

Abstract
Disruption of vulnerable atherosclerotic plaques often leads to myocardial infarction and stroke, the leading causes of morbidity and mortality in the United States. A diagnostic method that detects high-risk atherosclerotic plaques at early stages could prevent these sequelae.
The abundance of immune cells in the arterial wall, especially inflammatory Ly-6Chi monocytes and foamy macrophages, is indicative of plaque inflammation, and may be associated with plaque vulnerability. Hence, we sought to develop a new method that specifically targets these immune cells to offer clinically-relevant diagnostic information about cardiovascular disease.
We combine ultra-selective nanoparticle targeting of Ly-6Chi monocytes and foamy macrophages with clinically-viable photoacoustic imaging (PAI) in order to precisely and specifically image inflamed plaques ex vivo in a mouse model that mimics human vulnerable plaques histopathologically. Within the plaques, high-dimensional single-cell flow cytometry (13-parameter) showed that our nanoparticles were almost-exclusively taken up by the Ly-6Chi monocytes and foamy macrophages that heavily infiltrate plaques. PAI identified inflamed atherosclerotic plaques that display ~6-fold greater signal compared to controls (P<0.001) six hours after intravenous injection of ultra-selective carbon nanotubes, with in vivo corroboration via optical imaging.
Our highly selective strategy may provide a targeted, non-invasive imaging strategy to accurately identify and diagnose inflamed atherosclerotic lesions.
Keywords: carbon nanotubes, photoacoustic imaging, atherosclerosis, diagnostic imaging, immunoimaging, flow cytometry
1. INTRODUCTION
Vascular inflammation, in which circulating monocytes infiltrate the arterial wall and differentiate into foamy macrophages, plays a key role in plaque atherogenesis[1,2]. Monocyte/macrophage cell numbers approximately scale with plaque severity[3], are primarily recruited from the circulation[4], and they contribute to plaque destabilization[5]. Thus, their presence in a given vascular region is a leading proxy for the presence of an inflamed plaque that may be vulnerable to disruption and clinical sequelae[6–8].
Circulating monocytes can be subdivided into at least two functionally distinct subsets, namely Ly-6Clo and Ly-6Chi monocytes[9,10]. Inflammatory Ly-6Chi monocytes and the foamy macrophages to which they give rise, but not Ly-6Clo monocytes, are recognized as key components in the atherosclerotic inflammatory response[11]. The presence of these inflammatory cells in an atherosclerotic plaque contributes to the vulnerable plaque phenotype[7,8].
It has become clear that simple anatomic imaging of atherosclerotic plaques is insufficient, as the vast majority of adults have lesions, but only a subset of these go on to cause a clinical event. Hence, targeting foamy macrophages and their Ly-6Chi monocyte precursors in plaque with a highly selective imaging agent might provide clinically relevant biological information and allow for the identification of high-risk inflamed plaques, which are associated with lesion instability[6,8,12,13]. Specifically imaging the presence of cells that promote high plaque instability and morbidity could i) allow identification of inflammation and high-risk vascular regions, ii) eventually predict likelihood of plaque rupture, and iii) guide and monitor therapeutic response to plaque-stabilizing therapy[14].
Prior work has shown targeting and imaging of macrophages by contrast injection and uptake of iron and other nanoparticles[14–17] for MRI (magnetic resonance imaging) and PET (positron emission tomography) of plaques[17]. For instance, PET using 18F-FDG (18F-fluorodeoxyglucose), a glucose analogue that monitors cellular metabolism, has shown some promise in imaging macrophages in atherosclerosis. Others have applied optical imaging modalities, such as optical coherence tomography (OCT)[3], ultrasonic technology, such as intravascular ultrasound (IVUS)[18], or anatomic assessment, such as computed tomography (CT)[19].
However, these approaches are not highly selective in imaging foamy macrophages and their precursor Ly-6Chi monocytes, and the contrast, if used, is relatively indiscriminately taken up by a broad spectrum of phagocytic cells. This can decrease diagnostic accuracy and specificity. Additionally, these approaches are associated with radiation, are indiscriminate, and/or low resolution. Instead, selective targeting of Ly-6Chi monocytes and foamy macrophages is expected to lead to more accurate, specific biological imaging of inflammatory atherosclerotic plaques that may be vulnerable to rupture[20,21]. This strategy allows a more direct delineation of inflammatory plaque, based on the presence of inflammatory cells, and requires no ionizing radiation.
In recent work, we demonstrated unprecedented selective targeting of carbon nanotubes to circulating Ly-6Chi monocytes in mice[22]. We quantitatively showed that single-walled carbon nanotubes (SWNTs) are selectively taken up by Ly-6Chi monocytes, and not by any other peripheral blood cells, including Ly-6Clo monocytes, granulocytes, and lymphocytes, establishing highly selective targeting of Ly-6Chi monocytes in living subjects[22]. Therefore, because Ly-6Chi monocytes give rise to foamy macrophages in atherosclerotic plaques[11], here we tested whether we could use these highly selective SWNTs to specifically image atherosclerotic plaques in mice. Polyethylene glycol (PEG)-coated SWNTs such as those used herein are biocompatible and non-toxic based on murine and non-human primate studies[23,24], display relatively long circulation times, clear from mice through the liver, spleen, and kidneys[25], and do not significantly affect the monocytes which take them up[22].
Our primary goal is to provide optimal contrast for clinically-translatable photoacoustic imaging (PAI) of inflammatory atherosclerotic plaques in patients. Unlike typical optical modalities, where light must travel into and back out of the tissue, PAI applies laser light into tissues where it is absorbed and causes thermal expansion, with the resultant ultrasonic emission from the tissue used to generate images. We selected PAI as our plaque imaging technique because of its capacity for cellular-scale imaging, its lack of ionizing radiation, and because it is translatable to human medicine—either externally (e.g., in the case of carotid artery imaging, as we perform here) or intravascularly, as part of a combined intravascular photoacoustic (IVPA) and intravascular ultrasound (IVUS) imaging strategy; moreover, SWNTs serve as excellent contrast agents for PAI[17]. This work dynamically pairs PAI in parallel with high-dimensional flow cytometry studies to determine the cellular basis of the signal over time (Figure 1).
