenome (13), and reciprocally, it is controlled by the epigenome, as we initially demonstrated for the establishment of breast epithelial polarity (14). Thus, the architecture within which the epigenome is exposed to risk factors is likely to contribute to the transcriptomic response.
The nuclear origins of risk
Easily accessible in the blood, the DNA is currently at the center of risk stratification, especially in light of the contributions of SNPs identified via genome-wide association studies (GWAS). Initially, 170 breast cancer susceptibility loci were identified (5), accounting for 40% of heritability explained by all common variants on genome-wide SNP arrays. The SNPs are often within regulatory regions, like enhancers and sequences for transcription factor (TF) binding, hence potentially affecting gene transcription. The addition of functional studies to GWAS are revealing potential molecular mechanisms of risk through transcription profiles and redistribution of proteins like CCCTC-binding factor (CTCF) that affect chromosomal architecture (15). CTCF is a major organizer of higher order chromatin via anchoring and insulation of DNA loops and interactions with chromatin remodeling proteins, histone modifying enzymes and TFs.
It is important to link SNPs associated with cancer risk with specific molecular mechanisms to identify targets for interventions, especially since genetic variants confer risks that depend on breast cancer subtypes, at least estrogen receptor negative (ER−) versus ER positive (ER+) (5). Considering the subtypes of cancers in risk stratification underlies the importance of focusing on subgroups within the general population in which certain forms of breast cancer are preponderant. The subtyping of cancers in relation to molecular pathways and individuals is part of molecular pathological epidemiology (16). Since cancer subtypes are distinguishable via gene transcription patterns, themselves enabled by epigenetic modifications that respond to environmental exposures, an understanding of epigenetics is imperative to improve disease management.
The investigation of the epigenetic risk of cancer is challenging. The results of epigenome-wide association studies (EWAS) depend on the sources of normal tissues because the epigenome is not only tissue-specific but also condition-specific. Normal looking tissue adjacent to tumors is subject to field effect and its confounding impact, and breast tissue donated by cancer-free women needs to be accompanied with sufficient information to estimate cancer risk. Ultimately, the identified risk markers will have to be present in the blood or saliva for routine assessment. For this reason, DNA methylation, a stable epigenetic mark on circulating DNA, is a major target of investigation (17–19). However, most reports highlight different sets of genes when looking downstream of epigenetic changes associated with risk. This apparent heterogeneity in possible gene targets is most likely the illustration of the interaction between the individuals or groups of individuals and their specific environment, which makes it difficult to pinpoint the pathways to cancer onset.
Nevertheless, there are commonalities in epigenetic changes on a global chromatin organization scale. Higher mean DNA methylation at CpG islands and epigenetic age-acceleration have been associated with an increased risk of breast cancer (20). Interestingly, classes of risk modulators like aging, environmental exposure and diet trigger few but major changes in the tissue microenvironment that are all conducive to global alterations in chromatin. Tissue aging is linked to oxidative stress, in part generated by the accumulation of reactive oxygen species in the extracellular matrix (ECM) (21); increased body mass index (BMI) is associated with high leptin production by the adipose tissue, leading to a proinflammatory microenvironment and oxidative stress; mammographic density is related to increased ECM stiffness. Combining the different physiological and molecular consequences of epidemiologically confirmed risks might lead to additive effects on the epigenome.
In this short-review, using the example of breast cancer, we are placing epigenetic alterations in the multiparametric consideration of risk escalation. The integration of the epigenome in the notion of risk is discussed via evidence of the interplay between genetic and epigenetic alterations, the plasticity of higher order chromatin organization and the emerging investigations of how given risk factors specifically target epigenetic pathways.
INTERPLAY BETWEEN GENETIC AND EPIGENETIC ALTERATIONS TO DEFINE CANCER RISK
Tissue architecture is capable of taming cancer cells even in the presence of a plethora of genetic alterations (10). This seminal demonstration was performed in 3D cell culture that permits the production of specific tissue architectures. Induction of partial acinus differentiation leading to the quiescence of cancer cells was accompanied with a redistribution of epigenetic domains in the cell nucleus towards patterns observed in the phenotypically normal epithelium (22). Assuming that differentiation characterized by a specific tissue architecture controls higher order chromatin organization and its epigenetic structures, there might be protective epigenome features associated with normal differentiation that are progressively disrupted by the accumulation of risk factors.
Influence of SNPs on gene transcription control
Candidate causal variants (CCV) are noncoding in majority and primarily located in regulatory regions of gene transcription (23,24). Functional testing of the variants of interest involves mapping to epigenomic datasets that identify histone marks, like H3K27ac for enhancers (25). The combination of epigenetic mark H3K27ac, mediator complex subunits (MED) and epigenetic reader bromodomain containing protein 4 (BRD4) has been used to define super-enhancers that contribute to cell identity. These marks provided higher enrichment (i.e. lower P values compared to those expected by chance) of cancer specific-risk SNPs than binding profiles of TFs involved in cancer onset (26). By confirming a relation between enhancer activity and CCVs, these results bridge epigenetic and genetic fields for cancer risk assessment.
