

Abstract
Chronic infection with Helicobacter pylori increases risk of gastric diseases including gastric cancer. Despite development of a robust immune response, H. pylori persists in the gastric niche. Progression of gastric inflammation to serious disease outcomes is associated with infection with H. pylori strains which encode the cag Type IV Secretion System (cag T4SS). The cag T4SS is responsible for translocating the oncogenic protein CagA into host cells and inducing pro-inflammatory and carcinogenic signaling cascades. Our previous work demonstrated that nutrient iron modulates the activity of the T4SS and biogenesis of T4SS pili. In response to H. pylori infection, the host produces a variety of antimicrobial molecules, including the iron-binding glycoprotein, lactoferrin. Our work shows that apo-lactoferrin exerts antimicrobial activity against H. pylori under iron-limited conditions, while holo-lactoferrin enhances bacterial growth. Culturing H. pylori in the presence of holo-lactoferrin prior to co-culture with gastric epithelial cells, results in repression of the cag T4SS activity. Concomitantly, a decrease in biogenesis of cag T4SS pili at the host-pathogen interface was observed under these culture conditions by high-resolution electron microscopy analyses. Taken together, these results indicate that acquisition of alternate sources of nutrient iron plays a role in regulating the pro-inflammatory activity of a bacterial secretion system and present novel therapeutic targets for the treatment of H. pylori-related disease.
Keywords: Helicobacter pylori, lactoferrin, innate immunity, glycobiology, bacterial pathogenesis, antimicrobial, host-pathogen interactions, virulence, toxin secretion, iron homeostasis
TOC
Helicobacter pylori infection results in enhanced host production of lactoferrin which participates 554 in nutritional immunity and can also serve as a source of nutrient iron for this pathogen, which 555 can repress a major oncogenic virulence factor, the cag T4SS.
Introduction
The nutritional requirement for transition metals in a variety of biological processes is a feature of all kingdoms of life. Most bacterial pathogens require iron as a cofactor for cellular processes including DNA replication, respiration, electron transport, stress responses, and cellular division. The host deploys a variety of strategies to limit available iron, effectively starving invading pathogens in a process termed, “nutritional immunity” [1,2]. Nutritional immunity in a vertebrate host includes numerous proteins such as calprotectin, calgranulin C, ferritin, transferrin, hemoglobin, and lactoferrin.
Lactoferrin, or lactotransferrin, is a 80 kDA protein expressed by chromosome 3 in humans [3]. When translated, lactoferrin is a single polypeptide chain comprised of 692 amino acids, which is folded into two symmetrical lobes [4] and contains two high affinity iron binding sites but can also bind other transition metals [5]. As lactoferrin across mammalian species possesses the ability to bind iron and share 99% sequence homology, the heterogeneity of the biological properties of lactoferrins, such as the ability to modulate the immune system, is a result of its unique glycosylation patterns [6]. In humans, lactoferrin is expressed in large amounts in breast milk and secondary granules of neutrophils, and also by mucosal epithelial cells in the mucus [7]. Additionally, lactoferrin is found on neutrophil extracellular traps during infections [8]. Lactoferrin has antimicrobial activity against a wide range of bacteria, viruses, and fungi [9]. The protein largely controls infections by its iron-chelating activity as part of host nutritional immunity [1]. Lactoferrin is a positively charged molecule and can interact with negatively charged membranes, resulting in depolarization and perforation of the pathogen surface, ultimately perturbing basic bacterial processes [10]. Unlike transferrin, the structure of lactoferrin also confers the ability to survive digestion within the human gastric tract, making it an attractive candidate for oral supplementation [11].
Helicobacter pylori is a Gram-negative bacterial pathobiont which is a member of the Epsilonproteobacteria. H. pylori infects nearly half of the world’s population and exploits the human stomach as its primary reservoir [12]. H. pylori infection occurs primarily in people living within developing nations or with lower socio-economic backgrounds [13]. While the majority of people colonized by this bacterium remain asymptomatic, pathogen persistence within the stomach increases risk of negative outcomes, including peptic and duodenal ulcer, gastritis, dysplasia, mucosa-associated lymphoid tissue (MALT) lymphoma, and invasive gastric cancer [14]. H. pylori infection is the main cause of chronic gastritis and peptic ulcer disease [15]. In fact, H. pylori is the first carcinogenic bacterium identified, as chronic inflammation from persistence is epidemiologically linked to adenocarcinoma of the distal stomach [16].
Chronic inflammation and adverse disease outcomes such as gastric cancer have been linked to H. pylori strain variations. Strains harboring the oncogenic cag pathogenicity island-encoded type IV secretion system (cag T4SS) are associated with enhanced risk of disease outcomes [17]. The cag T4SS is a macromolecular nanomachine that penetrates cell membranes to inject substrates, such as peptidoglycan and the oncogenic effector molecule CagA, into host epithelial cells [18]. This translocation of effector proteins results in multiple consequences for host cell biology, including cytoskeletal rearrangements, induction of carcinogenic cell-signaling cascades, alteration of DNA methylation, perturbation of metal homeostasis, and secretion of proinflammatory molecules [18–21]. Persistence of the microbe and its use of the T4SS eventuates in chronic inflammation and gastric intestinal diseases associated with H. pylori infections.
H. pylori infections are frequently associated with iron deficiency anemia [22]. In fact, the pathogen is able to impair iron absorption by the host [23]. Iron deficiency is linked to acceleration of H. pylori–induced carcinogenesis in rodents and humans [24]. Our previous work revealed that nutrient iron modulates activity of the T4SS and biogenesis of T4SS pili. Given that lactoferrin, an iron chelating antimicrobial molecule, is commonly expressed during bacterial infections, we sought to understand dynamics between lactoferrin and H. pylori during microbial-host interactions.
