

Abstract
Bumped kinase inhibitors that target Cryptosporidium parvum calcium-dependent protein kinase 1 have been well established as potential drug candidates against cryptosporidiosis. Recently, BKI-1649, with a 7H-pyrrolo[2,3-d]pyrimidin-4-amine, or “pyrrolopyrimidine”, central scaffold, has shown improved efficacy in mouse models of Cryptosporidium at substantially reduced doses compared to previously explored analogs of the pyrazolopyrimidine scaffold. Here, two pyrrolopyrimidines with varied sunstituent groups, BKI-1812 and BKI-1814, were explored in several in vitro and in vivo models and show improvements in potency over the previously-utilized pyrazolopyrimidine bumped kinase inhibitors while maintaining equivalent results in other key properties, such as toxicity and efficacy, with their pyrazolopyrimidine isosteric counterparts.
Keywords: Cryptosporidium, cryptosporidiosis, bumped kinase inhibitors, pyrrolopyrimidines, pyrazolopyrimidines
Graphical Abstract
Cryptosporidium has been reported to be one of the major causes of child mortality and developmental delay among several resource-limited regions throughout the world, including Sub-Saharan Africa and Southeast Asia1–3. In the United States, several outbreaks happen each year, including a major outbreak that infected over 400,000 in the Milwaukee, Wisconsin area in 19934. Despite its prevalence, only one drug has been approved for use by the United States Food and Drug Administration (FDA). However that drug, nitazoxanide, has shown limited efficacy, fails to clear infection in immune-compromised patients, and is not approved for use in infants < 1 year-old5–7. Despite great progress in recent years in developing tools to advance the research and treatment of cryptosporidiosis8–9, as well as drug development efforts that have progressed through a variety of possible drug targets and classes10–18, a new clinical treatment has still not been found.
ATP-competitive inhibtors that target calcium-dependent protein kinase 1 (CDPK1) from Cryptosporidium parvum have shown promise as anti-parasitic agents19. CDPK1 is essential for Cryptosporidium parvum survival, but has no analogous protein in mammals, making it a promising target for treatment that may avoid toxic side effects in both humans and livestock. One class of drugs that specifically targets CDPK1 are bumped kinase inhibitors (BKIs) that inhibit cell proliferation in apicomplexan parasites by interfering with invasion and egress from mammalian host cells20–22.
A library consisting of several hundred BKIs has been developed and tested for activity against Cryptosporidium in vitro and in vivo13–14, 23–30. Most of these compounds possess either a 1H-pyrazolo[3,4-d]pyrimidin-4-amine (pyrazolopyrimidine, PP) or a 5-aminopyrazole-4-carboxamide (AC) central scaffold (Figure 1) with substituent groups at the C3 position that occupy a, enlarged hydrophobic pocket adjacent to the glycine gatekeeper residue of CDPK119, 29. Medicinal chemistry efforts around both scaffolds have yielded compounds that are potent against Cryptosporidium in vitro as well as in vivo mouse, piglet, and calf models13–14, 28, 30. However, some of these compounds have exhibited adverse side effects, including specific cardiovascular issues, such as inhibition of the human ether-à-go-go-related gene (hERG) channel that may lead to fatal arrhythmia31–32.
Figure 1.
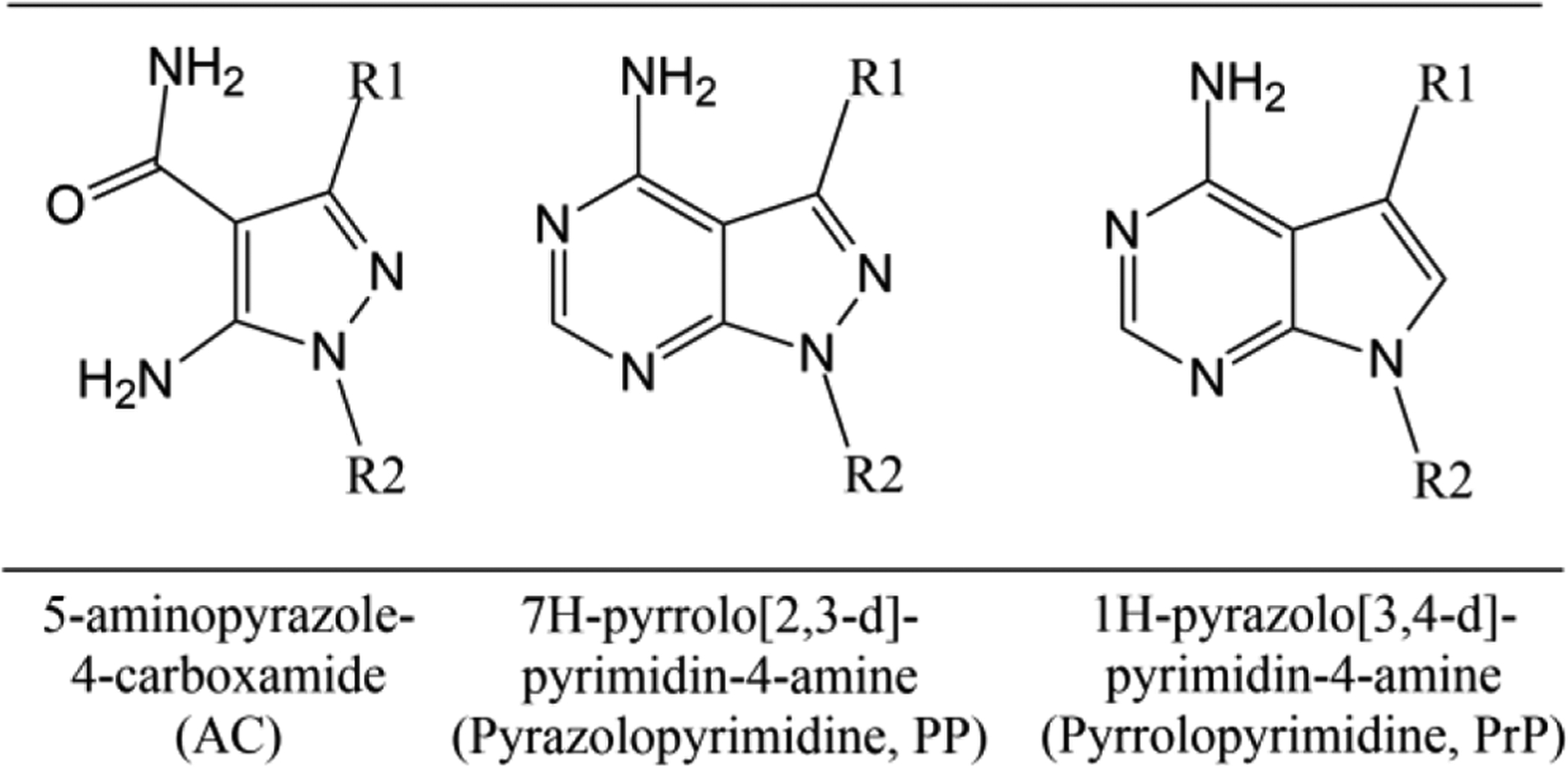
Central scaffolds of bumped kinase inhibitors that target calcium-dependent protein kinase 1 in Cryptosporidium.
Exploration of the chemistry and structure-activity relationship of a small modification to the PP scaffold, replacing nitrogen with carbon in the central scaffold, yielded a new scaffold, 7H-pyrrolo[2,3-d]pyrimidin-4-amine (pyrrolopyrimidine, PrP) (Figure 1), for use in the development of additional BKIs33.
