

Abstract
Mesenchymal stem/stromal cells (MSCs) have broad application prospects for regenerative medicine due to their self‐renewal, high plasticity, ability for differentiation, and immune response and modulation. Interest in turning MSCs into clinical applications has never been higher than at present. Many biotech companies have invested great effort from development of clinical grade MSC product to investigational new drug (IND) enabling studies. Therefore, the growing demand for publication of MSC regulation in China necessitates various discussions in accessible professional journals. The National Medical Products Administration has implemented regulations on the clinical application of MSCs therapy. The regulations for MSCs products as drug have been updated in recent years in China. This review will look over the whole procedure in allogeneic MSC development, including regulations, guidance, processes, quality management, pre‐IND meeting, and IND application for obtaining an approval to start clinical trials in China. The review focused on process and regulatory challenges in the development of MSCs products, with the goal of providing strategies to meet regulatory demands. This article describes a path for scientists, biotech companies, and clinical trial investigators toward the successful development of MSC‐based therapeutic product.
Keywords: adult stem cells, cell transplantation, cellular therapy, clinical translation, mesenchymal stem cells
Significance statement.
The present study is based on scientificity and compliance of mesenchymal stem/stromal cells (MSCs) products development. This article focuses on the whole procedure of allogeneic MSC medicine development in China, including regulations, guidance, processes, quality management, pre‐investigational new drug (IND) meeting, and IND application for obtaining an approval to start clinical trials. The results of this study will be valuable for researchers who are interested in developing MSCs products and other cell therapy products.
1. INTRODUCTION
Mesenchymal stem/stromal cells (MSCs) have been derived from a broad subset of human tissue sources, such as bone marrow, 1 , 2 adipose, 3 dental pulp, 4 umbilical cord, 5 , 6 placenta, 7 , 8 amniotic membrane, 9 , 10 endometrial tissue, 11 , 12 umbilical cord blood, peripheral blood, 13 , 14 and limbus, 15 and so forth. Over the last 20 years the MSC has become one of the most promising candidate cells used for therapeutic applications because of their role in anti‐inflammation, pro‐angiogenesis, immune modulation, and ability in multilineage differentiation under certain conditions. MSCs are immunoprivileged and need no matching for HLA before their administration. It is relatively easy to scale up for expansion from a small amount of starting biomaterial. In vivo studies have well demonstrated their potency, efficacy, and stability in various models to support potential clinical application. 16 , 17 , 18 There are more than 800 registered MSC clinical studies worldwide 19 and more than 55 000 publications readily available on MSCs. 20 Currently there is an increasing need to understand the regulatory issues that have led to the development of the MSC as a medical drug product.
The majority of MSCs products meet the minimal characterization criteria for MSCs as defined by the International Society for Cellular Therapy (ISCT) in 2006, which includes expression of a certain set of cluster of differentiation (CD) markers (ie, CD105, CD73, CD90), lack of expression of certain other markers that are usually associated with cells of hematopoietic lineage (ie, CD45, CD34, CD14, CD19, and HLA‐DR), and tri‐lineage potential differentiation. 21 These markers are short of specificity when characterizing an MSC cell product for a given indication. When considering these cell markers during MSC product development, the identity and the purity need to be defined, although most regulatory authorities in the world do not require a pure population of MSCs used as clinical product.
MSC products processed in culture expansion are regulated in the United States under Section 351 of the Public Health Service Act (PHS Act; 42 U.S.C. 262) and require an investigational new drug (IND) application from the Food and Drug Administration (FDA) to be tested in clinical trials.
In the European Union, MSC‐based medicinal product development and authorization is regulated through the European Medicines Agency (EMA). The Committee for Advanced Therapies (CAT) has been established at EMA, which is in charge of the certification and evaluation procedures of MSCs.
According to the current law and regulation in China, the use of MSCs in clinical study requires approval in one of two ways. In one, the Chinese Ministry of Health has implemented regulations on the clinical application of MSCs therapy based on approval from an ethics committee review and qualified designated hospital. In the other way, if MSCs are intended to be developed as a medical drug, then the clinical trial using MSCs products requires an IND approval from the National Center of Drug Evaluation (CDE) based on National Medical Products Administration (NMPA) regulations. An IND application must be filed by the applicant and provide comprehensive information that includes development of cellular and manufacturing process, quality standards, quality control, stability studies of MSCs products, efficacy studies, safety and toxicity studies, and the clinical trial plan and proposals. MSCs product must be manufactured under Good Manufacturing Practice (GMP) conditions according to IND regulations. 22 This review will provide an overview of the regulatory issues and procedures that facilitate manufacturers of MSCs products to gain approval for IND application.
2. OUTLINE OF MSCs MEDICINE DEVELOPMENT
In general, development of MSCs product includes several critical stages, including the following (Figure 1):
Basic research stage includes MSCs isolation, purification, cell characteristics analysis, cell culture medium development, expansion, and mechanisms of action (MOA) for proposed indications.
Pharmacy mainly includes chemistry manufacturing control, product testing and release, and stability programs, and so forth.
Pharmacology tries to define targeted indications, solidify anticipated mechanism of action, define route of administration, identify biologically active dose, determine efficacy, demonstrate safety and pharmacokinetics, and so forth.
Toxicology should consider a single dose toxicity test, repeated administration toxicity test, tumorigenicity research, genetic toxicity, reproductive toxicity, antigenicity test, immunotoxicity test, and local tolerance, and so forth.
IND application includes pre‐IND meeting, IND application filing, and IND submission.
Clinical trials include exploratory clinical trials and confirmatory clinical trials.
FIGURE 1.
