

Abstract
Age-related pathological alterations of the vasculature have a critical role in morbidity and mortality of older adults. In epidemiological studies age is the single most important cardiovascular risk factor that dwarfs the impact of traditional risk factors. In order to develop novel therapeutic interventions for prevention of age-related vascular pathologies it is crucial to understand the cellular and molecular mechanisms of vascular aging. In this review, shared molecular mechanisms of aging are considered in terms of their contribution to the pathogenesis of macro- and microvascular diseases associated with old age. The role of cellular senescence in development of vascular aging phenotypes is highlighted and potential interventions to prevent senescence and to eliminate senescent cells for prevention of vascular pathologies are presented. The evidence supporting a role for inter-organ communication and circulating pro-geronic and anti-geronic factors in vascular aging is discussed.
Keywords: geroscience, senescence, endothelial dysfunction, microcirculation, atherosclerosis
Condensed abstract:
Age-related pathological alterations of the vasculature have a critical role in morbidity and mortality of older adults. In this review, shared molecular mechanisms of aging are considered in terms of their contribution to the pathogenesis of macro- and microvascular diseases associated with old age. The role of cellular senescence in development of vascular aging phenotypes is highlighted and potential interventions to eliminate senescent cells for prevention of vascular pathologies are presented. The evidence supporting an important role for inter-organ communication and circulating pro-geronic and anti-geronic factors in orchestration of vascular aging processes is discussed.
Introduction: a man is as old as his arteries – and microvessels
The famous seventeenth-century physician Thomas Sydenham, known as the “English Hippocrates,” observed, “a man is as old as his arteries.” Modern medicine concurs: cardiovascular and cerebrovascular diseases are the leading causes of serious long-term disability and mortality among older adults in the developed world(1). Importantly, in population-based studies the effects of conventional risk factors (e.g., hypertension, hypercholesterolemia, etc.) on the prevalence of cardiovascular and cerebrovascular diseases are dwarfed by the single most important risk factor for these diseases: advanced aging (1).
In addition to recognized role of vascular aging processes in the genesis of age-related macrovascular diseases (e.g., atherosclerotic diseases of the large arteries (1)), there is also increasing appreciation of the importance of age-related microvascular pathologies (2). Aging causes multifaceted structural and functional microcirculatory impairment, which has deleterious effects on tissue oxygenation, nutrient delivery, waste removal and thus negatively impacts multiple organ functions (2). Aging also impairs local regulation of microvascular perfusion (e.g., by impairing endothelium-mediated, flow-induced arteriolar vasodilation), angiogenesis) and promotes pathological structural remodeling of the microvascular network, resulting in microvascular rarefaction (3). By altering the phenotype of endothelial cells, smooth muscle cells and pericytes, microvascular aging also negatively impacts the function of the immune system (e.g., white blood cell adhesion and chemotaxis) and endocrine system (e.g., insulin signaling), and impairs barrier function and stem cell physiology (e.g., altering stem cell niches and impacting endothelial cell-stem cell interactions during homing and engraftment) (2). Aging also alters the secretory phenotype of cells in the microcirculatory network, which results in deleterious changes in the production of a wide range of trophic factors, cytokines, chemokines, lipid mediators, exosomal factors, micropeptides and/or gasotransmitters(2). All of the aforementioned age-related microcirculatory alterations contribute to the age-related complex changes in the local humoral and cellular environment in the tissues. Aging-induced microvascular functional, structural and phenotypic alterations play a critical role in age-related dysfunction of multiple organ systems and contribute significantly to the pathogenesis of a diverse spectrum of age-related diseases (e.g., heart failure, Alzheimer’s disease, vascular cognitive impairment, age-related macular degeneration [AMD], sarcopenia and kidney disease)(2). The emerging concept is that shared molecular and cellular mechanisms underlie both age-related macrovascular and microvascular pathologies, as well as other diseases associated with old age (Central Illustration).
Central Illustration. Multiple shared mechanisms of aging contribute to the pathogenesis of diverse age-related diseases in each organ system, including the vasculature, simultaneously.
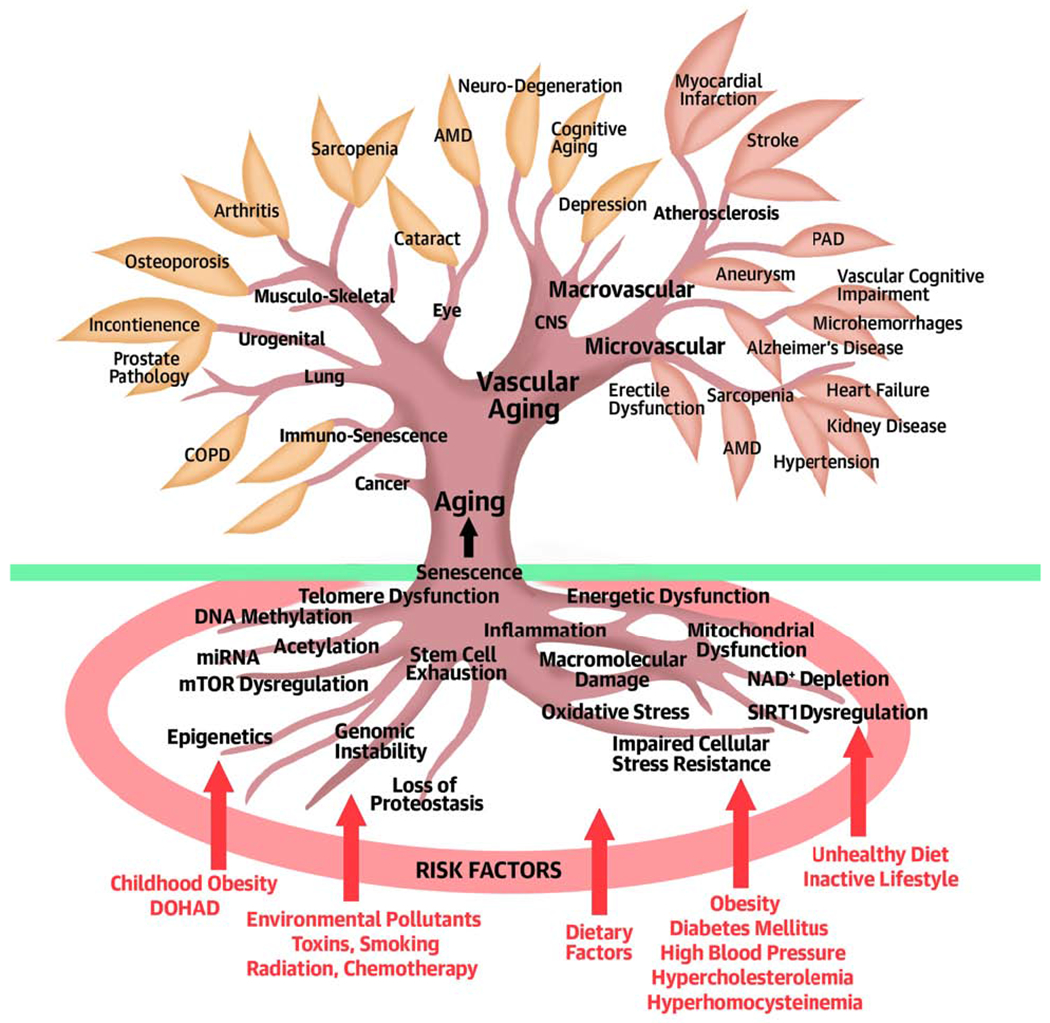
Consequences of vascular aging lead to the genesis of micro- and macrovascular pathologies, ranging from atherosclerotic vascular diseases to Alzheimer’s disease. Conventional risk factors (purple) promote age-related cardiovascular and cerebrovascular pathologies by exacerbating one or more fundamental molecular and cellular (cell autonomous and non-cell autonomous) aging processes (roots). Clinical disciplines, biogerontology and public health research separately focus on the individual age-related diseases (leaves), the mechanisms of aging (roots) and the risk factors, respectively. Geroscience is an integrative scientific field that considers the interaction of all of these levels.
