

Abstract
Background
Gastric nitric oxide (NO) production in response to Helicobacter pylori via inducible nitric oxide synthase (iNOS) is suggested as a biomarker of inflammation and cytotoxicity. The aim of this study was to investigate relationships between gastric [NO], immunological biomarkers and histopathology.
Materials and methods
Esophagogastroduodenoscopy was done in 96 dyspepsia patients. Luminal [NO] was measured by chemiluminescence. Biopsies were taken from gastric antrum and corpus for culture and histopathology. H. pylori IgG was detected by immunoblot assay. Biobanked plasma from 76 dyspepsia patients (11 H. pylori positives) was analyzed for 39 cytokines by multiplexed ELISA.
Results
H. pylori-positive patients had higher [NO] (336 ± 26 ppb, mean ± 95% CI, n = 77) than H. pylori-negative patients (128 ± 47 ppb, n = 19) (P < 0.0001). Histopathological changes were found in 99% of H. pylori-positive and 37% of H. pylori-negative patients. Histopathological concordance was 78–100% between corpus and antrum. Correlations were found between gastric [NO] and severity of acute, but not chronic, inflammation. Plasma IL-8 (increased in H. pylori positives) had greatest difference between positive and negative groups, with eotaxin, MIP-1β, MCP-4, VEGF-A, and VEGF-C also higher (P < 0.004 to P < 0.032). Diagnostic odds ratios using 75% cut-off concentration were 7.53 for IL-8, 1.15 for CRP, and 2.88 for gastric NO.
Conclusions
Of the parameters tested, increased gastric [NO] and circulating IL-8 align most consistently and selectively in H. pylori-infected patients. Severity of mucosal inflammatory changes is proportional to luminal [NO], which might be tied to IL-8 production. It is proposed that IL-8 be further investigated as a blood biomarker of treatment outcomes.
Keywords: Nitric oxide, Helicobacter pylori, inflammation, cytokines, biomarkers
Introduction
A troubling feature of Helicobacter pylori (H. pylori) is its persistence within the stomach via a combination of host, environmental, and bacterial factors (1, 2). Attachment of H. pylori to epithelial cells mediates gastric mucosal inflammation, including infiltration of neutrophils and monocytes into the gastric mucosa (3, 4). Gene and protein expressions of inducible nitric oxide (NO) synthase (iNOS, a product of NOS2 gene) are increased in gastric tissue in H. pylori infection (5). Simultaneous production of NO and O2 radicals presumably leads to nearly instantaneous combination, yielding highly reactive peroxynitrite (ONOO–). The extent to which NO itself can be detected and measured in real time as a feature of H. pylori infection in vivo is, therefore, unclear. Previous studies exploring cytokine panels of blood protein and tissue RNA concentration demonstrated the production of many immune signaling molecules in H. pylori infection (6, 7), albeit with somewhat different outcomes. For example, interleukins (IL)-6 and IL-8 are considered primary signals in infection but did not reach significance compared to controls across both these studies. IL-6 is thought to drive C-reactive protein (CRP) production, which is routinely measured in clinical chemistry. However, CRP has been shown to not be associated with H. pylori infection (8). Further investigations are needed to build concensus on human in vivo pathophysiology in H. pylori infection as to whether NO is detectable in the gastric lumen, whether detected NO is traceable to iNOS, which circulating cytokines are most likely tied to NO and H. pylori under different conditions (e.g. acute vs chronic inflammation), and also how cytokines relate to CRP.
The aim of this study was to assess relationships between H. pylori infection and direct measurements of gastric luminal gaseous NO concentration [NO] with inflammatory-associated immunological and histopathological findings. Separately, a 39-plex inflammatory biomarker panel was used to determine which plasma markers were most significantly associated with H. pylori infection. Correlation analysis was performed across all plasma markers.
Materials and methods
Study subjects
In a first study group, 96 adult patients (56 women and 40 men, mean age 39, range 17–86 years) referred for routine diagnostic upper gastrointestinal endoscopy, were included for NO measurement and biopsy investigations. Inclusion criteria were age 17–90 years with dyspeptic symptoms, either as postprandial distress or epigastric pain syndrome. Exclusion criteria were peptic ulcer disease, history of gastric surgery, use of proton pump inhibitors or non-steroidal antiinflammatory drugs, and ongoing upper gastrointestinal bleeding or delayed gastric emptying (defined as remaining gastric contents at endoscopy after a 4-h fasting period). In a second study group, inflammatory biomarker analysis was carried out on plasma samples obtained from a local biobank of patients referred for dyspepsia and upper gastrointestinal symptoms (11 H. pylori-positive and 65 H. pylori-negative patients; 46 women and 30 men, mean age 54, range 19–84 years). The same inclusion and exclusion criteria were applied to both groups.
Endoscopy clinical protocol
Patients were fasted overnight. Endoscopy was then carried out between 8:00 and 11:00 am the following day. All patients were offered surface anesthesia of the gullet by lidocaine (Xylocaine spray; 10 mg/mL, Astra Zeneca, Södertälje, Sweden) and intravenous midazolam administered at a dose of 0.06 mg/kg (1 mg/mL, Algol Pharma, Espoo, Finland).
Nitric oxide measurements
Luminal [NO] was measured as the initial step of each endoscopy session. Gastric air (50 mL) was withdrawn using a 60 mL syringe with a suction catheter through the working channel of the endoscope. The sampled air was immediately injected into a chemiluminescence analyzer (CLD 700, Eco Physics, Dürnten, Switzerland) for the analysis of the peak gaseous [NO]. The chemiluminescence method for gas analysis of [NO] relies on the measurement of light produced by the gas-phase titration of NO and ozone. NO is unstable, oxidizing to NO2 in the presence of ozone. This reaction produces a quantity of light for each NO molecule oxidized, which is detected by a photomultiplier tube. Because volumes of sample gas and excess ozone are controlled, light intensity in the reaction chamber is proportional to [NO]. Lower detection limit of [NO] was one part per billion (ppb). Analyzer calibration was done routinely using nitrogen with NO at known concentrations spanning 10–10,000 ppb (AGA AB, Lidingö, Sweden).
