

Abstract
Loss-of-function mutations in the Thanatos-associated domain-containing apoptosis-associated protein 1 (THAP1) gene cause partially penetrant autosomal dominant dystonia type 6, DYT6. However, the neuronal abnormalities that promote the resultant motor dysfunctions remain elusive. Studies in humans show that some non-manifesting DYT6 carriers have altered cerebello-thalamo-cortical function with subtle but reproducible tremor. Here, we uncover that Thap1 heterozygote mice have action tremor that rises above normal baseline values even though they do not exhibit overt dystonia-like twisting behavior. At the neural circuit level, we show using in vivo recordings in awake Thap1+/− mice that Purkinje cells have abnormal firing patterns and that cerebellar nuclei neurons, which connect the cerebellum to the thalamus, fire at a lower frequency. Although the Thap1+/− mice have fewer Purkinje cells and cerebellar nuclei neurons, the number of long-range excitatory outflow projection neurons is unaltered. The preservation of interregional connectivity suggests that abnormal neural function rather than neuronal loss instigates the network dysfunction and the tremor in Thap1+/− mice. Accordingly, we report an inverse correlation between the average firing rate of cerebellar nuclei neurons and tremor power. Our data show that cerebellar circuitry is vulnerable to Thap1 mutations and that cerebellar dysfunction may be a primary cause of tremor in non-manifesting DYT6 carriers and a trigger for the abnormal postures in manifesting patients.
Keywords: cerebellum, Purkinje cells, cerebellar nuclei, Thap1, DYT6, dystonia, tremor
Introduction
Dystonia is a severe motor disorder characterized by intermittent or continuous muscle contractions that result in abnormal, often repetitive, postures (Lungu et al., 2020). Dystonia is considered a heterogeneous disorder with different forms classified based on the affected body parts, age at onset, and underlying etiologies (Ozelius & Bressman, 2011; Fernández-Alvarez & Nardocci, 2012). Several genes are responsible for early onset primary dystonia; although different forms of dystonia are defined by a core set of neurological symptoms, tremor can also arise from mutations in the same genes. Mutations in TOR1A, which encodes the torsinA protein, cause primary torsion DYT1 (Ozelius et al., 1997) whereas loss-of-function mutations in THAP1, which encodes the THAP1 protein, cause DYT6 (Fuchs et al., 2009; Müller, 2009; Defazio, 2010; Ozelius & Bressman, 2011; Klein, 2014). DYT1 and DYT6 mutations are autosomal dominant with incomplete penetrance. Whereas DYT6 shows about 60% penetrance for the primary dystonia symptoms, three recent independent studies reported that upon detailed clinical examination, non-manifesting siblings or children from manifesting probands also have tremor (Zittel et al., 2010; LeDoux et al., 2012; Nikolov et al., 2019). Most symptomatic DYT1 and DYT6 patients experience minimal to no improvement after treatment, with surgical and electrical stimulation therapies providing only partial relief (Groen et al., 2010; Brüggemann et al., 2015; Fox & Alterman, 2015; Ahn et al., 2019). The development of additional therapeutic options for patients with early onset primary dystonia is, in part, impeded by the lack of symptomatic mouse models that carry disease-causing mutations. Here, we have focused our investigation on the neural substrates that drive tremor, a partially penetrant but critical symptom of dystonia.
The presence of tremor in specific cases of dystonia raises the question whether those patients have the same neural network deficits as other dystonia patients with more severe manifestations. Several lines of evidence motivate this question. Neuroimaging studies showed that both DYT1 and DYT6 non-manifesting carriers and manifesting patients have hypermetabolism in multiple regions of the motor cortex compared to control (no mutation) participants (Carbon et al., 2004; Carbon & Eidelberg, 2009). Furthermore, DYT1 and DYT6 carriers have a decrease in volume of the superior cerebellar peduncle, which connects the cerebellum to a large number of forebrain regions (Carbon et al., 2008), and reduced cerebellar outflow and cerebellothalamocortical connectivity (Argyelan et al., 2009; Niethammer et al., 2011). A recent study confirmed the presence of abnormal cerebellar function in a patient with DYT6 (Nikolov et al., 2019). And while Thap1 heterozygote mice (Thap1+/−) have no overt dystonia-like symptoms, they do have a reduction in cerebellar output neurons (Ruiz et al., 2015), suggesting that the cerebellum is sensitive to Thap1 loss-of-function mutations. These data are consistent with the heavy expression of Thap1 in the cerebellum (Zhao et al., 2013). In particular, RNA and protein expression indicate high levels of expression in developing and adult Purkinje cells (Zhao et al., 2013), and in situ hybridization data from the Allen Brain Atlas also indicate moderate to high levels of transcript in granular layer interneurons that are likely the large Golgi cells, as well as expression in all three major divisions of the cerebellar nuclei (Lein et al., 2007; Zhao et al., 2013). THAP1 expression in Purkinje cells and cerebellar nuclei neurons is of particular interest to the current work as these neurons and their connections are thought to play a major role in tremor pathogenesis (Pan et al., 2020; Brown et al., 2020a; Nietz et al., 2020).
In this study, we tested whether abnormal cerebellar function promotes neural network deficits that manifest as tremor in Thap1+/− mutant mice in which overt dystonia-like symptoms are lacking. We investigated the response of cerebellar neurons in a Thap1 loss-of-function model by performing single-unit in vivo electrophysiology recordings of Purkinje cells, the primary computational neurons in the cerebellar cortex, and of cerebellar nuclei neurons, which are the primary output neurons of the cerebellum. We also quantified cell number in a subpopulation of cerebellar nuclei neurons that are associated with tremor. By comparing our data from Thap1+/− mice to mouse models that have overt dystonia-like behaviors, we argue that the cerebellum may play multiple roles in the expression of different dystonia-associated behaviors.
Methods
Ethical approval:
Husbandry, housing, euthanasia, and experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine (protocol number: AN-5996) according to the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines.
Mice:
We used heterozygote and control littermates that were generated from Thap1+/− to control crosses (Ruiz et al., 2015). Pups were ear punched before weaning in order to minimalize stress on the animals. Tissue from the ear punches was used for PCR genotyping. We used Thap1+/− heterozygotes since the Thap1−/− homozygotes are embryonic lethal (Ruiz et al., 2015). Experimenters were blinded to the genotype of the mice when obtaining the data. We used mice of both sexes as previous studies suggested a gender bias in THAP1 dystonia (LeDoux et al., 2012) and cerebellar controls (Mercer et al., 2016). Nevertheless, in our experiments we did not observe a significant interaction effect between sex and genotype on any of the variables that we tested in this study and therefore combined all the data together (p-values of 2-way ANOVA interaction between sex and genotype are indicated in relevant sections of the results). Mice between 6 weeks and 6 months of age were used for the experiments. We intentionally included mice from multiple litters, both sexes, a range of ages as previously stated, and where possible included data that were generated from at least two experimenters to enhance the robustness and reproducibility of our findings across multiple biological and contextual variations (Voelkl et al., 2020). Additional experiments would be necessary to fully determine whether sex and age differences impact the cellular and network changes reported in this set of studies. After experiments, animals were anesthetized using isoflurane and then perfused as described below.
Tremor measurement and assessment:
We used a custom-built tremor monitor to analyze tremor power in our mice (Figure 1A) (Brown et al., 2020a; van der Heijden et al., 2020). This tremor monitor consists of a box that is suspended by eight bungy cords attached to each corner of the box and four vertical poles. We mounted an accelerometer at the bottom of the box that detects movements of the box that are caused by movements, including tremor, of the mouse. Therefore, the mouse is allowed to move freely within the 5 inches (length) by 4.5 inches (width) box arena, which enables us to track the moment-to-moment motor behavior changes in each mouse tested. The signals from the accelerometer were transformed to an electrical signal that in turn were digitized using a Brownlee amplifier. The recorded signals were recorded and analyzed using Spike2 software. We defined tremor using a power spectrum across naturally occurring tremor frequencies (0-30 Hz) using a fast Fourier transform (FFT) with Hanning window. We centered our recordings around 0 mV and down sampled our signal to produce frequency bins aligned to whole numbers. We defined the first 120 seconds (two minutes) as habituation period and analyzed the sequential 180 second (three minutes) in our recording for our final tremor power analysis. This approach provided us with one power spectrogram for each mouse, averaged over three minutes. We defined the peak power and average power in the 0-30 Hz power spectrogram for each animal and then performed a two-tailed t-test with the genotype as the independent variable on these values. We reported all values as mean and accepted a p-value smaller 0.05 as statistically significant.
Figure 1: Thap1+/− mice have a pathophysiological tremor.
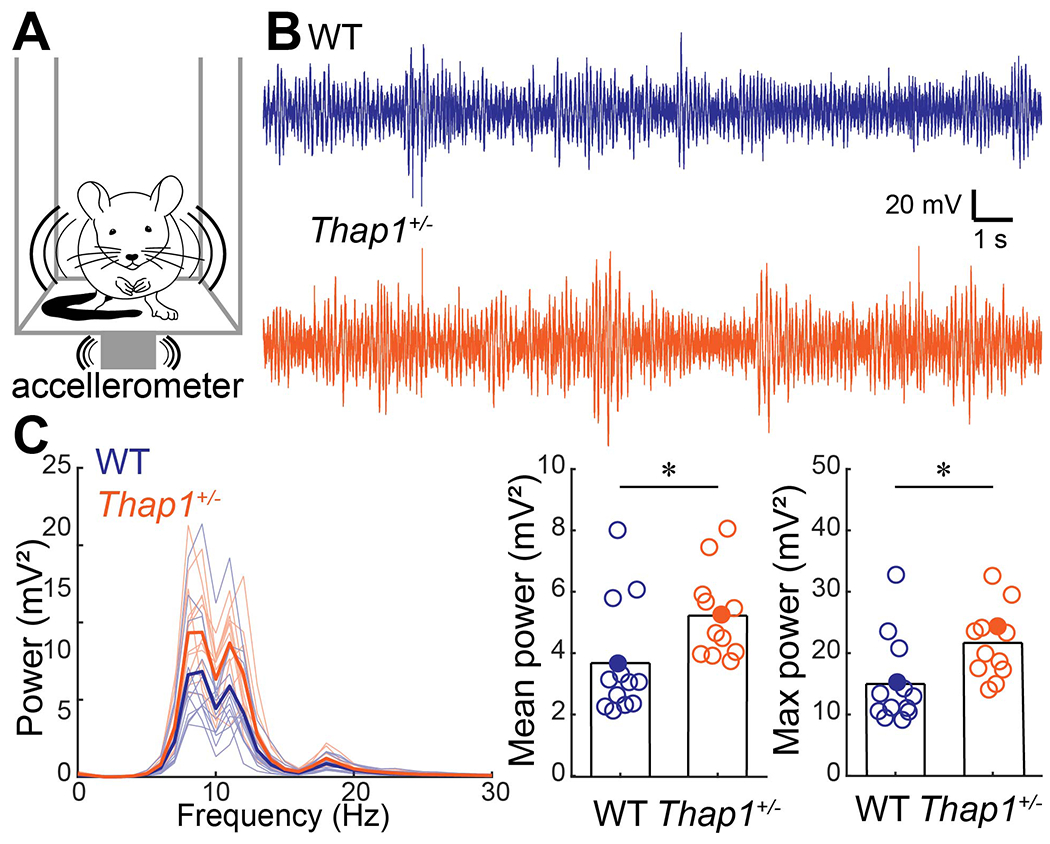
A. Schematic of tremor monitor with accelerometer attached to bottom of a box that is held in the air by eight elastic springs. B. Example traces of tremor signals obtained by the tremor monitor. Examples from filled circles in (C). C. Power spectrum of tremor signals for control (blue) and Thap1+/− (orange) mice (left). Thick line is the group mean, thin lines represent individual mice. Mean tremor power (0-30 Hz) (middle) and max power (right) were significant higher in Thap1 mice. Definitions of summary statistics: Bar height represents the mean of all animals. Control: N=13 mice; Thap1+/: N=12 mice. Student’s t-test: mean power, p=0.0265; max power, p=0.0145.