Figure 1. Schematic of experimental design and timeline.
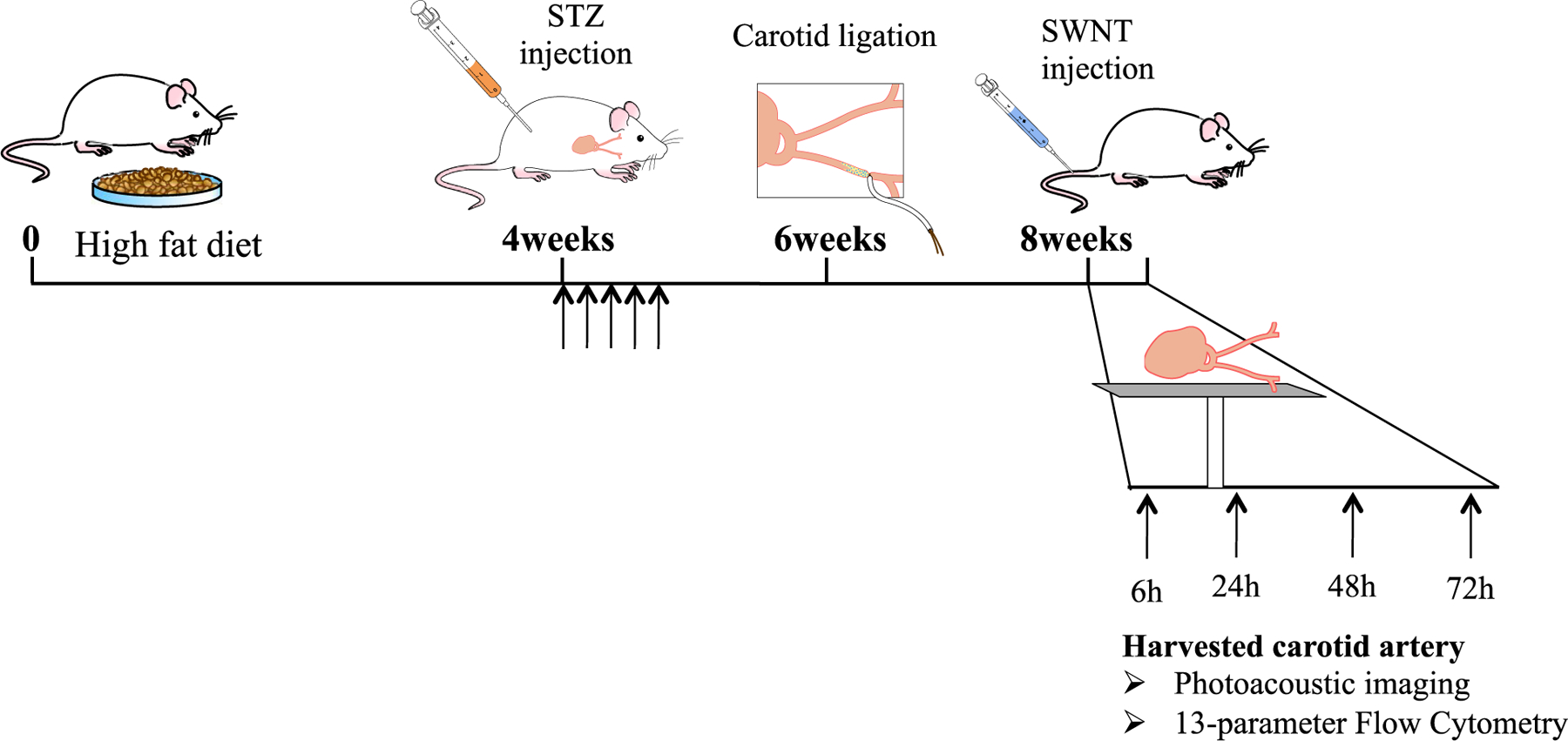
Mice were fed a high fat diet and given injections to induce diabetes prior to carotid artery ligation in order to generate a model of atherosclerotic plaque that is fairly similar to human disease. Afterward, a combination of photoacoustic imaging and biological characterization using FACS was performed.
Here, we show the ability to exploit PAI to detect an inflamed plaque phenotype on the basis of injectable carbon nanotube contrast agents that selectively target inflammatory monocytes and foamy macrophages.
2. RESULTS
2.1. Carotid-ligated diabetic mice develop atherosclerotic plaques enriched with inflammatory macrophages and monocytes
The immune cell component of human atherosclerotic lesions is very heterogeneous, including lymphocytes and myeloid cells (Figure S1). Among myeloid cells, inflammatory (Ly-6Chi) monocytes and foamy macrophages are known to be enriched in atherosclerotic lesions and to play major roles in the pathogenesis of atherosclerotic plaques[11]. Our carotid-ligated diabetic mouse model (Figure 1) develops macrophage-rich carotid plaques[29,30], which we analyzed for the specific monocyte/macrophage subtypes found in human disease. We processed the diseased carotids into single cell suspensions and analyzed the several immune and non-immune cell subsets using 13-parameter high-dimensional flow cytometry (Hi-D FACS). Immune cells were identified using the pan-hematopoietic marker CD45 (clone 30-F11), which is expressed by all immune cell lineages, including lymphocytes, myeloid cells, and granulocytes (i.e. CD45+). The remaining non-immune cells that make-up the arterial tissue, including endothelial cells and fibroblasts, were identified by the lack of expression of CD45 on their cell surface (i.e. CD45−). We found that, similar to human atherosclerotic lesions[31], mouse carotids were enriched with inflammatory monocytes (CD45+, Ly-6Chi, CD11b+ cells) and foamy macrophages (larger cells with CD45+, CD11bhi, F4/80+, and CD80/CD86+) (Figure 2 and S1). In addition, we show that atherosclerotic plaques also trigger the infiltration of B and T lymphocytes (defined as CD19+ and CD5hi, respectively) (Figure 2 and S1). Hence, our in vivo carotid-ligation mouse model provides a robust model with which to study inflamed atherosclerotic lesions.