Significant advances in geroscience in the 21st century have led to the identification of evolutionarily conserved pathways responsible for regulation of lifespan and health span (4). In the present review, the pathophysiological roles of these fundamental aging processes in the vascular system are considered in terms of their contribution to both micro- and macrovascular aging phenotypes and the pathogenesis of age-related vascular diseases (Central Illustration). The interconnectedness between the cellular and molecular mechanisms of vascular aging and the interaction among these aging processes, disease-specific pathways and cardiovascular risk factors is emphasized. We also provide an overview of emerging experimental evidence, which supports the existence and significance of diverse secreted/circulatory factors derived from distal organs that modulate vascular aging processes.
Our emerging understanding of vascular aging processes enable the identification of novel targets for therapeutic intervention to reverse the deleterious consequences of vascular aging and to improve cardiovascular and cerebrovascular health in older adults. In the present review we discuss the potential therapeutic benefits of a new class of treatment: prevention of cellular senescence and elimination of senescent cells. Related reviews in the same issue discuss in detail the vasoprotective effects of interventions that target sirtuin-regulated pathways and autophagy. For an overview of emerging therapies targeting other major processes involved in vascular aging (Table 1) and the effects of preventive measures already available and in clinical use (e.g., physical exercise, dietary regimens, smoking cessation, inhibitors of the renin-angiotensin system, statins) the interested reader is referred to recent comprehensive reviews (2,5).
Table 1.
Mechanisms of vascular aging
| Cellular and molecular mechanisms of aging(2) | Putative role in vascular pathologies(2) | Preclinical evidence | Translational evidence | Potential target for intervention (examples(5)) |
|---|---|---|---|---|
| Oxidative and nitrative stress | Atherogenesis, inflammation, endothelial dysfunction, blood flow↓, ECM remodeling, hypertension, microhemorrhages, aneurysm formation | yes | yes | yes (antioxidants, peroxynitrite scavengers, upstream activators of antioxidant pathways) |
| Impaired oxidative stress resistance (including Nrf2 dysfunction)(35) | Impaired response to injury, exacerbated effects of vascular risk factors (hypertension, metabolic diseases, smoking), inflammation, atherogenesis, aneurysm formation, microvascular damage, impaired angiogenesis | yes | ? | yes (Nrf2 activators, caloric restriction) |
| Chronic low grade sterile inflammation (NF-κB activation, cytokine dysregulation, DAMPs) | Atherogenesis, paracrine effects on tissue function (including stem cell niche impairment), barrier dysfunction, white blood cell extravasation | yes | yes | yes (inhibitors of NF-κB, upstream activators of anti-inflammatory pathways) |
| Mitochondrial dysfunction | Impaired endothelial vasomotor, transport and barrier functions(36), inflammation(37), atherogenesis(38) (39) | yes | ? | yes (mitochondria-targeted antioxidants(27)) |
| NAD+ depletion(40) | Endothelial dysfunction(30), cellular energetics↓, impaired angiogenesis, atherogenesis (?) | yes | yes | yes (nicotinamide mononucleotide(30), nicotinamide riboside, nicotinamide; PARP1 inhibitors(31)) |
| SIRT1 dysregulation | Endothelial dysfunction, inflammation, atherogenesis, microvascular dysfunction(9,41) | yes | ? | yes (SRT1720(9)) |
| mTOR dysregulation | Microvascular rarefaction, inflammation, atherosclerosis, vasomotor dysfunction(29), blood-brain barrier disruption(42) | yes | ? | yes (rapamycin, rapalogs(29)) |
| AMPK dysregulation | Endothelial dysfunction, vascular inflammation | yes | yes | yes (metformin(28)) |
| Δ DNA methylation | Vascular inflammation, aneurysm formation(43) | yes | ? | ? |
| miRNA dysregulation | Angiogenesis↓, atherogenesis(2) | yes | ? | ? |
| Loss of proteostasis (ubiquitin-proteasome↓, lysosome-autophagy system↓. | atherosclerosis, vascular inflammation(44,45), Alzheimer’s disease, amyloidosis | yes | ? | ? |
| Apoptosis↓, necroptosis↓ | Microvascular rarefaction, inflammation (?)(6), aneurysms (?) | yes | ? | ? |
| Progenitor cell exhaustion | Microvascular rarefaction (?), impaired angiogenesis and collateralization(?)(46) | yes | ? | ? |
| Genomic instability | Endothelial dysfunction, increased vascular stiffness, increased presence of senescence cells, hypertension(47), atherosclerosis(48) | yes | ? | ? |
| Cellular senescence↑(10) (15) | Atherogenesis, Angiogenesis↑, endothelial dysfunction, inflammation↑, blood brain barrier dysfunction, microvascular rarefaction | yes | yes | yes (senolytics) |
Shared mechanisms of macro- and micro-vascular aging
The goal of geroscience research is to develop drugs and interventions for prevention and treatment of a range of chronic diseases of old age as a class, by targeting shared mechanisms of aging (4). It is expected that this strategy will also result in revolutionary novel interventions preventing, attenuating and reversing age-related vascular pathologies (5).