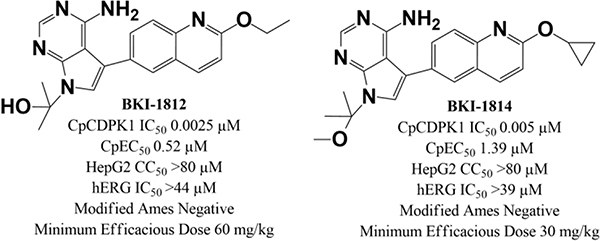
One PrP, BKI-1649 (Figure 2) was shown to be efficacious in C. parvum (Cp) infected mice at significantly lower doses than many PP BKIs require14. BKI-1649 itself was found to be toxic at higher concentrations, as it accumulated in plasma after multiple doses and caused abortions or stillbirths in mice when dosed at therapeutic levels27. It therefore could not be advanced as a possible treatment candidate. Additional PrP compounds were produced and tested against C. parvum CDPK1 (CpCDPK1) and C. parvum in vitro. All were shown to be equally or more efficacious in vitro than their respective PP isosteric counterparts with matching constituent groups. Two PrP BKIs-1812 and –1814 (Figure 2)33, were selected for further testing in multiple in vitro and in vivo models to determine if safety and efficacy of these PrP BKIs would circumvent some of the shortcomings observed within the PP series of BKIs.
Figure 2.
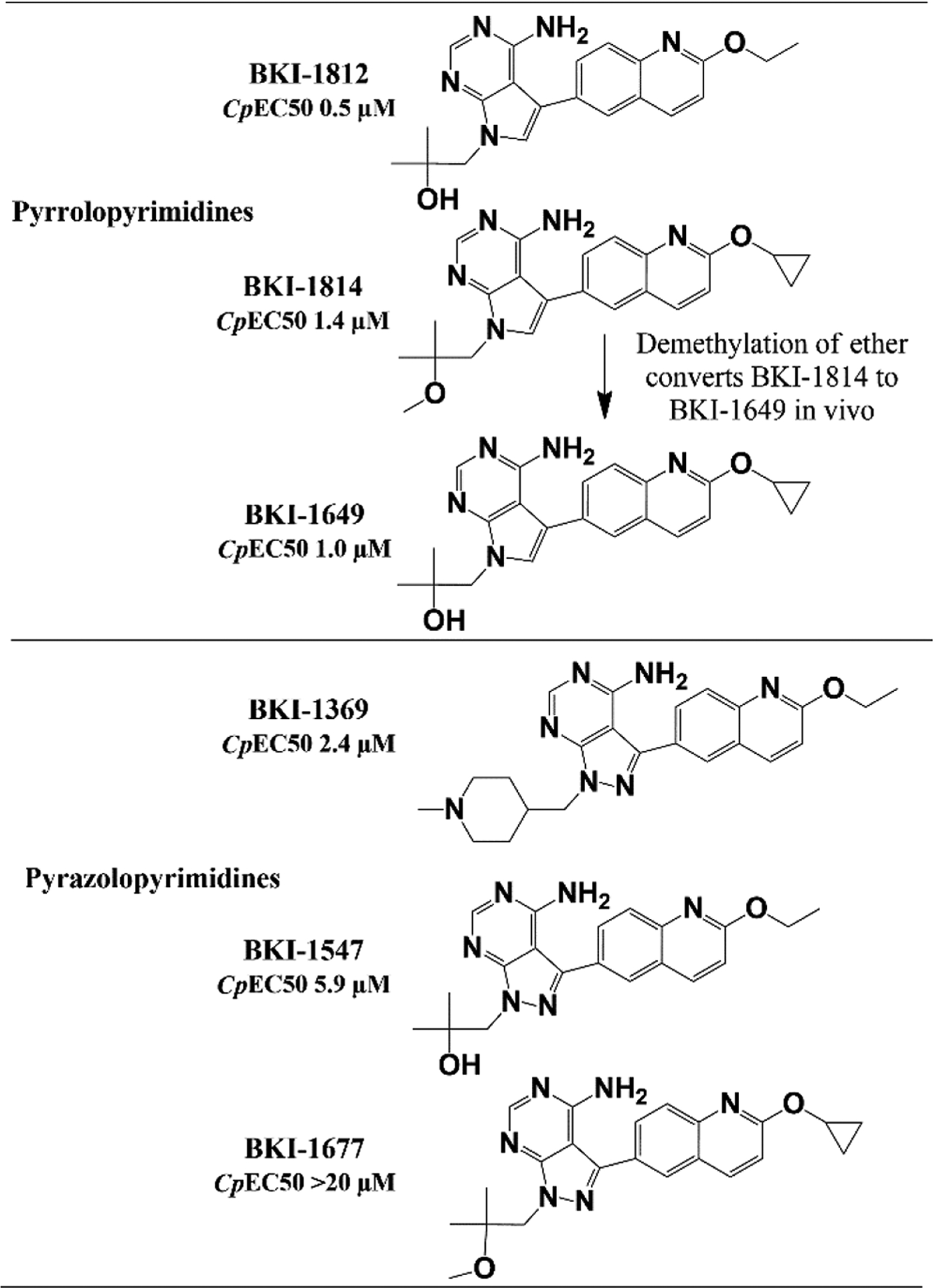
Chemical structures of pyrrolopyrimidines BKIs-1812, –1814, and –1649 and pyrazolopyrimidines BKIs-1369, –1547, and –1677.
RESULTS
In vitro tests for 50% effective concentration (EC50) against C. parvum, 50% inhibitory concentration (IC50) against recombinant CpCDPK1 protein, IC50 against the mammalian tyrosine kinase c-Src (a potential off-target liability in mammals), 50% cytotoxic concentration (CC50) against both CRL-8155 and HepG2 cells, and solubility at pH 2.0 and 6.5 were all previously reported for these compounds (Table 1)33.
Table 1.
In vitro properties of pyrrolopyrimidines, BKI-1812, BKI-1814, and BKI-1649.
| BKI-1812 | BKI-1814 | BKI-1649 | |
|---|---|---|---|
|
CpCDPK1 IC50 (μM) |
0.0025 | 0.005 | 0.0022 |
|
C. parvum EC50 (μM) |
0.52 | 1.39 | 1.03 |
|
CRL-8155 CC50 (μM) |
>80 | >80 | >40 |
|
HepG2 CC50 (μM) |
>80 | >80 | >40 |
|
Solubility
pH 2.0 (μM) |
>100 | >100 | >100 |
|
Solubility
pH 6.5 (μM) |
96.5 | >100 | 83 |
|
hERG IC50 (μM) |
>44 | >39 | >30 |
| Modified Ames | Negative | Negative | ND* |
| In vitro mononucleus genotoxicity | Negative | Negative | ND* |
| Plasma Protein Binding Mouse | 85.0% | 99.9% | 94% |
| Plasma Protein Binding Rat | 91.5% | 94.7% | 90% |
| Plasma Protein Binding Human | 99.9% | 99.9% | 56% |
ND = not done
Additional in vitro testing was conducted for both BKIs to determine their suitability for in vivo testing, including a modified Ames mutagenesis test, in vitro micronucleus genotoxicity test, hERG IC50, and plasma protein binding (Table 1). All of these in vitro test results were compatible with safety in vivo. A kinome panel was also run to screen for off-target activity against human kinases (Table 2). Although BKI-1812 had an IC50 value of 20 nM against binding to protein kinase D3 (Prkcn, or PKD3) and an IC50 of 40 nM against receptor-interacting serine/threonine-protein kinase 2 (RIPK2), these are not clear safety signals that would exclude its consideration for development. In contrast, BKI-1814 had far less activity against the panel of 80 mammalian protein kinases tested.
Table 2.