Materials and Methods
Bacterial strains, cell lines, and culture conditions
H. pylori strain 7.13 (a gerbil-adapted clinical isolate with a functional cag-T4SS), as well as an isogenic ΔcagE mutant was used for these studies [25, 26]. Bacteria were grown on tryptic soy agar plates supplemented with 5% sheep blood (blood agar plates), or in brucella broth supplemented with cholesterol at 37°C in ambient air containing 5% carbon dioxide. AGS human gastric adenocarcinoma cells (ATCC) were cultured to 60–70% confluency in RPMI medium supplemented with 2 mM L-glutamine, 10% FBS, and 10 mM HEPES buffer at 37°C in ambient air containing 5% carbon dioxide.
Mongolian gerbil model of chronic H. pylori gastric infection
The Mongolian gerbil model of chronic H. pylori infection was utilized as previously described [25]. All procedures were approved by the Vanderbilt University Medical Center Institutional Animal Care and Use Committee. Mongolian gerbils, aged 8 to 10 weeks (Charles River Laboratories) were challenged via orogastric gavage with two doses of 109 colony forming units (CFU) of H. pylori strain 7.13 in 0.5 mL of Brucella broth plus 10% fetal bovine serum over a 48 hour period. Infections were allowed to progress for 3 months post-inoculation, animals were euthanized, and gastric tissues were collected at necropsy. Gastric contents were gently removed, while each stomach was washed with phosphate-buffered saline and sections extending from the esophagogastric squamo-columnar junction to the proximal duodenum were prepared for immunohistochemical analyses.
Immunohistochemical analysis of lactoferrin in gastric tissues
Gastric tissues from 3 uninfected and 4 H. pylori infected gerbils were fixed in 10% buffered formalin and embedded in paraffin. Tissues were cut into 5 μm sections. After quenching with 0.03% hydrogen peroxide, antigen retrieval was performed by applying a heat-induced epitope retrieval solution (Universal De-Cloaker; Biocare Medical, Concord, CA) and using a pressure cooker at 121°C for 20 min. Samples were allowed to cool at room temperature prior to blocking with 10% normal goat serum in 0.1 M PBS (pH 7.4). Primary rabbit polyclonal antibody to lactoferrin (ThermoFisher) was applied for 1 h. Detection of primary antibody was performed using a rabbit horseradish peroxidase (HRP)-polymer system for 30 min and developed with 3,3’-diaminobenzidine tetrahydrochloride (H-DAB). Sections were counterstained with hematoxylin, rinsed, alcohol-dehydrated, and mounted with Cytoseal XYL before light microscopy analysis was performed. Micrographs were collected at a magnification of 100x and 400x. Micrograph images were analyzed with the ImageJ IHC toolbox plugin to quantify H-DAB staining by color detection, converted to 8-bit format, and densitometry quantification was carried out as previously described [27].
Preparation of lactoferrin isoforms
Iron-bound (holo-) or unbound (apo-) lactoferrin was prepared as previously described [8,28,29]. Briefly, a 10 mM stock of recombinant lactoferrin (Sigma Aldrich) was dialyzed against either 0.1 M sodium citrate-bicarbonate buffer (pH 8.2) alone to generate apo-lactoferrin, or 0.1 M sodium citrate-bicarbonate buffer (pH 8.2) containing 70 mM ferric chloride to generate holo-lactoferrin. Both apo- (apo-Lf) and holo-lactoferrin (holo-Lf) were dialyzed against 1X phosphate buffered saline (PBS) containing Chelex Resin (Sigma Aldrich) to remove any unbound iron from the samples.
Bacterial growth assays
For analysis of bacterial growth, H. pylori strain 7.13 were sub-cultured in brucella broth plus cholesterol referred to as “medium alone” or medium supplemented with increasing concentrations of 2, 2’-dipyridyl (DIP) alone or with 250 μM ferric chloride or 250 μg/mL of either apo- or holo-lactoferrin isoforms. Bacterial growth was evaluated by spectrophotometric reading of cell density as a measurement at 600 nm (OD600).
AGS co-culture assays
For co-culture assays, H. pylori strain 7.13 or the ΔcagE isogenic mutant, were sub-cultured in brucella broth supplemented with cholesterol referred to as “medium alone” [30] or medium supplemented with 100 μg/mL purified apo-Lf or holo-Lf, 100 M dipyridyl, 100 μM ferric chloride or combinations of these chemicals. Bacteria were grown at 37°C in ambient air containing 5% carbon dioxide overnight before being applied to AGS cells at a multiplicity of infection (MOI) of 100:1 (as determined by OD600 reading and plate counts of viable CFU/mL) for 4–6 hours of co-culture.
Evaluation of secreted IL-8
AGS cells were co-cultured with H. pylori as described above and co-culture supernatants were collected and centrifuged at 8,000 × g to remove cellular debris. Secreted IL-8 was evaluated by Quantikine human IL-8 sandwich ELISA (R&D Systems) per manufacturer’s instructions. IL-8 values were compared to those derived from co-culture samples from WT bacteria grown in medium alone as previously described [27,30].
High-resolution FEG-SEM of cag T4SS
Field emission gun scanning electron microscopy (FEG-SEM) of H. pylori cag T4SS at the host-pathogen interface was performed as previously described [30]. Briefly, co-cultures of H. pylori and AGS cells were grown on poly-L-lysine-treated coverslips. Samples were fixed with 2.0% paraformaldehyde, 2.5% glutaraldehyde in 0.05 M sodium cacodylate buffer (Electron Microscopy Sciences) for 1 h at room temperature. Samples were washed three times with 0.05 M sodium cacodylate buffer before secondary fixation with 1% osmium tetroxide (Electron Microscopy Sciences). Samples were sequentially dehydrated by washing with increasing concentrations of ethanol before being dried with a carbon dioxide critical point dryer (Tousimis), mounted on aluminum SEM stubs and sputter-coated with 20 nm of gold-palladium. A thin line of colloidal silver paint was applied at the sample edge to dissipate charging during FEG-SEM imaging. Samples were visualized with an FEI Quanta 250 FEG-SEM at high vacuum, and micrographs were analyzed with Image J software.