Figure 2. 13-parameter High-Dimensional flow cytometry data of diseased artery analyzed in single cell suspension.
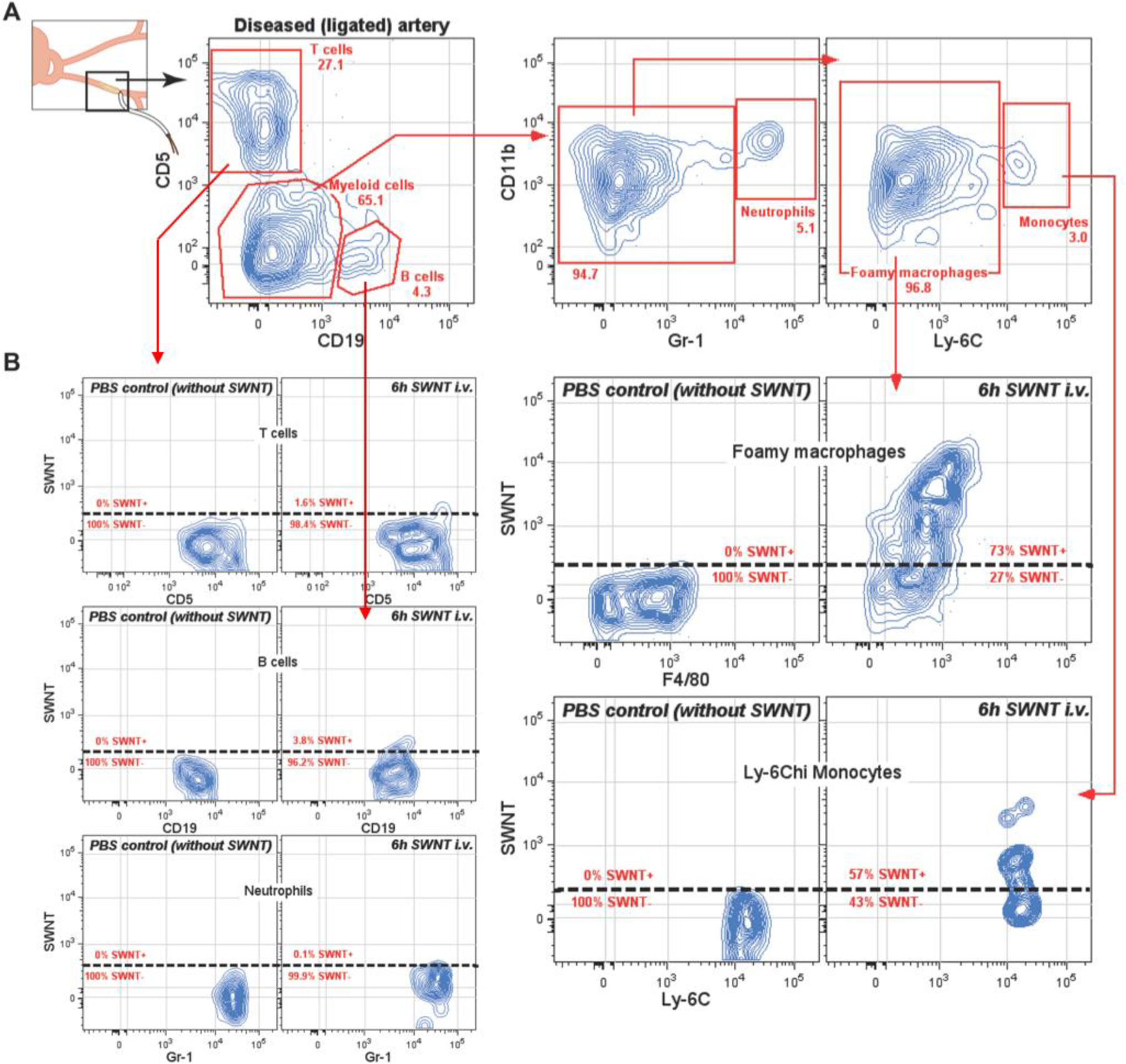
A) SWNT-Cy5.5 are selectively taken up by foamy macrophages and Ly-6Chi monocytes present in ligated diseased arteries (atheroma plaques). More than 70% of total foamy macrophages selectively take up SNWT-Cy5.5 6h after intravenous injection. B) Lymphocytes (T and B cells) and neutrophils, present within the diseased artery, do not take up SWNT-Cy5.5.
2.2. Single-walled carbon nanotubes, injected intravenously, selectively target inflammatory macrophages and monocytes within the atherosclerotic plaques
We recently showed that circulating inflammatory monocytes (Ly-6Chi) selectively took up SWNTs injected intravenously (i.v.) in a non-atherosclerotic cancer model[22]. Because inflammatory monocytes infiltrate and accumulate in atherosclerotic lesions we sought to determine whether infiltrating Ly-6Chi monocytes and the resident foamy macrophages would selectively pick up SWNTs, specifically marking sites of atherosclerotic lesions in vivo. We show that 6h after i.v. injection of SWNTs (time-point chosen based on prior SWNT studies[22,32]) into carotid-ligated mice only foamy macrophages and inflammatory Ly-6Chi monocytes picked up SWNTs in plaque, as determined by FACS using fluorescent dye Cy5.5 tagged onto SWNTs (Figure 2) and validated by histology using confocal microscopy (Figure S4). More than 70% of total foamy macrophages (FSChi, CD11b+, F4/80+), and more than 50% of total inflammatory monocytes (Ly-6Chi, CD11b+) internalized SWNTs 6h after i.v. injection. In contrast, other phagocytic cells present in atherosclerotic lesions, such as neutrophils (defined as Gr-1hi, CD11b+), minimally picked up SWNTs (<2%). Similarly, T and B lymphocytes, present in the diseased artery, failed to internalize SWNTs (<2% and <4%, respectively) (Figure 2). Non-immune cells (CD45−) also failed to internalize significant amounts of SWNTs in the diseased artery (Figure S2). Hence, we show that SWNT-Cy5.5 injected i.v. very selectively targets foamy macrophages and inflammatory monocytes, but not neutrophils, lymphocytes, and non-immune cells that accumulate in atherosclerotic lesions developed in vivo.