The consequences of risk-associated genetic variants on gene transcription control are multiple; however, all the mechanisms involved seem to alter chromatin interactions. There might be changes in chromatin-TF interactions as revealed for FOXA1, at distal regulatory elements, which leads to allele-specific gene expression (27). FOXA1 is labeled as pioneer TF since it can interact with compact DNA and make it accessible to other factors. Here, the cistromes of FOXA1 were enriched with risk-associated SNPs. FOXA1 was also among a cluster of four risk-TF that share regulatory mechanisms and would favor ER+ tumors (28). Such mechanistic studies have required new computational methodologies with the combination of epigenomics and genotype imputation with cistromics (i.e., the investigation of cis-acting targets of trans-acting factors), as well as grouping putative target genes of TF into regulons and assessing enrichments in risk loci to identify regulatory clusters. The impact of SNPs has also been abundantly identified for single genes. For instance, SNPs associated with a decrease in risk were identified within the promoter of TERT where they reduce promoter activity, without the involvement of distant regulatory elements (29). However, CCVs often modulate DNA looping. Locus 8p12 CCVs of the enhancer of the tumor suppressor gene DUSP4 either reduce its activity via looping to the gene's promoter region or prevent looping to the promoter (30).
SNPs associated with breast cancer risk are also found in the distribution maps of H3K4me1, a histone modification found in regulatory regions of genes (27). A similar observation has been made for many diseases, hence confirming that an alteration of enhancer function is one of the mechanisms associated with genetic risk.
Other categories of genes affected by variants associated with cancer risk include DNA integrity checkpoints, which would trigger further loss of homeostasis. For instance, SNPs in a distal enhancer of the intergenic region 11q13 decrease the expression of estrogen-regulated long noncoding (lnc)RNAs CUPID1&2 by reducing chromatin looping between the enhancer and the gene bidirectional promoter. Normally, these lncRNAs are regulating the choice of pathways used for double-strand break repair (31).
It is not surprising that SNPs impact transcription control, these alterations residing mostly in regulatory regions of the DNA. However, the accumulating evidence that SNPs are related to downstream modifications in a cell type specific manner, as shown by the examples of FOXA1, BRD4, H3K4me1 and DNA methylation supports the essential role that the epigenome might play in cancer risk.
Epigenetic marks of risk
DNA methylation at specific CpGs is envisioned as a source for risk markers since these alterations might be found in circulating DNA. However, it is important to establish that these methylation patterns exist in the breast at risk for cancer. Nested case-control studies from a prospective cohort of patients with ER+ breast cancer revealed that methylation changes in the CpGs of genes in the normal appearing epithelium mapped metabolic processes linked to fatty acids, although there was no difference in global methylation between cases (cancer developed a few years later in the contralateral breast) and controls. Three genes with methylation changes were validated in breast tissue and blood and might provide markers to improve risk stratification for the development of sporadic breast cancer (32). It was also observed that women at higher risk for breast cancer based on the Gail model are more likely to present methylation of tumor suppressor genes APC and RASSF1 in the breast tissue (33). The interest in a possible marker may be strengthened by additional studies that pinpoint this marker under different conditions, as shown for APC, since the possible protection of increasing parity is associated with decreased methylation of this gene in the breast (34). Like with SNPs, epigenetic modifications that influence DNA integrity may contribute to cancer risk. For instance, hypermethylation of the ATM gene detected in the blood stream was associated with breast cancer risk and proposed to be included in risk stratification studies (35).
The partnership between epigenetic modifications and SNPs highlighted by integrative bioinformatics analysis is useful to further refine cancer risk. For instance, the effect of SNPs on methylation may be defined as methylation quantitative traits loci (36). This method was used to identify prognostic gene signatures in breast cancer with the aim of reducing false positives via multiple sources pointing to the same genes. These CpG-SNP pairs might be particularly useful in risk stratification for primary prevention approaches.
From a perspective standpoint, the combination of genetic variants and epigenetic marks might be particularly useful for risk information on specific cancer subtypes. In a study with women of European and African ancestry, 53 genetic variants were associated with ER+ breast cancer and 37 variants were associated with ER− cancers (25). Risk variants found in exons of multi-exonic ncRNA genes have been linked to specific cancer subtypes (37). Some of these ncRNAs are expressed depending on risk variants, and the promoters of many of these RNAs loop to regions that contain risk variants, which suggests that genetic variants control these RNAs. The inclusion of epigenetic reader BRD4, that appears to identify risk loci and is cell type specific (26), might enable the identification of the cellular origin of risk for cancer subtypes and open new directions for epigenetic intervention on select cell populations (38). Overall, the flexibility of the epigenome makes it an attractive compartment to identify mechanisms by which specific factors lead to risk escalation for different cancer subtypes.
EPIGENETIC ESCALATION OF RISK
The multiplicity of epigenetic modifications occurring within higher order chromatin organization suggests the possibility of an epigenetic escalation of risk. Timing is particularly important in risk escalation for breast cancer, with windows of susceptibility that account for acute exposure leading to increased risk or protection over a defined period of life (e.g. childhood, pregnancy, menopause) and through chronic impact (e.g. hormonal, aging). Thus, exposure to environmental factors and lifestyle will have distinct influences throughout life.