Microbiology
At endoscopy, biopsies were taken for culture, one from gastric antrum and one from corpus. Blood was drawn and plasma prepared for use in serological diagnosis as previously described (9–11). H. pylori IgG antibodies were detected by Western blot (Helicoblot 2.1, MP Biomedicals Germany GmbH, Eschwege, Germany) according to manufacturers’ instructions.
Histopathology
At endoscopy, two biopsies were taken from antrum and two from corpus that were placed in separate vials containing 4% buffered formalin. Hematoxylin–eosin and Giemsa staining were performed. The presence of the parameters acute inflammation, chronic inflammation, H. pylori, mucosal atrophy, intestinal metaplasia, and pseudopyloric metaplasia was individually classified into grade 1 (with one or more foci having one on the above parameters) or grade 2 (where a parameter was present across the entire biopsy section) as reported previously (12). Grading was made by two observers (CAR and ASR) blinded to H. pylori status and [NO], as well as to morphological findings during endoscopy. Grading of each subject’s set of biopsies was used to calculate histopathological scores in biopsy pairs from antrum and corpus. Histopathological scores were calculated by summation of the grades of each biopsy, multiplied by number of biopsies displaying that specific finding. Maximum score was 8.
Classification of patients
H. pylori status of each patient was assessed individually and classified based on culture, immunoblot, and histopathology. Confirmation by culture was in all subjects scored as positive H. pylori status.
Cytokines and chemokines
Using the 76 biobanked plasma samples (46 women and 30 men, mean age 54, range 19–84 years), H. pylori-positive patients (n = 11) were compared with H. pylori-negative patients (n = 65). Chemokines and cytokines were quantified using a human cytokine 39-plex ELISA kit (Meso Scale Diagnostics, Rockville, MD, USA). Analytes were as follows: CRP, basic fibroblast growth factor (bFGF), eotaxin, eotaxin-3, Fms-related receptor tyrosine kinase 1 (Flt-1), granulocyte-macrophage colony-stimulating factor (GM-CSF), intercellular adhesion molecule 1 (ICAM-1), interferon (INF)-g, interferon inducible protein (IP)-10, IL-1a, IL-1b, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12p70, IL-12/IL-23p40, IL-13, IL-15, IL-16, IL-17A, macrophage-derived chemokine (MDC), macrophage inflammatory protein (MIP)-1a, MIP-1b, monocyte chemotactic protein (MCP)-1, MCP-4, placental growth factor (PIGF), serum amyloid A (SAA), thymus activation-regulated chemokine (TARC), tumor necrosis factor (TNF)-a, TNF-b, tyrosine-protein kinase receptor (Tie-2), vascular cell adhesion protein 1 (VCAM-1), vascular endothelial growth factor (VEGF-A), VEGF-C, and VEGF-D. Samples were analyzed in duplicate with electrochemiluminescence.
Statistical analysis
Statistical analyses for significant differences in biochemical variables between H. pylori-positive and H. pylori-negative patients were performed using Student’s t-test or Mann–Whitney U-test for data that were not normally distributed. For histopathology, comparisons were made with the Mann–Whitney U-test. Analysis for simple linear parametric correlations used Pearson’s method (P < 0.05 was deemed significant). Data were similarly assessed by Spearman’s rank method to account for outliers and non-parametric or curvilinear correlations. Diagnostic odds ratio (DOR) was calculated using the 75% percentile cut-off according to the formula DOR = (true positive/false negative)/(false positive/true negative). Histopathological results were regarded as gold standard (13).
Ethics
This study was approved by the Ethics Committee North at Karolinska University Hospital, Solna (Dnr 97-034; 1997-06-18 and 99-119; 1999-11-24). Verbal and written informed consents were obtained before performing endoscopy. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the institution’s human research committee.
Results
Endoscopic findings
In the group of dyspeptic patients undergoing endoscopy, 77 were H. pylori-positive patients (mean age 37, range 17–61 years), whereas 19 were H. pylori-negative patients (mean age 39, range 34–86 years). All patients had a similar clinical picture of functional dyspepsia with either epigastric pain syndrome or postprandial fullness syndrome.
H. pylori-positive patients had higher gastric luminal [NO] than H. pylori-negative subjects (336 ± 26 vs 128 ± 47 ppb; P < 0.0001) (Figure 1).
Figure. 1.
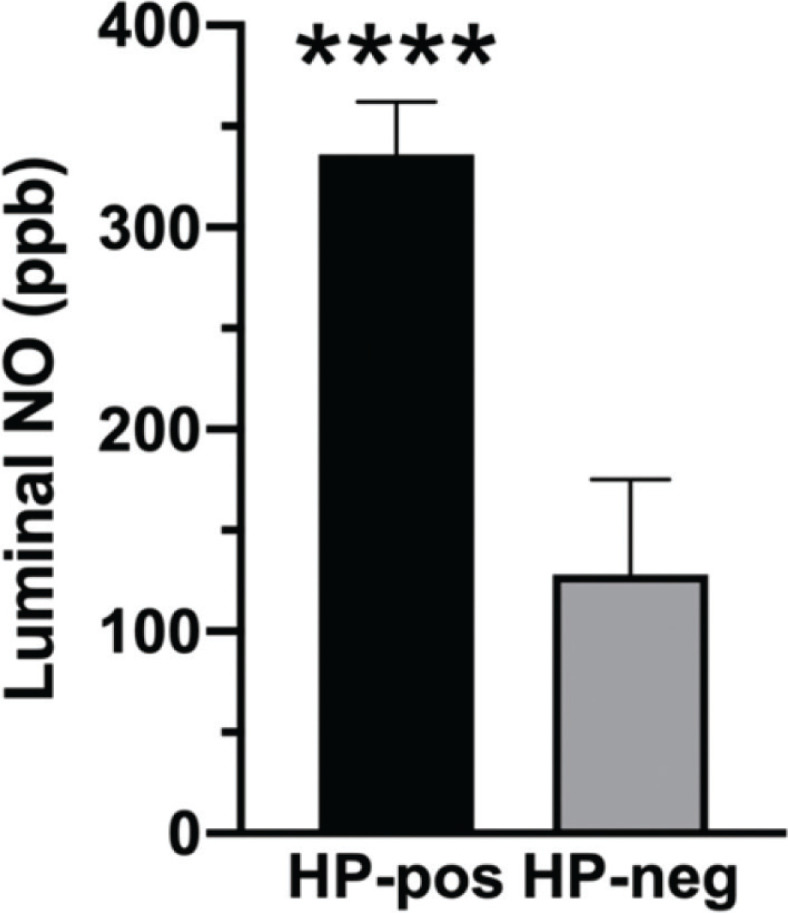
Luminal [NO] in the stomach of Helicobacter-positive (HP-pos, n = 77) as compared with Helicobacter-negative (HP-neg, n = 19) patients.