Horizontal ladder:
We let mice walk on a custom-built horizontal ladder. This ladder consists of round, horizontal rods, spaced 1 cm apart, walled off by two plexiglass plates. We used high-frame vides to record mice walking over this ladder and quantified the total number of miss-steps by front- and hind paws per run over a 30-centimeter-long section of the ladder. Miss-steps were determined as steps where the mouse would miss a rod and the paw dipped below the level of the rods and counted by an observer that was blinded to the genotype of the mouse. The data were compared using a Two-tailed t-test and a p-value smaller than 0.05 was considered statistically significant.
Head plate surgery:
In order to perform in vivo electrophysiology in awake animals, we first performed surgery to attach a head plate and make a craniotomy above the cerebellum. The head plate was secured to the skull to stabilize the head during recordings. These surgeries are described in detail in several previous publications (White et al., 2016; White & Sillitoe, 2017; Brown et al., 2020a). In brief, we provided preemptive analgesics (buprenorphine: 0.6 mg/kg subcutaneous; meloxicam: 4 mg/kg subcutaneous) and maintained mice under continuous anesthesia using nasally-delivered isoflurane gas (2-3%). All surgeries were performed on a stereotaxic platform (David Kopf Instruments) according to sterile surgery techniques that were approved by the IACUC at Baylor College of Medicine. We shaved fur from the head and used a scalpel blade to make an incision in the skin over the midline of the skull. We then mounted a custom head plate over bregma using C and B Metabond Adhesive Luting Cement (Parkell), and we placed a vertical piece of wire within the Metabond on top of bregma that could later be used to identify specific stereotaxic coordinates. We then placed a skull-screw over the left cerebellum. Over the right cerebellum (6.4 mm caudal and 1.3 mm lateral from bregma), we made a circular craniotomy about 2 mm in diameter. We placed a custom 3D-printed chamber over the craniotomy, filled the chamber with antibiotic ointment, and then closed it off with a custom 3D-printed screw top. Finally, we used dental cement (dental cement powder #525000; solution #526000; A-M Systems) to firmly attach the craniotomy-chamber, skull screw, and head plate to each other and the skull. Animals were placed in a clean cage on a small animal warmer while recovering from anesthesia, received additional analgesics for a minimum of three days after surgery, and were monitored for stress and pain throughout the experimental period, during which time neurons were recorded.
In vivo electrophysiological recordings in awake animals:
We performed in vivo recordings from Purkinje cells and cerebellar nuclei cells in awake, head-fixed animals. Single-unit extracellular recordings were performed according to the protocols detailed in several previous publications (White et al., 2016; White & Sillitoe, 2017; Brown et al., 2020a). In brief, we placed the mice on a rotating foam wheel and stabilized their heads by screwing the head plate to a frame. We allowed the animals to acclimate to the recording set-up for a minimum of thirty minutes prior to recording from neurons. For the recordings, after acclimation, we replaced the antibiotic ointment from the chamber with sterile physiological saline solution. Tungsten electrodes were manipulated using a motorized micromanipulator (MP-225; Sutter Instrument Co) that allowed us to precisely move the electrode in all axes and record neurons using stereotaxic coordinates to place the electrodes in the brain and accurately guide their penetration to the required recording depths. Coordinates of brain penetration for cells included in this study ranged from 0.09 to 2.5 mm lateral from bregma (mean = 1.2 mm; SD = 0.6 mm) and ranged from 5.0 to 7.0 mm caudal from bregma (mean = 6.2 mm; SD = 0.5 mm). These coordinates allowed us to easily access the central cerebellar lobules, but because of the folded architecture of the cerebellum, we were also able to record from some anterior lobules IV-V as well. The location of craniotomy was over the vermis/paravermis areas. Targeting of these medial-lateral cerebellar regions and these lobules was ideal because of their involvement in ongoing motor behavior, particularly with respect to movement of the limbs and torso. The neuronal signals were then amplified and bandpass (0.3-13 kHz) filtered (ELC-03XS amplifier, NPI Electronic Instruments) before being digitized (CED Power 1401, CED). All in vivo electrophysiology signals were recorded and analyzed using Spike2 software (CED).
Quantitative analyses of electrophysiological recordings:
We quantified Purkinje cells as cells that were recorded 0.4-2.6 mm from the brain surface (range = 0.4-2.6 mm; mean = 1.7 mm; SD: 0.6 mm) and had visible complex spikes (see Figure 2 for examples) and quantified cerebellar nuclei cells as cells that were 2.5-4.3 mm from the brain surface (range = 2.6-4.3 mm; mean = 3.4 mm; SD: 0.5 mm). Our single-unit in vivo recordings prevent us from controlling for sampling of certain molecular or functional subgroups of Purkinje cells and nuclei cells. We avoid sampling biases by recording from a large number of randomly encountered neurons (17-28 neurons per genotype per cell type). We analyzed neurons as described in our previous studies (Brown et al., 2020a; van der Heijden et al., 2020). First, we spike-sorted cells using Spike2 software. For Purkinje cells, we distinguished complex spikes from simple spikes. We only included cells from which we had obtained at least 50 seconds of high-quality continuous recordings (PC: range = 50-200 s; mean = 123 s; SD = 43 s; CN: range = 50-202 s; mean = 141 s; SD = 44 s), and there was no statistical difference in recording length between control and Thap1+/− cells (PC: WT = 124.5±48 s; Thap1+/− = 121.1±42 s; t-test, p=0.806; CN: WT = 152.5±45 s; Thap1+/− = 142.8±44 s; t-test, p=0.426). We analyzed a maximum of six cells for each neuronal type per mouse to avoid bias to single mice (PC: WT N=5 animals, n=18 cells; Thap1+/− N=6 animals, 23 cells; CN: WT N=6 animals, n=17 cells; Thap1+/− N=6 animals; 28 cells). After sorting spikes from in vivo recordings, we used the spike timing to identify firing parameters using custom code in MATLAB. We defined firing frequency as total number of spikes divided by recording time (spikes/s). We further analyzed the global (CV) and local (CV2) regularity of the inter spike intervals (ISI) between two adjacent spikes (in seconds): CV = stdev(ISI)/mean(ISI), and CV2 = mean(2*|ISIn-ISIn-1|/(ISIn+ISIn-1)). We used Linear Mixed Models (LMM) with mouse genotype as a fixed variable and mouse number as a random variable and accepted a p-value smaller 0.05 as statistically significant.
Figure 2: Purkinje cells in Thap1+/− mice fire more regularly than control Purkinje cells.
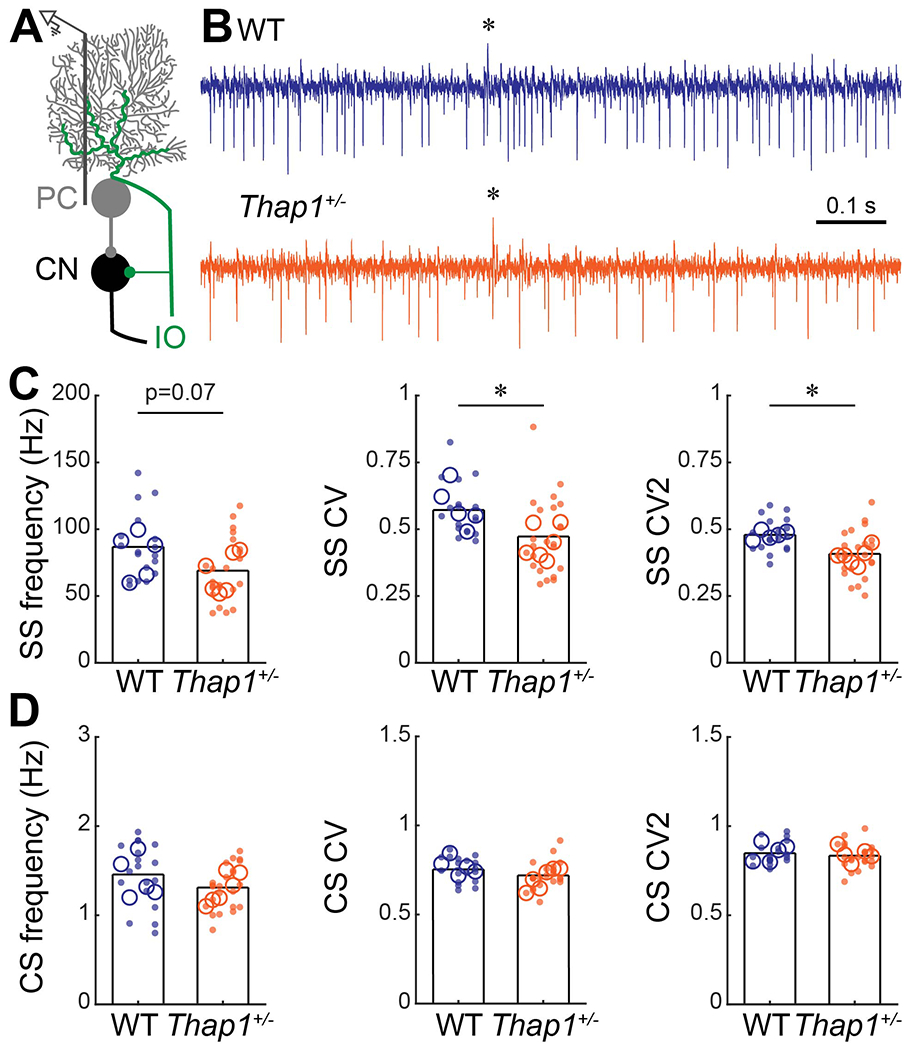
A. Schematic of extra-cellular recording from Purkinje cells in the cerebellar circuit. PC = Purkinje cell, CN = cerebellar nuclei, IO = inferior olive. B. Examples of electrophysiological recordings from Purkinje cells recorded from a control (top, blue) and Thap1+/− (bottom, orange) mouse. * = climbing fiber induced complex spike. Example traces are from cells that best represent the mean Frequency, CV, and CV2 for their genotype. C. Frequency, and global (CV) and local (CV2) irregularity of simple spike firing patterns. Purkinje cells in Thap1+/− mice firing significantly less irregular (more regular) than those in control mice. Frequency, p=0.0750; CV, p=0.0141; CV, p=0.0043 D. Frequency, and global (CV) and local (CV2) irregularity of complex spike firing patterns. No differences were observed between control mice and Thap1+/− mutants. Frequency, p=0.2708; CV, p=0.1204; CV, p=0.4640.
Definitions of summary statistics: Each large circle represents the mean for one mouse, whereas each dot represents one Purkinje cell. Bar height represents the mean of all cells for each genotype. Control: N=5 mice, n=18 cells; Thap1+/−: N=6 mice, n=23 cells. *p<0.05 as determined by a linear mixed model (LMM) with genotype as a fixed variable and mouse number as a random variable.