In vitro inhibition of human kinases.
| BKI-1812 | BKI-1814 | |
|---|---|---|
| IC50 (μM) | ||
| Prkcn | 0.0198 | 0.326 |
| RIPK2 | 0.0375 | 0.203 |
| Aurora2 | 0.284 | 0.405 |
| MAP4K5 | 0.584 | >10 |
| SIK1 | 1.01 | 4.82 |
| Blk | 1.11 | >10 |
| DDR1 | 1.38 | >10 |
| Lck | 1.48 | 4.2 |
| Flt1 | 2.07 | 6.68 |
| Aurora1 | 2.33 | 2.91 |
| IRAK4 | 2.54 | >10 |
| MEK1 | 2.69 | 3.5 |
| MAP4K3 | 2.83 | >10 |
| MST1 | 2.94 | 4.62 |
| CDK8/Cyclin C | 3.7 | >10 |
| TNK2 | 3.71 | >10 |
| RET | 4.02 | 5.18 |
| MINK1 | 4.3 | >10 |
| FGR | 4.39 | >10 |
| MAP4K1 | 4.42 | >10 |
| MAP4K2 | 4.59 | 8.16 |
| MAP4K4 | 4.79 | >10 |
| CSF1R | 5.14 | 6.11 |
| ErbB2 | 6.6 | >10 |
| HCK | 7.04 | >10 |
| STK33 | 7.96 | >10 |
| CSK | 8.75 | >10 |
| TYRO3 | >10 | 6.32 |
| All others tested | >10 | >10 |
Several pharmacokinetic (PK) parameters of BKI-1812 were previously reported for a single 25 mg/kg oral dose in adult female BALB/c mice33. This same assay was performed with a single 25 mg/kg oral dose of BKI-1814 to compare the two compounds. BKI-1814 had much lower oral exposure, with maximum concentration (Cmax) and area-under-the-curve representing total exposure (AUC) values of approximately 10-fold lower and a clearance rate that was 10-fold higher than those for BKI-1812 (Figure 3).
Figure 3.
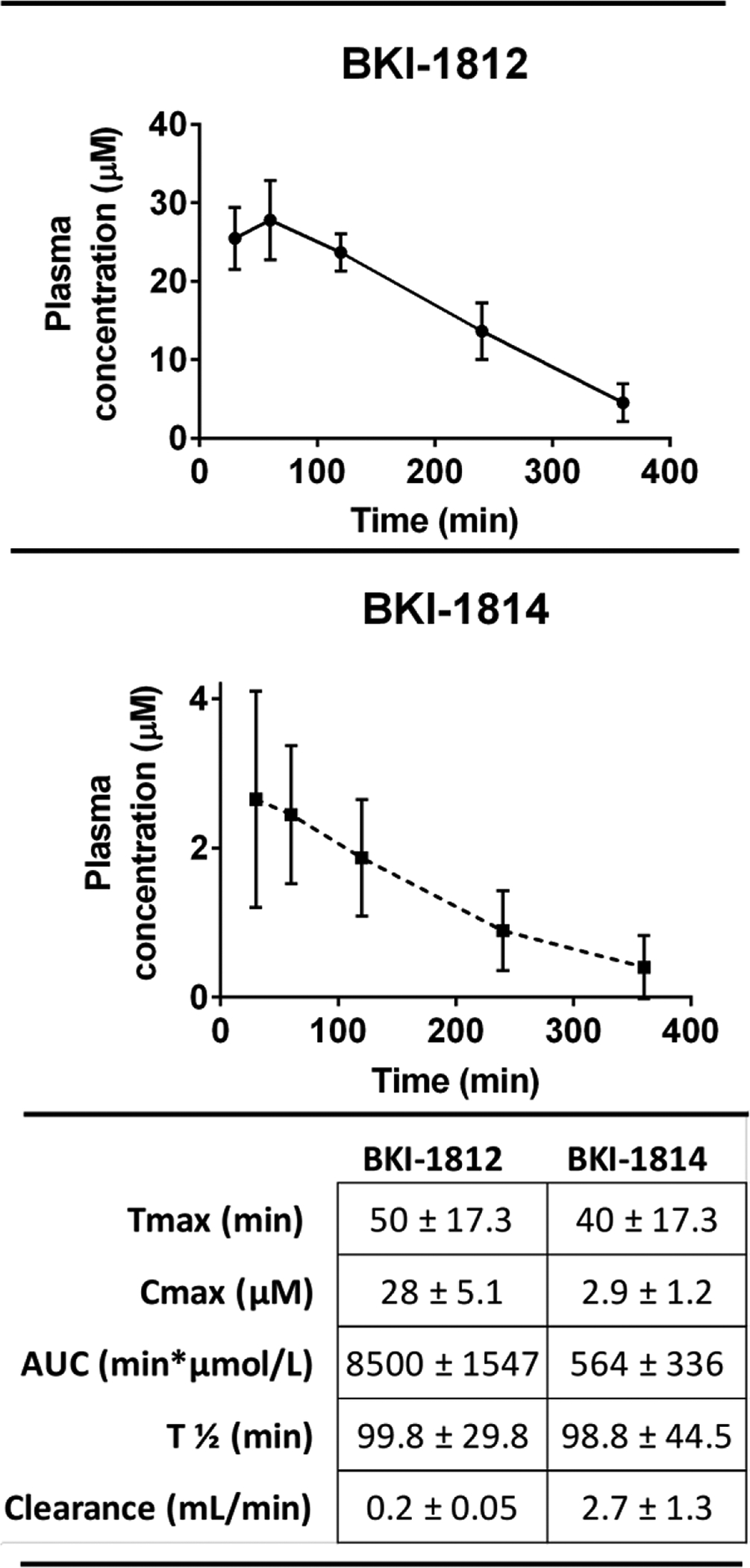
Pharmacokinetics of a single, 25 mg/kg oral dose in uninfected adult female BALB/c mice (n=3) for BKI-1812 and BKI-1814.
However, the time at maximum concentration (Tmax) and half-life (T½) values were similar for both compounds. Despite its relatively lower oral exposure, BKI-1814 was still tested in the mouse efficacy model as previous studies have suggested that in vivo efficacy of some BKI compounds is poorly predicted by high oral exposures14. Indeed, modeling predicts that many BKIs are delivered to cryptosporidium-infected enterocyte from the luminal surface and not from the bloodstream.
To compare the effects of BKI-1812 and BKI-1814 in vivo given their differential oral PK profiles, both compounds were tested as a dose titration in adult female interferon-γ knockout (IFN-γ KO) mice infected with nanoluciferase (Nluc) expressing C. parvum. Groups (n=3/dose) were given oral doses of 60 mg/kg, 30 mg/kg, and 15 mg/kg of BKIs once daily for 5 days beginning on Day 6 post-infection (PI), and infection levels were monitored up to Day 20 PI (Figure 4).
Figure 4.
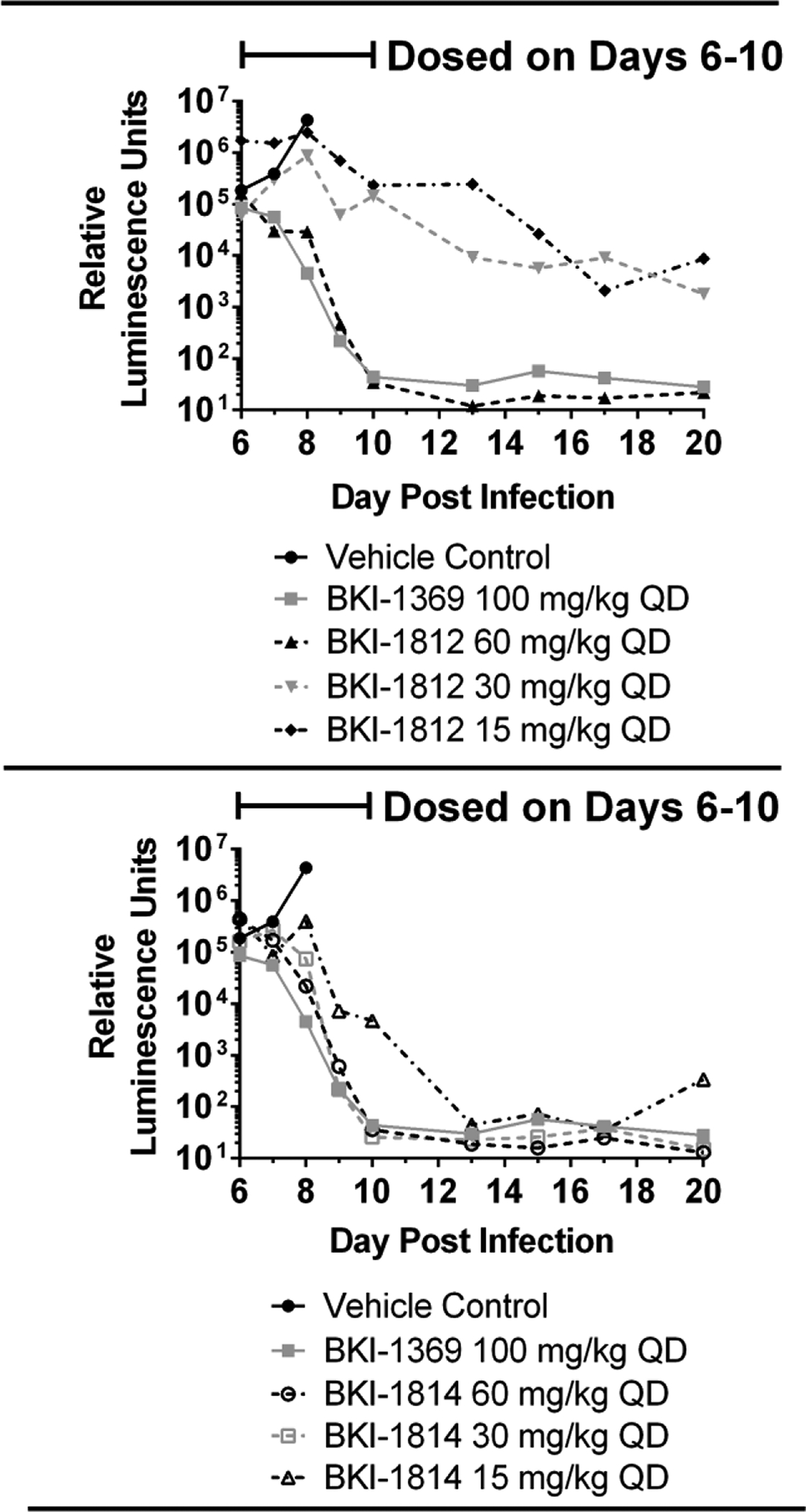
Dose titration of BKI-1812 and BKI-1814 in adult female IFN-γ KO mice (n=3) infected with nanoluciferase-expressing Cryptosporidium parvum. Blood was sampled on Day 9 post infection for LC-MS/MS analysis of plasma concentrations, as indicated in Table 3.