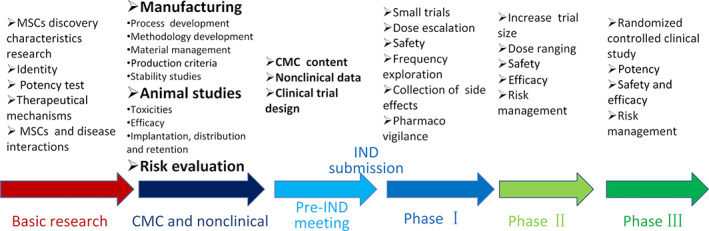
Content and flow chart of mesenchymal stem/stromal cells (MSCs) products development. Process development, formulation selection, process validation, analytical method development and validation, different release and stability specification, nonclinical studies, investigational new drug (IND) application, phase I, phase II, phase III, new drug application (NDA), and commercialization. CMC, chemistry, manufacturing, and controls
3. REGULATIONS AND GUIDANCE APPLICABLE TO MSCs PRODUCTS
The development of all medicinal products for human use is subject to legal requirements that a biotech company must comply with. In the United States, MSC‐ and other cell‐based medicinal products fall under the remit of the Center for Biologics Evaluation and Research (CBER), specifically the Office of Cellular Tissue and Gene Therapies (OCTGT), who regulate such products as human cells, tissue, and cellular and tissue‐based products under Part 1271 of CFR Title 21. In the European Union, MSC‐ and other cell‐based medicinal products fall under the remit of the advanced therapy medicinal products (ATMP) including that somatic cell therapy medicinal products, gene therapy medicinal products, and tissue‐engineering products. In Japan, since 2014, human cell‐, gene‐, or tissue‐derived medicinals have been regulated under the Pharmaceuticals and Medical Devices Act (PMD Act). 23 In China, it is guided by the Drug Administration Law of the People's Republic of China, which was first issued by the Chinese People's Congress in 1984. The current version of this legal document was revised in 2019. 24 Subsequently NMPA was formed according to this law. NMPA is responsible to formulate drug quality standards, drug management regulations, safety supervision, and management. NMPA has established the Center of Drug Evaluation (CDE) for drugs review and evaluation. 24
All cell therapies in China are regulated by technical guidelines for research and evaluation of the cell therapy products by NMPA in 2017. It includes stem cell product, immune cell product, and gene edited cell product. In addition, NMPA has renewed a biological product appendix to the “Good Manufacturing Practice for Drugs” on April 26, 2020. Moreover, NMPA has drafted the cell therapy Products Appendix on the “Pharmaceutical Good Manufacturing Practice (Revised in 2010)” (draft for comments). Meanwhile, on August 4, 2020 NMPA also has drafted the “Technical Guidelines for Clinical Trials of Human‐derived Stem Cells and Derived Cell Therapeutics (Draft for Comment).” Both current Chinese Pharmacopoeia (http://www.baidu.com/link?url=Uh0CVEwAL0kfvPk7i8o0YYBMwg6WPuhUJg70ByiMfyAypukqOgXMn0xKQQK_KCP8RvvwWADB‐byk14hCLEY06YdzIAPzcS8SVPPoqejIeNcnxF42CGgRwGiikICstUvt) and other national drugs standards need to be complied in new drug development. Furthermore, NMPA was approved by International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) as a new regulation member in 2017 and all guidelines issued by ICH will be approved by NMPA. Therefore, the guidelines of ICH can be referenced by pharmaceutical research and development enterprise. Further reference regarding cell therapies could refer to the regulations and guidance documents from the US Food and Drug Administration (FDA), the European Medicines Agency (EMA) in the European Union, Parenteral Drug Association (PDA) in the US, current Good Tissue Practice (GTP) in the US, and International Society for Pharmaceutical Engineering (ISPE).
4. CHEMISTRY, MANUFACTURING, AND CONTROLS
4.1. GMP compliant quality management system establishment and implementation
GMP is a quality assurance system to ensure that MSC products are consistently produced and controlled to state‐of‐the‐art quality standards appropriate to clinical use. Therefore, clinical grade MSCs should follow GMP guidelines to be manufactured in pharmaceutical quality according to IND regulations. 22 GMP principles significantly contribute to providing consumers and patients with products with a consistent quality and high safety level. To meet the principles, manufacturers must establish, implement, and maintain a pharmaceutical quality system. 25 The pharmaceutical quality system should oversee all activities, from control of incoming raw materials, manufacturing, testing, staff, equipment, labeling, review of production records, error detection and correction, and authorization for the final release of the product. 26 To identify and implement appropriate product quality improvements, process improvement, variability reduction, innovations and pharmaceutical quality system enhancement, and increasing the ability to fulfill quality needs consistently. Quality risk management can be useful for identifying and prioritizing areas for continual improvement. 27
The manufacture of MSCs products relies upon well‐trained staff that must be qualified for the job post requirement. It is an important element of GMP quality system. The expectation of China GMP is that staff involved with all aspects of manufacturing and management will have adequate education, ongoing training and periodic assessment, and experience to perform their assigned duties. People in responsible positions should have specific duties and adequate authority to carry out their responsibilities. 25 In addition, the Cell Therapy Products Appendix draft of the “Pharmaceutical Good Manufacturing Practice (Revised in 2010)” (draft for comments) presented clearly higher and detailed qualification requirement for key staff including the head of product manufacturing, the head of quality control and qualified person. 28
The air cleanliness level of the production environment and facilities of MSCs products should be compatible with the products and production operations, and the manufacturing environment and facilities should not cause pollution to raw materials, intermediates, and finished products. And the manufacturing environment and facilities of MSCs products should be easily and effectively cleaned and disinfected. In addition, manufacturing environment and facilities of MSCs products must be continuously monitoring. Furthermore, cleaning, disinfection, maintaining and monitoring operations of manufacturing environment and facilities of MSCs products must be regularly validated and continuously controlled. 25 Manufacturing equipment should be designed, located and maintained to suit its intended purpose and utilization. Manufacturing, measuring, weighing, recording and control equipments should be maintained, calibrated and checked at defined intervals by appropriate methods and qualified institutions. 25
Compliance with GMP is primarily available to the CDE through the applicant's documentation system. This must address all aspects of operations and quality managements so that an audit can be performed that follows all activities related to manufacturing and quality control of the product. In addition, Chapter 8 Pharmaceutical Good Manufacturing Practices (Revised in 2010) points out that documentation is a fundamental element of a quality assurance system. Manufacturers must have written management procedures, quality standards, technical guidelines, production prescriptions and process regulations, standard operating procedures and records with correct content. Enterprises should establish management and operating procedures for document management and systematically design, formulate, review, approve and issue documents. The quality management department shall review documents related to this specification. The content of the document should be consistent with the relevant requirements of the drug production license and drug registration, and help to trace the history of each batch of products. 25
4.2. Materials management
Materials refer to substances or materials used to manufacture MSCs products, including cells, balanced salt solutions, proteases, serum, culture media, cytokines, various additives, cryopreservation reagents, genetic modification materials, excipients, culture flasks, cell cryopreservation bags, and so forth. Manufacturing materials are directly related to the quality of MSCs products. Therefore, material management must be based on a good and standardized quality assurance and evaluation system including risk assessment, qualification audit for key materials manufacturers and suppliers, development and improvement of quality release testing, and so forth. The source, composition, use, dosage and quality control of materials shall be clear and reasonable. When selecting materials, consideration should be given to the necessity, rationality and safety, and develop verification of process removal effect and safety risk assessment, and the materials residual should be tested for release when necessary. It is quite important to use materials that have been approved for use in humans or meet pharmacopoeia standards. For biological origin materials, such as fetal bovine serum and human albumin, comprehensive exogenous factor testing should be carried out, and the knowledge of new exogenous factors should be taken into account in technological development. The self‐use of products should strictly prevent possible risks of the spread of foreign factors. 29 The information of materials management should be provided in the chemistry, manufacturing, and controls (CMC) section of the IND application and CDE reviewers will determine the suitability for manufacturing.