Immunohistochemistry:
Tissue processing and immunohistochemistry were performed as described in our previous publications (Brown et al., 2020a; van der Heijden et al., 2020). In brief, we anesthetized mice and cardiac perfused them with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde (4% PFA). We then dissected the brain out of the skull and post-fixed brain tissue in 4% PFA overnight. Concomitantly, we cryoprotected the tissue in a sucrose gradient (10% → 20% → 30% sucrose in PBS) until the tissue sank and then froze the tissue in optimal cutting temperature (OCT) solution. We stored the tissue at −80 °C until cut in 40-micron floating sections. We stored cut sections in PBS at 4 °C until we stained for goat (gt)-α-RAR-related orphan receptor α (RORα; 0.8 μg/mL; Santa Cruz Biotechnology; #sc-6062), rabbit (rb)-α-carbonic anhydrase (Car8; 0.2 μg/mL; Santa Cruz Biotechnology, #sc-67330) and rb-α-Inositol trisphosphate receptor1 (IP3R1; 0.4 μg/mL; Santa Cruz Biotechnology; #sc-6093) (Figure 3), or mouse(ms)-Neurofilament Heavy (NFH; 1 μg/mL; Biolegend; #801701) and rb-α-Neuronal Nuclei (NeuN; 1 μg/mL; Abcam; #ab104225) (Figure 5). In brief, we immunostained our sections as follows: blocked for two hours in blocking solution (10% normal goat serum, 0.1% Triton-X in PBS (PBS-T)); primary antibody in blocking buffer overnight at concentrations mentioned above; washed three times in PBS-T; secondary antibody in PBS-T (2 μg/mL); washed three times in PBS-T; mounted on electrostatically-coated slides with hard-set DAPI containing mounting medium. We used the following secondary antibodies: donkey (dk)-α-rabbit IgG Alexa Fluor 555 (Thermo Fisher Scientific; #A31572) and dk-α-goat IgG Alexa Fluor 488 (Thermo Fisher Scientific; #A32814) (Figure 3), or dk-α-rabbit IgG Alexa Fluor 555 (Thermo Fisher Scientific; #A31572) and dk-α-mouse IgG Alexa Fluor 647 (Thermo Fisher Scientific; #A32787) (Figure 5). We imaged the stained sections using Leica DPC365FX and DMC2900 cameras that were mounted onto a Leica DM4000 B LED microscope. Cell numbers were counted and normalized to the width of the field of view (PC, Figure 3) or the area of the field of view (as seen in all other samples shown in Figure 3 and Figure 5) with this number corresponding to the images used for analysis in ImageJ software. Gross Purkinje cell morphology was assessed by averaging the average soma size of five Purkinje cells per section and measuring the height of the molecular layer in ImageJ (used as a measure of Purkinje cell dendrite length and defined as the distance from the start of the primary dendrites at the very top of the somata to the pial surface directly above). We obtained the measurements from the midline of dorsal lobule V and VI using two to five sections per animal (n) and three or four animals per genotype (N). As for the in vivo electrophysiology, we used LMM with genotype as a fixed variable and mouse number as a random variable, and we accepted a p-value smaller 0.05 as statistically significant.
Figure 3: Thap1+/− mutant mice have a reduction in the number of Purkinje cells.
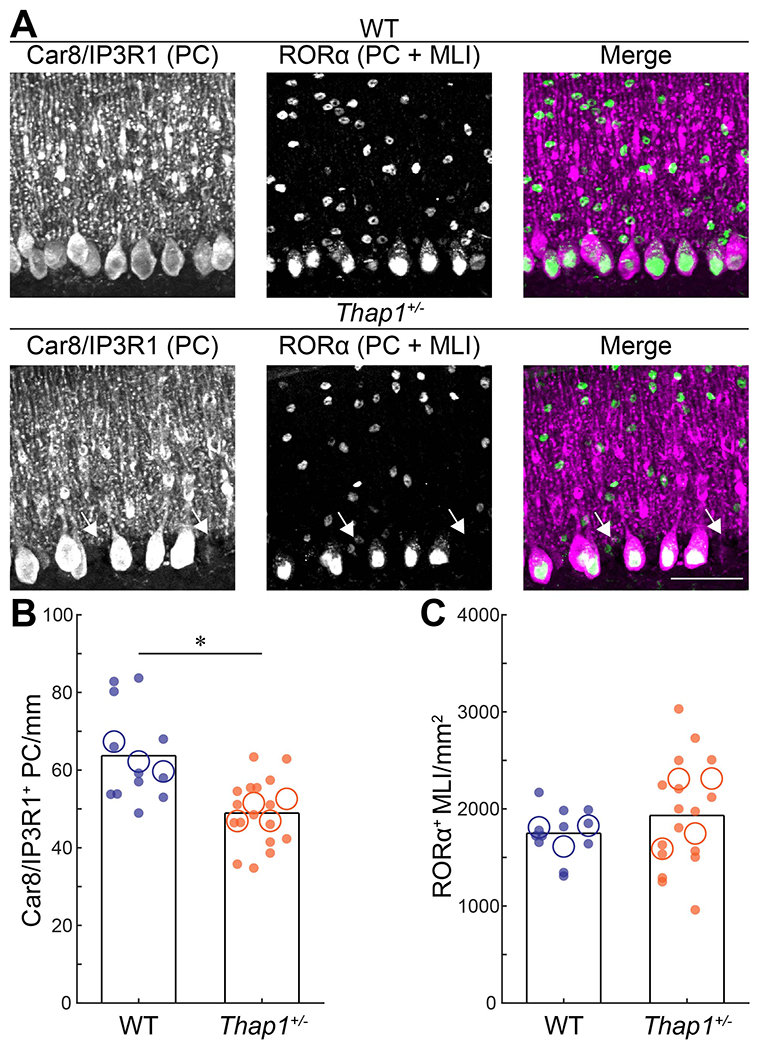
A. Representative images of Purkinje cells (PC) and molecular layer interneurons (MLIs) with a combination of Car8/IP3R1 (magenta) and RORα (green) in control (top) and Thap1+/− (bottom) mice. Arrows pointing at missing Purkinje cells. B. Neural density of Car8/IP3R1+ (PC) cells/mm, p=0.0005. C. Neural density of RORα cells/mm2, p=0.3275.
Definitions of summary statistics: Each large circle represents the mean for one mouse, whereas each dot represents one field of view. Bar height represents the mean of based on all cells for each genotype. Control: N=3 mice, n=12 sections; Thap1+/−: N=4 mice, n=17 sections. *p<0.05 as determined by a linear mixed model (LMM) with genotype as a fixed variable and mouse number as a random variable. All images were taken at same magnification, scalebar = 50 μm.
Figure 5: Thap1+/− mutants have a reduced number of cerebellar nuclei neurons but the number of excitatory projection neurons is preserved.
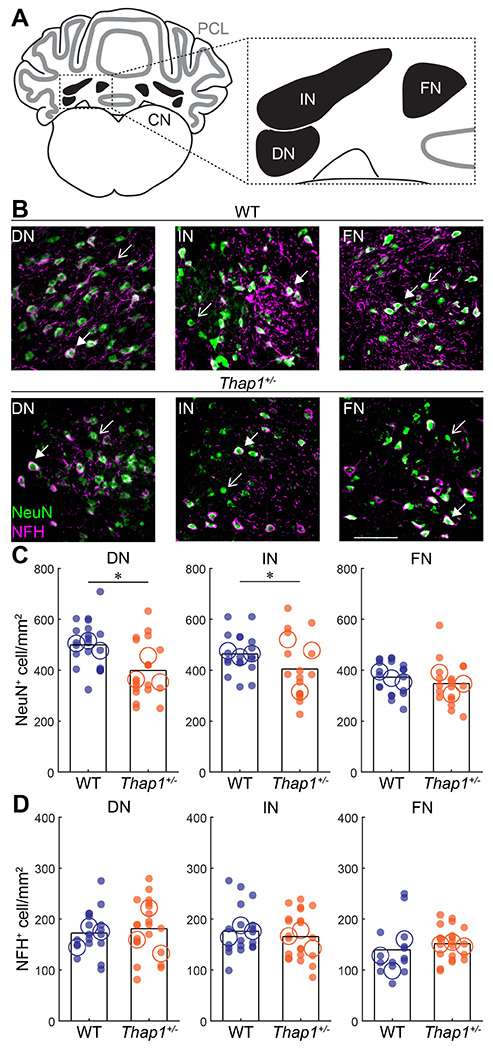
A. Schematic of a coronal section of the cerebellum with the Purkinje cell layer in gray and cerebellar nuclei in black. Inset shows the anatomical location of the dentate nucleus (DN), interposed nucleus (IN), and fastigial nucleus (FN). B. Representative images of DN, IN, and FN nuclei stained for the neuronal marker NeuN (green) and marker for large, excitatory projection neurons with NFG (magenta) in control (top) and Thap1+/− (bottom) mice. Open arrowheads indicate NeuN+/NFH− neurons; closed arrowheads indicate NeuN+/NFH+ neurons. C. Neural density of NeuN+ cells/mm2 in the DN, IN, and FN. A reduction in neuron number was observed in the DN and IN of Thap1+/− mice. DN, p=0.0129; IN, p=0.0206; FN, p=0.5279. D. Neural density of NFH+ cells/mm2 in the DN, IN, and FN is unaffected in Thap1+/− mice. DN, p=0.8447; IN, p=0.4772; FN, p=0.3679.
Definitions of summary statistics: Each large circle represents the mean for one mouse, whereas each dot represents one field of view. Bar height represents the mean of all cells for each genotype. Control: N=3 mice, n=15-20 sections; Thap1+/−: N=3 mice, n=17-22 sections. *p<0.05 as determined by a linear mixed model (LMM) with genotype as a fixed variable and mouse number as a random variable. All images were taken at same magnification, scale bar = 100 μm.
Results
Thap1+/− mice have a pathophysiological level of tremor
A subpopulation of DYT6 patients have dystonic tremors, although mild tremors have also been reported in THAP1 loss-of-function mutation carriers that did not exhibit the other common motor symptoms of dystonia (Zittel et al., 2010; LeDoux et al., 2012; Nikolov et al., 2019). To investigate whether heterozygote Thap1 mice (Thap1+/− are viable and live to adulthood) (Ruiz et al., 2015) display an enhanced tremor, we measured tremor power in Thap1+/− mutants and compared their levels to littermate controls using a custom-made tremor monitor (Brown et al., 2020a) (Figure 1A–B). Compared to the controls, we found that Thap1+/− mutants had an increase in tremor power within the frequency range that is typical of normal physiological tremor (8-12 Hz). The 8-12 Hz range has a particular relevance to cerebellar function and dysfunction since olivocerebellar connectivity (Kuo et al., 2019), which normally operates at sub-threshold levels within this range, is also associated with tremor, albeit at a higher power (White & Sillitoe, 2017; Brown et al., 2020a). We found no significant interaction between genotype and sex on either mean or max tremor power (2-way ANOVA, interaction between sex and genotype: mean power, p=0.4862; max power, p=0.5706). Accordingly, we grouped both sexes together in our analysis and we found that mean tremor power (0-30 Hz) and max tremor power were significantly higher in the Thap1+/− mice (Figure 1C) (t-test: mean power, p=0.0265; max power, p=0.0145). We also confirmed that Thap1+/− mice do not exhibit dystonia-like symptoms by observing their ambulatory activity in the home cage. Furthermore, the Thap1+/− mice did not have obvious abnormalities in motor activity as measured by the number of miss-steps that the mice take when walking on a 30-centimeter-long horizontal ladder (Miss steps: WT: 3.3±2.3 miss-steps per run (N=7 mice, 3 runs per mouse), Thap1+/−: 3.4±2.9 miss-steps per run (N=6 mice, 3 runs per mouse), t-test: p=0.9447). The data showed that, similar to some non-manifesting DYT6 carriers, Thap1+/− mutant mice have a statistically significant increase in tremor that does not impair gross motor performance (Ruiz et al., 2015), but was nevertheless stronger than what is normal for control mice. Our data generated in mice are therefore consistent with the clinical presentation of human neurological symptoms reported in a subpopulation of non-manifesting DYT6 carriers.