These results were compared to a negative control group dosed with vehicle only and a positive control group dosed with 100 mg/kg of BKI-1369, a PP BKI that is non-toxic in mice and has been shown to reduce infection quickly in this model27. The 60 mg/kg dose of BKI-1812 reduced the infection levels to below the average background signals by the fourth dose, while the 30 mg/kg and 15 mg/kg doses only partially reduced the infection levels by approximately 2 to 2.5 log units below their peaks. Both the 60 mg/kg and 30 mg/kg doses of BKI-1814 reduced the infection to below background by the fourth dose, while 15 mg/kg reduced the infection to below background after the fifth dose. Mice for all dosing groups of BKI-1812 and BKI-1814 appeared healthy and maintained their weights throughout the experiment until Day 20 PI. This was a marked improvement over the vehicle-only control mice, which rapidly lost weight, became dehydrated, and were euthanized by Day 8 PI per animal ethics protocol due to a ≥15% weight loss.
Blood was taken on the fourth day of dosing (Day 9 PI) to determine the plasma exposure after reaching steady state for each dose group (Table 3). During the analysis of BKI-1814, a second major peak was observed on the LC-MS/MS that matched the mass to charge ratio (m/z) and retention time of BKI-1649, a metabolic demethylation is likely to convert BKI-1814 to BKI-1649 (Figure 2). Due to this observation, all the plasma samples for the IFN-γ KO mouse efficacy study were reanalyzed by LC-MS/MS to determine concentrations of both the parent compound, BKI-1814, and its metabolite, BKI-1649 (Table 3).
Table 3.
Mean ± standard deviation plasma concentrations of BKIs –1812, –1814, and –1649. Taken from the 4th day (day 9 PI) of dosing C. parvum infected adult female IFN-γ KO mice.
| BKI-1812 | 60 mg/kg | 30 mg/kg | 15 mg/kg |
|---|---|---|---|
| Time sampled | Plasma concentration (μM) | ||
| predose | 15.7 ± 11.7 | 2.1 ± 0.2 | 1.5 ± 0.3 |
| 1 h post dose | 258.9 ± 22.5 | 111.4 ± 71 | 49.4 ± 26.3 |
| 2 h post dose | 250.5 ± 48.4 | 115.7 ± 65.9 | 37.4 ± 19.3 |
| 4 h post dose | 170.1 ± 110.8 | 54.6 ± 31.7 | 14.1 ± 9.2 |
| BKI-1814 | 60 mg/kg | 30 mg/kg | 15 mg/kg |
| Time sampled | Plasma concentration (μM) | ||
| predose | 1.83 ± 1.4 | 0.03 ± 0.06 | 0.24 ± 0.2 |
| 0.5 h post dose | 17.5 ± 10.7 | 7.1 ± 2.5 | 1.68 ± 0.6 |
| 1 h post dose | 19.5 ± 9.8 | 6.88 ± 1.5 | 2.46 ± 0.5 |
| 2 h post dose | 18.2 ± 10.2 | 5.86 ± 1.6 | 2.25 ± 0.8 |
| BKI-1649* | 60 mg/kg | 30 mg/kg | 15 mg/kg |
| Time sampled | Plasma concentration (μM) | ||
| predose | 346 ± 78 | 113 ± 31 | 76 ± 1.2 |
| 0.5 h post dose | 340 ± 177 | 110 ± 65 | 79 ± 44 |
| 1 h post dose | 345 ± 188 | 111 ± 66 | 82 ± 46 |
| 2 h post dose | 360 ± 199 | 134 ± 81 | 93 ± 51 |
BKI-1649 formed in vivo by demethylation of ether on BKI-1814
To further evaluate the safety of these compounds in regards to effects on cardiovascular function, each was dosed in an in vivo rat cardiovascular safety pharmacology assay (three back-to-back, ascending doses of each compound, each dose continuously administered intravenously for 30 minutes). BKI-1812 showed biologically-relevant increases of 18% in heart rate and 23% in cardiac contractility at the highest dose tested, 30 mg/kg (Table 4). Plasma levels at this dose were sampled and determined by LC-MS/MS to be 188 μM. BKI-1814 showed no biologically-relevant changes to any of the measured parameters in this assay up to a maximum plasma concentration of 15.3 μM following a 30 mg/kg dose (Table 4).
Table 4.
Cardiovascular effects of intravenous infusion in anesthetized rats (n=3). A change of ± 15% is considered biologically relevant for mean arterial pressure (MAP) or heart rate (HR). A change of ± 20% is considered biologically relevant for cardiac contractility (dP/dt@50). Plasma concentration values are listed as mean ± SEM. % values are listed as mean change relative to vehicle.
| Dose (mg/kg) | Plasma Concentration (μM) | MAP (%) | HR (%) | dP/dt@50 (%) | |
|---|---|---|---|---|---|
| BKI-1812 | 3 | 1.59 ± 0.42 | −2 | 5 | 5 |
| 10 | 11.3 ± 0.5 | 4 | 13 | 10 | |
| 30 | 188 ± 44.1 | −5 | 18 | 23 | |
| BKI-1814 | 3 | 1.74 ± 0.04 | −3 | −3 | 4 |
| 10 | 6.67 ± 0.37 | −4 | −5 | 6 | |
| 30 | 15.3 ± 0.04 | −6 | −9 | 16 |
DISCUSSION
In vitro results suggest that both BKI-1812 and BKI-1814 show promise for further development in the search for superior drugs against cryptosporidiosis. Both are negative in the Ames and IVMN assays and demonstrate no signs of cytotoxicity in the mammalian cells tested33. Both were also sufficiently soluble at pH values representative of those in the stomach and small intestine, suggesting the compounds would stay reasonably soluble and available for absorption from the lumen and transport to the site of infection within the enterocytes13. Plasma protein binding is high for both of these BKIs, but it is unclear how this may relate to free levels in the fecal stream delivered to enterocytes. As Cryptosporidium infection is typically localized to enterocytes, the percentage of plasma binding may only be a concern in the context of toxic side effects due to systemic exposure. As such, the high degree of protein binding should be beneficial, acting as a buffer and protecting major organs in mammalian hosts from exposure-related toxicity. In the two C. parvum mouse efficacy experiments, it was necessary to sacrifice the vehicle control animals after Day 8 post-infection because the mice had lost >20% of their baseline body weight and appeared very ill. Thus, there were few days over which the BKI-treated animals could be compared. However, even the lower doses of BKI-1812 that did not appear to greatly reduce the parasite excretion, they prevented the weight loss and apparent illness seen in controls.