Lifelong risk of aging
Aging is the strongest risk factor that is likely responsible for the majority of postmenopausal breast cancers. It is promoted in part by an imbalance in anti- and pro-oxidative factors leading to chronic oxidative stress (21). Differences in breast aging, in light of its epigenome, compared to the person's chronological age have been linked with heightened breast cancer risk (20,39). The epigenetic clock is based on DNA methylation at 353 CpG sites calculated by elastic net regression or 385 CpG sites giving an estimate of mitotic division (9,39). Intrinsic epigenetic age acceleration (i.e. the difference between epigenetic [biological] and chronological ages) is the risk factor associated with aging; its elevation by one unit has been linked with a 4% increase odds of developing breast cancer, with age acceleration detected 10 years prior to diagnosis (20). Logically, epigenetic aging interferes with the addition and the removal of methyl groups and it is likely to modify the impact of other influential factors.
Aside from the epigenetic clock, DNA methylation in breast tissues of aging women has been shown to be enriched at enhancer regions and binding sites for chromatin remodelers Myc and CTCF (40). Moreover, hypomethylation represented 30.7% of the modified CpG sites and was identified primarily at binding sites for transcriptional activators c-Fos and Stat-3. The CpG regions in tissues of aging women only partially overlapped with those used to estimate tissue age (only 3 and 17 of the 787 CpGs identified in this study were found in the two epigenetic clock CpG lists), demonstrating a discrepancy between chronological (person's age) and biological (tissue age) that might be important for disease risk. The fact that these CpG regions were further modulated in cancer led to the proposition that they are involved in risk (40). This study performed on a limited number of individuals (100) highlights the preponderance of aging in DNA methylation changes associated with breast cancer risk. Strikingly, no CpG sites were significantly modified for their methylation level in relation to BMI, parity and family history that are factors involved in risk modulation. Whether these other risk factors act on different epigenetic pathways will be important to determine for risk escalation associated to the combination of different risk factors.
Accelerated tissue aging may have consequences on the impact of TFs since methylation usually prevents their binding to DNA (41). Moreover, the expression of TF changes with age hence, so does their binding to regulatory SNPs. Age-interacting SNPs have been analyzed via a novel SNiPAge concept to better understand age-associated phenotypes (9). In the breast, 536 interaction triplets (TF-SNP-gene) linked to aging were detected based on TFs that show reduction in expression with age. Overall, the SNPs identified were significantly related to breast cancer and located in open chromatin, often in regulatory regions like enhancers and promoters, as shown by epigenetic marks H3K27ac and H3K4me3, respectively. Interestingly, these SNPs were enriched in regions that negatively regulate the stress response, highlighting a potential means for the accumulation of alterations leading to increased risk, since an altered response to stress decreases DNA repair capabilities.
The participation of SNPs in premature aging and breast cancer onset is not a new observation. In 2007, it was reported that one SNP as well as combinations of multiple SNPs in the WRN gene controlling life span and aging were associated with breast cancer risk. Very interestingly, the level of significance increased if there was a longer time between onset of menarchy and first pregnancy, which confirms the additive effects of two different risk factors (here aging and a reproductive pattern that influences estrogen impact) (42). Also, a rare homozygous CC genotype in WRN was associated with increased breast cancer risk in premenopausal women (43).
SNPs associated with aging and their biological functions appear to include DNA methylation that is an essential modulator of tumorigenesis (9). However, the exact contribution of these SNPs to epigenetic aging remains to be understood. In the case of the classical WRN gene alteration, differentially methylated regions are different from those identified in epigenetic aging. These regions are enriched in genes that control transcription factor activity and sequence-specific DNA binding to promoters, possibly leading to transcriptional misregulation (44). Thus, instead of furthering standard epigenetic aging, SNPs might bring cells further away from their normal homeostasis via epigenetic changes that complement those linked to accelerated aging.
Epigenetic risk and life events
The nongenetic factors that contribute to risk stratification according to epidemiologists encompass BMI, reproductive history and mammographic density (45). These factors might have a determinant impact on a specific period in life, and all appear to induce epigenetic changes. Here we are discussing these factors in light of the major physiological disturbances with which they are associated, namely metabolic disturbances (e.g. pro-inflammatory, pro-oxidative).
Diet and metabolism
The metabolism has been implicated in the development of cancers, including breast cancer and metabolic pathways are modulated by aging, notably via the relation TF-SNPs (9). High BMI is both the result and a source of metabolic imbalance leading to pro-inflammatory and oxidative conditions (21); it acts as a breast cancer risk inducer among postmenopausal women and a mediator of aggressive breast cancers among premenopausal women. High BMI has been associated with DNA methylation changes in breast tissue and blood (46,47). Moreover, free fatty acids linked to obesity influence ERα and mTOR pathways, which could impact gene transcription (48). Importantly, an increase in BMI appears to precipitate epigenetic aging as shown for the liver (49) and can be related to an increase in age based on DNA methylation in breast tissues (50). LINE-1 hypomethylation has been proposed to mediate more than 80% of the effects of BMI on breast cancer risk (51). Thus, high metabolic disturbance associated with BMI might contribute to loss of normal epigenetic balance via both the accumulation of epigenetic aging alterations and specific changes in transcription control.
In addition to metabolic disorders, specific components of diets have been studied early on for their impact on the epigenome or at least their relation to epigenetic changes (52); however, attempting to link the degree of methylation of CpGs, nutrient level and breast cancer risk is a difficult task. The threshold at which the concentration of the nutrient studied has biological relevance for cancer risk, as it was discussed for Vitamin D, is paramount to determine. Such considerations are essential for elements like the vitamin D/VDR complex known to control gene transcription involved in cancer risk and the immune system (53) and for which there is still a debate regarding its exact role in breast cancer risk.