Purkinje cell firing patterns have an abnormally high regularity in Thap1+/− mice
There are several lines of evidence showing cerebellar involvement in DYT6 dystonia, including data demonstrating that the cerebellum was malfunctional in a DYT6 patient (Nikolov et al., 2019) and that non-manifesting DYT6 carriers have abnormal connectivity in the cerebello-thalamo-cortical pathway (Argyelan et al., 2009; Niethammer et al., 2011). In regards to dystonia phenotypes in general, there are compelling arguments that the cerebellum is a main instigator (LeDoux & Lorden, 2002; Pizoli et al., 2002; Neychev et al., 2008; Calderon et al., 2011; Luna-Cancalon et al., 2014; Fremont et al., 2015, 2017; Washburn et al., 2019). Moreover, altering cerebellar function in mice not only induces severe dystonia-like behaviors, but it also triggers a pathophysiological level of tremor (White & Sillitoe, 2017). Based on these previous studies, we postulated that cerebellar function may also be affected in Thap1 mutant mice. To test whether cerebellar neurons in the Thap1+/− mice have abnormal functional activity, we performed single-unit in vivo recordings on Purkinje cells since they form the sole output of the cerebellar cortex, and they execute the essential computations required for proper movement (Figure 2A). Purkinje cells fire two distinct types of action potentials: simple spikes and complex spikes. Simple spikes are intrinsically generated action potentials (Figure 2B). Simple spike frequency, defined here as the number of spikes over a pre-determined period of time, can be modulated by cerebellar afferent input and by interneurons in the cerebellar cortex (Palmer et al., 2010; Galliano et al., 2013; Brown et al., 2019). Complex spikes are generated by input from climbing fibers that originate in the inferior olive of the brainstem (Davie et al., 2008; Palmer et al., 2010) and can be recognized by an initial large amplitude spike that is followed by three to five smaller spikelets (“*” in Figure 2B). Therefore, simple spikes and complex spikes not only inform about Purkinje cell firing, but also how the surrounding circuit drives this cerebellar cortical output. We recorded Purkinje cell activity in awake control mice and Thap1+/− mutants to investigate whether either type of action potential was affected by Thap1 heterozygosity (Figure 2B). The anterior and central lobules of the vermis/paravermis regions were targeted for the recordings. Purkinje cells in these regions of the cerebellum are critical for ongoing motion, and modulation of their activity can drive tremor (Brown et al., 2020a). Making a craniotomy over these cerebellar regions not only provided access to specific Purkinje cells, but also to their topographically connected cells that are located downstream in the cerebellar nuclei (Sillitoe et al., 2012). We found no significant interaction between genotype and sex on Purkinje cell firing rate or regularity (2-way ANOVA, interaction between sex and genotype: SS frequency, p=0.5344; SS CV, p=0.1570; SS CV2, p=0.5388; CS frequency, p=0.3822; CS CV, p=0.1085; CS CV2, p=0.5552). We therefore grouped the neuronal recordings from male and female mice together. We found that Purkinje cells in the Thap1+/− mutant mice have a small decrease in simple spike firing rate, although the effect was not statistically significant. However, they have a significant decrease in global (CV) and local (CV2) inter-spike interval variability (Figure 2C; Linear Mixed Model (LMM): Frequency, p=0.0750; CV, p=0.0141; CV, p=0.0043). We did not observe any differences in the frequency or regularity of complex spikes (Figure 2D; LMM: Frequency, p=0.2708; CV, p=0.1204; CV2, p=0.4640). These results show that the loss of Thap1 does not eliminate the neurophysiological function of Purkinje cells, but instead it alters their ability to fire specific kinds of neuronal signals. This means that THAP1 protein normally supports intrinsic Purkinje cell activity or their responses to the afferent activity provided by their innervating inhibitory interneurons (Brown et al., 2019) and excitatory granule cells (Galliano et al., 2013), which altogether ultimately results in setting the proper regularity of Purkinje cell firing.
Purkinje cell number is decreased in Thap1+/− mutant mice
A mild reduction in Purkinje cell number was observed in a neuropathological report of DYT6 cases and non-manifesting DYT6 carriers (Prudente et al., 2013; Paudel et al., 2016). In addition, reduced spontaneous firing rates of Purkinje cells are observed in mouse models of Purkinje cell degeneration and these firing changes can even precede progressive cell loss (Hourez et al., 2011; Hansen et al., 2013; Cook et al., 2021). Furthermore, loss of stellate cell function, a type of inhibitory interneuron residing in the molecular layer of the cerebellar cortex, results in an increase of local Purkinje cell firing regularity (Brown et al., 2019). Therefore, we sought to investigate whether the abnormal firing pattern of Purkinje cell activity is accompanied by a reduction in the number of Purkinje cells or molecular layer interneurons (MLIs). To identify Purkinje cells, we stained for two reliable Purkinje cell markers, Car8 and IP3R1, that together mark all adult Purkinje cells (Figure 3A, magenta). We also stained for RORα, a nuclear receptor that is expressed in Purkinje cells and MLIs (Figure 3A, green). We found a reduced number of Purkinje cells in Thap1+/− mice compared to control mice (Figure 3B; LMM: p=0.0005), but we did not observe a difference in MLI number (Figure 3C; LMM: p=0.3275). We did not find any changes in gross Purkinje cell morphology as measured by soma size or dendrite length (assessed by measuring molecular layer thickness) (Soma size: WT: 277±70 μm2 (N=3 mice, n=12 sections, 5 cells per section), Thap1+/−: 299±51 μm2 (N=4 mice, n=17 sections, 5 cells per section), LMM: p=0.4775; Dendrite length: WT: 0.17±0.05 mm (N=3 mice, n=12 sections), Thap1+/−: 0.18±0.06 (N=4 mice, n=17 sections), LMM: p=0.4286). Thus, the changes in Purkinje cell firing activity may be early signs of cellular pathophysiology and these functional changes are likely independent of molecular layer interneuron number. Based on these data, we predicted that the structural and functional impact of cerebellar cortical defects, specifically in the Purkinje cells, would be translated at the circuit level and reflected as changes in cerebellar output function.
Cerebellar nuclei neurons have abnormally low firing frequencies in Thap1+/− mice
Purkinje cells project primarily onto cerebellar nuclei neurons, which in turn form the major connections between the cerebellar cortex and the thalamus, brainstem, and spinal cord (Beckinghausen & Sillitoe, 2019). Each of these regions are required for smooth movement, and each one has been implicated in dystonia (Cammarota et al., 1995; McNaught et al., 2004; Neychev et al., 2008, 2011; Blood et al., 2012; Zhang et al., 2017; DeSimone et al., 2019). Notably, cerebellar nuclei cells have abnormal firing patterns in mouse models of dystonia (Calderon et al., 2011; Fremont et al., 2014, 2017; White & Sillitoe, 2017). In addition to Purkinje cells, THAP1 protein is also expressed in all three divisions of the cerebellar nuclei; the fastigial (most medial), interposed (middle), and dentate (most lateral) (Allen Brain Atlas). We postulated that in Thap1+/− mice, the changes in Purkinje cell function (Figure 2) and reduced Purkinje cell number (Figure 3), or the loss of gene function in both Purkinje cells and cerebellar nuclear neurons, could lead to functional defects in the cerebellar nuclei. Therefore, we recorded cerebellar nuclei cells in vivo in Thap1+/− mice to test whether they also have abnormal firing activity (Figure 4A–B). We focused our recordings on the interposed nucleus because this cerebellar nucleus is essential for ongoing movement (Bracha et al., 1999; Low et al., 2018; Becker & Person, 2019), rhythmic stimulation of the interposed nucleus can cause tremor (Brown et al., 2020a), and deep brain stimulation targeted to the interposed nucleus can alleviate tremor and dystonia in mouse models (White & Sillitoe, 2017; Brown et al., 2020a). We recorded from a wide range of neurons that are likely a mix of excitatory and inhibitory nuclei neurons (Uusisaari et al., 2007; Özcan et al., 2020). We found no significant interaction between genotype and sex on nuclei cell firing rate or regularity (2-way ANOVA, interaction between sex and genotype: frequency, p=0.5418; CV, p=0.8575; CV2, p=0.3145). As for Purkinje cells, we grouped the recordings from male and female mice together and found that the action potentials in the interposed nuclear neurons of Thap1+/− mice had a significantly lower firing frequency compared to control mice. However, we did not observe a change in the global (CV) or local (CV2) interspike interval in mutant cells compared to cells recorded in the control mice (Figure 4C; LMM: Frequency, p=0.0153; CV, p=0.8870; CV, p=0.7360). Thus, the interposed cerebellar nucleus, which controls ongoing motor behavior during normal movement and drives tremor in disease, is defective in the Thap1+/− mice.
Figure 4: Cerebellar nuclei cells in Thap1+/− mice fire slower than control nuclei cells.
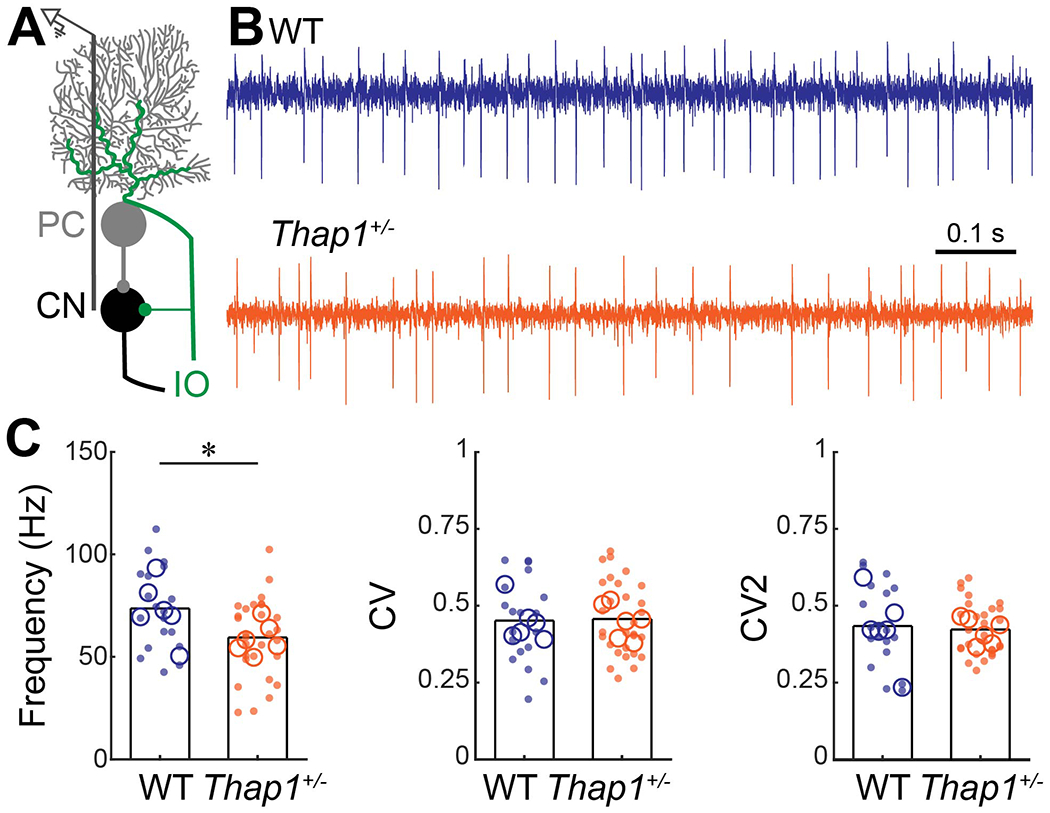
A. Schematic of extra-cellular recording from cerebellar nuclei cells in the cerebellar circuit. PC = Purkinje cell, CN = cerebellar nuclei, IO = inferior olive. B. Examples of electrophysiological recordings from cerebellar nuclei cells recorded from a control (top, blue) and Thap1+/− (bottom, orange) mouse traces are from cell that best represent the mean Frequency, CV, and CV2 for their genotype. C. Frequency, and global (CV) and local (CV2) irregularity of cerebellar nuclei firing patterns. Cerebellar nuclei cells in Thap1+/− mice fire significantly slower than control cerebellar nuclei cells. Frequency, p=0.0153; CV, p=0.8870; CV, p=0.7360.