Statistical analyses
Statistical analysis of IL-8 secretion, immunohistochemistry, and pilus quantifications were performed using paired Student’s t test or one-way ANOVA. Analysis of growth data was performed using two-way ANOVA. All data analyzed in this work were derived from at least three separate biological replicates or two biological replicates with multiple technical replicates. Statistical analyses were performed using GraphPad Prism Software and Microsoft Excel.
Results
Lactoferrin production is enhanced in gastric mucosa in response to H. pylori infection
Our previous work demonstrated that antimicrobial proteins are produced in response to H. pylori infection of the gastric niche [26,29]. Additionally, we demonstrated that micronutrient iron influences H. pylori virulence and cognate disease outcomes. We, therefore, hypothesized that iron-binding antimicrobial proteins, such as lactoferrin, which is expressed by epithelial cells at mucosal surfaces, could play an important role in the host-pathogen dialogue during H. pylori gastric infection. could play an important role in the host-pathogen dialogue during H. pylori infection. First, we sought to determine if lactoferrin was produced in gastric tissues in response to H. pylori infection. To test this, we utilized a gerbil model of chronic H. pylori infection as previously described [24,29]. Gastric tissues were collected from uninfected negative control animals and H. pylori-infected animals at three months post-infection and evaluated by immunohistochemical techniques using a polyclonal rabbit antibody to lactoferrin. Microscopical analyses (Figure 1, panels A-D) revealed that lactoferrin (visualized by H-DAB as a dark brown stain) localized to the gastric epithelia proximal to the lumen of the stomach in uninfected animals (Figure 1, panels A and C), but in H. pylori-infected tissues, lactoferrin was present in higher abundance and deeper within tissues including gastric pits and as far down as the muscularis (Figure 1, panels B and D). Quantification of H-DAB stain (Figure 1, panel E) reveals a 9.3-fold increase in lactoferrin within H. pylori-infected gastric tissues compared to uninfected tissues; a result that was statistically significant (P=0.0148, Student’s t test with Welch’s correction, N=3–4 animals per condition).
Figure 1.
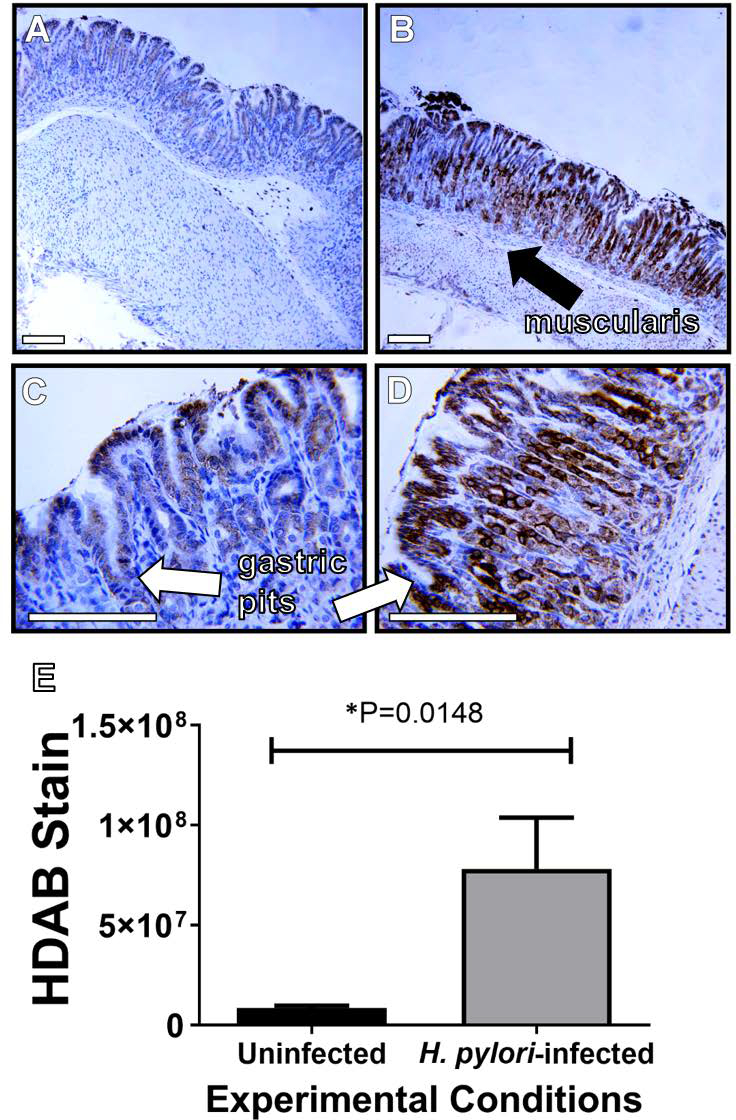
Lactoferrin is elevated in H. pylori-infected gastric tissues compared to uninfected gastric tissues. Immunohistochemical (IHC) and microscopical evaluation of gerbil gastric tissue specimens reveals elevated lactoferrin levels (indicated by the dark brown stain) associated with H. pylori chronic infection (B and D) compared to uninfected negative controls (A and C). Micrographs were collected at 100X (A and B) and 400X magnification (magnification bars indicate 100 μm) and representative micrographs are shown (N=3–4). Healthy, uninfected gastric tissue produces lower levels of lactoferrin, largely associated with gastric epithelia proximal to the lumen of the stomach. Infected tissues produce lactoferrin at higher levels and deeper into tissues towards gastric pits (white arrows) and muscularis (black arrows). H-DAB stain quantification (panel E) reveals that lactoferrin levels are significantly higher in H. pylori-infected gastric tissues compared to uninfected control tissues (*P=0.0148, Student’s t test with Welch’s correction).