4.2.1. Biomaterials
Biomaterials used for isolation and culture of MSCs are all from donors and signed in informed consent that was approved by hospital medical research ethics committees. Each donor is screened and tested for human pathogens. Especially for the presence of human immunodeficiency virus (HIV), hepatitis B virus, and hepatitis C virus should be analyzed. All starting materials and reagents required for the culture expansion are required to be analyzed to certify that they were sterile and endotoxin free. 30
The procedures for cell isolation and culture expansion should be developed under GMP condition and validated to establish master cell bank. Quality control needs to implement during these procedures to ensure cells stored in mast cell bank have been tested and monitored for sterility, mycoplasma, and endotoxin. Thus, develop clear specifications and requirements, such as the cell surface markers, optimized culture condition, cell passage number, growth characteristics, and so forth. In principle, for cells primary isolated from biomaterials that are suitable for establishing mask cell bank, following cell test, preservation, and manufacture are planned. The level of the cell bank can be considered comprehensively according to the cell's own characteristics, production status and clinical application; and the inspection standard of the cell bank should be established, and the inspection should meet the basic requirements of safety, quality controllability and/or effectiveness. 30
4.2.2. Excipients
The use, dosage and quality of excipients in MSCs products should be developed and verified to prove the necessity, safety and rationality of their use. It is advisable to select excipients that are approved for use in humans. Otherwise, comprehensive research and evaluation are required. In addition, there is a better choice to apply key excipients related review and approve during product IND application. 31
4.2.3. Containers and closure systems
In order to avoid unexpected changes in product quality due to storage, manufacturers should accomplish safety assessments and compatibility studies on the packaging containers and closed systems used for the samples storage and/or finished products in the manufacturing process of MSCs products. To illustrate their rationality of packaging containers and closed systems use, air tightness research and frozen storage adaptability research should also be accomplished. 29
The manufacturing process of MSCs products may involve the steps of short‐term direct contact between the sample and the container, such as the collected tissue or cells, the cells in the preparation process, and infusion of the finished product. The manufacturers should conduct a safety assessment and compatibility study between contact container system and MSCs products, and so forth. 29 In addition, it is recommended to apply direct contact container and closure systems related review and approve during IND application. 31
4.3. CMC in GMP workshop
Since cells have the ability to survive in vivo, autonomously proliferate, or/and differentiate, their clinical research and quality control should fully consider the above basic characteristics, while MSCs products should meet the general requirements of drug quality management, and the whole manufacturing process of clinical samples is reasonable comply with the basic principles and related requirements of the “Pharmaceutical Good Manufacturing Practice.” Special attention should be paid to personnel, environment, equipments and other requirements during the manufacturing process and quality control. The manufacturing of MSCs products should establish a whole‐process or lifecycle quality assurance and control system; the manufacturing process should fulfill strict process verification and establish clear key control points. Manufacturing materials should be controlled strictly and standard operation procedure of the MSCs products manufacturing site should be established and validated to avoid raw materials and production possible external source pollution or cross‐contamination during manufacturing. Effective isolation measures should also be taken to prevent the confusion of products from different donors or different expected products. 29
4.3.1. Manufacturing
The manufacturing process of MSCs products refers to the process of in vitro operations from the donor obtaining cells to the completion of the cell input into the recipient. Manufacturers must perform process development and verification to prove the process feasibility and robustness. Each step of manufacturing process is controlled to assure that the finished product meets all quality attributes including specifications. Process validation is defined as the collection and evaluation of data, from the process design stage through commercial production, which established scientific evidence that a process is capable of consistently delivering quality product. Process validation involves a series of activities taking place over the lifecycle of the MSCs products and process. 32 The process designation should avoid the cells unexpected or abnormal changes, and meet the requirements of eliminating related impurities. It is quite necessary to establish technical standards, process standard operation procedure, key process control parameters, internal control indicators and waste standards, and monitor the entire manufacturing process. Manufacturers should continue to optimize the manufacturing process, reduce the unintended effects of physical, chemical or biological effects on cell characteristics, and reduce impurity, such as proteases, nucleases, and the use of selective inhibitors. If there is discontinuous production in the manufacturing process, the storage conditions and duration of the cells should be developed and verified. A closed or semi‐closed preparation process is recommended to reduce the risk of contamination and cross‐contamination.
The monitoring of the entire manufacturing process includes the monitoring of critical process parameters (CPP) and the achievement of process control indicators. Manufacturers should clarify the key manufacturing steps in process control and formulate the limited range of sensitive parameters based on the understanding of the overall process and the accumulated experience of the products to avoid process drift. When necessary, the quality control of the cells in the manufacturing process can also be carried out. The quality control and the cell release detection in the process are combined and complementary to each other to achieve the control of the overall process and product quality. 29 Because of donor material variability and may contain infectious disease pathogens, and the process is likely to cause contamination, special attentions should be paid to the prevent microbial contamination during the entire manufacturing process of MSCs products. 28
Dosage form, prescription and prescription process of the MSCs product should be determined according to the clinical medication requirements and the stability of the MSCs product. Some MSCs products need to undergo steps such as the transformation of the physical state of the product components, the conversion of the container, the filtration and cleaning, the combination with other structural materials, and the adjustment of the drug dosage before administration. The determination of these process steps should also be developed and validated, and strictly implemented in actual products manufacture and applications. 29 Manufacture information regarding the components and materials used in the manufacturing of the MSCs should be provided, with a summary of the testing performed on these materials. A list of all components used in the manufacture should be provided. For allogeneic cells, information on the donor screening and testing conducted should show that all donors were qualified according to donor standards. 30 The tissue source and MSCs isolation, culturing for large scale expansion and collection must be described in detail. The cell bank section should provide information regarding the history, source, derivation, and characterization, including testing to establish the safety, identity, purity, and stability of the cells. 22
All reagents information used in cell manufacturing process must be listed in the IND. These include basic cell medium, cytokines, growth factors, antibiotics, and fetal bovine serum, buffers, and protease, and so forth. These reagents may have an influence on the safety, potency, and purity of the final product and are often a source of adventitious agents, so their source, quality, qualification, and removal from the final product should be controlled and demonstrated. Certificate of analysis (COA) of manufacturers, quality criteria and COA of applicant must be one of most important information for materials management. Excipients which are present in the final product should similarly be listed. 29 , 33 Manufacturing procedures should be described in detail, with schematics of the isolation, passage, purification, large scale expansion, collection, preparation and subpackage of the product provided with in‐process and final product testing identified within these schematics. In addition, preparation of the cells, process timing and intermediate storage, and the final formulation should be described in detail. 22 , 29
4.3.2. Product testing and release
The quality research of MSCs products should perform on samples that selected from representative product batches and appropriate manufacture stage samples throughout development and scale‐up activities. Quality research should cover various cell analysis including function, purity and safety, other related research projects can be added according to the characteristics and attributes of the product. Manufacturers must establish quality standards and quality control strategies for MSCs products. The MSCs products should comply with the drug quality standards approved by the NMPA. The drug quality standards approved by the NMPA are the drug registration standards. The drug registration standards should comply with the general technical requirements of the current “Chinese Pharmacopeia” and should not be lower than the provisions of the current “Chinese Pharmacopoeia.” If the test items or indicators of the declared registered varieties do not apply to the current “Chinese Pharmacopoeia,” the manufacturers should provide sufficient and reasonable supporting data. It is recommended to adopt a mechanism that combines quality inspection of intermediate samples and final product release test. 22
The analysis procedures and methods of MSCs products must be verified and validated systematically. Applicants should submit analytical procedures and methods verification and validation data to support the documentation of the identity, strength, quality, purity, and potency of MSCs cell bank and MSCs products. For intended purpose, the typical validation characteristics of analytical procedures and methods should consider the followings, accuracy, precision (including repeatability and intermediate precision), specificity, robustness, detection limit, quantitation limit, linearity and range. The analysis report should include principle, equipment, operating parameters, reagents, reference standards and materials, acceptance criteria, sample preparation, stability, standards preparation, test procedure, system suitability, calculations, data analysis and conclusion. It is highly recommended to revalidated if there are any key changes of the drug materials, manufacturing process, the legal analytical procedures, national standards and self‐developed analytical procedure during MSCs product's life cycle. In addition, analytical methods in comparability are requested when manufactures propose to substitute a validated analytical procedure with alternative analytical procedure or when an analytical procedure is transferred from one laboratory to the other. 33 , 34 , 35 At present, the main challenge of analytical procedures validation is the serious shortage of standards and reference reagents in China and even in worldwide. Therefore, for solving the problem of serious shortage of standards and reference reagents, close collaboration between manufacturers of MSCs products and institutions of standards and reference reagents is quite necessary and urgent.