2.3. Photoacoustic imaging selectively identifies atherosclerotic plaque regions containing inflammatory macrophages and monocytes
Because of the selectivity of targeting to inflammatory cells based on FACS studies, we measured whether the delivered SWNTs were detectable in the plaques using PAI (Figure 3, S4, S5, and S7). We evaluated PAI signal by imaging excised arteries in phantoms at time-points from 6h to 72h after single i.v. injections of SWNTs (N=3 mice per time-point, in addition to non-ligated control mice). Each time-point represents carotid arteries harvested from different mice. The ligated arteries (left arteries) at the 6h time-point displayed higher signal than all other conditions, including both the controls (right arteries and control mice) and experimental conditions (i.e., other time-points) as well as in ligated arteries 3h post-injection (Figure S7–S9). The 6h time-point displayed at least 6-fold higher signal than both the controls and other time-points (Figure 3 and S3). At the earlier 3h time-point many fewer plaque leukocytes (essentially only inflammatory monocytes and macrophages based on 18-parameter flow cytometry, Figure S8) engulfed SWNTs at 3h compared to the 6h p.i. timepoint (Figure S9). The 24h time-point appeared to trend somewhat higher than the signal in other conditions. To test the significance, we computed the ratios of left (L) and right (R) (diseased and healthy control, respectively) arteries at all time-points. We found there is a significant decrease over time after 6h and that 6h is the maximal time-point (P<0.001) using a Jonckheere-Terpstra test. Moreover, using a non-parametric multivariable model, we showed that the L/R ratio decreases significantly over time (P=0.008), revealing that earlier time-points improved signal and that 6h was the optimal time-point tested to maximize PAI signal. Hence, PAI reliably identifies sites of atherosclerotic plaques by detecting SWNTs present in foamy macrophages and inflammatory monocytes, which are enriched only in the atherosclerotic lesions but not in healthy control arteries. We validated the PAI results using fluorescence imaging of the exteriorized arteries ex vivo via the Maestro imaging system (Figure S5A) and in vivo (mouse neck region) via Near-Infrared (NIR)-II imaging of intrinsic SWNT fluorescence (Figure S5B), both measured 6h after SWNT injection. We also imaged Cy5.5 fluorescence from SWNT-Cy5.5 in vivo in mice using fluorescence molecular tomography (FMT), which efficiently removes surface-weighted signal to support clear deep tissue imaging (Figure S5C). Both ex vivo and in vivo fluorescence imaging data confirm the PAI data in that the diseased artery (i.e., ligated) displays higher fluorescence signal intensity than the control (i.e., non-ligated) artery and compared with non-SWNT-injected mice.
Figure 3. Photoacoustic imaging of mouse carotid arteries.
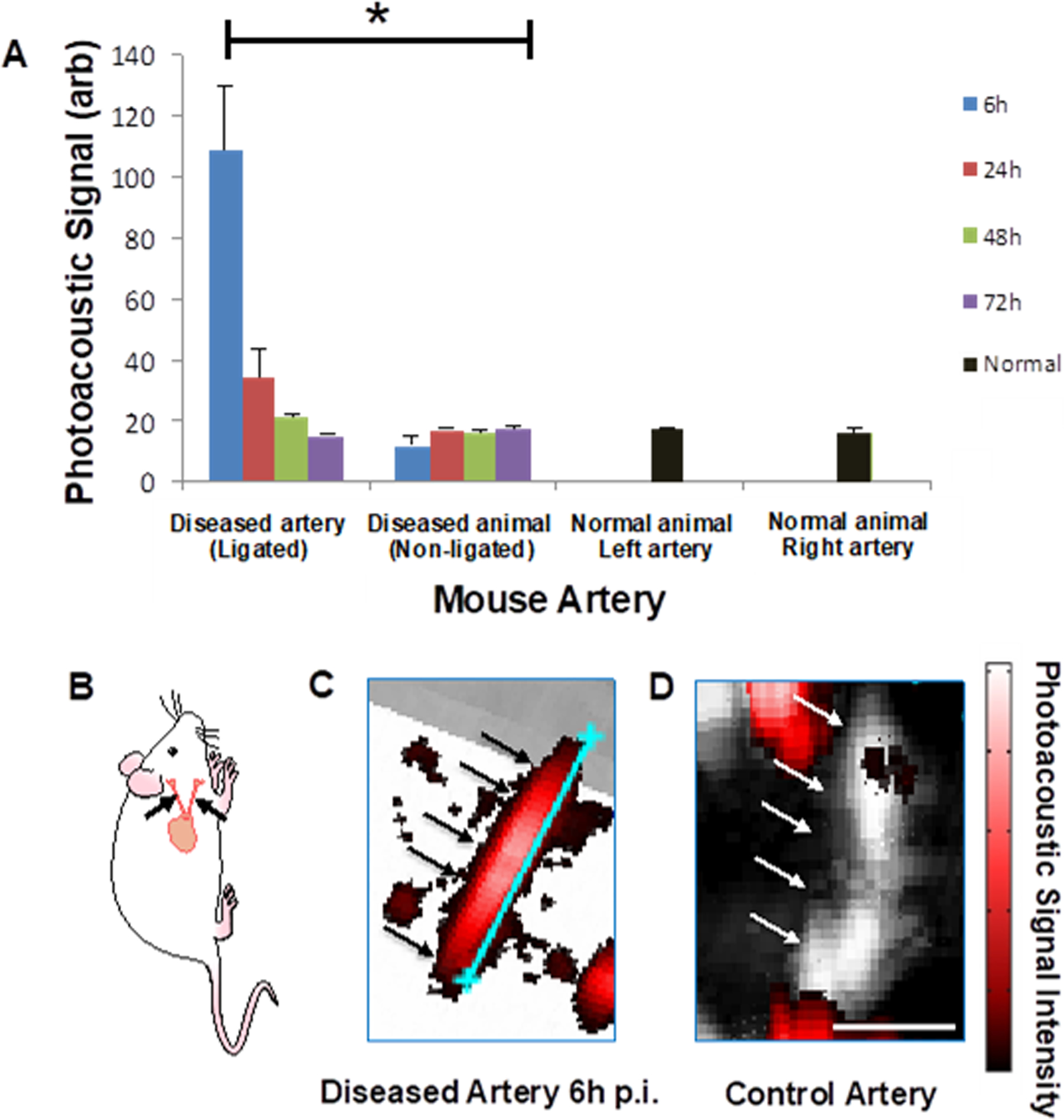
A) Mouse carotid arteries were imaged at multiple time-points after injection of SWNTs (6, 24, 48, and 72h). PAI intensity of a region of carotid plaques was quantified at each time-point. The signal was significantly higher at 6h post-injection compared to any other time-point and compared with controls. Arteries from diseased mice (‘diseased’) and normal mice (‘normal’) were measured with PAI; the ‘left side’ corresponds to the ligated artery side, although no ligation was performed on the normal mice. Each timepoint represents an average of at least N=3 animals; error bars are S.E.M. B) Schematic of a mouse including the carotid arteries (black arrows) which are removed and imaged with PAI in (C-D). C-D) Red signal is photoacoustic, with intensity scale at right. The black/white signal represents ultrasonic signal and black and white arrows designate the arteries. Shown is an example of a diseased artery at 6h post-injection with the blue scale bar showing the artery length = 5.2 mm (C), with PAI signal overwhelming the underlaid ultrasonic signal, and an example of a control artery in a mouse receiving SWNTs but without carotid ligation with scale bar 1.44 mm (D). Very little photoacoustic signal is measured, as the artery shows almost entirely ultrasonic signal (shown in black and white). Signal observed outside the arteries is from tiny air bubbles that may arise in the phantom. * P<0.001.
