

Abstract
Ovarian cancer is one of the leading causes of gynecological malignancy-related deaths. The underlying molecular development mechanism has however not been elucidated. In this study, we used bioinformatics to reveal critical molecular and biological processes associated with ovarian cancer. The microarray datasets of miRNA and mRNA expression profiles were downloaded from the Gene Expression Omnibus (GEO) database. Besides, we performed target prediction of the identified differentially expressed miRNAs. The overlapped differentially expressed genes (DEGs) were obtained combined with miRNA targets predicted and the DEGs identified from the mRNA dataset. The Cytoscape software was used to design a regulatory network of miRNA-gene. Moreover, the overlapped DEGs in the network were subjected to enrichment analysis to explore the associated biological processes. The molecular protein-protein interaction (PPI) network was used to identify the key genes among the DEGs of prognostic value for ovarian cancer, and the genes were evaluated via Kaplan-Meier curve analysis. A total of 186 overlapped DEGs were identified. Through miRNA-gene network analysis, we found that miR-195-5p, miR-424-5p, and miR-497-5p highly exhibited targeted association with overlapped DEGs. The three miRNAs are critical in the regulatory network and act as tumor suppressors. The overlapped DEGs were mainly associated with protein metabolism, histogenesis, and development of the reproductive system and ocular tissues. The PPI network identified 10 vital genes that promote tumor progression. Survival analysis found that CEP55 and CCNE1 may be associated with the prognosis of ovarian cancer. These findings provide insights to understand the pathogenesis of ovarian cancer and suggest new candidate biomarkers for early screening of ovarian cancer.
1. Introduction
Ovarian cancer is one of the leading causes of deaths resulting from gynecological malignancies. The latest statistics indicate that about 295,414 new cases of ovarian cancer were reported globally in 2018 [1]. The overall 5-year survival rate of ovarian cancer is below 45% mainly because distant metastasis occurs earlier before diagnosis. Biomarkers such as CA125 are currently in clinical use; however, they are unspecific, and ultrasound examinations cannot identify early cases [2, 3]. Therefore, there is an urgent need to reexcavate new diagnosed biomarkers for ovarian cancer and reidentify the associated key molecules. This will be vital in devising strategies to manage ovarian cancer at prevention and control levels.
In recent years, the extensive application of expression profiles has accumulated enormous omics data, which is dependent on in-depth interpretation. Following relevant researches in the past three years, a few reports on the use of expression profiles linked with bioinformatics to discover key genes of ovarian cancer have been published [4–7]. However, most of the research groups selected similar microarray profiles thereby may cause lower accuracy and gave false-positive results. The miRNAs are noncoding RNAs that bound to complementary sequences in the mRNA via base pairing. This promotes mRNA silencing and negatively regulates downstream gene expression [8]. Many studies have found that miRNA disorders can occur in nearly all types of tumors thereby affecting target gene expression [9, 10]. A number of studies have evaluated that miRNA expression profiles in patients with ovarian cancer can be used as molecular markers of malignant tumors. Functional experiments related to ovarian cancer have confirmed that many miRNAs have cancer-promoting or antitumor effects, and miRNAs can inhibit the translation process of target mRNAs to participate in the regulation of many cell processes related to ovarian cancer [11–13].
Therefore, our study adopted integrated miRNA and mRNA microarray expression profiles for joint analysis. Through bioinformatics, we constructed regulatory networks to identify key molecules and biological processes associated with ovarian cancer. This provides a scientific and accurate theoretical basis to elucidate the mechanism of ovarian cancer onset.
2. Materials and Methods
2.1. Data Sources
We searched for the microarray expression profiles of ovarian cancer in the GEO (a public functional genomics database) by limiting the sample size to more than 10 and compared tumor tissue with normal tissue for the experiment type. GSE83693 and GSE36668 were the two eligible profile datasets. The former was a miRNA profile that included 4 normal tissues and 16 tumor tissues whereas the latter was an mRNA profile that included 4 normal tissues and 8 tumor tissues.