The relative contribution of cell autonomous and non-autonomous mechanisms to systemic aging in human subjects, laboratory animals and different model organisms is hotly debated. The model for vascular aging that we have recently proposed(2) predicts that a range of shared and interconnected cell-autonomous molecular mechanisms of aging contribute both to the genesis of vascular aging phenotypes and age-related pathologies of other organ systems (Central Illustration). These mechanisms of vascular aging are listed in Table 1. In general, many of these mechanisms are related to spontaneous, stochastic damage that activate evolutionarily conserved cellular programs (e.g., senescence, inflammatory responses, etc.) and the pathways determining the resilience of the cells to such damage/stress (e.g., loss of proteostasis, impaired Nrf2-driven antioxidant response and DNA repair, etc.). For a detailed discussion of the roles of these cellular and molecular mechanisms in age-related vascular pathologies the interested reader is referred to recent reviews (2). In the present review the role of increased cellular senescence, a specific cell-autonomous mechanism of vascular aging, , and its potential therapeutic considerations are discussed in more detail (Figure 1).
Figure 1. Conceptual model for the causes and pathological consequences of cellular senescence in vascular aging.
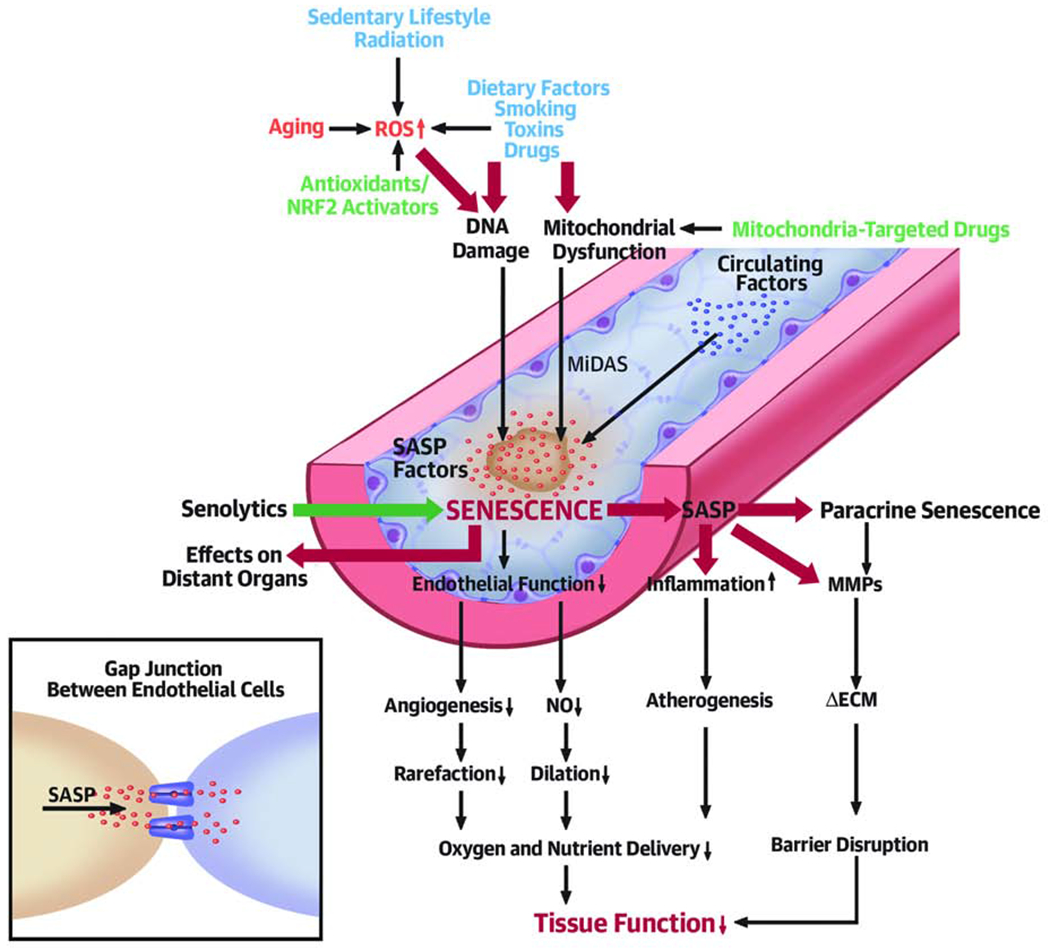
The model predicts that increased presence of senescent cells in the aged vasculature and their proinflammatory secretome (SASP: senescence-associated secretory phenotype) contributes to impaired angiogenesis and microvascular rarefaction, impaired vasodilation, chronic inflammation, pathological remodeling of the extracellular matrix (ECM), barrier disruption, and/or atherogenesis, all of which contribute to age-related tissue dysfunction. It is predicted that signals that induce senescence may be transmitted between cells (vis gap junctions). Secretion of SASP factors may also induce paracrine senescence and/or impair the function of neighboring cells. The model predicts that in aging increased ROS, DNA damage and/or mitochondrial dysfunction promote cellular senescence in the vasculature (MiDAS: mitochondrial dysfunction-associated senescence). Exogenous stressors/risk factors (blue) exacerbate vascular aging by induction of senescence. Interventions for targeting senescence-related mechanisms include prevention of ROS-mediated DNA damage (by antioxidants and Nrf2 activators), mitochondria-targeted treatments and removal of senescent cells by senolytic treatment.