The kinome panels revealed several human kinases that are affected by both BKIs at IC50 values for binding of less than 10 μM, but only a small fraction that reach sub-micromolar IC50 values. Both showed activity against RIPK2, Prkcn, and serine/threonine-protein kinase aurora 2 (Aurora 2), with BKI-1812 also acting on mitogen-activated protein kinase kinase kinase kinase 5 (MAP4K5). RIPK2 has a biological role in promoting inflammation-related immune responses. While some drugs that inhibit RIPK2 have been investigated as a treatment for inflammation and autoimmune diseases, such as Crohn’s disease, it can also potentially limit the inflammatory response to bacterial infections34. However, it is uncertain whether this control of inflammation may be beneficial in the case of cryptosporidiosis since gut inflammation associated with pathogenic infections has been shown to lead to environmental enteropathy (EE)35. EE causes sustained impairment of growth and development due to malnutrition and repeated enteric infections, which lead to damage to the gut lining and dysbiosis, even after the pathogen has been cleared35. Prkcn, commonly referred to as PKCn or PKD3, plays a role in multiple biological pathways, including cell cycle regulation, programmed cell death, and gene expression36. Aurora 2 kinase has been shown to play a role in mitosis and is essential for cell proliferation37. MAP4K5 expression has been studied in pancreatic cancer, with a low expression of MAP4K5 correlating with decreased survival among patients38. Consequently, inhibition of any one of these three kinases has the potential to increase the likelihood of oncogenesis. Therefore, extended safety studies would be required to determine if these liabilities translate to increased risk of cancer in vivo. However, a short duration of 5 days dosing needed for cryptosporidiosis therapy may alleviate some concerns about these effects.
Other concerns for BKI-1812 and BKI-1814 arise when observing their PK properties at efficacious doses. BKI-1812 reached a high Cmax during the efficacy study, achieving 250 μM in plasma in mice dosed with 60 mg/kg. This concentration exceeds the levels that led to increases in heart rate and cardiac contractility in rats and is high enough to be of concern to inhibit mammalian kinases. Even at a dose of 15 mg/kg, plasma levels of BKI-1812 reached a Cmax value of 50 μM, allowing for less than a four-fold difference between this treatment and levels that led to changes in cardiovascular function in rats, and it could also be of concern for inhibition of mammalian kinases. For BKI-1814, plasma levels at the efficacious doses of 30 mg/kg and 15 mg/kg remain well below acceptable levels in the rat cardiovascular assay. However, BKI-1814 is quickly metabolized into high plasma concentrations of BKI-1649. Given BKI-1649’s aforementioned abortifacient toxic side effects in pregnant mice, this leaves no room to establish a safety window between an efficacious dose of BKI-1814 and the onset of toxicity brought on by high levels of BKI-1649. It is also possible that BKI-1814 is metabolized into BKI-1649 at similar levels in the gut as are seen in plasma. Given that BKI-1649 is efficacious at such low doses14, this could mean that the BKI-1649 that was metabolized from BKI-1814 was driving the efficacy and not the parent compound itself.
CONCLUSION
Although these two particular BKIs have properties that likely disqualify them from further consideration for human treatment, there are promising signs that the PrP scaffold may show some improvement over the more extensively explored PP BKIs in terms of potency, though this improvement is inconsistent and only results from certain pairings of constituent groups on the scaffolds. Additionally, other factors, such as hERG inhibition and cytotoxicity are equivalent between the PP and PrP isoteric counterparts when the same substituent groups are present. Mindful of these comparisons, it is worth examining both the PP and PrP scaffolds for any given set of equivalent constituent groups to determine whether the PrP variant would provide improved results. Overall, the data from these studies serve as a strong proof-of-concept for the continued development of the PrP central scaffold as inhibitors of CpCDPK1 for the treatment of Cryptosporidium.
METHODS
Single dose pharmacokinetics.
Methods for PK analysis of mouse plasma concentrations of BKI by LC-MS/MS analysis have been previously described14, 28, 39. Briefly, three female BALB/c mice (10–12 weeks old) were used in each group. Each group was administered an oral dose via gavage of an individual BKI dissolved in 3% ethanol/7% Tween 80/90% saline at a dose of 25 mg/kg body weight. Blood samples were taken by tail bleeding into heparinized tubes at designated time points and centrifuged to obtain plasma. The plasma samples were frozen at −20°C.
Plasma protein binding.
Methods for in vitro plasma protein binding using dialysis membranes have been previously described40. Briefly, a dialysis membrane sheet (MW cutoff 3.5 kDa) (HTDialysis, LLC, Gales Ferry, CT) was soaked for 1 h in distilled water and then in 20% ethanol for 30 min. The membrane was clamped between two Teflon plates containing a row of opposing wells. Test compound in DMSO was added to 0.12 mL of serum to a concentration of 9 μM, a small aliquot was taken as a 100% recovery standard, and the solution was placed on one side of the membrane. The well on the other side of the membrane was charged with an equal volume of DPBS, and the plate was placed on an orbital shaker for 18 hours at 37°C. An aliquot was taken from each side of the membrane. One-fourth volume of acetonitrile was added to each aliquot, and the samples were centrifuged to precipitate protein. Test compound in the supernatants was quantified by LC/MS analysis to determine the concentration on each side of the membrane and the total recovery of test compound from the device. A control dialysis was carried out with dialysis buffer on both sides of the membrane and test compound on one side to ensure that equilibration across the membrane was achieved. The fraction of compound bound to protein was calculated as bound/(unbound + bound).
Rat cardiovascular screening.
Methods for rat cardiovascular screening have been previously described41. Briefly, male Sprague-Dawley rats (325–375 g, Charles River, Indianapolis, IN) were anesthetized with the long-acting barbiturate Inactin (100 mg/kg i.p.). A catheter (PE50) was placed in the femoral artery for measurement of mean arterial blood pressure (MAP) and heart rate (HR). A 3F Micro-Tip catheter (Millar Instruments, Houston, TX) was advanced into the left ventricle to measure dP/dt50, a recognized index of cardiac contractility. An additional catheter (PE50) was placed in the femoral vein for administering escalating i.v. doses of drugs (dosing volume 1 mL/kg/30 min). After a 30 min baseline or equilibration period, each compound was tested using a dose-escalating protocol consisting of three 30 min escalating i.v. infusions. All studies described were acute studies, with 20% N,N-dimethylacetamide (DMA)/40% propylene glycol (PG)/40% polyethylene glycol (PEG-400) used as a formulation for acute drug administration for iv infusion at a constant rate with step-wise ascending drug concentrations encompassing therapeutic and projected supratherapeutic drug exposures. Hemodynamic data (mean arterial pressure, heart rate, and cardiac contractility) was monitored continuously, acquired every 10 s, and recorded every 5-min using a PONEMAH Physiology Platform (version number 5.2, Data Sciences International, St Paul, MN). Blood samples (each 0.25 mL volume) were obtained from the femoral artery at the end of each infusion period to assess drug exposures
C. parvum efficacy in mice.
Methods for Nluc expressing C. parvum in an infected IFN-γ KO mouse efficacy model have been previously described14. Briefly, female interferon-γ knock-out (IFN-γ KO) mice (B6.129S7-Ifngtm1Ts/J, Jackson Laboratories), aged 8–10 weeks, were infected by oral gavage (PO) with 1,000 to 10,000 UGA1 Nluc expressing C. parvum oocysts in 0.1 mL DPBS. Beginning on Day 6 PI, mice were dosed PO with BKI suspended in 0.2 mL oral vehicle (3% ethanol/7% Tween 80/90% saline) or vehicle only once daily for 5 days. Mice were moved to clean cages after each dose and fecal collection. Feces was collected daily and weighed from each group during dosing and then twice weekly out to 21 days PI. Each fecal sample was checked for luminescence9 on day of collection and the relative luminescence unit readings were normalized to fecal sample weights.