2.2. Data Processing
2.2.1. Differential Expression
For the mRNA dataset, the probe ID was converted to the corresponding gene name following its platform annotation file. The “limma” package of R language was used to analyze the DEGs [14], where adj.p.val < 0.05 and the absolute value of log2FC > 2 were defined as a statistically significant expression. Further, we used the R package “http://org.Hs.eg.db/” to convert the gene name to the corresponding gene ID [15] and eventually performed the subsequent enrichment analysis. The miRNA dataset was processed using the same method and standard.
2.2.2. miRNA Target Prediction
The Funrich software 3.1.3 was used to predict the downstream targets of the identified differentially expressed miRNAs [16]. The predicted gene list was intersected with the DEGs identified from the mRNA dataset. Thus, the overlapped DEGs were generated for subsequent regulatory networks, used to identify the key genes, and for functional enrichment analysis and so on.
2.2.3. Regulatory Network
The miRNAs negatively regulate target genes; therefore, the combinations of matching miRNA-gene were screened to construct regulatory networks and were visualized using the Cytoscape 3.7.1 software.
2.2.4. Key Genes
The string database was used to predict the interaction network between proteins encoded by the DEGs [17]. Then, the cytohubba module in the Cytoscape software was used to identify key genes [18]; here, the source data obtained from the network file was generated via the string database. The MCC algorithm of the module was selected to identify the top 10 key genes.
2.2.5. Functional Enrichment Analysis
The R package “clusterProfiler” was used to perform the GO and KEGG enrichment annotation of the overlapped DEGs [15]. GO annotation was grouped into three subcategories: molecular function (MF), biological process (BP), and cellular components (CC). KEGG is a comprehensive database that integrates genomic, chemical knowledge, and system functional information, which we used for enrichment annotation of gene pathways. The p value cutoff = 0.05 of the R package's parameter was considered statistically significant.
2.2.6. Subsistence Analysis
The TCGA database aided in the diagnosis, treatment, and prevention of tumors through a shared mechanism. The cbioportal is a visual analytics platform developed based on the TCGA. To further evaluate the pathogenesis of key genes we obtained, the cbioportal was adopted to identify the association of key genes with the survival prognosis of ovarian cancer [19].
2.2.7. Expression Level Verification
GEPIA2 is an updated version of GEPIA for analyzing the RNA sequencing expression data of 9,736 tumors and 8,587 normal samples from the TCGA and the GTEx projects, using a standard processing pipeline [20]. GEPIA2 provides customizable functions such as tumor/normal differential expression analysis, profiling according to cancer types or pathological stage, similar gene detection, correlation analysis, and dimensionality reduction analysis. Through this database, we verified whether the expression levels of candidate genes in our study were consistent.
3. Results
3.1. Differential Expression
A total of 53 differentially expressed miRNAs were identified using the GSE83693 dataset, out of which 35 were downregulated, while 18 were upregulated. Besides, 680 differentially expressed mRNAs were identified using the GSE36668 dataset, out of which 239 and 441 mRNAs were downregulated and upregulated, respectively. The differentially expressed molecules are shown in Figure 1.
Figure 1.
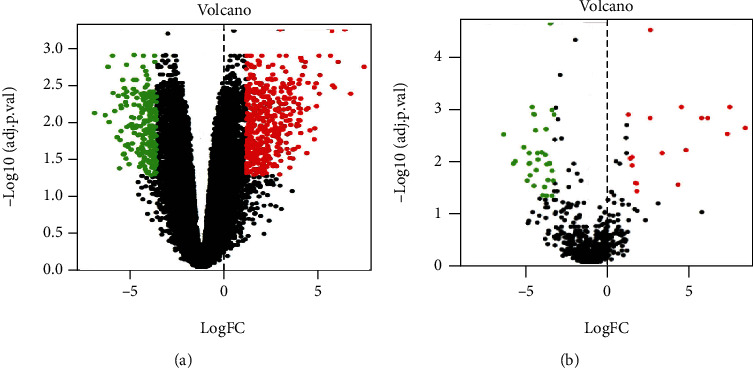
Volcanic map distribution of differential expression: (a) differentially expressed miRNA map; (b) differentially expressed gene map. Horizontal axis: log2(FC); vertical axis: -log10(adj.P.Val). The FC represents the fold change in expression of tumor samples compared to normal samples, and the adj.P.Val represents the calibrated p value. Green color indicates that differential expression is downregulated; red color indicates that differential expression is upregulated.