****P < 0.0001. Data are mean ± SEM.
Among H. pylori-positive patients, 76 out of 77 were found to have histopathological changes of the mucosa, whereas in the H. pylori-negative control group, only 7 of 19 patients had histopathological changes. Concordance in histopathology between paired biopsies from corpus and antrum was high; 78–100% of the paired biopsies received the same histological grade. In gastric antrum, histopathological changes of H. pylori-infected patients showed not only acute and chronic inflammations (P <0.004) with infiltration of lymphocytes and plasma cells but also structural changes with mucosal atrophy and metaplasia approaching significance (P < 0.087), as compared to non-infected antrum. Mucosal inflammatory and atrophic changes showed stratefication at scoring 2 with moderate inflammation. In corpus, histopathology showed inflammation (P < 0.001) as well as structural changes with mucosal atrophy and metaplasia (P < 0.029). Again, acute inflammatory and atrophic changes were moderate with stratefication at scoring 2, whereas chronic inflammation was more severe and scored 4 (Table 1).
Table 1.
Histopathological scoring of the gastric antrum (top) and corpus (bottom) in Helicobacter-positive and Helicobacter-negative patients.
| Antrum |
Acute inflammation |
Chronic inflammation |
Mucosal atrophy |
Intestinal metaplasia |
Pseudopyloral metaplasia |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Score |
H. pylori
|
H. pylori
|
H. pylori
|
H. pylori
|
H. pylori
|
|||||
| +ve | −ve | +ve | −ve | +ve | −ve | +ve | −ve | +ve | −ve | |
| 0 | 24 | 18 | 12 | 15 | 52 | 16 | 71 | 18 | 71 | 17 |
| 1 | 5 | 0 | 3 | 2 | 8 | 1 | 2 | 0 | 2 | 0 |
| 2 | 44 | 1 | 37 | 1 | 15 | 1 | 4 | 1 | 4 | 2 |
| 3 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 4 | 4 | 0 | 18 | 1 | 2 | 1 | 0 | 0 | 0 | 0 |
|
| ||||||||||
| Total | 77 | 19 | 77 | 19 | 77 | 19 | 77 | 19 | 77 | 19 |
| Corpus |
Acute inflammation |
Chronic inflammation |
Mucosal atrophy |
Intestinal metaplasia |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Score |
H. pylori
|
H. pylori
|
H. pylori
|
H. pylori
|
||||||
| +ve | −ve | +ve | −ve | +ve | −ve | +ve | −ve | |||
| 0 | 9 | 18 | 3 | 16 | 28 | 17 | 65 | 18 | ||
| 1 | 8 | 0 | 1 | 0 | 13 | 0 | 5 | 0 | ||
| 2 | 55 | 1 | 24 | 3 | 33 | 2 | 6 | 0 | ||
| 3 | 1 | 0 | 9 | 0 | 2 | 0 | 1 | 0 | ||
| 4 | 4 | 0 | 40 | 0 | 1 | 4 | 0 | 1 | ||
|
| ||||||||||
| Total | 77 | 19 | 77 | 19 | 77 | 19 | 77 | 19 | ||
H. pylori: Helicobacter pylori; +ve: positive; −ve: negative.
Figure 2 shows [NO] relative histopathology scores. In H. pylori positives, a correlation was found between gastric luminal [NO] and acute inflammation scores (Figure 2a). This trend was not observed in scores of chronic histopathological changes (Figure 2b). Conversely, luminal [NO] was lower in the presence of mucosal atrophy (Figure 2c). In relative controls, [NO] was elevated to a similar extent in those with the presence of acute or chronic inflammation, mucosal atrophy, and intestinal or pseudopyloral metaplasia (Figure 2d).
Figure. 2.

Luminal [NO] in antrum in relation to histopathology scores. (a) Luminal NO concentration relative acute inflammation. Number of patients were [score(n)]: 0(5); 1–2(25); 3–4(40); 5–8(7). (b) Luminal [NO] relative chronic inflammation. Number of patients were [score(n)]: 0(3); 1–2(4); 3–4(25); 5–8(45). (c) Luminal [NO] relative mucosal atrophy. Number of patients were [score(n)]: 0(23); 1–2(35); 3–4(16); 5–8(3). Scores in A, B, and C are for H. pylori-positive group only. Data are mean ± SEM. (d) Histopathology by positive clinical finding independent of score. In histopathology, more than one positive finding can occur in the same visual field. Data are mean ± SEM, left to right, n = 36, 67, 12, 14, 15, and 12). Control is H. pylori-negative by histopathology. ***P < 0.001, ****P < 0.0001.
Blood sample findings
In the study group from which biobanked plasma was analyzed for circulating cytokines and chemokines, IL-8 was elevated the most in H. pylori-positive patients, with eotaxin, MIP-1β, MCP-4, VEGF-A, and VEGF-C also higher (P < 0.004 to P < 0.032) (Table 2). DOR values using the 75% percentile cut-off for true positives were 7.53 for IL-8 as compared with 1.15 for CRP and 2.88 for gastric [NO].