It has also become apparent in recent years that cell-autonomous mechanisms alone(4) are inadequate to explain all aspects of vascular aging. The hierarchical regulatory cascade for vascular aging also involves modulation of cell-autonomous cellular and molecular aging processes by systemic/circulating factors (2). In addition, age-related cell autonomous changes also cause non-cell autonomous consequences (e.g., release of paracrine mediators from senescent cells) that affect vascular aging (2). According to the model proposed, circulating progeronic factors and anti-geronic factors derived from the central nervous system, endocrine organs, the immune system, the adipose tissue and other organs (including the gastrointestinal tract) orchestrate microvascular and macrovascular aging processes (Figure 2). An important prediction of the model is that conventional risk factors promote age-related cardiovascular and cerebrovascular pathologies by exacerbating one or more fundamental molecular and cellular (cell autonomous and non-cell autonomous) aging processes (Central Illustration).
Figure 2. Regulation of vascular aging by pro-geronic and anti-geronic circulating factors.
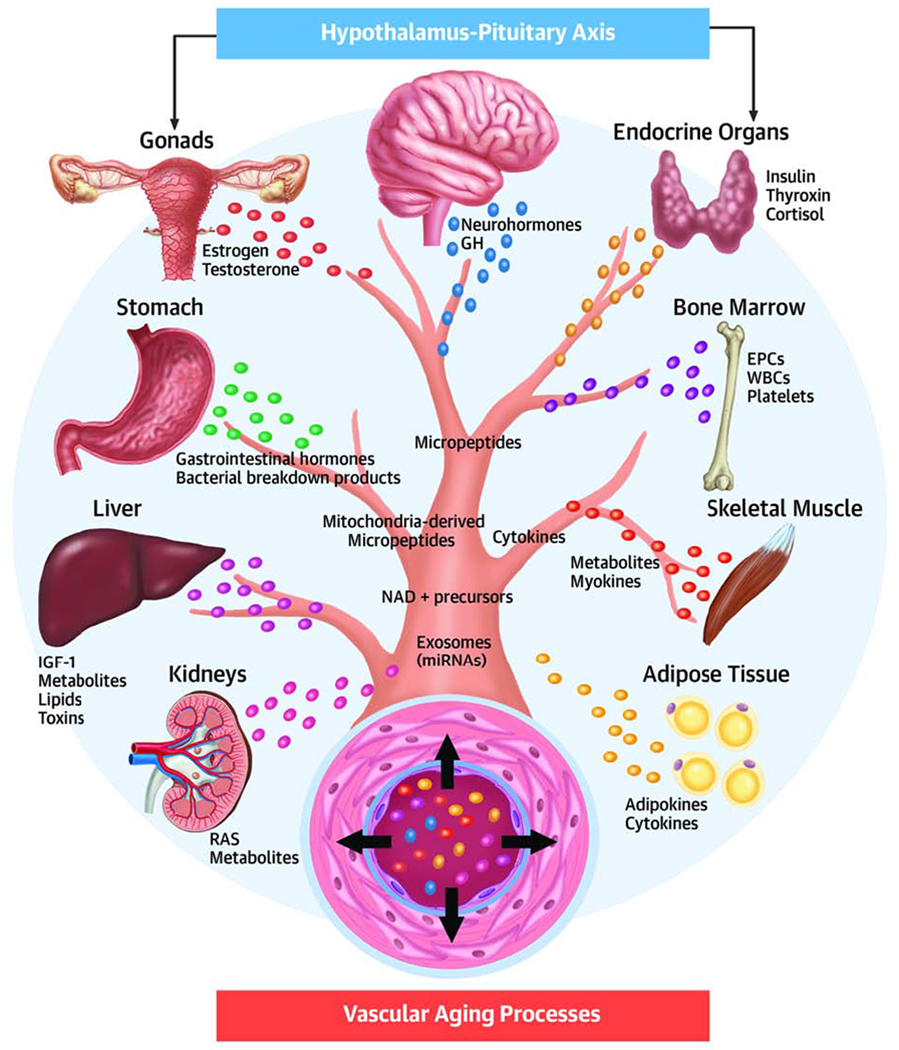
The model depicts the role of inter-organ communication in the hierarchical regulatory cascade for vascular aging. Cell-autonomous cellular and molecular aging processes in the vascular wall are modulated by circulating pro-geronic (e.g., inflammatory cytokines) and anti-geronic factors (e.g., IGF-1, mediators of caloric restriction, estrogen) derived from the central nervous system, endocrine organs, the adipose tissue and other organs. RAS: renin-angiotensin system; MMPs: matrix metalloproteinases; DAMPs: damage-associated molecular patterns; EPCs: endothelial progenitor cells; GH: growth hormone.
