

Summary
Crimean-Congo Hemorrhagic Fever Virus (CCHFV) is a World Health Organization priority pathogen. CCHFV infections cause a highly lethal hemorrhagic fever for which specific treatments and vaccines are urgently needed. Here, we characterize the human immune response to natural CCHFV infection to identify potent neutralizing antibodies (nAbs) targeting the viral glycoprotein. Competition experiments showed that these nAbs bind six distinct antigenic sites in the Gc subunit. These sites were further delineated through mutagenesis and mapped onto a prefusion model of Gc. Pairwise screening identified combinations of non-competing nAbs that afford synergistic neutralization. Further enhancements in neutralization breadth and potency were attained by physically linking variable domains of synergistic nAb pairs through bispecific antibody (bsAb) engineering. Although multiple nAbs protected mice from lethal CCHFV challenge in pre- or post-exposure prophylactic settings, only a single bsAb, DVD-121-801, afforded therapeutic protection. DVD-121-801 is a promising candidate suitable for clinical development as a CCHFV therapeutic.
Graphical Abstract
In Brief:
By isolating monoclonal antibodies against Crimean-Congo Hemorrhagic Fever Virus glycoproteins from human survivors, Fels et al. were able to identify combinations of synergistic neutralizing antibodies and engineer bispecific antibodies that provide therapeutic protection
Introduction
Crimean-Congo hemorrhagic fever virus (CCHFV) is a tick-borne virus in the family Nairoviridae (order Bunyavirales) of segmented single-strand RNA viruses that can cause severe hemorrhagic disease in humans. Case-fatality rates of 5–30% are typically observed in CCHF outbreaks but have been reported to be as high as 80% (Spengler et al., 2018, 2019). Carried mainly by ticks of the Hyalomma genus, CCHFV is the most prevalent tick-borne virus that causes human disease and is endemic in countries across Europe, Asia, and Africa (Bente et al., 2013; Messina et al., 2015; Spengler et al., 2019). Human infection typically results from the bite of an infected tick or contact with blood from infected livestock (Bente et al., 2013), but nosocomial infection has also been reported (Conger et al., 2015; Smego et al., 2004). Despite the widespread geographic distribution of CCHFV and the severity of CCHF disease, no specific medical countermeasures are currently available; the broad-spectrum antiviral, ribavirin, is used off-label in some regions, but evidence for its efficacy is controversial (Johnson et al., 2018). In 2017, the World Health Organization declared CCHF a Blueprint Priority Disease (Mehand et al., 2018).
CCHFV harbors a tripartite, negative-strand RNA genome with the nucleoprotein, glycoprotein precursor, and RNA-dependent RNA polymerase encoded by the small (S), medium (M), and large (L) RNA segments, respectively. The glycoprotein precursor is co- and post-translationally processed to generate two structural glycoproteins, Gn and Gc, which mediate cell attachment and membrane fusion, as well as three secreted, putatively non-virion–associated (“nonstructural”) glycoproteins, GP38, GP85, and GP160 (Zivcec et al., 2016) (Figure S1A). Circulating strains of CCHFV have been classified into seven distinct clades based on phylogenetic comparison of their M segments (Carroll et al., 2010) with most confirmed reports occurring in Turkey, where clade V strains predominate (Bente et al., 2013; Spengler et al., 2018).
Previous work has shown that the lack of serum antibodies correlates with fatality in human CCHF patients, suggesting that antibodies may play a role in protection from lethal CCHFV infection (Ergonul et al., 2006; Shepherd et al., 1989). Thus far, monoclonal antibodies (mAbs) and neutralizing mAbs (nAbs) against CCHFV have only been isolated from immunized mice (Bertolotti-Ciarlet et al., 2005). Three Gc-specific nAbs from this panel demonstrated cross-neutralizing activity against multiple strains (Bertolotti-Ciarlet et al., 2005; Zivcec et al., 2017); however, these nAbs failed to provide protection in mouse models of CCHF (Bertolotti-Ciarlet et al., 2005; Golden et al., 2019). By contrast, mAbs targeting the Gn polyprotein precursor (pre-Gn) and/or GP38 lacked neutralizing activity, but a few nonetheless afforded pre-exposure protection in mice (Bertolotti-Ciarlet et al., 2005; Golden et al., 2019; Mishra et al., 2020; Zivcec et al., 2017). Together, these data indicate that existing recombinant mAbs may have prophylactic utility against CCHFV but suggest that new mAbs that enhance prophylactic potency and afford therapeutic protection are needed.
Recent studies with other highly virulent and lethal viruses (e.g. Ebola virus) have shown that mAbs derived from survivors of natural infection can be effective as therapeutics, even when administered after the onset of severe disease (Bornholdt et al., 2019; Caskey et al., 2015; Corti et al., 2015; De Benedictis et al., 2016; Mire et al., 2017; Mulangu et al., 2019; Wec et al., 2019). Moreover, characterization of human mAbs targeting viral pathogens can illuminate specific targets for vaccine design and enhance outbreak preparedness (Bloom et al., 2017; Correia et al., 2014; Quiroz et al., 2019; Rappuoli et al., 2016; Sesterhenn et al., 2020; Steichen et al., 2019). Here, we mined the B cell repertoires of four CCHF-convalescent donors to discover and characterize mAbs specific for the CCHFV Gn/Gc glycoproteins. We identified broadly neutralizing mAbs (bnAbs) that recognize distinct epitopes in Gc and could protect mice from lethal CCHFV challenge in pre-exposure or post-exposure prophylactic settings. However, neither these bnAbs nor GP38-specific mAbs from this and previous work afforded protection when administered 24 h after viral challenge, either alone or in combination (Bertolotti-Ciarlet et al., 2005; Golden et al., 2019). To generate antibodies with enhanced antiviral potency, we engineered and evaluated bispecific antibodies (bsAbs) bearing variable domains from synergistic bnAbs and identified one bsAb that could provide therapeutic protection against CCHFV with a single dose. Our findings inform the development of broadly protective CCHF immunotherapeutics and the rational design of vaccines tailored to elicit highly potent bnAbs.
Results
Isolation of recombinant Gn/Gc-reactive monoclonal antibodies from CCHF-convalescent donors
We collected blood samples from four CCHF-convalescent Ugandan donors ranging between 2–46 months post-infection (Table S1). To isolate glycoprotein-reactive B cells from peripheral blood, we designed a recombinant polyprotein bait (rGn/Gc) retaining the ectodomains of the polyprotein precursor but lacking the transmembrane domains and endodomains. Further, sequences corresponding to the mature Gn and Gc ectodomains were fused together with a flexible Gly-Ser linker (Voss et al., 2010), and proteolytic cleavage between the GP38 and Gn domains was enhanced by replacement of the SKI-I/S1P-like cleavage site with that of the Golgi proprotein convertase, furin (Figure S1A). Sera from the CCHF-convalescent donors and previously characterized murine mAbs targeting GP38 and Gc (Bertolotti-Ciarlet et al., 2005; Golden et al., 2019) bound to rGn/Gc (Figure S1B–C). Binding assays using anti-CCHFV mAbs that bind conformation-dependent epitopes (Bertolotti-Ciarlet et al., 2005) suggest that this construct recapitulates conformational features of CCHFV Gc (Figure S1C). Surprisingly, despite cleavage of the rGn/Gc polyprotein to liberate the mucin-like and GP38 domains (Figure S1D), rGn/Gc was still recognized by the GP38-specific mAbs (Figure S1C). Consistent with this, follow-up analysis of rGn/Gc by mass spectrometry-based peptide mapping and intact mass analysis identified the presence of GP38 in purified preparations (Figure S1E). Incomplete furin cleavage and/or purification may explain the presence of GP38 in these samples. Alternatively, or in addition, it is conceivable that a non-covalent association between GP38 and Gn/Gc accounts for their co-purification in our rGn/Gc preparations. Indeed, GP38 was recently shown to be present on the envelopes of CCHFV virus-like particles (Golden et al., 2019).
We evaluated the CCHFV glycoprotein-specific memory B cell response in peripheral blood by fluorescently labeling B cell surface markers (CD19, CD20, IgM, IgD, and CD27) and staining with rGn/Gc probes (Figure 1A, S1F), which showed that 0.3–1% of class-switched B cells (CD19/CD20+IgM−IgD−) were rGn/Gc-reactive (Figure 1A). Approximately 450 (Donor 1) or 180 (Donors 5, 6, 7) rGn/Gc-reactive B cells were single-cell sorted for antibody cloning, sequencing, and production in an engineered strain of S. cerevisiae (Bornholdt et al., 2016; Tiller et al., 2008). Although we sorted all rGn/Gc-reactive B cells regardless of antibody isotype or CD27 surface expression, index sorting revealed that most (83–98%) rGn/Gc-reactive mAbs derived from class-switched B cells (Figure 1B, S1G). Approximately half of the isolated rGn/Gc-reactive B cells from Donors 1 and 5 displayed low levels of CD27 surface expression (Figure 1B, S1G), consistent with an increasing CD27− resting memory B cell population in peripheral blood over time (Andrews et al., 2019).
Figure 1. Isolated CCHFV rGn/Gc-specific mAbs represent diverse lineages.
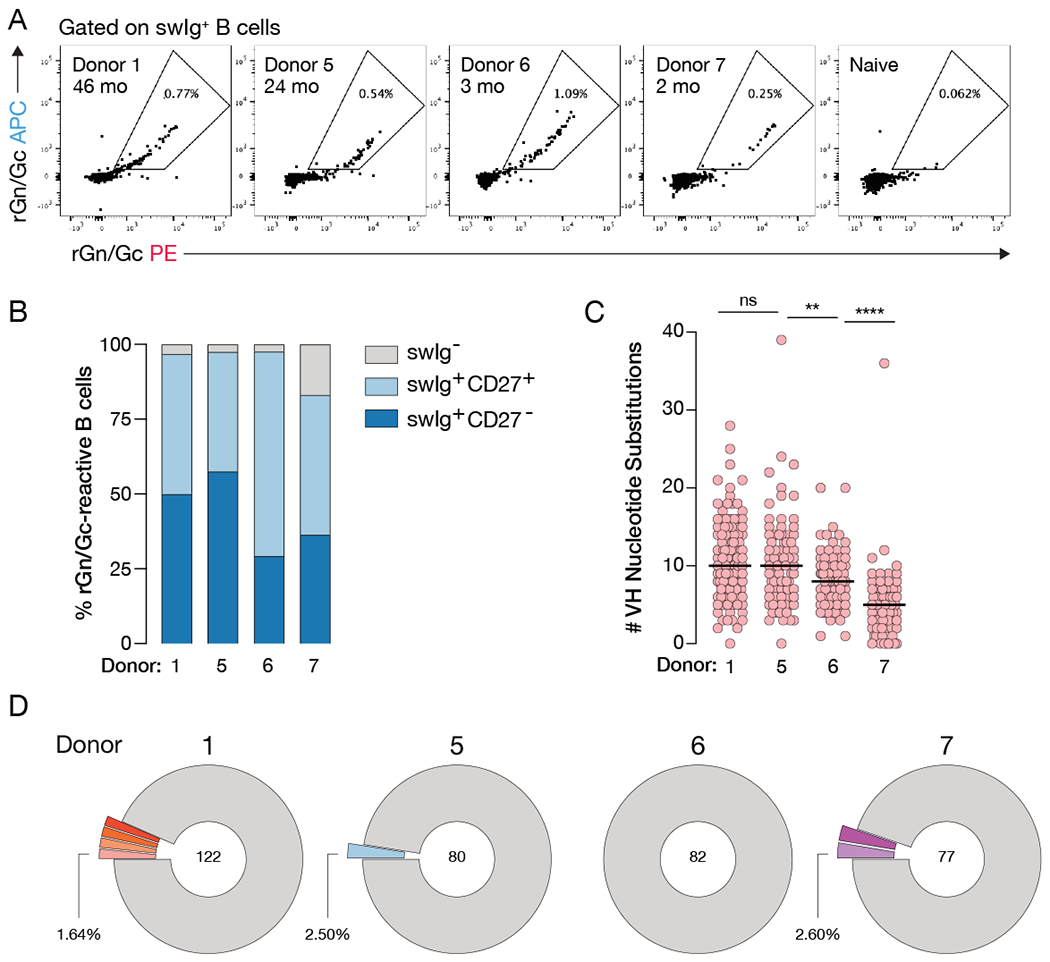
(A) Flow cytometric analysis of rGnGc-specific B cells in four Ugandan donors. Flow plots are gated on CD14/CD16/CD3/PI/IgM/IgD− CD19/CD20+ rGn/Gc+ B cells. Inset denotes donor ID and approximate time between infection and blood draw. (B) Frequency of B cell subsets that encoded the 361 antibody genes described throughout, swIg = class-switched immunoglobulin (IgM−/IgD−). (C) Load of somatic hypermutation as determined by the number of VH nucleotide substitutions away from the predicted VH germline. Statistical comparison was made by the Mann-Whitney test (**** P < 0.0001, ** P < 0.01, ns = not significant). See also Table S1. (D) Clonal analysis of isolated antibody genes. Clones that encode the same germline genes, CDR3 lengths, and have >80% nucleotide sequence homology within CDRH3 are considered to be within the same lineage. Colored and expanded slices represent lineages for which more than one clone was identified. Unique clones are shown as a single gray segment. Numbers in the center of the pie represent the total number of mAbs with binding verified by biolayer interferometry. See also Figure S1.
The median levels of somatic hypermutation (SHM) in the variable region of the heavy chain (5–10 nucleotide substitutions) are similar to those observed in antibodies induced by other primary viral infections or vaccination (Bornholdt et al., 2016; Goodwin et al., 2018; Rogers et al., 2017; Wec et al., 2020a). On average, SHM levels were higher in the mAbs cloned from donors who experienced longer intervals between infection and blood draw (Figure 1A, 1C, Table S1), suggesting that affinity maturation likely continues for several months following the period of active viral replication, as previously reported in the context of Ebola virus infection and yellow fever vaccination (Davis et al., 2019; Wec et al., 2020a) (Figure 1C). Analysis of paired heavy and light chain sequences showed that the rGn/Gc-reactive B cells isolated from all four donors were clonally diverse, with only ~0–7% per donor belonging to expanded clonal lineages (Figure 1D). Altogether, we generated a panel of 361 rGn/Gc-reactive mAbs from four donors at different times post-infection and largely from clonally diverse, class-switched B cells.
Most rGn/Gc-reactive mAbs bind conserved epitopes in Gc
To identify which component of rGn/Gc is targeted by each mAb, we first tested binding to IbAr10200-derived rGn/Gc and Gc antigens using biolayer interferometry. Most of the mAbs (>80%) reacted with both rGn/Gc and Gc (Figure 2A, 2D), indicating that they target epitopes in the Gc subunit. The mAbs that reacted with rGn/Gc, but not Gc, were further examined for their capacity to recognize recombinant Gn and GP38. Surprisingly, none reacted with either in-house or commercially produced rGn (Figure S2A). Instead, most of these mAbs bound rGP38 (Figure 2D, S2A).
Figure 2. mAbs mainly target conserved antigenic sites in Gc.