Parity and inflammation
Certain diets and environmental stresses like pollutants have a proinflammatory effect in the tissue microenvironment that is considered abnormal and the source of disorders like cancers. However, a natural consequence of parity in the breast is the elevation of the expression of genes related to inflammation (54). This inflammatory effect is limited in time, but a slight increase in breast cancer risk has been documented within two years following pregnancy. The epigenetic origin of this phenomenon is being progressively unraveled. Genes active during cancer development and coding for proteases MMP9 and calpains are targeted by p65/p300 during mammary gland involution (55). The p65/p300 complex is associated with the NF-kappaB inflammatory pathway, the activity of which has been known to be tightly controlled by the balance between histone acetyl transferases and histone deacetylases (56). In addition to modifications in the ECM, Calpain 1 modulates preadipocytes through chromatin remodeling, which suggests a profound impact on tissues that control mammary homeostasis. However, parity is also associated with hypermethylation of CpG islands of FOXA1 (57), which might account for the ‘delayed’ protective effects of increasing parity. The TF FOXA1 facilitates chromatin binding of ERα and downstream transcriptional activation. Thus, it is envisioned that by favoring epigenetic silencing of FOXA1, parity would attenuate the estrogen impact on the mammary gland and decrease breast cancer risk over a long period.
Breast density and ECM stiffness
High mammographic density has been recognized as an important element for risk stratification, and SNPs associated with increased breast density and breast cancer risk have been identified in GWAS meta-analysis (58). High mammographic density is partially linked to increased ECM stiffness (via stromal collagen (59), which is one of the consequences of an inflammatory condition (60,61). Thus, breast density is related to epigenetic mechanisms of risk via inflammatory pathways. Few studies have been performed on the direct epigenetic impact of ECM stiffness. However, 3D cell culture revealed an effect on chromatin organization, notably via nuclear wrinkling and increased heterochromatin bundles at the nuclear periphery (8). There was also a significant decrease in acetylated H4 and an increase in acetylated H3. The change in nuclear morphometry was related to histone deacetylase (HDAC) activity. Increased stiffness also induced chromatin opening, rendering regulatory elements in the genome more accessible to certain TFs, and primarily Sp1, a regulator of malignant transformation and interactor of chromatin modifying enzymes like class I HDACs 3 and 8 (8). This study brings another dimension to epigenetic risk escalation via a global reorganization of chromatin that affects certain categories of TFs. It is expected that a change of this magnitude would alter the impact on the epigenome of any additional risk factor, especially since ECM stiffness is influenced early and later in life by pollutants and nutrition, respectively (62,63).
Overall, many of the risk factors may be related to central metabolic pathways in cells like inflammatory and oxidative pathways for which epigenetic mechanisms are being progressively understood. Nevertheless, other effects specific to each risk factor ought to be studied for a possible deleterious influence on the epigenome that would add to that of inflammatory and oxidative pathways.
CONCLUSIONS
Placing the information highlighted in this short-review in perspective, risk escalation depends on timing of exposure and the combination of genetic and epigenetic alterations. It is recognized that prepuberty is a window of susceptibility, notably because of the epigenetic status of the mammary cells that are primed but not fully differentiated, making them more susceptible to undergo protective or deleterious permanent epigenetic changes. Risk escalation might also depend on ancestry. Many risk loci are common among women of European, Asian and African ancestry (25). However, only a small portion of variants common to African and European ancestries have the same directionality, and in the same risk regions, variants with significance in terms of risk might be different (25). Moreover, increased epigenetic aging has been associated with race (40). Ethnic difference in age-associated diseases is suggested by the genomic proximity of ethnicity-associated SNPs and SNPs linked with aging (9). Whether these relationships could explain differences in thresholds for the effective impacts of nutrients and BMI levels on risk among ethnic groups remains to be determined.
Measurable markers are necessary for risk stratification. A multiparameter, medical biology approach to risk stratification should combine genetic and epigenome-related markers, since reaching a risk level necessary for tumor onset is likely to entail the accumulation of nuclear changes. We propose that risk escalation in the cell nucleus encompasses the reciprocal influence of genetic and epigenetic alterations on DNA integrity and the targeting of different features of chromatin organization (Figure 1).
Figure 1.

Cancer risk escalation in the cell nucleus. It is essential to study changes in the epigenome and its related parameters that are associated with cancer risk in the normal tissue of origin. For the breast, the luminal epithelium is organized as a monolayer (internal to the layer of myoepithelial cells- not shown) within which cell heterogeneity is represented by different phenotypes and the presence of a small percentage of progenitor cells. These cells have a different organization of chromatin and its epigenetic content that might be detectable via a different shape and size of the nucleus. The source of this tissue might be from reduction mammoplasty (left) or from biopsies from normal-appearing tissue adjacent to tumors (right). The association of certain epigenetic alterations with risk is sometime confirmed because of an increase or an extension of these alterations in the same DNA regions as cancer develops and progresses. The breast epithelium may be reproduced in 3D cell culture to enable the mechanistic investigation of risk factors. The nucleus (pale blue magnified circle) of an epithelial cell at risk may display different types of chromatin alterations, like DNA methylation at CpG sites (often increased), changes in chromatin loops and anchorage/condensation. The presence of SNPs associated with risk at DNA regulatory regions (e.g. super enhancers marked by BRD4, TF binding regions) modifies gene transcription via a direct impact on ligand binding or through changes in chromatin looping. Depending on the risk factors (red bubbles) one or more effect on chromatin has been identified so far such as changes in DNA methylation, TF binding or expression, and chromatin compaction. Importantly, risk factors may also feed into others’ effects and could strengthen the extent of alterations as shown by the dashed red arrows. There are at least three possible measurable means (written in blue) to build up risk and that could help identify epigenome-related thresholds to cancer onset: DNA methylation profiles, genetic alterations and gene expression profiles. Moreover, SNPs and DNA methylation influence each other leading to the accumulation of genetic alterations. As illustrated in the ‘risk escalation’ box, how the risk is built in the chromatin is likely to influence the speed at which the epigenome loses control of normal differentiation and the resulting subtype of cancer. The blood stream is an important compartment to routinely measure cancer risk because of the presence of circulating DNA, ncRNAs and exosomes. Circulating DNA and ncRNAs might be specific or nonspecific indicators of risk, whereas exosomes might be traceable to a specific tissue at risk. The combination of different measurable parameters might be necessary to identify a risk of cancer in a specific tissue and how the risk was built (i.e., the epigenetic pathways altered, the type of cells involved) so that interventions may be tailored to the origin(s) of the increase in risk.
