

Abstract
Danger signals, or damage-associated molecular patterns (DAMPs), instigate mitochondrial innate immune responses wherein Mitochondrial Antiviral Signaling protein (MAVS) functions as a key platform molecule to mediate them. The role of MAVS in the pathogenesis of idiopathic pulmonary fibrosis (IPF), however, has not been identified yet. A possibility whether the MAVS signaling can be modulated by currently existing drugs has not been explored, either. Here, using an established model of pulmonary fibrosis, we demonstrate that MAVS plays as a critical mediator of multiple DAMPs signaling pathways and the consequent lung fibrosis after bleomycin-induced injury in vivo. After bleomycin injury, the expression of MAVS was mainly observed in macrophages. In addition, multimeric MAVS aggregation, a key event of MAVS signaling activation, was significantly increased and persisted in bleomycin-injured lungs. Interestingly, a proapoptotic BH3 mimetic ABT-263 attenuated the expression of MAVS and its signaling and, consequently, the development of experimental pulmonary fibrosis. In contrast, the therapeutic effects of Pirfenidone or Nintedanib, two approved drugs for IPF treatment, were not related to the modulation of MAVS or its signaling. Importantly, multimeric MAVS aggregation was significantly increased in lungs from the patients with IPF as well. In conclusion, MAVS may play an important role in the development of pulmonary fibrosis, and targeting MAVS with BH3 mimetics may provide a novel therapeutic strategy for IPF, a major unmet disorder.
Introduction
Idiopathic pulmonary fibrosis (IPF) is defined as a specific form of chronic, progressive, fibrosing interstitial pneumonia of unknown cause and is characterized by the histopathologic and/or radiologic pattern of usual interstitial pneumonia (UIP) [1]. IPF is a fatal lung disease. Patients of this disorder are destined to suffer an unpredictable decline of lung function with a lower survival rate and often die from respiratory insufficiency within 2–5 years of diagnosis [1–3]. As such, it is a major unmet medical need in human health and requires better mechanistic understanding of pathogenesis to develop novel disease-modifying therapeutics.
Mitochondrial function and behavior have been highlighted to be fundamental to the physiology of cellular and organismal health; consequently, “mitochondrial dysfunction” has been implicated in a wide range of diseases that encompass all aspects of health and disease [4–7]. In accordance with the recent evolution of our understanding of the mitochondrial role in human health, increasing attention has been paid to the functional roles of mitochondrial molecules and their underlying mechanisms by which they may contribute to the pathogenesis of IPF [8–10].
During tissue injury or damage responses, mitochondrial functions are influenced responding to intracellular perturbations. The alteration of mitochondrial functions, in turn, modulates various intracellular signaling in order to execute appropriate cellular functions [11]. Mitochondrial antiviral signaling protein (MAVS) represents such an example and functions as a platform molecule to mediate mitochondrial innate immune signaling [12–14]. The role of MAVS contributing to IPF pathogenesis wherein dysregulated tissue damage responses play an important role, however, has not been identified yet. In addition, a possibility whether the MAVS signaling can be modulated by currently existing drugs in order to offer a novel therapeutic strategy has not been explored, either.
Here, we demonstrate that MAVS plays a critical role in the development of experimental pulmonary fibrosis after bleomycin-induced lung injury in vivo, an established mammalian model of IPF. Bleomycin-induced fibrotic responses and multiple DAMPs signaling pathways were significantly attenuated via a MAVS-dependent manner in murine lungs in vivo. In addition, bleomycin-induced cellular senescence was significantly attenuated in MAVS-deficiency. Intriguingly, the BH3 (B-cell lymphoma 2 (Bcl-2) Homology 3) mimetic ABT-263 induced a significant reduction of the MAVS signaling and, consequently, attenuated the development experimental pulmonary fibrosis in our model. In contrast, the therapeutic effects of Pirfenidone or Nintedanib, respectively, the currently approved drugs known to decelerate IPF progression, were not related to the MAVS signaling. Finally, human studies revealed a significant activation of MAVS in lungs from the patients with IPF compared to those of controls. Taken together, these data suggest that MAVS and its mitochondrial innate immune signaling, which can be modulated by BH3 mimetics, is activated and may play an important role in the pathogenesis of IPF.
Materials and Methods
Animals and experimental design
Wild type (WT, Mavs+/+) (from Jackson Laboratories) and Mavs−/− (from Dr. Z. J. Chen, University of Texas) were all kept on C57BL/6J background and bred at Yale University Animal Facility. All animal experiments were approved by the Yale Animal Care and Use Committee (YACUC). An established mammalian model of idiopathic pulmonary fibrosis (IPF) was utilized as described in previous publications [15, 16]. Briefly, to induce a pulmonary fibrosis, mice were treated with bleomycin (Pfizer, #NDC 61703-0332-18) with phosphate buffered saline (PBS) by intra-tracheal administration or oropharyngeal aspiration, respectively. At designated time points after bleomycin administration, mice were euthanized with urethane by intraperitoneally injection, hearts were perfused with PBS, and lungs were harvested for further analyses. For BAL collection, the trachea was cannulated and lavaged two times with 0.9 ml PBS. Samples were centrifuged at 1,500 rpm for 5 min, and the cell-free supernatants were collected for ELISA assay. The cell pellets were recovered in 200 μl sterile PBS, and total cell count of BAL was done using automated cell count using a Coulter Analzer (Beckman Coulter, Brea, CA).
Therapeutic drugs treatment.
To test the therapeutic efficacy of various existing drugs in our modeling of IPF, ABT-263 (40 mg/kg; MedChemExpress), pirfenidone (40 mg/kg; MedChemExpress), nintedanib (40 mg/kg; MedChemExpress) or vehicle controls (5% tween-80), respectively, were administered by intra-peritoneal injection at days 8, 10, 12, 14, 16, 18 and 20 after bleomycin administration, and the mice were sacrificed at day 21.
Human sample preparation
For the evaluation and comparison of MAVS aggregation in the patient with IPF or healthy controls, the lung tissue samples were obtained from Dr. Ivan Rosas’ laboratory. Human lung explants were obtained from donations of patients who sign informed consent and undergo lung transplantation at the Brigham and Women’s Hospital, or donor organs provided by the New England Organ Bank and/or the National Disease Research Interchange (NDRI). The study’s protocol was approved by the Partners Healthcare Institutional Board Review (IRB Protocol # 2011P002419). Briefly, lungs were sliced and washed with cold sterile PBS several times. Visible airway structures, vessels, blood clots and mucin were removed. Tissues were minced mechanically into small pieces (<5 mm) and then incubated for 45 minutes in 37°C within the digestion medium, which consist of 30 U/ml elastase (Elastin Products Company, Owensville, MO), 0.2 mg/ml DNAse I (Sigma, St. Louis, MO), 0.3 mg/ml liberase (Roche, Basel, Switzerland) and 1% Penicillin/Streptomycin diluted in DMEM/F12 medium (Lonza, Basel, Switzerland). Digested tissues were filtered using a metal strainer (Sigma). Unfiltered tissues were incubated a second time in digestion medium for 30 minutes or less, followed by repeat filtration and addition of 10% FBS to stop the enzymatic reaction. Flow-through from both filtrations was combined and centrifuged at 600G, 4°C for 10 minutes. The pellet was resuspended in red cell lysis buffer (VWR, Radnor, PA) for <5 minutes in 37°C and centrifuged again. The pellet was resuspended in DMEM/F12 medium and filtered using a 100 μm strainer (Fisher Scientific, Waltham, MA). Freezing medium (10% FBS and 10% DMSO in DMEM/F12) was then added to the filtrate. Cell suspensions were aliquoted and stored in liquid nitrogen for future SDD-AGE or BN-PAGE applications.
Statistical analysis
All statistical analysis was executed using the Prism (GraphPad, version 6) software program. Comparisons between two groups were performed with Student t test (unpaired). For the multiple comparisons, 2-way ANOVA was used. The significance of survival rate was analyzed with the log-rank test. Values are expressed as mean ± SEM, SD or Min to Max. Statistical significance was defined at a level of P less than 0.05.
Additional experimental methods were described in Supplementary Information; these methods include (a) Sircol assay, (b) Flow cytometric analysis, (c) Histology, immunohistochemistry and immunofluorescence, (d) Isolation of murine alveolar macrophages, (e) ELISA, (f) qRT-PCR, (g) Western blot analysis, (h) Quantification of cell-free double-stranded DNA, (i) Mitochondrial isolation, (j) Blue Native-Polyacrylamide Gel Electrophoresis (BN-PAGE) and Semi-Denaturing Detergent-Agarose Gel Electrophoresis (SDD-AGE), (k) In vitro cell culture, and (l) Isolation and culture of mouse lung fibroblasts and mouse embryonic fibroblasts.
Results
MAVS plays a crucial role in lung fibrosis after bleomycin administration in vivo.