Holo-lactoferrin promotes H. pylori growth under iron limiting conditions
Our previous work has shown that different isoforms of lactoferrin, either the iron-bound form (holo-lactoferrin) or the unbound form (apo-lactoferrin) exert different effects on bacterial cells [8]. Additionally, work from Senkovich and colleagues demonstrated that H. pylori can bind lactoferrin and utilize it as an iron source when grown in a chemically defined medium [29]. We hypothesized that H. pylori could exploit holo-lactoferrin as a source of nutrient iron under conditions of iron sequestration similar to those imposed by the vertebrate host during infection. To test this, H. pylori was grown in the presence of the synthetic iron chelator, 2, 2’-dipyridyl (DIP) (Figure 2). At a concentration of 250 μM, DIP inhibited H. pylori growth 58.2% after 48 hours of incubation compared to the medium alone negative control. Addition of 250 μM of ferric chloride (Fe) to DIP-treated cultures restored bacterial growth to 109% compared to medium alone, a result that was significantly higher than DIP-treated cultures (P<0.0001, two-way ANOVA). Similarly, addition of holo-lactoferrin to DIP-treated cultures restored bacterial growth to 105% compared to medium alone, a result that was significantly higher than DIP-treated cultures (P<0.0001, two-way ANOVA). Conversely, addition of apo-lactoferrin to DIP-treated cultures was unable to restore bacterial growth, and cultures were indistinguishable from DIP-treatment alone. Addition of apo-lactoferrin resulted in a 56% growth phenotype compared to medium alone, a result that was statistically significant (P<0.0001, two-way ANOVA).
Figure 2.

H. pylori can utilize holo-lactoferrin (holo-Lf) as an alternate source of nutrient iron under iron-limiting conditions. H. pylori growth in the presence of the synthetic iron chelator, dipyridyl (DIP), is attenuated. However, supplementation with an exogenous source of iron, such as ferric chloride (Fe) rescues growth. Similarly, supplementation with lactoferrin saturated with iron or holo-Lf enhances H. pylori growth in conditions where iron availability is low. Conversely, supplementation with lactoferrin lacking bound iron or apo-lactoferrin (apo-Lf) does not restore H. pylori growth under these conditions. (N=3, a, b, c = P<0.0001, comparing medium alone to medium supplemented with DIP + apo-Lf, and d, e, f = P<0.0001, comparing medium alone to medium supplemented with DIP, two-way ANOVA).
High-resolution FEG-SEM reveals H. pylori cag T4SS pilus deployment is abrogated by exposure to holo-lactoferrin
The cag T4SS is a macromolecular machine that engages host cells to deliver effector molecules, and upon host contact an extracellular organelle, commonly referred to as the “cag T4SS pilus” is elaborated, which can be visualized using high resolution microscopy [24, 26, 27, 30]. Detection of the extracellular organelle (commonly referred to as the T4SS pilus) by electron microscopy is a proxy for studying the assembly of the cag T4SS at the host-pathogen interface. Conditions of iron availability influence H. pylori virulence, including activity of the cag T4SS. Our previous work demonstrated that iron can repress biogenesis and activity of the cag T4SS [24]. Therefore, we hypothesized that lactoferrin-associated iron could affect elaboration of the cag T4SS. To test this, we employed high-resolution FEG-SEM analyses as previously described to visualize the cag T4SS at the host-pathogen interface [24, 26, 27, 30]. Bacteria were grown in culture medium alone or supplemented with either ferric chloride, apo-, or holo-lactoferrin prior to co-culture with human gastric epithelial cells. SEM analyses revealed that exposure to ferric iron or holo-lactoferrin repressed formation of cag T4SS pili at the bacterial-host interface (Figure 3A). Quantification of cag T4SS pili per cell (derived from direct counts of more than 60 cells per experimental condition) revealed an average of 5 pili per cell in medium alone conditions (Figure 3B). Addition of either ferric chloride (Fe) or holo-lactoferrin (holo-Lf) resulted in an 87% and 84% reduction in mean pili per cell, respectively (P<0.0001, one-way ANOVA compared to medium alone). Analysis of percentage of piliated cells (Figure 3C) indicates that exposure to ferric chloride results in a 63% inhibition compared to medium alone (P<0.05, one-way ANOVA). Exposure to holo-lactoferrin results in a 65% inhibition of piliated cells compared to medium alone (P<0.05, one-way ANOVA), and a 66% inhibition compared to cultures treated with apo-lactoferrin (P<0.05, one-way ANOVA).
Figure 3.