The essential participation of the epigenome in risk escalation is linked to the possibility of increasing or decreasing the degree of DNA methylation at CpGs of regulatory regions depending on risk and protective factors. Moreover, in light of studies with the type of microenvironmental alterations that the risk factors generate, changes in gene expression leading to tumor onset rely heavily on DNA methylation, as well as alterations in DNA looping and chromatin packing.
Many more investigations are necessary to understand how the combination of major risk factors builds up genetic and epigenetic alterations, not only to identify risk markers but also to design targeted interventions to decrease cancer risk with a precision prevention approach. For instance, knowing the specific epigenetic pathway responsible for changes in DNA methylation is paramount to apply proper preventive treatment (17).
Models for such investigations need to be carefully selected. The organization of the epigenome of the noncancerous cells of the tissue of origin appears to determine the mutation patterns of cancers for this tissue (64). This finding means that there is a continuity in the accumulation of risk from cancer onset to progression via an interplay between the genome and the epigenome. It also suggests that it is paramount for in vitro models of cancer risk with human cells to reproduce proper tissue architecture since it is responsible for higher order chromatin organization (22). These models ought to include the type of cells (e.g. luminal progenitor) and the differentiation stages corresponding to specific windows of risk susceptibility, since these elements appear essential to determine the types of breast cancer (at least ER– or ER+) (65) for which there are also corresponding SNPs and epigenetic changes (66).
Future investigations ought to determine how epigenome-related parameters specific for an increased risk in a specific organ might be measurable in blood samples. The organization of the epigenome appears specific to each tissue; thus, tissue-specific risk markers, if present in the blood through free DNA or exosomes, are within reach.
DATA AVAILABILITY
There are no unpublished data from the author reported in this short-review.
ACKNOWLEDGEMENTS
Due to limitations in the number of references allowed, the author apologizes if additional important citations are missing. SL is part of the International Breast Cancer & Nutrition (IBCN) network.
FUNDING
Pays-de-la-Loire Connect Talent to SL.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lelièvre S.A., Bellanger M., Seewaldt V., Talhouk R.S., Terry M.B. Perspectives in primary prevention research for breast cancer: a focus on gene-environment interactions. Front. Med. (Lausanne). 2020; 7:621959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hüsing A., Fortner R.T., Kühn T., Overvad K., Tjønneland A., Olsen A., Boutron-Ruault M.C., Severi G., Fournier A., Boeing H. et al. Added value of serum hormone measurements in risk prediction models for breast cancer for women not using exogenous hormones: results from the EPIC cohort. Clin. Cancer Res. 2017; 23:4181–4189. [DOI] [PubMed] [Google Scholar]
- 3. Mavaddat N., Pharoah P.D.P., Michailidou K., Tyrer J., Brook M.N., Bolla M.K., Wang Q., Dennis J., Dunning A.M., Shah M. et al. Prediction of breast cancer risk based on profiling with common genetic variants. Natl. Cancer Inst. 2015; 107:djv036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee A., Mavaddat N., Wilcox A.N., Cunningham A.P., Carver T., Hartley S., Babb de Villiers C., Izquierdo A., Simard J., Schmidt M.K. et al. BOADICEA: a comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 2019; 21:1708–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mavaddat N., Michailidou K., Dennis J., Lush M., Fachal L., Lee A., Tyrer J.P., Chen T.H., Wang Q., Bolla M.K. et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 2019; 104:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang P., Kitchen-Smith I., Xiong L., Stracquadanio G., Brown K., Richter P.H., Wallace M.D., Bond E., Sahgal N., Moore S. et al. Germline and somatic genetic variants in the p53 pathway interact to affect cancer risk, progression, and drug response. Cancer Res. 2021; 81:1667–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brentnall A.R., van Veen E.M., Harkness E.F., Rafiq S., Byers H., Astley S.M., Sampson S., Howell A., Newman W.G., Cuzick J. et al. A case-control evaluation of 143 single nucleotide polymorphisms for breast cancer risk stratification with classical factors and mammographic density. Int. J. Cancer. 2020; 146:2122–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stowers R.S., Shcherbina A., Israeli J., Gruber J.J., Chang J., Nam S., Rabiee A., Teruel M.N., Snyder M.P., Kundaje A. et al. Matrix stiffness induces a tumorigenic phenotype in mammary epithelium through changes in chromatin accessibility. Nat. Biomed. Eng. 2019; 3:1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang K., Basu M., Malin J., Hannenhalli S. A transcription-centric model of SNP-age interaction. PLoS Genet. 2021; 17:e1009427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weaver V.M., Petersen O.W., Wang F., Larabell C.A., Briand P., Damsky C., Bissell M.J. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J. Cell Biol. 1997; 137:231–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang F., Weaver V.M., Petersen O.W., Larabell C.A., Dedhar S., Briand P., Lupu R., Bissell M.J. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:14821–14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bissell M.J., Weaver V.M., Lelièvre S.A, Wang F., Petersen O.W., Schmeichel K.L. Tissue structure, nuclear organization, and gene expression in normal and malignant breast. Cancer Res. 1999; 59:1757–1763s. [PubMed] [Google Scholar]