LC-MS/MS Analysis.
All LC-MS/MS analytes were measured with an Acquity ultra-performance liquid chromatography (UPLC) system in tandem with a Xevo TQ-S micro mass spectrometer (Waters, Milford, MA, USA). PK calculations of Cmax, Tmax, AUC, clearance, and T½ were performed using Pharsight Phoenix WinNonlin software (Certara, St. Louis, MO).
Additional methods.
Methods for hERG channel effects assessment by automated electrophysiological patch clamp42. in vitro micronucleus genotoxicity assay (IVMN)43, the 24-well modified Ames mutagenesis assay44, and kinome profiling45, have been previously described in detail.
ACKNOWLEDGMENTS
We thank Geno De Hostos and Robert Choy (PATH Drug Solutions), for their consultation in the drug development pathway, and Gail M. Freiberg (AbbVie) for distributing compounds and tracking them for the hERG inhibition, kinome inhibition, Ames and IVMN in vitro results and in vivo rat cardiotoxicity of the BKIs.
Funding Sources
This work was supported by the US Department of Veterans Affairs Biomedical Laboratory Research and Development Career Development Award (grant number BX002440); National Institute of Allergy and Infectious Diseases, National Institutes of Health (grant numbers R21AI123690, R01AI089441, R01AI111341, R01A1112427, and R01HD080670); and the US Department of Agriculture, National Institute of Food and Agriculture (grant number 2014-06183).
Footnotes
ANIMAL ETHICS All animal experiments conducted at the University of Washington, USA, and AbbVie, Inc. were approved by the respective Institutional Animal Care and Use Committees at these institutions. All animals used in these studies were handled in strict accordance with practices made to minimize suffering.
Publisher's Disclaimer: Disclaimer
Publisher's Disclaimer: The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the US Department of Agriculture National Institute of Food and Agriculture
Potential Conflicts of Interest
We have no conflicts of interest to report except that W.V.V. is the president of and owns stock in ParaTheraTech Inc., a small animal health company that is developing BKIs as potential animal therapeutics. W.V.V. helped to plan the experiments and edited the paper. He did not carry out the experiments or do the initial interpretation.
REFERENCES
- 1.Kotloff KL; Nataro JP; Blackwelder WC; Nasrin D; Farag TH; Panchalingam S; Wu Y; Sow SO; Sur D; Breiman RF; Faruque AS; Zaidi AK; Saha D; Alonso PL; Tamboura B; Sanogo D; Onwuchekwa U; Manna B; Ramamurthy T; Kanungo S; Ochieng JB; Omore R; Oundo JO; Hossain A; Das SK; Ahmed S; Qureshi S; Quadri F; Adegbola RA; Antonio M; Hossain MJ; Akinsola A; Mandomando I; Nhampossa T; Acacio S; Biswas K; O’Reilly CE; Mintz ED; Berkeley LY; Muhsen K; Sommerfelt H; Robins-Browne RM; Levine MM (2013) Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382, 209–222. DOI: 10.1016/s0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 2.Checkley W; White AC Jr.; Jaganath D; Arrowood MJ; Chalmers RM; Chen XM; Fayer R; Griffiths JK; Guerrant RL; Hedstrom L; Huston CD; Kotloff KL; Kang G; Mead JR; Miller M; Petri WA Jr.; Priest JW; Roos DS; Striepen B; Thompson RC; Ward HD; Van Voorhis WA; Xiao L; Zhu G; Houpt ER (2015) A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet. Infect. Dis 15, 85–94. DOI: 10.1016/s1473-3099(14)70772-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Platts-Mills JA; Babji S; Bodhidatta L; Gratz J; Haque R; Havt A; McCormick BJ; McGrath M; Olortegui MP; Samie A; Shakoor S; Mondal D; Lima IF; Hariraju D; Rayamajhi BB; Qureshi S; Kabir F; Yori PP; Mufamadi B; Amour C; Carreon JD; Richard SA; Lang D; Bessong P; Mduma E; Ahmed T; Lima AA; Mason CJ; Zaidi AK; Bhutta ZA; Kosek M; Guerrant RL; Gottlieb M; Miller M; Kang G; Houpt ER (2015) Pathogen-specific burdens of community diarrhoea in developing countries: a multisite birth cohort study (MAL-ED). Lancet Glob. Health 3, e564–e575. DOI: 10.1016/s2214-109x(15)00151-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mac Kenzie WR; Hoxie NJ; Proctor ME; Gradus MS; Blair KA; Peterson DE; Kazmierczak JJ; Addiss DG; Fox KR; Rose JB; Davis JP (1994) A massive outbreak in Milwaukee of cryptosporidium infection transmitted through the public water supply. N. Engl. J. Med 331, 161–167. DOI: 10.1056/NEJM199407213310304. [DOI] [PubMed] [Google Scholar]
- 5.Amadi B; Mwiya M; Musuku J; Watuka A; Sianongo S; Ayoub A; Kelly P (2002) Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: a randomised controlled trial. Lancet 360, 1375–1380. DOI: 10.1016/s0140-6736(02)11401-2. [DOI] [PubMed] [Google Scholar]
- 6.Rossignol JF; Ayoub A; Ayers MS (2001) Treatment of diarrhea caused by Cryptosporidium parvum: a prospective randomized, double-blind, placebo-controlled study of Nitazoxanide. J. Infect. Dis 184, 103–106. DOI: 10.1086/321008. [DOI] [PubMed] [Google Scholar]
- 7.Sparks H; Nair G; Castellanos-Gonzalez A; White AC Jr. (2015) Treatment of Cryptosporidium: What We Know, Gaps, and the Way Forward. Curr. Trop. Med. Rep 2, 181–187. DOI: 10.1007/s40475-015-0056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Love MS; Beasley FC; Jumani RS; Wright TM; Chatterjee AK; Huston CD; Schultz PG; McNamara CW (2017) A high-throughput phenotypic screen identifies clofazimine as a potential treatment for cryptosporidiosis. PLoS Neglected Trop. Dis 11, e0005373. DOI: 10.1371/journal.pntd.0005373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vinayak S; Pawlowic MC; Sateriale A; Brooks CF; Studstill CJ; Bar-Peled Y; Cipriano MJ; Striepen B (2015) Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature 523, 477–480. DOI: 10.1038/nature14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baragana B; Forte B; Choi R; Nakazawa Hewitt S; Bueren-Calabuig JA; Pisco JP; Peet C; Dranow DM; Robinson DA; Jansen C; Norcross NR; Vinayak S; Anderson M; Brooks CF; Cooper CA; Damerow S; Delves M; Dowers K; Duffy J; Edwards TE; Hallyburton I; Horst BG; Hulverson MA; Ferguson L; Jimenez-Diaz MB; Jumani RS; Lorimer DD; Love MS; Maher S; Matthews H; McNamara CW; Miller P; O’Neill S; Ojo KK; Osuna-Cabello M; Pinto E; Post J; Riley J; Rottmann M; Sanz LM; Scullion P; Sharma A; Shepherd SM; Shishikura Y; Simeons FRC; Stebbins EE; Stojanovski L; Straschil U; Tamaki FK; Tamjar J; Torrie LS; Vantaux A; Witkowski B; Wittlin S; Yogavel M; Zuccotto F; Angulo-Barturen I; Sinden R; Baum J; Gamo FJ; Maser P; Kyle DE; Winzeler EA; Myler PJ; Wyatt PG; Floyd D; Matthews D; Sharma A; Striepen B; Huston CD; Gray DW; Fairlamb AH; Pisliakov AV; Walpole C; Read KD; Van Voorhis WC; Gilbert IH (2019) Lysyl-tRNA synthetase as a drug target in malaria and cryptosporidiosis. Proc. Natl. Acad. Sci. U. S. A 