Definitions of summary statistics: Each large circle represents the mean for one mouse, whereas each dot represents one nuclei cell. Bar height represents the mean of all cells for each genotype. Control: N=6 mice, n=17 cells; Thap1+/−: N=6 mice, n=28 cells. *p<0.05 as determined by a linear mixed model (LMM) with genotype as a fixed variable and mouse number as a random variable.
Projection neurons are preserved in the interposed and dentate cerebellar nuclei of Thap1+/− mice
Next, we set out to determine whether the abnormalities in cerebellar firing frequency were accompanied by overt changes in the cellular make-up of the cerebellar nuclei (Figure 5A). Each cerebellar nucleus consists of excitatory projection neurons that are intermingled with inhibitory projection and local interneurons. The excitatory nuclei neurons have a higher firing frequency (Özcan et al., 2020) and a larger cell body (Uusisaari et al., 2007) compared to the inhibitory neurons. A previous study found that Thap1+/− mice have a decreased number of cells in the dentate (most lateral) cerebellar nucleus without a change in average cell size (Ruiz et al., 2015). To investigate whether this change in cell number can help explain the defects we observed in neuronal activity and whether the lower cell number encompasses all three divisions of the cerebellar nuclei, we stained for the neuronal marker Neuronal Nuclei (NeuN). In addition, to identify whether pathological defects in the high-frequency, excitatory projection neurons could account for the observed decrease in firing frequency of cerebellar nuclei recordings, we quantified the number of excitatory neurons by staining for Neurofilament Heavy (NFH) that marks the large excitatory projection neurons in the cerebellum (Figure 5B) (Fujita et al., 2020). We stained, imaged, and counted NeuN+ and NFH+ neurons in the dentate, interposed, and fastigial nucleus. We found a marked reduction in the density of NeuN+ cells in the dentate and interposed nuclei but not in the fastigial nucleus of Thap1+/− mice (Figure 5C; LMM: dentate, p=0.0129; interposed, p=0.0206; fastigial, p=0.5279). We did not find a difference in the density of NFH+ neurons in any of the cerebellar nuclei (Figure 5D; LMM: dentate, p=0.8447; interposed, p=0.4772; fastigial, p=0.3679). These results indicate that the decrease in the number of neurons in the cerebellar nuclei does not include the high-frequency, excitatory neurons. Thus, at the gross anatomy level, the excitatory long-range projection neurons that send axons out of the cerebellum are present and intact in Thap1+/− mice.
Tremor accompanies the low frequency firing of cerebellar nuclei neurons
The data from our anatomical and electrophysiological experiments suggested that the abnormal function of cerebellar neurons rather than lower cell number may be the cause of neural network wide abnormalities that cause tremor in Thap1+/− mice and non-manifesting DYT6 carriers. In line with this hypothesis, a more severe network deficit would be predicted to lead to a stronger tremor along with overt dystonia. Indeed, we found a statistically significant, inverse correlation between the max tremor power and the mean firing frequency of cerebellar nuclei neurons (n=2-6 cells per mouse) that were recorded from the same mouse (Figure 6A) (LM: p=0.0389). Interestingly, we also found that one Thap1+/− mouse that had high cerebellar nuclei firing frequency also presented with a low tremor intensity and one control mouse with a low cerebellar nucleus firing frequency had a high tremor intensity. Based on these analyses, the statistical correlation between tremor power and cerebellar nuclei firing frequency may be a marker for tremor, independent of genotype.
Figure 6: Reduced firing frequency of cerebellar neurons correlates with tremor power.
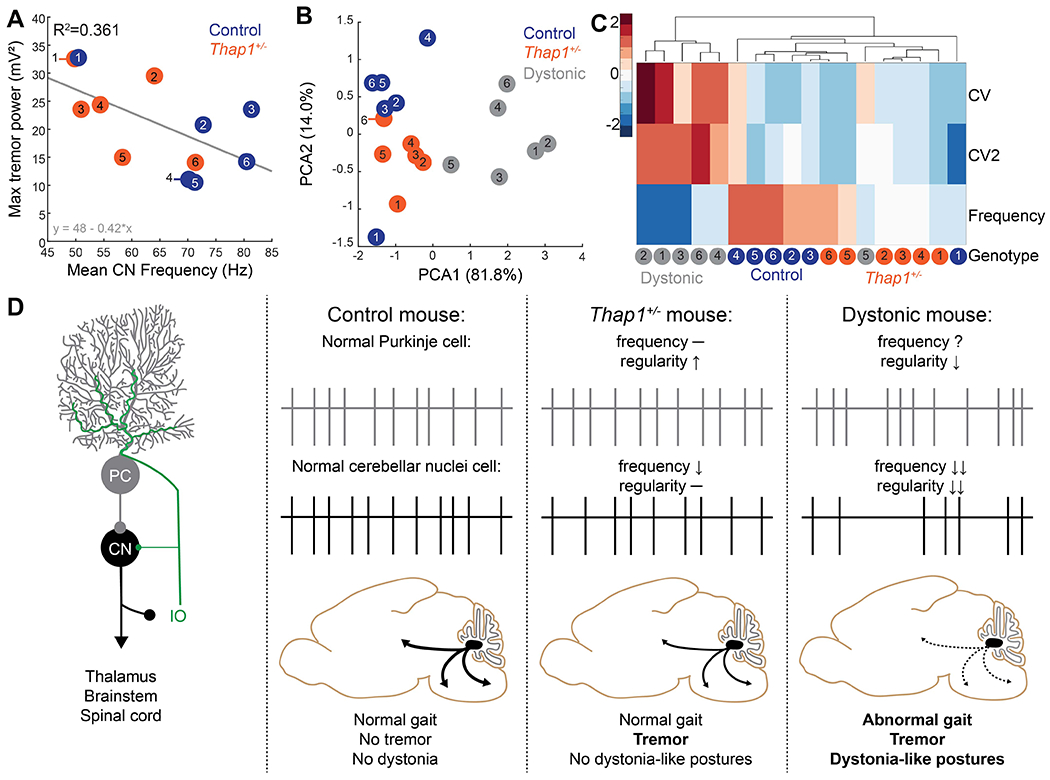
A. Correlation between max tremor power and mean cerebellar nuclei (CN) firing frequency of mice in this study (Figure 4). There is a significant inverse correlation between tremor power and CN firing frequency as determined using a linear model, p=0.0389. B. Principle component analysis on average firing frequency and global (CV) and local (CV2) regularity of cerebellar nuclei neurons recorded in control (blue), Thap1+/− mice (orange), and Ptf1aCre/+;Vglut2fl/fl mice (dystonic, grey). Data were reanalyzed from previous publication (White & Sillitoe, 2017). C. Unbiased cluster analysis of average firing frequency and global (CV) and local (CV2) regularity of cerebellar nuclei neurons recorded in control (blue), Thap1+/− mice (orange), and Ptf1aCre/+;Vglut2fl/fl mice (dystonic, grey). Each column represents the average value for one mouse. Values are normalized within each parameter with dark blue representing the lowest values and dark red representing the highest values. For A-C, each dot represents the average for one mouse from two to six recorded cerebellar nuclei cells. D. Model for how cerebellar network abnormalities can lead to motor dysfunction.
Irregular firing of cerebellar nuclei neurons may specifically drive dystonia-like postures
Deficits in the firing patterns of cerebellar neurons, specifically the cerebellar nuclei neurons, have been observed in a growing list of mouse models that manifest with dystonia-like symptoms. Given the changes in cerebellar nuclei properties we observed in Thap1+/− mice, we next wanted to test whether the network deficits in Thap1+/− mice, which only have tremor, reflect a subset of the phenotype observed in other mouse models for dystonia. We compare the firing activity of the Thap1+/− cerebellar nuclei to in vivo electrophysiology data obtained from Ptf1aCre/+;Vglut2fl/fl mice. The Ptf1aCre/+;Vglut2fl/fl mice have a striking motor phenotype that consists of tremor and severe dystonia-like postures (White & Sillitoe, 2017). First, we performed a principle component analysis on the main firing features (frequency, CV, and CV2) of cerebellar nuclei from control, Thap1+/−, and Ptf1aCre/+;Vglut2fl/fl mice (Figure 6B). We found that severely dystonic Ptf1aCre/+;Vglut2fl/fl mice formed a distinct cluster from the control and Thap1+/− mice. Next, to test whether a specific electrophysiological feature that we measured might contribute to this distinction, we performed an unbiased cluster analysis (Figure 6C). We found that while firing frequency was relatively low in both Thap1+/− mice and Ptf1aCre/+;Vglut2fl/fl mice, only Ptf1aCre/+;Vglut2fl/fl mice showed a relatively high global (CV) and local (CV2) inter spike interval irregularity compared to control mice. Thus, we attribute the irregular firing, rather than the decreased firing frequency, as the likely cause of dystonia-like twisting postures (Figure 6D).
Discussion
In this study, we investigated tremor severity and cerebellar network function in Thap1+/− mice to better understand the neural network deficits in DYT6 patients and non-manifesting carriers. We found that Thap1+/− mice have increased power of physiological tremor, Purkinje cells have highly regular activity, and cerebellar nuclei cells fire at slow rates. Thap1+/− mice also have a reduction in the number of Purkinje cells, and neuron number in the cerebellar nuclei is lower than controls, although the number of fast-firing, excitatory, thalamus-projecting neurons is unaffected. Finally, we found an inverse correlation between average cerebellar nuclei neuron firing rate when comparing the values in each mouse to its own maximum tremor power. These results support the proposed involvement of the cerebellum in DYT6 (Ruiz et al., 2015; Nikolov et al., 2019). The data also provide a cellular substrate for the abnormal cerebello-thalamo-cortical network deficits found in both non-manifesting DYT6 carriers and manifesting patients (Argyelan et al., 2009; Niethammer et al., 2011). Furthermore, our results show that specific cerebellar network deficits may promote pathophysiological levels of tremor even when dystonia (twisting, muscle over- or co-contraction) and other motor impairments are either mild or not present at all (Figure 6D) (Argyelan et al., 2009; Zittel et al., 2010; LeDoux et al., 2012).