- 13. Lelièvre S.A., Chittiboyina S. Microphysiological systems to study microenvironment-cell nucleus interaction: importance of tissue geometry and heterogeneity. Microphysiol. Syst. 2018; 2:12. [Google Scholar]
- 14. Plachot C., Lelièvre S.A. DNA methylation control of tissue polarity and cellular differentiation in the mammary epithelium. Exp. Cell Res. 2004; 298:122–132. [DOI] [PubMed] [Google Scholar]
- 15. Tehranchi A.K., Myrthil M., Martin T., Hie B.L., Golan D., Fraser H.B Pooled ChIP-Seq links variation in transcription factor binding to complex disease risk. Cell. 2016; 165:730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ogino S., Lochhead P., Chan A.T., Nishihara R., Cho E., Wolpin B.M., Meyerhardt J.A., Meissner A., Schernhammer E.S., Fuchs C.S. et al. Molecular pathological epidemiology of epigenetics: emerging integrative science to analyze environment, host, and disease. Mod. Pathol. 2013; 26:465–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duforestel M., Nadaradjane A., Bougras-Cartron G., Briand J., Olivier C., Frenel J.S., Vallette F.M., Lelièvre S.A., Cartron P.F. Glyphosate primes mammary cells for tumorigenesis by reprogramming the epigenome in a TET3-dependent manner. Front. Genet. 2019; 10:885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kresovich J.K., Xu Z., O’Brien K.M., Shi M., Weinberg C.R., Sandler D.P., Taylor J.A. Blood DNA methylation profiles improve breast cancer prediction. Mol Oncol. 2021; https://febs.onlinelibrary.wiley.com/doi/pdf/10.1002/1878-0261.13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gillman A.S., Helmuth T., Koljack C.E., Hutchison K.E., Kohrt W.M., Bryan A.D. The effects of exercise duration and intensity on breast cancer-related DNA methylation: a randomized controlled trial. Cancers (Basel). 2021; 13:4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ambatipudi S., Horvath S., Perrier F., Cuenin C., Hernandez-Vargas H., Le Calvez-Kelm F., Durand G., Byrnes G., Ferrari P., Bouaoun L. et al. DNA methylome analysis identifies accelerated epigenetic ageing associated with postmenopausal breast cancer susceptibility. Eur. J. Cancer. 2017; 75:299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chittiboyina S., Bai Y., Lelièvre S.A. Microenvironment-cell nucleus relationship in the context of oxidative stress. Front. Cell Dev. Biol. 2018; 6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chandramouly G., Abad P.C., Knowles D.W., Lelièvre S.A. The control of tissue architecture over nuclear organization is crucial for epithelial cell fate. J. Cell Sci. 2007; 120:1596–1606. [DOI] [PubMed] [Google Scholar]
- 23. Fachal L., Aschard H., Beesley J., Barnes D.R., Allen J., Kar S., Pooley K.A., Dennis J., Michailidou K., Turman C. et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat. Genet. 2020; 52:56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beesley J., Sivakumaran H., Moradi Marjaneh M., Lima L.G., Hillman K.M., Kaufmann S., Tuano N., Hussein N., Ham S., Mukhopadhyay P. et al. Chromatin interactome mapping at 139 independent breast cancer risk signals. Genome Biol. 2020; 21:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feng Y., Rhie S.K., Huo D., Ruiz-Narvaez E.A., Haddad S.A., Ambrosone C.B., John E.M., Bernstein L., Zheng W., Hu J.J. et al. Characterizing genetic susceptibility to breast cancer in women of African ancestry. Cancer Epidemiol. Biomarkers Prev. 2017; 26:1016–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zuber V., Bettella F., Witoelar A.PRACTICAL Consortium CRUK GWAS BCAC Consortium TRICL Consortium PRACTICAL Consortium CRUK GWAS BCAC Consortium TRICL Consortium Andreassen O.A., Mills I.G., Urbanucci A. Bromodomain protein 4 discriminates tissue-specific super-enhancers containing disease-specific susceptibility loci in prostate and breast cancer. BMC Genomics. 2017; 18:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cowper-Sal lari R., Zhang X., Wright J.B., Bailey S.D., Cole M.D., Eeckhoute J., Moore J.H., Lupien M. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nat. Genet. 2012; 44:1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Castro M.A., de Santiago I., Campbell T.M., Vaughn C., Hickey T.E., Ross E., Tilley W.D., Markowetz F., Ponder B.A., Meyer K.B. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat. Genet. 