DISCUSSION
Here we define a novel and highly specific diagnostic technology to image atherosclerotic plaques in diseased arteries of atherogenic mice. These findings have the potential to improve current methods used to identify atherosclerotic lesions, especially inflamed plaques, which may be indicative of vulnerability and subsequent rupture in patients. Our novel method employs the injection of SWNTs, which are selectively taken up into Ly-6Chi inflammatory monocytes and foamy macrophages, two of the primary culprits behind atherogenesis and plaque vulnerability[5,6,33]. This ultra-selective uptake into only the inflammatory monocytes and macrophages results in the accumulation of large amounts of SWNTs within the atherosclerotic plaques, but not within healthy arteries. The extensive localized accumulation of SWNTs allowed us to image the inflamed arterial plaque using photoacoustic imaging technology with very high contrast 6h after injection. Other approaches such as MR-based smart probes have yielded considerably lower contrast enhancement factors (~3.5-fold compared to their control group, also in ex vivo animal experiments)[34], which may be challenging to translate to patients.
Unlike commonly-used angiographic and ultrasound-based methods which identify vascular stenosis (poor predictors of plaque inflammation[35,36]) or molecular lesion composition (e.g. lipid or calcium content[37,38]), our method provides cellular information regarding the presence and burden of inflammatory monocytes and macrophages. Selective targeting of immune cell subsets has long been sought[14,21] for diagnostic purposes such as that described here. Ly-6Chi monocytes are a critical subset in atherosclerosis for both its pathogenesis and the identification of an inflamed and potentially unstable plaque. This, and their enriched accumulation in inflamed plaques, makes them ideal for targeted imaging.
Thus, a crucial advantage of this approach is that it specifically identifies plaques containing the key inflammatory cell types associated with vascular inflammation that may indicate vulnerability to plaque progression and disruption. By linking the cellular component of the plaque responsible for its pathogenesis with its identification, we can optimize diagnostic accuracy. We have shown that we can accurately identify inflamed plaques, which may support guided therapy. Moreover, identification of the sites of inflammatory atherosclerotic plaque can be followed by either site-specific interventions if sufficiently inflamed (e.g., drug-eluting stents) and/or intensification of systemic treatments such as statin therapy and other anti-inflammatory agents currently in development[39,40]. Our approach may in the future serve as a predictive tool for the therapeutic efficacy of such anti-inflammatory agents. Moreover, given the selectivity of targeting we show here and the pathogenicity of the cellular subsets targeted, we recently tested and demonstrated that our SWNTs could be used as an atherosclerotic plaque therapeutic. By selectively delivering inhibitory molecules to the macrophages in diseased plaque, the pathogenicity of apoptotic debris in the core was reversed by blocking the pro-inflammatory axes and restoring macrophage efferocytosis, leading to plaque amelioration[41,42]. Thus, by equipping SWNTs with anti-inflammatory drug molecules, we developed a selective cellular therapeutic strategy. Combined with our current data, SWNTs might serve as a dual diagnostic/therapeutic agent (theranostic) for simultaneous monocyte/macrophage-selective inflammatory plaque identification and therapy.