Our findings add to a growing body of work showing compelling evidence that cerebellar deficits contribute to a multi-focal dysfunctional neural network, which ultimately causes dystonia (Shakkottai et al., 2017). We compared our data with those of five other mouse models with dystonia-like symptoms from which cerebellar neurons were recorded (Table 1). In addition to our previously described genetic model, we included three models of RNA-knock down for dystonia-associated genes in the cerebellum (TorsinA, DYT1 (Fremont et al., 2017); Scge, DYT11 (Washburn et al., 2019); ATP1α3, DYT12 (Fremont et al., 2015)) and a pharmacological model with cerebellar infusion of ouabain, an ATP1α3 antagonist (Calderon et al., 2011; Fremont et al., 2014). Apart from the Thap1+/− mice used in this study, all mouse models in Table 1 have cerebellar-specific manipulations. It is therefore a reasonable prediction to expect various alterations to cerebellar function, but it is the convergence in the type of cerebellar deficits that gives insights into what type of cerebellar dysfunctions causes dystonia. In agreement with several previous observations, all models that showed decreased regularity (increased irregularity) of cerebellar nuclei firing patterns had overt dystonia-like symptoms whereas all models with reported tremor had a decrease in nuclei firing. Based on these findings, we propose a model in which Thap1 loss-of-function mutations, in Thap1+/− mice and pre- or asymptomatic DYT6 carriers, results in decreased cerebellar nuclei firing frequencies and tremor, and that overt dystonia-like features only emerge if irregularity in the firing of cerebellar nuclei also co-occurs (Figure 6D). Based on these data, we predict that the neural networks in Thap1+/− mice may have an increased sensitivity to pharmacological manipulations, such as ouabain injections into the cerebellum, which have been shown to induce strong dystonia-like symptoms in otherwise normal mice (Pizoli et al., 2002; Calderon et al., 2011; Fremont et al., 2014; Pelosi et al., 2017).
Table 1: Comparison of Thap1+/− mutant mice with other mouse models of dystonia.
Summary of cerebellar neuron dysfunction across mouse models of dystonia with phenotypic manifestations showing that low firing frequency is present in all models with reporter tremor, whereas reduced firing regularity is observed in all mouse models manifesting with dystonia-like postures.
| Mouse model: | Thap1+/− | Ptf1aCre/+; Vglut2fl/fl | TorsinA KD | Sgce KD | ATP1α3 KD | Cerebellar ouabain |
|---|---|---|---|---|---|---|
| Dystonia type: | DYT6 | − | DYT1 | DYT11 | DYT12 | DYT12 |
| PC firing | F –; R ↑ | F –; R – | F ↓; R ↓ | F –; R ↓ | F –; R – | F ↑; R ↓ |
| CN firing | F ↓; R – | F ↓; R ↓ | F ↓; R ↓ | F ↓; R ↓ | F ↑; R ↓ | F ↑; R ↓ |
| Dystonia | N | Y | Y | Y | Y | Y |
| Tremor | Y | Y | NR | NR | NR | NR |
| Gait | N | Y | Y | Y | Y | Y |
KD= RNA Knock Down; F= firing frequency; R= firing regularity; N= not observed; Y= observed; NR= not reported.
Our observation of decreased firing frequency of neurons in the cerebellum of Thap1+/− mice is in agreement with previous human neuroimaging studies showing reduced cerebello-thalamo-cortical signaling in both DYT6 non-manifesting carriers and manifesting patients (Carbon et al., 2004; Argyelan et al., 2009; Carbon & Eidelberg, 2009; Niethammer et al., 2011). The behavior of neurons in Thap1+/− mice is also consistent with a mouse model of DYT1 that carries a human-specific mutation without displaying overt dystonia-like symptoms (Uluğ et al., 2011). Previous studies showed that the molecular pathophysiology of DYT1 and DYT6 dystonia also converges (Kaiser et al., 2010; Zakirova et al., 2018; Frederick et al., 2019) and that the TOR1A and THAP1 proteins are direct binding partners (Gavarini et al., 2010). Since the pathophysiology of DYT1 and DYT6 converges at the functional and the molecular levels, perhaps preclinical studies, such as those making use of mouse genetic models, could be cautiously extrapolated to inform about childhood onset primary torsion dystonia in humans.
Thap1 encodes a ubiquitously expressed transcription factor and loss-of-function mutations in Thap1 result in widespread changes in RNA transcription (Zakirova et al., 2018; Frederick et al., 2019). Some of the transcriptional changes found in both studies occur in genes encoding potassium channels (Kcnc1, Kcnj10), voltage gated calcium channels (Cacna1b, Cacna1e), and membrane transporters (Slc38a1, Slc39a9) that all have the potential to change the intrinsic excitability of cerebellar Purkinje and nuclei cells. Furthermore, several studies found that mutations in genes are associated with dystonia (Cacna1b, Cacna1e) and ataxia (Car8, Grid2, Grik2, Kcnc1, Kcnj10) (Kaya et al., 2011; Utine et al., 2013; Groen et al., 2015; Guzmán et al., 2017; Nicita et al., 2018; Helbig et al., 2018; Park et al., 2019). Since these molecular analyses were performed using bulk cerebellar tissue and because Thap1 is universally expressed in the cerebellar circuit, including Purkinje cells, cerebellar nuclei cells, and interneurons in the cerebellar cortex, the observed molecular changes could therefore drive changes in the intrinsic excitability and firing patterns of all cerebellar neurons. Additionally, a lower density of Purkinje cells could potentially decrease ephaptic coupling between neighboring cells and might lead to increased regularity of firing patterns (Han et al., 2018, 2020). Likewise, in addition to changes in the intrinsic firing patterns, cerebellar nuclei firing is likely influenced by the abnormal activity of Purkinje cells, especially since Purkinje cells form the predominant input onto the nuclei cells. The relative contributions of changes in intrinsic excitability and abnormal inputs on the alterations in nuclei firing are hard to disentangle as the precise relationship between Purkinje cell function on cerebellar nuclei population firing activity is still an outstanding question in the field (Lang & Blenkinsop, 2011). For instance, how a Purkinje cell single-unit alone or its zonally organized cohort (with synchronous firing capabilities) influences their target nuclear neurons is unclear, and whether these properties are modified in disease remains a possibility. Altogether, the changes we have observed in cerebellar function likely arise from the dysregulation of many genes that are essential to normal cerebellar physiology, but are also central to causing the abnormal cell number and aberrant cellular firing activity of multiple components in the cerebellar circuit. Further studies could use conditional genetics to test the contribution of Thap1 loss-of-function models to understand the molecular and functional changes in specific cerebellar cell-types.
Our work shows that Thap1+/− mice are a powerful model for mimicking behaviors typically observed in the types of dystonia involving mutations that cause partially penetrant forms of the disease. Of particular relevance are the forms of dystonia in which the core symptoms are accompanied by a clinically distinct motor phenotype, such as tremor. Fortunately, in human patients, tremor phenotypes can be monitored in a high-throughput manner during a neurological exam or preclinical testing of new therapies. Thus, Thap1+/− mice are not only a valuable model to understand the neural network deficits in DYT6 but could also be an informative preclinical model for testing therapeutic approaches. The motor symptoms of DYT6 patients are often refractory to medication, although there are positive responses to deep brain stimulation (DBS) of the internal segment of the globus pallidus (GPi) (Brüggemann et al., 2015; Ahn et al., 2019). However, there is a significant variation in responsiveness between DYT6 patients, and GPi DBS does not eliminate all symptoms (Groen et al., 2010; Krause et al., 2015). Importantly, cerebellar DBS improves mobility in a mouse model of dystonia (White & Sillitoe, 2017) and a patient with acquired dystonia (Horisawa et al., 2019). A recent study also reported a reduction in the severity of dystonia as well as improvements in posture, gait, and pain after cerebellar DBS in a patient with acquired hemidystonia (Brown et al., 2020b). Given that the phenotype of Thap1+/− mice and the symptoms in DYT6 patients highlight the cerebellum as a possible site of convergence for multiple neural pathways that instigate dystonia, and that cerebellar neuromodulation is relatively safe and well tolerated (França et al., 2018; Miterko et al., 2019), we postulate that the cerebellum could serve as an effective therapeutic target for DYT6 (Nicholson et al., 2019).
The origin of variability for how dystonia-related symptoms are expressed remains an issue of major concern. It is unclear why some patients express the common features of dystonia-related behavior, and it is equally unclear why certain patients have more severe behavioral consequences compared to others (Lerner et al., 2013). The differences could conceivably arise and reside at the level of the individual responsible genes (Zorzi et al., 2018) and/or the neural circuitry that is affected (Hanekamp & Simonyan, 2020). During cerebellar development, it has been suggested that genetic modifiers can alter the impact of genes that are essential for morphogenesis (Sillitoe & Joyner, 2007). For instance, based on the genetic background and the related modifiers of those alleles, loss of engrailed1 (En1) can have polar opposite effects, to the extremes that on the C57BL/6 background, En1 null mice have a cerebellum, whereas on the 129/S1 background strain the entire cerebellum is eliminated if En1 is deleted (Bilovocky et al., 2003; Murcia et al., 2007). Therefore, why do not all carriers with dystonia-causing mutations develop twisting and postural abnormalities or tremor? We hypothesize that primary genetic mutations paired with linked genetic alterations, such as epigenetics, cis and trans gene regulatory mechanisms, and even protein level interactions, all intersect with the formation of critical neural circuits that together determine how dystonia severity is expressed. However, the mechanism for when and how such mechanisms intersect during development remain unresolved. We propose that the cerebellum may be a central locus that facilitates such anatomical, genetic, and molecular interactions.
Key points.
Thap1 loss-of-function mutations cause partially penetrant dystonia type 6 (DYT6)
Some non-manifesting DYT6 mutation carriers have tremor and abnormal cerebello-thalamo-cortical signaling
We show that Thap1 heterozygote mice have action tremor, a reduction in cerebellar neuron number, and abnormal electrophysiological signals in the remaining neurons
These results underscore the importance of Thap1 levels for cerebellar function
These results uncover how cerebellar abnormalities contribute to different dystonia-associated motor symptoms
Translational Perspective:
Dystonia is a movement disorder characterized by involuntary, repetitive muscle contractions that can cause abnormal movements of varying severity. DYT6 is a form of partially penetrant, early onset dystonia caused by dominant loss-of-function mutations in the THAP1 gene. It is currently unclear what neural mechanisms underlie the differences in motor function between manifesting and non-manifesting patients. Here, we studied Thap1+/− mice, a genetic model of DYT6. We found that these mice do not exhibit dystonia-associated twisting postures but do have a tremor, which is observed in some non-manifesting DYT6 patients. We show that the tremor is accompanied by abnormal electrophysiological activity in Purkinje and cerebellar nuclei cells, the latter of which connect to other regions that also control movement. We compared the function of cerebellar nuclei cells in Thap1+/− mice to the cerebellar nuclei in a mouse model that does have severe dystonia-like twisting. The cerebellar nuclei cells in both models fired less frequently, but only the cells in the severe dystonia model fired less regularly. These data suggest that irregular firing of cerebellar nuclei neurons may drive the postural changes in dystonia while decreased firing frequency is related to the tremor. Together, we show that cerebellar circuit function is sensitive to Thap1 levels and different features of cerebellar dysfunction might drive dystonia in multi-symptomatic DYT6 patients but mainly tremor in mildly symptomatic cases. Future studies may uncover whether normalizing cerebellar circuit function using deep brain stimulation or transcranial magnetic stimulation of the cerebellar nuclei restores movement in DYT6 patients.