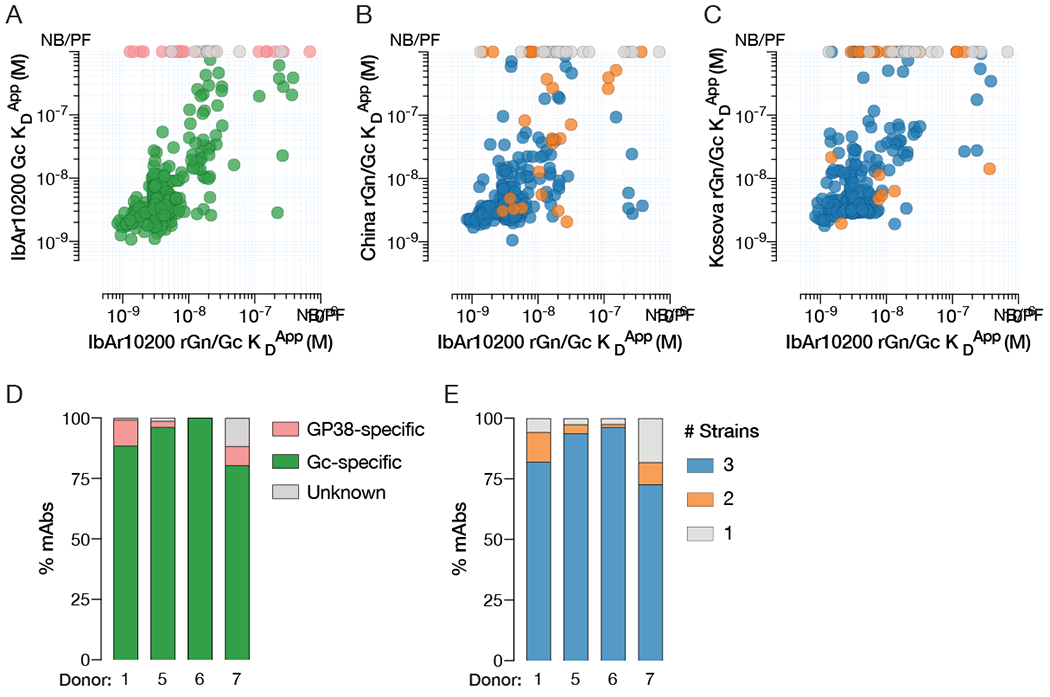
Apparent KD values (KDApp) of mAbs binding IbAr10200 rGn/Gc (x-axis) and IbAr10200 Gc (A), China rGn/Gc (B), or Kosova Hoti rGn/Gc (C) (y-axis), as determined by biolayer interferometry. NB/PF denotes mAbs with no binding or poor fits. Each circle represents a single mAb and is color coded as in D and E. (D) Frequency of mAbs from each donor that bind to rGn/Gc broken down by subunit. (E) Cross-reactivity profiles of antibodies isolated from each donor showing the number of strains (IbAr10200, China, and Kosova Hoti) bound by mAbs. Binding was determined by > 0.1 nm response in the BLI assay. See also Figure S2.
We next assessed mAb cross-reactivity by measuring binding with rGn/Gc antigens derived from IbAr10200, China (79121M18), and Kosova Hoti strains, representing clades III, I, and V, respectively (Figure 2B–C, 2E). Concordant with the high degree of Gc sequence similarity among these strains (~89-95% identity, Figure S2B), the majority of the Gc-specific mAbs recognized all three rGn/Gc antigens tested (Figure 2E). We also identified seven cross-reactive GP38-specific mAbs, six of which recognized the same conserved antigenic site on GP38 (Figure S2C). Further, five of these six cross-reactive, GP38-specific antibodies shared Vλ3-21 germline usage without being clonally related, suggesting convergent recognition of this antigenic site (Figure S2D). In summary, the majority of rGn/Gc-reactive antibodies isolated from CCHF-convalescent donors recognize conserved epitopes in Gc with a smaller subset targeting sites in GP38.
mAbs recognize six distinct antigenic sites in Gc with the majority targeting the fusion loops
To determine the number and frequency of mAbs targeting distinct antigenic sites within Gc, we used a previously described yeast-based assay to test the entire panel of rGn/Gc-reactive mAbs for competition with seven antigen-binding fragments (Fab) that bind different epitopes in Gc (ADI-36121, ADI-36122, ADI-36124, ADI-36125, ADI-36136, and ADI-36193) and GP38 (ADI-36120) (Figure S3A) (Gilman et al., 2016). The majority of Gc-reactive mAbs from each donor could be assigned to one of six different competition groups (Figure 3). For clarity, these competition groups were designated Antigenic Sites 1–6 according to frequency (Site 1 > Site 6) (Figure 3A–C). mAbs that competed with ADI-36125 (Site 2) often also competed with ADI-36121, ADI-36122, or ADI-36193, suggesting that these epitopes are in close proximity. Accordingly, we designated these Antigenic Sites 2a–d (Figure 3).
Figure 3. Distribution of antigenic sites targeted and mapping of epitopes.
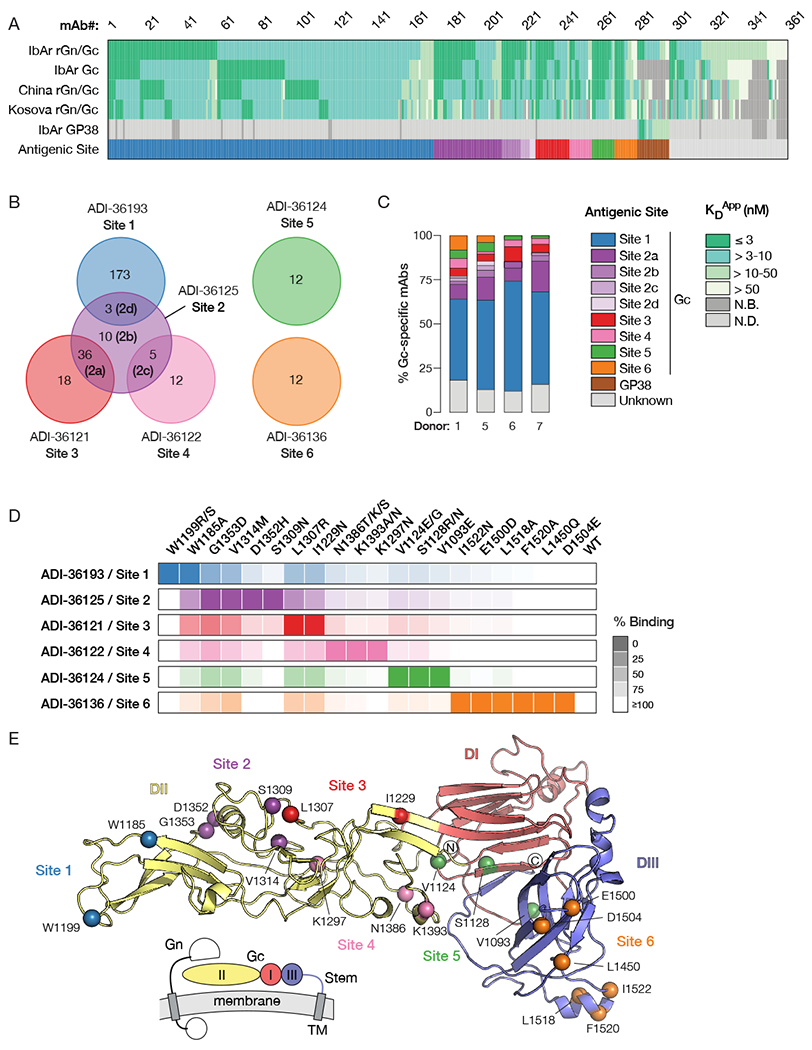
(A) Summary of mAb binding data together with targeted antigenic sites as determined by binding of IgG to rGn/Gc with pre-complexed competitor or to rGP38 by BLI. Each column denotes the antigen reactivity of an individual mAb, colored by KDApp (see legend in panel C). Each of the top five rows indicates the test antigen. The last row denotes the antigenic site, color coded according to the legend in panel C. (B) Distribution of targeted antigenic sites by Gc-specific antibodies for which a targeted site was determined in panel A. Named mAbs (e.g. ADI-36193) shown with each group represent the Fab used in the competition assay. mAbs for which the targeted site could not be readily defined (“Unknown”) or that bound GP38 are not shown. (C) Distribution of targeted antigenic sites by Gc-specific mAbs across all donors. mAbs with “Unknown” antigenic sites include those with weak affinities and/or weak competition (<10-fold reduction in binding in the presence of competitive Fab). (D) Heat map depicting the magnitude of loss of binding of single mutations compared to wild-type Gc on the surface of yeast. Darker shades indicate greater loss of binding. Shown are mutations that reduce binding of mAbs by >90%. (E) Homology model of CCHFV Gc based on Maporal virus Gc with an associated schematic illustrating the approximate domain organization in relation to the viral membrane. The stem region and transmembrane domain are not visualized in this model. Residues at which mutations reduce binding by >90% are colored as in D. See also Figure S3 and S4.
To elucidate the location of the antigenic sites within Gc, the mAbs that defined each of the six antigenic sites in competitive binding assays were used for epitope mapping studies by employing mutagenized Gc libraries displayed on the surface of yeast (Mata-Fink et al., 2013). In the first and second rounds of fluorescence-activated cell sorting, mutants that lost binding to a particular antibody were isolated. In the third round of sorting, a mixture of antibodies targeting different antigenic sites were used for positive selection, thereby selecting mutants that did not disrupt the global conformation of Gc (Figure 3D, S4A). After sequencing single clones, mutants with a single amino acid mutation were displayed on the surface of yeast and assayed for the binding of antibodies targeting all six antigenic sites to validate the identified mutations (Figure 3D, S4B–C). Comparison of binding relative to wild-type determined whether each antibody was sensitive to the isolated variants. In total, 2—6 positions were identified to be important for the specific binding of each antibody (Figure 3D, S4B).
Current evidence indicates that the Gc subunits of orthobunyaviruses, phleboviruses, and hantaviruses are Class II viral membrane fusion proteins structurally homologous to the fusion subunits of alphaviruses, rubiviruses, and flaviviruses (DuBois et al., 2013; Guardado-Calvo et al., 2016; Kuhn et al., 2002; Lescar et al., 2001, Hellert et al., 2019), strongly suggesting that nairovirus Gc is also a Class II fusion protein. Concordantly, we were able to generate a homology model based on the Gc subunit of the hantavirus, Maporal virus (Figure 3E). To identify the location of the antigenic sites on the structure of CCHFV Gc, we mapped the mutations onto this homology model. Site 1, which is targeted by about half (41–62%) of the antibodies, encompasses the highly conserved fusion loops. Site 2a mAbs mapped to the fusion loop-proximal region and display convergent sequence features suggestive of a public antibody response. Specifically, they are characterized by (i) the usage of similar germline genes; (ii) relatively short CDRH3s; (iii) elevated polyreactivity; and (iv) similar somatic mutations both within and across donors (Figure S3B–G). The Site 3 mAb ADI-36121 targets residues modeled as domain II. The mAbs targeting Sites 4 and 5 mapped to domain II and domain I, respectively. Site 6 was the only antigenic site mapping to domain III and binding of the Site 6-targeting mAb ADI-36145 to recombinant domain III was verified by biolayer interferometry (Figure S4D).
Identification of mAbs with neutralizing activity
We used transcription- and entry-competent virus-like particles (tecVLPs) to evaluate the neutralizing potential of CCHFV Gn/Gc-reactive mAbs under biosafety level 2 conditions. As previously described, tecVLPs contain all structural proteins of the authentic virus and a minigenome expressing a Nano-Glo luciferase reporter gene. They also recapitulate the behavior of authentic CCHFV in studies of cell entry and inhibition with small molecules and mAbs (Zivcec et al., 2015). To identify mAbs with neutralizing potential against a clinically relevant strain, we initially screened a single concentration of each mAb (35 nM) against tecVLPs bearing Oman Gn/Gc. This clade IV strain of CCHFV has been endemic in Oman for at least 20 years and its incidence has been rising steadily (Al-Abri et al., 2019). Our screens revealed a broad range of neutralizing activity among mAbs derived from each donor. We prioritized potent nAbs, affording reductions of tecVLP reporter activity >90%, for further analysis (Figure 4A). A breakdown of nAb potency by targeted antigenic site showed that potent neutralization could be achieved by binding some, but not all, antigenic sites in Gc. Among these, fusion-loop targeting (Site 1) and domain II-targeting (Site 3) nAbs displayed the highest median potencies (~70-80% neutralization), with the most potent nAbs belonging to the latter group (Figure 4B). None of the GP38-specific mAbs afforded potent neutralization, consistent with previous reports (Figure 4B) (Golden et al., 2019; Zivcec et al., 2017).
Figure 4. Distribution of neutralization potency by antigenic sites targeted, and breadth of neutralization by candidate nAbs.
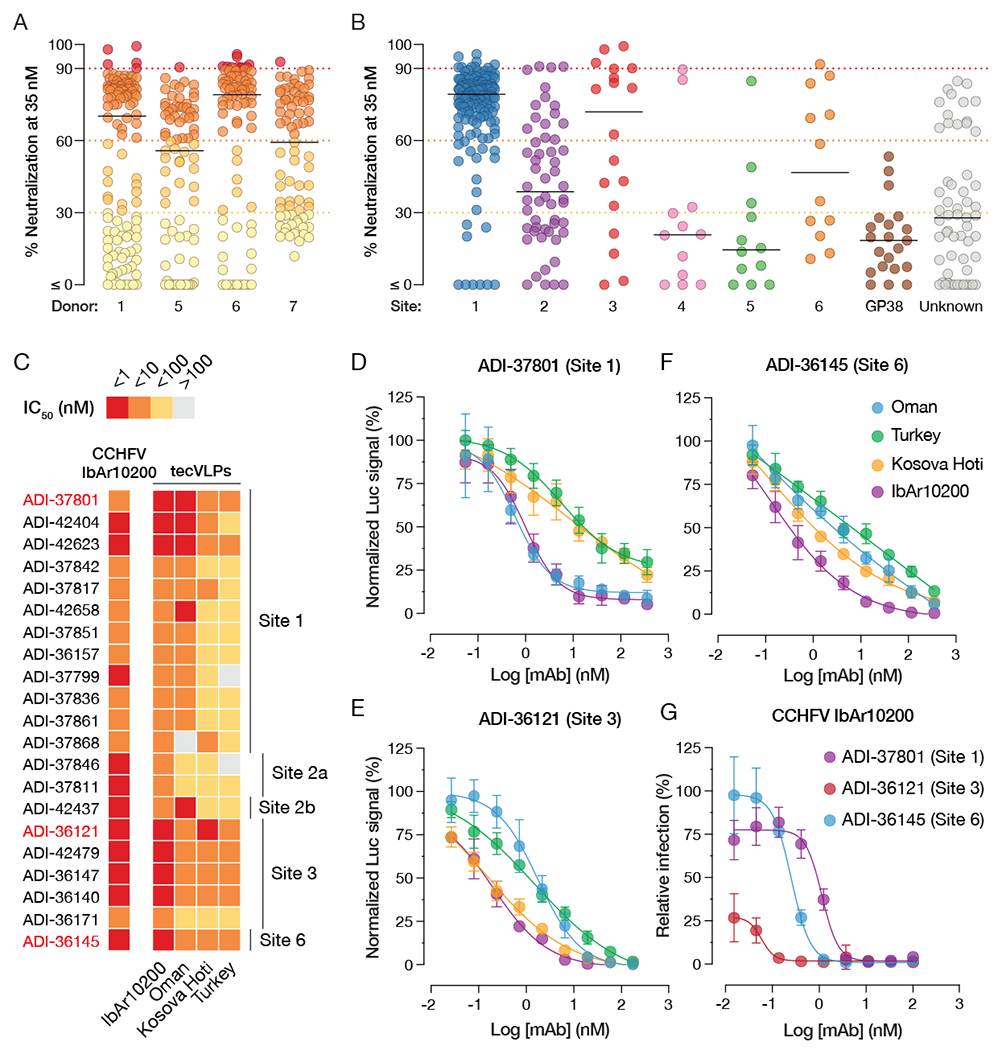
(A) Neutralization of Oman tecVLPs at 35 nM mAb, as measured by reduction in luciferase activity in Vero target cells compared to no-antibody treatment. Each circle represents a different mAb. (B) Breakdown of mAb neutralizing activity by targeted antigenic site. (C) Heatmap showing relative or absolute neutralization activity (IC50) of 21 nAbs selected for further characterization. Relative IC50 values are shown for sigmoidal curves, whereas absolute IC50 values are provided for curves with poorly defined plateaus. See Figure S5 for all neutralization curves. (D-F) Neutralization curves of ADI-37801 (D), ADI-36121 (E), and ADI-36145 (F) against tecVLPs bearing Gn/Gc proteins from four CCHFV strains. (G) Neutralization curves of ADI-37801, ADI-36121, and ADI-36145 against authentic CCHFV-IbAr10200. All tecVLP neutralization assays were performed using Vero target cells, while neutralization of authentic CCHFV IbAr10200 was assayed in Vero-E6 cells. n = 6, from two independent experiments for all neutralization curves.