Table 2.
Cytokine and chemokine plasma concentrations in Helicobacter-positive (n = 11) and Helicobacter-negative (n = 65) subjects. Data are median ± SD. Statistics are Mann–Whitney ranked sum test. Units are ng/L (see footnotes for exceptions). Significant differences are in bold.
| Cytokine/chemokine | Lower limit of quantification (LLOQ)2 | Helicobacter pylori |
P | |
|---|---|---|---|---|
| Positive | Negative | |||
| CRP1 | 27.6 | 1.95 ±9.72 | 1.01 ±5.85 | 0.36 |
| bFGF | 2.60 | 3.27 ±4.26 | 2.29 ±12.72 | 0.44 |
| Eotaxin | 12.3 | 92.81 ± 52.11 | 70.40 ± 43.02 | 0.022 |
| Eotaxin-3 | 10.2 | 14.72 ±8.5 | 16.23 ±23.66 | 0.39 |
| Flt-1 | 10.0 | 41.66 ±9.11 | 36.29 ±12.46 | 0.44 |
| GM-CSF | 0.842 | 0.15 ±0.10 | 0.29 ±0.35 | 0.11 |
| ICAM-11 | 15.0 | 0.22 ±0.08 | 0.22 ±0.11 | 0.84 |
| IFN-γ | 1.76 | 3.44 ±3.05 | 3.98 ±4.54 | 0.50 |
| IP-10 | 1.37 | 213.6 ±962.74 | 182.10 ±367.3 | 0.79 |
| IL-1α | 2.85 | 4.67 ±4.25 | 4.77 ±6.22 | 0.72 |
| IL-1β3 | 0.646 | 0.08 ±0.05 | 0.11 ±0.59 | 0.75 |
| IL-23 | 0.890 | 0.28 ±0.18 | 0.17 ±0.58 | 0.53 |
| IL-4 | 0.218 | 0.08 ±0.07 | 0.06 ±0.46 | 0.74 |
| IL-5 | 4.41 | 0.66 ±0.64 | 0.58 ±0.55 | 0.85 |
| IL-6 | 0.633 | 0.46 ±1.03 | 0.49 ±0.65 | 0.71 |
| IL-7 | 0.546 | 5.7 ±6.52 | 2.11 ±2.08 | 0.08 |
| IL-8 | 0.591 | 5.87 ±2.11 | 2.52 ±3.08 | 0.004 |
| IL-10 | 0.298 | 0.34 ±0.30 | 0.30 ±0.83 | 0.91 |
| IL-12p70 | 1.22 | 0.30 ±16.4 | 0.27 ±0.70 | 0.74 |
| IL-12/IL-23p40 | 1.32 | 87.41 ±54.51 | 94.22 ±80.78 | 0.99 |
| IL-13 | 4.21 | 0.94 ±0.52 | 1.09 ±2.01 | 0.66 |
| IL-15 | 0.774 | 1.20 ±0.44 | 1.25 ±0.63 | 0.68 |
| IL-16 | 19.1 | 116.28 ±23.59 | 108.70 ±51.75 | 0.51 |
| IL-17A | 3.19 | 4.77 ±2.65 | 4.28 ±4.53 | 0.99 |
| MDC | 88.3 | 829.31±360.99 | 559.42 ±290.62 | 0.12 |
| MIP-1α | 13.8 | 19.95 ±6.93 | 16.15 ±7.74 | 0.11 |
| MIP-1β | 1.02 | 94.63 ±38.93 | 63.85 ±105.08 | 0.014 |
| MCP-1 | 1.09 | 61.99 ±107.01 | 39.65± 48.06 | 0.33 |
| MCP-4 | 5.13 | 81.84 ±93.43 | 56.60 ±42.09 | 0.023 |
| PIGF | 1.50 | 5.04 ±1.04 | 4.42 ±1.94 | 0.38 |
| SAA1 | 54.0 | 2.22 ±26.67 | 2.61 ±16.36 | 0.55 |
| TARC | 3.32 | 45.69 ±167.40 | 31.85 ±102.53 | 0.42 |
| TNF-α | 0.690 | 1.87 ±0.67 | 1.67 ±1.08 | 0.65 |
| TNF-β | 0.465 | 0.42 ±0.11 | 0.47 ±0.34 | 0.28 |
| Tie-2 | 396 | 850.89 ±169.84 | 816.00 ±219.69 | 0.43 |
| VCAM-11 | 37.6 | 0.22 ±0.10 | 0.28±0.14 | 0.30 |
| VEGF-A | 5.00 | 29.33±149.01 | 18.68±42.32 | 0.027 |
| VEGF-C | 146 | 110.53 ±500.44 | 56.10 ±230.04 | 0.032 |
| VEGF-D | 67.1 | 2167.99 ±1289.84 | 1833.37 ±718.70 | 0.24 |
CRP: C-reactive protein; bFGF: basic fibroblast growth factor; GM-CSF: granulocyte-macrophage colony-stimulating factor; ICAM-1: intercellular adhesion molecule 1; IP, interferon inducible protein; IFN-g: interferon-gamma; IL, interleukins; Flt-1, Fms-related receptor tyrosine kinase 1; MDC: macrophage-derived chemokine; MIP: macrophage inflammatory protein; MCP: monocyte chemotactic protein; PIGF: placental growth factor; SAA: serum amyloid A; TARC: thymus activation-regulated chemokine; TNF: tumor necrosis factor; VCAM-1: vascular cell adhesion protein 1; VEGF: vascular endothelial growth factor.
Plasma concentrations (but not LLOQ [lower limit of quantification]) are mg/L due to their comparatively high concentrations.
LLOQ values are taken from product certificate of analysis and were consistent with calculations from experimental results. Plasma concentrations below LLOQ (e.g. VEGF-C) were above lower limit of detection, but must be considered unreliable.
Many samples were below detection limit, in which case no value was obtained.