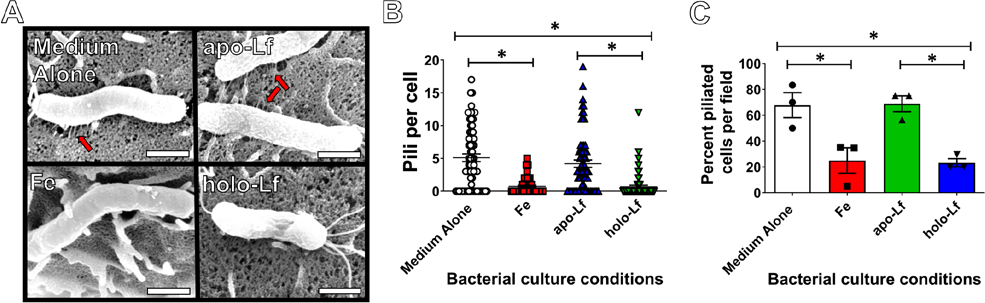
High resolution scanning electron microscopy analysis of H. pylori cag T4SS. Bacteria were grown in culture medium alone or supplemented with either ferric chloride, apo-, or holo-lactoferrin prior to co-culture with human gastric epithelial cells. A) SEM analyses revealed that exposure to ferric iron (Fe) or holo-lactoferrin (holo-Lf) repressed the formation of cag T4SS pili at the bacterial-host interface, whereas bacterial exposure to medium alone (Medium Alone) or apo-lactoferrin (apo-Lf) results in pili biogenesis. B) Quantification of cag T4SS pili per cell and C) analysis of percentage of piliated cells indicates that exposure to ferric chloride or holo-lactoferrin results in inhibition of piliation compared to medium alone (P<0.05, one-way ANOVA). Magnification bar indicates 500 nm. Red arrows indicate cag T4SS pili. (N=3 biological replicates, *P<0.05, One-way ANOVA).
H. pylori induction of human gastric epithelial cell IL-8 secretion is perturbed by bacterial exposure to holo-lactoferrin
Because holo-lactoferrin repressed elaboration of the cag T4SS, we hypothesized holo-lactoferrin would also repress function of the cag T4SS. One consequence of an active cag T4SS is induction of proinflammatory signaling cascades that ultimately lead to host cell IL-8 secretion [24, 26, 27, 30]. Therefore, we tested our hypothesis by collecting supernatants from co-cultures and analyzed them for IL-8 secretion using a sandwich ELISA (Figure 4). H. pylori exposed to ferric chloride prior to co-culture with AGS cells elicited 62% less IL-8 compared to bacteria grown in medium alone, a result that was statistically significant (P=0.00077, Student’s t test). Interestingly, co-cultures containing bacteria grown in the presence of holo-lactoferrin exhibited a 52% decrease in IL-8 secretion compared to bacteria grown in medium alone (P=0.0190, Student’s t test). Samples exposed to holo-lactoferrin were statistically indistinguishable from samples exposed to ferric chloride (P=0.2523, Student’s t test), whereas bacteria exposed to apo-lactoferrin induced significantly more IL-8 than those exposed to ferric iron (P=0.0049, Student’s t test). Together, these results indicate that holo-lactoferrin pretreatment mimics ferric chloride pretreatment by repressing the activity of the cag T4SS.
Figure 4.
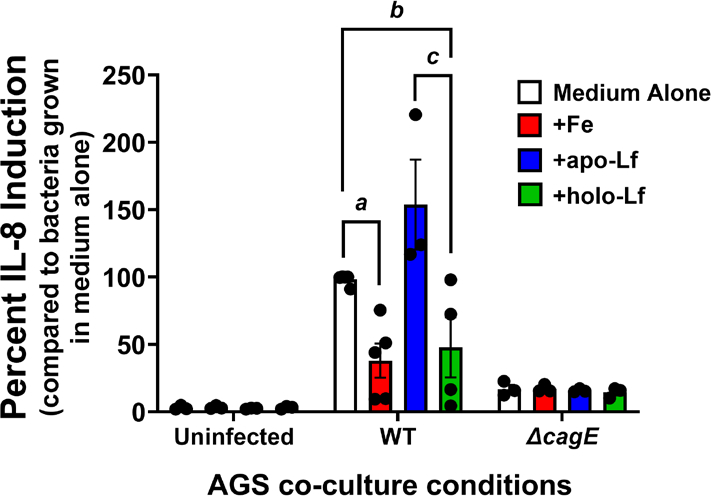
Analysis of human gastric epithelial cell IL-8 response to the H. pylori cag T4SS. H. pylori was grown in culture medium alone or supplemented with either ferric chloride, apo-, or holo-lactoferrin prior to co-culture with human gastric epithelial cells. ELISA analysis of IL-8 secretion (a proxy for cag T4SS activity) revealed that exposure to ferric iron (Fe) or holo-lactoferrin (holo-Lf) repressed the activity of the cag T4SS, whereas bacterial exposure to medium alone (Medium Alone) or apo-lactoferrin (apo-Lf) results in robust IL-8 secretion, indicative of cag T4SS activity. (N=3–5 biological replicates, a,b,c=P<0.05, One-way ANOVA).
Discussion
Lactoferrin is expressed during H. pylori infections
Our previously published work, and the work of others, has indicated that lactoferrin is produced in response to bacterial infection of host tissues [8,31]. Our work in this manuscript demonstrates that H. pylori infection induces a robust increase in lactoferrin within gastric tissues of infected gerbils. Similarly, Doğan and colleagues previously showed that lactoferrin levels are higher in gastric tissues derived from H. pylori-positive patients compared to patients who tested negative for H. pylori. Additionally, lactoferrin has been observed to be elevated in gastric juices derived from H. pylori-positive patients compared to H. pylori-negative patients [32]. Taken together, these results demonstrate that lactoferrin is a component of the innate immune system’s antimicrobial response to H. pylori infection. These data also underscore the utility of lactoferrin as a potential biomarker for H. pylori infection and associated gastritis.