Candidate mAbs display potent cross-clade neutralization
A challenge to the development of broadly protective immunotherapeutics against CCHF is the existence of multiple, geographically distinct viral clades with divergent Gn/Gc sequences (Figure S2B). Strains responsible for the majority of CCHF cases in humans belong to clades III, IV, and V (Bente et al., 2013; Hoogstraal, 1979; Lukashev et al., 2016). Accordingly, we examined the capacity of selected nAbs targeting 5 different antigenic sites to neutralize not only Oman tecVLPs (Clade IV) but also tecVLPs bearing Gn/Gc proteins from the prototypic Clade III CCHFV strain IbAr10200, and the virulent Clade V strains Turkey and Kosova Hoti. Most of the nAbs potently neutralized IbAr10200 tecVLPs, with IC50 values in the sub-nanomolar or single-digit-nanomolar range. Largely similar results were observed against Oman tecVLPs, with the exception that nAbs targeting Site 2a (adjacent to the fusion loops) displayed lower activity against this strain (Figure 4, S5). Most nAbs targeting Sites 1, 3, and 6 retained broad neutralizing activity against the Clade V tecVLPs (Kosova Hoti and Turkey), albeit with somewhat reduced potency (Figure 4C–F). The Site 3 nAb ADI-36121 was especially noteworthy in its capacity to neutralize Kosova Hoti tecVLPs with sub-nanomolar potency. Based on these findings, we down-selected to three nAbs, ADI-37801 (Site 1/fusion loops), ADI-36121 (Site 3/domain II), and ADI-36145 (Site 6/domain III). The same set of nAbs was active against authentic CCHFV IbAr10200, with potencies equivalent to or surpassing those observed against tecVLPs (Figure 4G).
ADI-37801 displays potent synergistic neutralization when combined with ADI-36121 or ADI-36145
Although antiviral monotherapies have demonstrated clinical efficacy (Mulangu et al., 2019; The IMpact-RSV Study Group, 1998), and in the case of palivizumab, been licensed by regulatory agencies, antibody monotherapies against viral agents may elicit viral neutralization escape variants that reduce their efficacy or render them inactive (Caskey et al., 2015; Klein et al., 2012; Webster and Laver, 1980). To mitigate this risk and identify nAb cocktails with enhanced efficacy and/or breadth, we next explored the synergistic potential of our lead nAbs. Specifically, we measured the activities of equimolar mixtures of non-competing nAb pairs relative to those of the individual nAbs and performed combination-index (CI) analysis of the resulting neutralization curves using the well-established method of Chou and Talalay (Chou and Talalay, 1984; Li et al., 1998; ter Meulen et al., 2006; Zwick et al., 2001). ADI-36121 (Site 3/domain II) and ADI-36145 (Site 6/domain III) lacked synergistic activity (CI ≥ 1) against Oman tecVLPs (Figure 5A, 5D). In contrast, combinations of ADI-37801 (Site 1/fusion loops) with either ADI-36121 or ADI-36145 synergistically neutralized Oman tecVLPs (CI < 1) over a broad range of nAb concentrations (Figure 5B–C, 5E–F). Similar results were observed with IbAr10200, Turkey, and Kosova Hoti tecVLPs, although the effect was smaller for IbAr10200 than for the other strains (Figure 5G). Synergistic neutralization also extended to other nAbs targeting Site 1, indicating that it is a general property of Gc fusion loop binders and not specific to ADI-37801 (Figure S6).
Figure 5. Synergistic neutralization potential of lead nAb candidates.
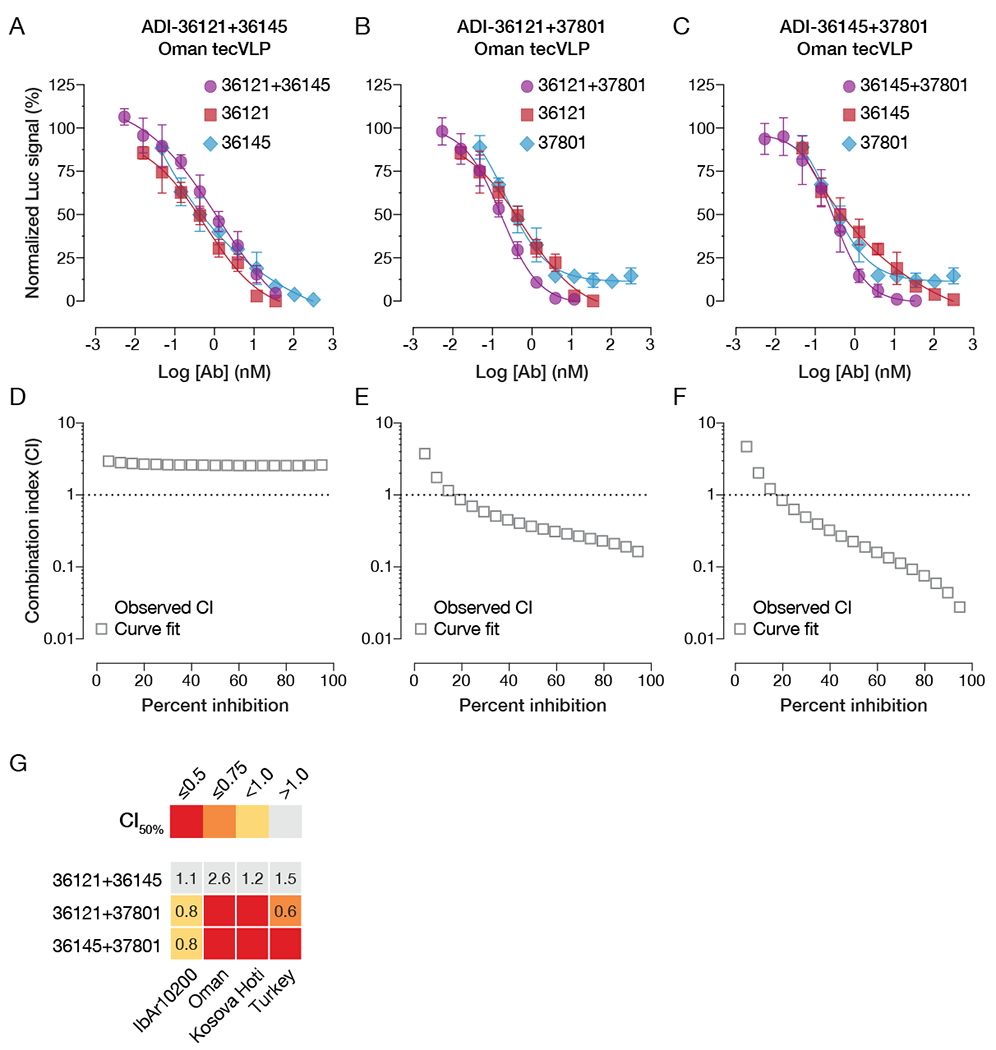
(A-C) Neutralization curves of individual nAbs and their 1:1 combinations. (A) ADI-36121 and ADI-36145, (B) ADI-36121 and ADI-37801, (C) ADI-36145 and ADI-37801. (D-F) Combination-index (CI) analyses of neutralization curves in panels A–C to determine additive (CI ~ 1) or synergistic effects (positive, CI < 1; negative, CI >> 1). (G) Summary of CI values at 50% neutralization for nAb combinations against tecVLPs carrying divergent Gn/Gc proteins. Synergistic neutralization was assayed in Vero target cells. n = 6, from two independent experiments for all neutralization curves. See also Figure S6.
Lead nAbs and nAb combinations afford prophylactic but not therapeutic protection against CCHFV challenge
We evaluated the prophylactic and therapeutic potential of our lead nAbs and nAb combinations in two distinct immunocompromised rodent models of lethal CCHFV challenge. First, we benchmarked the prophylactic efficacy of ADI-36121, ADI-36145, and ADI-37801 against a chimerized version of the previously described GP38-specific mouse mAb 13G8 (c13G8) (Mishra et al., 2020). We treated type I interferon α/β R−/− (IFNAR1-KO) mice (Bereczky et al., 2010; Zivcec et al., 2013) with 1 mg of each mAb per animal (~50 mg/kg), as well as with combinations of the Gc-specific nAbs that afforded synergistic neutralization and exposed them to CCHFV-IbAr10200 at 24 h post-treatment. Essentially complete protection was observed with all mAbs and mAb combinations tested (Figure 6A), with associated prevention of weight loss (Figure 6B). ADI-36121 and ADI-36145 also protected STAT1−/− (STAT1-KO) mice (Bente et al., 2010; Bowick et al., 2012) against CCHFV-Turkey challenge when administered at 30 min postexposure (Figure 6C–D), as shown previously for c13G8 (Mishra et al., 2020).
Figure 6. Protective efficacy of lead nAbs and nAb combinations in two murine models of lethal CCHFV challenge.
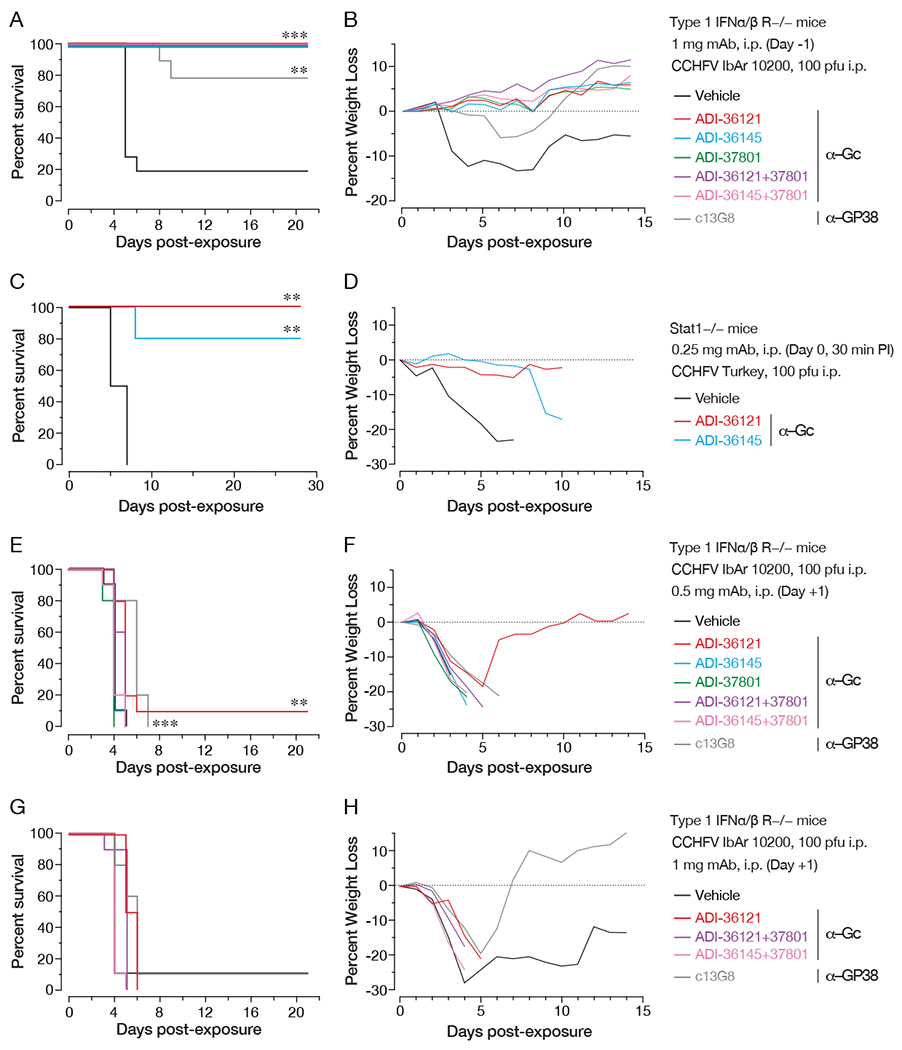
(A-B) Type I IFN α/β/ R−/− (IFNAR1-KO) mice were treated with the indicated mAbs or mAb combinations 1 day prior to challenge with CCHFV IbAr10200. (n = 10 mice per group) (A) Survival curves (vehicle versus test mAb) were compared by Mantel-Cox test (*** P < 0.001, ** P < 0.01). (B) Associated mean weight loss data are shown. (C-D) Stat1−/− mice were challenged with CCHFV Turkey/2004 and then treated with single doses of the indicated mAbs or vehicle at 30 min post-exposure. (n = 5 mice per group) (C) Survival curves (vehicle versus test mAb) were compared by Mantel-Cox test (*** P < 0.001, ** P < 0.01). (D) Associated mean weight loss data are shown. (E-H) IFNAR1-KO mice were exposed to CCHFV IbAr10200 and treated with the indicated mAbs or mAb combinations at 1 day post-challenge. (n = 10 mice per group) (E, G) Survival curves (vehicle versus test mAb) were compared by Mantel-Cox test (*** P < 0.001, ** P < 0.01). (F, H) Associated mean weight loss data are shown.
Having demonstrated pre-exposure and post-exposure prophylactic protection, we next tested the same set of mAbs in a therapeutic setting (24 h post-challenge). Neither individual Gc-specific nAbs nor their combinations afforded measurable protection against CCHFV-IbAr10200 when dosed at 0.5 mg (Figure 6E–F) or 1 mg (Figure 6G–H) per animal (25 or 50 mg/kg, respectively). Further, although two 1-mg doses of 13G8 administered at days 1 and 4 post-challenge afforded partial protection in the same murine model (Golden et al., 2019), we observed no significant protection with single doses of c13G8 (Figure 6E–H). Taken together, these findings indicate that human nAbs targeting CCHFV Gc can be at least as efficacious as non-neutralizing mAbs targeting GP38 in prophylactic and pre-exposure settings. However, we also show that single doses of currently available mAbs cannot protect against CCHFV challenge in a therapeutic setting, at least in the highly stringent rodent models presently in use.
Engineered dual-variable domain IgGs provide enhanced synergistic neutralization and potent therapeutic protection
We postulated that the synergistic activity of our most potent nAb pairs (ADI-36121+ADI-37801, ADI-36145+ADI-37801) could be further amplified through avidity effects by physically linking their corresponding variable domains into bispecific antibodies (Wec et al., 2016). Accordingly, we engineered four dual-variable domain immunoglobulins (DVD-Igs) to bear these pairs of variable domains in both possible configurations (i.e., as “inner” or “outer” variable domains) (Figure 7A). The DVD-Igs were purified from ExpiCHO cells and size-exclusion chromatography indicated the preparations were largely monodisperse and of expected molecular weights (Figure S7A–D). The capacity of both combining sites in each DVD-Ig to recognize rGn/Gc was confirmed by biolayer interferometry through two-phase experiments in which binding to rGn/Gc in the presence of the respective parental monospecific mAbs was assayed. The DVD-Igs were able to bind rGn/Gc in the presence of each monospecific mAb (Figure S7E–H) but not in the presence of both mAbs (Figure S7I–J) indicating that both sets of variable domains recognized their expected epitopes. In neutralization studies, DVD-121-801 was more potent than the equimolar mixture of its parental nAbs against all four tecVLP preparations bearing divergent Gn/Gc proteins (Figure 7B–E). DVD-145-801 also exhibited enhanced potency, but only against Oman and Kosova Hoti tecVLPs; it resembled the mixture of its parental nAbs in its activity against IbAr10200 and Turkey tecVLPs (Figure 7F–I). Interestingly, the relative positions of the two sets of variable domains in these two DVD-Igs was crucial to their enhanced activity: neither of their counterparts bearing the same variable domains in the reverse configuration (DVD-801-121 and DVD-801-145; Figure 7A) afforded enhanced neutralization against Oman tecVLPs (Figure 7B, 7F).
Figure 7. Neutralization activity and protective efficacy of engineered bsAbs.