As the research progresses, process‐related information should be gradually accumulated, and analytical procedures should be improved to meet the quality control requirements of each stage. Quality control should generally consider identity (cell morphology, phenotype, cell markers, etc), biological efficacy, purity, impurities, cell number (viable cell number, functional cell number, etc), cell growth activity, cell cycle, cell doubling time, clone formation rate, and general testing, (such as sterility, mycoplasma, endotoxin, expression and activity of telomerase, etc). Cell characteristics research should base on the cell types, that covers cell morphology, cell surface markers, gene expression, cell differentiation, surface marker expression, potency test, qualitative and quantitative measurement of secretome, and residual amount detection of ingredients which added during process. 29 , 33
Analytical procedures for functional research of MSCs products should be established based on the nature, characteristics and intended utilization of cells. To establish reasonable and effective analytical procedures on biological efficacy, the mechanism of action should be consistent with clinical indications. In terms of cell purity, it is recommended to test cell subpopulations and viability. Impurities research should include in cell processing (such as proteases, animal serum protein, recombinant protein and cytokines, etc) as well as final cell product (such as dead cell residues and other possible biodegradation products, etc). Safety‐related research should be considered based on the characteristics of cell source and process, and can be selected for exogenous factors, the possibility of cell malignant transformation, genetic stability, tumorigenesis and tumorigenicity. 29 , 33
Relevant research imitates the cell administration process should be developed and improved. If the operation (cell recovery, products dilution, etc) is carried out in a research center of hospital, applicants should establish the standard operation procedures and give clinical researchers good training. It is highly recommended to carry out the standardized process validation test to verify final product quality after completing the operation before administration process, such as cell morphology, cell phenotype, multilineage differentiation potency, cells number and viable ratio, appearance, potency, identity, sterility, mycoplasma, endotoxin, other exogenous substances, and so forth. 35 Potency measurements are used to demonstrate that only product lots that meet defined specifications or acceptance criteria are administered during all phases of clinical investigation and following market approval. 36 In general, for cellular and gene therapy products, there is no single test that can adequately measure those product attributes that predicts clinical efficacy. Therefore, it is necessary and advisable for developing several attributes test for potency evaluation of cellular and gene therapy products.
Release testing should be performed on each manufactured product lot. Specifications for the final product should be clearly set up in the IND, usually in a tabular format. The final release testing data should be available preferably prior to administration of the MSCs product in clinic. All products must be trackable and identifiable from product collection to administration and carry appropriate labeling. 33
4.4. Stability studies of MSCs products
The manufacturing of MSCs products is recommended to adopt a continuous process. For samples that need to be temporarily stored during the production process, stability studies should be carried out to support their storage conditions and storage period. Stability testing must be performed to show that the final cell product is sufficiently stable over the time required in the study (from final release to administration) and an expiration date for final product should be indicated. The basic principles of MSCs product stability research can be based on a reasonable research plan according to the product's own characteristics, clinical drug needs, and storage, packaging and transportation conditions. Research should be carried out using representative cell samples and storage conditions. 37 In addition, final product transportation proposals should also be developed and improved, and the stability proposals should be validated. Furthermore, products preparation procedure before administration also should be validated to confirm the final products quality. 29 , 33
5. PHARMACOLOGY PART IN IND FILING
The pharmacology section presented in the IND application must support the proposed clinical trial. The objectives of the nonclinical program should be to establish the biological plausibility of the proposed therapeutic product and identify biologically active dose levels. 29
5.1. Pharmacodynamics
Pharmacodynamics research should adopt reliable methods to verify the basic treatment mechanism of MSCs products and determine biological effect markers. The trial design should consider factors such as mechanism of action on MSC product, the length of the disease cycle, administration procedure, together combined with the characteristics and survival time of the cell in vivo. It is recommended to complete the pharmacodynamics research of MSCs products using both relevant in vitro and in vivo models. 29
5.2. Pharmacokinetics
Pharmacokinetic studies should be able to clarify in vivo processes of cells and the accompanying biological behaviors. Appropriate animal models should be selected according to the types and characteristics of MSCs products. Generally, both male and female models should be considered. According to research purpose and clinical value of indicators, establish a suitable biological analytical procedure and perform the necessary analytical validation. Pharmacokinetic studies should focus on the proliferation of target cells, the expression and/or secretion of bioactive molecules in the body, and the interaction with host tissues. In general, pharmacokinetic research mainly includes distribution, migration and homing of cells, cell differentiation, apoptosis, aging, and so forth. 29
6. TOXICOLOGY PART IN IND FILING
The toxicology research and evaluation of MSCs products should comply with the “Pharmaceutical Non‐clinical Trial Good Laboratory Practice” (GLP). In addition, for certain studies carried out under non‐GLP conditions, the impact of non‐GLP on the reliability and integrity of test results and the overall safety evaluation of MSCs products should be explained and evaluated. 29
6.1. Safety pharmacology studies
The active substances secreted by MSCs may have impact in multiple systems in our body including central nervous system, cardiovascular system, respiratory system, and so forth. MSCs products may also affect vital organ function. Therefore, safety pharmacological tests could identify undesirable pharmacodynamics properties of factors that may have relevance to human safety and should be considered for MSCs products. In addition, if potential risks are found in toxicity tests, relevant safety pharmacological tests should be supplemented for further evaluation. 29
6.2. Single dose toxicity test
The single‐dose toxicity test can obtain the dose‐response relationship between the dose and the systemic or local toxicity, which is helpful to understand the target organ of toxicity, and can also provide a certain reference for the dosage design of repeated‐dose toxicity test. Since MSCs products can play or induce long‐term effects, the observation time of a single administration should consider the survival time of cells, which should generally be longer than the conventional observation time of a single administration toxicity test. 29 At present, for MSCs product, the observation period of single dose toxicity test is 1 month.
6.3. Repeated administration toxicity studies
The test design should include the basic elements of conventional toxicology research, and be in conjunction with the particularity of MSCs product in order to obtain as much safety information as possible. According to the different characteristics of MSCs product, animal species that can produce the biological activity of MSCs product is used for repeated dose toxicity studies. If nonclinical studies can be carried out without related species, animal experiments on non‐related species may also be valuable for evaluating manufacturing process, safety of the full prescription, and nontarget effects. Referring to the “Technical Guidelines for Toxicity Research on Repeated Drug Administration,” multiple dose levels need to be designed, including the proposed clinical dose range and the maximum feasible dose, and the prescription composition and manufacturing process. Besides conventional observation indicators, appropriate observation indicators should be selected based on product characteristics such as behavioral testing, neurological function testing, cardiac function evaluation, ectopic proliferative lesions (such as tumor), biomarkers, active molecules secretion, anti‐MSCs antibody level, immune response, and interaction with host tissues. 29 , 38 As for the repeated administration toxicity test duration, ICH guideline recommends for 6 months on rodent model and for 9 months on non‐rodent model. 38
6.4. Immunogenicity and immunotoxicity studies
MSCs products need to be studied for their potential immunogenicity. For immunogenicity studies, please refer to the latest version of the technical research guidelines. 29 , 39 In addition, attention should be paid to the immunotoxicity induced by MSCs products.