In these studies, we questioned whether MAVS, a novel adaptor molecule of mitochondrial innate immune signaling, may have a functional role in the pathogenesis in a mammalian model of experimental pulmonary fibrosis. When 1.5 unit amount (U)/kilogram of body weight (kg) of bleomycin was administered to male and female mice, respectively, via intra-tracheal (IT) delivery, the bleomycin-induced tissue injury responses revealed striking difference between two groups of C57BL/6J wild type (WT) or MAVS null mutants (−/−) mice. Specifically, all mice from WT controls were dead while 9 of 14 (64%) mice survived among Mavs−/− group (Figure 1a). With 1.0 U/kg of bleomycin dose, more than half of WT mice were dead while all Mavs−/− mice survived (Figure 1b). With 0.4 U/kg of bleomycin dose via IT delivery, we obtained about 3-fold increase of total lung collagen contents, which is comparable to known publications (Figure 1c) [8, 9]. Hence, 0.4 U/kg of bleomycin was administered via IT delivery for all the following in vivo experiments, if not specified. Bleomycin-induced pulmonary fibrosis and loss of body weight, respectively, were significantly ameliorated in the lung from Mavs−/− mice compared to those of WT controls (Figure 1c and d). These observations were confirmed by histologic evaluations (Figure 1e and Figure S1a). A similar level of the reduction of fibrotic response was observed between male and female groups of mice (data not shown). Immunohistochemistry (IHC) and immunofluorescence evaluations demonstrated significant staining of MAVS molecule after bleomycin administration and the staining was observed most prominently in macrophages but not restricted to this cell population (Figure 1f, g and S1a). Flow cytometric evaluation revealed that CD45+F4/80+ cells occupied about 80 % of the lung cells which were stained with anti-MAVS antibody after bleomycin treatment (Figure 1h). The specificity of MAVS staining in our imaging studies as well as our flow cytometric evaluation were presented (Figure S1b and c). The identification of specific cell types and the gating strategy of our flow cytometric evaluation were undertaken following the methods as reported in the literatures (Fig S1d) [17, 18]. To determine whether the bleomycin-induced tissue injury in vivo is altered in the presence or absence of MAVS, the quantification of Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and the evaluation of the active form of caspase-3, a marker of apoptotic cell death, were undertaken in the lungs of WT and MAVS−/− mice. The lungs were obtained at the time point of Day 3 and Day 5, respectively, after the administration of bleomycin in vivo. Indeed, these results revealed that the bleomycin-induced tissue injury in vivo was not attenuated in MAVS deficiency (Fig S1e – g). Overall, these data reveal that MAVS is expressed mainly in pulmonary macrophages and plays an important pathogenic role in a mammalian IPF model.
Figure 1. MAVS plays a crucial role in lung fibrosis after bleomycin administration in vivo.
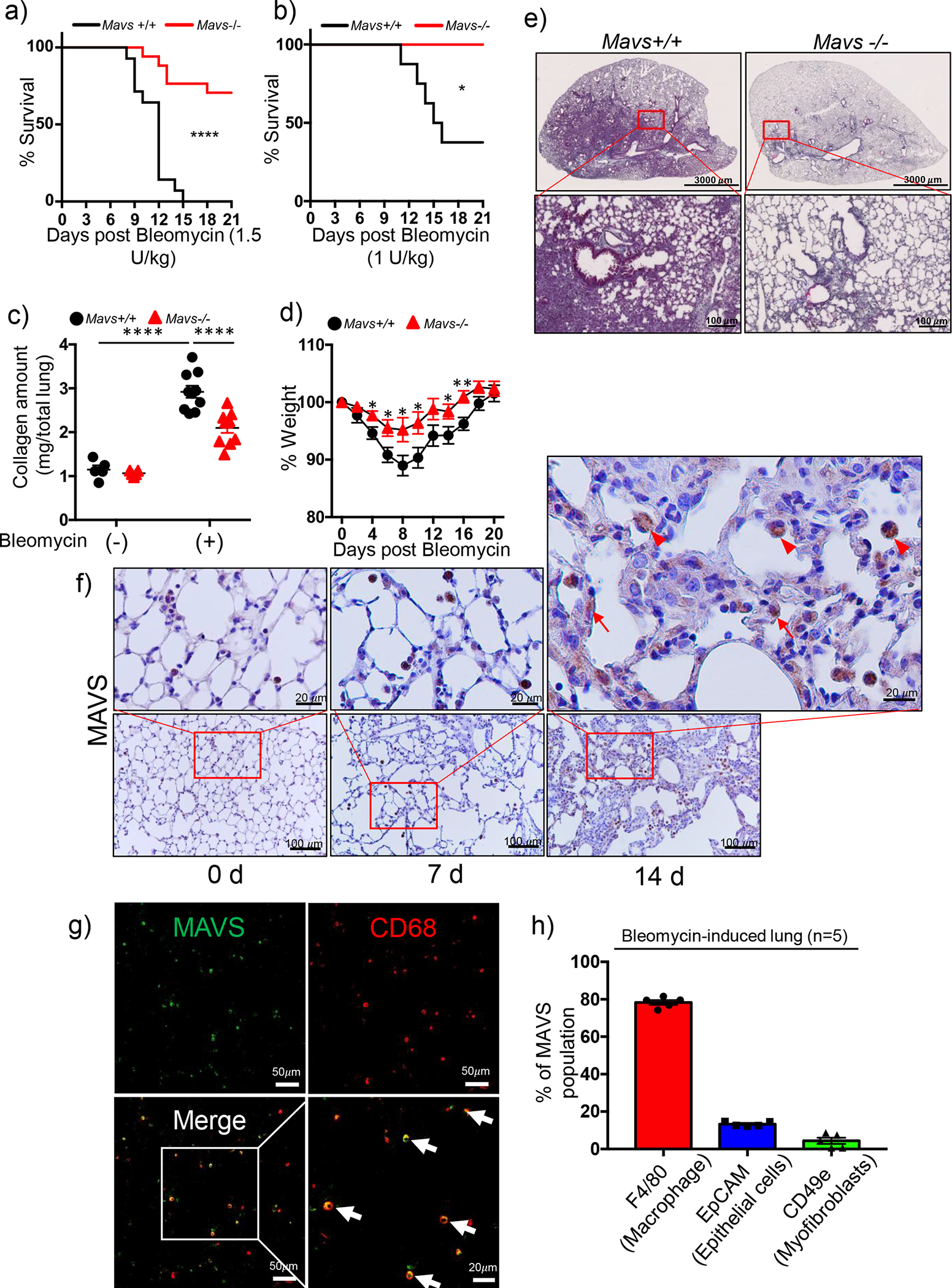
The indicated dose of bleomycin was administered through intra-tracheal (IT) route to C57BL/6J wild type (Mavs+/+) and MAVS null mutant (Mavs−/−) mice. (a and b) Survival rates were monitored during the course of the experiment after (a) 1.5 U/kg (n=14 per each of both groups) and (b) 1.0 U/kg bleomycin administration (n=8 per Mavs+/+, n=7 per Mavs−/− group). (c-g) Saline (bleomycin -) or 0.4 U/kg of bleomycin (+) was administered through IT route to Mavs+/+ and Mavs−/− mice. (c) Collagen contents of total lung tissues obtained from the mice sacrificed at day 21 after bleomycin administration were measured (n=5 per each of both saline groups, n=10 per each of both bleomycin-administered Mavs+/+ and Mavs−/− mice groups, respectively). (d) Body weight changes were monitored during the course of the experiment. (e) Representative histologic findings are presented after Masson’s trichrome staining of collagen on lung sections at day 21 after bleomycin administration (Scale bars: 3,000 μm (top panels), 100 μm (lower panels)). (f) Immunohistochemistry analysis of MAVS on lung sections at indicated time points after saline or bleomycin administration. Representative images of the staining are presented (n=5 per each of the groups). The red boxed regions are magnified for closer observation. Arrows point to alveolar epithelial cells; Arrowheads indicate macrophages. (g) Representative images (Scale bars: 50 μm) from the immunofluorescence analysis of MAVS staining (green) on lung sections at 14 d after bleomycin administration are presented (n=5). CD68 antibody (red) was used for the immunofluorescence staining of macrophages. The white boxed region is magnified for closer observation (Scale bars: 20 μm). (h) The percentage of each cell populations among the MAVS-stained lung cells from bleomycin-administered lungs at day 14 was determined by flow cytometry (n=5). Specific surface markers were used to identify lung cell populations; macrophages (CD45+F4/80+), epithelial cells (CD45−CD31−CD49e−EpCAM+) and myofibroblasts (CD45−CD31−EpCAM−CD49e+). Data are the mean ± SEM. Statistical significance was determined using the log-rank (Mantel-Cox) (a and b) or 2-way ANOVA with Tukey’s multiple comparisons test (c) or multiple t test (d). *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
MAVS-dependent fibrogenic responses in bleomycin-induced pulmonary fibrosis
Next, the experiments were undertaken to determine the role of MAVS in regulating TGF-β signaling, often regarded as the master regulator of tissue fibrosis, as well as the consequent fibrogenic responses. Initially, the bleomycin-induced induction of total and active TGF-β seemed to be attenuated but didn’t reach statistical significance in the lung from Mavs−/− mice compared to those of WT controls (Figure 2a and b). For accurate determination of these statistical significance, additional experiments were undertaken with increased numbers of mice. Indeed, the results revealed that the bleomycin-induced induction of total and active TGF-β, respectively, were significantly attenuated in MAVS deficiency in vivo (Figure 2c and d). In addition, when their gene expressions were evaluated for the molecules involved in TGF-β superfamily signaling, the mRNA expressions of connective tissue growth factor (Ctgf) and interleukin (IL)-13 (Il13), respectively, were significantly attenuated in MAVS deficiency (Fig S2a – f). Both Ctgf and Il13 have also been identified to regulate the phosphorylation status of Smad2/3, a critical event in TGF-β super family signaling [19–21]. Intra-alveolar inflammatory response was not significantly altered between the two groups (Figure S2g). Additional evaluations of molecular markers which are known to be differentially expressed in macrophages during distinct types of classical, alternative and fibrotic/reparative activations, respectively, didn’t reveal specific differences between the two groups, either (Figure S2h). Importantly, Western blot evaluation of the activation status of SMAD-2/3, canonical markers of TGF-β distal signaling molecules, revealed the marked attenuation in MAVS deficiency (Figure 2e). Moreover, well-known marker molecules of tissue fibrotic responses were significantly altered via a MAVS-dependent manner. Specifically, the molecular expressions of Fibronectin, Alpha-smooth muscle actin (α-Sma) and Collagen 1α (Col1α), respectively, which were induced after bleomycin injury, were markedly attenuated in MAVS-deficient lungs (Figure 2f – i). These observations were further validated by the densitometry evaluations of these molecules (Fig S2g – j). In addition, IHC evaluation of Col1α confirmed the above findings, revealing that bleomycin injury-induced expression of Col1α was markedly attenuated in lung tissues from Mavs−/− mice (Figure 2j). Collectively, these data suggest that MAVS-dependent and independent distinct mechanisms exist for the pathogenesis of pulmonary fibrosis in our model.