H. pylori utilizes lactoferrin as an iron source during chronic infection
Previous work demonstrated that peptides derived from lactoferrin, such as lactoferricin, have antimicrobial activity against H. pylori. In vitro culture studies indicate lactoferricin has rapid, dose-dependent bactericidal activity against H. pylori, indicating peptic cleavage of lactoferrin could result in enhanced antimicrobial activity in the gastric niche. Interestingly, pre-saturation of lactoferrin with iron ablated its antimicrobial activity against H. pylori [33]. Our results demonstrate that H. pylori can utilize lactoferrin as a source of nutrient iron under conditions where iron availability is low. Previous reports by Senkovich et al., demonstrated that H. pylori can bind to both apo- and holo-lactoferrin and that the utility of lactoferrin as a nutrient source for H. pylori is dependent on the level of iron saturation in a chemically defined medium [29]. Similarly, Husson and colleagues showed that 30% saturated lactoferrin from pooled lactosera samples was capable of stimulating H. pylori growth under iron-limited conditions [34]. Our work using recombinant lactoferrin in the apo- and holo-isoforms further confirms that H. pylori can utilize the holo- isoform as a source of nutrient iron under conditions of iron sequestration. Although our work and the work of others has demonstrated that H. pylori can bind holo-lactoferrin and use it as a nutrient source, the mechanism by which iron is liberated from lactoferrin remains unclear. H. pylori cells have not been observed to synthesize siderophores, however they do produce a variety of siderophore receptors and iron acquisition functions, including lactoferrin and transferrin receptors which are highly expressed in iron-deficient conditions [28], which could aid in this process in vivo.
Lactoferrin and other nutritional immunity proteins inhibits H. pylori virulence
In addition to altering bacterial growth, lactoferrin alters H. pylori cell biology [33]. Exposure to peptic-digested lactoferrin products such as lactoferricin resulted in a significant decrease in H. pylori urease activity; an enzyme required to overcome the acidic environment in the gastric niche. Furthermore, our work has shown that several antimicrobial proteins produced by the innate immune system can repress H. pylori virulence via nutritional immunity strategies targeting transition metals. Specifically, S100A-family proteins such as calgranulin C (S100A12) [27, 35] and calprotectin (S100A8/A9) [30] can repress H. pylori growth, viability, and cag T4SS biogenesis and activity. These proteins, like lactoferrin, are produced as part of the antimicrobial repertoire the innate immune system deploys to control bacterial infections. Thus, our current work, together with our previous work, indicate that S100A12, S100A8/A9, and lactoferrin can repress H. pylori virulence. Our group has also demonstrated that lactoferrin possesses antimicrobial properties against other bacterial pathogens such as Streptococcus agalactiae (Group B Streptococcus) [28] and Acinetobacter baumannii [36] through the conserved immune strategy of nutritional immunity.
Iron acquired from iron-bound lactoferrin represses cag T4SS
Our previous work has also shown that nutrient iron represses the cag T4SS and influences disease outcome [24]. Our current work supports this framework, as apo- isoforms of lactoferrin were unable to repress elaboration of cag T4SS pili as well as the activity of the cag T4SS. Conversely, holo-lactoferrin can repress H. pylori virulence by participating in the repression of the proinflammatory and oncogenic cag T4SS, although the direct mechanism of this regulation remains obscure. Similarly, Huynh and colleagues observed that lactoferrin and desferrioxamine (an iron chelator) enhanced gastric inflammation, underscoring the role that iron availability plays in H. pylori virulence [37]. It is tantalizing to speculate that when host iron levels are low, lactoferrin produced in response to infection is likely to be in the apo- isoform, and conversely, when iron is high lactoferrin is more likely to be in the holo- isoform. Our previous work showed that when a host is iron deficient disease progression such as carcinogenesis is enhanced, and when iron supplementation is used, it decreases carcinogenesis. Additionally, unbiased proteomics experiments revealed that exposure to iron-deficient conditions enhanced H. pylori production of cag pathogenicity island components including CagA, and that the regulation of this was likely independent of the ferric uptake regulator (FUR) [24].
Lactoferrin as a potential chemotherapeutic strategy to treat H. pylori infections
Because of its antimicrobial utility against bacterial infections, lactoferrin has piqued interest of researchers and clinicians as a potential chemotherapeutic strategy [38,39]. Lactoferrin has been shown in several studies to enhance efficacy of levofloxacin-based triple therapy against H. pylori and to aid in control of H. pylori infection when adsorbed to hydroxyapatite nanocrystals [40,41]. However, other studies have shown that lactoferrin, when given as a single dose, was unable to convert patient urea breath tests from positive to negative. Additionally, there was no consistent change in urea breath test count to indicate suppression of H. pylori in the gastric niche [42]. Our results, together with the results of others, indicate that the particular isoform of lactoferrin is important for antimicrobial activity, and these salient differences could account for the controversy in the literature. Thus, supplementation with lactoferrin could be effective in improving eradication rates of anti-H. pylori therapy, but specific attention should be paid to both the iron-binding status of the lactoferrin molecule as well as host iron status.
Conclusions
In conclusion, we propose a model of the interaction of H. pylori with lactoferrin (Figure 5) in which H. pylori infection of the gastric niche results in host production of lactoferrin. Apo-lactoferrin lacks iron and, therefore, cannot be utilized as a source of nutrient iron under iron-limited conditions, resulting in elaboration of the cag T4SS at the host interface. H. pylori cag T4SS translocates proinflammatory substrates, including the oncogenic CagA cytotoxin, into host gastric epithelial cells. As a consequence of the activity of the H. pylori cag T4SS, host cells upregulate production of the proinflammatory cytokine and chemokine interleukin 8 (IL-8). In contrast, holo-lactoferrin can be utilized as a source of nutrient iron for H. pylori in conditions where iron availability is low. Holo-lactoferrin represses the elaboration of the H. pylori cag T4SS and represses the proinflammatory activity of this toxin secretion system, resulting in decreased IL-8 secretion by host cells.
Figure 5.