3.2. Overlapped DEGs and Regulatory Networks
According to the DEGs of mRNA dataset, combined with the target genes of differentially expressed miRNA predicted by the Funrich software, the 186 overlapped DEGs' network files were obtained (attachment S1 and S2 shown). The network files were imported into the Cytoscape software for visual analysis (Figure 2). It was found that miR-195-5p, miR-424-5p, and miR-497-5p were in the hub core of network regulation, of which number of target genes were the most. All three key miRNAs had 16 target DEGs.
Figure 2.
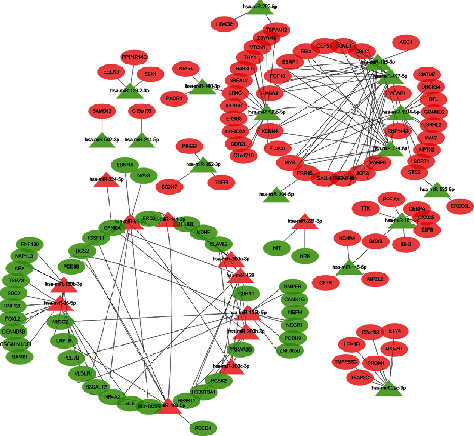
The regulation network of miRNA-gene. The ovals represent differential target genes; the triangles represent differential miRNAs; the lines represent the existence of targeted regulatory relationships. The red color shows upregulation while the green color shows downregulation.
3.3. Identifying the Key Genes
The results showing the identified key genes in the overlapped DEGs via the string database and cytohubba module are shown in Table 1. All the key genes were overexpressed differential genes, of which expressions were consistent with the GEPIA2 database. The top 10 key genes were screened following the latest MCC algorithm, namely, TTK, CEP55, KIT, DTL, E2F8, SOX9, ERCC6L, KIF18B, THY1, and CCNE1. Notably, TTK, CEP55, and KIT showed the highest scores and were at the key core of the network (Figure 3).
Table 1.
Function of the top 10 key genes at the hub core of the PPI network.
| No | Gene | Full name | Gene functions |
|---|---|---|---|
| 1 | TTK | TTK protein kinase | Related to cell proliferation, encoding key protein for mitotic checkpoints. Abnormal mitotic spindles are produced when expression is abnormal, resulting in tumor occurring possibly. |
| 2 | CEP55 | centrosomal protein 55 | Playing roles in mitosis and cytokinesis. Related pathways include DNA damage and cytoskeleton signaling. |
| 3 | KIT | KIT proto-oncogene | Encoded protein is a type III transmembrane receptor; genetic variation is related to gastrointestinal stromal tumors and mast cells. |
| 4 | DTL | denticleless E3 ubiquitin protein | The homologue of E3 ubiquitin protein ligase, which maybe degrade PDCD4 and promote tumor development. It may be a therapeutic target for ovarian epithelial cancer. |
| 5 | E2F8 | E2F transcription factor 8 | A member of family encoding transcription factors, which regulate cell cycle-related gene expression and is involved in the promotion of a variety of tumors |
| 6 | SOX9 | SRY-box transcription factor 9 | Participated in identifying specific sequences. Related to bone deformity, nodular atypical hyperplasia and other diseases. |
| 7 | ERCC6L | excision repair 6 like | Members of family encoding protein belong to DNA transport enzymes and are necessary genes for mitotic sister chromatid isolation; involved in cell proliferation. |
| 8 | KIF18B | kinesin family member 18B | A member of the kinetin family, which constitutes the main positive end of microtubule depolymerization in mitotic cells, ensuring that the spindle is centered. |
| 9 | THY1 | Thy-1 cell surface antigen | Encoding cell surface glycoproteins and proteins, involving in adhesion and communication of multiple cell types, which promotes nasopharyngeal carcinoma. |
| 10 | CCNE1 | cyclin E1 | Encoded protein belongs to cell cycle family. Overexpression of gene is observed in many tumors, causing chromosomal instability and may promote tumorigenesis. |
Note: The annotations in this table referred to the clear conclusions in NCBI (https://www.ncbi.nlm.nih.gov/gene), and relevant scattered reports weren't included in a single literature.
Figure 3.