6.5. Tumorigenicity/carcinogenicity studies
The tumorigenicity/carcinogenic risk of MSCs products depends on the differentiation status of cells, the changes in growth kinetics caused by the cell culture procedure used in production process, inducing or enhancing the possibility of tumor formation in the host and target patient population, and so forth, need to be comprehensively considered based on the above characteristics. At present, there is no scientific consensus on how to select animal models for tumorigenicity/carcinogenicity studies. The tumorigenicity/carcinogenicity studies should use the clinically intended products for testing. The tumorigenicity/carcinogenicity studies need to ensure that the cells can survive in the body for a long time to investigate whether there is potential tumor formation. The following aspects should be considered carefully in experimental design: (a) Appropriate control group (such as positive control, blank control), in addition, it is recommended there are three different MSCs cell dose group; (b) Each group must have enough animals to make the analysis of tumor incidence meet the statistical requirements, and it is recommended there are 20 mice for each group; (c) Need including the maximum feasible dose, and it is recommended there are 10 times dosage for the highest group more than proposed clinical dosage; (d) The test substance should reach the planned clinical treatment site, and both subcutaneous and clinical route of administration should be considered; (e) A sufficiently long test period, in general, 6 months was recommended. In addition, due to immune rejection, tumorigenicity/carcinogenicity studies of human‐derived MSCs products can be considered using immunodeficiency rodent models NOD/SCID, NPG, NSG, NOG, and so forth. 29 , 40 , 41 , 42
6.6. Reproductive toxicity studies
The evaluation of reproductive and developmental toxicity of MSCs products mainly depends on the characteristics of the MSCs product, clinical indications and the population to be used clinically, and should be analyzed in detail according to specific conditions. 29 , 43
6.7. Genetic toxicity studies
For human‐derived MSCs products, if the product has a direct interaction with DNA or other genetic material, a genotoxicity test is required. 29 , 44
6.8. Special safety studies
According to the characteristics and clinical application of MSCs products, evaluation of local tolerance, tissue compatibility and tolerance to secreted substances should also be considered.
7. CLINICAL TRIAL CONSIDERATIONS IN IND
When MSCs products enter the stage of clinical trials, researchers should follow the requirements of the “Pharmaceutical Clinical Trials Good Clinical Practice” (GCP), Technical guidelines for clinical trials of human‐derived stem cells and their derived cell therapy products (draft for comments) (Released in July 2020 by the Biological Products Clinical Department of CDE, NMPA), ICH harmonized Tripartite Guideline for Good Clinical Practice (E6‐R1). 45 , 46 , 47 In principle, the clinical trial content should include clinical safety evaluation, pharmacokinetics, pharmacodynamics, dose exploration and confirmatory clinical trials. In addition, the specific clinical study design can be adjusted as selected indicators, according to the nature of different MSCs products. 29 , 47
7.1. Indicator selection
The therapeutic potential of MSCs products from their several properties, including their ability to (a) differentiate into various cell lineages, (b) secrete soluble factors crucial for survival and proliferation, (c) modulate immune response, and (d) migrate to the injury site. 23 The properties of MSCs are the main factors for considering to select the indicator. In addition, investigators usually select the appropriate indicator according to in vitro and animal model pharmacodynamic. Furthermore, indicators reported in papers, registered in clinical trials institutes, approved by some Center of Drug Evaluation also be recommended to refer for new indicator selection.
7.2. Subject's protection
The purpose of the subject selection process is to make sure that the expected risks and potential benefits to the research subjects are carefully evaluated, while achieving the scientific purpose of the research. For MSCs products with high‐risk characteristics such as long‐lasting or permanent effects and invasive administration, patients who are expected to benefit from the treatment should be selected for the trial. The subject choice may affect the risks and benefits of clinical trials. The possible risks and benefits of subjects should be fully stated in the clinical trial proposals and informed consent form. 29 , 47
7.3. Pharmacodynamics
In early clinical trials, the main purpose is usually to evaluate the safety of the product, and the secondary purpose is to initially evaluate the efficacy of the product, that is, pharmacodynamic evaluation. Evaluation indicators are short‐term effects or long‐term outcomes that may indicate potential effectiveness. In the activity evaluation of MSCs products, it can include special indicators such as gene expression, cell implantation, morphological changes, and other biomarkers. It can also include changes in immune function, or various types of physiological responses. If the purpose of using MSCs products is to correct the biological functions of functionally defective or damaged cells or tissues, functional testing of MSCs products should be performed. If the intended utilization of the MSCs product is to repair/immunize/replace cells/tissues and is expected to function for life, the relevant structural/histological test indicators should be tested as potential pharmacodynamic markers, including microscopy, histological testing, imaging technology or enzyme activity index testing, and so forth. 29 , 47
7.4. Pharmacokinetics
The in vivo process research of MSCs products should be carried out as much as possible. In clinical trials, the research requirements, possible methods and feasibility should be discussed, and attention should be paid to the detection of cell viability, proliferation and differentiation capabilities, in vivo distribution/migration and related biology during the expected activity process of MSCs products. If multiple administrations of MSCs products are required, the expected survival time and corresponding functions of the MSCs products in the body should be considered when designing the clinical protocol. 29 , 47 However, it is actually quite difficult to implement pharmacokinetics because of methodological limitation.
7.5. Dosage exploration
One of the purposes of early clinical trials is to explore the effective dose range of MSCs products. If possible, the maximum tolerated dose should also be determined. The dosage of the MSCs product should be determined based on the results obtained in the product quality control research and nonclinical research, and the biological efficacy of the product should be comprehensively considered. Some MSCs products will exist in the subject for a long time. Therefore, the first human trial should adopt a single‐dose regimen. The repeated dose clinical trial can be performed after considering fully both understanding of the product's toxicity and duration of action. The setting of the dose increase should take into account the risks and activities related to the dose change in the preclinical data and any existing clinical data. At the same time, the specific safety risks of MSCs products should be fully considered, and sufficient dosing intervals and follow‐up times should be set to observe acute and subacute adverse events. Although the dosage of MSCs products may depend on the individual conditions of the patient, the evidence of dosage exploration studies provided by early clinical trials is still an important basis for determining dosage in confirmatory clinical trials. 29 , 47
7.6. Clinical efficacy
Usually, the confirmatory clinical trial should be carried out in the target indication population, there should be sufficient sample size, reasonable control, selection of clinically significant endpoint indicators to be reached and the statistical methods used to address them in detail. And the method of endpoint measurement must be determined. The statisticians should calculate the appropriate sample size that is required to answer the questions with sufficient statistical power. In addition, the clinical trial should be able to provide a clinical dosing regimen that can produce the expected therapeutic effect, the duration of efficacy, and the benefits and risks in the target population. The previously validated or generally recognized indicators can be used as a surrogate end point, which should have clinical significance and be related to the efficacy of the treatment. If the efficacy of MSCs product depends on the long‐term maintenance of the biological activity of the input cells, the clinical trial observation time should be designed according to the expected biological activity of MSCs product, and a long‐term patient follow‐up plan should be provided. 29 , 47
7.7. Clinical safety