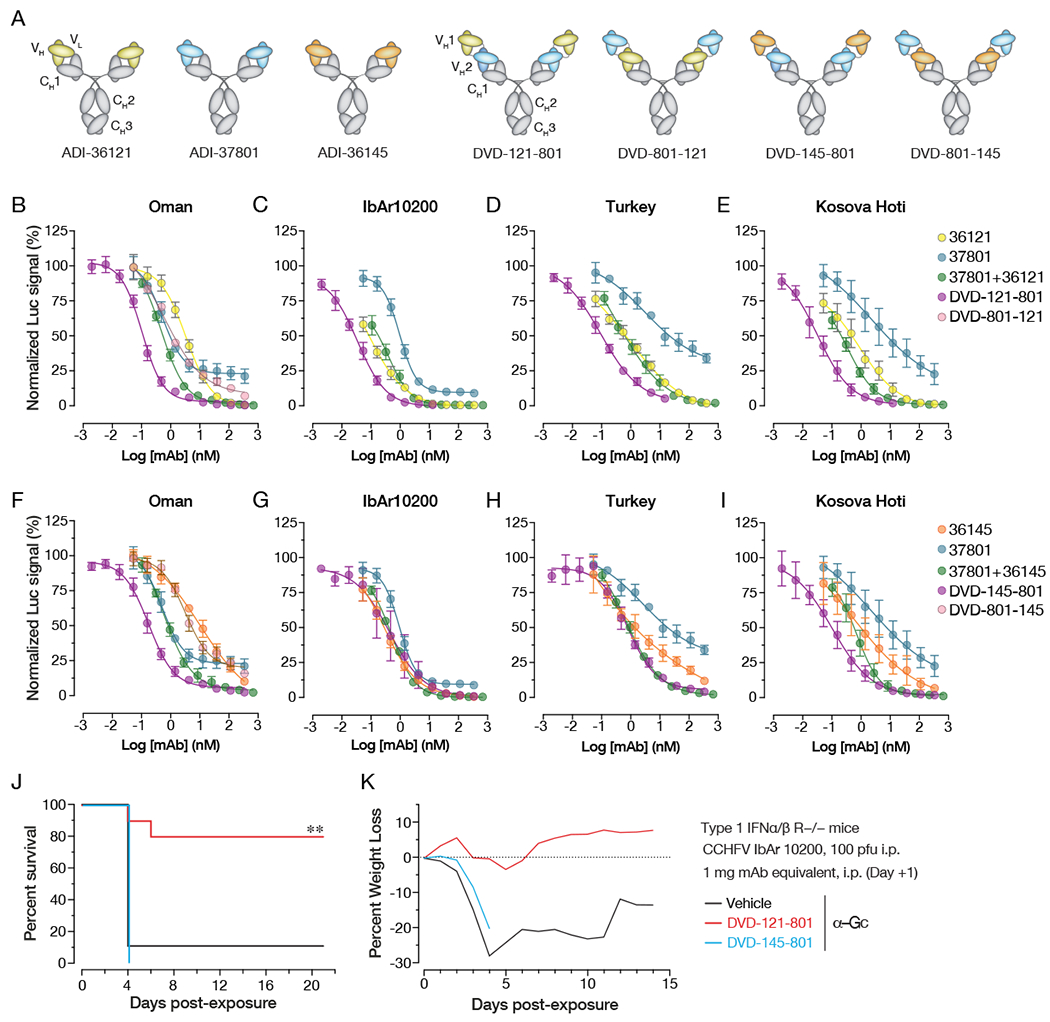
(A) Schematic illustration of candidate nAb IgG1s and dual-variable domain IgGs (DVD-Igs) derived from them by combining IgG1 variable domains. (B-I) Neutralization curves of the indicated nAbs, nAb combinations, and bsAbs against against tecVLPs bearing Gn/Gc proteins from (B, F) Oman, (C, G) IbAr10200, (D, H) Turkey, (E, I) Kosova Hoti strains. (J,K) IFNAR1-KO mice were exposed to CCHFV IbAr10200 and treated with the indicated bsAbs at 1 day post-challenge. (n = 10 mice per group) (J) Survival curves (vehicle versus test mAb) were compared by Mantel-Cox test (*** P < 0.001, ** P < 0.01). (K) Associated mean weight loss data are shown. See also Figure S7.
Finally, we sought to test if the enhanced in vitro activity of DVD-121-801 and DVD-145-801 would translate to improved therapeutic efficacy in vivo. To this end, we challenged IFNAR1-KO mice with CCHFV-IbAr10200 and then treated animals with DVD-121-801 and DVD-145-801 (equimolar to 1 mg mAb) as part of the study described above (Figure 6G–H) that also included nAb and nAb-combination arms. Strikingly, DVD-121-801 afforded essentially complete protection whereas DVD-145-801 provided no benefit (Figure 7J–K). This result was concordant with the enhanced neutralizing activity of the former bsAb, but not the latter, against IbAr10200 tecVLPs (Figure 7C, 7G). We thus identified a single bsAb that combines two synergizing bnAbs that target distinct sites in Gc domain II to achieve 10–100-fold enhanced neutralization potency relative to its component nAbs. A single dose of this bsAb afforded therapeutic protection in a stringent model of lethal CCHFV infection.
Discussion
Previous work suggests that monoclonal antibody-based treatments can help fulfill the urgent and currently unmet need for medical countermeasures against CCHF disease in humans. mAbs recognizing the CCHFV glycoproteins pre-Gn/GP38 and Gc have been isolated previously through experimental vaccination of mice (Bertolotti-Ciarlet et al., 2005). These studies also found that, although mAbs targeting Gc could neutralize viral infection, only non-neutralizing mAbs targeting GP38 could confer protection in mouse models of CCHFV challenge through an unknown mechanism (Bertolotti-Ciarlet et al., 2005; Golden et al., 2019). Because GP38 sequences are more divergent among different CCHFV clades than those of the Gn/Gc glycoprotein complex, it is unclear if GP38-targeting mAbs can form the sole basis of broadly protective CCHF therapeutics. As a case in point, mAb 13G8 lacked cross-protective activity against relatively closely related viral strains (Golden et al., 2019). Herein, we characterized the human memory B cell response to natural infection in CCHF survivors from Uganda and described fully human nAbs that can target Gn/Gc from a geographically diverse set of CCHFV isolates. We identified synergistic combinations of Gc-specific nAbs and demonstrated that a single dose of a bsAb incorporating one such combination could afford therapeutic protection against lethal CCHFV infection.
Our analysis of 361 mAbs isolated from rGn/Gc-specific human memory B cells identified high-affinity binders to Gc or GP38 but none that could be explicitly mapped to Gn. For some Bunyavirales members, such as phleboviruses, Gn has been suggested as the main nAb target (Hao et al., 2020; Wang et al., 2019) whereas for others, such as hantaviruses, both Gn and Gc were found to be targeted (Duehr et al., 2020; Rissanen et al., 2020). By contrast, immunization with recombinant ectodomain forms of CCHFV Gc, but not Gn, induced robust nAbs in mice (Kortekaas et al., 2015). We speculate that our failure to isolate CCHFV Gn-specific mAbs and their paucity in the literature reflect the topological arrangement of these subunits on the viral particle and the attendant immunodominance of Gc over Gn. Thus, the nairovirus Gn/Gc complex may more closely resemble its orthobunyavirus counterpart in quaternary organization (Gc>>Gn in surface exposure) than it does the phlebovirus or hantavirus Gn/Gc complexes (Hellert et al., 2019; Serris et al., 2020). Alternatively, it is conceivable that the Gn component of our rGn/Gc probe is occluded or improperly folded, and/or that there are significant differences in antigenicity between IbAr10200 Gn and the unknown infecting strain(s) (Figure S2B). Unfortunately, no validated Gn-specific reagents, including conformation-sensitive control antibodies, are currently available to evaluate these hypotheses.
Our epitope-binning studies uncovered six distinct antigenic sites in Gc that we fine-mapped by mutagenesis (Figure 3D). Strikingly, mapping of the antigenic sites onto a homology-based structural model of Gc (Figure 3E), showed that Site 1, corresponding to the Gc fusion loops, represented ~50% of mAbs isolated from each donor, indicating that its targeting is a shared immunological solution in humans. This apparent immunodominance of the fusion loops is concordant with similar observations for some flavivirus E proteins (Beltramello et al., 2010; Crill et al., 2007; Stettler et al., 2016; Wec et al., 2020a). Site 1 mAbs afforded neutralization with moderate-to-high potency and breadth; however, they typically failed to fully neutralize tecVLP entry. Quaternary Gn/Gc interactions in the viral envelope that partially and/or transiently occlude the fusion loops and render them unavailable for mAb binding may account for this un-neutralized fraction. Interestingly, similar nAb potencies against Gn/Gc from different CCHFV strains could be accompanied by quantitative differences in un-neutralized fraction (compare ADI-37801 curves in Figure 7B, 7F vs. 7C, 7G), raising the possibility that sequence-dependent differences in Gn/Gc organization and/or structural dynamics exist. Similar observations have been made for dengue virus (DENV), a class II fusion protein-bearing flavivirus (Austin et al., 2012; Cherrier et al., 2009; Dowd et al., 2015; Lok et al., 2008; Zhang et al., 2013). In contrast to DENV, however, for which cross-reactive fusion loop-targeting mAbs generally lack neutralizing activity and can promote enhancement of infection (Dejnirattisai et al., 2010), we found that the fusion loop-directed mAbs elicited by natural CCHFV infection possess both neutralizing activity and some degree of neutralization breadth. In the case of DENV, this has fueled efforts to design glycoprotein-based immunogens that can steer the antibody response away from the immunodominant fusion loops (Rouvinski et al., 2017). Our work suggests instead that CCHFV vaccines may benefit from targeting the fusion loops together with other conserved sites in Gc domains II and III, such as Sites 3 and 6, that afford potent neutralization (Figure 4C) and can synergize with the Site I/fusion loop binders (Figure 5B–C, 5E–F). We anticipate that the structural elucidation of CCHFV Gn/Gc in its apo and antibody-bound forms will enhance our understanding of the mechanistic basis of neutralization mediated by mAbs recognizing specific antigenic sites and inform the development of next-generation vaccines that elicit antibodies to these sites.
To enhance neutralization potency and breadth and mitigate the risk of viral mutational escape (Gilchuk et al., 2020; Keeffe et al., 2018; Wec et al., 2019), we evaluated combinations of non-competing nAbs. Only combinations of Site 3/domain II or Site 6/domain III binders with Site 1/fusion loop binders afforded synergistic neutralization, characterized by Chou-Talalay combination index (CI) scores <1 and improvements in both neutralization IC50 and un-neutralized fraction. The site-specificity of nAb synergy suggested that it did not arise solely from the simultaneous engagement of two non-overlapping functional epitopes (Diamant et al., 2015). Instead, we speculate that these synergies reflect cooperative binding. Specifically, the Site 3 and Site 6 nAbs may trap “open” Gc conformers in which the fusion loops are more exposed for recognition by Site 1 nAbs. The further improvements in neutralization IC50 with bsAbs may reflect increases in binding avidity of the physically linked variable domains to these pairs of sites, which may be further magnified by the tetravalent DVD format used herein (Diamant et al., 2015; Jakob et al., 2013; Wec et al., 2016). The reduced potency of the alternate DVD-Ig configurations bearing the fusion loop-binding domains as outer domains (DVD-801-121 and DVD-801-145) presumably reflects structural constraints that reduce the efficiency of bivalent and/or tetravalent bsAb engagement with Gc. An understanding of these constraints awaits structural elucidation of the supramolecular organization of CCHFV Gn/Gc in intact viral particles.
Single doses of candidate nAbs targeting Gc domains II and III afforded prophylactic protection against virulent CCHFV strains from two distinct viral clades. That these studies were independently performed in two BSL-4 laboratories using genetically distinct immunocompromised murine models attests to the robustness of our findings. However, single doses of all tested nAbs, synergistic nAb combinations, and non-neutralizing GP38-specific mAbs failed to protect mice in a more stringent therapeutic setting. Strikingly, a single bsAb, DVD-121-801, combining variable domains from the synergizing nAbs ADI-36121 (Site 3/domain II) and ADI-37801 (Site 1/fusion loops), protected mice when administered 24 h post-viral challenge, concordant with its enhanced neutralization potency against tecVLPs bearing Gn/Gc from the cognate challenge strain. DVD-121-801 is thus an antibody-based treatment demonstrated to afford therapeutic protection against lethal CCHFV challenge with a single dose, and it is a lead candidate for further evaluation in murine and nonhuman primate models of CCHFV challenge (Cross et al., 2020; Haddock et al., 2018).
Limitations of the study
In this study, potent and protective antibodies targeting CCHFV Gc were generated and characterized. However, due to the nature of the human antibody response to natural CCHFV infection and/or the properties of the sorting antigen used, no antibodies targeting Gn were identified. The characteristics, and indeed the existence, of such antibodies therefore remain largely unknown.
Our understanding of the interactions between mAbs or DVDs and CCHFV Gc is currently limited by a lack of structural information regarding Gn/Gc and their supramolecular organization on the surface of viral particles. Structural studies of this kind will greatly advance our understanding of the mechanistic underpinnings of neutralization and will aid in the precise mapping of antigenic sites.
Finally, while stringent immunocompromised rodent models of CCHFV infection are useful in down-selection of mAbs, studies in larger animal models that more faithfully recapitulate human CCHF are needed to advance a lead candidate for therapeutic use in humans.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Requests for reagents and further information should be directed to the lead contact, Kartik Chandran (kartik.chandran@einsteinmed.org).
Materials availability
DVD-Igs and mAbs generated in this study will be made available to researchers upon request.
Data and code availability
This study did not generate any new datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Vero cells, a spontaneously immortalized cell line isolated from the kidney of an adult female African grivet monkey (RRID:CVCL_0059), were obtained from the American Type Culture Collection (ATCC). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher) supplemented with 2% fetal bovine serum (Atlanta Biologicals), 1% GlutaMAX (Thermo Fisher), and 1% penicillin-streptomycin (Thermo Fisher). BSR-T7 cells (RRID:CVCL_RW96), generated by stable T7 RNA polymerase expression in BHK-21 cells, were a kind gift from K.-K. Conzelmann. The parent cell line (RRID: CVCL_1915) was isolated from the kidney of a 1-day old male golden hamster. BSR-T7 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher) supplemented with 10% fetal bovine serum (Atlanta Biologicals), 1% GlutaMAX (Thermo Fisher), and 1% penicillin-streptomycin (Thermo Fisher). Vero E6, a spontaneously immortalized cell line isolated from the kidney of an adult female African grivet monkey (RRID: CVCL_0574) were obtained from ATCC. Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher) supplemented with 2% fetal bovine serum (Atlanta Biologicals), 1% GlutaMAX (Thermo Fisher), and 1% penicillin-streptomycin (Thermo Fisher). All cell lines were maintained in a humidified 37°C incubator supplied with 5% CO2. Cell lines were not authenticated following purchase.
Virus-like particles
Amino acid sequences of the IbAr10200, Oman-199809166, Kosova Hoti, and Turkey2004 strain GPCs used to generate tecVLPs were derived from GenBank M-segment sequences with accession numbers NC_005300, KR864901, EU037902, and ALT31694 respectively. These sequences were inserted into the pCAGGS-GPC plasmid and used to generate tecVLPs as described by Zivcec and colleagues (Zivcec et al., 2015).
Viral strains
The authentic CCHFV strains CCHFV-Turkey2004 and CCHFV-IbAr10200 were used in this study.
Animal models
4–8-week-old female 129S6/SvEv-Stat1tm1Rds (STAT-1−/−) mice (Bente et al., 2010; Bowick et al., 2012) (Taconic) and 5–8-week old male and female Type 1 interferon α/β receptor knockout mice (Type 1 IFNα/β R−/−) (Bereczky et al., 2010; Zivcec et al., 2013) (Jackson Labs) were used in animal challenge experiments. Animals were provided food and water ad libitum and housed in individual ventilated cages.
Murine challenge studies were conducted under IACUC-approved protocols in compliance with the Animal Welfare Act, PHS Policy, and other applicable federal statutes and regulations relating to animals and experiments involving animals. The facilities where these studies were conducted (USAMRIID and UTMB) are accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International (AAALAC), and adhere to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 2013. Euthanasia criteria were defined as follows: mouse displays severely hunched posture, inability or reluctance to move, appears weak (staggering when moving around cage), or has labored breathing.