Exacerbation of vascular aging processes by cardiovascular and cerebrovascular risk factors
From the standpoint of geroscience, conventional risk factors promote cardiovascular and cerebrovascular pathologies by inducing an ‘accelerated vascular aging’ phenotype, which account for the inter-individual variability in the rate of development and progression of age-related vascular diseases (Central Illustration). Each conventional cardiovascular and cerebrovascular risk factor exacerbates one or more fundamental cellular and molecular aging processes in the vasculature (2). Accordingly, hypercholesterolemia, hypertension, high fat diet consumption, hyperhomocysteinemia, obesity and diabetes mellitus promote increased inflammatory status, oxidative/nitrosative stress, mitochondrial dysfunction, endothelial apoptosis, macromolecular damage and/or increased senescence in the vascular wall (2,6). Smoking, environmental pollutants (e.g., diesel exhaust particles) and other toxicants promote oxidative/nitrosative stress, inflammation, induce DNA damage, and promote cellular senescence in the vasculature(2). Sedentary lifestyle (and the consequentially altered hemodynamic environment, including decreased shear stress) is associated with increased inflammatory status and oxidative stress(2). Whole brain radiation therapy of tumor patients promotes DNA damage and induces cellular senescence, which results in an accelerated cerebromicrovascular aging phenotype contributing to the genesis of cognitive decline(7,8). Importantly, a better mechanistic understanding of the interaction between cardiovascular risk factors and specific aging processes will enable the development of innovative combination treatments for prevention in elderly patients at risk for cardiovascular and cerebrovascular pathologies. It is expected that in the future preventive measures already available and in clinical use that target conventional risk factors (e.g., physical exercise, dietary regimens, smoking cessation, inhibition of the renin – angiotensin-aldosterone system, statins) will be combined with interventions that increase the resilience of the vasculature to cellular stresses (e.g., by boosting the endogenous cell survival pathways, including SIRT1(9) and Nrf2 regulated pathways(6)) and target the cellular and molecular aging processes contributing to the pathogenesis of vascular diseases. One caveat to keep in mind is that therapies that increase microvascular blood supply and/or stimulate prosurvival pathways in transformed cells may also promote tumor growth in older individuals.
Elimination of senescent cells: an emerging approach for prevention of age-related vascular pathologies
Cellular senescence is regarded as a fundamental aging process characterized by irreversible growth arrest, functional impairment and profound pro-inflammatory secretome changes (10) (Figure 1). Various endogenous and exogenous stressors (e.g., reactive oxygen and nitrogen species, DNA damage, mitochondrial dysfunction, telomere dysfunction, paracrine signals) can exacerbate cellular senescence in aging (11,12). Strong preclinical data show that depletion of p16INK4A expressing senescent cells can significantly extend lifespan and health span in mouse models (11). These data support the concept that increased presence of senescent cells have important roles in age-related physiological decline and vulnerability to diseases. In previous studies senescent cells have been implicated in the pathogenesis of a wide range of age-related diseases, including chronic obstructive pulmonary disease, sarcopenia, liver fibrosis, obesity, diabetes mellitus, chronic kidney disease, Alzheimer’s disease, Parkinson’s disease, cataracts, AMD, diabetic retinopathy, cardiac fibrosis, heart failure, osteoporosis, osteoarthritis and cancer(13). There is also growing evidence that both aging and pathophysiological conditions associated with accelerated vascular aging associate with increased presence of senescent cells in the vasculature (14,15). Though the ratio of senescent cells is usually low, multiple mechanisms have been identified by which they may impair vascular function and promote development of age-related vascular pathologies (Figure 1). Senescent vascular cells exhibit increased production of ROS and acquire a senescence-associated secretory phenotype (SASP), which is characterized by increased production of inflammatory cytokines and chemokines and altered synthesis of lipid mediators (16). There are multiple ways by which senescent cells may affect the function and phenotype of neighboring cells in the vascular system. Endothelial cells are connected by gap junctions and function as a syncytium. Thus, it is possible that signals that induce senescence may be transmitted between cells (Figure 1). Secretion of SASP factors may also induce paracrine senescence and/or impair the function of neighboring cells. Through the aforementioned mechanisms senescent cells may contribute to endothelial dysfunction (14), impaired barrier function, heightened inflammatory status and pathological remodeling of arteries and/or microvessels in aging (17). In senescent cells the biosynthesis of components of the extracellular matrix (ECM), secretion of ECM degrading matrix metalloproteinases (MMPs) and expression of growth factors that regulate remodeling of the ECM are also altered. Thus, it is also possible that presence of senescent cells in the arterial wall may also contribute to decreased elasticity, increased stiffness and impaired resilience of the vascular wall to mechanical damage. Replicative senescence may also be potentially important for age-related impairment of regenerative and angiogenic capacity of the microvascular endothelial cells. These hypotheses should be experimentally tested. Proof-of-concept for the important pathological role of cellular senescence in accelerated vascular aging has been provided by investigations using a mouse model of y-irradiation-induced, DNA damage-dependent senescence(16) showing that experimentally induced senescence in cells of the neurovascular unit promotes dysregulation of cerebral blood flow, neurovascular dysfunction, disruption of the blood brain barrier, microvascular rarefaction and cognitive deficits, mimicking the brain aging phenotype (7). Studies on genetic murine models of accelerated cellular senescence (e.g., BubR1H/H mice) extend these findings demonstrating that induction of senescence in vascular endothelial cells and pericytes associates with blood brain barrier disruption(18). Importantly, increased presence of senescent cells has been shown in advanced atherosclerotic lesions(10). Elimination of senescent cells in Ldlr−/−mice by genetic and pharmacological approaches exerts anti-atherogenic effects, implicating senescent cells both in exacerbation of vascular inflammatory processes(19) and plaque instability (10). Future studies also should investigate the possibility of paracrine transmission of senescence from vascular cells to perivascular and parenchymal cells (11).