2016; 48:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helbig S., Wockner L., Bouendeu A., Hille-Betz U., McCue K., French J.D., Edwards S.L., Pickett H.A., Reddel R.R., Chenevix-Trench G. et al. Functional dissection of breast cancer risk-associated TERT promoter variants. Oncotarget. 2017; 8:67203–67217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Glubb D.M., Shi W., Beesley J., Fachal L., Pritchard J.L., McCue K., Barnes D.R., Antoniou A.C., Dunning A.M., Easton D.F. et al. Candidate causal variants at the 8p12 breast cancer risk locus regulate DUSP4. Cancers (Basel). 2020; 12:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Betts J.A., Moradi Marjaneh M., Al-Ejeh F., Lim Y.C., Shi W., Sivakumaran H., Tropée R., Patch A.M., Clark M.B., Bartonicek N. et al. Long noncoding RNAs CUPID1 and CUPID2 mediate breast cancer risk at 11q13 by modulating the response to DNA damage. Am. J. Hum. Genet. 2017; 101:255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ennour-Idrissi K., Dragic D., Issa E., Michaud A., Chang S.L., Provencher L., Durocher F., Diorio C. DNA methylation and breast cancer risk: an epigenome-wide study of normal breast tissue and blood. Cancers (Basel). 2020; 12:3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lewis C.M., Cler L.R., Bu D.W., Zöchbauer-Müller S., Milchgrub S., Naftalis E.Z., Leitch A.M., Minna J.D., Euhus D.M. Promoter hypermethylation in benign breast epithelium in relation to predicted breast cancer risk. Clin. Cancer Res. 2005; 11:166–172. [PubMed] [Google Scholar]
- 34. Euhus D.M., Bu D., Milchgrub S., Xie X.J., Bian A., Leitch A.M., Lewis C.M. DNA methylation in benign breast epithelium in relation to age and breast cancer risk. Cancer Epidemiol. Biomarkers Prev. 2008; 17:1051–1059. [DOI] [PubMed] [Google Scholar]
- 35. Tang Q., Cheng J., Cao X., Surowy H., Burwinkel B. Blood-based DNA methylation as biomarker for breast cancer: a systematic review. Clin. Epigenetics. 2016; 8:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shilpi A., Bi Y., Jung S., Patra S.K., Davuluri R.V. Identification of genetic and epigenetic variants associated with breast cancer prognosis by integrative bioinformatics analysis. Cancer Inform. 2017; 16:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moradi Marjaneh M., Beesley J., O’Mara T.A., Mukhopadhyay P., Koufariotis L.T., Kazakoff S., Hussein N., Fachal L., Bartonicek N., Hillman K.M. Non-coding RNAs underlie genetic predisposition to breast cancer. Genome Biol. 2020; 21:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Casey A.E., Sinha A., Singhania R., Livingstone J., Waterhouse P., Tharmapalan P., Cruickshank J., Shehata M., Drysdale E., Fang H. et al. Mammary molecular portraits reveal lineage-specific features and progenitor cell vulnerabilities. J. Cell Biol. 2018; 217:2951–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson K.C., Houseman E.A., King J.E., Christensen B.C. Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res. 2017; 19:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maurano M.T., Wang H., John S., Shafer A., Canfield T., Lee K., Stamatoyannopoulos JA. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep. 2015; 12:1184–1195. [DOI] [PubMed] [Google Scholar]
- 42. Ding S.L., Yu J.C., Chen S.T., Hsu G.C., Shen C.Y. Genetic variation in the premature aging gene WRN: a case-control study on breast cancer susceptibility. Cancer Epidemiol. Biomarkers Prev. 2007; 16:263–269. [DOI] [PubMed] [Google Scholar]
- 43. Zins K., Frech B., Taubenschuss E., Schneeberger C., Abraham D., Schreiber M. Association of the rs1346044 polymorphism of the Werner syndrome gene RECQL2 with increased risk and premature onset of breast cancer. Int. J. Mol. Sci. 2015; 16:29643–29653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maierhofer A., Flunkert J., Oshima J., Martin G.M., Poot M., Nanda I., Dittrich M., Müller T., Haaf T. Epigenetic signatures of Werner syndrome occur early in life and are distinct from normal epigenetic aging processes. Aging Cell. 2019; 18:e12995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shieh Y., Scott C.G., Jensen M.R., Norman A.D., Bertrand K.A., Pankratz V.S., Brandt K.R., Visscher D.W., Shepherd J.A., Tamimi R.M. et al. Body mass index, mammographic density, and breast cancer risk by estrogen receptor subtype. Breast Cancer Res. 2019; 21:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wilson L.E., Harlid S., Xu Z., Sandler D.P., Taylor J.A. An epigenome-wide study of body mass index and DNA methylation in blood using participants from the Sister Study cohort. Int. J. Obes. (Lond). 2017; 41:194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hair B.Y., Xu Z., Kirk E.L., Harlid S., Sandhu R., Robinson W.R., Wu M.C., Olshan A.F., Conway K., Taylor J.A. et al. Body mass index associated with genome-wide methylation in breast tissue. Breast Cancer Res. Treat. 2015; 151:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Madak-Erdogan Z., Band S., Zhao Y.C., Smith B.P., Kulkoyluoglu-Cotul E., Zuo Q., Santaliz Casiano A., Wrobel K., Rossi G., Smith R.L. et al. Free fatty acids rewire cancer metabolism in obesity-associated breast cancer via estrogen receptor and mTOR signaling. Cancer Res. 2019; 79:2494–2510. [DOI] [PubMed] [Google Scholar]