The safety monitoring of MSCs products should run through the entire process of product development. All safety issues arising in nonclinical studies should be analyzed and solution proposals should be proposed, especially in the absence of corresponding animal models or lack of homologous animal models to predict the safety of humans and animals in the context of physiological differences. In early trials, its main purpose is to evaluate safety. Based on risk considerations, other subjects should be included on a case‐by‐case basis after the safety of the first subject is fully exposed and evaluated. General monitoring of safety evaluation usually includes symptom records and routine clinical examinations. The specific monitoring items depend on a variety of factors, such as the nature of MSCs product and its mechanism action, the research population, the results of animal studies, and any relevant clinical trial experience including being published papers. Besides general inspections and monitoring for expected and unexpected safety issues, it is also necessary to evaluate specific expected safety issues for MSCs products. Such as acute or delayed infusion reactions, cytokine release syndrome, autoimmune reactions, graft failure or MSCs product inactivation, graft vs host reaction, associated malignant diseases, transmission of donor infectious diseases, and so forth. In addition, applicants must collect all adverse events in clinical trials and report to CDE as required of ICH E2A: clinical safety data management: definitions and standards for expedited reporting (http://www.cde.org.cn/ichWeb/guideIch/downloadAtt/1/4f620036ec6954c3eae5890c22ac2ab3) and ICH E2B(R3): implementation guide for electronic transmission of individual case safety reports (ICSRs) E2B(R3) data elements and message specification (http://www.cde.org.cn/ichWeb/guideIch/downloadAtt/1/cce6da0f334b083a752a888dac60df4a). 48 , 49
In confirmatory clinical trials and post‐marketing phases of MSCs products, besides general symptom records and routine clinical examinations, attention should also be paid to changes in some important biological processes, including immune response, immunogenicity, infection, malignant transformation, and so forth. Since the pharmacological activity of MSCs products may be slow or delayed, regardless of whether the subject has received the entire treatment plan, the safety and pharmacological activity should be continuously monitored. For products that are expected to have long‐term activity, patients should be followed up to determine the long‐term efficacy and fully expose the safety issues related to the MSCs product. The duration of follow‐up should be able to provide preliminary evidence of effectiveness and the duration of the MSCs product activity, and consideration should be given to factors such as whether the product causes late‐onset safety issues. 29 , 47
Based on risk management, it is recommended to carry out clinical safety studies of repeated dosing products. The possibility of repeated dose should be considered when determining the maximum safe dose. In the clinical trials of MSCs products, there is considerable uncertainty in the frequency or severity of adverse reactions. Therefore, the clinical trial plan should include stopping standards, risk assessment plans, and an independent data and safety monitoring committee. 29 , 47
7.8. Trial stop rules
NMPA recommends that clinical trial programs of MSCs products should include trial stop rules so that the risks and the number of subjects can be controlled at all times. Trial stop rules usually stipulate the severity or frequency of events (such as specific medical events or deaths related to the indication or mode of administration). And enrollment and administration will be suspended until the situation is assessed. Based on the evaluation results, the clinical trial protocol may be revised to reduce the safety risks of the subjects. The revisions generally include the revision of the entry criteria (eg, the exclusion of subjects who are at higher risk of specific adverse events), or the reduction of doses and times, the adjustment of product formulation or administration mode, or the improvement of subject safety monitoring programs. After the trial plan is adjusted and improved, the trial may be resumed. 47
7.9. Risk management plan
When formulating a risk management plan, the conventional pharmacovigilance and product traceability should be elaborated. Furthermore, the possible differences in drug delivery, individualized preparation, special treatment or adjuvant therapy that could results in efficacy and safety difference of MSCs products should be considered comprehensively. As part of risk management, standardized and feasible standard operating procedures should be formulated and clinical investigators should receive systematic training and passed the assessment. 29 , 47
MSCs products may require specific long‐term studies to monitor specific safety issues, including failure. Long‐term safety issues such as infection, immunogenicity/immunosuppression, and malignant transformation should be evaluated. And sufficient follow‐up time is needed to evaluate MSCs products safety. As high‐risk MSCs products, patients should be followed up for long enough, or even for life in early stage. The follow‐up interval can be extended or shortened in late clinical stage according to increasing experience of MSCs products application. 29 , 47
8. PRE‐IND MEETING
A pre‐IND meeting with CDE is highly recommended for any innovative medicines including MSCs products. The pre‐IND meeting is a type II meeting and to solve major technical problems before submitting the first clinical trial application. The technical problems mainly including whether the existing research data supports the proposed clinical trial, whether the risk of clinical trial subjects is controllable, and so forth. The meeting request should submit enough information including clinical trial proposal, pharmaceutical, and nonclinical research information. Applicants should prepare and submit qualified “Communication Meeting Application Form” and “Communication Meeting Materials” through the “Applicant Window” on the website of the Drug Evaluation Center, and the form of communication should be indicated when applying. The forms of communication include: face‐to‐face meetings, video meetings, teleconferences, or written responses. And applicants are encouraged to communicate with the Drug Evaluation Center through telephone conferences. 22 , 50 However, the authors suggest that face‐to‐face meetings communication is the most effective choice for innovative medicines.
CDE will decide whether communicate application meets the requirement and the form of communication that applicant submitting. CDE will grant usually a pre‐IND meeting within 60 days after receipting a communication meeting application. Meetings are typically most productive when questions are focused and specific. Questions should be as precise as possible and include a foreword and background with a brief explanation of the context and purpose of the question. Opened and new questions should be avoided as much as possible. Typically, pre‐IND meetings are scheduled for 60 and occasionally 90 minutes. 50
The meeting minutes should be written in accordance with the requirements of the “Communication and Exchange Meeting Minutes Template.” If the applicant and CDE reach an agreement, they should state their common views. If the applicant and CDE have not reached an agreement, they should state their respective views, and authors recommend that applicant should formulate the following plan and proposals and gain the approval of CDE for responding to disagreement views as soon as possible. The meeting minutes shall be finalized no later than 30 days after the end of the meeting. The meeting minutes are encouraged to be formed on the spot. The meeting minutes will be uploaded to the communication system by the CDE project management staff within 2 days after the final draft, and the applicant can check it through the applicant window. The minutes of the meeting mainly include two parts: consensus and disagreement, and are archived as important documents. 50
9. IND SUBMISSION
The submission of the IND application will follow the pre‐IND meeting and meeting minutes signed. Meanwhile, applicant must fully address any issues raised at the pre‐IND meeting to be successful. The cell therapy products should be filed according to guideline documents of organization of the common technical document of the registration of pharmaceuticals for human use since February, 2018. 51 Once the IND application was received, the CDE has 5 days to check the completeness of the submitted materials to decide if the IND application will be accepted according to guidelines for acceptance and review of registration of biological products (type II therapeutic biological products, draft for Comments). 52 , 53 CDE will grant one acceptance number and issue acceptance notice to applicant following acceptation of the IND application. After the IND accepted, CDE will carry out professional review and evaluation.
10. CDE ACTION ON IND APPLICATION
After accepting of IND application, CDE will organize pharmacy, pharmacology, toxicology, medicine and other technical personnel to review and evaluate for IND application materials. Reviewers may have questions, provide non hold comments, or place the IND on clinical hold within that 60‐day time limit. Typically, CDE will have comments and/or questions and will contact the applicant prior to the 60‐day deadline. Being responsive to the agency at this point will help the application clear within the 60‐day time frame. 22 And applicant should respond comprehensively these questions in 5 days. CDE may apply a clinical hold to stop the clinical investigation from proceeding until identified issues are comprehensively addressed. 22 Several key sections in the IND are the clinical protocol with appropriate eligibility, inclusion and exclusion criteria for subjects, investigation endpoints, follow‐up cycle, stopping rules and dosing justification (starting dose/dose escalation), dosing frequency and schedules, the process and qualification studies, and safety, pharmacology and toxicology studies. For holding of clinical trial application, applicant can apply clinical trial resume by resubmitting registration IND application materials after accomplishing supplementary researches.