Correlation analysis for plasma cytokines was done with combined data from H. pylori positives and negatives. For 39 analytes, there are 741 comparison pairs per test. Pearson’s and Spearman’s methods gave similar results. Complete data from Pearson’s method are available in Supplementary Table 1. Numerous significant correlations were found. Terms like ‘moderate’ or ‘strong’ are arbitrary. By setting a lower cutoff for moderate correlations as R > 0.50 or <−0.50 (i.e. antiparallel) as well as P <0.05, Pearson’s test identified 45 comparisons meeting this criteria, whereas Spearman’s test identified 42. There was substantial overlap in which same analyte pairs correlated. By setting a cutoff for strong correlations as R > 0.66 or < −0.66 (i.e. antiparallel) as well as P < 0.001, Pearson’s test identified 19 comparisons meeting this criteria, whereas Spearman’s test identified 6. Importantly, IL-8 did not correlate even by the moderate criteria with any other analyte in either test. Hence, by the criteria above, IL-8 was an independent measure. For comparison, IL-6 (believed to drive acute phase liver proteins) had consistent moderate or strong correlations with CRP and SAA across both tests. In both tests, IL-8 vs CRP correlation was close to 0, with P ~ 0.5 (i.e. no correlation). It can, therefore, be argued that IL-8 is a more reliable indicator of H. pylori infection than CRP. Since samples were stored many years in a biobank, it is likely that IL-8 is also sufficiently stable for use as biomarker of infection.
Discussion
Many researchers have assumed increased NO elaboration based on the expression of different NOS isoforms (14, 15). Higher gastric [NO] was, indeed, found in Helicobacter-infected subjects.
NO has a dual role: constitutive at low concentrations for maintaining physiological functions (16) and inducible during inflammatory reactions, even reaching cytotoxic concentrations (17). Basal mucosal NO production originates from nNOS and eNOS present in the gastric mucosa. Higher concentrations result from upregulated iNOS in gastric epithelial cells (18) as well as neutrophils and macrophages (19). In support of this, luminal [NO] measurement in the gastrointestinal tract has proven useful in inflammatory bowel disease, infective gastroenteritis, and celiac sprue, where similar inflammatory mechanisms are thought to occur (20–22).
Elevated [NO] is generated by upregulated iNOS triggered by inflammation or tissue injury in gastric epithelial cells (15, 18, 23). This has also been reported for neutrophils and macrophages exposed to H. pylori (19), consistent with NO as an acute proinflammatory agent (24). The inducibility of iNOS seems, however, to be reversible. Increased iNOS expression in H. pylori-associated chronic gastritis diminishes and returns to baseline once H. pylori is eradicated (17).
Some researchers have found lower luminal [NO] using a similar chemiluminescence measurement technique (25–27). Differences in results may be due to difficulties in the sampling techniques with dilution of gaseous samples. In the present study, luminal suctions were carried out in a standardized manner but still may involve a certain degree of aerial dilution. Swallowed air during the intubation procedure may have further diluted the NO samples. Taken together, these two factors may have mitigated the [NO] issues in this study. The H. pylori infection in publications of Fändriks et al. (25) and Lundberg et al. (26) may have reached a broader state of chronicity and mucosal atrophy with extended chronic gastritis being less capable of producing NO, but still harboring H. pylori. However, the normal distribution of the NO data, together with the marked differences between H. pylori-positive and H. pylori-negative patients speaks in favor of the NO measurements being representative of an inflammatory disease process.
Other authors have also found higher gastric [NO] in H. pylori-infected than non-infected subjects with an associated prognosis of chronic gastritis and atrophy of the corpus. The latter being known to increase gastric pH (15), but with no histopathological investigation to confirm this. Likewise, Shiotani et al. (12) found elevated pH and increased nitrites in gastric juice during H. pylori-infection. This correlated with degree and spread of gastritis and atrophy. Otherwise, Shiotani et al. (27) in another study reported low gastric [NO] with increased nitrites in gastric juice as a result of an elevated pH in gastric juice in patients with long-standing Helicobacter infection (27).
The association between the presence of H. pylori and continuous disease process is complicated. A disconnection between the rate of NO synthesis and augmented iNOS expression has been described with a similar NO formation in H. pylori infected and non-infected antral mucosa despite elevated iNOS activity in the H. pylori infected. This finding was explained by unique ability of H. pylori to produce a competitive NOS inhibitor down-regulating the L-arginine/NO-pathway to evade host defense mechanisms (15). Gobert et al. reported that H. pylori expresses the gene rocF that encodes arginase, which depletes arginine, effectively dampening NO production by iNOS in macrophages (28).
Positive trends were found between [NO] and the score of acute inflammation in the mucosa of antrum and corpus, as well as to serum concentrations of anti-H. pylori IgG, but with no statistical significance. This might be due to the successive development of an atrophic mucosa not capable of producing NO. Another reason for these results being disparate from two other human studies (25, 29) could be the younger age of our present patients. The age factor might well influence the results due to the paucity of the development of gastric atrophy in young compared to old individuals. This is supported by the histopathological findings in our patients, in which chronic gastritis in antrum was more frequent than in corpus in younger patients. This implies that acid secretion was still normal, and intragastric reduction of nitrite/nitrate to NO was preserved. This is also in agreement with data showing a trend toward lower [NO] among H. pylori-infected patients with mucosal atrophy (12).
H. pylori infection has been reported to induce proinflammatory cytokines and chemokines, such as IL-1β, IL-6, IL-8, IL-23, and TNF-α, where the IL-8 secreted from gastric mucosal epithelium was the major chemokine driving inflammatory response (30). In the present study, IL-8 had strongest significance (P = 0.004) and largest fold difference (2.8-fold higher in H. pylori positives) among analytes. IL-8 also had the highest DOR, including by comparison to NO or CRP. It is possible for iNOS and IL-8 to coordinate acute gastric inflammation in a common signaling pathway. Released NO can activate oxidant sensitive transcription factors. NO donors can induce increased concentrations of IL-8 and nitrite as well as a time-dependent mRNA expression of IL-8 in cell lines, indicating signaling between the l-arginase/NO pathway and IL-8 that is activated by Helicobacter (31).