While a number of imaging approaches have potential for characterizing atherosclerotic plaque, PAI allows biological information to be acquired from within plaques at high depths of penetration (up to ~5 cm) while maintaining the fairly high spatial resolution enabled by the optical component of PAI. Previously, PAI has been used to image intrinsic signal via a spectroscopic technique that analyzes lipid content[37], but this approach is not highly sensitive nor specific. Conversely, other PAI approaches have employed nanomaterials as contrast agents[17,43]. For instance, untargeted gold nanorods[44] demonstrated PAI signal in atherosclerotic rabbit arteries. Similarly, VCAM-1-targeted gold nanoshells were directed to atherosclerotic plaques in ApoE−/− mice to yield PAI contrast[45]. These studies demonstrate the potential of PAI, but do not display the cellular selectivity of the nanoparticles that provides the excellent PAI signal contrast in the plaques that we observed. Moreover, our SWNTs produce high PAI signal that is linearly quantitative in concentration (Figure S6), are stable in serum for at least a month (Figure S10), and are not toxic[23,41], supporting their potential clinical utility.
While our study highlights the efficacy of using SWNTs to image inflamed plaque, one of its limitations includes the murine model itself. While we have studied this model extensively[13,16,28] and we show that these inflamed atherosclerotic plaques recapitulate the cellular content of human vulnerable plaques, our model does not spontaneously lead to plaque disruption/thrombosis, which is a known challenge of most animal models. Because of the lack of such models, while we have not proven the capability to predict disease vulnerability/severity with the current study, our data reveal the ability to accurately identify inflamed plaques. A second limitation is that, in our experiments, PAI was performed with exteriorized carotid arteries ex vivo. However, our in vivo results obtained using two different optical imaging modalities provide strong evidence to support translating our current proof-of-concept PAI studies into living subjects. These strategies, in vivo FMT imaging of Cy5.5 on SWNTs and NIR-II imaging of intrinsic SWNT fluorescence (Figure S5B–5C), consistently corroborate the high SWNT uptake (based on flow cytometry) and PAI signal in the carotid artery. Finally, in view of the high uptake of SWNTs by inflammatory monocytes and macrophages in the plaques within relatively short time periods, it is likely that the signal observed arises from a combination of monocytes trafficking to the plaque and uptake by cells resident in the plaque (e.g., in situ uptake by foamy macrophages). Given the strong signal enhancement detected, follow-up studies will include directly imaging the carotids in living subjects. Future goals of this work include in vivo murine PAI studies, translation of the contrast agent for noninvasive carotid artery imaging in patients, a combined IVPA/IVUS platform to intravascularly image plaque-selective nanoparticle contrast using PAI, and eventually a theranostic nanomaterial capable of not only highly selective diagnostic imaging, but also precision treatment.
4. Materials and Methods
4.1. Single-walled carbon nanotubes
Biocompatible SWNTs conjugated with a fluorescent dye were prepared as previously reported[22,25–27]. Briefly, an aqueous solution of Hipco SWNTs (Raw, Lot# R0513, Unidym, Sunnyvale, CA) were combined with DSPE-PEG5000-Amine (NOF Corp), sonicated for 1h, then centrifuged at 24,000 g for 6h, yielding PEGylated SWNTs. Filtration through 100 kDa filters (Millipore) removed excess coating polymer. For conjugation of Cy5.5 to SWNTs, Cy5.5-NHS (Invitrogen) was mixed with the SWNT solution at pH 7.4 for 2h. Excess dye was removed via centrifugal filtrations through 100 kDa filters and then washed away by distilled water. SWNTs are approximately 2 nm width X 200 nm in length ([22] and Figure S11).
SWNT concentrations were established spectrophotometrically (using a DU 640 from Beckman Coulter, Fullerton, CA) with an extinction coefficient of 7.9 × 106 mol−1·cm−1 at 808 nm as described previously[22]. They were further analyzed for Cy5.5 content (~ 20 Cy5.5 dyes per SWNT).
4.2. Carotid-ligation model
Eight-week-old male FVB mice were fed a high-fat diet (D12109, Research Diets, Inc., New Brunswick, NJ, USA) for 1 month, and then diabetes was induced by 5 daily intraperitoneal injections of streptozotocin (STZ, 40 mg/kg, Sigma-Aldrich)[16,28]. Two weeks after the initiation of STZ injection, the left common carotid artery was ligated below the bifurcation. The study protocol was approved by the Stanford University Administrative Panel on Laboratory Animal Care (10051) and the Michigan State University IACUC (AAALAC International).
4.3. Photoacoustic imaging
SWNT-Cy5.5 (400 nM in 180 µL) was injected into mice via tail vein. A VisualSonics Vevo LAZR system (VisualSonics Inc, Toronto, Canada) was used to evaluate photoacoustic signal from the mouse arteries at timepoints 6 h, 24 h, 48 h, and 72 h post-injection. For PAI, carotid artery samples were immersed in 1% agarose gel in de-ionized water and placed in a culture dish, which was affixed to the bottom of a plastic box. Once dry, the plastic box was filled with de-ionized water for use as the photoacoustic coupling agent. The box was placed in the photoacoustic chamber, and photoacoustic signals were excited by a pulsed (20 Hz, <10 ns pulse-width), tunable Nd:YAG laser (OPOTEK Inc., Carlsbad, CA, USA). Three-dimensional signals were visualized as a maximal intensity projection. B-mode ultrasound images at 16 MHz were acquired at the same time as photoacoustic signal. Acquired images were evaluated with a region-of-interest analysis using Vevo software and a custom Matlab program.