11. MOVING TO A CLINICAL TRIAL
Clinical trial is last but not least step for MSCs products development. Safety and efficacy of MSCs therapy must be verified and conformed in clinical trials. When an IND becomes active, investigators and clinicians can carry out clinical trial according to trial proposals approved by CDE. The applicant shall formulate the corresponding clinical trial plan before carrying out the subsequent clinical trial, which shall be carried out after review and approval by the ethics committee, and submit the corresponding clinical trial plan and supporting information. The applicant shall regularly submit a safety update report during the trial on the website of the CDE. 48 , 49 The safety update report during the trial should be submitted once a year and within 2 months after every 1 year after being approved. For suspicious and unexpected serious adverse reactions and other potentially serious safety risk information during clinical trials, the applicant shall promptly report to the CDE in accordance with relevant requirements. 22 , 47
12. FUTURE PERSPECTIVE
Translation of MSCs from the research and development stage into therapeutic application in the clinic via regulatory authority is demanding. Understanding overall pathways for regulatory issue could help to establish a clear framework for the generation and compilation of the required data in IND application. In China, early communication with CDE is advisable.
It is challenging work to make a consistent, efficacious MSC product in regulation. The inherent characteristic of MSC for disease intervention is that they are living cells. Furthermore, the heterogeneous population of MSCs conducted in clinical trials may result in a variable outcome. Optimization of manufacture process along with the development and validation of potency assays could take long time. In addition, biological variability, raw materials standard, characterization, reagents, aseptic processing, and cryopreservation are all essential and necessary for examination during development.
The identification of relevant MOA will be of crucial importance in determining the acceptability of a degree of heterogeneity, since MSC activity in a specific clinical application should help inform selection of an ideal MSC population, whether this may be a defined heterogeneous preparation or a specified subset. The majority of MSCs product candidates developed currently are heterogeneous population expanded under various protocols. As such MSC products may generate various features from in vitro (cellular and biochemical) and in vivo (animal and clinical) studies. Product needs to ensure meeting labeling requirements and complying with applicable biologics and cGMP regulations. It is required by CDE to submit product data used during all phases clinical study to ensure the quality, identity, purity, strength, and stability.
It is very important for potency assay development as it correlates to a therapeutic dose and product efficacy. Potency assay measures biologic activity for a MSC product, and also verifies the comparability of different lots of a product or between different MSC products. Potency assay data need to demonstrate a biological relevance to proposed clinical application. In case potency assay is not available, a surrogate test could be considered if correlation with functionality has been demonstrated, for instance transcriptome and secretome analysis. The FDA has published a guidance document with recommendation for developing potency assay for cellular and gene therapy products. 36
Before submitting an IND application, there are many key translational steps to be completed including process development and improvement, analytical procedures development and verification, GMP compliance workshop establishment and validation, quality risk evaluation and management, pharmaceutical quality system construction and implementation, and so forth. All these development steps are linked to each other and are quite critical for MSCs product transition into a clinic trial. Therefore, MSCs products development and IND application is a long and complex process.
In general, the IND application process for MSCs products covers compliance process of product preparation, quality testing, quality management, and so forth. The scientific aspect of MSCs product development is mainly resolved by the basic research at the drug discovery stage, while the compliance MSCs products development is mainly dealing with reference regulations and guidelines of national and NMPA for product development process. From the perspective of MSC therapeutic company, early regulatory strategy development is advisable. Communicating and cooperating closely with regulatory authority throughout the MSC product development will make the regulatory pathway straightforward.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
Qinjun Zhao designed and drafted the manuscript. Zhibo Han wrote sections of the manuscript. Jiajun Wang and Zhongchao Han critically revised the manuscript and contributed to writing the final manuscript.
Zhao Q, Han Z, Wang J, Han Z. Development and investigational new drug application of mesenchymal stem/stromal cells products in China. STEM CELLS Transl Med. 2021;10(S2):S18‐S30. 10.1002/sctm.21-0083
Contributor Information
Jialun Wang, Email: jack.wang@health-biotech.com.
Zhongchao Han, Email: hanzhongchao@hotmail.com.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created and analyzed in this study.
REFERENCES
- 1. Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143‐147. [DOI] [PubMed] [Google Scholar]
- 2. Au P, Tam J, Fukumura D, Jain RK. Bone marrow‐derived mesenchymal stem cells facilitate engineering of long‐lasting functional vasculature. Blood. 2008;111(9):4551‐4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zuk PA, Zhu M, Ashjian P, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13(12):4279‐4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Huang GT, Gronthos S, Shi S. Mesenchymal stem cells derived from dental tissues vs. those from other sources: their biology and role in regenerative medicine. J Dent Res. 2009;88(9):792‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sarugaser R, Lickorish D, Baksh D, Hosseini MM, Davies JE. Human umbilical cord perivascular (HUCPV) cells: a source of mesenchymal progenitors. Stem Cells. 2005;23(2):220‐229. [DOI] [PubMed] [Google Scholar]
- 6. Lu LL, Liu YJ, Yang SG, et al. Isolation and characterization of human umbilical cord mesenchymal stem cells with hematopoiesis‐supportive function and other potentials. Haematologica. 2006;91(8):1017‐1026. [PubMed] [Google Scholar]
- 7. Fukuchi Y, Nakajima H, Sugiyama D, Hirose I, Kitamura T, Tsuji K. Human placenta‐derived cells have mesenchymal stem/progenitor cell potential. Stem Cells. 2004;22(5):649‐658. [DOI] [PubMed] [Google Scholar]
- 8. Matikainen T, Laine J. Placenta—an alternative source of stem cells. Toxicol Appl Pharmacol. 2005;207(2 suppl):544‐549. [DOI] [PubMed] [Google Scholar]
- 9. Díaz‐Prado S, Muiños‐López E, Hermida‐Gómez T, et al. Human amniotic membrane as an alternative source of stem cells for regenerative medicine. Differentiation. 2011;81(3):162‐171. [DOI] [PubMed] [Google Scholar]
- 10. Toda A, Okabe M, Yoshida T, Nikaido T. The potential of amniotic membrane/amnion‐derived cells for regeneration of various tissues. J Pharmacol Sci. 2007;105(3):215‐228. [DOI] [PubMed] [Google Scholar]
- 11. Schwab KE, Hutchinson P, Gargett CE. Identification of surface markers for prospective isolation of human endometrial stromal colony‐forming cells. Hum Reprod. 2008;23(4):934‐943. [DOI] [PubMed] [Google Scholar]
- 12. Xiang L, Chan RW, Ng EH, et al. Nanoparticle labeling identifies slow cycling human endometrial stromal cells. Stem Cell Res Ther. 2014;5(4):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tondreau T, Meuleman N, Delforge A, et al. Mesenchymal stem cells derived from CD133‐positive cells in mobilized peripheral blood and cord blood: proliferation, Oct4 expression, and plasticity. Stem Cells. 2005;23(8):1105‐1112. [DOI] [PubMed] [Google Scholar]
- 14. Peters R, Wolf MJ, van den Broek M, et al. Efficient generation of multipotent mesenchymal stem cells from umbilical cord blood in stroma‐free liquid culture. PLoS One. 2010;5(12):e15689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo P, Sun H, Zhang Y, et al. Limbal niche cells are a potent resource of adult mesenchymal progenitors. J Cell Mol Med. 2018;22(7):3315‐3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Galipeau J, Sensébé L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. 2018;22(6):824‐833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krueger T, Thorek D, Denmeade SR, et al. Concise review: mesenchymal stem cell‐based drug delivery: the good, the bad, the ugly, and the promise. Stem Cells Translational Medicine. 2018;7(9):651‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown C, McKee C, Bakshi S, et al. Mesenchymal stem cells: cell therapy and regeneration potential. J Tissue Eng Regen Med. 2019;13(9):1738‐1755. [DOI] [PubMed] [Google Scholar]
- 19. Moll G, Ankrum JA, Kamhieh‐Milz J, et al. Intravascular mesenchymal stromal/stem cell therapy product diversification: time for new clinical guidelines. Trends Mol Med. 2019;25(2):149‐163. [DOI] [PubMed] [Google Scholar]
- 20. Pittenger MF, Discher DE, Péault BM, Phinney DG, Hare JM, Caplan AI. Mesenchymal stem cell perspective: cell biology to clinical progress. NPJ Regen Med. 2019;4:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315‐317. [DOI] [PubMed] [Google Scholar]
- 22. Drug Registration Management Measures; 2020 edition (in Chinese). https://www.nmpa.gov.cn/xxgk/fgwj/bmgzh/20200330180501220.html. Accessed July 1, 2020.