Elevated eotaxin suggests an eosinophilic component of H. pylori inflammatory response. Eosinophils may cross the endothelium to penetrate into gastric inflammatory tissue by a regulated process through a coordinated interaction between networks involving chemokine eotaxin-1, eosinophil adhesion molecules, and adhesion receptors on the endothelium (32). Elevated plasma MCP-4 concentrations in H. pylori-positive patients are consistent with its production at the infection site. MCP-4 induction has been identified in complex tissue proteomes (33). Furthermore, MIP-1β has previously been shown to be upregulated in Helicobacter-infected monocytes (34), which, together with IL-8, may be associated with neutrophil infiltration characteristic of H. pylori-infected mucosa (35). To this end, VEGF concentrations, primarily of the A and C subtypes considered to be endothelial cell specific, were elevated. Together with IL-8, VEGF has previously been found to be increased in gastric epithelial cells during H. pylori infection, being important for vascular remodeling in gastric epithelial cells (36). VEGF has been suggested to enhance healing of gastric injury in an iNOS-dependent manner independently of stimulated angiogenesis (37). In addition to stimulating angiogenesis to promote restoration of connective tissue and microvessels in injured mucosa, effects through iNOS downregulation exert a therapeutic effect on mucosal lesions.
Although CRP, a clinical marker of acute inflammation, averaged about double that of the control group, significance was not reached due to skewed spread in CRP concentrations. However, strong correlations between IL-6 and CRP were found, consistent with the concept that IL-6 drives CRP production. This correlation was found with this panel in Crohn’s disease patients (38). H. pylori infection did not consistently elevate IL-6, whereas IL-8 apparently cannot replace IL-6 to drive CRP production.
In the short term, elevated [NO] in H. pylori-infected patients is beneficial for the host. This benefit may be reduced with more profound tissue injury. l-Arginine-dependent NO production in neutrophils and macrophages increases when cocultured with H. pylori (19, 39), consistent with the present results. However, chronically elevated NO in chronic gastritis (Figure 2, panels b and d) might contribute to gastric adenocarcinoma, as by immunosuppressive nitrotyrosine production in cytotoxic T cells (40).
In conclusion, the results showed significant increases of gastric [NO] and circulating IL-8 in H. pylori-infected patients compared with those without H. pylori. Severity of mucosal inflammatory changes, however, is not unambiguously revealed by luminal [NO] alone, thus requiring histopathological assessment.
Strengths, limitations, and clinical significance
This exploratory study had limited sample size. Age, BMI, smoking, alcohol consumpton, and other data that could be relevant for cytokine concentrations were unavailable. NO data were obtained from different patients than IL-8, VEGF, CRP, and other circulating inflammatory biomarkers; correlation analyses between NO and IL-8 were not possible. IL-8 can easily be measured with good accuracy, which is clinically important. Successful H. pylori treatment requires not only eradication of H. pylori but would also escape opportunistic secondary infections, many of which elevate IL-8. This study indicates that IL-8 may have clinical chemistry utility in H. pylori treatment follow-ups and outcomes.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge proofreading assistance from Richard R. Wathen.
Disclosure statement
The authors declare that there are no conflicts of interest.
Funding
This study was supported by the Swedish Research Council (7916), the Stockholm County Research funding and ALF strategic funds (ALF-899261) to PMH and DLW. Funding from OE and Edla Johansson’s Science Foundation 2017 and 2018 as well as Selanders Foundation 2020 and 2021 to DLW are gratefully appreciated.
Notes on Contributors
Hiwa K. Saaed, PhD, senior lecturer, College of Pharmacy, University of Sulaimani-Iraq, Kurdistan Region, Iraq. Here, Dr. Saaed obtained his 5 year BSc in pharmacy and the higher diploma degree in clinical pharmacy as well as an MSc and a PhD in clinical pharmacology and toxicology. During this study, he was a guest researcher at the Gastroenterology and Hepatology Unit, Department of Medical Sciences, Uppsala University, Uppsala, Sweden.
Lisa Chiggiato, MSc, biologist, Gastroenterology and Hepatology Unit, Department of Medical Sciences, Uppsala University, Uppsala, Sweden.
Dominic-Luc Webb, BSc, PhD, associate professor of experimental gastroenterology, researcher and supervisor at Gastroenterology and Hepatology Unit, Department of Medical Sciences, Uppsala University, Uppsala, Sweden.
Ann-Sofie Rehnberg, MD, PhD, specialist in gastroenterology, Department of Gastroenterology, Karolinska University Hospital, Stockholm, Sweden
Carlos A. Rubio, MD, PhD, pathologist, specialist in gastroenterological histopathology, Karolinska Institutet, Stockholm, Sweden
Ragnar Befrits, MD, PhD, associate professor, specialist in gastroenterology, at the time of the study senior consultant, Department of Gastroenterology, Karolinska University Hospital, Stockholm, Sweden.
Per M. Hellström, MD, PhD, study lead, professor of gastroenterology, Gastroenterology and Hepatology Unit, Department of Medical Sciences, Uppsala University, Uppsala, Sweden.