Figure 2. MAVS-dependent and independent fibrogenic responses in bleomycin-induced pulmonary fibrosis.
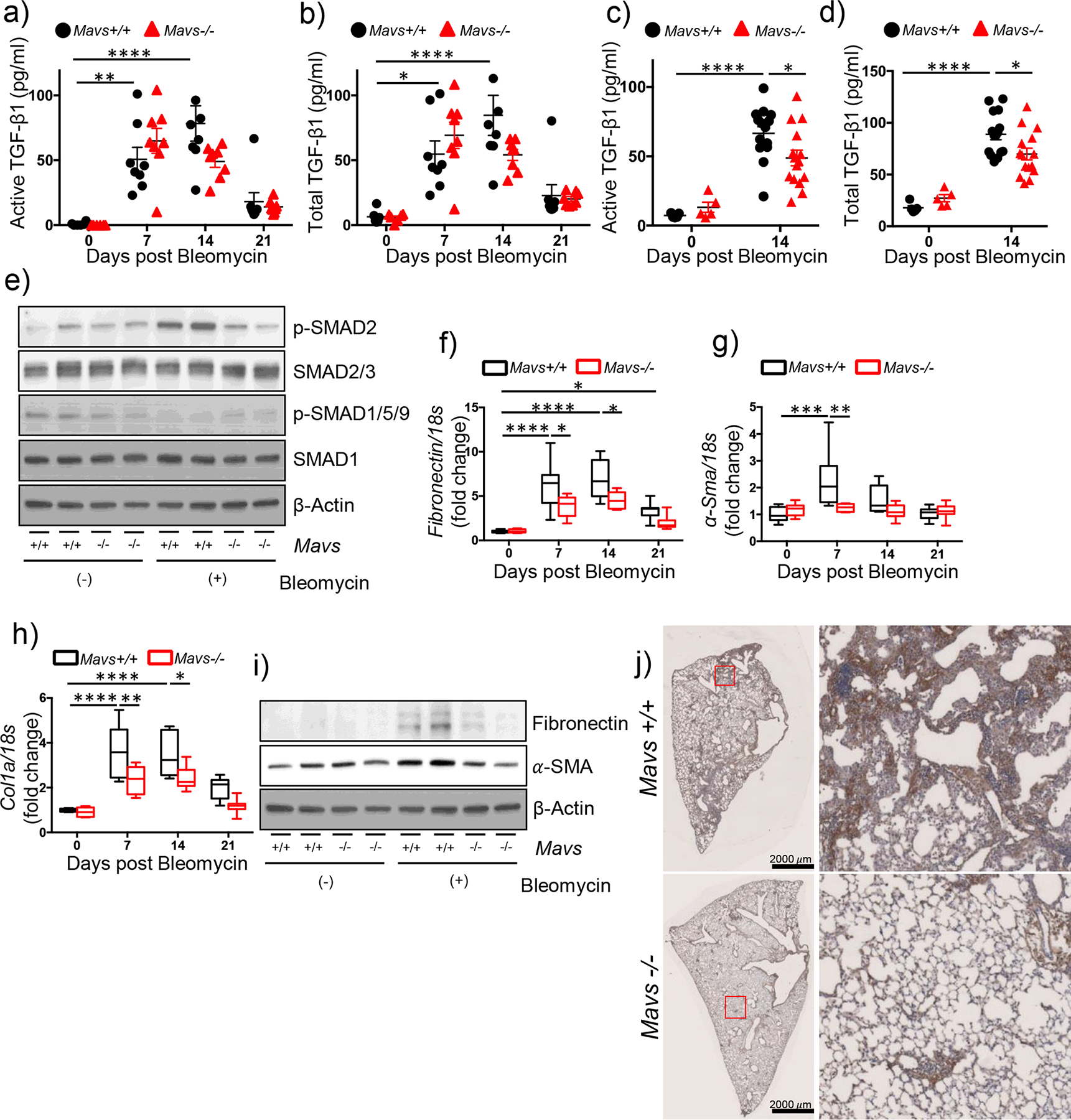
(a-d) Saline (bleomycin -) or bleomycin (+) was administered to wild type (Mavs+/+) and MAVS null mutant (Mavs−/−) mice. (a) Active and (b) total TGF-β1 levels at indicated time points after bleomycin administration were measured from the bronchoalveolar lavage (BAL) fluid by ELISA (n=6 per saline groups, n=8 per bleomycin-administered groups). (c) Active and (d) total TGF-β1 levels at day 14 after bleomycin administration were measured from the BAL fluid by ELISA. (n=5 per saline groups, n=15 per bleomycin-administered groups). (e) Western blot analysis of p-SMAD2, SMAD2/3, p-SMAD1/5/9, and SMAD1 expressions in whole lung tissue lysates at day 14 after saline or bleomycin administration. β -Actin was used as a loading control. (f-h) qRT-PCR analysis of the expression of (f) Fibronectin, (g) Alpha-smooth muscle actin (α-SMA) and (h) Collagen 1α (Col1α) mRNAs, respectively, in lung tissues at day 14 after saline or bleomycin administration (n=6 per saline group, n=8 per bleomycin-administered group) are illustrated. (i) Western blot analysis of Fibronectin and α-SMA expressions in lung tissue lysates day 14 after saline or bleomycin administration are presented. β-Actin was used as a loading control. (j) Representative images from immunohistochemistry analysis of Col1α on lung sections at day 14 after bleomycin administration are presented (n=5 per each of the groups). Scale bars: 2,000 μm. The red boxed regions are magnified on the right for closer observation. Data are the mean ± SEM or Min to Max. Statistical significance was determined using the 2-way ANOVA with Tukey’s multiple comparisons test (a-d and f-h). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
MAVS amplifies multiple DAMPs signaling during fibrotic phase after bleomycin injury
The above findings intrigued us to investigate how MAVS may play a crucial role in the development of experimental pulmonary fibrosis although the induction of total and active TGF-β are not significantly altered. Paying our attention to the fact that MAVS determines even organismal death or survival after severe bleomycin injury, experiments were undertaken to investigate whether DAMPs signaling is regulated via a MAVS-dependent manner after bleomycin injury. Indeed, multiple molecules related to various DAMPs signaling pathways were induced via a time-kinetic manner after bleomycin injury in vivo (Figure 3a – g). It is noteworthy that the measurement of cell-free dsDNA amount in bronchoalveolar lavage (BAL) fluid revealed the significant increase after bleomycin injury in vivo (Figure 3g). Interestingly, among these multiple molecules, dsDNA, cGAS and STING, respectively, were significantly attenuated via a MAVS-dependent manner after bleomycin injury in vivo (Figure 3h – i). Recently, polymerization of STING was identified as a critical event of its activation [22]. Indeed, its evaluation by the application of semi-denaturing detergent agarose gel electrophoresis (SDD-AGE) revealed a significant induction of STING aggregation at day 14, a time-point often regarded as the peak of fibrogenesis in our IPF murine modeling (Figure 3j). Intriguingly, the bleomycin-induced STING aggregation was markedly attenuated in MAVS-deficient lungs (Figure 3j). Similar observations were further accentuated in a higher dose (1.0 U/kg) of bleomycin injury in vivo. Specifically, the expression of additional molecules of DAMPs signaling pathways including Sting, Hmgb1, Nlrp3 and purinergic receptor P2Y2 (P2ry2) molecules was significantly attenuated in MAVS-deficient lungs with the higher dose (1.0 U/kg) (Figure S3a – d). In accordance with this, bleomycin injury-induced induction of dsDNA amount from BAL fluid as well as cGAS protein and STING aggregation were also significantly attenuated via a MAVS-dependent manner in this high dose (1.0 U/kg) (Figure S3e – g). Taken together, these results suggest that multiple DAMPs signaling pathways, which are induced or activated by bleomycin injury in vivo and persist during the fibrotic phase of IPF murine modeling, are critically regulated via a MAVS-dependent manner.
Figure 3. MAVS amplifies multiple DAMPs signaling during fibrotic phase.