METHOD DETAILS
Patient recruitment and ethics statement
CCHF convalescent donors were recruited through the Uganda Virus Research Institute, Entebbe, Uganda. All donors had documented clinical history of CCHF infection in Agago and Nakaseke districts, Uganda, ranging from 2013 to 2017. The study was approved by the Helsinki committees of Uganda Virus Research Institute (UVRI), Entebbe, Uganda (reference number GC/127/13/01/15); Soroka Hospital, Beer Sheva, Israel (protocol number 0263-13-SOR); and the Ugandan National Council for Science and Technology (UNCST) (registration number HS1332). Written informed consent, as well as a personal health questionnaire, was completed for each donor who participated in this study. Study participants were adults, or minors with parental consent, and were not related. We confirm that all experiments were performed in accordance with the relevant guidelines and regulations.
Generation of recombinant Gn/Gc, Gn, GP38, and Gc antigens
rGn/Gc:
A construct containing CCHFV strain IbAr10200 GPC residues 1 to 1579 with an internal deletion of residues 691-1040 replaced with a 29-amino acid glycine/serine linker, fusing Gn to Gc was expressed in stably transfected Schneider 2 (S2) insect cells. The N-terminal MLD (GPC residues 1 to 247) was cleaved at the native furin cleavage site (GPC residues 244 to 247), and GP38 was also cleaved via an introduced furin cleavage site, replacing the native SKI-1 site (residues 516 to 519). The resulting Gn/Gc fusion protein was secreted and harvested from clarified S2 culture supernatant via a C-terminal double strep-tag II sequence using affinity chromatography (5 ml. StrepTrap™ HP column, Cytiva) using phosphate buffered saline (PBS) supplemented with 150 mM NaCl and eluted using the same buffer with 2.5 mM desthiobiotin. The Gn/Gc protein was then purified by size-exclusion chromatography (SEC) using an S200 column (Cytiva) in PBS supplemented with 150 mM NaCl.
rGn:
A construct containing CCHFV strain IbAr10200 GPC residues 1 to 690 was expressed in stably transfected Schneider 2 (S2) insect cells. The N-terminal MLD (GPC residues 1–247) was cleaved at the native furin cleavage site (GPC residues 244–247), and GP38 was also cleaved from Gn via an introduced furin cleavage site, replacing the native SKI-1 site (residues 521–524). The resulting Gn protein was secreted and harvested from clarified S2 culture supernatant via a C-terminal double strep-tag II sequence using affinity chromatography (5 mL StrepTrap™ HP column, Cytiva) using phosphate buffered saline (PBS) supplemented with 150 mM NaCl and eluted using the same buffer with 2.5 mM desthiobiotin.
rGc:
A construct containing CCHFV strain IbAr10200 GPC residues 1–1579 with an internal deletion of residues 520–1039, fusing Gc directly to the MLD and GP38 was expressed in stably transfected Schneider 2 (S2) insect cells. The N-terminal MLD (GPC residues 1–247) was cleaved at the native furin cleavage site (GPC residues 244–247), and GP38 was also cleaved from Gn via an introduced furin cleavage site at the C-terminus of GP38 (residue 519) prior to the Gc sequence. The resulting Gc protein was secreted and harvested from clarified S2 culture supernatant via a C-terminal double strep-tag II sequence using affinity chromatography (5 mL StrepTrap™ HP column, Cytiva) using phosphate buffered saline (PBS) supplemented with 150 mM NaCl and eluted using the same buffer with 2.5 mM desthiobiotin.
rGP38:
GP38 was expressed in stably transfected Schneider 2 (S2) insect cells and the protein construct consisted of GPC residues 1–519 of CCHFV strain IbAr10200. The N-terminal MLD (GPC residues 1–247) was cleaved at the native furin cleavage site (GPC residues 244–247), and secreted GP38 was purified from clarified supernatant via streptavidin affinity chromatography utilizing a double C-terminal Strep-tag-II sequence and eluted into phosphate buffered saline (PBS) supplemented with 2.5mM desthiobiotin followed by size-exclusion chromatography (SEC) into PBS.
For experiments detailed below using biotinylated rGn/Gc or GP38, the proteins were biotinylated with EZ-Link Sulfo-NHS-LC Biotin (Thermo Fisher Scientific) for 14–16 h at 4°C followed by desalting using a Zeba spin column (Thermo Fisher Scientific).
Amino acid sequences of the IbAr10200, China-79121M18, and Kosova Hoti strain GPCs used to generate recombinant CCHFV glycoproteins were derived from GenBank M-segment sequences with accession numbers NC_005300, GU477493, and ABW04159 respectively.
rGn/Gc ELISA for serum reactivity
rGn/Gc serum reactivity ELISAs were performed in Greiner high-binding half-area plates. Individual wells were coated with 250 ng of purified, recombinant Gn/Gc diluted in phosphate-buffered saline (PBS) (pH 7.4). Wells were washed three times with PBS supplemented with 0.05% Tween 20 (PBST) and then blocked with 2% bovine serum albumin (BSA) in PBS. Five-fold serial dilutions of CCHF patient serum, control serum, or PBS were then added to the blocked wells, followed by incubation for 1 h at ambient temperature. Samples were prepared in duplicate. After 3 washes with PBST, bound rGn/Gc-specific antibody was detected with an anti-human Fc secondary antibody conjugated to horseradish peroxidase (HRP). KPL SureBlue TMB microwell peroxidase was used as the HRP substrate, and the reaction was stopped with an equal volume of 1M sulfuric acid. The absorbance at 450 nm was measured with a Perkin Elmer EnVision multimode plate reader.
Analysis of rGn/Gc by SDS-PAGE
Following affinity purification, IbAr10200 rGn/Gc was analyzed by SEC using an S200 10/300GL column as described above. The two major peaks resolved by gel filtration were collected separately and analyzed on a 4-20% Biorad mini-PROTEAN TGX Stain-free SDS-PAGE gel (BioRad, Cat#4568096). For sample preparation, 2.5 μg of protein was diluted with 2x Laemmeli Sample Buffer (BioRad, Cat#1610737) to a final concentration of 1x and either loaded directly, or reduced with 10 mM DTT and boiled prior to loading. The gel was run at 200V for 30 min, then imaged directly with a BioRad ChemiDoc MP imager.
rGn/Gc intact mass analysis
Gn/Gc protein was buffer-exchanged with 50 mM sodium phosphate buffer (pH 7.5) using 3 kDa MWCO spin filter (Amicon Ultra, Millipore) to remove the desthiobiotin buffer. N-deglycosylation was performed by adding PNGaseF Prime (N-Zyme Scientifics) to the sample and incubating overnight at 37°C. The deglycosylated rGn/Gc was desalted off-line using C4 Ziptip (Millipore) and analyzed by MALDI-TOF in the positive linear mode using UI trail extreme MALDI-TOF/TOF mass spectrometer (Bruker).
Protein identification by peptide mapping
The rGn/Gc sample was buffer-exchanged with 50 mM NH4HCO3 using 3 kDa MWCO spin filter (Amicon Ultra, Millipore) to remove the desthiobiotin buffer. The sample was then dried down by vacuum centrifugation and then re-suspended in 8 M Urea/ 5 mM DTT and incubated at room temperature for 1 h. This was followed by alkylation with 20 mM iodoacetamide in 50 mM NH4HCO3 for 30 min in the dark at room temperature. The sample was diluted with 50 mM NH4HCO3 down to 1.4 M urea final concentration and digested with Trypsin Gold (Promega) overnight at 37°C. The peptide digests were desalted using C18 HLB Oasis resin (Waters) and analyzed by nanoLC-ESI-MS/MS using a nanoRSLC UPLC system (Dionex, Thermo Scientific) equipped with a C-18 reverse phase column connected to an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific). Protein identification was performed by automated protein database search using Proteome Discoverer v 2.4 (Thermo Scientific) against the rGn/Gc protein sequence. Identified spectra were filtered for <1% false discovery rate.
Single B cell sorting
IbAr10200 rGn/Gc streptamers were generated by complexing IbAr10200 rGn/Gc with Strep-Tactin PE (IBA Lifesciences) or Strep-Tactin APC (IBA Lifesciences) (1.4 μg rGn/Gc, 1.65 μL streptactin, PBS to a total of 20 uL) for at least 20 min on ice and in the dark prior to staining. Amounts were calculated such that upon diluting the streptamers in the staining solution, the B cells were stained with 25 nM of each protein/fluorophore combination. PBMCs from CCHFV convalescent donors or a healthy donor control were stained with PerCP-Cy5.5 labeled anti-human CD3 (BioLegend), CD8 (BioLegend), CD14 (Invitrogen), CD16 (BioLegend), PE-Cy7 labeled anti-human CD19 and CD20 (BioLegend), and 25 nM PE and APC streptamers and incubated for 1 hour on ice in the dark. PBMCs were additionally stained with AF488 labeled anti-human IgM (BioLegend), BV421 labeled anti-human IgD (BioLegend), BV510 labeled anti-human CD27 (BioLegend), and propidium iodide (Sigma) and incubated for 30 min on ice in the dark. The PBMCs were loaded onto a BD FACS Fusion (BD Biosciences) for analysis and sorting. Total rGn/Gc-specific B cells (CD19/CD20+ cells) were single cell sorted into 96-well tissue culture plates containing 20 μL per well of lysis buffer [5 μL of 5X first strand cDNA buffer (Invitrogen), 0.625 μL of NP-40 (Thermo Scientific), 0.25 μL RNaseOUT (Invitrogen), 1.25 μL 0.1M dithiothreitol (Invitrogen), and 12.875 μL dH2O]. Plates were briefly centrifuged before storage at −80°C.
Amplification and cloning of antibody variable genes
Antibody variable genes were amplified based on previously described methods (Tiller et al., 2008). IgH, Igκ, and Igλ variable genes were reverse transcribed (Superscirpt III, Thermo Fisher) prior to two rounds of PCR using a mixture of IgG-, IgA-, and IgM-specific primers and HotStarTaq Plus polymerase (Qiagen). The primers in the second round of PCR contain homologous ends to a digested expression plasmid for cloning by homologous recombination in an engineered strain of S. cerevisiae using the lithium acetate chemical transformation method (Gietz and Woods, 2006). A combination of 10 μL of unpurified variable heavy chain PCR product, 10 μL of unpurified variable light chain PCR product, and 200 ng of digested plasmid were used for transformation into 107 yeast cells by resuspending the cells in a mixture of polyethylene glycol 3350 (240 uL, 50% w/v, Sigma), lithium acetate (36 uL, 1M, Sigma), boiled salmon sperm DNA (10 uL, Invitrogen), water (67 uL), and the unpurified PCR products and digested plasmids. After transformation and plating, single yeast colonies were picked for sequencing and IgG production.
Expression and purification of IgGs and Fab fragments
mAbs used in biolayer interferometry assays and neutralization studies were produced as soluble, full-length IgG, in S. cerevisiae as previously detailed (Wec et al., 2020b). Supernatant was harvested by centrifugation and IgG was purified by Protein A-affinity chromatography. IgG bound to Protein A agarose was eluted with 200 mM acetic acid / 50 mM NaCl (pH 3.5) into 1/8th volume 2M HEPES buffer (pH 8.0) and subsequently buffer-exchanged into PBS (pH 7.0). Fab fragments used for competition assays were generated from full-length IgG by 2 h of papain digestion, which was terminated by iodoacetamide addition. Fab was purified from the mixture by passing the solution over Protein A agarose to remove molecules with intact Fc, followed by purification using CaptureSelect IgG-CH1 affinity resin (Thermo Fisher). The Fab fragments were eluted with 200 mM acetic acid / 50 mM NaCl (pH 3.5) into 1/8th volume 2M HEPES (pH 8.0) and buffer-exchanged into PBS (pH 7.0).
Murine antibodies 13G8, 5A5, 6B12, 10E11, 7F5, 8F10, 11F6, 12A9, 30F7, 13G5, 1H6, 3E3, and 11E7 were obtained from BEI Resources. c13G8 (chimeric 13G8 with human constant regions) as well as Gc-targeting mAbs and DVDs used for animal challenges were constructed and purified at Mapp Biopharmaceutical. Antibodies were expressed transiently in ExpiCHO cells (Thermo Fisher Scientific) and purified from cell supernatants using a GE MabSelect SuRe LX protein A affinity chromatography column on an AKTA pure fast protein liquid chromatography (FPLC) system. The antibodies were eluted using a glycine elution buffer at pH 2.2 and neutralized with 2M Tris base to a pH of ~7.
Biolayer interferometry binding analysis
Biolayer interferometry (BLI) kinetic measurements were acquired at 25°C with a ForteBio Octet HTX instrument (Sartorius) using Phosphate-buffered saline with 0.1% BSA (PBSF). For binding analyses to rGn/Gc, IgG (100 nM in PBSF) were captured with anti-human IgG Fc capture (AHC) sensors (Sartorius). For binding experiments with Gc, IgG (100 nM in PBSF) were captured with with Protein A (ProA) sensors (biotinylated Protein A captured to a streptavidin sensor tip (ForteBio) (Sartorius). After capture of IgG, the sensors were soaked in PBSF for a minimum of 30 min. Baseline was recorded for 60 s in PBSF before dipping the sensors into the antigen solution (100 nM in PBSF) for 180s (association) followed by PBSF for 180s (dissociation). For binding studies with GP38, biotinylated GP38 (100 nM in PBSF) was loaded onto streptavidin sensors (ForteBio, Cat #: 18-5021), soaked in PBSF (30 min), exposed to IgG in solution (100 nM in PBSF, 180s), and then dipped into PBSF (dissociation, 180s). For binding studies with murine antibodies, either murine IgG was loaded onto anti-mouse Fc capture (AMC, ForteBio) sensors, or biotinylated antigen (Gn/Gc, Gc, or GP38) was loaded onto streptavidin sensors and the assays were performed as described above. Binding was determined by a response of > 0.1 nm upon exposure to the antigen or IgG. The binding curves were fit to a 1:1 binding model using the ForteBio Data Analysis Software version 11.1.3.10. Irrelevant, anti-hen egg lysozyme human IgG or murine IgG isotype controls were used as negative controls in these assays.
Antibody competition assays
Biotinylated CCHFV rGn/Gc IbAr10200 (5 nM, 15 nM, or 50 nM) was incubated with a twentyfold excess of anti-CCHFV Fab (100 nM, 300 nM, or 1 μM respectively) for 30 min at room temperature before mixing with yeast expressing monoclonal anti-CCHFV rGn/Gc IgG. After washing with PBSF to remove unbound antigen, the bound antigen was detected using streptavidin Alexa Fluor 633 (Invitrogen) and antibody light chain was detected using Goat F(ab’)2 anti-human kappa FITC and Goat F(ab’)2 anti-human lambda FITC (SouthernBiotech). The samples were analyzed by flow cytometry using FACSCanto II (BD Biosciences). The amount of antigen bound was normalized to the light chain FITC level. Competition level was determined by the fold reduction in normalized antigen binding in the presence of a competitor Fab compared to antigen binding in the absence of competition: Fold Reduction = (AF633 MFI/FITC MFI)No-competition / (AF633 MFI/FITC MFI)Competition. Antibodies with greater than tenfold reduction were considered to be in competition with the precomplexed Fab.
Display of CCHFV Gc on the surface of yeast
A plasmid was designed whereby CCHFV Gc (IbAr10200, Gc amino acid residues [from GenBank Accession: NC_005300] 1041-1579) was inserted into a plasmid with an N-terminal HA tag - G4S linker and C-terminal G4S linker - HA tag - (G4S)2 linker - Aga2. Yeast carrying the plasmid encoding CCHFV Gc (see below for transformation details) were grown overnight in synthetic complete (SC) media minus tryptophan, 4% dextrose, 0.1 M sodium phosphate, pH 6.3). The following day, yeast were inoculated into a new culture at a density of 0.2 OD. Once the yeast had grown to ~1.0 OD, the yeast were pelleted (2200 x g, 3 minutes) and the pellet was resuspended with the same volume of galactose-containing media (SC media minus tryptophan, 2% galactose, 2% raffinose, 0.01 M Sodium phosphate, 0.13 M NaCl, pH 6.0). The cultures were then shaken at 16 °C for 36-48 hours. After 36-48 hours, the yeast were pelleted and washed once with cold PBSF (PBS + 0.1% BSA) before staining for flow cytometry (see below).