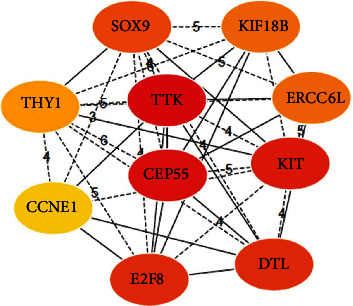
Identifying the key DEGs. When the red color appears darker, a higher score is noted, indicating a highly significant biometric significance.
3.4. Functional Enrichment Annotation
The results of GO enrichment analysis using the “clusterProfiler” package are shown in Figure 4. In BP, the DEGs were significantly enriched in developing the reproductive structure, remodeling of the reproductive system, positive regulation of protein catabolism, retinal morphogenesis, and differentiation pathways of the ocular photoreceptor cell. In MF, DEGs were enriched in DNA-binding transcriptional activation. Of note, since we set the filtering parameter at (p value cutoff = 0.05), KEGG analysis did not enrich the entries with coherent biological meaning.
Figure 4.
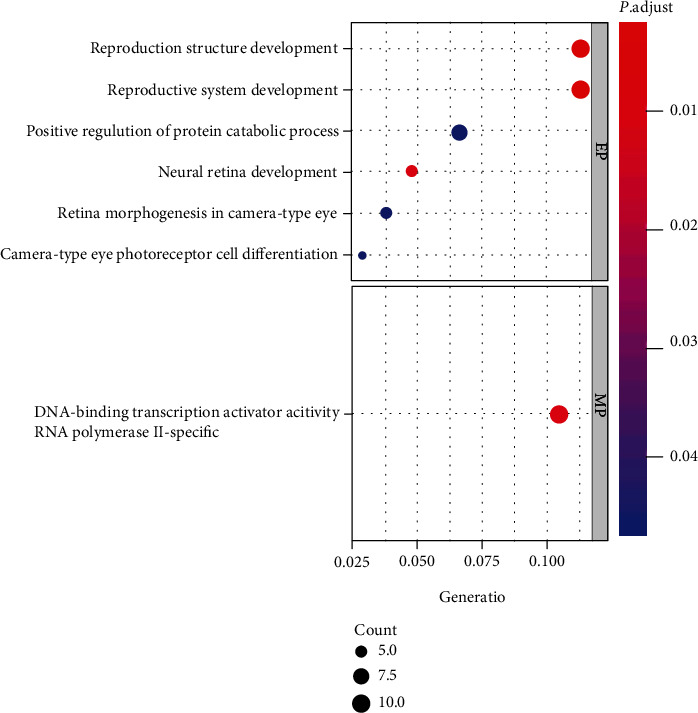
Bubble chart of GO enrichment. Horizontal axis: the proportion of genes; vertical axis: enrichment items. The color of the point corresponds to the value of p adjust, while the size of the point corresponds to the number of DGEs under the GO entry.
3.5. Survival Analysis
Through cbioportal analysis of how the genes were correlated with prognosis (Figure 5), we observed improved overall survival rate of ovarian cancer patients with CEP55 variation, with a statistically significant difference (p = 0.012). Moreover, patients with CCNE1 variation showed poorer survival prognosis compared to nonvariant tumor patients (p = 2.397e − 6). However, no significant differences were observed in the survival analysis of other key genes between the two tumor groups.
Figure 5.