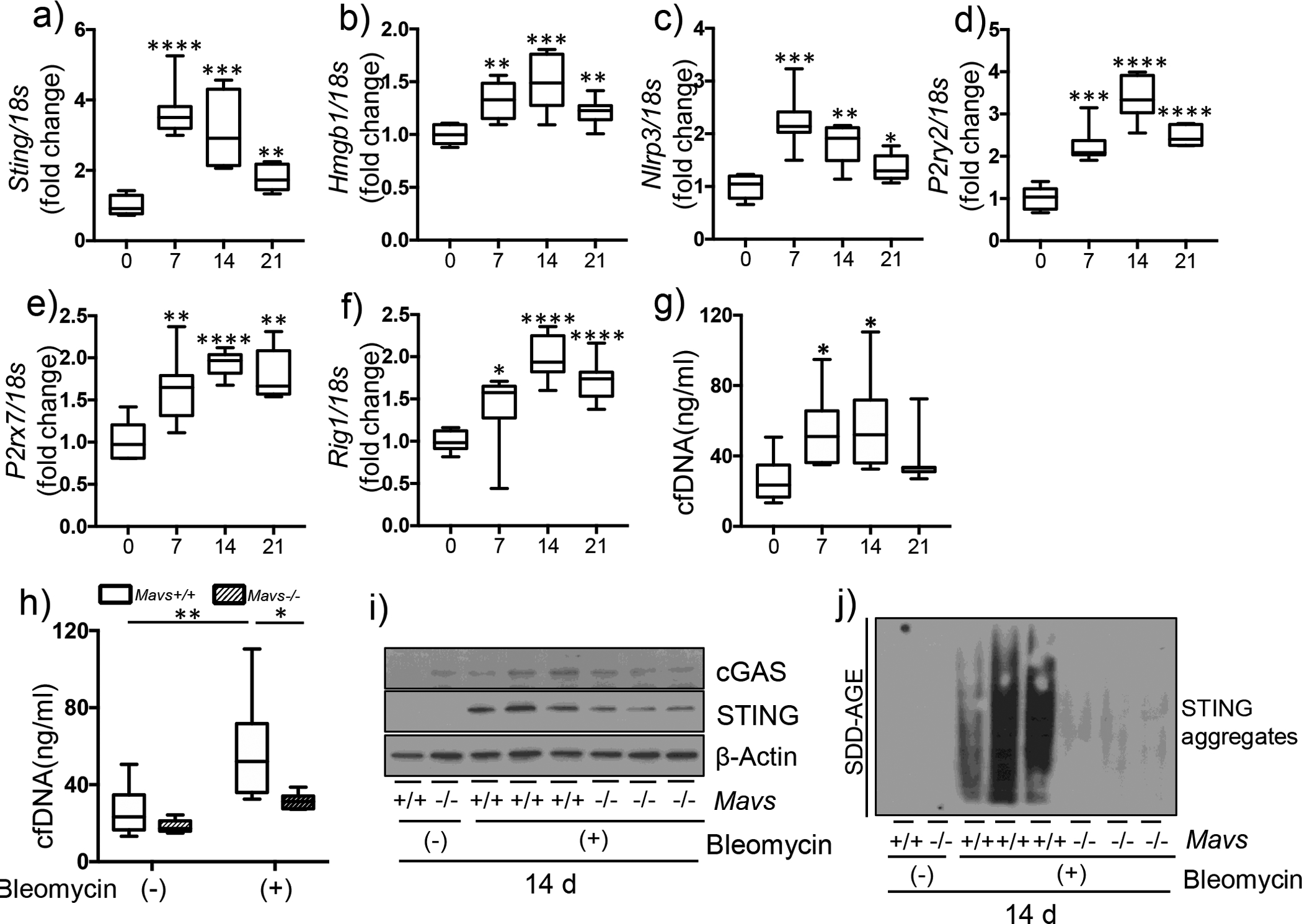
(a-g) Saline (bleomycin -) or bleomycin (+) was administered to wild type mice. (a-f) From whole lung tissues at the indicated time points after bleomycin administration, the expression levels of (a) Sting, (b) Hmgb1, (c) Nlrp3, (d) P2ry2, (eE) P2rx7, and (f) Rig-I mRNAs, respectively, were evaluated by qRT-PCR. (g) The amount of cell-free dsDNA (cfDNA) in BAL fluid was evaluated by fluorometric assay. (h-j) Saline (bleomycin -) or bleomycin (+) was administered to wild type (Mavs+/+) and MAVS null mutant (Mavs−/−) mice. (h) The results of cfDNA amount from BAL fluid, and (i) Western blot evaluations for cGAS and STING proteins from whole lung tissue lysates, respectively, at day 14 after bleomycin administration are presented. β-Actin was used as a loading control. (j) The result of STING aggregation is presented. Lung tissue lysates at day 14 after bleomycin administration were separated by semi-denaturating detergent agarose gel electrophoresis (SDD-AGE) and detected with STING antibody. Data are the mean Min to Max. Statistical significance was determined using unpaired t test (a-g) or 2-way ANOVA with Tukey’s multiple comparisons test (h). *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
A proapoptotic BH3 mimetic ABT-263 inhibits MAVS and lung fibrosis in vivo.
While we were undergoing experiments to define the role of apoptosis in the regulation of DAMPs signaling in our system, surprisingly, we observed a previously unidentified effect of proapoptotic BH3 mimetics. Specifically, BH3 mimetic drugs could reduce the MAVS expression. In murine lung epithelial-12 (MLE 12) cells, when several BH3 mimetics including ABT-263 (a Bcl-2 and Bcl-xl inhibitor), ABT-199 (a Bcl-2 inhibitor) and A1155483 (a Bcl-xl inhibitor), respectively, were treated after bleomycin injury in vitro, we noticed that the expression of MAVS molecule was significantly reduced (Figure 4a and b). In addition, the ABT-263-induced reduction of MAVS was not related to the decrease of mitochondrial proteins; the components of mitochondrial oxidative phosphorylation (OXPHOS) were not altered in our experiments (Figure 4b). A similar finding was observed when we expanded the evaluation to different types of primary cells including murine embryonic fibroblasts (MEFs), peritoneal macrophages (PMs), as well as primary mouse lung fibroblasts (MLF) cells, respectively (Figure 4c – e). Inspired by these observations, a pharmacologic approach in vivo was undertaken in order to evaluate potential therapeutic effects of ABT-263 in our murine pulmonary fibrosis model (Figure 4f). Specifically, from day 8 after bleomycin injury in vivo, ABT-263 (40mg/kg of body weight) was treated by intraperitoneal administration every other day and the fibrotic responses were evaluated. Indeed, the bleomycin-induced increase of total lung collagen contents was significantly attenuated with ABT-263 treatment in vivo (Figure 4f and S4). In line with this, Western blot evaluation revealed that the activation status of SMAD-2/3, the expression of Fibronectin, as well as the expression of α-SMA, respectively, were significantly ameliorated with ABT-263 treatment in vivo (Figure 4g). These observations were supported by in vitro experiments. In MEF, MLF and normal human lung fibroblasts (NHLF) cells, respectively, TGF-β1-induced activation of SMAD-2/3 was significantly reduced after ABT-263 treatment in vitro (Figure 4h). Overall, these results provide compelling evidence that the BH3 mimetic ABT-263 can attenuate the expression of MAVS and, consequently, the development of experimental pulmonary fibrosis.
Figure 4. A BH3 mimetic ABT-263 attenuates the expression of MAVS and lung fibrosis.
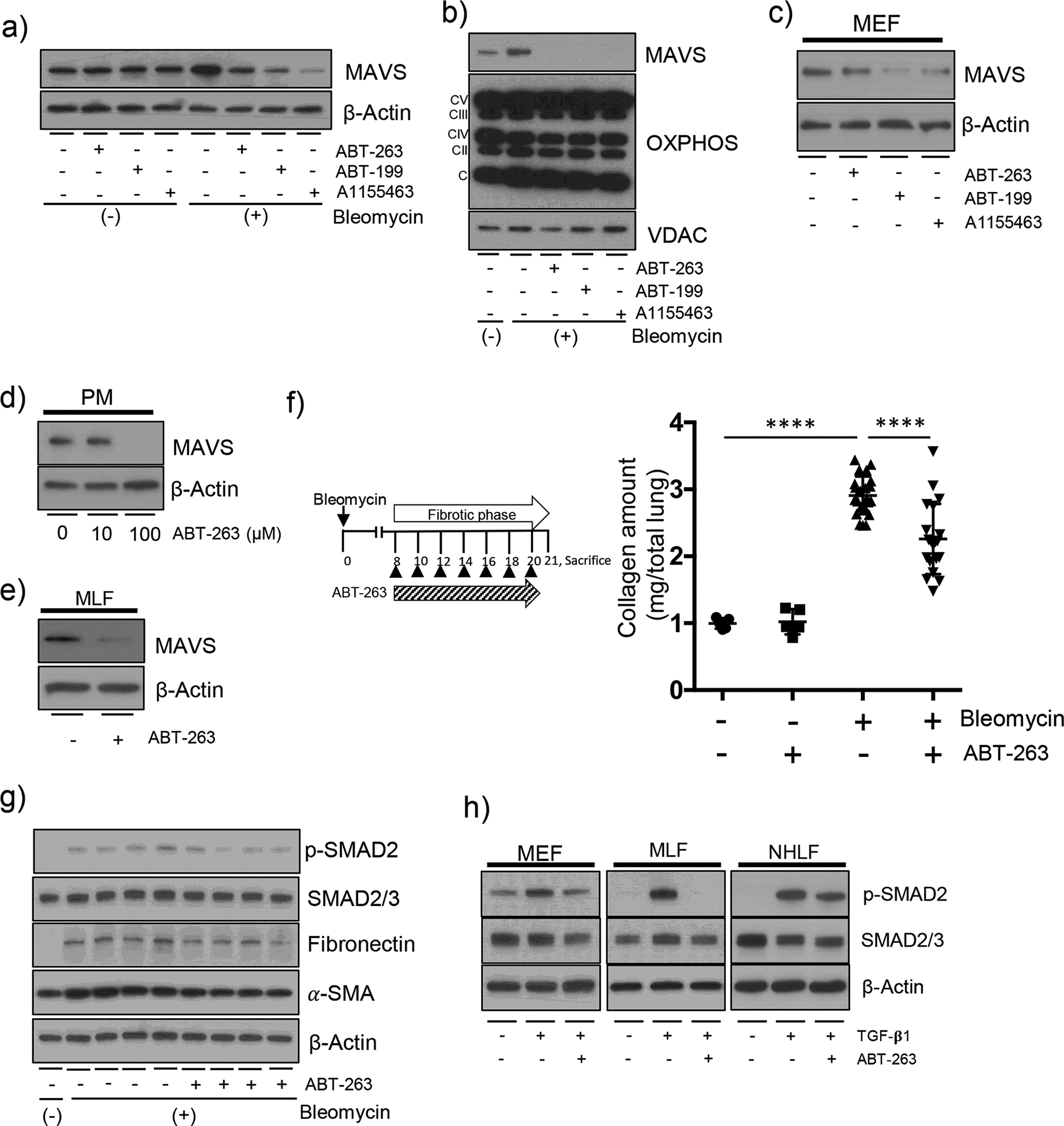
(a-e) The indicated BH3 mimetics with 100 μM amounts, respectively, were treated for 3 h. (a) MLE-12 cells were treated with the indicated BH3 mimetics after 10 mU/ml bleomycin treatment for 3 days. The expression of MAVS was evaluated by Western blot analysis. (b) The mitochondrial fractions of MLE-12 cells treated with the indicated BH3 mimetics after 10 mU/ml bleomycin treatment for 3 days, respectively. The expression of MAVS and Oxidative phosphorylation (OXPHOS) complexes were evaluated by Western blot analysis. Voltage-dependent anionic channel (VDAC) protein was evaluated as loading controls of mitochondria. (c) The mouse embryonic fibroblasts (MEF) cells were treated with the indicated BH3 mimetics. The expression of MAVS was evaluated by Western blot analysis. (d) Primary peritoneal macrophages (PM) were isolated from the peritoneal cavity of wild type mice. The cells were treated with 10 and 100 μM ABT-263. The expression of MAVS was evaluated by Western blot analysis. (e) Primary murine lung fibroblasts (MLF) were isolated from wild type murine lungs. The cells were treated with ABT-263. The expression of MAVS was evaluated by Western blot analysis. (f) The Scheme of the experimental approach, and evaluation results of the total lung collagen contents from wild type murine lungs are presented. The mice were administered with bleomycin (+) and treated after day 8 with ABT-263 (40 mg/kg, every 2 d, i.p.), and sacrificed at day 21. Each dot indicates the individual mouse used for the experiment. (n=5 per each of both control groups, n=23 per bleomycin only treatment group, n=20 per bleomycin+ABT-263 treatment group, respectively). (g) Western blot evaluations for p-SAMD2, SMAD2/3, Fibronectin and a-SMA proteins from whole lung tissue lysates, respectively, at day 14 after bleomycin administration are presented. (n=5 per each of the groups). (h) After stimulation with 20 ng/ml TGF-β1 for 24 h, MEF, MLF and human normal lung fibroblasts (NHLF) were treated with 100 μM ABT-263 for 3 h. Then, the expression of p-SMAD2, SMAD2/3, respectively, were evaluated by Western blot analysis. For panels (a), (c-e) and (g-h), β-Actin was used as a loading control. Data are the mean ± SEM. Statistical significance was calculated using the 2-way ANOVA with Tukey’s multiple comparisons test (f) ****, P < 0.0001.