CCHFV Gc library construction
Alanine Scanning.
Alanine scan libraries across the whole length of CCHFV IbAr10200 Gc were constructed by using pools of eight overlapping 60-mer oligonucleotides (Integrated DNA Technologies) each encoding sequential alanine mutations in the center of each oligonucleotide. Each pool was used to mutagenize eight sequential individual amino acids to alanine with the QuikChange Multi Site-Directed Mutagenesis Kit (Agilent) according to the manufacturer’s directions. The resulting plasmid pools were transformed into chemically competent E. coli in 96-well plates (NEB, Cat#C2987P). After overnight growth of E. coli, the plasmid pools were isolated by a 96-well miniprep kit (Macherey-Nagel, Cat#740616.4). Isolated plasmid pools were then transformed chemically as described above (Gietz and Woods. 2006). For sorting, pools of eight mutations from each well were combined to ~160 amino acid stretches (~20 pools, 4 alanine scanning libraries) for ease of downstream sequencing.
Random Mutagenesis.
As previously reported (Mata-Fink et al., 2013), PCR with an error-prone polymerase (Agilent, GeneMorph II Random Mutagenesis Kit) was used to generate a randomly mutagenized Gc library. Amplification of 500 ng of target DNA in 25 cycles was used to keep the mutation rate low. The resulting product was combined with digested plasmid and electroporated into yeast (strain EBY100, ATCC:MYA-4941), resulting in a maximal diversity of ~1.6 × 107 as determined by the number of colonies that grew on agar plates lacking tryptophan after electroporation.
Flow cytometric sorting of mutant Gc libraries
Negative Selection (first and second rounds).
Displayed wild-type IbAr10200 Gc was used as a control to draw the gates for negative selection of mutant libraries. All staining steps were performed on ice and in the dark when fluorophores were present. Wild-type Gc or Gc mutant libraries were stained with an anti-CCHFV rGn/Gc IgG at 10 nM in PBSF. After washing twice with PBSF, wild-type Gc or Gc mutant libraries were stained with an anti-HA APC antibody (BioLegend, Clone: 16B12, Dilution: 1:100) and Goat F(ab’)2 anti-human IgG PE (SouthernBiotech, Dilution: 1:100). Clones displaying loss-of-binding mutants were sorted and cultured in SC media containing dextrose (see above) for subsequent rounds of selectio.
Positive Selection (third round).
Wild-type Gc or Gc mutant libraries were stained with five non-competing antibodies and detected as described above. Displayed wild-type Gc was used as a control to draw the gates for the positive selection of mutant libraries. Mutant clones stained as similar levels relative to wild-type were sorted and plated on agar plates lacking tryptophan. Between 16-32 clones were picked for each library for sequencing. Validation of loss of binding (see below) was performed on clones that contained only single mutations.
Flow cytometric analysis of single Gc mutants
Yeast carrying plasmid encoding a single Gc mutation were induced in deep 96-well plates as described above. 36-48 hours after induction, the yeast were washed with PBSF and stained as described above at antibody concentrations of 1 nM or 10 nM. Bound antibody was detected as above. Antibody binding was normalized to the amount of HA tag expression on the surface of the yeast (PE signal / APC signal). Background was determined by staining with an irrelevant anti-hen egg lysozyme antibody followed by the same secondary stain. To determine loss of binding, the normalized binding score was compared to wild-type as a percentage for each antibody tested: (Mutant binding - Mutant background) / (Wild-type binding - Wild-type background) x 100%. Each Gc mutant clone was tested against a panel of antibodies targeting non-overlapping epitopes to ensure loss of binding was specific for a given antibody and antigenic site.
Homology modeling onto hantavirus Gc
Based on sequence and secondary structure alignment, a homology model for CCHFV Gc was obtained using the structure of Maporal virus Gc in prefusion conformation (PDB ID: 6y62) as a template via SWISS-MODEL in the expasy web server (Waterhouse et al., 2018).
Polyreactivity assay
Antibody polyreactivity was tested as described previously (Xu et al., 2013). 2 x 106 IgG-presenting yeast were pelleted in a 96-well plate before resuspension with biotinylated (ThermoFisher Scientific Cat #A39257) soluble membrane and cytosolic protein preparations from Chinese hamster ovary cells and incubated on ice for 20 min. Cells were washed twice with cold PBSF before staining with 50 μL of Extravadin-R-PE (Sigma), goat F(ab’)2 anti-human kappa-FITC (SouthernBiotech), goat F(ab’)2 anti-human lambda-FITC (SouthernBiotech), and propidium iodide (Sigma) and incubating on ice for 20 min. After two additional washes with cold PBSF, cells were resuspended in 50 μL of PBSF to be analyzed by a FACSCanto II (BD Biosciences) with an HTS sample injector. Polyreactivity was determined by the fluorescence intensity of the PE and FITC signals and normalized to three control antibodies with low, medium, and high levels of polyreactivity.
Generation of tecVLPs
Amino acid sequences for the IbAr10200, Oman, Kosova Hoti, and Turkey strain GPCs were derived from GenBank M-segment sequences with accession numbers NC_005300, KR864901, EU037902, and ALT31694, respectively. Transcription- and entry-competent virus-like particles bearing CCHFV Gn/Gc were generated as described by Zivcec and colleagues (Zivcec et al., 2015). Briefly, BSR-T7 cells were cultured in Dulbecco’s modified eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (Atlanta Biologicals), 1% penicillin-streptomycin (Life Technologies) and 1% GlutaMax (Life Technologies), prior to transfection with plasmids encoding the CCHFV nucleoprotein (NP), glycoprotein complex (GPC), polymerase (L), as well as T7 polymerase and a minigenome expressing Nano-Glo Luciferase. 10 h post-transfection, cells were washed with phosphate-buffered saline in order to remove residual Luciferase-expressing plasmid, before supplying fresh DMEM growth media. 48 h post-transfection, tecVLP-containing supernatants were collected, clarified by low-speed centrifugation, and finally pelleted by ultracentrifugation at 30,000 × g for 2.5 h. Pelleted tecVLPs were then resuspended in plain DMEM before storage at −80°C prior to use in any infection experiments.
Screening of mAb panel for neutralizing activity
Infectivity of Oman tecVLPs in the presence of CCHFV-specific mAbs was assessed in Vero cells, maintained in DMEM (Life Technologies) supplemented with 2% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 1% penicillin-streptomycin (Life Technologies) and 1% GlutaMax (Life Technologies). Prior to infection, a set amount of Oman tecVLPs was incubated with each mAb at concentrations of 35 or 350 nM for 1 h at 4°C. The amount of tecVLPs was determined empirically, in order to achieve a maximal luciferase signal in the target cells of >500-fold over background. Luciferase signal was assayed in target cells ~14 h post-infection using the Nano-Glo Luciferase assay system (Promega). The signal for each mAb tested was then normalized to a no antibody control.
Neutralization assays against tecVLPs carrying divergent Gn/Gc
Neutralization by select candidate mAbs against tecVLPs carrying divergent Gn/Gc were assessed in Vero cells, maintained as above. Antibodies were diluted to starting concentrations of 350, 175 or 125 nM, before making 3-fold dilutions in complete DMEM. Empirically determined amounts of CCHFV Gn/Gc VLPs, determined as above, were then added and incubated together with the antibodies for 1 h at 4°C. Antibody:tecVLP mixtures were then added to Vero cells and infection allowed to proceed for ~14 h before Nano-Glo Luciferase activity was assayed. Absolute or relative IC50 values for each mAb were derived from inhibition curves using non-linear regression (variable slope, 4 parameters; GraphPad Prism).
Synergistic neutralization and combination index analysis
In order to assess synergistic neutralization, mAbs were combined at a 1:1 molar ratio before performing neutralization assays as described above. Neutralization curves of individual mAbs, and the combinations thereof, were then used to derive combination index values in accordance with the process described for CompuSyn (Chou, 2006; Chou and Talalay, 1984). Briefly, values were log transformed and analyzed using simple linear regression. From the slope and median effect dose, CI values were then calculated using the Combination index equation.
Neutralization assays against authentic CCHFV
CCHFV-IbAr10200 was incubated with serial 3-fold dilutions of mAbs for 1 h at 37°C in a humidified incubator. The antibody-virus mixture was applied to monolayers of Vero-E6 cells in a 96-well plate at final multiplicity of infection of 3 and incubated for 1 hour at 37°C in a humidified incubator. Infection media was then removed, and cells were washed once with 1X PBS, followed by addition of fresh cell culture media. Culture media was removed 24 h post infection and cells were washed once with 1X PBS. PBS was removed and plates were submerged in formalin fixing solution, then permeabilized with 0.2% Triton-X for 10 min at room temp and treated with blocking solution. Infected cells were detected by sequential incubation with CCHFV-specific antibody 9D5 (BEI, NR40270) and secondary detection antibody (goat anti-mouse) conjugated to AlexaFluor 488. Infected cells were enumerated using Operetta high content imaging instrument and data analysis was performed using the Harmony software (Perkin Elmer). IC50 values for each mAb were derived from inhibition curves using nonlinear regression analysis (variable slope, 4 parameters; GraphPad Prism).
DVD-Ig cloning and expression
pMAZ-IgH and pMAZ-IgL vectors encoding DVD-Igs were subcloned with outer variable domains for both the heavy and light chains N-terminally linked via “ASTKGP” and “TVAAP” linkers, respectively, as previously described (Wu et al., 2007). All sequences were confirmed by Sanger sequencing. Heavy and light chain vectors were co-transfected into ExpiCHO-STM cells (Gibco) as per manufacturer’s instructions. Cells were incubated in a humidified shaking incubator (37°C, 8% CO2, 125 rpm), fed on day 1 and harvested on day 8 according to manufacturer’s standard protocol. Clarified supernatant (4000 rpm, 15 min) was incubated with 1ml protein A resin (GoldBio) for 2 h at 4°C. Antibodies were purified using the Gentle Antibody Elution System (Thermo Scientific) and buffer exchanged into 150 mM HEPES [pH7.4], 200 mM NaCl.
Biolayer interferometry of DVD-Igs
Binding properties of DVD-Igs were determined using the OctetRed™ system (ForteBio). Parental IgG was initially loaded onto anti-human IgG Fc biosensors (ForteBio). Biosensors were then allowed to equilibrate in solution containing CCHFV (IbAr10200 strain) rGn/Gc before being transferred into a solution with DVD-Ig.
Murine challenge studies
Prophylactic Turkey/2004 Study:
4- to 8-week-old female 129S6/SvEv-Stat1tm1Rds (STAT-1−/−) mice (Taconic) were exposed to CCHFV-Turkey2004 through intraperitoneal (IP) injection of viral stocks diluted in Hanks balanced salts medium with 2% fetal bovine serum (FBS) for a final challenge dose of 100 PFU. Thirty min after challenge, all treatment groups were administered 0.25 mg/mouse of the indicated mAb IP. Animals were then observed for clinical scoring, temperature, and weight change. Mice were monitored daily and euthanized upon reaching institutionally approved endpoint score criteria or study endpoint (28 days postinfection).
Prophylactic IbAr10200 Study:
5–8-week old male and female Type 1 interferon α/β receptor knockout mice (Type 1 IFNα/β R−/−) (Jackson Labs) were treated IP with 1 mg of indicated mAb or mAb cocktail or an equivalent volume (200 μL) of phosphate buffered saline (PBS) vehicle. Twenty-four h post-treatment, mice were exposed IP to 100 plaque forming units (pfu) of CCHFV-IbAr10200. Animals were observed daily for clinical signs of disease and morbidity for 28 days. Daily observations were increased to a minimum of twice daily while mice were exhibiting signs of disease. Moribund mice were humanely euthanized on the basis of IACUC-approved criteria.
Therapeutic IbAr10200 Study:
6–9-week old male and female Type 1 interferon α/β receptor knockout mice (Type 1 IFNα/β R−/−) (Jackson Labs) were exposed IP to 100 plaque forming units (pfu) of CCHFV-IbAr10200. Mice were treated IP with 1 mg of indicated mAb or mAb cocktail, molar equivalent dose of bsAb, or an equivalent volume (200 μL) of phosphate buffered saline (PBS) vehicle 24 h post-exposure. Animals were observed daily for clinical signs of disease and morbidity for 28 days. Daily observations were increased to a minimum of twice daily while mice were exhibiting signs of disease. Moribund mice were humanely euthanized on the basis of IACUC-approved criteria.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details for each experiment can be found in the respective figure legends and in the results section. These include the number of replicates (n), measures of precision, and the statistical test used. Levels of somatic hypermutation in figure 1 were compared using a Mann-Whitney test. Dose-response neutralization curves were analyzed using a 4 parameter logistic model. Survival curves in figures 7 and 8 were compared by a Mantel-Cox (log-rank) test. All statistical analyses were carried out in GraphPad Prism.
Supplementary Material
Supplemental Figure Legends
Figure S1. Generation of a recombinant Gn/Gc bait protein for B cell sorting, related to Figure 1. (A) Schematic depicting the wild-type IbAr10200 CCHFV glycoprotein precursor (top) and the designed recombinant Gn/Gc protein (bottom). Numbers correspond to full-length glycoprotein amino acid precursor numbering. Ectodomains, transmembrane (TM) domains, and endodomains were predicted using the TMHMM server v. 2.0. (B) ELISA data showing sera reactivity towards rGn/Gc from convalescent CCHF donors. (C) Apparent KD measurements from BLI-based binding assays using a panel of mouse mAbs that target GP38 or Gc. SA = streptavidin; AMC = anti-mouse IgG Fc capture. (D) SDS-PAGE analysis of rGnGc under non-reduced (left) and reduced (right) conditions. (E) Sequence of rGn/Gc with peptides identified by LC-MS highlighted in red. (F) Representative gating strategy to sort single B cells specific for rGn/Gc. (G) Flow-cytometric analysis of rGn/Gc-specific B cells for IgM, IgD and CD27.
Figure S2. Binding specificities of non-Gc-reactive mAbs, related to Figure 2. (A) Response units from BLI-based binding assays using in-house produced Gn, commercially produced Gn, and GP38 with a panel of mAbs that bind rGn/Gc, but not Go. ADI-36121, which binds Go, is included as a control, n.d. = no data. (B) Matrices showing the conservation levels (percent identity determined by Clustal2.1) of the M segment (full-length glycoprotein precursor, left) or Gc ectodomain (IbAr10200 glycoprotein precursor amino acid residues 1041–1598) (right). (C) Analysis of cross-reactivity, ADI-36120 competition, and Vλ3-21 germline usage from the panel of mAbs shown in panel A. (D) CDR sequences of cross-reactive, GP38-specific mAbs with the Vλ3-21 germline. Red letters indicate amino acids that are predicted to be mutated from germline.
Figure S3. Characteristics of Site 2-targeting mAbs, related to Figure 3. (A) Cross-competition of the mAbs used to identify antigenic sites. (B) VH germline gene usage broken down by targeted Gc antigenic site. (C) Germline gene pairing of mAbs targeting Site 2. (D) CDRH3 amino acid lengths comparing Site 2-targeting mAbs to other Gc-specific mAbs. (E) Comparison of CDRH3 amino acid lengths. Statistical comparison was performed using the Mann-Whitney test (**** P < 0.0001, ** P < 0.01). Bars represent median CDRH3 amino acid length. (F) Polyreactivity scores between Site-2 targeting mAbs and all others. Statistical comparison was performed using Fisher’s exact test (**** P < 0.0001). (G) CDR amino acid sequences of mAbs sharing the VH3-21/VK1-27 germline gene pair from all four donors. Red letters indicate amino acids that are predicted to be mutated from germline.
Figure S4. Epitope mapping using yeast surface display, related to Figure 3.
(A) A representative example of the yeast display selection strategy to map the epitope of ADI-36122 is shown. In the first and second rounds, yeast mutants were selected for loss of binding for an antibody of interest. In the third round, a pool of antibodies that do not compete with the antibody of interest were used to select mutants that maintain proper folding of other epitopes. Wild-type Gc was used as a control for gating purposes. (B) Extended version of Figure 3D to include all antibodies used in selections and all isolated single mutants. Darker shades indicate a greater loss of binding relative to wild-type. (C) On-yeast titrations of the mAbs used to define antigenic sites demonstrating specific loss of binding. Data is shown as the ratio of antibody binding to displayed Gc. (D) Biolayer interferometry sensor-grams showing the binding kinetics of CCHFV Gc DIII to ADI-36145 IgG.