ORCID
Hiwa K. Saaed https://orcid.org/0000-0003-1078-514X
Lisa Chiggiato https://orcid.org/0000-0003-2784-8880
Dominic-Luc Webb https://orcid.org/0000-0002-6979-9194
Carlos A. Rubio https://orcid.org/0000-0003-3879-5729
Per M. Hellström https://orcid.org/0000-0001-8428-0772
References
- 1.Testerman TL, McGee DJ, Mobley HLT. Adherence and colonization. In: Mobley HL, Mendz GL, Hazell SL, directeurs. Helicobacter pylori: physiology and genetics. [En ligne]. Washington, DC: ASM Press; 2001. Available from: http://www.ncbi.nlm.nih.gov/books/NBK2437/ [cited 15 January 2021]. [PubMed] [Google Scholar]
- 2.Bugaytsova JA, Chernov YA, Gideonsson P, Björnham O, Henriksson S, Mendez M, et al. Adaption of Helicobacter pylori to chronic infection and gastric disease by pH-responsive BabA-mediated adherence. Cell Host Microbe 2017;21:376–89. doi: 10.1016/j.chom.2017.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nielsen H, Andersen LP. Activation of human phagocyte oxidative metabolism by Helicobacter pylori. Gastroenterology 1992;103:1747–53. doi: 10.1016/0016-5085(92)91430-C [DOI] [PubMed] [Google Scholar]
- 4.Dixon MF. Pathology of gastritis and peptic ulceration. In: Mobley HL, Mendz GL, Hazell SL, directeurs. Helicobacter pylori: physiology and genetics. [En ligne]. Washington, DC: ASM Press; 2001. Available from: http://www.ncbi.nlm.nih.gov/books/NBK2461/ [cited 15 January 2021]. [PubMed] [Google Scholar]
- 5.Gobert AP, Wilson KT. The immune battle against Helicobacter pylori infection: NO offense. Trends Microbiol 2016;24:366–76. doi: 10.1016/j.tim.2016.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris PR, Weber HC, Wilcox CM, Jensen RT, Smith PD. Cytokine gene profile in gastric mucosa in Helicobacter pylori infection and Zollinger-Ellison syndrome. Am J Gastroenterol 2002;97:312–18. doi: 10.1111/j.1572-0241.2002.05463.x [DOI] [PubMed] [Google Scholar]
- 7.Khaiboullina SF, Abdulkhakov S, Khalikova A, Safina D, Martynova EV, Davidyuk Y, et al. Serum cytokine signature that discriminates Helicobacter pylori positive and negative juvenile Gastroduodenitis. Front Microbiol 2016;7:1916. doi: 10.3389/fmicb.2016.01916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim TJ, Pyo JH, Lee H, Baek SY, Ahn SH, Min YW, et al. Lack of association between Helicobacter pylori infection and various markers of systemic inflammation in asymptomatic adults. Korean J Gastroenterol 2018;72:21–7. doi: 10.4166/kjg.2018.72.1.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tindberg Y, Bengtsson C, Bergstrom M, Granstrom M. The accuracy of serologic diagnosis of Helicobacter pylori infection in school-aged children of mixed ethnicity. Helicobacter 2001;6:24–30. doi: 10.1046/j.1523-5378.2001.00005.x [DOI] [PubMed] [Google Scholar]
- 10.Hoang TTH, Wheeldon T-U, Bengtsson C, Phung DC, Sorberg M, Granstrom M. Enzyme-linked immunosorbent assay for Helicobacter pylori needs adjustment for the population investigated. J Clin Microbiol 2004;42:627–30. doi: 10.1128/JCM.42.2.627-630.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sörberg M, Engstrand L, Ström M, Jönsson K-Å, Jörbeck H, Granström M. The diagnostic value of enzyme immunoassay and immunoblot in monitoring eradication of Helicobacter pylori. Scand J Infect Dis 1997;29:147–51. doi: 10.3109/00365549709035875 [DOI] [PubMed] [Google Scholar]
- 12.Shiotani A, Iishi H, Kumamoto M, Nakae Y. Helicobacter pylori infection and increased nitrite synthesis in the stomach. Dig Liver Dis 2004;36:327–32. doi: 10.1016/j.dld.2003.11.029 [DOI] [PubMed] [Google Scholar]
- 13.Parikh R, Mathai A, Parikh S, Chandra Sekhar G, Thomas R. Understanding and using sensitivity, specificity and predictive values. Indian J Ophthalmol 2008;56:45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kai H, Ito M, Kitadai Y, Tanaka S, Haruma K, Chayama K. Chronic gastritis with expression of inducible nitric oxide synthase is associated with high expression of interleukin-6 and hypergastrinaemia. Aliment Pharmacol Ther 2004;19:1309–14. doi: 10.1111/j.1365-2036.2004.01965.x [DOI] [PubMed] [Google Scholar]
- 15.Von Bothmer C, Edebo A, Lönroth H, Olbe L, Pettersson A, Fändriks L. Helicobacter pylori infection inhibits antral mucosal nitric oxide production in humans. Scand J Gastroenterol 2002;37:404–8. doi: 10.1080/003655202317316024 [DOI] [PubMed] [Google Scholar]
- 16.Webb D-L, Rudholm-Feldreich T, Gillberg L, Halim MdA, Theodorsson E, Sanger GJ, et al. The type 2 CCK/gastrin receptor antagonist YF476 acutely prevents NSAID-induced gastric ulceration while increasing iNOS expression. Naunyn Schmiedebergs Arch Pharmacol 2013;386:41–9. doi: 10.1007/s00210-012-0812-5 [DOI] [PubMed] [Google Scholar]
- 17.Calatayud S, Barrachina D, Esplugues JV. Nitric oxide: relation to integrity, injury, and healing of the gastric mucosa. Microsc Res Tech 2001;53:325–35. doi: 10.1002/jemt.1100 [DOI] [PubMed] [Google Scholar]
- 18.Kim JM, Kim JS, Jung HC, Song IS, Kim CY. Up-regulation of inducible nitric oxide synthase and nitric oxide in Helicobacter pylori-infected human gastric epithelial cells: possible role of interferon-gamma in polarized nitric oxide secretion. Helicobacter 2002;7:116–28. doi: 10.1046/j.1083-4389.2002.00068.x [DOI] [PubMed] [Google Scholar]