Therapeutic effects of Pirfenidone or Nintedanib are not related to MAVS.
To date, Pirfenidone and Nintedanib are the only approved drugs known to decelerate the disease progression of IPF [23, 24]. However, it has not been known whether these drugs can modulate MAVS signaling in vitro or in vivo. When in vitro experiments were undertaken to address this issue, these two drugs failed to reduce the expression of MAVS while ABT-263 can significantly reduce it (Figure 5a and b). Overall mitochondrial proteins such as the OXPHOS components were not altered in our experiments after the treatment of these drugs (Figure 5b). Next, when the therapeutic effects of these drugs in our IPF modeling system were compared, the bleomycin-induced increase of total lung collagen contents as well as semi-quantitative severity scores of the microscopic changes were significantly ameliorated after treatment with these three drugs, respectively (Figure 5c, d and Figure S5). No statistical difference was noted between the three groups (Figure 5c and d, * note: Two mice were dead during the fibrotic phase in the Nintedanib treatment group). In line with this, Western blot evaluation revealed that the activation status of SMAD-2/3, the expression of Fibronectin, as well as the expression of α-SMA, respectively, were significantly ameliorated with the treatment of these three drugs, respectively (Figure 5e). Overall, these data suggest that the pharmacologic effects of Pirfenidone or Nintedanib, respectively, for the treatment of IPF are not directly related to MAVS.
Figure 5. Therapeutic effects of Nintedanib or Pirfenidone, two approved drugs, are not related to MAVS.
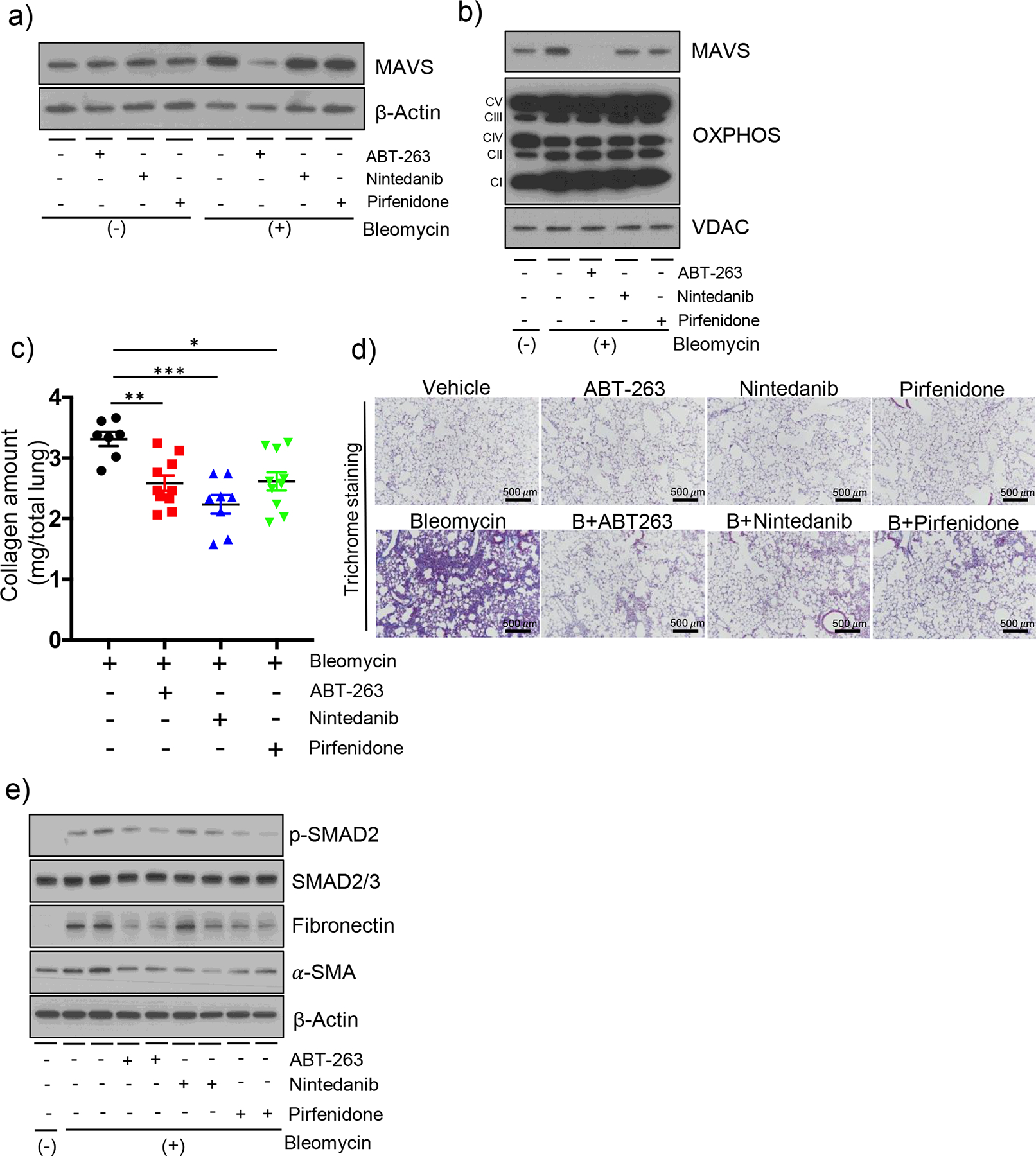
(a-b) MLE-12 cells were treated with ABT-263 (100 μM), Nintedanib (100 μM) and Pirfenidone (100 μM) for 3 h after 10 mU/ml bleomycin treatment for 3 days. The expression of MAVS from (a) whole cell lysates, and (b) mitochondrial fractions, respectively, was evaluated by Western blot analysis. In panel (b), the expression of OXPHOS complexes and VDAC protein as loading controls of mitochondria are presented. (c) The evaluation results of the total lung collagen contents and (d) the representative images of Masson’s trichrome staining evaluation are presented. C57BL/6J wild type mice were administered with bleomycin (+) and treated after day 8 with ABT-263 (40 mg/kg, every 2 d, i.p.), Nintedanib (40 mg/kg, every 2 d, i.p.) and Pirfenidone (40 mg/kg, every 2 d, i.p.), and sacrificed at day 21. Each dot indicates the individual mouse used for the experiment. (n=10 per bleomycin treatment group (3-mice dead), n=10 per Bleomycin+ABT-263 treatment group, n=10 per Bleomycin+Nintedanib treatment group (2-mice dead) and n=10 per Bleomycin+Pirfenidone treatment group, respectively). (Scale bars: 500 μm). (e) Western blot evaluations for p-SAMD2, SMAD2/3, Fibronectin and a-SMA proteins from whole lung tissue lysates, respectively, at day 14 after bleomycin administration are presented. For panels (a) and (e), β-Actin was used as a loading control. Data are the mean ± SEM. Statistical significance was calculated using the 2-way ANOVA with Tukey’s multiple comparisons test (c) *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Bleomycin-induced cellular senescence is inhibited in MAVS deficiency.