Figure S5. Breadth of neutralization for candidate nAbs, related to Figure 4. (A-R) Individual neutralization curves against IbAr10200, Oman, Kosova Hoti, and Turkey tecVLPs for the indicated mAbs. All tecVLP neutralization assays were performed using Vero target cells, n = 6, from two independent experiments for all neutralization curves.
Figure S6. Synergistic neutralization by Site 1-targeting mAbs, related to Figure 5. (A-C) Neutralization of Oman tecVLPs by the indicated nAbs and their 1:1 combinations. (D-F) Combination index (CI) analyses of the neutralization curves in panels A-C. Synergistic neutralization was assayed using Vero target cells, n = 6, from two independent experiments for all neutralization curves.
Figure S7. Biophysical and binding properties of dual-variable domain Igs (DVD-Igs), related to Figure 7. (A-D) Size-exclusion chromatography traces of the indicated DVD-Igs (E-J) Two-phase binding experiments for the DVD-Igs by BLI. Each probe bearing the indicated IgG was sequentially dipped in analyte solutions containing rGn/Gc and then the indicated DVD-Ig. In panels I and J, the presence of both parental mAbs block binding by the corresponding DVD-Ig.
Highlights (each 85 chars or less including spaces):
361 monoclonal antibodies against CCHFV glycoproteins isolated from human survivors
Potent and broad neutralizers targeting six antigenic sites in Gc identified
Specific combinations of non-competing antibodies afford synergistic neutralization
Bispecific antibody combining synergistic antibodies confers therapeutic protection
Acknowledgements
This manuscript is dedicated to the memory of our dear colleague and co-author, Dr. Leslie Lobel, whose vision and energy helped to initiate and drive this project. We thank Isabel Gutierrez, Estefania Valencia, and Laura Polanco for laboratory management and technical assistance. All IgGs, but not bsAbs, were sequenced by Adimab’s Molecular Core, and yeast-expressed mAbs and Fabs were produced by Adimab’s High Throughput Expression group. BLI binding experiments with IgGs were performed by Adimab’s Protein Analytics group. We thank Scott J. Garforth and the Macromolecular Therapeutics Development Facility for their support with size-exclusion chromatographic analyses of the DVD-Igs. We thank Christina F Spiropoulou, Éric Bergeron, and Marko Zivcec at the Centers for Disease Control and Prevention for kindly providing the plasmids and protocols necessary to generate tecVLPs. We thank members of all of our groups and the Prometheus consortium for their feedback on preliminary versions of the manuscript. Research was supported by NIAID of the National Institutes of Health under award number U19AI142777 (Centers of Excellence in Translational Research) to K.C., L.M.W., J.M.D., Z.A.B., J.R.L., A.S.H., and L.Z. Z.A.B, D.M.A, C.L.M, L.Z., R.W.C. and T.W.G were also supported by NIH grant R01AI132246. A.S.W. was also supported by NIH training grant T32AI070117 to the Albert Einstein College of Medicine. E.K.N. was also supported by a DAAD (Deutscher Akademischer Austauschdienst, German Academic Exchange Service) fellowship. S.S. was supported by the Leukemia Research Foundation for the Hollis Brownstein New Investigator Research Grant, AFAR for the Sagol Network GerOmics award for aging research, Einstein-Montefiore for the SARS-CoV-2 (COVID-19) grant, and the Basic Biology of Aging 2020 award sponsored by the Nathan Shock Institute for Aging Research. F.A.R. acknowledges funding from Institut Pasteur, CNRS and Labex IBEID (grant ANR-10-IHUB-0002), as well as support of JH via the GIS IBiSA (Infrastructures en biologie santé et agronomie), grant ANR-13-ISV8-0002-01 and the Région Ile de France. The content is solely the responsibility of the authors and does not necessarily represent the official views of our institutions or funders.
Footnotes
Conflicts of Interest
K.C. is a scientific advisory board member of Integrum Scientific, LLC and Biovaxys Technology Corporation, LLC. K.C. and J.R.L. are scientific advisory board members of the Pandemic Security Initiative of Celdara Medical, LLC. N.T.P. and L.M.W. are employees and shareholders of Adimab, LLC. D.P.M. is a shareholder of Adimab, LLC. Z.A.B, D.M.A, C.L.M, and L.Z. are shareholders and employees of Mapp Biopharmaceutical, Inc. Mapp Biopharmaceutical, Inc. has filed for a patent related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ADDITIONAL RESOURCES
None.
Bibliography
- Al-Abri SS, Hewson R, Al-Kindi H, Al-Abaidani I, Al-Jardani A, Al-Maani A, Almahrouqi S, Atkinson B, Al-Wahaibi A, Al-Rawahi B, et al. (2019). Clinical and molecular epidemiology of Crimean-Congo hemorrhagic fever in Oman. PLoS Negl. Trop. Dis 13, e0007100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SF, Chambers MJ, Schramm CA, Plyler J, Raab JE, Kanekiyo M, Gillespie RA, Ransier A, Darko S, Hu J, et al. (2019). Activation Dynamics and Immunoglobulin Evolution of Pre-existing and Newly Generated Human Memory B cell Responses to Influenza Hemagglutinin. Immunity 51, 398–410.e5. [DOI] [PubMed] [Google Scholar]
- Austin SK, Dowd KA, Shrestha B, Nelson CA, Edeling MA, Johnson S, Pierson TC, Diamond MS, and Fremont DH (2012). Structural basis of differential neutralization of DENV-1 genotypes by an antibody that recognizes a cryptic epitope. PLoS Pathog. 8, e1002930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramello M, Williams KL, Simmons CP, Macagno A, Simonelli L, Quyen NTH, Sukupolvi-Petty S, Navarro-Sanchez E, Young PR, de Silva AM, et al. (2010). The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 8, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bente DA, Alimonti JB, Shieh W-J, Camus G, Ströher U, Zaki S, and Jones SM (2010). Pathogenesis and immune response of Crimean-Congo hemorrhagic fever virus in a STAT-1 knockout mouse model. J. Virol 84, 11089–11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bente DA, Forrester NL, Watts DM, McAuley AJ, Whitehouse CA, and Bray M (2013). Crimean-Congo hemorrhagic fever: history, epidemiology, pathogenesis, clinical syndrome and genetic diversity. Antiviral Res. 100, 159–189. [DOI] [PubMed] [Google Scholar]
- Bereczky S, Lindegren G, Karlberg H, Akerström S, Klingström J, and Mirazimi A (2010). Crimean-Congo hemorrhagic fever virus infection is lethal for adult type I interferon receptor-knockout mice. J. Gen. Virol 91, 1473–1477. [DOI] [PubMed] [Google Scholar]
- Bertolotti-Ciarlet A, Smith J, Strecker K, Paragas J, Altamura LA, McFalls JM, Frias-Stäheli N, García-Sastre A, Schmaljohn CS, and Doms RW (2005). Cellular localization and antigenic characterization of crimean-congo hemorrhagic fever virus glycoproteins. J. Virol 79, 6152–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom DE, Black S, and Rappuoli R (2017). Emerging infectious diseases: A proactive approach. Proc Natl Acad Sci USA 114, 4055–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornholdt ZA, Turner HL, Murin CD, Li W, Sok D, Souders CA, Piper AE, Goff A, Shamblin JD, Wollen SE, et al. (2016). Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 351, 1078–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornholdt ZA, Herbert AS, Mire CE, He S, Cross RW, Wec AZ, Abelson DM, Geisbert JB, James RM, Rahim MN, et al. (2019). A Two-Antibody Pan-Ebolavirus Cocktail Confers Broad Therapeutic Protection in Ferrets and Nonhuman Primates. Cell Host Microbe 25, 49–58.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowick GC, Airo AM, and Bente DA (2012). Expression of interferon-induced antiviral genes is delayed in a STAT1 knockout mouse model of Crimean-Congo hemorrhagic fever. Virol. J 9, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SA, Bird BH, Rollin PE, and Nichol ST (2010). Ancient common ancestry of Crimean-Congo hemorrhagic fever virus. Mol. Phylogenet. Evol 55, 1103–1110. [DOI] [PubMed] [Google Scholar]
- Caskey M, Klein F, Lorenzi JCC, Seaman MS, West AP, Buckley N, Kremer G, Nogueira L, Braunschweig M, Scheid JF, et al. (2015). Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature 522, 487–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrier MV, Kaufmann B, Nybakken GE, Lok S-M, Warren JT, Chen BR, Nelson CA, Kostyuchenko VA, Holdaway HA, Chipman PR, et al. (2009). Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J. 28, 3269–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou T-C (2006). Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev 58, 621–681. [DOI] [PubMed] [Google Scholar]
- Chou T-C, and Talalay P (1984). Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul 22, 27–55. [DOI] [PubMed] [Google Scholar]
- Conger NG, Paolino KM, Osborn EC, Rusnak JM, Günther S, Pool J, Rollin PE, Allan PF, Schmidt-Chanasit J, Rieger T, et al. (2015). Health care response to CCHF in US soldier and nosocomial transmission to health care providers, Germany, 2009. Emerging Infect. Dis 21, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia BE, Bates JT, Loomis RJ, Baneyx G, Carrico C, Jardine JG, Rupert P, Correnti C, Kalyuzhniy O, Vittal V, et al. (2014). Proof of principle for epitope-focused vaccine design. Nature 507, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti D, Zhao J, Pedotti M, Simonelli L, Agnihothram S, Fett C, Fernandez-Rodriguez B, Foglierini M, Agatic G, Vanzetta F, et al. (2015). Prophylactic and postexposure efficacy of a potent human monoclonal antibody against MERS coronavirus. Proc Natl Acad Sci USA 112, 10473–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crill WD, Trainor NB, and Chang G-JJ (2007). A detailed mutagenesis study of flavivirus cross-reactive epitopes using West Nile virus-like particles. J. Gen. Virol 88, 1169–1174. [DOI] [PubMed] [Google Scholar]
- Cross RW, Prasad AN, Borisevich V, Geisbert JB, Agans KN, Deer DJ, Fenton KA, and Geisbert TW (2020). Crimean-Congo hemorrhagic fever virus strains Hoti and Afghanistan cause viremia and mild clinical disease in cynomolgus monkeys. PLoS Negl. Trop. Dis 14, e0008637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CW, Jackson KJL, McElroy AK, Halfmann P, Huang J, Chennareddy C, Piper AE, Leung Y, Albariño CG, Crozier I, et al. (2019). Longitudinal analysis of the human B cell response to ebola virus infection. Cell 177, 1566–1582.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejnirattisai W, Jumnainsong A, Onsirisakul N, Fitton P, Vasanawathana S, Limpitikul W, Puttikhunt C, Edwards C, Duangchinda T, Supasa S, et al. (2010). Cross-reacting antibodies enhance dengue virus infection in humans. Science 328, 745–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Benedictis P, Minola A, Rota Nodari E, Aiello R, Zecchin B, Salomoni A, Foglierini M, Agatic G, Vanzetta F, Lavenir R, et al. (2016). Development of broad-spectrum human monoclonal antibodies for rabies post-exposure prophylaxis. EMBO Mol. Med 8, 407–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamant E, Torgeman A, Ozeri E, and Zichel R (2015). Monoclonal Antibody Combinations that Present Synergistic Neutralizing Activity: A Platform for Next-Generation Anti-Toxin Drugs. Toxins (Basel) 7, 1854–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd KA, DeMaso CR, and Pierson TC (2015). Genotypic differences in dengue virus neutralization are explained by a single amino acid mutation that modulates virus breathing. MBio 6, e01559–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBois RM, Vaney M-C, Tortorici MA, Kurdi RA, Barba-Spaeth G, Krey T, and Rey FA (2013). Functional and evolutionary insight from the crystal structure of rubella virus protein E1. Nature 493, 552–556. [DOI] [PubMed] [Google Scholar]
- Duehr J, McMahon M, Williamson B, Amanat F, Durbin A, Hawman DW, Noack D, Uhl S, Tan GS, Feldmann H, et al. (2020). Neutralizing Monoclonal Antibodies against the Gn and the Gc of the Andes Virus Glycoprotein Spike Complex Protect from Virus Challenge in a Preclinical Hamster Model. MBio 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, and Woods RA (2006). Yeast transformation by the LiAc/SS Carrier DNA/PEG method. Methods Mol. Biol 313, 107–120. [DOI] [PubMed] [Google Scholar]
- Ergonul O, Celikbas A, Baykam N, Eren S, and Dokuzoguz B (2006). Analysis of risk-factors among patients with Crimean-Congo haemorrhagic fever virus infection: severity criteria revisited. Clin. Microbiol. Infect 12, 551–554. [DOI] [PubMed] [Google Scholar]
- Gilchuk P, Murin CD, Milligan JC, Cross RW, Mire CE, Ilinykh PA, Huang K, Kuzmina N, Altman PX, Hui S, et al. (2020). Analysis of a therapeutic antibody cocktail reveals determinants for cooperative and broad ebolavirus neutralization. Immunity 52, 388–403.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman MSA, Castellanos CA, Chen M, Ngwuta JO, Goodwin E, Moin SM, Mas V, Melero JA, Wright PF, Graham BS, et al. (2016). Rapid profiling of RSV antibody repertoires from the memory B cells of naturally infected adult donors. Sci. Immunol 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden JW, Shoemaker CJ, Lindquist ME, Zeng X, Daye SP, Williams JA, Liu J, Coffin KM, Olschner S, Flusin O, et al. (2019). GP38-targeting monoclonal antibodies protect adult mice against lethal Crimean-Congo hemorrhagic fever virus infection. Sci. Adv 5, eaaw9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin E, Gilman MSA, Wrapp D, Chen M, Ngwuta JO, Moin SM, Bai P, Sivasubramanian A, Connor RI, Wright PF, et al. (2018). Infants Infected with Respiratory Syncytial Virus Generate Potent Neutralizing Antibodies that Lack Somatic Hypermutation. Immunity 48, 339–349.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardado-Calvo P, Bignon EA, Stettner E, Jeffers SA, Pérez-Vargas J, Pehau-Arnaudet G, Tortorici MA, Jestin J-L, England P, Tischler ND, et al. (2016). Mechanistic Insight into Bunyavirus-induced Membrane Fusion from Structure-Function Analyses of the Hantavirus Envelope Glycoprotein Gc. PLoS Pathog. 12, e1005813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddock E, Feldmann F, Hawman DW, Zivcec M, Hanley PW, Saturday G, Scott DP, Thomas T, Korva M, Avšič-Županc T, et al. (2018). A cynomolgus macaque model for Crimean-Congo haemorrhagic fever. Nat. Microbiol 3, 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao M, Zhang G, Zhang S, Chen Z, Chi X, Dong Y, Fan P, Liu Y, Chen Y, Song X, et al. (2020). Characterization of Two Neutralizing Antibodies against Rift Valley Fever Virus Gn Protein. Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellert J, Aebischer A, Wernike K, Haouz A, Brocchi E, Reiche S, Guardado-Calvo P, Beer M, and Rey FA (2019). Orthobunyavirus spike architecture and recognition by neutralizing antibodies. Nat. Commun 10, 879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogstraal H (1979). The epidemiology of tick-borne Crimean-Congo hemorrhagic fever in Asia, Europe, and Africa. J. Med. Entomol 15, 307–417. [DOI] [PubMed] [Google Scholar]
- Jakob CG, Edalji R, Judge RA, DiGiammarino E, Li Y, Gu J, and Ghayur T (2013). Structure reveals function of the dual variable domain immunoglobulin (DVD-Ig™) molecule. MAbs 5, 358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S, Henschke N, Maayan N, Mills I, Buckley BS, Kakourou A, and Marshall R (2018). Ribavirin for treating Crimean Congo haemorrhagic fever. Cochrane Database Syst. Rev 6, CD012713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeffe JR, Van Rompay KKA, Olsen PC, Wang Q, Gazumyan A, Azzopardi SA, Schaefer-Babajew D, Lee YE, Stuart JB, Singapuri A, et al. (2018). A Combination of Two Human Monoclonal Antibodies Prevents Zika Virus Escape Mutations in Non-human Primates. Cell Rep. 25, 1385–1394.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F, Halper-Stromberg A, Horwitz JA, Gruell H, Scheid JF, Bournazos S, Mouquet H, Spatz LA, Diskin R, Abadir A, et al. (2012). HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature 492, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortekaas J, Vloet RPM, McAuley AJ, Shen X, Bosch BJ, de Vries L, Moormann RJM, and Bente DA (2015). Crimean-Congo Hemorrhagic Fever Virus Subunit Vaccines Induce High Levels of Neutralizing Antibodies But No Protection in STAT1 Knockout Mice. Vector Borne Zoonotic Dis. 15, 759–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, et al. (2002). Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108, 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Wengler G, and Rey FA (2001). The fusion glycoprotein shell of semliki forest virus. Cell 105, 137–148. [DOI] [PubMed] [Google Scholar]