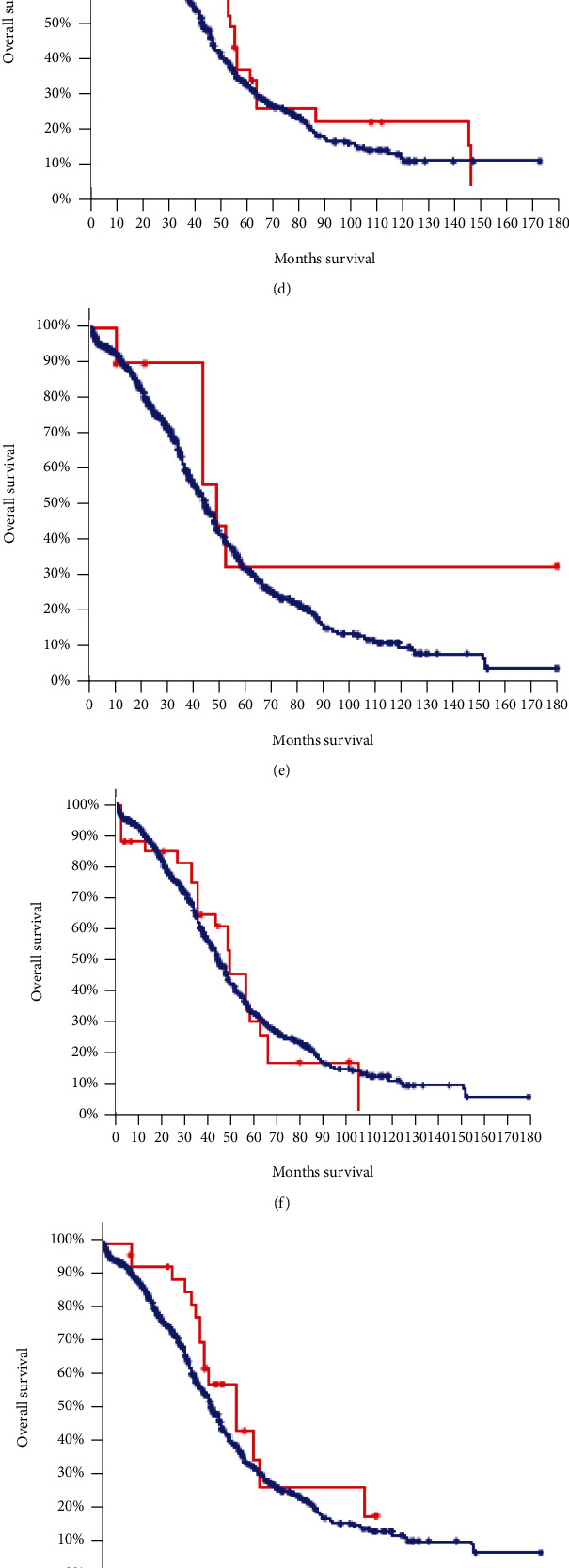
Kaplan-Meier survival curve. The red represents the ovarian cancer group with target gene mutation; blue represents the ovarian cancer group without target gene mutation; p value < 0.05 is considered statistically significant.
4. Discussion
Ovarian cancer is a common gynecological malignancy. However, the molecular mechanism by which ovarian cancer is associated with pathogenicity has not been fully elucidated. Notably, BRCA is one of the currently identified gene that has a key role in ovarian cancer [21]. The BRCA mutation frequency of ovarian cancer ranges from 3% to 27%; the gene test provides precise guidelines for preventing, diagnosing, and treating ovarian cancer [22]. However, there is a need to identify other novel molecules to jointly screen for most of the remaining cancer cases.
Current researches [10, 23, 24] indicate that besides genes, the dysregulated expression of noncoding RNAs such as miRNA can widely mediate various types of malignant tumors. Therefore, to improve the prediction accuracy, our study identified overlapped DEGs based on integrated miRNA and mRNA expression profiles of ovarian cancer. We constructed the miRNA-gene regulatory network to identify three key miRNAs (miR-195-5p, miR-424-5p, and miR-497-5p) as tumor suppressors based on the principle of complementary binding of miRNAs to the target mRNAs, negatively regulating genes. Moreover, the 10 key genes were predicted and screened by visualizing the overlapped DEGs in the network using the Cytoscape software, whereby TTK, CEP55, and KIT were at the center of the molecular network. By querying the NCBI database, we found that majority of the 10 genes were associated with specific tumorigenesis that mainly involves mitosis and cell proliferation (Table 1). Additionally, the survival curve analysis revealed that CEP55 and CCNE1 should be potential prognostic genes. The biological processes involved in overlapped DEGs were enriched through the R package. It was found that 22.6% (24/106) and 10.4% (11/106) of the DEGs were significantly enriched in biological processes of the reproductive system and DNA transcription activation function, respectively. These results concur with the actual functions and roles of ovarian tissue thereby justifying the reliability of our study. The abnormal expression of these differential miRNAs and genes was likely to mediate the occurrence and development of ovarian cancer. Besides, for the three key miRNAs discovered as tumor suppressors, we conducted an experimental literature search for recently published reports. Notably, Luo et al. [25] identified that the expression of miR-195-5p was significantly reduced in 40 breast cancers through qPCR experiments. Also, he identified CCNE1 was as the direct target of miR-195-5p in a dual-luciferase reporter assay. Elsewhere, Kong et al. [26] found that miR-195-5p played a tumor-suppressive role in endometrial cancer through similar methodologies. Moreover, Liu et al. [27] identified that miR-424-5p directly targeted CCNE1 to inhibit epithelial ovarian cancer through in vitro experiments. Notably, we obtained similar findings on the miRNA expression and target prediction. In addition, Liu et al. [28] constructed a lentiviral miR-497-5p system, and through qRT-PCR, she verified that its overexpression enhanced cell apoptosis of ovarian cancer. Similarly, we reported that miR-497-5p as a tumor suppressor molecule.