- 49. Horvath S., Erhart W., Brosch M., Ammerpohl O., von Schönfels W., Ahrens M., Heits N., Bell J.T., Tsai P.C., Spector T.D. et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:15538–15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sehl M.E., Henry J.E., Storniolo A.M., Horvath S., Ganz P.A. The effects of lifetime estrogen exposure on breast epigenetic age. Cancer Epidemiol. Biomarkers Prev. 2021; 30:1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Awada Z., Bouaoun L., Nasr R., Tfayli A., Cuenin C., Akika R., Boustany R.M., Makoukji J., Tamim H., Zgheib N.K. et al. LINE-1 methylation mediates the inverse association between body mass index and breast cancer risk: a pilot study in the Lebanese population. Environ. Res. 2021; 197:111094. [DOI] [PubMed] [Google Scholar]
- 52. Teegarden D., Romieu I., Lelièvre S.A. Redefining the impact of nutrition on breast cancer incidence: is epigenetics involved. Nutr. Res. Rev. 2012; 25:68–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. O’Brien K.M., Sandler D.P., Xu Z., Kinyamu H.K., Taylor J.A., Weinberg C.R. Vitamin D, DNA methylation, and breast cancer. Breast Cancer Res. 2018; 20:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Asztalos S., Gann P.H., Hayes M.K., Nonn L., Beam C.A., Dai Y., Wiley E.L., Tonetti D.A. Gene expression patterns in the human breast after pregnancy. Cancer Prev. Res. (Phila.). 2010; 3:301–311. [DOI] [PubMed] [Google Scholar]
- 55. Zaragozá R., García-Trevijano E.R., Lluch A., Ribas G., Viña J.R. Involvement of Different networks in mammary gland involution after the pregnancy/lactation cycle: Implications in breast cancer. IUBMB Life. 2015; 67:227–238. [DOI] [PubMed] [Google Scholar]
- 56. Choi K.C., Jung M.G., Lee Y.H., Yoon J.C., Kwon S.H., Kang H.B., Kim M.J., Cha J.H., Kim Y.J., Jun W.J. et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009; 69:583–592. [DOI] [PubMed] [Google Scholar]
- 57. Ghosh S., Gu F., Wang C.M., Lin C.L., Liu J., Wang H., Ravdin P., Hu Y., Huang T.H., Li R. Genome-wide DNA methylation profiling reveals parity-associated hypermethylation of FOXA1. Breast Cancer Res. Treat. 2014; 147:653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sieh W., Rothstein J.H., Klein R.J., Alexeeff S.E., Sakoda L.C., Jorgenson E., McBride R.B., Graff R.E., McGuire V., Achacoso N. et al. Identification of 31 loci for mammographic density phenotypes and their associations with breast cancer risk. Nat. Commun. 2020; 11:5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huo C.W., Chew G., Hill P., Huang D., Ingman W., Hodson L., Brown K.A., Magenau A., Allam A.H., McGhee E. et al. High mammographic density is associated with an increase in stromal collagen and immune cells within the mammary epithelium. Breast Cancer Res. 2015; 17:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sturgeon K.M., Schweitzer A., Leonard J.J., Tobias D.K., Liu Y., Cespedes Feliciano E., Malik V.S., Joshi A., Rosner B., De Jonghe B.C. Physical activity induced protection against breast cancer risk associated with delayed parity. Physiol. Behav. 2017; 169:52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sun X., Glynn D.J., Hodson L.J., Huo C., Britt K., Thompson E.W., Woolford L., Evdokiou A., Pollard J.W., Robertson S.A. et al. CCL2-driven inflammation increases mammary gland stromal density and cancer susceptibility in a transgenic mouse model. Breast Cancer Res. 2017; 19:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Binder A.M., Corvalan C., Pereira A., Calafat A.M., Ye X., Shepherd J., Michels K.B. Prepubertal and pubertal endocrine-disrupting chemical exposure and breast density among chilean adolescents. Cancer Epidomiol. Biomarkers Prev. 2018; 27:1491–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mishra G.D., dos Santos Silva I., McNaughton S.A., Stephen A., Kuh D. Energy intake and dietary patterns in childhood and throughout adulthood and mammographic density: results from a British prospective cohort. Cancer Causes Control. 2011; 22:227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Polak P., Karlić R., Koren A., Thurman R., Sandstrom R., Lawrence M., Reynolds A., Rynes E., Vlahoviček K., Stamatoyannopoulos J.A. et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015; 518:360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rotunno M., Sun X., Figueroa J., Sherman M.E., Garcia-Closas M., Meltzer P., Williams T., Schneider S.S., Jerry D.J., Yang X.R. et al. Parity-related molecular signatures and breast cancer subtypes by estrogen receptor status. Breast Cancer Res. 2014; 16:R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ralph D.A., Zhao L.P., Aston C.E., Manjeshwar S., Pugh T.W., DeFreese D.C., Gramling B.A., Shimasaki C.D., Jupe E.R. Age-specific association of steroid hormone pathway gene polymorphisms with breast cancer risk. Cancer. 2007; 109:1940–1948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
There are no unpublished data from the author reported in this short-review.