Cellular senescence has recently emerged as a key driving force of IPF pathogenesis [25, 26]. In addition, ABT263 has been identified to mediate anti-fibrotic effects via its selective removal of senescent cells due to the induction of their apoptotic cell death, called senolysis [25]. Thus, experiments were undertaken to further determine how ABT263-induced senolytic effect might be related to the molecular function of MAVS in cellular senescence. From MEF cells, bleomycin-induced cellular senescence, which was evaluated by the measurement of SA-β-Gal activity, was significantly attenuated after treatment with ABT-263 (Figure 6a and b). As expected, the reduction of cellular senescence was related to the induction of apoptosis and the consequent decrease of cell viability via a dose-dependent manner (Figure 6c, d and data not shown). Notably, a significant reduction of the MAVS protein expression was observed in association with the ABT263-induced senolysis (Figure 6d). Similar results were obtained from MLE12 cells, further confirming the reduction of MAVS in association with ABT263-mediated senolysis (Figure S6a – d). Next, in order to clarify whether the inhibitory effect of ABT263 on regulation of MAVS protein expression is mainly related to its selective removal of senescent cells (senolysis) in the setting of cellular senescence or whether ABT263 may mediate its inhibitory effect on MAVS via a senolysis-independent mechanism, further experiments with no stimulation of bleomycin were undertaken, eliminating the bleomycin-induced cellular senescence. Interestingly, after treatment with ABT263, a significant reduction of MAVS protein was observed from all three different cell types tested in this setting (Figure 6e – g). Finally, to define the functional role of MAVS which may play in bleomycin-induced cellular senescence in vitro, when SA-β-Gal activity was evaluated and compared in the presence or absence of MAVS, the bleomycin-induced cellular senescence was significantly attenuated in MAVS deficiency (Figure 6h & i). Intriguingly, the level of apoptotic cell death or cell viability was not significantly different in the presence or absence of MAVS (Figure 6j and data not shown), revealing that the reduction of bleomycin-induced cellular senescence in MAVS deficiency cannot be attributed to senolysis. Rather, these results demonstrate that the bleomycin-induced cellular senescence is inhibited in MAVS deficiency. The functional significance of MAVS in the regulation of bleomycin-induced senescence was also evaluated in our in vivo model. When the gene expressions of known senescence markers were evaluated, P16INK4a, P19ARF and P21Cip1 mRNAs, respectively, were significantly induced in our murine pulmonary fibrosis model (Figure S6e–g). Indeed, the bleomycin injury-induced mRNA expressions of P16INK4a and P19ARF, respectively, were significantly attenuated in a MAVS-dependent manner (Figure 6k and l). In accordance with the time-kinetic patterns of its mRNA expressions, the P16INK4a protein expression was significantly induced, reaching the peak of its expression at day 14 after bleomycin injury in vivo (figure S6h). Importantly, the induction of P16INK4a protein was significantly reduced in MAVS deficiency at day 14 and day 21, respectively, after bleomycin administration in vivo (Figure 6m, S6i). Collectively, these results suggest that, in addition to its known senolytic effect, ABT263 may exert its anti-fibrotic effects by the reduction of MAVS. In addition, MAVS may play a functional role in the induction of bleomycin-induced cellular senescence.
Figure 6. Bleomycin-induced cellular senescence is inhibited in MAVS deficiency in vitro/vivo.
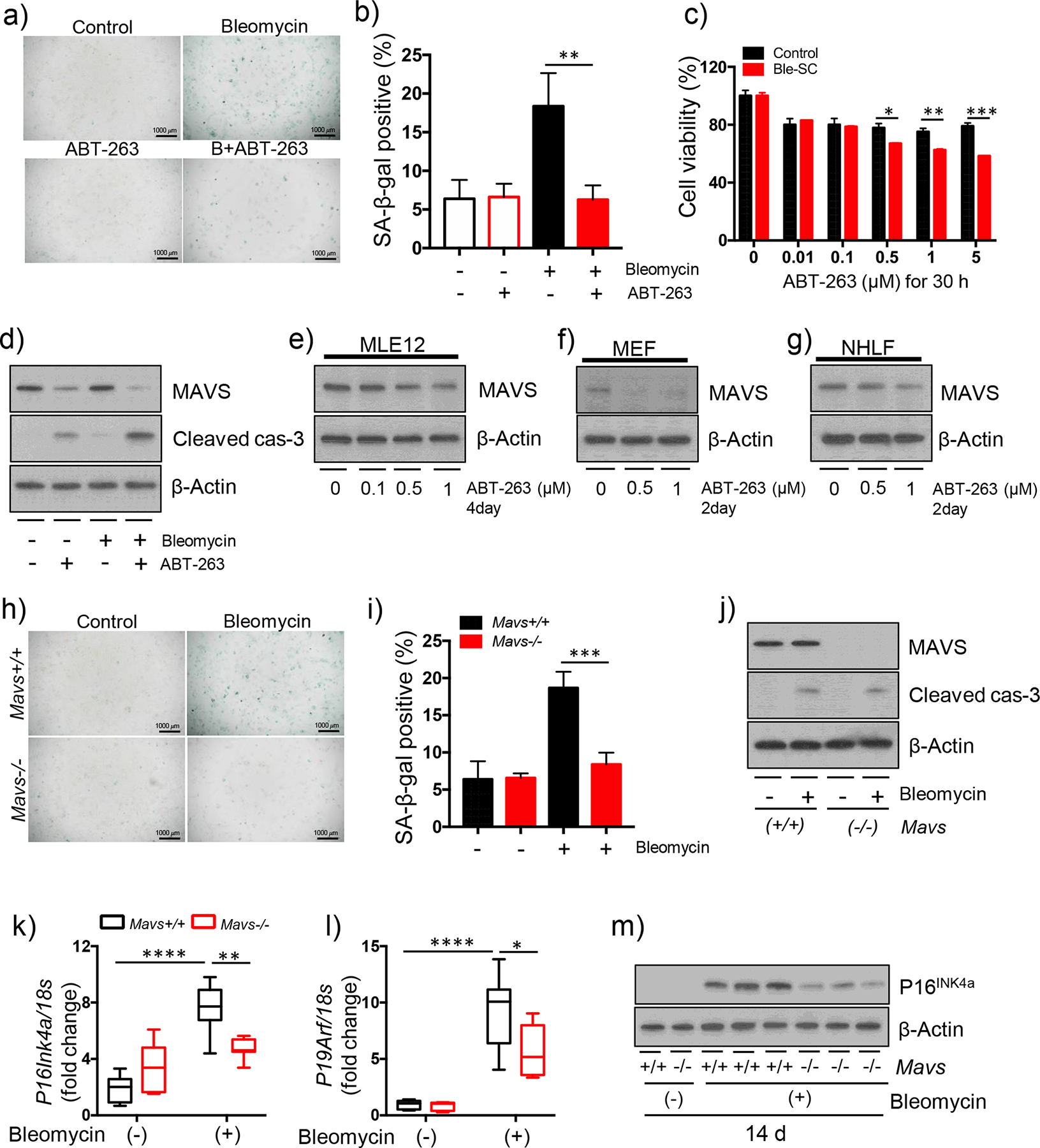
(a-d) MEF cells were treated with indicated concentration of ABT-263 for 30 h after 50 mU/ml bleomycin treatment. (a) The result of SA-β-Gal staining and (b) quantification of SA-β-gal-positive cells are presented (Scale bars: 100 μm). (c) Cell viability was measured by XTT assay. (d) The Western blot results of MAVS and Cleaved caspase-3 (cas-3) proteins are presented. (e) MLE-12, (f) MEF and (g) NHLF cells were treated with the indicated concentration of ABT-263 for indicated time points, respectively. The expression of MAVS was evaluated by Western blot analysis. (h-i) Mavs+/+ and Mavs−/− MEF cells were treated with 1 μM ABT-263 for 30 h after 50 mU/ml bleomycin treatment. (h) The result of SA-β-Gal staining and (i) quantification of SA-β-gal-positive cells are presented (Scale bars: 100 μm). (j) Mavs+/+ and Mavs−/− MEF cells were treated with 50 mU/ml bleomycin treatment for 30 h. The Western blot results of MAVS and Cleaved caspase-3 (cas-3) proteins are presented. (k-m) Saline (bleomycin -) or bleomycin (+) was administered to wild type (Mavs+/+) and MAVS null mutant (Mavs−/−) mice. From whole lung tissues at day 14 after bleomycin administration, the expression levels of (k) P16INK4a and (l) P19ARF were evaluated by qRT-PCR, respectively. (m) The Western blot result of P16INK4a protein is presented. For panels (d), (e), (f), (g) (j) and (m), β-Actin was used as a loading control. Data are the mean ± SD. Statistical significance was calculated using the 2-way ANOVA with Tukey’s multiple comparisons test *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Multimeric aggregation of MAVS, a critical event of MAVS signaling, is significantly activated in humans as well as in mice
Because prion-like multimeric aggregation of MAVS molecules on mitochondrial outer membrane was identified as a critical event for its proper signaling [27], experiments were undertaken to evaluate this in our system. Indeed, SDD-AGE as well as blue native-polyacrylamide gel electrophoresis (BN-PAGE) evaluation revealed that bleomycin injury induces a significant MAVS aggregation in vivo (Fig 7a). This observation could be observed from mitochondrial fraction and whole lung lysates as well (Figure 7a). Surprisingly, this phenomenon persisted even at day 14 after bleomycin administration. An additional finding was noted in our experiments. Although the expression of MAVS was shown to be markedly enhanced after bleomycin administration in IHC evaluation (Figure 1e), the absolute amount of MAVS protein in the lung tissues seemed to be decreased during bleomycin-induced fibrogenesis in vivo (Figure 7a). We interpret the data as additional evidence for supporting the multimeric aggregation of MAVS during fibrogenesis. Specifically, the strong positive staining of MAVS obtained from IHC evaluation as demonstrated in Figure 1E may reflect the multimeric aggregation of MAVS which is not related to its molecular induction after bleomycin injury in vivo. Next, when the experiments were undertaken whether the bleomycin-induced multimeric aggregation of MAVS in vivo can be modulated by ABT-263, Pirfenidone or Nintedanib, respectively, intriguingly, the multimeric aggregation of MAVS was markedly attenuated only after ABT-263 treatment (Figure 7b and c). Among multiple drugs tested, all BH3 mimetics could ameliorate the multimeric aggregation of MAVS (Figure 7d). In contrast, Pirfenidone or Nintedanib, respectively, failed to ameliorate it (Figure 7d). Finally, in order to determine the clinical relevance of the above finding observed in murine disease modeling, the experiment was undertaken to test whether MAVS aggregation is observed in the lung tissues from human patients with IPF. Indeed, significant increase of MAVS aggregation was observed from 11 patients among 13 specimens from IPF patients, while only two specimens showed MAVS aggregation among 9 controls, demonstrating a compelling statistical significance (Figure 7e and f, **p < 0.01). Overall, these data demonstrate that persistent MAVS aggregation, a key event of MAVS signaling activation, is observed in lungs from the patients with IPF as well as in our mammalian model of IPF, and, importantly, can be therapeutically targeted by BH3 mimetics.
Figure 7. Multimeric aggregation of MAVS, a critical event of MAVS signaling, is significantly activated in humans as well as in mice.