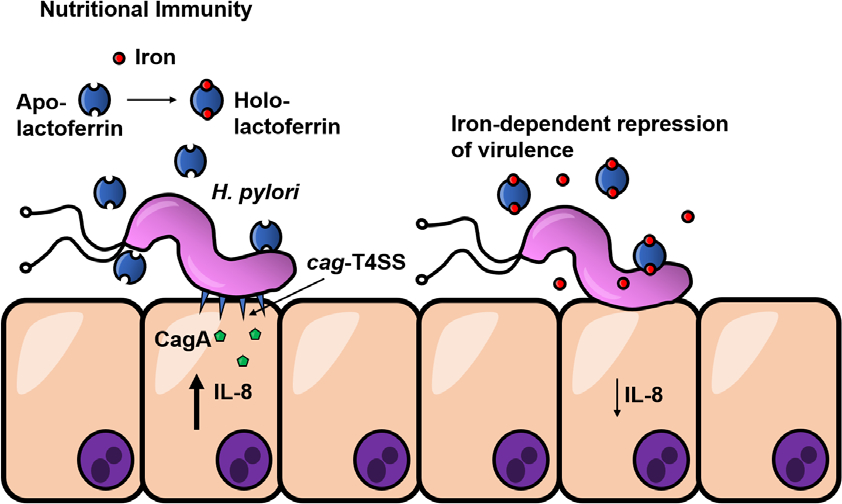
Conceptual model of the interaction of H. pylori with lactoferrin. H. pylori infection of the gastric niche results in production of lactoferrin. Apo-lactoferrin lacks iron and, therefore, cannot be utilized as a source of nutrient iron under iron-limited conditions, resulting in the elaboration of the cag T4SS at the host interface. The H. pylori cag T4SS translocates proinflammatory substrates, including the oncogenic CagA cytotoxin, into host gastric epithelial cells. As a consequence of the activity of the H. pylori cag T4SS, host cells upregulate production of the proinflammatory cytokine and chemokine interleukin 8 (IL-8). Holo-lactoferrin can be utilized as a source of nutrient iron for H. pylori in conditions where iron availability is low. Holo-lactoferrin represses the elaboration of the H. pylori cag T4SS and represses the proinflammatory activity of this toxin secretion system, resulting in decreased IL-8 secretion by host cells.
Acknowledgements
This work has been funded primarily by a Career Development Award IK2BX001701 (to J.A.G) from the Office of Medical Research, Department of Veterans Affairs and by the National Institutes of Health grant R01 HD090061 (to J.A.G.), and by NIH T32 HL007411–36S1 (supporting J.L.), Childhood Infections Research Program T32-AI095202 (supporting K.P.H.), 2T32AI112541–06 (supporting J.F.), K08AI151100 (supporting R.S.D.), and R35GM133602 (to S.D.T.). Additional funding from the National Science Foundation Award Numbers 1547757 and 1400969, and NIH grant GM05551 (to S.M.D.) supported this work. Core services, including use of the Cell Imaging Shared Resource and the Tissue Morphology Subcore, were performed through both Vanderbilt University Medical Center’s Digestive Disease Research Center, supported by NIH grant P30DK058404 Core Scholarship, and the Vanderbilt Institute for Clinical and Translational Research program, supported by the National Center for Research Resources, grant UL1RR024975–01, and the National Center for Advancing Translational Sciences grant 2UL1TR000445–06, 5UL1TR002243–03.
References
- [1].Hood MI, Skaar EP, Nat. Rev. Microbiol 2012, 10, 525–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lu J, Francis J, Doster RS, Haley KP, Craft KM, Moore RE, Chambers SA, Aronoff DM, Osteen K, Damo SM, Manning S, Townsend SD, Gaddy JA, ACS Infect. Dis. 2020, DOI 10.1021/acsinfecdis.0c00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yi HM, Li H, Peng D, Zhang HJ, Wang L, Zhao M, Yao KT, Ren CP, Oncol. Res 2006, DOI 10.3727/000000006783981008. [DOI] [PubMed] [Google Scholar]
- [4].Anderson BF, Baker HM, Norris GE, Rice DW, Baker EN, J. Mol. Biol. 1989, 209, Structure of human lactoferrin: Crystallographic s. [DOI] [PubMed] [Google Scholar]
- [5].Appelmelk BJ, An YQ, Geerts M, Thijs BG, De Boer HA, MacLaren DM, De Graaff J, Nuijens JH, Infect. Immun 1994, 62, 2628–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Karav S, German JB, Rouquié C, Le Parc A, Barile D, Int. J. Mol. Sci 2017, 18, DOI 10.3390/ijms18040870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].García-Montoya IA, Cendón TS, Arévalo-Gallegos S, Rascón-Cruz Q, Biochim. Biophys. Acta 2012, 1820, 226–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kothary V, Doster RS, Rogers LM, Kirk LA, Boyd KL, Romano-Keeler J, Haley KP, Manning SD, Aronoff DM, Gaddy JA, Front. Cell. Infect. Microbiol 2017, 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wiesner J, Vilcinskas A, Virulence 2010, 1, 440–64. [DOI] [PubMed] [Google Scholar]
- [10].Ulvatne H, Haukland HH, Olsvik O, Vorland LH, FEBS Lett. 2001, 492, 62–5. [DOI] [PubMed] [Google Scholar]
- [11].Troost FJ, Steijns J, Saris WHM, Brummer RJM, J. Nutr 2001, DOI 10.1093/jn/131.8.2101. [DOI] [PubMed] [Google Scholar]
- [12].Cover TL, Blaser MJ, Gastroenterology 2009, DOI 10.1053/j.gastro.2009.01.073. [DOI] [Google Scholar]
- [13].Mandeville KL, Krabshuis J, Ladep NG, Mulder CJJ, Quigley EMM, Khan SA, World J Gastroenterol. 2009, DOI 10.3748/wjg.15.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Stolte M, Lancet 1992, DOI 10.1016/0140-6736(92)90645-J. [DOI] [Google Scholar]
- [15].Blaser MJ, in J. Infect. Dis, 1999. [DOI] [PubMed] [Google Scholar]