116, 7015–7020. DOI: 10.1073/pnas.1814685116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buckner FS; Ranade RM; Gillespie JR; Shibata S; Hulverson MA; Zhang Z; Huang W; Choi R; Verlinde C; Hol WGJ; Ochida A; Akao Y; Choy RKM; Van Voorhis WC; Arnold SLM; Jumani RS; Huston CD; Fan E (2019) Optimization of Methionyl tRNA-Synthetase Inhibitors for Treatment of Cryptosporidium Infection. Antimicrob. Agents Chemother 63, e02061–18. DOI: 10.1128/aac.02061-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hennessey KM; Rogiers IC; Shih HW; Hulverson MA; Choi R; McCloskey MC; Whitman GR; Barrett LK; Merritt EA; Paredez AR; Ojo KK (2018) Screening of the Pathogen Box for inhibitors with dual efficacy against Giardia lamblia and Cryptosporidium parvum. PLoS Neglected Trop. Dis 12, e0006673. DOI: 10.1371/journal.pntd.0006673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang W; Hulverson MA; Choi R; Arnold SLM; Zhang Z; McCloskey MC; Whitman GR; Hackman RC; Rivas KL; Barrett LK; Ojo KK; Van Voorhis WC; Fan E (2019) Development of 5-Aminopyrazole-4-carboxamide-based Bumped-Kinase Inhibitors for Cryptosporidiosis Therapy. J. Med. Chem 62, 3135–3146. DOI: 10.1021/acs.jmedchem.9b00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hulverson MA; Vinayak S; Choi R; Schaefer DA; Castellanos-Gonzalez A; Vidadala RSR; Brooks CF; Herbert GT; Betzer DP; Whitman GR; Sparks HN; Arnold SLM; Rivas KL; Barrett LK; White AC Jr.; Maly DJ; Riggs MW; Striepen B; Van Voorhis WC; Ojo KK (2017) Bumped-Kinase Inhibitors for Cryptosporidiosis Therapy. J. Infect. Dis 215, 1275–1284. DOI: 10.1093/infdis/jix120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janes J; Young ME; Chen E; Rogers NH; Burgstaller-Muehlbacher S; Hughes LD; Love MS; Hull MV; Kuhen KL; Woods AK; Joseph SB; Petrassi HM; McNamara CW; Tremblay MS; Su AI; Schultz PG; Chatterjee AK (2018) The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. U. S. A 115, 10750–10755. DOI: 10.1073/pnas.1810137115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jumani RS; Bessoff K; Love MS; Miller P; Stebbins EE; Teixeira JE; Campbell MA; Meyers MJ; Zambriski JA; Nunez V; Woods AK; McNamara CW; Huston CD (2018) A Novel Piperazine-Based Drug Lead for Cryptosporidiosis from the Medicines for Malaria Venture Open-Access Malaria Box. Antimicrob. Agents Chemother 62, e01505–17. DOI: 10.1128/aac.01505-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lunde CS; Stebbins EE; Jumani RS; Hasan MM; Miller P; Barlow J; Freund YR; Berry P; Stefanakis R; Gut J; Rosenthal PJ; Love MS; McNamara CW; Easom E; Plattner JJ; Jacobs RT; Huston CD (2019) Identification of a potent benzoxaborole drug candidate for treating cryptosporidiosis. Nat. Commun 10, 2816. DOI: 10.1038/s41467-019-10687-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manjunatha UH; Vinayak S; Zambriski JA; Chao AT; Sy T; Noble CG; Bonamy GMC; Kondreddi RR; Zou B; Gedeck P; Brooks CF; Herbert GT; Sateriale A; Tandel J; Noh S; Lakshminarayana SB; Lim SH; Goodman LB; Bodenreider C; Feng G; Zhang L; Blasco F; Wagner J; Leong FJ; Striepen B; Diagana TT (2017) A Cryptosporidium PI(4)K inhibitor is a drug candidate for cryptosporidiosis. Nature 546, 376–380. DOI: 10.1038/nature22337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy RC; Ojo KK; Larson ET; Castellanos-Gonzalez A; Perera BG; Keyloun KR; Kim JE; Bhandari JG; Muller NR; Verlinde CL; White AC Jr.; Merritt EA; Van Voorhis WC; Maly DJ (2010) Discovery of Potent and Selective Inhibitors of Calcium-Dependent Protein Kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med. Chem. Lett 1, 331–335. DOI: 10.1021/ml100096t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kieschnick H; Wakefield T; Narducci CA; Beckers C (2001) Toxoplasma gondii attachment to host cells is regulated by a calmodulin-like domain protein kinase. J. Biol. Chem 276, 12369–12377. DOI: 10.1074/jbc.M011045200. [DOI] [PubMed] [Google Scholar]
- 21.Lourido S; Tang K; Sibley LD (2012) Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J. 31, 4524–4534. DOI: 10.1038/emboj.2012.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ojo KK; Larson ET; Keyloun KR; Castaneda LJ; Derocher AE; Inampudi KK; Kim JE; Arakaki TL; Murphy RC; Zhang L; Napuli AJ; Maly DJ; Verlinde CL; Buckner FS; Parsons M; Hol WG; Merritt EA; Van Voorhis WC (2010) Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat. Struct. Mol. Biol 17, 602–607. DOI: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnold SLM; Choi R; Hulverson MA; Schaefer DA; Vinayak S; Vidadala RSR; McCloskey MC; Whitman GR; Huang W; Barrett LK; Ojo KK; Fan E; Maly DJ; Riggs MW; Striepen B; Van Voorhis WC (2017) Necessity of Bumped Kinase Inhibitor Gastrointestinal Exposure in Treating Cryptosporidium Infection. J. Infect. Dis 216, 55–63. DOI: 10.1093/infdis/jix247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castellanos-Gonzalez A; Sparks H; Nava S; Huang W; Zhang Z; Rivas K; Hulverson MA; Barrett LK; Ojo KK; Fan E; Van Voorhis WC; White AC Jr. (2016) A Novel Calcium-Dependent Kinase Inhibitor, Bumped Kinase Inhibitor 1517, Cures Cryptosporidiosis in Immunosuppressed Mice. J. Infect. Dis 214, 1850–1855. DOI: 10.1093/infdis/jiw481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castellanos-Gonzalez A; White AC Jr.; Ojo KK; Vidadala RS; Zhang Z; Reid MC; Fox AM; Keyloun KR; Rivas K; Irani A; Dann SM; Fan E; Maly DJ; Van Voorhis WC (2013) A novel calcium-dependent protein kinase inhibitor as a lead compound for treating cryptosporidiosis. J. Infect. Dis.s 208, 1342–8. DOI: 10.1093/infdis/jit327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang W; Choi R; Hulverson MA; Zhang Z; McCloskey MC; Schaefer DA; Whitman GR; Barrett LK; Vidadala RSR; Riggs MW; Maly DJ; Van Voorhis WC; Ojo KK; Fan E (2017) 5-Aminopyrazole-4-Carboxamide-Based Compounds Prevent the Growth of Cryptosporidium parvum. Antimicrob. Agents Chemother 61, e00020–17. DOI: 10.1128/aac.00020-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hulverson MA; Choi R; Arnold SLM; Schaefer DA; Hemphill A; McCloskey MC; Betzer DP; Muller J; Vidadala RSR; Whitman GR; Rivas KL; Barrett LK; Hackman RC; Love MS; McNamara CW; Shaughnessy TK; Kondratiuk A; Kurnick M; Banfor PN; Lynch JJ; Freiberg GM; Kempf DJ; Maly DJ; Riggs MW; Ojo KK; Van Voorhis WC (2017) Advances in bumped kinase inhibitors for human and animal therapy for cryptosporidiosis. Int. J. Parasitol 47, 753–763. DOI: 10.1016/j.ijpara.