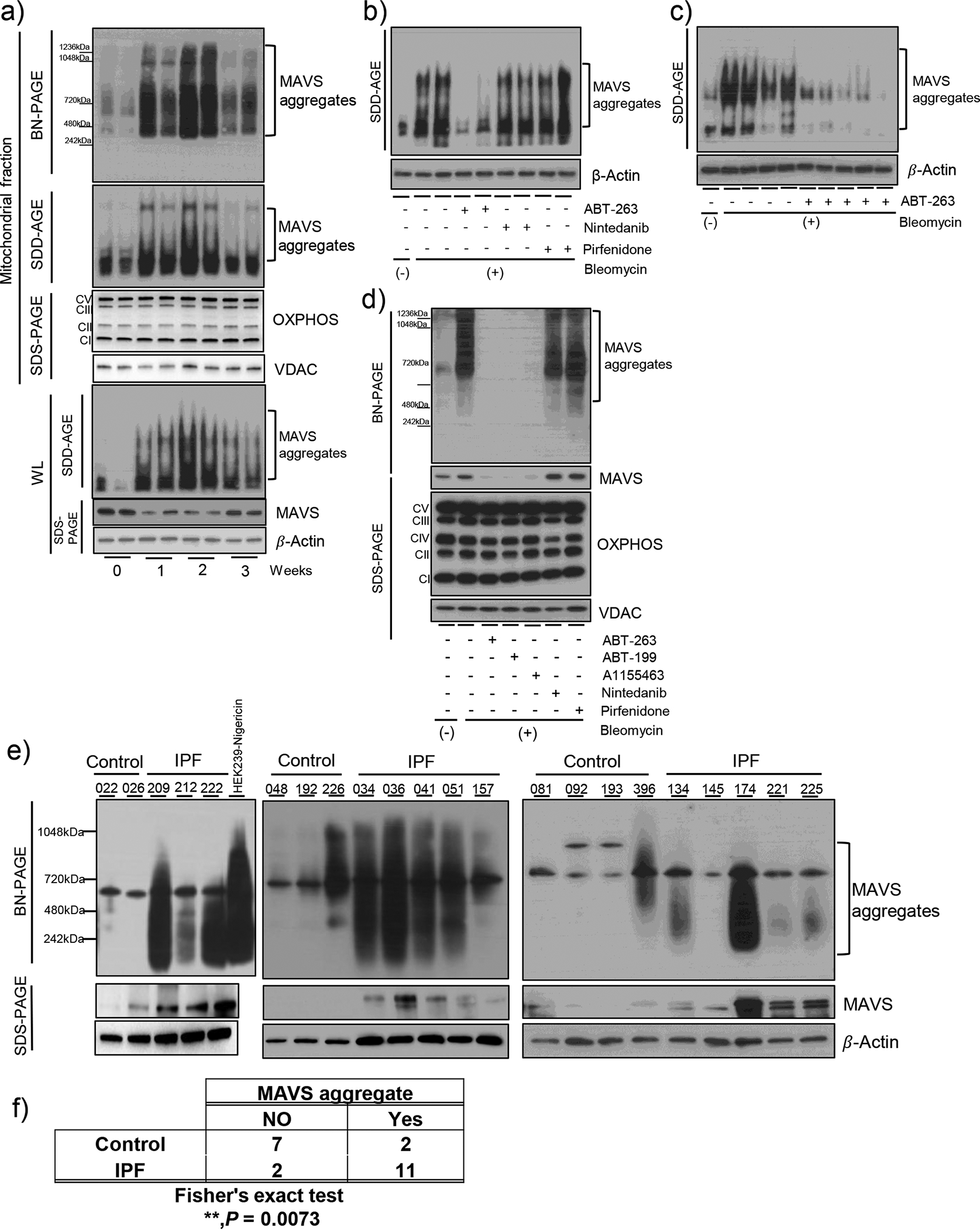
(a) Saline (bleomycin -) or bleomycin (+) was administered to wild type (Mavs+/+) mice. The multimeric MAVS aggregation from whole lung tissue lysates (WL) and mitochondrial fractions (MF) at indicated time points was evaluated by blue native (BN)-PAGE and SDD-AGE. (b-c) Wild type mice were administered with bleomycin (+) and treated after day 8 with ABT-263 (40 mg/kg, every 2 d, i.p.), Nintedanib (40 mg/kg, every 2 d, i.p.) and Pirfenidone (40 mg/kg, every 2 d, i.p.). The multimeric MAVS aggregation in WL at day 14 was evaluated by SDD-AGE. (d) The mitochondrial fractions of MLE-12 cells treated with 100 μM ABT-263, ABT-199, A-1155463, Nintedanib and Pirfenidone, respectively, for 3 h after 10 mU/ml bleomycin treatment for 3 days. The multimeric MAVS aggregation and expression of MAVS and OXPHOS complexes were evaluated by BN-PAGE and SDS-PAGE, respectively. VDAC protein was evaluated as loading controls of mitochondria for panels (a) and (e). (e) The multimeric MAVS aggregation was evaluated and compared by the application of BN-PAGE from lungs obtained from the patients with IPF or healthy controls. (n=9 per normal group, n=13 per IPF group; each number indicates an individual subject). The cell lysate from human embryonic kidney 293 cells treated with nigericin was used as a positive control in order to evaluate MAVS aggregation. β-Actin was used as a loading control for panels (a), (b), (c) and (e). (f) Fisher’s exact test was applied to determine statistical significance of the above result presented in panel D. **, P < 0.01.
Discussion
The current studies are novel as they demonstrate that (i) MAVS, a key adaptor of mitochondrial innate immune signaling, is significantly activated after fibrogenic injury; (ii) the activation of MAVS is associated with various DAMPs signaling pathways, especially, cGAS-STING innate immune signaling; (iii) proapoptotic BH3 mimetics can reduce the MAVS expression as well as its multimeric aggregation; and most importantly, (iv) the multimeric aggregation of MAVS, a key event during MAVS signaling activation, is markedly enhanced in lungs from the patients with IPF. Overall, these studies suggest that MAVS plays a critical pathogenic role in experimental pulmonary fibrosis.
As the first mitochondria-localized protein to be linked to innate immunity [12], MAVS has been identified to be involved in multiple biological phenomena including mitochondrial innate immune signaling, apoptosis, autophagy and metabolic functions [28–30]. Given that recent studies are continuously highlighting the functional roles of mitochondria and their underlying mechanisms by which mitochondrial dysfunction contributes to the pathogenesis of IPF [6, 31], the current studies emphasize mitochondrial significance by illuminating the role of MAVS-mediated DAMPs signaling in fibrotic tissue injury/damage responses.
Danger theory has been implicated in the pathogenesis of many chronic disorders for which effective therapeutics are not available, thus remaining major unmet medical needs [32–34]. IPF is such an example. Intriguingly, MAVS, a molecule mostly studied in the context of viral PAMPs signaling, is significantly activated by bleomycin-induced injury which is obviously not related to viral infection. In addition, persistent MAVS aggregation is observed in the lungs from IPF patients. Fibrogenic tissue injury may drive fibrosis by inducing various endogenous DAMPs signaling which could be converged to MAVS. A potential significance of MAVS signaling in the pathogenesis of non-viral injury-induced chronic diseases was reported in another publication [35].
The phosphorylation state of receptor-regulated Smads (R-Smads) such as Smad2/3 determines the most critical event in TGF-β super family signaling (reviewed in Reference [36]). Importantly, multiple TGF-β-dependent and -independent complex regulatory mechanisms have been identified to ensure their phosphorylation status as a dynamic and tightly controlled event (reviewed in References [36, 37]). Our current study adds additional knowledge to this important research area, by revealing the regulation of R-Smads phosphorylation via a MAVS-dependent manner. In addition, it has rarely been reported whether MAVS may play a role in cellular senescence. By demonstrating that bleomycin injury-induced cellular senescence is significantly attenuated in a MAVS-dependent manner, our study may provide additional mechanistic insight of how MAVS contributes to the development of pulmonary fibrosis. The significance of these novel observations should be explored in future studies, considering that cellular senescence has emerged as a crucial driving force of IPF pathogenesis [25, 26, 38].
In line with our studies, a recent study demonstrated a potential efficacy of targeting myofibroblast antiapoptotic proteins with BH3 mimetic drugs in skin fibrosis [39]. In addition, the pharmacologic effects of BH3 mimetics that can selectively remove senescent cells, called senolysis, are recently being highlighted to have therapeutic potentials for the treatment of age-associated diseases [40, 41]. Regarding this, our finding that BH3 mimetics can attenuate the MAVS expression in vitro and in vivo might open up new vistas of the underlying mechanisms by which BH3 mimetics function as novel therapeutics. Specifically, when combined with the inhibitory role of MAVS in regulating multiple DAMPs signaling as well as cellular senescence, the current study enables us to speculate that BH3 mimetics might take its antifibrotic effects by inhibiting MAVS and the consequent DAMPs signaling pathways for the treatment of fibrosis, in addition to their known senolytic effects [40]. Or the senolytic effects of BH3 mimetics might be linked to their inhibitory function on MAVS through an intricate regulatory network. Currently, these questions about senescence or non-senescence impact of BH3 mimetics on the regulation of fibrosis, especially in relation to their inhibitory function on MAVS, have rarely been explored. Our novel observations cast intriguing mechanistic questions to explore therapeutic potentials BH3 mimetics may have for the treatment of fibrotic disorders including IPF in humans.
To date, Pirfenidone and Nintedanib are the only approved drugs known to decelerate the disease progression of IPF [23, 24]. Pirfenidone is known to mediate its antifibrotic effects via its anti-oxidant, anti-fibrotic and anti-inflammatory properties [42]. Nintedanib is a multi-tyrosine kinase inhibitor and is known to inhibit the receptor kinases of platelet-derived growth factor (PDGF), fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF), which are all thought to play an important role in the pathogenesis of IPF [24]. However, their modulatory effects on the expression of MAVS or its-mediated DAMPs signaling have not been explored yet. Although this question has not been investigated in a comprehensive manner, our studies suggest that therapeutic effects of Pirfenidone or Nintedanib, respectively, for the treatment of IPF, are not related to the modulation on MAVS and its signaling.