It is expected that therapeutic strategies that increase the stress resilience of vascular cells would prevent vascular senescence. The Nrf2-dependent homeostatic antioxidant defense pathway plays a central role in vascular stress resilience by regulation of both cellular DNA repair and elimination of ROS. Importantly, genetic depletion of Nrf2 exacerbates age-related vascular senescence(20). Treatment with pharmacological activators of Nrf2, including resveratrol, which attenuates ROS-induced DNA damage is expected to prevent ROS-induced senescence in vascular cells(2). Importantly, senescent cells can also be eliminated pharmacologically(21). There are several experimental senolytic strategies extant, including treatment with dasatinib, the polyphenols quercetin and fisetin and the Bcl-2/Bcl-XL inhibitor navitoclax (ABT263), which result in effective and selective removal of senescent cells in a number of organs, including the vasculature(10,21). Initial studies demonstrate that chronic treatment with senolytic drugs can attenuate atherogenesis(10) and improve endothelial function in aged mice(14). Future studies should test the protective effects of a wide range of translationally relevant senolytic treatments in animal models of various age-related macro- and microvascular pathologies. The exact cause of increased cellular senescence in the vasculature, including the role of increased oxidative stress, impaired stress resilience pathways and increased DNA damage should be further explored. The contribution of exogenous stressors (e.g., toxicants, dietary factors) and the role of factors released from senescent cells residing in other organs (e.g., the adipose tissue(12)) in older individuals towards induction of vascular senescence should also be elucidated. The potential unwanted/unexpected side effects of senolytic treatments should also be considered. For example, it should be methodically tested whether senescence cell removal may cause temporary microvascular damage (e.g., blood brain barrier disruption). Future studies also should explore variations in the senescent phenotype within the vascular cells. Studies using single cell sequencing methods will be quite informative in that regard. Finally, new biomarkers for senescence are needed that could be used in translational studies. These may include analysis of a wide range of SASP factors and exosomes derived from senescent cells, among others.
Regulation of vascular aging by pro-geronic and anti-geronic circulating factors
Increasing evidence suggests that organismal aging is associated with complex changes in inter-organ communication, which play a critical role in orchestrating/modulating cellular and molecular aging processes in the vasculature (Figure 2). Factors derived from the brain, the endocrine system, immune system, the adipose tissue and other organs (including the gastrointestinal tract and the symbiotic microbiota) can alter the rate of vascular aging. Our proposed model predicts that circulating pro-geronic factors (whose production increases with age and which impair vascular homeostasis; e.g., inflammatory mediators) and anti-geronic factors (which reverse/prevent development of aging phenotypes; e.g., IGF-1, mediators of caloric restriction, vasoprotective hormones) orchestrate cellular and molecular aging processes in the entire vascular system, including macrovascular and microvascular endothelial and smooth muscle cells, pericytes and cells of the neurovascular unit (including astrocytes). Changes in the balance of these circulating factors result in generalized functional alterations in both the large vessels and the microcirculation affecting structure and vasomotor, barrier, secretory and transport functions of the vasculature, promoting adverse structural remodeling and the development of a spectrum of age-related vascular pathologies.
In that regard, age-related changes in vasoprotective endocrine factors are of great significance, including altered interactions among the brain (hypothalamus), anterior pituitary gland, their target organs (gonads, liver) and the vasculature. Age-related changes in the gonadal and growth-hormone/insulin-like growth factor (GH/IGF-I) axes are associated with significant decreases in circulating levels of GH, IGF-1 and estrogens, which contribute to impairment of endothelial vasodilation, impaired autoregulation of cerebral blood flow, pathological vascular remodeling, atherogenesis, impaired vascular stress resilience, impaired angiogenic processes and microvascular rarefaction (2,3).
The critical role of circulating pro- and anti-geronic factors on cellular aging phenotypes was demonstrated by investigations using mouse models of heterochronic parabiosis (when a young mouse is surgically joined to an aged partner connecting their circulatory systems (22)) and mice with heterochronic blood apheresis(23) (which enables heterochronic blood exchange between young and old mice without sharing other organs). There is preliminary evidence that circulating anti-geronic factors derived from young mice can rejuvenate both endothelial function in large arteries (Ungvari, Csiszar, Huffman and Tarantini, unpublished observation 2019) and microvascular network architecture in aged heterochronic parabionts (22). Pro-geronic circulating factors, whose levels increase with age, may also contribute to impairment of vascular homeostasis(23). There is initial evidence that inflammatory cytokines (e.g. TNFα) may serve as pro-geronic circulating factors(2). Importantly, many of these factors are secreted by senescent cells in distant organs (e.g. the adipose tissue(12,13)).
Further evidence to demonstrate a key role of circulating anti-geronic factors orchestrating vascular aging processes is derived from investigations on animal models of calorie restriction, a dietary regimen, which extends health span and/or lifespan. Calorie restriction in rodents is associated with vascular rejuvenation, including rescue of endothelial function and attenuation of oxidative stress and inflammation(2). Importantly, in vitro treatment of endothelial cells in culture with sera derived from caloric restricted rodents and non-human primates recapitulates cellular rejuvenating effects (including anti-inflammatory and pro-angiogenic effects) observed in vivo in caloric restricted animals(24,25).
The exact nature of the anti-geronic and pro-geronic circulating factors responsible for regulation of vascular aging processes and the vascular rejuvenating effects observed in the aforementioned studies, is a focus of current investigations. Figure 2 depicts potential candidates whose role can be inferred from indirect evidence. In addition to proteins, peptides, steroid hormones and other lipid mediators the roles of micropeptides, metabolites, NAD+ precursors, bacterial breakdown products and circulating exosomes, which contain many types of biomolecules, including cellular proteins, miRNAs and mRNAs, should be elucidated. Future studies should identify cellular origins of newly discovered circulating pro-geronic and anti-geronic factors that modulate vascular aging processes and determine their specific pathogenic roles in atherogenesis, endothelial dysfunction, blood brain barrier disruption and microvascular pathologies. Further studies are also warranted to better understand the role of circulating cells (immunocytes, endothelial precursor cells, platelets) in vascular rejuvenation. Human studies are needed to confirm that the role of these circulating factors in regulation of vascular aging is not species-specific and identify relevant pathological conditions that alter their levels in the circulation. Mechanistic studies characterizing the cellular effects of circulating pro-geronic and anti-geronic factors in the vascular cells are warranted. Pathways modulating cellular energy metabolism, including mitochondrial pathways and cellular nutrient sensing pathways, emerge as critically important areas for understanding vascular aging. Importantly, many putative circulating pro-geronic and anti-geronic factors (from TNFα to IGF-1 and mediators of caloric restriction) appear to modulate cellular energetics and mitochondrial function in vascular cells (2).