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaefer DA; Betzer DP; Smith KD; Millman ZG; Michalski HC; Menchaca SE; Zambriski JA; Ojo KK; Hulverson MA; Arnold SL; Rivas KL; Vidadala RS; Huang W; Barrett LK; Maly DJ; Fan E; Van Voorhis WC; Riggs MW (2016) Novel Bumped Kinase Inhibitors Are Safe and Effective Therapeutics in the Calf Clinical Model for Cryptosporidiosis. J. Infect. Dis 214, 1856–1864. DOI: 10.1093/infdis/jiw488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z; Ojo KK; Vidadala R; Huang W; Geiger JA; Scheele S; Choi R; Reid MC; Keyloun KR; Rivas K; Siddaramaiah LK; Comess KM; Robinson KP; Merta PJ; Kifle L; Hol WG; Parsons M; Merritt EA; Maly DJ; Verlinde CL; Van Voorhis WC; Fan E (2014) Potent and selective inhibitors of CDPK1 from T. gondii and C. parvum based on a 5-aminopyrazole-4-carboxamide scaffold. ACS Med. Chem. Lett 5, 40–44. DOI: 10.1021/ml400315s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S; Ginese M; Beamer G; Danz HR; Girouard DJ; Chapman-Bonofiglio SP; Lee M; Hulverson MA; Choi R; Whitman GR; Ojo KK; Arnold SLM; Van Voorhis WC; Tzipori S (2018) Therapeutic Efficacy of Bumped Kinase Inhibitor 1369 in a Pig Model of Acute Diarrhea Caused by Cryptosporidium hominis. Antimicrob. Agents Chemother 62, e00147–18. DOI: 10.1128/aac.00147-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ojo KK; Eastman RT; Vidadala R; Zhang Z; Rivas KL; Choi R; Lutz JD; Reid MC; Fox AM; Hulverson MA; Kennedy M; Isoherranen N; Kim LM; Comess KM; Kempf DJ; Verlinde CL; Su XZ; Kappe SH; Maly DJ; Fan E; Van Voorhis WC (2014) A specific inhibitor of PfCDPK4 blocks malaria transmission: chemical-genetic validation. J. Infect. Dis 209, 275–284. DOI: 10.1093/infdis/jit522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vidadala RS; Rivas KL; Ojo KK; Hulverson MA; Zambriski JA; Bruzual I; Schultz TL; Huang W; Zhang Z; Scheele S; DeRocher AE; Choi R; Barrett LK; Siddaramaiah LK; Hol WG; Fan E; Merritt EA; Parsons M; Freiberg G; Marsh K; Kempf DJ; Carruthers VB; Isoherranen N; Doggett JS; Van Voorhis WC; Maly DJ (2016) Development of an Orally Available and Central Nervous System (CNS) Penetrant Toxoplasma gondii Calcium-Dependent Protein Kinase 1 (TgCDPK1) Inhibitor with Minimal Human Ether-a-go-go-Related Gene (hERG) Activity for the Treatment of Toxoplasmosis. J. Med. Chem 59, 6531–6546. DOI: 10.1021/acs.jmedchem.6b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vidadala RSR; Golkowski M; Hulverson MA; Choi R; McCloskey MC; Whitman GR; Huang W; Arnold SLM; Barrett LK; Fan E; Merritt EA; Van Voorhis WC; Ojo KK; Maly DJ (2018) 7 H-Pyrrolo[2,3- d]pyrimidin-4-amine-Based Inhibitors of Calcium-Dependent Protein Kinase 1 Have Distinct Inhibitory and Oral Pharmacokinetic Characteristics Compared with 1 H-Pyrazolo[3,4- d]pyrimidin-4-amine-Based Inhibitors. ACS Infect. Dis 4, 516–522. DOI: 10.1021/acsinfecdis.7b00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canning P; Ruan Q; Schwerd T; Hrdinka M; Maki JL; Saleh D; Suebsuwong C; Ray S; Brennan PE; Cuny GD; Uhlig HH; Gyrd-Hansen M; Degterev A; Bullock AN (2015) Inflammatory Signaling by NOD-RIPK2 Is Inhibited by Clinically Relevant Type II Kinase Inhibitors. Chem. Biol. (Oxford, U. K.) 2015, 22, 1174–1184. DOI: 10.1016/j.chembiol.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerrant RL; Leite AM; Pinkerton R; Medeiros PH; Cavalcante PA; DeBoer M; Kosek M; Duggan C; Gewirtz A; Kagan JC; Gauthier AE; Swann J; Mayneris-Perxachs J; Bolick DT; Maier EA; Guedes MM; Moore SR; Petri WA; Havt A; Lima IF; Prata MM; Michaleckyj JC; Scharf RJ; Sturgeon C; Fasano A; Lima AA (2016) Biomarkers of Environmental Enteropathy, Inflammation, Stunting, and Impaired Growth in Children in Northeast Brazil. PloS One 11, e0158772. DOI: 10.1371/journal.pone.0158772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y; Li J; Zhang J; Yu Z; Yu S; Wu L; Wang Y; Gong X; Wu C; Cai X; Mo L; Wang M; Gu J; Chen L (2017) Oncogenic Protein Kinase D3 Regulating Networks in Invasive Breast Cancer. Int. J. Biol. Sci 13, 748–758. DOI: 10.7150/ijbs.18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolanos-Garcia VM (2005) Aurora kinases. Int. J. Biochem. Cell Biol 37, 1572–1577. DOI: 10.1016/j.biocel.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 38.Wang OH; Azizian N; Guo M; Capello M; Deng D; Zang F; Fry J; Katz MH; Fleming JB; Lee JE; Wolff RA; Hanash S; Wang H; Maitra A (2016) Prognostic and Functional Significance of MAP4K5 in Pancreatic Cancer. PloS One 11, e0152300. DOI: 10.1371/journal.pone.0152300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ojo KK; Pfander C; Mueller NR; Burstroem C; Larson ET; Bryan CM; Fox AM; Reid MC; Johnson SM; Murphy RC; Kennedy M; Mann H; Leibly DJ; Hewitt SN; Verlinde CL; Kappe S; Merritt EA; Maly DJ; Billker O; Van Voorhis WC (2012) Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J. Clin. Invest 122, 2301–2305. DOI: 10.1172/jci61822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tatipaka HB; Gillespie JR; Chatterjee AK; Norcross NR; Hulverson MA; Ranade RM; Nagendar P; Creason SA; McQueen J; Duster NA; Nagle A; Supek F; Molteni V; Wenzler T; Brun R; Glynne R; Buckner FS; Gelb MH (2014) Substituted 2-phenylimidazopyridines: a new class of drug leads for human African trypanosomiasis. J. Med. Chem 57, 828–835. DOI: 10.1021/jm401178t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banfor PN; Gintant GA; Lipari JM; Zocharski PD (2016) A novel intravenous vehicle for preclinical cardiovascular screening of small molecule drug candidates in rat. J. Pharmacol. Toxicol. Methods 82, 62–67. DOI: 10.1016/j.vascn.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 42.Danker T; Moller C (2014) Early identification of hERG liability in drug discovery programs by automated patch clamp. Front. Pharmacol 5, 203. DOI: 10.3389/fphar.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nicolette J; Diehl M; Sonders P; Bryce S; Blomme E (2011) In vitro micronucleus screening of pharmaceutical candidates by flow cytometry in Chinese hamster V79 cells. Environ. Mol. Mutagen 52, 355–362. DOI: 10.1002/em.20631. [DOI] [PubMed] [Google Scholar]
- 44.Fluckiger-Isler S; Kamber M (2012) Direct comparison of the Ames microplate format (MPF) test in liquid medium with the standard Ames pre-incubation assay on agar plates by use of equivocal to weakly positive test compounds. Mutat. Res 747, 36–45. DOI: 10.1016/j.mrgentox.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 45.Goedken ER; Argiriadi MA; Banach DL; Fiamengo BA; Foley SE; Frank KE; George JS; Harris CM; Hobson AD; Ihle DC; Marcotte D; Merta PJ; Michalak ME; Murdock SE; Tomlinson MJ; Voss JW (2015) Tricyclic covalent inhibitors selectively target Jak3 through an active site thiol. J. Biol. Chem 290, 4573–4589. DOI: 10.1074/jbc.M114.595181. [DOI] [PMC free article] [PubMed] [Google Scholar]