Acknowledgements:
This work was supported by Baylor College of Medicine (BCM), Texas Children’s Hospital, The Hamill Foundation, BCM IDDRC U54HD083092 (Neurovisualization Core), the National Institutes of Neurological Disorders and Stroke (NINDS) R01NS089664 and R01NS100874 to RVS and NINDS R01NS081282 to MEE.
Author profile
Dr. Meike van der Heijden earned a BSc from the University of Amsterdam before pursuing a PhD in Neuroscience at Baylor College of Medicine, where she now works as a postdoctoral fellow. Her research focusses on understanding the neuropathological mechanisms of dystonia. She uses a multi-disciplinary approach in which she combines mouse genetics, behavioral analyses, in vivo electrophysiology, and classic anatomical techniques. She aims to uncover how functional changes in cerebellar neurons drive the manifestation of dystonia-associated symptoms. Her overarching goal is to uncover key cellular nodes in the dystonic neural network and to identify neural substrates that could be used as therapeutic targets to restore motor behavior in patients.
Data availability:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Bibliography
- Ahn JH, Kim AR, Kim NKD, Park W-Y, Kim JS, Kim M, Park J, Lee J-I, Cho JW, Cho KR & Youn J (2019). The Effect of Globus Pallidus Interna Deep Brain Stimulation on a Dystonia Patient with the GNAL Mutation Compared to Patients with DYT1 and DYT6. J Mov Disord 12, 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argyelan M, Carbon M, Niethammer M, Ulug AM, Voss HU, Bressman SB, Dhawan V & Eidelberg D (2009). Cerebellothalamocortical connectivity regulates penetrance in dystonia. J Neurosci 29, 9740–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker MI & Person AL (2019). Cerebellar control of reach kinematics for endpoint precision. Neuron 103, 335–348.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckinghausen J & Sillitoe RV (2019). Insights into cerebellar development and connectivity. Neurosci Lett 688, 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilovocky NA, Romito-DiGiacomo RR, Murcia CL, Maricich SM & Herrup K (2003). Factors in the genetic background suppress the engrailed-1 cerebellar phenotype. J Neurosci 23, 5105–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood AJ, Kuster JK, Woodman SC, Kirlic N, Makhlouf ML, Multhaupt-Buell TJ, Makris N, Parent M, Sudarsky LR, Sjalander G, Breiter H, Breiter HC & Sharma N (2012). Evidence for altered basal ganglia-brainstem connections in cervical dystonia. PLoS One 7, e31654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracha V, Kolb FP, Irwin KB & Bloedel JR (1999). Inactivation of interposed nuclei in the cat: classically conditioned withdrawal reflexes, voluntary limb movements and the action primitive hypothesis. Exp Brain Res 126, 77–92. [DOI] [PubMed] [Google Scholar]
- Brown AM, Arancillo M, Lin T, Catt DR, Zhou J, Lackey EP, Stay TL, Zuo Z, White JJ & Sillitoe RV (2019). Molecular layer interneurons shape the spike activity of cerebellar Purkinje cells. Sci Rep 9, 1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, White JJ, van der Heijden ME, Zhou J, Lin T & Sillitoe RV (2020a). Purkinje cell misfiring generates high-amplitude action tremors that are corrected by cerebellar deep brain stimulation. Elife; DOI: 10.7554/eLife.51928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EG, Bledsoe IO, Luthra NS, Miocinovic S, Starr PA & Ostrem JL (2020b). Cerebellar deep brain stimulation for acquired hemidystonia. Mov Disord Clin Pract (Hoboken) 7, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüggemann N et al. (2015). Short- and long-term outcome of chronic pallidal neurostimulation in monogenic isolated dystonia. Neurology 84, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon DP, Fremont R, Kraenzlin F & Khodakhah K (2011). The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci 14, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarota A, Gershanik OS, García S & Lera G (1995). Cervical dystonia due to spinal cord ependymoma: involvement of cervical cord segments in the pathogenesis of dystonia. Mov Disord 10, 500–503. [DOI] [PubMed] [Google Scholar]
- Carbon M & Eidelberg D (2009). Abnormal structure-function relationships in hereditary dystonia. Neuroscience 164, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon M, Kingsley PB, Tang C, Bressman S & Eidelberg D (2008). Microstructural white matter changes in primary torsion dystonia. Mov Disord 23, 234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon M, Su S, Dhawan V, Raymond D, Bressman S & Eidelberg D (2004). Regional metabolism in primary torsion dystonia: effects of penetrance and genotype. Neurology 62, 1384–1390. [DOI] [PubMed] [Google Scholar]
- Cook AA, Fields E & Watt AJ (2021). Losing the beat: contribution of purkinje cell firing dysfunction to disease, and its reversal. Neuroscience 462, 247–261. [DOI] [PubMed] [Google Scholar]
- Davie JT, Clark BA & Häusser M (2008). The origin of the complex spike in cerebellar Purkinje cells. J Neurosci 28, 7599–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defazio G (2010). The epidemiology of primary dystonia: current evidence and perspectives. Eur J Neurol 17 Suppl 1, 9–14. [DOI] [PubMed] [Google Scholar]
- DeSimone JC, Archer DB, Vaillancourt DE & Wagle Shukla A (2019). Network-level connectivity is a critical feature distinguishing dystonic tremor and essential tremor. Brain 142, 1644–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Alvarez E & Nardocci (2012). Update on pediatric dystonias: etiology, epidemiology, and management. Degener Neurol Neuromuscul Dis 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MD & Alterman RL (2015). Brain stimulation for torsion dystonia. JAMA Neurol 72, 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- França C, de Andrade DC, Teixeira MJ, Galhardoni R, Silva V, Barbosa ER & Cury RG (2018). Effects of cerebellar neuromodulation in movement disorders: A systematic review. Brain Stimulat 11, 249–260. [DOI] [PubMed] [Google Scholar]
- Frederick NM, Shah PV, Didonna A, Langley MR, Kanthasamy AG & Opal P (2019). Loss of the dystonia gene Thap1 leads to transcriptional deficits that converge on common pathogenic pathways in dystonic syndromes. Hum Mol Genet 28, 1343–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont R, Calderon DP, Maleki S & Khodakhah K (2014). Abnormal high-frequency burst firing of cerebellar neurons in rapid-onset dystonia-parkinsonism. J Neurosci 34, 11723–11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont R, Tewari A, Angueyra C & Khodakhah K (2017). A role for cerebellum in the hereditary dystonia DYT1. Elife; DOI: 10.7554/eLife.22775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremont R, Tewari A & Khodakhah K (2015). Aberrant Purkinje cell activity is the cause of dystonia in a shRNA-based mouse model of Rapid Onset Dystonia-Parkinsonism. Neurobiol Dis 82, 200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB & Ozelius LJ (2009). Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet 41, 286–288. [DOI] [PubMed] [Google Scholar]
- Fujita H, Kodama T & du Lac S (2020). Modular output circuits of the fastigial nucleus for diverse motor and nonmotor functions of the cerebellar vermis. Elife; DOI: 10.7554/eLife.58613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliano E, Gao Z, Schonewille M, Todorov B, Simons E, Pop AS, D’Angelo E, van den Maagdenberg AMJM, Hoebeek FE & De Zeeuw CI (2013). Silencing the majority of cerebellar granule cells uncovers their essential role in motor learning and consolidation. Cell Rep 3, 1239–1251. [DOI] [PubMed] [Google Scholar]
- Gavarini S, Cayrol C, Fuchs T, Lyons N, Ehrlich ME, Girard J-P & Ozelius LJ (2010). Direct interaction between causative genes of DYT1 and DYT6 primary dystonia. Ann Neurol 68, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen JL, Andrade A, Ritz K, Jalalzadeh H, Haagmans M, Bradley TEJ, Jongejan A, Verbeek DS, Nürnberg P, Denome S, Hennekam RCM, Lipscombe D, Baas F & Tijssen MAJ (2015). CACNA1B mutation is linked to unique myoclonus-dystonia syndrome. Hum Mol Genet 24, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groen JL, Ritz K, Contarino MF, van de Warrenburg BP, Aramideh M, Foncke EM, van Hilten JJ, Schuurman PR, Speelman JD, Koelman JH, de Bie RMA, Baas F & Tijssen MA (2010). DYT6 dystonia: mutation screening, phenotype, and response to deep brain stimulation. Mov Disord 25, 2420–2427. [DOI] [PubMed] [Google Scholar]
- Guzmán YF, Ramsey K, Stolz JR, Craig DW, Huentelman MJ, Narayanan V & Swanson GT (2017). A gain-of-function mutation in the GRIK2 gene causes neurodevelopmental deficits. Neurol Genet 3, e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K-S, Chen CH, Khan MM, Guo C & Regehr WG (2020). Climbing fiber synapses rapidly and transiently inhibit neighboring Purkinje cells via ephaptic coupling. Nat Neurosci 23, 1399–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K-S, Guo C, Chen CH, Witter L, Osorno T & Regehr WG (2018). Ephaptic coupling promotes synchronous firing of cerebellar purkinje cells. Neuron 100, 564–578.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanekamp S & Simonyan K (2020). The large-scale structural connectome of task-specific focal dystonia. Hum Brain Mapp 41, 3253–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen ST, Meera P, Otis TS & Pulst SM (2013). Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet 22, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden ME, Lackey EP, Işleyen FS, Brown AM, Perez R, Lin T, Zoghbi HY & Sillitoe RV (2020). Maturation of Purkinje cell firing properties relies on granule cell neurogenesis. BioRxiv; DOI: 10.1101/2020.05.20.106732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig KL et al. (2018). De Novo Pathogenic Variants in CACNA1E Cause Developmental and Epileptic Encephalopathy with Contractures, Macrocephaly, and Dyskinesias. Am J Hum Genet 103, 666–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horisawa S, Arai T, Suzuki N, Kawamata T & Taira T (2019). The striking effects of deep cerebellar stimulation on generalized fixed dystonia: case report. J Neurosurg 1–5. [DOI] [PubMed] [Google Scholar]
- Hourez R, Servais L, Orduz D, Gall D, Millard I, de Kerchove d’Exaerde A, Cheron G, Orr HT, Pandolfo M & Schiffmann SN (2011). Aminopyridines correct early dysfunction and delay neurodegeneration in a mouse model of spinocerebellar ataxia type 1. J Neurosci 31, 11795–11807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser FJ, Osmanoric A, Rakovic A, Erogullari A, Uflacker N, Braunholz D, Lohnau T, Orolicki S, Albrecht M, Gillessen-Kaesbach G, Klein C & Lohmann K (2010). The dystonia gene DYT1 is repressed by the transcription factor THAP1 (DYT6). Ann Neurol 68, 554–559. [DOI] [PubMed] [Google Scholar]