- 23. Squillaro T, Peluso G, Galderisi U. Clinical trials with mesenchymal stem cells: an update. Cell Transplant. 2016;25(5):829‐848. [DOI] [PubMed] [Google Scholar]
- 24. Drug Administration Law of the People's Republic of China; 2019 edition (in Chinese). https://www.nmpa.gov.cn/xxgk/fgwj/flxzhfg/20190827083801685.html. Accessed December 1, 2019.
- 25. Pharmaceutic Good Manufacturing Practice; Revised in 2010 (in Chinese). https://www.nmpa.gov.cn/xxgk/fgwj/bmgzh/20110117120001434.html. Accessed March 1, 2011.
- 26. Q10 Pharmaceutical Quality System. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/1/0. Accessed July 4, 2010.
- 27. Rockhold FW, Enas GG. 10 years with ICH E10: choice of control groups. Pharm Stat. 2011;10(5):407‐409. [DOI] [PubMed] [Google Scholar]
- 28. Pharmaceutic Good Manufacturing Practice; Revised in 2010. Appendix: Cell Therapy Product (in Chinese). https://www.cfdi.org.cn/resource/news/11931.html. Accessed November 28, 2019.
- 29. Technical Guidelines for Research and Evaluation of Cell Therapy Products (For Trial Implementation) (in Chinese). https://www.nmpa.gov.cn/directory/web/nmpa/xxgk/zhcjd/zhcjdyp/20171222145901282.html. Accessed December 31, 2018.
- 30. Current Good Tissue Practice (CGTP) and Additional Requirements for Manufacturers of Human Cells, Tissues and Cellular and Tissue‐Based Products (HCTPs) Questions and Answers. https://www.fda.gov/media/82724/download. Accessed May 25, 2005.
- 31. Announcement of the General Administration on Matters Concerning the Association Review and Approval of Pharmaceutical Packaging Materials, Excipients and Drugs (2016 No. 134) (in Chinese). https://www.nmpa.gov.cn/directory/web/nmpa/xxgk/ggtg/qtggtg/20160810115701940.html. Accessed August 10, 2016.
- 32. Process‐Validation—General‐Principles‐and‐Practices. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/process‐validation‐general‐principles‐and‐practices. Accessed January, 2011.
- 33. Stem Cell Preparation Quality Control and Preclinical Research Guidelines (For Trial Implementation) (in Chinese). https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20150731120001226.html. Accessed July 31, 2015.
- 34. Q2(R1) Validation of Analytical Procedures Text and Methodology. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/1/0. Accessed November, 2005.
- 35. Analytical Procedures and Methods Validation for Drugs and Biologics. https://www.fda.gov/media/87801/download. Accessed July, 2015.
- 36. Final Guidance for Industry Potency Tests for Cellular and Gene Therapy Products. https://www.fda.gov/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/default.htm. Accessed November 30, 2017.
- 37. Q1A(R2) Stability Testing of New Drug Substances and Products. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/1/0. Accessed January, 2011.
- 38. S4 Duration of Chronic Toxicity Testing in Animals (Rodent and Non Rodent Toxicity Testing). http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/2/1. Accessed February 6, 2003.
- 39. S8 Immunotoxicity Studies for Human Pharmaceuticals. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/2/1. Accessed September 2, 1998.
- 40. S1B Testing for Carcinogenicity of Pharmaceuticals. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/2/1. Accessed September 15, 2005.
- 41. S1C(R2) Dose Selection for Carcinogenicity Studies of Pharmaceuticals. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/2/1. Accessed July 16, 1997.
- 42. S1A Need for Carcinogenicity Studies of Pharmaceuticals. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/2/1. Accessed March 11, 2008.
- 43. S5(R3) Detection of Reproductive and Developmental Toxicity for Human Pharmaceuticals. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/2/1. Accessed November 29, 1995.
- 44. S2(R1) Guidance on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/2/1. Accessed November, 2005. [PubMed]
- 45. E6(R1) Guideline for Good Clinical Practice. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/3/0. Accessed November 9, 2011.
- 46. Standards for Quality Management of Drug Clinical Trials (in Chinese). https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20200426162401243.html. Accessed June 10, 1996.
- 47. Technical Guidelines for Clinical Trials of Human Stem Cells and Derived Cell Therapy Products (Draft for Comment) (in Chinese). http://www.cde.org.cn/news.do?method=largeInfo&id=6cf5b4fbef3256d9. Accessed July 1, 2020.
- 48. E2A: Clinical Safety Data Management: Definitions and Standards for Expedited Reporting. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/3/0. Accessed August 24, 2020.
- 49. E2B(R3): Implementation Guide for Electronic Transmission of Individual Case Safety Reports (ICSRs) E2B(R3) Data Elements and Message Specification. http://www.cde.org.cn/ichWeb/guideIch/toGuideIch/3/0. Accessed October 27, 1994.
- 50. Guidance of Drug Research and Development and Technical Review Communication Management (in Chinese). https://www.nmpa.gov.cn/yaopin/ypggtg/ypqtgg/20181008172601715.html. Accessed November 10, 2016.
- 51. Announcement of the State Food and Drug Administration on the Application of the Second‐Level Guidelines of the International Human Drug Registration Technical Coordination Committee (2018 No. 10) (in Chinese). https://www.nmpa.gov.cn/zhuanti/ypqxgg/ggzhcfg/20180125175101846.html. Accessed December 11, 2020.
- 52. The State Food and Drug Administration Publicly Solicits Opinions on 7 Documents Including the “Registration Classification and Application Data Requirements for Biological Products (Draft for Comments)” (in Chinese). https://www.nmpa.gov.cn/zhuanti/ypzhcglbf/ypzhcglbfzhqyj/20200430164901807.html. Accessed June 7, 2018.
- 53. Appendix 3. Guidelines for Acceptance and Review of Registration of Biological Products (Part II Biological Products for Treatment) (Draft for Comments) (in Chinese). https://www.nmpa.gov.cn/zhuanti/ypqxgg/ggzhcfg/20171130203401214.html. Accessed April 30, 2020.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created and analyzed in this study.