- Li A, Katinger H, Posner MR, Cavacini L, Zolla-Pazner S, Gorny MK, Sodroski J, Chou TC, Baba TW, and Ruprecht RM (1998). Synergistic neutralization of simian-human immunodeficiency virus SHIV-vpu+ by triple and quadruple combinations of human monoclonal antibodies and high-titer anti-human immunodeficiency virus type 1 immunoglobulins. J. Virol 72, 3235–3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok S-M, Kostyuchenko V, Nybakken GE, Holdaway HA, Battisti AJ, Sukupolvi-Petty S, Sedlak D, Fremont DH, Chipman PR, Roehrig JT, et al. (2008). Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat. Struct. Mol. Biol 15, 312–317. [DOI] [PubMed] [Google Scholar]
- Lukashev AN, Klimentov AS, Smirnova SE, Dzagurova TK, Drexler JF, and Gmyl AP (2016). Phylogeography of Crimean congo hemorrhagic fever virus. PLoS ONE 11, e0166744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata-Fink J, Kriegsman B, Yu HX, Zhu H, Hanson MC, Irvine DJ, and Wittrup KD (2013). Rapid conformational epitope mapping of anti-gp120 antibodies with a designed mutant panel displayed on yeast. J. Mol. Biol 425, 444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehand MS, Millett P, Al-Shorbaji F, Roth C, Kieny MP, and Murgue B (2018). World health organization methodology to prioritize emerging infectious diseases in need of research and development. Emerging Infect. Dis 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messina JP, Pigott DM, Golding N, Duda KA, Brownstein JS, Weiss DJ, Gibson H, Robinson TP, Gilbert M, William Wint GR, et al. (2015). The global distribution of Crimean-Congo hemorrhagic fever. Trans. R. Soc. Trop. Med. Hyg 109, 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Meulen J, van den Brink EN, Poon LLM, Marissen WE, Leung CSW, Cox F, Cheung CY, Bakker AQ, Bogaards JA, van Deventer E, et al. (2006). Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 3, e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mire CE, Cross RW, Geisbert JB, Borisevich V, Agans KN, Deer DJ, Heinrich ML, Rowland MM, Goba A, Momoh M, et al. (2017). Human-monoclonal-antibody therapy protects nonhuman primates against advanced Lassa fever. Nat. Med 23, 1146–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra AK, Moyer CL, Abelson DM, Deer DJ, El Omari K, Duman R, Lobel L, Lutwama JJ, Dye JM, Wagner A, et al. (2020). Structure and Characterization of Crimean-Congo Hemorrhagic Fever Virus GP38. J. Virol 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulangu S, Dodd LE, Davey RT, Tshiani Mbaya O, Proschan M, Mukadi D, Lusakibanza Manzo M, Nzolo D, Tshomba Oloma A, Ibanda A, et al. (2019). A randomized, controlled trial of ebola virus disease therapeutics. N. Engl. J. Med 381, 2293–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz JA, Malonis RJ, Thackray LB, Cohen CA, Pallesen J, Jangra RK, Brown RS, Hofmann D, Holtsberg FW, Shulenin S, et al. (2019). Human monoclonal antibodies against chikungunya virus target multiple distinct epitopes in the E1 and E2 glycoproteins. PLoS Pathog. 15, e1008061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappuoli R, Bottomley MJ, D’Oro U, Finco O, and De Gregorio E (2016). Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. J. Exp. Med 213, 469–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissanen I, Stass R, Krumm SA, Seow J, Hulswit RJG, Paesen GC, Hepojoki J, Vapalahti O, Lundkvist Å, Reynard O, et al. (2020). Molecular rationale for hantavirus neutralization by a reservoir host-derived monoclonal antibody. BioRxiv. [Google Scholar]
- Rogers TF, Goodwin EC, Briney B, Sok D, Beutler N, Strubel A, Nedellec R, Le K, Brown ME, Burton DR, et al. (2017). Zika virus activates de novo and cross-reactive memory B cell responses in dengue-experienced donors. Sci. Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouvinski A, Dejnirattisai W, Guardado-Calvo P, Vaney M-C, Sharma A, Duquerroy S, Supasa P, Wongwiwat W, Haouz A, Barba-Spaeth G, et al. (2017). Covalently linked dengue virus envelope glycoprotein dimers reduce exposure of the immunodominant fusion loop epitope. Nat. Commun 8, 15411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serris A, Stass R, Bignon EA, Muena NA, Manuguerra J-C, Jangra RK, Li S, Chandran K, Tischler ND, Huiskonen JT, et al. (2020). The hantavirus surface glycoprotein lattice and its fusion control mechanism. Cell 183, 442–456.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesterhenn F, Yang C, Bonet J, Cramer JT, Wen X, Wang Y, Chiang C-I, Abriata LA, Kucharska I, Castoro G, et al. (2020). De novo protein design enables the precise induction of RSV-neutralizing antibodies. Science 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd AJ, Swanepoel R, and Leman PA (1989). Antibody response in Crimean-Congo hemorrhagic fever. Rev. Infect. Dis 11 Suppl 4, S801–6. [DOI] [PubMed] [Google Scholar]
- Smego RA, Sarwari AR, and Siddiqui AR (2004). Crimean-Congo hemorrhagic fever: prevention and control limitations in a resource-poor country. Clin. Infect. Dis 38, 1731–1735. [DOI] [PubMed] [Google Scholar]
- Spengler JR, Bente DA, Bray M, Burt F, Hewson R, Korukluoglu G, Mirazimi A, Weber F, and Papa A (2018). Second International Conference on Crimean-Congo Hemorrhagic Fever. Antiviral Res. 150, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler JR, Bergeron É, and Spiropoulou CF (2019). Crimean-Congo hemorrhagic fever and expansion from endemic regions. Curr. Opin. Virol 34, 70–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steichen JM, Lin Y-C, Havenar-Daughton C, Pecetta S, Ozorowski G, Willis JR, Toy L, Sok D, Liguori A, Kratochvil S, et al. (2019). A generalized HIV vaccine design strategy for priming of broadly neutralizing antibody responses. Science 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stettler K, Beltramello M, Espinosa DA, Graham V, Cassotta A, Bianchi S, Vanzetta F, Minola A, Jaconi S, Mele F, et al. (2016). Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353, 823–826. [DOI] [PubMed] [Google Scholar]
- The IMpact-RSV Study Group (1998). Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. The IMpact-RSV Study Group. Pediatrics 102, 531–537. [PubMed] [Google Scholar]
- Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, and Wardemann H (2008). Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J. Immunol. Methods 329, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss JE, Vaney M-C, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, and Rey FA (2010). Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468, 709–712. [DOI] [PubMed] [Google Scholar]
- Wang Q, Ma T, Wu Y, Chen Z, Zeng H, Tong Z, Gao F, Qi J, Zhao Z, Chai Y, et al. (2019). Neutralization mechanism of human monoclonal antibodies against Rift Valley fever virus. Nat. Microbiol 4, 1231–1241. [DOI] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster RG, and Laver WG (1980). Determination of the number of nonoverlapping antigenic areas on Hong Kong (H3N2) influenza virus hemagglutinin with monoclonal antibodies and the selection of variants with potential epidemiological significance. Virology 104, 139–148. [DOI] [PubMed] [Google Scholar]
- Wec AZ, Nyakatura EK, Herbert AS, Howell KA, Holtsberg FW, Bakken RR, Mittler E, Christin JR, Shulenin S, Jangra RK, et al. (2016). A “Trojan horse” bispecific-antibody strategy for broad protection against ebolaviruses. Science 354, 350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wec AZ, Bornholdt ZA, He S, Herbert AS, Goodwin E, Wirchnianski AS, Gunn BM, Zhang Z, Zhu W, Liu G, et al. (2019). Development of a Human Antibody Cocktail that Deploys Multiple Functions to Confer Pan-Ebolavirus Protection. Cell Host Microbe 25, 39–48.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wec AZ, Haslwanter D, Abdiche YN, Shehata L, Pedreño-Lopez N, Moyer CL, Bornholdt ZA, Lilov A, Nett JH, Jangra RK, et al. (2020a). Longitudinal dynamics of the human B cell response to the yellow fever 17D vaccine. Proc Natl Acad Sci USA 117, 6675–6685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wec AZ, Wrapp D, Herbert AS, Maurer DP, Haslwanter D, Sakharkar M, Jangra RK, Dieterle ME, Lilov A, Huang D, et al. (2020b). Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, McCarthy D, Zhu R-R, Santora L, et al. (2007). Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol 25, 1290–1297. [DOI] [PubMed] [Google Scholar]
- Xu Y, Roach W, Sun T, Jain T, Prinz B, Yu T-Y, Torrey J, Thomas J, Bobrowicz P, Vásquez M, et al. (2013). Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng. Des. Sel 26, 663–670. [DOI] [PubMed] [Google Scholar]
- Zhang X, Sheng J, Plevka P, Kuhn RJ, Diamond MS, and Rossmann MG (2013). Dengue structure differs at the temperatures of its human and mosquito hosts. Proc Natl Acad Sci USA 110, 6795–6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivcec M, Safronetz D, Scott D, Robertson S, Ebihara H, and Feldmann H (2013). Lethal Crimean-Congo hemorrhagic fever virus infection in interferon α/β receptor knockout mice is associated with high viral loads, proinflammatory responses, and coagulopathy. J. Infect. Dis 207, 1909–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivcec M, Metcalfe MG, Albariño CG, Guerrero LW, Pegan SD, Spiropoulou CF, and Bergeron É (2015). Assessment of Inhibitors of Pathogenic Crimean-Congo Hemorrhagic Fever Virus Strains Using Virus-Like Particles. PLoS Negl. Trop. Dis 9, e0004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivcec M, Scholte FEM, Spiropoulou CF, Spengler JR, and Bergeron É (2016). Molecular Insights into Crimean-Congo Hemorrhagic Fever Virus. Viruses 8, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivcec M, Guerrero LIW, Albariño CG, Bergeron É, Nichol ST, and Spiropoulou CF (2017). Identification of broadly neutralizing monoclonal antibodies against Crimean-Congo hemorrhagic fever virus. Antiviral Res. 146, 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwick MB, Wang M, Poignard P, Stiegler G, Katinger H, Burton DR, and Parren PW (2001). Neutralization synergy of human immunodeficiency virus type 1 primary isolates by cocktails of broadly neutralizing antibodies. J. Virol 75, 12198–12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure Legends
Figure S1. Generation of a recombinant Gn/Gc bait protein for B cell sorting, related to Figure 1. (A) Schematic depicting the wild-type IbAr10200 CCHFV glycoprotein precursor (top) and the designed recombinant Gn/Gc protein (bottom). Numbers correspond to full-length glycoprotein amino acid precursor numbering. Ectodomains, transmembrane (TM) domains, and endodomains were predicted using the TMHMM server v. 2.0. (B) ELISA data showing sera reactivity towards rGn/Gc from convalescent CCHF donors. (C) Apparent KD measurements from BLI-based binding assays using a panel of mouse mAbs that target GP38 or Gc. SA = streptavidin; AMC = anti-mouse IgG Fc capture. (D) SDS-PAGE analysis of rGnGc under non-reduced (left) and reduced (right) conditions. (E) Sequence of rGn/Gc with peptides identified by LC-MS highlighted in red. (F) Representative gating strategy to sort single B cells specific for rGn/Gc. (G) Flow-cytometric analysis of rGn/Gc-specific B cells for IgM, IgD and CD27.
Figure S2. Binding specificities of non-Gc-reactive mAbs, related to Figure 2. (A) Response units from BLI-based binding assays using in-house produced Gn, commercially produced Gn, and GP38 with a panel of mAbs that bind rGn/Gc, but not Go. ADI-36121, which binds Go, is included as a control, n.d. = no data. (B) Matrices showing the conservation levels (percent identity determined by Clustal2.1) of the M segment (full-length glycoprotein precursor, left) or Gc ectodomain (IbAr10200 glycoprotein precursor amino acid residues 1041–1598) (right). (C) Analysis of cross-reactivity, ADI-36120 competition, and Vλ3-21 germline usage from the panel of mAbs shown in panel A. (D) CDR sequences of cross-reactive, GP38-specific mAbs with the Vλ3-21 germline. Red letters indicate amino acids that are predicted to be mutated from germline.
Figure S3. Characteristics of Site 2-targeting mAbs, related to Figure 3. (A) Cross-competition of the mAbs used to identify antigenic sites. (B) VH germline gene usage broken down by targeted Gc antigenic site. (C) Germline gene pairing of mAbs targeting Site 2. (D) CDRH3 amino acid lengths comparing Site 2-targeting mAbs to other Gc-specific mAbs. (E) Comparison of CDRH3 amino acid lengths. Statistical comparison was performed using the Mann-Whitney test (**** P < 0.0001, ** P < 0.01). Bars represent median CDRH3 amino acid length. (F) Polyreactivity scores between Site-2 targeting mAbs and all others. Statistical comparison was performed using Fisher’s exact test (**** P < 0.0001). (G) CDR amino acid sequences of mAbs sharing the VH3-21/VK1-27 germline gene pair from all four donors. Red letters indicate amino acids that are predicted to be mutated from germline.
Figure S4. Epitope mapping using yeast surface display, related to Figure 3.
(A) A representative example of the yeast display selection strategy to map the epitope of ADI-36122 is shown. In the first and second rounds, yeast mutants were selected for loss of binding for an antibody of interest. In the third round, a pool of antibodies that do not compete with the antibody of interest were used to select mutants that maintain proper folding of other epitopes. Wild-type Gc was used as a control for gating purposes. (B) Extended version of Figure 3D to include all antibodies used in selections and all isolated single mutants. Darker shades indicate a greater loss of binding relative to wild-type. (C) On-yeast titrations of the mAbs used to define antigenic sites demonstrating specific loss of binding. Data is shown as the ratio of antibody binding to displayed Gc. (D) Biolayer interferometry sensor-grams showing the binding kinetics of CCHFV Gc DIII to ADI-36145 IgG.
Figure S5. Breadth of neutralization for candidate nAbs, related to Figure 4. (A-R) Individual neutralization curves against IbAr10200, Oman, Kosova Hoti, and Turkey tecVLPs for the indicated mAbs. All tecVLP neutralization assays were performed using Vero target cells, n = 6, from two independent experiments for all neutralization curves.
Figure S6. Synergistic neutralization by Site 1-targeting mAbs, related to Figure 5. (A-C) Neutralization of Oman tecVLPs by the indicated nAbs and their 1:1 combinations. (D-F) Combination index (CI) analyses of the neutralization curves in panels A-C. Synergistic neutralization was assayed using Vero target cells, n = 6, from two independent experiments for all neutralization curves.
Figure S7. Biophysical and binding properties of dual-variable domain Igs (DVD-Igs), related to Figure 7. (A-D) Size-exclusion chromatography traces of the indicated DVD-Igs (E-J) Two-phase binding experiments for the DVD-Igs by BLI. Each probe bearing the indicated IgG was sequentially dipped in analyte solutions containing rGn/Gc and then the indicated DVD-Ig. In panels I and J, the presence of both parental mAbs block binding by the corresponding DVD-Ig.
Data Availability Statement
This study did not generate any new datasets or code.