Conclusively, this study purposed to reveal the key molecules of ovarian cancer by analyzing the integrated miRNA-mRNA expression profiles. Notably, the identified key miRNAs or genes require in-depth experimental verification through in vitro studies. Nevertheless, the bioinformatics is a reliable method to predict the expression profiles by narrowing the scope of in vitro experiments and saving valuable resources. In the future, we believe that global researchers will be able to instantly reveal key molecules of many complex and diverse tumors using the tumor big data strategy which is dependent on computational biology.
Acknowledgments
The authors were grateful to all colleagues of Foshan Women and Children Hospital Affiliated to Southern Medical University.
Data Availability
All datasets generated for this study are included in the article.
Ethical Approval
Ethical approval is not applicable.
Disclosure
This manuscript [29] had been released as a preprint at Cancer Biology (Chao Li et al.).
Conflicts of Interest
The authors declare no conflict of interest.
Authors' Contributions
The research idea was derived from XK Yang, and C Li designed the research. ML Ou and XD Zhu participated in data collection. ZT Hong and C Li analyzed the data. Then, C Li wrote the paper and revised it. All the authors helped with article review, and they had read and approved the final manuscript.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R. L., Torre L. A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a Cancer Journal for Clinicians . 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Zheng L.-E., Qu J.-Y., He F. The diagnosis and pathological value of combined detection of HE4 and CA125 for patients with ovarian cancer. Open Medicine . 2016;11(1):125–132. doi: 10.1515/med-2016-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore R. G., Miller M. C., Steinhoff M. M., et al. Serum HE4 levels are less frequently elevated than CA125 in women with benign gynecologic disorders. American Journal of Obstetrics and Gynecology . 2012;206(4):351.e1–351.e8. doi: 10.1016/j.ajog.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lv B.-W., Qian J., Wang J., et al. miR-455-5p expression in epithelial ovarian cancer and functional analysis of its target genes. Journal of Practical Oncology . 2019;33(2):115–121. [Google Scholar]
- 5.Guo S.-M., Li H.-M. Using bioinformatics to discover the HADH and SRC as potential therapeutic target genes for ovarian cancer. Clinical Medicine Electronic Journal . 2017 [Google Scholar]
- 6.Yang X., Zhu S., Li L., et al. Identification of differentially expressed genes and signaling pathways in ovarian cancer by integrated bioinformatics analysis. Oncotargets and Therapy . 2018;Volume 11:1457–1474. doi: 10.2147/OTT.S152238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng H., Gu Z.-Y., Li Q., Liu Q. H., Yang X. Y., Zhang J. J. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. Journal of Ovarian Research . 2019;12(1):p. 35. doi: 10.1186/s13048-019-0508-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindsay M. A., Griffiths-Jones S., Dalmay T. Mechanism of miRNA-mediated repression of mRNA translation. Essays in Biochemistry . 2013;54:29–38. doi: 10.1042/bse0540029. [DOI] [PubMed] [Google Scholar]
- 9.Lujambio A., Lowe S. W. The microcosmos of cancer. Nature . 2012;482(7385):347–355. doi: 10.1038/nature10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rupaimoole R., Calin G. A., Lopez-Berestein G., Sood A. K. miRNA deregulation in cancer cells and the tumor microenvironment. Cancer Discovery . 2016;6(3):235–246. doi: 10.1158/2159-8290.CD-15-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yokoi A., Matsuzaki J., Yamamoto Y., et al. Integrated extracellular microRNA profiling for ovarian cancer screening. Nature Communications . 2018;9(1):1–10. doi: 10.1038/s41467-018-06434-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Resnick K., Alder H., Hagan J. P., Richardson D. L., Croce C. M., Cohn D. E. The detection of differentially expressed microRNAs from the serum of ovarian cancer patients using a novel real-time PCR platform. Gynecologic Oncology . 2009;112(1):55–59. doi: 10.1016/j.ygyno.2008.08.036. [DOI] [PubMed] [Google Scholar]