Perspectives
The aforementioned studies established the paradigm of the plasticity of vascular aging, demonstrating that vascular aging phenotypes can be reversed (6,14,15,26). The concept presented here (Table 1, Central Illustration) implies that several interrelated cell-autonomous cellular and molecular aging processes, also modulated by systemic/circulating factors, contribute to vascular aging. Thus, we predict that effective interventional strategies will use combination treatments targeting multiple vascular aging processes simultaneously. We propose that senolytic strategies (14) can be combined with treatments that prevent senescence induction (e.g., free radical and peroxynitrite scavengers, Nrf2 activators(6)), exert anti-inflammatory effects mitigating the impact of the SASP factors(26), improve mitochondrial function(27), activate AMPK(28) and sirtuin pathways(9), inhibit mTOR(29) and/or restore cellular NAD+ levels(30,31). Anti-aging treatments that prove to be effective in preclinical studies should be also tested in translational studies(26). Importantly, a wide range of relevant vascular endpoints should be studied, from large artery health to microvascular physiology of the brain, heart, skeletal muscle (e.g. intermittent claudication), eye (e.g. AMD), kidney and ear (e.g. microvascular contributions to tinnitus). Critical areas of vascular aging research include mechanistic investigations into the microvascular contributions to cognitive decline, neurodegeneration and heart failure as well as complex geriatric syndromes such as frailty in older adults. Interventions that improve the microcirculation are expected to exert pleiotropic therapeutic effects in these syndromes. Future studies should investigate cellular heterogeneity in the aging vasculature and its role in focal development of micro- and macrovascular pathologies, ranging from amyloid angiopathy and perivascular development of amyloid plaques in Alzheimer’s disease (32), through atherosclerotic lesions to stroke, aneurysm and microhemorrhages (33). We also expect to see important breakthroughs in the near future in the fields at the intersection of microbiology, aging research and vascular biology. These will include better understanding of the role of the microbiota and the leaky gut in older adults in vascular pathologies and the contribution of infectious agents to the pathogenesis of age-related vascular diseases (including viruses with endothelial cell tropism, such as the human cytomegalovirus (34), fungi and bacterial pathogens penetrating the central nervous system though the lamina cribrosa). Public health research should shed light on the influence of the determinants of unsuccessful vascular aging (including genetic, environmental, dietary and socio-economic factors). Investigation of the mechanisms and consequences of macro- and microvascular aging evidently requires a multidisciplinary approach. We believe that universities with integrated translational geroscience programs bringing together expertise in cardiovascular and microvascular research, biogerontology, translational and public health research are the best positioned for this task.
Highlights.
Shared molecular mechanisms of aging promote macro- and microvascular pathologies associated with old age.
Elimination of senescent cells is a promising approach for prevention of vascular diseases.
Circulating pro-geronic and anti-geronic factors (e.g., IGF-1) regulate cell-autonomous processes of vascular aging.
Future studies should elucidate how conventional cardiovascular risk factors exacerbate molecular mechanisms of vascular aging.
Acknowledgment:
This work was supported by grants from the American Heart Association (ST), the Oklahoma Center for the Advancement of Science and Technology (to AC, ZU), the Presbyterian Health Foundation (to ZU, AC), the NIA-supported Geroscience Training Program in Oklahoma (T32AG052363), and the Cellular and Molecular GeroScience CoBRE (1P20GM125528).
Abbreviations:
- SASP
senescence-associated secretory phenotype
- AMD
Age-related macular degeneration
- ROS
Reactive oxygen species
- MMP
matrix metalloproteinase
- ECM
Extracellular matrix
- RAS
renin-angiotensin system
- DAMPs
damage-associated molecular patterns
- EPCs
endothelial progenitor cells
- GH
growth hormone
- IGF-1
Insulin-like growth factor-1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None
References
- 1.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation 2003;107:139–46. [DOI] [PubMed] [Google Scholar]
- 2.Ungvari Z, Tarantini S, Donato AJ, Galvan V, Csiszar A. Mechanisms of Vascular Aging. Circ Res 2018;123:849–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ungvari Z, Tarantini S, Kiss T et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol 2018;15:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013;153:1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alfaras I, Di Germanio C, Bernier M et al. Pharmacological Strategies to Retard Cardiovascular Aging. Circ Res 2016;118:1626–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearson KJ, Baur JA, Lewis KN et al. Resveratrol Delays Age-Related Deterioration and Mimics Transcriptional Aspects of Dietary Restriction without Extending Life Span. Cell Metab 2008;8:157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warrington JP, Ashpole N, Csiszar A, Lee YW, Ungvari Z, Sonntag WE. Whole brain radiation-induced vascular cognitive impairment: mechanisms and implications. J Vasc Res 2013;50:445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ungvari Z, Tarantini S, Hertelendy P et al. Cerebromicrovascular dysfunction predicts cognitive decline and gait abnormalities in a mouse model of whole brain irradiation-induced accelerated brain senescence. Geroscience 2017;39:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gano LB, Donato AJ, Pasha HM, Hearon CM Jr., Sindler AL, Seals DR. The SIRT1 activator SRT1720 reverses vascular endothelial dysfunction, excessive superoxide production, and inflammation with aging in mice. Am J Physiol Heart Circ Physiol 2014;307:H1754–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016;354:472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker DJ, Childs BG, Durik M et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker DJ, Wijshake T, Tchkonia T et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011;479:232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 2013;123:966–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roos CM, Zhang B, Palmer AK et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016;15:973–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossman MJ, Kaplon RE, Hill SD et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol 2017;313:H890–H895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ungvari Z, Podlutsky A, Sosnowska D et al. Ionizing radiation promotes the acquisition of a senescence-associated secretory phenotype and impairs angiogenic capacity in cerebromicrovascular endothelial cells: role of increased DNA damage and decreased DNA repair capacity in microvascular radiosensitivity. J Gerontol A Biol Sci Med Sci 2013;68:1443–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgan RG, Ives SJ, Lesniewski LA et al. Age-related telomere uncapping is associated with cellular senescence and inflammation independent of telomere shortening in human arteries. Am J Physiol Heart Circ Physiol 2013;305:H251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamazaki Y, Baker DJ, Tachibana M et al. Vascular Cell Senescence Contributes to Blood-Brain Barrier Breakdown. Stroke 2016;47:1068–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gardner SE, Humphry M, Bennett MR, Clarke MC. Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1alpha-Dependent Senescence-Associated Secretory Phenotype. Arterioscler Thromb Vasc Biol 2015;35:1963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fulop GA, Kiss T, Tarantini S et al. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience 2018;40:513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu M, Pirtskhalava T, Farr JN et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 2018;24:1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katsimpardi L, Litterman NK, Schein PA et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 2014;344:630–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rebo J, Mehdipour M, Gathwala R et al. A single heterochronic blood exchange reveals rapid inhibition of multiple tissues by old blood. Nat Commun 2016;7:13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Csiszar A, Labinskyy N, Jimenez R et al. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and SIRT1. Mech Ageing Dev 2009;130:518–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Csiszar A, Sosnowska D, Tucsek Z et al. Circulating factors induced by caloric restriction in the nonhuman primate Macaca mulatta activate angiogenic processes in endothelial cells. J Gerontol A Biol Sci Med Sci 2013;68:235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang M, Zhang L, Zhu W et al. Calorie Restriction Curbs Proinflammation That Accompanies Arterial Aging, Preserving a Youthful Phenotype. J Am Heart Assoc 2018;7:e009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossman MJ, Santos-Parker JR, Steward CAC et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018;71:1056–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sardu C, Paolisso P, Sacra C et al. Effects of Metformin Therapy on Coronary Endothelial Dysfunction in Patients With Prediabetes With Stable Angina and Nonobstructive Coronary Artery Stenosis: The CODYCE Multicenter Prospective Study. Diabetes Care 2019;42:1946–1955. [DOI] [PubMed] [Google Scholar]
- 29.Lin AL, Zheng W, Halloran JJ et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow Metab 2013;33:1412–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarantini S, Valcarcel-Ares MN, Toth P et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol 2019;24:101192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pacher P, Mabley JG, Soriano FG, Liaudet L, Komjati K, Szabo C. Endothelial dysfunction in aging animals: the role of poly(ADP-ribose) polymerase activation. Br J Pharmacol 2002;135:1347–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer EP, Ulmann-Schuler A, Staufenbiel M, Krucker T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer’s disease. Proc Natl Acad Sci U S A 2008;105:3587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toth P, Tarantini S, Springo Z et al. Aging exacerbates hypertension-induced cerebral microhemorrhages in mice: role of resveratrol treatment in vasoprotection. Aging Cell 2015;14:400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grahame-Clarke C, Chan NN, Andrew D et al. Human cytomegalovirus seropositivity is associated with impaired vascular function. Circulation 2003;108:678–83. [DOI] [PubMed] [Google Scholar]
- 35.Ungvari Z, Bailey-Downs L, Sosnowska D et al. Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of Nrf2-mediated antioxidant response Am J Physiol Heart Circ Physiol 2011;301:H363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tarantini S, Valcarcel-Ares NM, Yabluchanskiy A et al. Treatment with the mitochondrial-targeted antioxidant peptide SS-31 rescues neurovascular coupling responses and cerebrovascular endothelial function and improves cognition in aged mice. Aging Cell 2018;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ungvari ZI, Orosz Z, Labinskyy N et al. Increased mitochondrial H2O2 production promotes endothelial NF-kB activation in aged rat arteries. Am J Physiol Heart Circ Physiol 2007;293:H37–47. [DOI] [PubMed] [Google Scholar]
- 38.Botto N, Berti S, Manfredi S et al. Detection of mtDNA with 4977 bp deletion in blood cells and atherosclerotic lesions of patients with coronary artery disease. Mutat Res 2005;570:81–8. [DOI] [PubMed] [Google Scholar]
- 39.Yu E, Calvert PA, Mercer JR et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013;128:702–12. [DOI] [PubMed] [Google Scholar]
- 40.Csiszar A, Tarantini S, Yabluchanskiy A et al. Role of endothelial NAD+ deficiency in age-related vascular dysfunction. Am J Physiol Heart Circ Physiol 2019:in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen YX, Zhang M, Cai Y, Zhao Q, Dai W. The Sirt1 activator SRT1720 attenuates angiotensin II-induced atherosclerosis in apoE(−)/(−) mice through inhibiting vascular inflammatory response. Biochem Biophys Res Commun 2015;465:732–8. [DOI] [PubMed] [Google Scholar]
- 42.Van Skike CE, Jahrling JB, Olson AB et al. Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer’s disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol 2018;314:H693–H703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toghill BJ, Saratzis A, Harrison SC, Verissimo AR, Mallon EB, Bown MJ. The potential role of DNA methylation in the pathogenesis of abdominal aortic aneurysm. Atherosclerosis 2015;241:121–9. [DOI] [PubMed] [Google Scholar]
- 44.Wilck N, Ludwig A. Targeting the ubiquitin-proteasome system in atherosclerosis: status quo, challenges, and perspectives. Antioxid Redox Signal 2014;21:2344–63. [DOI] [PubMed] [Google Scholar]
- 45.Marfella R, Di Filippo C, Laieta MT et al. Effects of ubiquitin-proteasome system deregulation on the vascular senescence and atherosclerosis process in elderly patients. J Gerontol A Biol Sci Med Sci 2008;63:200–3. [DOI] [PubMed] [Google Scholar]
- 46.Hayek SS, MacNamara J, Tahhan AS et al. Circulating Progenitor Cells Identify Peripheral Arterial Disease in Patients With Coronary Artery Disease. Circ Res 2016;119:564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Durik M, Kavousi M, van der Pluijm I et al. Nucleotide excision DNA repair is associated with age-related vascular dysfunction. Circulation 2012;126:468–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gray K, Kumar S, Figg N et al. Effects of DNA damage in smooth muscle cells in atherosclerosis. Circ Res 2015;116:816–26. [DOI] [PubMed] [Google Scholar]