4.4. Flow cytometry
Carotid arteries (N=3 mice at each time point, 6 h, 24 h, 48 h, and 72 h post-SWNT injection, for both left (ligated) and right (non-ligated) arteries) were digested in an enzymatic solution (Collagenase-I and -XI, hyaluronidase-I, and DNase-I) to obtain single-cell suspensions. At each time-point, peripheral blood was also drawn from the tail vein into heparin-containing PBS solution. Red blood cells were lysed using ammonium-chloride-potassium (ACK) lysing buffer. Digested arteries and white blood cells were separately suspended in deficient RPMI-1640 medium containing 3% newborn calf serum (RPMI-1640 medium was obtained from Invitrogen and deficient in biotin, L-glutamine, phenol red, riboflavin, and sodium bicarbonate). Single-cell suspensions were pre-incubated with anti–CD16/CD32 mAb to block FcγRII/III receptors and stained on ice for 30 min with the following fluorochrome-conjugated mAbs in an 11-color staining combination: FITC-labeled anti-Ly-6C (AL-21); PE-labeled anti-IA/IE (M5/114.15.2) or AnnexinV; PE-Cy5-labeled anti-CD5 (53–7.3); PE-Cy5.5-labeled anti-CD19 (1D3) or anti-CD11c (N418); PE-Cy7-labeled anti-Gr-1 (RB6–8C5) or anti-CD11b (M1/70); APC-labeled anti-CD49b (DX5); APC-Cy7-labeled anti-CD11b (M1/70) or anti-CD45 (30-F11); Pacific Blue-labeled anti-F4/80 (BM8) or anti-Gr-1 (RB6–8C5); Pacific Orange-labeled anti-F4/80 (BM8), and biotin-labeled anti-CD11c (HL3), anti-CD80 (16–10A1), or anti-CD86 (GL-1). The Cy5.5-labeled SWNTs occupy the 730/745 nm bandpass filter off the red laser (633nm). Cells were then washed and stained again on ice for 15 min with streptavidin Qdot605 (Invitrogen) to reveal biotin-coupled antibodies. Antibodies were either purchased (Invitrogen and BD Pharmingen) or conjugated in our laboratory. After washing, stained cells were resuspended in 10 μg/mL propidium iodide (revealed in the PE-Texas Red channel) to exclude dead cells. Cells were analyzed on Stanford FACS facility instruments (Becton Dickinson LSRII). Data were collected for 20–200 × 105 cells. Data were analyzed with FlowJo software (TreeStar). To distinguish autofluorescent cells from cells expressing low levels of individual surface markers, we established upper thresholds for autofluorescence by staining samples with fluorescence-minus-one (FMO) control stain sets in which a reagent for a channel of interest is omitted. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supporting Information. Additional data are available from authors upon request.
4.5. Statistics
To assess the statistical significance of PAI across time-points and controls (left vs. right), we converted each time-point into a ratio of left signal/right signal. We then performed a Jonckheere-Terpstra test for trend, which indicates a significant difference over time in the disease group compared with the control group. We also developed a non-parametric multivariable model that was fit with the R package ‘Rfit’ and analyzed for the effects of disease and interaction of disease-by-time.
Supplementary Material
Acknowledgements
We gratefully thank Professor Eric Keyser (Stanford) for comments and critical reading; Megan Phillips and Jeffrey Waters for technical help; Mitul Desai for analytical support; and Drs. Ping Wang and Sihai Liu for support with animal surgeries. We gratefully acknowledge our funding sources, including the American Heart Association (AHA) Transformational Project Award 18TPA34230113 (BRS), NIH K99 CA160764 (BRS), National Institutes of Health (NIH) NIAID R01 AI123126 (EEBG), NIH P50 CA114747 (SSG), and Herzenberg Lab funds.
We dedicate this manuscript to the memory of Professor Sanjiv Sam Gambhir.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Disclosures
MVM has received MRI research support from GE Healthcare and is currently on partial leave of absence from Stanford and employed by Verily Life Sciences. NL is a co-founder and holds equity interest in 47 Incorporated.
REFERENCES
- [1].Ley K, Miller YI, Hedrick CC, Arterioscler. Thromb. Vasc. Biol 2011, 31, 1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gui T, Shimokado A, Sun Y, Akasaka T, Muragaki Y, Mediators Inflamm 2012, 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tearney GJ, Yabushita H, Houser SL, Aretz HT, Jang IK, Schlendorf KH, Kauffman CR, Shishkov M, Halpern EF, Bouma BE, Circulation 2003, 107, 113. [DOI] [PubMed] [Google Scholar]
- [4].Williams K, Zaitsev JW, Kim K, Nat Immunol 2020, 21, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Barascuk N, Skjøt-Arkil H, Register TC, Larsen L, Byrjalsen I, Christiansen C, Karsdal MA, BMC Cardiovasc. Disord 2010, 10, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Seneviratne A, Hulsmans M, Holvoet P, Monaco C, Cardiovasc. Res 2013, 99, 284. [DOI] [PubMed] [Google Scholar]
- [7].De Meyer I, Martinet W, De Meyer GRY, Br. J. Clin. Pharmacol 2012, 74, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Woollard KJ, Geissmann F, Nat. Rev. Cardiol 2010, 7, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ghosn EEB, Cassado AA, Govoni GR, Fukuhara T, Yang Y, Monack DM, Bortoluci KR, Almeida SR, Herzenberg LA, Herzenberg LA, Proc. Natl. Acad. Sci. U. S. A 2010, 107, 2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F, Science 2007, 317, 666. [DOI] [PubMed] [Google Scholar]