- 13.Gao J., Wu N., Liu X., et al. MicroRNA-142-3p inhibits cell proliferation and chemoresistance in ovarian cancer via targeting sirtuin 1. Experimental and Therapeutic Medicine . 2018;15(6):5205–5214. doi: 10.3892/etm.2018.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smyth G. K., Ritchie M., Thorne N., Wettenhall J. LIMMA: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor . Statistics for Biology and Health; 2005. [Google Scholar]
- 15.Yu G., Wang L.-G., Han Y., He Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics: a Journal of Integrative Biology . 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pathan M., Keerthikumar S., Ang C. S., et al. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics . 2015;15(15):2597–2601. doi: 10.1002/pmic.201400515. [DOI] [PubMed] [Google Scholar]
- 17.Szklarczyk D., Morris J. H., Cook H., et al. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic Acids Research . 2017;45(D1):D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chin C.-H., Chen S.-H., Wu H.-H., Ho C. W., Ko M. T., Lin C. Y. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Systems Biology . 2014;8(S4):p. S11. doi: 10.1186/1752-0509-8-S4-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao J., Aksoy B. A., Dogrusoz U., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signaling . 2013;6(269):p. pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang Z., Kang B., Li C., Chen T., Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Research . 2019;47(W1):W556–W560. doi: 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neff R. T., Senter L., Salani R. BRCAmutation in ovarian cancer: testing, implications and treatment considerations. Therapeutic Advances in Medical Oncology . 2017;9(8):519–531. doi: 10.1177/1758834017714993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alsop K., Fereday S., Meldrum C., et al. BRCAMutation frequency and patterns of treatment response inBRCAMutation–Positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. Journal of Clinical Oncology . 2012;30(21):2654–2663. doi: 10.1200/JCO.2011.39.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy K. B. MicroRNA (miRNA) in cancer. Cancer Cell International . 2015;15(1):p. 38. doi: 10.1186/s12935-015-0185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang W., Wan S., Yang Z., Teschendorff A. E., Zou Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics . 2018;34(3):398–406. doi: 10.1093/bioinformatics/btx622. [DOI] [PubMed] [Google Scholar]
- 25.Luo Q., Wei C., Li X., et al. MicroRNA-195-5p is a potential diagnostic and therapeutic target for breast cancer. Oncology Reports . 2014;31(3):1096–1102. doi: 10.3892/or.2014.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kong F., Ma J., Yang H., Yang, Wang, Ma Long non-coding RNA PVT1 promotes malignancy in human endometrial carcinoma cells through negative regulation of miR-195-5p. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research . 2018;1865(10):1479–1490. doi: 10.1016/j.bbamcr.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 27.Liu J., Gu Z., Tang Y., Hao J., Zhang C., Yang X. Tumour-suppressive microRNA-424-5p directly targets CCNE1 as potential prognostic markers in epithelial ovarian cancer. Cell Cycle . 2018;17(3):309–318. doi: 10.1080/15384101.2017.1407894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu C., Bordeaux A., Hettich S., Han S. MicroRNA-497-5p functions as a modulator of apoptosis by regulating metadherin in ovarian cancer. Cell Transplantation . 2020;29, article 0963689719897061 doi: 10.1177/0963689719897061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li C., Zhu X., Zhang L., Ou M., Hong Z. Integrated miRNA-mRNA Expression Profiles Revealing Key Molecules in Ovarian Cancer Based on Bioinformatics Analysis . Research Sqaure; 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All datasets generated for this study are included in the article.