- [16].Parsonnet J, in Environ. Health Perspect, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ding SZ, Goldberg JB, Hatakeyama M, Futur. Oncol 2010, DOI 10.2217/fon.10.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Posselt G, Backert S, Wessler S, Cell Commun. Signal. 2013, DOI 10.1186/1478-811X-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sugiyama T, Asaka M, Med. electron Microsc. Off. J. Clin. Electron Microsc. Soc. Japan 2004, 37, 149–157. [DOI] [PubMed] [Google Scholar]
- [20].Stein M, Ruggiero P, Rappuoli R, Bagnoli F, Front. Immunol 2013, DOI 10.3389/fimmu.2013.00328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Tan S, Noto JM, Romero-Gallo J, Peek RM, Amieva MR, PLoS Pathog. 2011, DOI 10.1371/journal.ppat.1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Monzón H, Forné M, Esteve M, Rosinach M, Loras C, Espinós JC, Viver JM, Salas A, Fernández-Bañares F, World J. Gastroenterol 2013, DOI 10.3748/wjg.v19.i26.4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ciacci C, Sabbatini F, Cavallaro R, Castiglione F, Di Bella S, Iovino P, Palumbo A, Tortora R, Amoruso D, Mazzacca G, Dig. Liver Dis 2004, DOI 10.1016/j.dld.2004.02.008. [DOI] [PubMed] [Google Scholar]
- [24].Noto JM, Gaddy JA, Lee JY, Piazuelo MB, Friedman DB, Colvin DC, Romero-Gallo J, Suarez G, Loh J, Slaughter JC, Tan S, Morgan DR, Wilson KT, Bravo LE, Correa P, Cover TL, Amieva MR, Peek RM, J. Clin. Invest 2013, DOI 10.1172/JCI64373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gaddy JA, Radin JN, Loh JT, Zhang F, Kay Washington M, Peek RM, Scott Algood HM, Cover TL, Infect. Immun 2013, DOI 10.1128/IAI.01271-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Haley KP, Blanz EJ, Gaddy JA, J. Vis. Exp 2014, DOI 10.3791/52122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Haley KP, Delgado AG, Piazuelo MB, Mortensen BL, Correa P, Damo SM, Chazin WJ, Skaar EP, Gaddy JA, Infect. Immun 2015, DOI 10.1128/IAI.00544-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lu, Francis JD, Guevara MA, Moore RE, Chambers SA, Doster RS, Eastman AJ, Rogers LM, Noble KN, Manning SD, Damo SM, Aronoff DM, Townsend SD, Gaddy JA, Chembiochem 2021, DOI 10.1002/cbic.202100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Senkovich O, Ceaser S, McGee DJ, Testerman TL, Infect. Immun 2010, DOI 10.1128/IAI.01258-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gaddy JA, Radin JN, Loh JT, Piazuelo MB, Kehl-Fie TE, Delgado AG, Ilca FT, Peek RM, Cover TL, Chazin WJ, Skaar EP, Scott Algood HM, PLoS Pathog. 2014, DOI 10.1371/journal.ppat.1004450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Doğan Y, Erkan T, Önal Z, Usta M, Doğusoy G, Çokuğraş FÇ, Kutlu T, Mediators Inflamm. 2012, 2012, 214581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nakao K, Imoto I, Gabazza EC, Yamauchi K, Yamazaki N, Taguchi Y, Shibata T, Takaji S, Ikemura N, Misaki M, Scand. J. Gastroenterol 1997, 32, 530–534. [DOI] [PubMed] [Google Scholar]
- [33].Yamazaki N, Yamauchi K, Kawase K, Hayasawa H, Nakao K, Imoto I, J. Infect. Chemother 1997, 3, 85–89. [Google Scholar]
- [34].Husson MO, Legrand D, Spik G, Leclerc H, Infect. Immun 1993, DOI 10.1128/iai.61.6.2694-2697.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jackson E, Little S, Franklin DS, Gaddy JA, Damo SM, J. Vis. Exp 2017, DOI 10.3791/55557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Avery TM, Boone RL, Lu J, Spicer SK, Guevara MA, Moore RE, Chambers SA, Manning SD, Dent L, Marshall D, Damo SM, Townsend SD, Gaddy JA, ACS Infect. Dis 2021, DOI 10.1021/acsinfecdis.1c00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Huynh HQ, Campbell MAF, Couper RTL, Tran CD, Lawrence A, Butler RN, Lett. Appl. Microbiol 2009, DOI 10.1111/j.1472-765X.2009.02557.x. [DOI] [PubMed] [Google Scholar]
- [38].Ciccaglione AF, Di Giulio M, Di Lodovico S, Di Campli E, Cellini L, Marzio L, J. Antimicrob. Chemother 2019, DOI 10.1093/jac/dky510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Fulgione A, Nocerino N, Iannaccone M, Roperto S, Capuano F, Roveri N, Lelli M, Crasto A, Calogero A, Pilloni AP, Capparelli R, PLoS One 2016, DOI 10.1371/journal.pone.0158646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zou J, Dong J, Yu XF, Helicobacter 2009, DOI 10.1111/j.1523-5378.2009.00666.x. [DOI] [Google Scholar]
- [41].Tong JL, Ran ZH, Shen J, Zhang CX, Xiao SD, Aliment. Pharmacol. Ther 2007, DOI 10.1111/j.1365-2036.2006.03179.x. [DOI] [PubMed] [Google Scholar]
- [42].Guttner Y, Windsor HM, Viiala CH, Marshall BJ, Aliment. Pharmacol. Ther 2003, DOI 10.1046/j.1365-2036.2003.01395.x. [DOI] [PubMed] [Google Scholar]