- [11].Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ, J. Clin. Invest 2007, 117, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kojima K, Volkmer Y, McKenna J, Nature 2016, 536, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Withana NP, Saito T, Ma X, Garland M, Liu C, Kosuge H, Amsallem M, Verdoes M, Ofori LO, Fischbein M, Arakawa M, Cheng Z, McConnell MV, Bogyo M, J. Nucl. Med 2016, 57, 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Weissleder R, Nahrendorf M, Pittet MJ, Nat. Mater 2014, 13, 125. [DOI] [PubMed] [Google Scholar]
- [15].Smith BR, Heverhagen J, Knopp M, Schmalbrock P, Shapiro J, Shiomi M, Moldovan NI, Ferrari M, Lee SC, Biomed. Microdevices 2007, 9, 719. [DOI] [PubMed] [Google Scholar]
- [16].Kosuge H, Terashima M, Uchida M, Sherlock S, Tsao PS, Young MJ, Douglas T, Dai H, McConnell MV, J. Cardiovasc. Magn. Reson 2009, 11, P151. [Google Scholar]
- [17].Smith BR, Gambhir SS, Chem. Rev 2017, 117, 901. [DOI] [PubMed] [Google Scholar]
- [18].Hu X-B, Zhang P-F, Su H-J, Yi X, Chen L, Rong Y-Y, Zhang K, Li X, Wang L, Sun C-L, Cai X-J, Li L, Song J-T, Dai X-M, Sui X-D, Zhang Y, Zhang M, Ultrasound Med. Biol 2011, 37, 1579. [DOI] [PubMed] [Google Scholar]
- [19].Antonopoulos AS, Sanna F, Sabharwal N, Thomas S, Oikonomou EK, Herdman L, Margaritis M, Shirodaria C, Kampoli A-M, Akoumianakis I, Petrou M, Sayeed R, Krasopoulos G, Psarros C, Ciccone P, Brophy CM, Digby J, Kelion A, Uberoi R, Anthony S, Alexopoulos N, Tousoulis D, Achenbach S, Neubauer S, Channon KM, Antoniades C, Sci. Transl. Med 2017, 9, eaal2658. [DOI] [PubMed] [Google Scholar]
- [20].Lameijer MA, Tang J, Nahrendorf M, Beelen RHJ, Mulder WJM, Expert Rev. Mol. Diagn 2013, 13, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Moghimi SM, Parhamifar L, Ahmadvand D, Wibroe PP, Andresen TL, Farhangrazi ZS, Hunter a C., J. Innate Immun 2012, 4, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Smith BR, Ghosn EEB, Rallapalli H, Prescher JA, Larson T, Herzenberg LA, Gambhir SS, S. B.R., G. E.E.B., R. H., P. J.A., L. T., H. L.A., G. S.S., Nat. Nanotechnol 2014, 9, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schipper ML, Nakayama-Ratchford N, Davis CR, Kam NWS, Chu P, Liu Z, Sun X, Dai H, Gambhir SS, Nat. Nanotechnol 2008, 3, 216. [DOI] [PubMed] [Google Scholar]
- [24].Alidori S, Akhavein N, Thorek DLJ, Behling K, Romin Y, Queen D, Beattie BJ, Manova-Todorova K, Bergkvist M, Scheinberg DA, McDevitt MR, Sci. Transl. Med 2016, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu Z, Cai W, He L, Nakayama N, Chen K, Sun X, Chen X, Dai H, Nat. Nanotechnol 2007, 2, 47. [DOI] [PubMed] [Google Scholar]
- [26].Smith BR, Kempen P, Bouley D, Xu A, Liu Z, Melosh N, Dai H, Sinclair R, Gambhir SS, Nano Lett 2012, 12, 3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhu X, Vo C, Taylor M, Smith BR, Mater. Horizons 2019, 6, 1094. [Google Scholar]
- [28].Jiang L, Tu Y, Kimura RH, Habte F, Chen H, Cheng K, Shi H, Gambhir SS, Cheng Z, J. Nucl. Med 2015, 56, 939. [DOI] [PubMed] [Google Scholar]
- [29].Terashima M, Ehara S, Yang E, Kosuge H, Tsao PS, Quertermous T, Contag CH, McConnell MV, Mol. Imaging Biol 2011, 13, 1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kosuge H, Sherlock SP, Kitagawa T, Terashima M, Barral JK, Nishimura DG, Dai H, McConnell MV, PLoS One 2011, 6, e14523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hilgendorf I, Swirski FK, Curr. Atheroscler. Rep 2012, 14, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Smith BR, Zavaleta C, Rosenberg J, Tong R, Ramunas J, Liu Z, Dai H, Gambhir SS, Nano Today 2013, 8, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Foster EJ, Gower GA, Stanhope RM, Havel KL, Simon PJ, Armstrong SI, Proc. Natl. Acad. Sci. (PNAS) 2013, 110, 13944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chaudhary JR, Roy R, Kanwar K, Walder RK, Kanwar K, J. Nanobiotechnology 2016, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bartels S, Franco AR, Rundek T, Perspect. Med 2012, 1, 139. [Google Scholar]
- [36].Wald DS, Bestwick JP, J. Med. Screen 2009, 16, 147. [DOI] [PubMed] [Google Scholar]
- [37].Zhang J, Yang S, Ji X, Zhou Q, Xing D, J. Am. Coll. Cardiol 2014, 64, 385. [DOI] [PubMed] [Google Scholar]
- [38].Lutter C, Joner M, Contin. Cardiol. Educ 2016, 2, 56. [Google Scholar]
- [39].Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, N. Engl. J. Med 2017, 377, 1119.28845751 [Google Scholar]
- [40].Everett BM, Pradhan AD, Solomon DH, Paynter N, Macfadyen J, Zaharris E, Gupta M, Clearfield M, Libby P, Hasan AAK, Glynn RJ, Ridker PM, Am. Heart J 2013, 166, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Flores AM; Hosseini-Nassab N; Jarr KU; Ye J; Zhu X; Wirka R; Koh AL; Tsantilas P; Wang Y; Nanda V; Kojima Y; Zeng Y; Lotfi M; Sinclair R; Weissman IL; Ingelsson E; Smith BR; Leeper NJ, Nat Nanotechnol 2020, 15, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhang BR, Hosseini-Nassab Y, Flores N, Kalashnikova A, Paluri I, Lotfi SLA, Leeper M, Smith NJ, Nano Res 2020.
- [43].Liang X, Deng Z, Jing L, Li X, Dai Z, Li C, Huang M, Chem. Commun. (Camb) 2013, 49, 11029. [DOI] [PubMed] [Google Scholar]
- [44].Yeager D, Karpiouk A, Wang B, Amirian J, Sokolov K, Smalling R, Emelianov S, J. Biomed. Opt 2012, 17, 106016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rouleau L, Berti R, Ng VWK, Matteau-Pelletier C, Lam T, Saboural P, Kakkar AK, Lesage F, Rhéaume E, Tardif J-C, Contrast Media Mol. Imaging 8, 27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.