- 19.Tsuji S, Kawano S, Tsujii M, Takei Y, Tanaka M, Sawaoka H, et al. Helicobacter pylori extract stimulates inflammatory nitric oxide production. Cancer Lett 1996;108(2):195–200. doi: 10.1016/S0304-3835(96)04410-2 [DOI] [PubMed] [Google Scholar]
- 20.Rubio CA. My approach to reporting a gastric biopsy. J Clin Pathol 2006;60:160–6. doi: 10.1136/jcp.2006.039008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herulf M, Ljung T, Hellström P. Increased luminal nitric oxide in inflammatory bowel disease as shown with a novel minimally invasive method. Scand J Gastroenterol 1998;33:164–9. doi: 10.1080/00365529850166897 [DOI] [PubMed] [Google Scholar]
- 22.Herulf M, Blomquist L, Ljung T. Increased rectal nitric oxide in coeliac disease after local challenge with gluten. Scand J Gastroenterol 2001;36:169–73. doi: 10.1080/00365520118744 [DOI] [PubMed] [Google Scholar]
- 23.Konturek PC, Konturek JW, Konturek SJ. Gastric secretion and the pathogenesis of peptic ulcer in the Helicobacter pylori infection. J Physiol Pharmacol 1996;47:5–19. [PubMed] [Google Scholar]
- 24.Iijima K, Shimosegawa T. Involvement of luminal nitric oxide in the pathogenesis of the gastroesophageal reflux disease spectrum: nitric oxide and esophagus. J Gastroenterol Hepatol 2014;29:898–905. doi: 10.1111/jgh.12548 [DOI] [PubMed] [Google Scholar]
- 25.Fändriks L, von Bothmer C, Åne A. Intragastric nitric oxide/nitrite in Helicobacter pylori -infected subjects. Scand J Gastroenterol 2001;36:347–50. doi: 10.1080/00365520119610 [DOI] [PubMed] [Google Scholar]
- 26.Lundberg JO, Weitzberg E, Lundberg JM, Alving K. Intragastric nitric oxide production in humans: measurements in expelled air. Gut 1994;35:1543–6. doi: 10.1136/gut.35.11.1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiotani A, Yanaoka K, Iguchi M, Saika A, Itoh H, Nishioka S. Helicobacter pylori infection reduces intraluminal nitric oxide in humans. J Gastroenterol 1999;34:668–74. doi: 10.1007/s005350050317 [DOI] [PubMed] [Google Scholar]
- 28.Gobert AP, McGee DJ, Akhtar M, Mendz GL, Newton JC, Cheng Y, et al. Helicobacter pylori arginase inhibits nitric oxide production by eukaryotic cells: a strategy for bacterial survival. Proc Natl Acad Sci U S A 2001;98:13844–9. doi: 10.1073/pnas.241443798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lundberg JO, Govoni M. Inorganic nitrate is a possible source for systemic generation of nitric oxide. Free Radic Biol Med 2004;37:395–400. doi: 10.1016/j.freeradbiomed.2004.04.027 [DOI] [PubMed] [Google Scholar]
- 30.Crabtree J. Gastric mucosal inflammatory responses to Helicobacter pylori. Aliment Pharmacol Ther 1996;10(Suppl. 1):29–37. doi: 10.1046/j.1365-2036.1996.22164003.x [DOI] [PubMed] [Google Scholar]
- 31.Seo JY, Yu J-H, Lim JW, Mukaida N, Kim H. Nitric oxide-induced IL-8 expression is mediated by NF-kappaB and AP-1 in gastric epithelial AGS cells. J Physiol Pharmacol Off J Pol Physiol Soc 2009;60(Sup7):101–6. [PubMed] [Google Scholar]
- 32.Bochner BS, Schleimer RP. The role of adhesion molecules in human eosinophil and basophil recruitment. J Allergy Clin Immunol 1994;94:427–38. doi: 10.1016/0091-6749(94)90195-3 [DOI] [PubMed] [Google Scholar]
- 33.Ellmark P, Ingvarsson J, Carlsson A, Lundin BS, Wingren C, Borrebaeck CAK. Identification of protein expression signatures associated with Helicobacter pylori infection and gastric adenocarcinoma using recombinant antibody microarrays. Mol Cell Proteomics 2006;5:1638–46. doi: 10.1074/mcp.M600170-MCP200 [DOI] [PubMed] [Google Scholar]
- 34.Maeda S, Akanuma M, Mitsuno Y, Hirata Y, Ogura K, Yoshida H, et al. Distinct mechanism of Helicobacter pylori-mediated NF-κB activation between gastric cancer cells and monocytic cells. J Biol Chem 2001;276:44856–64. doi: 10.1074/jbc.M105381200 [DOI] [PubMed] [Google Scholar]
- 35.Crabtree JE, Wyatt JI, Trejdosiewicz LK, Peichl P, Nichols PH, Ramsay N, et al. Interleukin-8 expression in Helicobacter pylori infected, normal, and neoplastic gastroduodenal mucosa. J Clin Pathol 1994;47:61–6. doi: 10.1136/jcp.47.1.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim G, Kim J-E, Kang M-J, Jang A-R, Kim YR, Kim S, et al. Inhibitory effect of 1tetradecanol on Helicobacter pyloriinduced production of interleukin8 and vascular endothelial growth factor in gastric epithelial cells. Mol Med Rep 2017;16:9573–8. doi: 10.3892/mmr.2017.7793 [DOI] [PubMed] [Google Scholar]
- 37.Dudar GK, D’Andrea LD, Di Stasi R, Pedone C, Wallace JL. A vascular endothelial growth factor mimetic accelerates gastric ulcer healing in an iNOS-dependent manner. Am J Physiol-Gastrointest Liver Physiol 2008;295:G374–81. doi: 10.1152/ajpgi.90325.2008 [DOI] [PubMed] [Google Scholar]
- 38.Al-Saffar AKh, Meijer CH, Gannavarapu VR, Hall G, Li Y, Diaz Tartera HO, et al. Parallel changes in Harvey-Bradshaw Index, TNF α, and intestinal fatty acid binding protein in response to infliximab in Crohn’s disease. Gastroenterol Res Pract 2017;2017:1–8. doi: 10.1155/2017/1745918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shapiro KB, Hotchkiss JH. Induction of nitric oxide synthesis in murine macrophages by Helicobacter pylori. Cancer Lett 1996;102:49–56. doi: 10.1016/0304-3835(96)04154-7 [DOI] [PubMed] [Google Scholar]
- 40.Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med 2005;201:1257–68. doi: 10.1084/jem.20042028 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.