Several issues remain unanswered in the current studies. Detailed mechanistic explanations are not provided about how endogenous DAMPs-mediated innate immune signaling pathways may contribute to the pathogenesis of IPF. The molecular action mechanism(s) by which proapoptotic BH3 mimetics function to reduce the MAVS expression and its aggregation has not been investigated, either. In addition, an interesting question is raised about whether synergistic therapeutic effects will be observed if a BH3 mimetic is combined together with Pirfenidone or Nintedanib, respectively, for the IPF treatment. Future studies will be required to explore these important questions.
Although bleomycin-induced experimental fibrosis modeling may not fully recapitulate the pathogenesis of human IPF, multimeric aggregation of MAVS was markedly observed in the lungs of the patients with IPF as well as the late fibrotic phase in our experimental modeling. We believe that a similar observation of multimeric MAVS aggregation in humans as well as in our murine model highlights a potential significance of MAVS in IPF pathogenesis. An enhanced understanding of MAVS and its signaling in fibrotic tissue injury or damage responses may bring a significant impact on overall fibrosis biology including IPF.
In conclusion, these studies demonstrate that MAVS plays an important role in the development of experimental pulmonary fibrosis in a mammalian model of IPF, and provide a rationale that targeting MAVS or its signaling may be considered as a feasible strategy for the regulation of pulmonary fibrosis.
Supplementary Material
Take home message:
This study provides proof-of-concept that MAVS may play a critical role in the development of pulmonary fibrosis and targeting MAVS or its signaling by proapoptotic BH3 mimetics may be considered as a feasible strategy for the treatment of IPF.
Acknowledgments
The authors thank Ms. Susan Ardito for excellent administrative assistance. These studies were supported by NHLBI R01HL130283 (MJK) and NIA R01AG053495 (MJK).
Footnotes
Conflict of interest
The authors declare no competing financial interests.
References
- 1.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr., Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ, Fibrosis AEJACoIP. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. American journal of respiratory and critical care medicine 2011: 183(6): 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol 2014: 9: 157–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutchinson JP, McKeever TM, Fogarty AW, Navaratnam V, Hubbard RB. Increasing global mortality from idiopathic pulmonary fibrosis in the twenty-first century. Annals of the American Thoracic Society 2014: 11(8): 1176–1185. [DOI] [PubMed] [Google Scholar]
- 4.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell 2012: 148(6): 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dela Cruz CS, Kang MJ. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mora AL, Bueno M, Rojas M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. The Journal of clinical investigation 2017: 127(2): 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Picard M, Wallace DC, Burelle Y. The rise of mitochondria in medicine. Mitochondrion 2016: 30: 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bueno M, Lai YC, Romero Y, Brands J, St Croix CM, Kamga C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, Duncan SR, Rojas M, Shiva S, Chu CT, Mora AL. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. The Journal of clinical investigation 2015: 125(2): 521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016: 44(3): 582–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ryu C, Sun H, Gulati M, Herazo-Maya JD, Chen Y, Osafo-Addo A, Brandsdorfer C, Winkler J, Blaul C, Faunce J, Pan H, Woolard T, Tzouvelekis A, Antin-Ozerkis DE, Puchalski JT, Slade M, Gonzalez AL, Bogenhagen DF, Kirillov V, Feghali-Bostwick C, Gibson K, Lindell K, Herzog RI, Dela Cruz CS, Mehal W, Kaminski N, Herzog EL, Trujillo G. Extracellular Mitochondrial DNA Is Generated by Fibroblasts and Predicts Death in Idiopathic Pulmonary Fibrosis. American journal of respiratory and critical care medicine 2017: 196(12): 1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandel NS. Evolution of Mitochondria as Signaling Organelles. Cell metabolism 2015: 22(2): 204–206. [DOI] [PubMed] [Google Scholar]
- 12.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005: 122(5): 669–682. [DOI] [PubMed] [Google Scholar]
- 13.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 2013: 153(2): 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zevini A, Olagnier D, Hiscott J. Crosstalk between Cytoplasmic RIG-I and STING Sensing Pathways. Trends Immunol 2017: 38(3): 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang D, Liang J, Hodge J, Lu B, Zhu Z, Yu S, Fan J, Gao Y, Yin Z, Homer R, Gerard C, Noble PW. Regulation of pulmonary fibrosis by chemokine receptor CXCR3. The Journal of clinical investigation 2004: 114(2): 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, de Castro JPW, DeIuliis G, Ahangari F, Woolard T, Aurelien N, Arrojo EDR, Gan Y, Graham M, Liu X, Homer RJ, Scanlan TS, Mannam P, Lee PJ, Herzog EL, Bianco AC, Kaminski N. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med 2018: 24(1): 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jurgensen HJ, Norregaard KS, Sibree MM, Santoni-Rugiu E, Madsen DH, Wassilew K, Krustrup D, Garred P, Bugge TH, Engelholm LH, Behrendt N. Immune regulation by fibroblasts in tissue injury depends on uPARAP-mediated uptake of collectins. J Cell Biol 2019: 218(1): 333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akamatsu T, Arai Y, Kosugi I, Kawasaki H, Meguro S, Sakao M, Shibata K, Suda T, Chida K, Iwashita T. Direct isolation of myofibroblasts and fibroblasts from bleomycin-injured lungs reveals their functional similarities and differences. Fibrogenesis Tissue Repair 2013: 6(1): 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Meyer C, Müller A, Herweck F, Li Q, Müllenbach R, Mertens PR, Dooley S, Weng HL. IL-13 induces connective tissue growth factor in rat hepatic stellate cells via TGF-β-independent Smad signaling. Journal of immunology (Baltimore, Md : 1950) 2011: 187(5): 2814–2823. [DOI] [PubMed] [Google Scholar]
- 20.Ramazani Y, Knops N, Elmonem MA, Nguyen TQ, Arcolino FO, van den Heuvel L, Levtchenko E, Kuypers D, Goldschmeding R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol 2018: 68–69: 44–66. [DOI] [PubMed] [Google Scholar]
- 21.Rodríguez-Vita J, Sánchez-López E, Esteban V, Rupérez M, Egido J, Ruiz-Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005: 111(19): 2509–2517. [DOI] [PubMed] [Google Scholar]
- 22.Ergun SL, Fernandez D, Weiss TM, Li L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell 2019: 178(2): 290–301 e210. [DOI] [PubMed] [Google Scholar]
- 23.Noble PW, Albera C, Bradford WZ, Costabel U, du Bois RM, Fagan EA, Fishman RS, Glaspole I, Glassberg MK, Lancaster L, Lederer DJ, Leff JA, Nathan SD, Pereira CA, Swigris JJ, Valeyre D, King TE, Jr. Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology 2016: 47(1): 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, Kolb M. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology 2015: 45(5): 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehmann M, Korfei M, Mutze K, Klee S, Skronska-Wasek W, Alsafadi HN, Ota C, Costa R, Schiller HB, Lindner M, Wagner DE, Günther A, Königshoff M. Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. The European respiratory journal : official journal of the European Society for Clinical Respiratory Physiology 2017: 50(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mora AL, Rojas M, Pardo A, Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nature reviews Drug discovery 2017: 16(11): 755–772. [DOI] [PubMed] [Google Scholar]
- 27.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011: 146(3): 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, Chen ZJ. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 2006: 24(5): 633–642. [DOI] [PubMed] [Google Scholar]
- 29.Sun X, Sun L, Zhao Y, Li Y, Lin W, Chen D, Sun Q. MAVS maintains mitochondrial homeostasis via autophagy. Cell discovery 2016: 2: 16024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li T, Li X, Attri KS, Liu C, Li L, Herring LE, Asara JM, Lei YL, Singh PK, Gao C, Wen H. O-GlcNAc Transferase Links Glucose Metabolism to MAVS-Mediated Antiviral Innate Immunity. Cell host & microbe 2018: 24(6): 791–803 e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malsin ES, Kamp DW. The mitochondria in lung fibrosis: friend or foe? Translational research : the journal of laboratory and clinical medicine 2018. [DOI] [PubMed] [Google Scholar]
- 32.Matzinger P The danger model: a renewed sense of self. Science 2002: 296(5566): 301–305. [DOI] [PubMed] [Google Scholar]
- 33.Dela Cruz CS, Kang MJ. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018: 41: 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellson CD, Dunmore R, Hogaboam CM, Sleeman MA, Murray LA. Danger-associated molecular patterns and danger signals in idiopathic pulmonary fibrosis. American journal of respiratory cell and molecular biology 2014: 51(2): 163–168. [DOI] [PubMed] [Google Scholar]
- 35.Kang MJ, Yoon CM, Kim BH, Lee CM, Zhou Y, Sauler M, Homer R, Dhamija A, Boffa D, West AP, Shadel GS, Ting JP, Tedrow JR, Kaminski N, Kim WJ, Lee CG, Oh YM, Elias JA. Suppression of NLRX1 in chronic obstructive pulmonary disease. The Journal of clinical investigation 2015: 125(6): 2458–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wrighton KH, Lin X, Feng XH. Phospho-control of TGF-beta superfamily signaling. Cell Res 2009: 19(1): 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003: 425(6958): 577–584. [DOI] [PubMed] [Google Scholar]
- 38.Kang MJ. Recent Advances in Molecular Basis of Lung Aging and Its Associated Diseases. Tuberc Respir Dis (Seoul) 2020: 83(2): 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lagares D, Santos A, Grasberger PE, Liu F, Probst CK, Rahimi RA, Sakai N, Kuehl T, Ryan J, Bhola P, Montero J, Kapoor M, Baron M, Varelas X, Tschumperlin DJ, Letai A, Tager AM. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Transl Med 2017: 9(420). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 2016: 22(1): 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Deursen JM. Senolytic therapies for healthy longevity. Science 2019: 364(6441): 636–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaefer CJ, Ruhrmund DW, Pan L, Seiwert SD, Kossen K. Antifibrotic activities of pirfenidone in animal models. European respiratory review : an official journal of the European Respiratory Society 2011: 20(120): 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.