- Kaya N, Aldhalaan H, Al-Younes B, Colak D, Shuaib T, Al-Mohaileb F, Al-Sugair A, Nester M, Al-Yamani S, Al-Bakheet A, Al-Hashmi N, Al-Sayed M, Meyer B, Jungbluth H & Al-Owain M (2011). Phenotypical spectrum of cerebellar ataxia associated with a novel mutation in the CA8 gene, encoding carbonic anhydrase (CA) VIII. Am J Med Genet B, Neuropsychiatr Genet 156B, 826–834. [DOI] [PubMed] [Google Scholar]
- Klein C (2014). Genetics in dystonia. Parkinsonism Relat Disord 20 Suppl 1, S137–42. [DOI] [PubMed] [Google Scholar]
- Krause P, Brüggemann N, Völzmann S, Horn A, Kupsch A, Schneider GH, Lohmann K & Kühn A (2015). Long-term effect on dystonia after pallidal deep brain stimulation (DBS) in three members of a family with a THAP1 mutation. J Neurol 262, 2739–2744. [DOI] [PubMed] [Google Scholar]
- Kuo S-H et al. (2019). Current opinions and consensus for studying tremor in animal models. Cerebellum 18, 1036–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang EJ & Blenkinsop TA (2011). Control of cerebellar nuclear cells: a direct role for complex spikes? Cerebellum 10, 694–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS & Lorden JF (2002). Abnormal spontaneous and harmaline-stimulated Purkinje cell activity in the awake genetically dystonic rat. Exp Brain Res 145, 457–467. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, Xiao J, Rudzińska M, Bastian RW, Wszolek ZK, Van Gerpen JA, Puschmann A, Momčilović D, Vemula SR & Zhao Y (2012). Genotype-phenotype correlations in THAP1 dystonia: molecular foundations and description of new cases. Parkinsonism Relat Disord 18, 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176. [DOI] [PubMed] [Google Scholar]
- Lerner RP, Niethammer M & Eidelberg D (2013). Understanding the anatomy of dystonia: determinants of penetrance and phenotype. Curr Neurol Neurosci Rep 13, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low AYT, Thanawalla AR, Yip AKK, Kim J, Wong KLL, Tantra M, Augustine GJ & Chen AI (2018). Precision of discrete and rhythmic forelimb movements requires a distinct neuronal subpopulation in the interposed anterior nucleus. Cell Rep 22, 2322–2333. [DOI] [PubMed] [Google Scholar]
- Luna-Cancalon K, Sikora KM, Pappas SS, Singh V, Wulff H, Paulson HL, Burmeister M & Shakkottai VG (2014). Alterations in cerebellar physiology are associated with a stiff-legged gait in Atcay(ji-hes) mice. Neurobiol Dis 67, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lungu C et al. (2020). Defining research priorities in dystonia. Neurology 94, 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaught KSP, Kapustin A, Jackson T, Jengelley T-A, Jnobaptiste R, Shashidharan P, Perl DP, Pasik P & Olanow CW (2004). Brainstem pathology in DYT1 primary torsion dystonia. Ann Neurol 56, 540–547. [DOI] [PubMed] [Google Scholar]
- Mercer AA, Palarz KJ, Tabatadze N, Woolley CS & Raman IM (2016). Sex differences in cerebellar synaptic transmission and sex-specific responses to autism-linked Gabrb3 mutations in mice. Elife; DOI: 10.7554/eLife.07596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miterko LN et al. (2019). Consensus paper: experimental neurostimulation of the cerebellum. Cerebellum 18, 1064–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller U (2009). The monogenic primary dystonias. Brain 132, 2005–2025. [DOI] [PubMed] [Google Scholar]
- Murcia CL, Gulden FO, Cherosky NA & Herrup K (2007). A genetic study of the suppressors of the Engrailed-1 cerebellar phenotype. Brain Res 1140, 170–178. [DOI] [PubMed] [Google Scholar]
- Neychev VK, Fan X, Mitev VI, Hess EJ & Jinnah HA (2008). The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain 131, 2499–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neychev VK, Gross RE, Lehéricy S, Hess EJ & Jinnah HA (2011). The functional neuroanatomy of dystonia. Neurobiol Dis 42, 185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson CL, Coubes P & Poulen G (2019). The Rationale for the Dentate Nucleus as a Target for Deep Brain Stimulation in Dystono-Dyskinetic Syndromes. Annals of Neurological Surgery. [DOI] [PubMed] [Google Scholar]
- Nicita F, Tasca G, Nardella M, Bellacchio E, Camponeschi I, Vasco G, Schirinzi T, Bertini E & Zanni G (2018). Novel Homozygous KCNJ10 Mutation in a Patient with Non-syndromic Early-Onset Cerebellar Ataxia. Cerebellum 17, 499–503. [DOI] [PubMed] [Google Scholar]
- Niethammer M, Carbon M, Argyelan M & Eidelberg D (2011). Hereditary dystonia as a neurodevelopmental circuit disorder: Evidence from neuroimaging. Neurobiol Dis 42, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nietz A, Krook-Magnuson C, Gutierrez H, Klein J, Sauve C, Hoff I, Christenson Wick Z & Krook-Magnuson E (2020). Selective loss of the GABAAα1 subunit from Purkinje cells is sufficient to induce a tremor phenotype. J Neurophysiol; DOI: 10.1152/jn.00100.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov P, Hassan SS, Aytulun A, Hartmann CJ, Kohlhase J, Schnitzler A, Albrecht P, Minnerop M & Groiss SJ (2019). Cerebellar Involvement in DYT-THAP1 Dystonia. Cerebellum; DOI: 10.1007/s12311-019-01062-0. [DOI] [PubMed] [Google Scholar]
- Özcan OO, Wang X, Binda F, Dorgans K, De Zeeuw CI, Gao Z, Aertsen A, Kumar A & Isope P (2020). Differential coding strategies in glutamatergic and gabaergic neurons in the medial cerebellar nucleus. J Neurosci 40, 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ & Bressman SB (2011). Genetic and clinical features of primary torsion dystonia. Neurobiol Dis 42, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF & Breakefield XO (1997). The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 17, 40–48. [DOI] [PubMed] [Google Scholar]
- Palmer LM, Clark BA, Gründemann J, Roth A, Stuart GJ & Häusser M (2010). Initiation of simple and complex spikes in cerebellar Purkinje cells. J Physiol (Lond) 588, 1709–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan M-K, Li Y-S, Wong S-B, Ni C-L, Wang Y-M, Liu W-C, Lu L-Y, Lee J-C, Cortes EP, Vonsattel J-PG, Sun Q, Louis ED, Faust PL & Kuo S-H (2020). Cerebellar oscillations driven by synaptic pruning deficits of cerebellar climbing fibers contribute to tremor pathophysiology. Sci Transl Med; DOI: 10.1126/scitranslmed.aay1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J et al. (2019). KCNC1-related disorders: new de novo variants expand the phenotypic spectrum. Ann Clin Transl Neurol 6, 1319–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudel R, Li A, Hardy J, Bhatia KP, Houlden H & Holton J (2016). DYT6 dystonia: A neuropathological study. Neurodegener Dis 16, 273–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelosi A, Menardy F, Popa D, Girault J-A & Hervé D (2017). Heterozygous Gnal Mice Are a Novel Animal Model with Which to Study Dystonia Pathophysiology. J Neurosci 37, 6253–6267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizoli CE, Jinnah HA, Billingsley ML & Hess EJ (2002). Abnormal cerebellar signaling induces dystonia in mice. J Neurosci 22, 7825–7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudente CN, Pardo CA, Xiao J, Hanfelt J, Hess EJ, Ledoux MS & Jinnah HA (2013). Neuropathology of cervical dystonia. Exp Neurol 241, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz M, Perez-Garcia G, Ortiz-Virumbrales M, Méneret A, Morant A, Kottwitz J, Fuchs T, Bonet J, Gonzalez-Alegre P, Hof PR, Ozelius LJ & Ehrlich ME (2015). Abnormalities of motor function, transcription and cerebellar structure in mouse models of THAP1 dystonia. Hum Mol Genet 24, 7159–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG et al. (2017). Current opinions and areas of consensus on the role of the cerebellum in dystonia. Cerebellum 16, 577–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe RV, Fu Y & Watson C (2012). Cerebellum. In The mouse nervous system, pp. 360–397. Elsevier. [Google Scholar]
- Sillitoe RV & Joyner AL (2007). Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Annu Rev Cell Dev Biol 23, 549–577. [DOI] [PubMed] [Google Scholar]
- Uluğ AM, Vo A, Argyelan M, Tanabe L, Schiffer WK, Dewey S, Dauer WT & Eidelberg D (2011). Cerebellothalamocortical pathway abnormalities in torsinA DYT1 knock-in mice. Proc Natl Acad Sci USA 108, 6638–6643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utine GE, Haliloğlu G, Salanci B, Çetinkaya A, Kiper PÖ, Alanay Y, Aktas D, Boduroğlu K & Alikaşifoğlu M (2013). A homozygous deletion in GRID2 causes a human phenotype with cerebellar ataxia and atrophy. J Child Neurol 28, 926–932. [DOI] [PubMed] [Google Scholar]
- Uusisaari M, Obata K & Knöpfel T (2007). Morphological and electrophysiological properties of GABAergic and non-GABAergic cells in the deep cerebellar nuclei. J Neurophysiol 97, 901–911. [DOI] [PubMed] [Google Scholar]
- Voelkl B, Altman NS, Forsman A, Forstmeier W, Gurevitch J, Jaric I, Karp NA, Kas MJ, Schielzeth H, Van de Casteele T & Würbel H (2020). Reproducibility of animal research in light of biological variation. Nat Rev Neurosci 21, 384–393. [DOI] [PubMed] [Google Scholar]
- Washburn S, Fremont R, Moreno-Escobar MC, Angueyra C & Khodakhah K (2019). Acute cerebellar knockdown of Sgce reproduces salient features of myoclonus-dystonia (DYT11) in mice. Elife; DOI: 10.7554/eLife.52101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ, Lin T, Brown AM, Arancillo M, Lackey EP, Stay TL & Sillitoe RV (2016). An optimized surgical approach for obtaining stable extracellular single-unit recordings from the cerebellum of head-fixed behaving mice. J Neurosci Methods 262, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JJ & Sillitoe RV (2017). Genetic silencing of olivocerebellar synapses causes dystonia-like behaviour in mice. Nat Commun 8, 14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakirova Z, Fanutza T, Bonet J, Readhead B, Zhang W, Yi Z, Beauvais G, Zwaka TP, Ozelius LJ, Blitzer RD, Gonzalez-Alegre P & Ehrlich ME (2018). Mutations in THAP1/DYT6 reveal that diverse dystonia genes disrupt similar neuronal pathways and functions. PLoS Genet 14, e1007169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Weinrich JAP, Russ JB, Comer JD, Bommareddy PK, DiCasoli RJ, Wright CVE, Li Y, van Roessel PJ & Kaltschmidt JA (2017). A Role for Dystonia-Associated Genes in Spinal GABAergic Interneuron Circuitry. Cell Rep 21, 666–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Xiao J, Gong S, Clara JA & Ledoux MS (2013). Neural expression of the transcription factor THAP1 during development in rat. Neuroscience 231, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zittel S, Moll CKE, Brüggemann N, Tadic V, Hamel W, Kasten M, Lohmann K, Lohnau T, Winkler S, Gerloff C, Schönweiler R, Hagenah J, Klein C, Münchau A & Schneider SA (2010). Clinical neuroimaging and electrophysiological assessment of three DYT6 dystonia families. Mov Disord 25, 2405–2412. [DOI] [PubMed] [Google Scholar]
- Zorzi G, Carecchio M, Zibordi F, Garavaglia B & Nardocci N (2018). Diagnosis and treatment of pediatric onset isolated dystonia. Eur J Paediatr Neurol 22, 238–244. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.